

Abstract
Limb-girdle muscular dystrophy type 2A (LGMD2A or LGMDR1) is a neuromuscular disorder caused by mutations in the calpain 3 gene (CAPN3). Previous experiments using adeno-associated viral (AAV) vector–mediated calpain 3 gene transfer in mice indicated cardiac toxicity associated with the ectopic expression of the calpain 3 transgene. Here, we performed a preliminary dose study in a severe double-knockout mouse model deficient in calpain 3 and dysferlin. We evaluated safety and biodistribution of AAV9-desmin-hCAPN3 vector administration to nonhuman primates (NHPs) with a dose of 3 × 1013 viral genomes/kg. Vector administration did not lead to observable adverse effects or to detectable toxicity in NHP. Of note, the transgene expression did not produce any abnormal changes in cardiac morphology or function of injected animals while reaching therapeutic expression in skeletal muscle. Additional investigation on the underlying causes of cardiac toxicity observed after gene transfer in mice and the role of titin in this phenomenon suggest species-specific titin splicing. Mice have a reduced capacity for buffering calpain 3 activity compared to NHPs and humans. Our studies highlight a complex interplay between calpain 3 and titin binding sites and demonstrate an effective and safe profile for systemic calpain 3 vector delivery in NHP, providing critical support for the clinical potential of calpain 3 gene therapy in humans.
INTRODUCTION
Genetic defects in the calpain 3 gene (CAPN3) lead to autosomal recessive limb-girdle muscular dystrophy type 2A or R1 (LGMD2A or LGMDR1 according to the new nomenclature; OMIM 253600), also called calpainopathy (1). The disorder was first identified in a population from the Réunion Island and later shown to be one of the most frequent types of LGMD, with prevalence reported between 10 and 70 per million individuals (2–4). The deficiency results in a slowly progressive muscle disorder with onset between the first and second decades of life and a mean age of loss of independent ambulation ~15 years later. Muscle weakness is remarkably symmetrical and predominant in the axial muscles of the trunk and proximal muscles of the lower limb (5). Most patients have a slightly reduced forced vital capacity, but although respiratory complications are possible, they are not a salient feature in primary calpainopathy. Although calpain 3 deficiency is not associated with cardiac involvement, cardiac dysfunction can coincidentally be present in some patients with LGMD2A (5, 6).
Calpain 3 is a member of the calpain family of non–lysosomal calcium–activated cysteine proteases. Although some isoforms are present in the lens of the eye and the brain, the classical full-length form of CAPN3 is primarily expressed in skeletal muscle (7). Expression in the heart has also been reported during development as well as in murine and human adult tissues, where expression is 100-fold lower than that found in skeletal muscle (8, 9). It has been observed that most of the protein within skeletal muscle is present as a dormant enzyme, probably through interaction with the giant protein titin, one of its partners (10, 11). Titin carries several CAPN3 binding sites in the I-band and M-line regions. Suppression of the activity of the protease through its interaction with titin has been demonstrated experimentally at the N2A region, a region of titin that undergoes tissue-specific alternative splicing (12). It was shown that calpain 3 self-activates by autolysis through the removal of an internal peptidic chain to free the catalytic site for substrate accessibility (13, 14). The precise function(s) of calpain 3 and the mechanism by which it causes LGMD2A are not yet fully understood, although some evidence indicates that the protein plays a role in cytoskeleton remodeling (15–17).
To date, there is still no cure for LGMD2A. The current standard of care for managing disease symptoms involves physical therapy focusing on contractures, pain management, use of orthosis, and occupational therapy. An annual respiratory assessment and biennial cardiac assessment are recommended.
To develop a therapeutic strategy for gene transfer for LGMD2A, we initiated a series of experiments using recombinant adeno-associated viral (rAAV) vectors, the current standard tool for gene transfer in skeletal muscle. After local injections of rAAV vectors expressing CAPN3 in skeletal muscle of a murine model deficient in calpain 3, we showed efficient CAPN3 transgene expression in skeletal muscles with correct localization at the sarcomere (18). This expression resulted in restoration of both calpain 3 proteolytic activity and muscle function. However, cardiac toxicity was detected when testing a systemic route of administration of these vectors and was confirmed to be caused by unregulated activity of the ectopically expressed calpain 3 in the heart (19). A second generation of vectors was then designed using new promoters and/or by introducing a sequence target of a cardiac-specific microRNA (miR-208a) in the 3′ untranslated region (3′UTR). These modifications suppress the cardiac toxicity while conserving the therapeutic effect in skeletal muscles (19).
Here, we evaluated the therapeutic effect of the rAAV vectors expressing CAPN3 and a double miR-208a target sequence under the control of the human desmin promoter in a double-knockout (dKO) mouse deficient in calpain 3 and dysferlin. This model was more severe than the calpain 3–deficient mouse model and therefore suitable for evaluating whether calpain 3 expression ameliorates its pathology. We also conducted a preclinical study in wild-type (WT) Macaca fascicularis (mfa) nonhuman primates (NHPs) to evaluate the biodistribution and safety of AAV9-mfa CAPN3 vectors with a thorough analysis of the heart. A single injection of calpain 3 at a dose of 3 × 1013 viral genomes (vg)/kg was well tolerated in NHP, and the amount of calpain 3 expressed corresponded to the therapeutic effect observed in the dKO mouse. We observed species- and tissue-specific differences after systemic administration of the calpain 3 vector, identifying distinct roles of the calpain 3 binding sites on titin in the regulation of calpain 3 proteolytic activity. Our study supports the use of AAV-mediated transfer of the calpain 3 gene in clinical studies in patients with LGMD2A.
RESULTS
A low amount of calpain 3 protein corrects pathology in a dKO mouse model
To circumvent the issue that calpain 3–deficient mouse models present a very mild pathology, we created a more severe dKO model based on the known interaction between calpain 3 and dysferlin. The latter protein is the cause of muscular dystrophy when mutated, presenting either as a proximal form (LGMD2B/LGMDR2) or as a distal form (Miyoshi myopathy). Whereas dysferlin plays an important role in cellular membrane repair (20), calpain 3 does not (21), suggesting that even if these proteins interact with each other, the pathways by which they intervene likely differ. We therefore hypothesized that combining both deficiencies would lead to a more severe phenotype. We created a dKO model, B6.129-DysfprmdCapn3tm2.1Gnt (dKO or BDC; for BlaJ deficient in calpain 3), by crossing the B6-Capn3tm2.1Gnt [CAPN3 KO; (22)] with the dysferlin-deficient model B6.A-Dysfprmd/J [BlaJ; (23)] (Fig. 1, A to C). To analyze whether the double deficiency results in a more severe phenotype, we characterized mice homozygous for both KO (Dysf−/−Capn3−/−; also referred to as HOHO) and mice homozygous for the dysferlin KO but heterozygous at the calpain 3 locus (Dysf−/−Capn3+/−; also referred to as HOHE). Histological analysis performed at 4 months of age indicated that the BDC HOHO and, to a smaller extent, the BDC HOHE were more affected than mice solely deficient in dysferlin (Fig. 1D) and a fortiori the mice deficient in calpain 3. The less calpain 3 expressed, the more deleterious the global condition.
Fig. 1. Molecular and histological validation of the dKO mouse model.
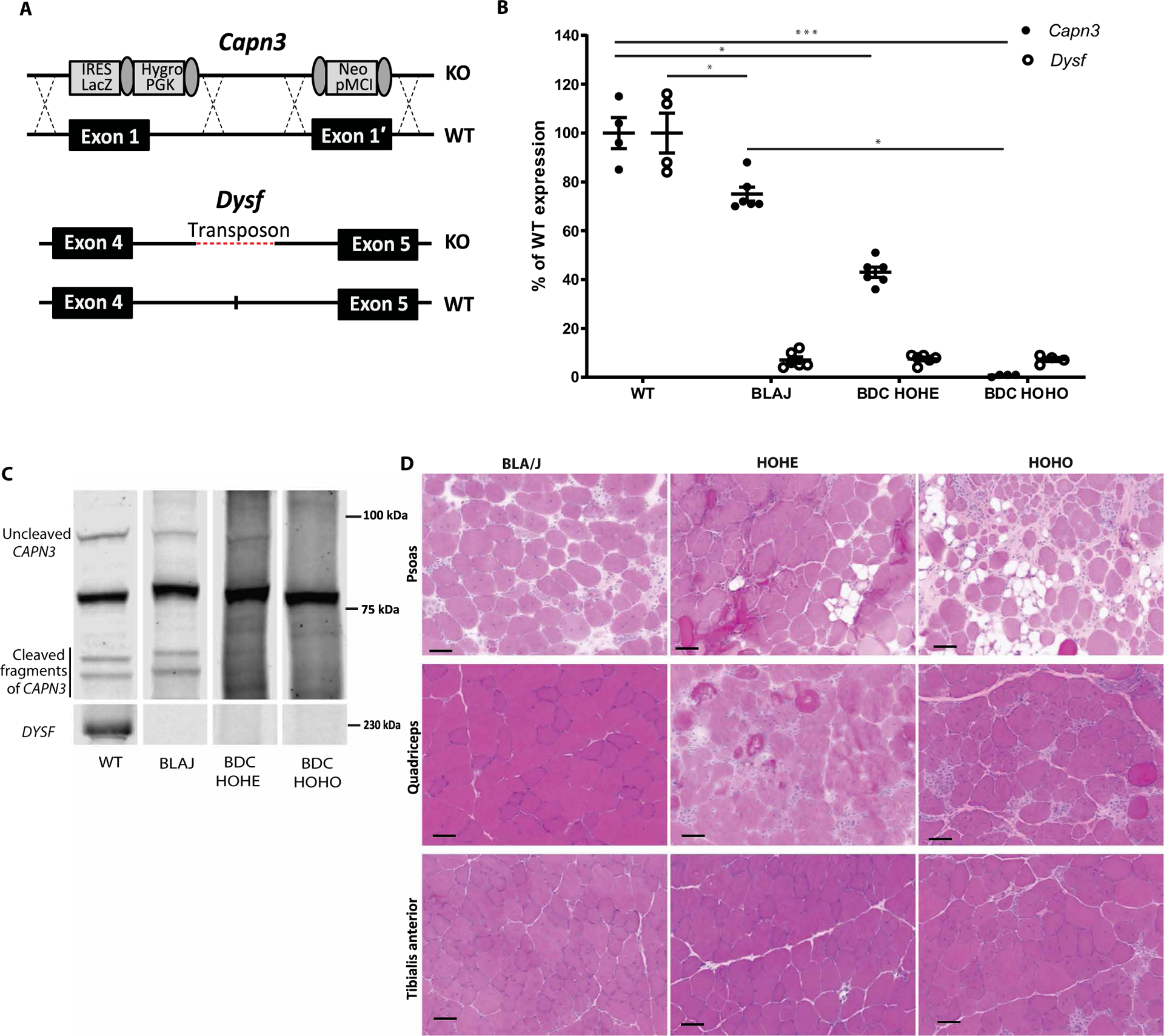
(A) Diagram of the genetic modification present in the C3 KO and Blaj mice. (B) mRNA expression of Calpain 3 and Dysferlin in mice according to their genotype. The measures were performed on gluteus, gastrocnemius, and soleus muscle. Statistical analyses were performed using nonparametric Kruskal-Wallis test and the post hoc multiple comparisons Dunn’s test (n = 4 to 6). *P < 0.05; ***P < 0.001. (C) Western blot of calpain 3 and dysferlin in skeletal muscle extracts according to the mouse genotype. (D) Histological sections of skeletal muscles (psoas, quadriceps, and TA) from each genotype. Scale bars, 50 μm. BLAJ, B6.A-Dysfprmd/J; BDC HOHE, Dysf−/−Capn3+/−−; BDC HOHO, Dysf−/−Capn3−/−.
This newly developed model (Dysf−/−Capn3−/−; HOHO) was then used for evaluation of an rAAV2/9 vector expressing a V5-tagged CAPN3 under control of the human desmin promoter: rAAV-hDes-mfaCAPN3-V5-SV40+2xmiR-208aT (rAAV-C3 with miRT). Because one of our goals was to use the same vector in a subsequent study in NHP, we used the coding sequence of the mfa calpain 3 (97.8% of identity and 100% of homology compared to the human sequence). We also introduced two copies of target sequence of the cardiac-specific miR-208a in the 3′UTR of the vector as we previously described in (19) (Fig. 2A). The gluteus muscles of 2.5- to 3-month-old BDC HOHO (Dysf−/−Capn3−/−) were injected with the vector (Fig. 2B). After 1 month, we validated the expression of the transgene by reverse transcription polymerase chain reaction (RT-PCR) (Fig. 2C). Muscles of injected animals demonstrated an improvement in myopathology compared to noninjected animals (Fig. 2D and fig. S1A). To evaluate the extent of restoration, we analyzed different dystrophic features by quantifying the expression of genes from pathways known to be involved in the dystrophic process. We evaluated muscle regeneration using myogenin, myoD, and Tmem8c (24); macrophage infiltration by Cd11b; and fibrosis by collagen VI, collagen 1, and fibronectin. After injection, the BDC HOHO mice showed a decrease in all examined markers (Fig. 2, E and F, and fig. S1B). These data indicate that the amount of calpain 3 expression attained led to a therapeutic effect in the dKO mice.
Fig. 2. Evaluating calpain 3 gene transfer in the Dysf−/−Capn3−/− dKO model.
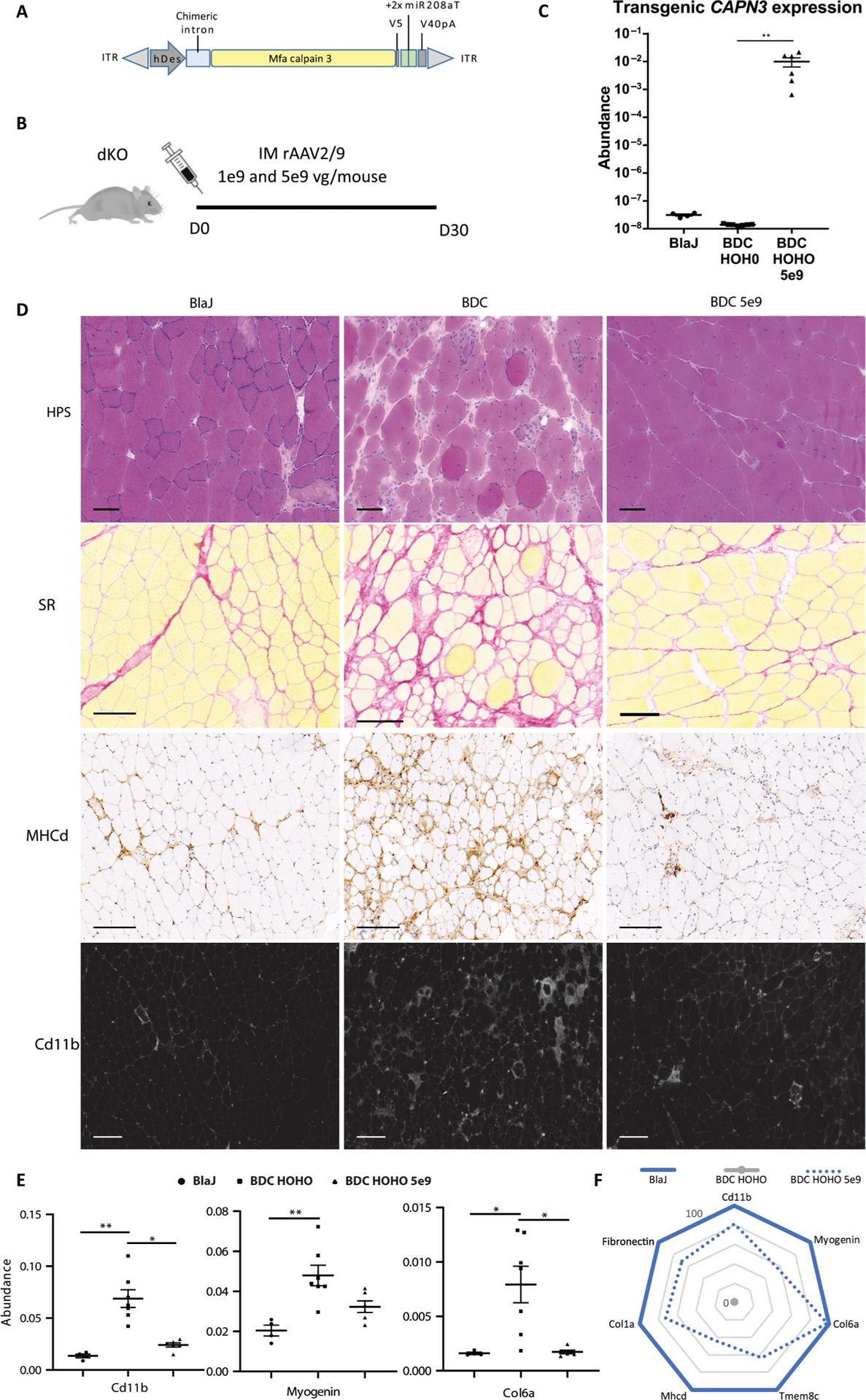
(A) The expression cassette used in this study includes a human desmin promoter, a chimeric β-globin intron, the mfa calpain 3 coding sequence fused with a V5 tag, and a simian SV40 polyadenylation signal. In addition, a tandem miR-208a target sequence was included in the 3′UTR of the vector (rAAV-C3+miRT). (B) Schematic representing design of the in vivo experiment (n = 4 to 7). IM, intramuscular; D, days. (C) Calpain 3 mRNA expression obtained by ddPCR in skeletal muscle extracts. Statistical analyses were performed using nonparametric Kruskal-Wallis test and the post hoc multiple comparison Dunn’s test (n = 4 to 7). **P < 0.01. (D) Representative gluteus muscle sections stained by hematoxylin phloxine saffron (HPS), Sirius red (SR), myosin heavy chain developmental form (MHCd), and CD11b. Scale bars, 150 μm. (E) Quantitative RT-PCR (qRT-PCR) quantification of genes representative of several dystrophic features: Cd11b for macrophage infiltrates, myogenin for regeneration, and collagen VI (Col6a) for fibrosis in muscles of Blaj, BDC HOHO, and injected BDC HOHO. The data are expressed as abundance normalized by Rplp0. Statistical analyses were performed using nonparametric Kruskal-Wallis test and the post hoc multiple comparisons Dunn’s test (n = 4 to 7) *P < 0.05; **P < 0.01. (F) Radar graph showing recovery score for each dystrophic markers. Values are expressed relative to those of BlaJ mice (set to 100%) and BDC HOHO (0%). Individual data are presented in fig. S1B.
AAV9 biodistribution favors cardiac targeting over skeletal muscle in primates
To initiate a preclinical study using calpain 3 AAV vectors, we first screened a group of fifty 2- to 3-year-old NHPs for AAV9 seronegativity. A series of dilutions of serum of cynomolgus monkeys (mfa) was analyzed using an enzyme-linked immunosorbent assay for the AAV9 capsid. The immunoglobulin antibody titer indicated that nine of these animals were seronegative (table S1). Sera of these animals were then used in a neutralizing antibody test based on an AAV9-luciferase transfection assay. We selected five primates of the seven that were negative for both tests for inclusion in a preclinical pilot study (table S1 and Fig. 3A), consisting of intravascular delivery of AAV vectors. In this experiment, we evaluated two rAAV2/9 vectors, rAAV-hDes-mfaCAPN3-V5SV40+2xmiR-208aT (rAAV-C3+miRT) and rAAV-hDes-mfaCAPN3-V5-SV40 (rAAV-C3), differing only by the presence of a tandem of target sequences of miR-208a (miR-208aT) in the former (Fig. 3A). The primates Perceval, Merlin, and Karadoc received rAAV-C3+miRT, and the primates Lancelot and Mordred received rAAV-C3 at the dose of 1 × 1014 vg per primate (~3 × 1013 vg/kg), the highest dose we could deliver in this study. The primates were clinically followed until the end of the experiments. No abnormal behavior, morbidity, or mortality was observed for any of the primates at any point during the experiment. Evaluation of the heart function and structure was performed by echocardiography before and at the conclusion of the experiment. Blood samples were collected before, at the midway point, and at the conclusion of the experiment for analysis of the biochemistry/hematology parameters and the expression of cardiac-specific markers.
Fig. 3. NHP biodistribution study and transgene expression.
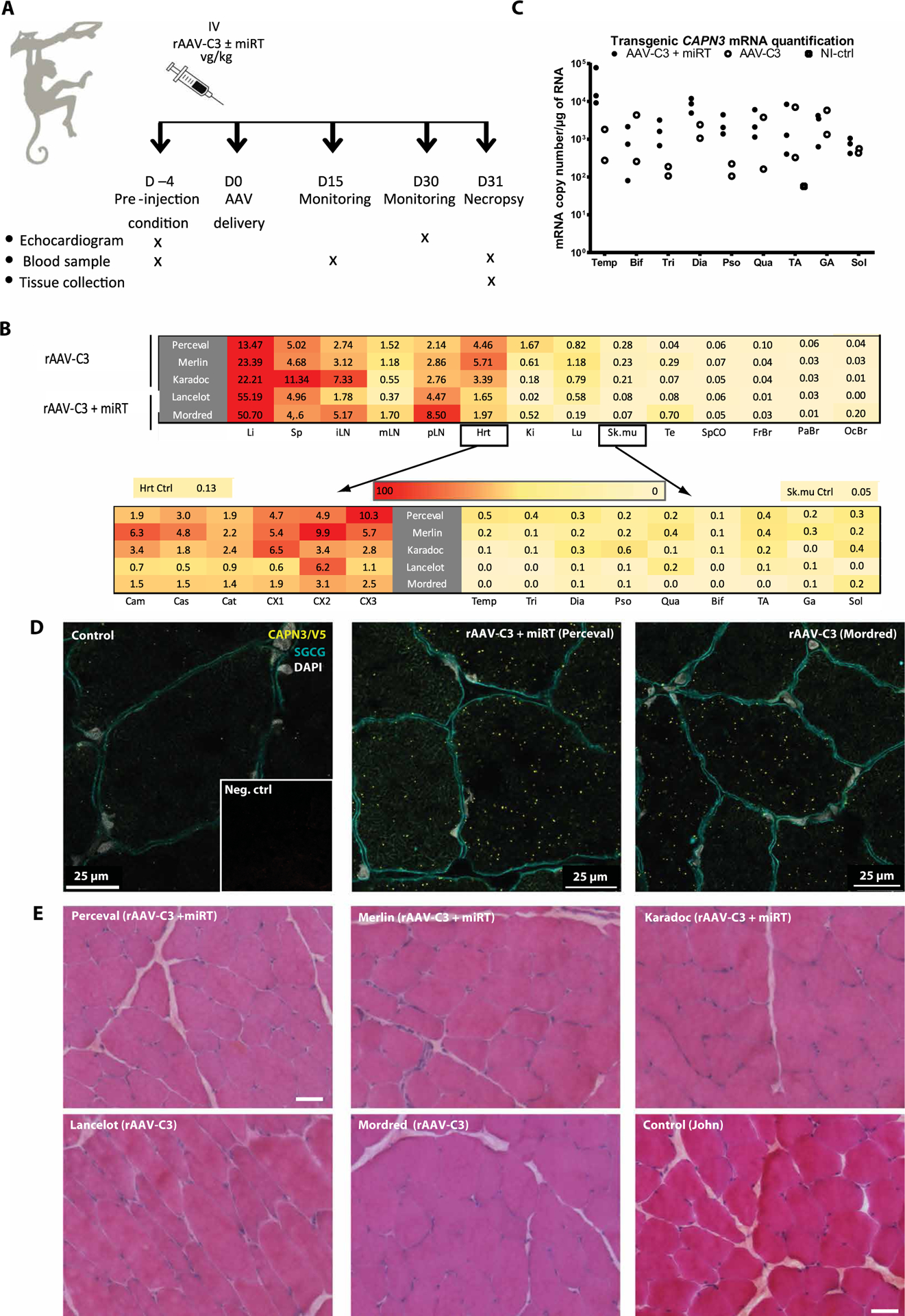
(A) Study design (n = 2 to 3). IV, intravenous injection. (B) Determination of vector copy number (VCN) per diploid genome in a range of tissues after systemic injection of rAAV-C3 or rAAV-C3+miRT sequences, represented by heat maps with a linear color table from red (high) to yellow (low). The values presented in the upper heat map for heart and skeletal muscle correspond to the mean of six different heart sections and the mean of all limb muscles, respectively, whose individual values are indicated in the lower heat maps. Li, liver; Sp, spleen; iLN, inguinal lymph nodes; mLN, mesenteric lymph nodes; pLN, popliteal lymph nodes; Hrt, heart; Ki, kidney; Lu, lung; Sk.mu, skeletal muscle; Te, testis; SpCO, spinal cord; FrBr, frontal brain section; PaBr, parietal brain section; OcBr, occipital brain section. (C) Expression of transgenic calpain 3 in a range of Macaca skeletal muscles (n = 2 to 3). Transgene expression was measured by RT-ddPCR and is presented in copies per microgram of RNA normalized to Rplp0. Temp, temporalis; Bif, biceps femoris; Tri, triceps brachii; Dia, diaphragm; Pso, psoas; Qua, quadriceps; GA, gastrocnemius; Sol, soleus. (D) Detection of transgenic calpain 3 using PLA. Red dots correspond to a specific amplification due to proximity ligation of two antibodies detecting epitopes present in our transgenic protein, specifically CAPN3 and V5 antibodies. Colabeling with γ-sarcoglycan (green) was performed to visualize muscle fibers, and 4′,6-diamidino-2-phenylindole (DAPI) is shown in blue. Inset: PLA negative control. Scale bars, 25 μm. (E) Sections of biceps femoris for all primates stained with HPS. Scale bars, 100 μm.
We first evaluated the antibody response to the AAV9 serotype using sera collected 4 days before AAV delivery (D−4) and at day 31 (D31) (table S2). As expected, the amount of anti-AAV9 immunoglobulin G (IgG) increased after the injection, confirming the delivery of the vectors and indicating the immunization of the primates for this serotype. We evaluated the biodistribution of rAAV9 by droplet digital PCR (ddPCR) using DNA extracted from the major organs (heart, liver, spleen, lymph nodes, kidney, lung, testis, spinal cord, and brain) and from a series of skeletal muscles [temporalis, triceps, diaphragm, psoas, quadriceps, biceps femoris, gastrocnemius, soleus, and tibialis anterior (TA)] (Fig. 3B). Hearts were split into six different sections: three for the atrium (left atrium and mitral valve, central part between both atria with sigmoid valve, and right atrium and tricuspid valve) and three for the ventricular parts (from the apex to the upper part of ventricles). The profile of biodistribution of both cassettes (rAAV-C3 or rAAV-C3+miRT) was similar, showing slightly lower transfer for the vector with the miR target sequence for most organs except the liver. As expected, filtering organs (liver, lymph nodes, spleen, lungs, and kidneys) presented the highest vector copy number (VCN) per diploid genome, with a mean of 0.3 to 55.19 VCN per diploid genome. However, the VCN in the liver was not associated with toxicity or inflammation in hepatocytes (fig. S2A). Samples from the nervous system (spinal cord and brain) and gonads showed the lowest VCN (mean of 0.01 to 0.7 VCN per diploid genome). The heart and skeletal muscles were in the middle range, with a higher VCN in the heart (mean of 3.4) compared to that found in the skeletal muscles (mean of 0.17).
NHP skeletal muscle expresses calpain 3 without toxicity after AAV–calpain 3 transfer
To evaluate the expression of transgenic calpain 3 in muscle tissues, we performed an analysis by RT-ddPCR using primers specific to the vector sequence. Samples from the BDC mice injected with the vectors revealed that all muscles from head to upper and lower limbs efficiently expressed both vectors. The expression varied from 149 to 6750 copies for rAAV-C3+miRT and from 29 to 734 copies for rAAV-C3 (Fig. 3C). rAAV-C3 led to lower mRNA expression (~1 log unit) compared to rAAV-C3+miRT, supported by reduced VCN (Fig. 3B). The mean value of CAPN3 mRNA per microgram of RNA in muscles, after delivery of 3 × 1013 vg/kg intravenously, was in a similar range to the expression observed with the intramuscularly delivered dose (5 × 109 vg) in the dKO study (~1 × 106 copies/μg of RNA) (fig. S2B). To investigate whether the human desmin promoter has a similar strength in primate and mouse muscle cells, we performed an in vitro experiment in which primary murine and primate skeletal muscle cells were transduced with a desmin–green fluorescent protein (GFP) lentivirus vector. GFP fluorescence-activated cell sorting (FACS) analysis indicated similar expression patterns, with a mean intensity of fluorescence slightly higher in primate than in the murine cells (fig. S2C). These observations suggest that the expression achieved in primates corresponds at minima to a dose that is therapeutic in the murine model.
For the quantitative measure of calpain 3 protein, we relied on the proximity ligation assay (PLA). The V5 tag in fusion with the transgenic calpain 3 protein allowed us to use an antibody specific for calpain 3 and one for the V5 tag to detect the transgenic protein while preventing detection of the endogenous protein. Dots corresponding to the presence of the transgenic protein were observed in biceps femoris muscles, showing that all muscle fibers presented a positive PLA signal with both vectors (Fig. 3D). There were no abnormalities detected by histological examination of the skeletal muscles (Fig. 3E) as expected considering previous experiments in mice (18, 19).
No cardiac toxicity was detected in hearts transduced with or without miRT
Similar to skeletal muscle, we analyzed the expression of the transgenic calpain in cardiac sections using RT-ddPCR and PLA. RT- ddPCR showed a substantial expression of transgenic calpain 3 with both vectors, with or without the miRT sequences, in all injected primates (Fig. 4A), indicating that the dose of the AAV9 vector injected in the primates was efficient at achieving expression of the transgene in the heart. In the six different sections of the heart, the number of copies per microgram of total RNA varied from 8 × 106 to 3.9 × 107, showing no preferential area of expression within the tissue. The PLA showed the presence of the transgenic protein in samples from all injected primates, confirming the capacity of the AAV9 to drive expression of a transgene in the heart of primates (Fig. 4B). Quantification of the specific signal was performed on heart sections (Fig. 4C); there was no statistically significant difference among groups. This observation contrasts with the observed higher RNA expression that may result from more efficient AAV transfer; the trend suggests that analysis of a larger number of animals could potentially be consistent with the previous observation that the cardiac-specific miR-208a leads to a reduction of translation (19).
Fig. 4. Cardiac expression and histological analyses after systemic delivery of calpain 3 in NHP.
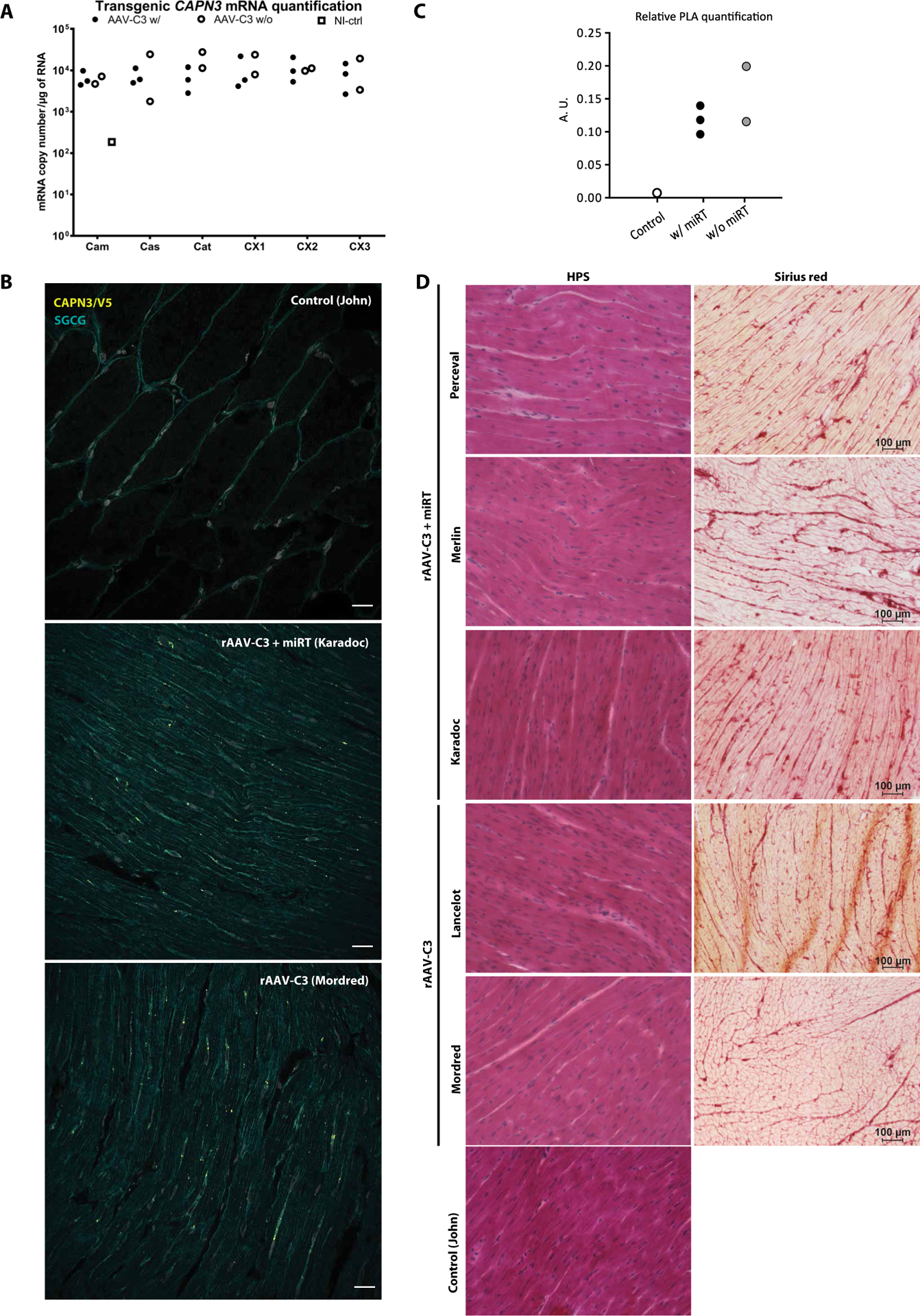
(A) Expression of transgenic calpain 3 per microgram of RNA in a range of heart sections measured by RT-ddPCR. Calpain 3 expression was normalized to Rplp0 and compared to the quantity of detected expression in a noninjected primate (total heart lysate). Cam, left atrium from mitral valve to aorta; Cas, central part of both atria including pulmonary valve and aorta; Cat, right atrium from vena cava to tricuspid valve; CX1–3, three sections taken between the apex and the top part of the ventricles. (B) Detection of the calpain 3 transgene using PLA. Red dots correspond to a specific amplification due to proximity ligation of two antibodies detecting epitopes present in our transgenic protein, specifically CAPN3 and V5 antibodies. Colabeling with γ-sarcoglycan was performed to visualize fibers (green). Scale bar, 25 μm. (C) Quantification of PLA. A.U., arbitrary unit. (D) Cardiac sections (CX3) of all injected primates stained by HPS or Sirius red. Scale bars, 100 μm.
Routine blood analysis was performed D−4, D+15, and D+30. No noticeable variation in blood parameters (hematology and biochemistry) associated with the transfer of the vectors or the expression of the transgenic protein was observed (table S3). In addition, several classical nonspecific (creatine kinase, lactate dehydrogenase, and myoglobin) and specific [troponin T and N-terminal fragment of brain natriuretic peptide (NT-proBNP)] cardiac biomarkers were measured in the serum at D4, D15, and D30 (Table 1). We also measured two cardiac miRs (miR-208a and miR-499) in the serum by RT-PCR (Table 2). No increases indicative of a cardiac impairment were observed during the experiment. To evaluate cardiac function, we performed two echocardiograms before injection (D−4) and the day before the study endpoint (D30). The global aspect of the heart was evaluated using the BiDimensional mode, potential dilatation using the Time-Move mode, and the blood flow through heart valves (pulmonary, aorta, and mitral valves) using pulsed Doppler. All primates presented with normal heart function (values within normal range for this species according to ONIRIS experience) at the two time points for both vectors (table S4A), with the absence of detectable abnormality of the myocardium and no ventricle dilatation. No noticeable difference was observed between D−4 and D30 (table S4B). Cryosections of the different parts of the heart stained using HPS or Sirius red revealed no abnormalities after injection of rAAV-C3 or rAAV-C3+miRT (Fig. 4D). Thus, no toxic or deleterious effects were observed for either of the vectors during physiological or histological testing.
Table 1. Evaluation of different serum cardiac biomarkers at D−4, D+15, and D+30.
CK, creatine kinase; AST, aspartate aminotransferase; LD, lactate dehydrogenase; NT-proBNP, N-terminal pro b-type natriuretic peptide. No substantial difference was observed between the pre- and post-injection conditions.
| Number | Day | CK (μkat/liter) | LDH (μkat/liter) | Myoglobin (ng/ml) | Troponin T (ng/ml) | NT-proBNP (ng/ml) |
|---|---|---|---|---|---|---|
| Perceval | D-4 | 7.68 | 5.01 | 34.93 | 2.3 | 4.5 |
| With mir | D15 | 10.66 | 9.9 | 79.99 | 2.3 | 6,0 |
| D30 | 12.89 | 5.79 | 21.11 | 2.8 | 2.9 | |
| Merlin | D-4 | 11.17 | 4.73 | 56.36 | 1.2 | 0.4 |
| With mir | D15 | 17.04 | 8.32 | 99.02 | 1.2 | 0.7 |
| D30 | 15.83 | 6.31 | 45.27 | 1.3 | 2.2 | |
| Karadoc | D-4 | 16.62 | 8.16 | 35.46 | 1.5 | 4.8 |
| With mir | D15 | 20.59 | 12.47 | 84.54 | 1.8 | 5.1 |
| D30 | 46.57 | 10.03 | 226.7 | 1.8 | 3.6 | |
| Lancelot | D-4 | 16.46 | 5.3 | 27.37 | 1.5 | 4.8 |
| Without mir | D15 | 7.03 | 6.26 | 71.56 | 1.3 | 3,0 |
| D30 | 14.85 | 5.24 | 60.22 | 1,0 | 5,0 | |
| Mordred | D-4 | 10.58 | 4.73 | 77.46 | 7.8 | 4.3 |
| Without mir | D15 | 5.67 | 7.5 | 76.96 | 5.3 | 2.9 |
| D30 | 17.96 | 6.78 | 57.08 | 3.7 | 3.8 |
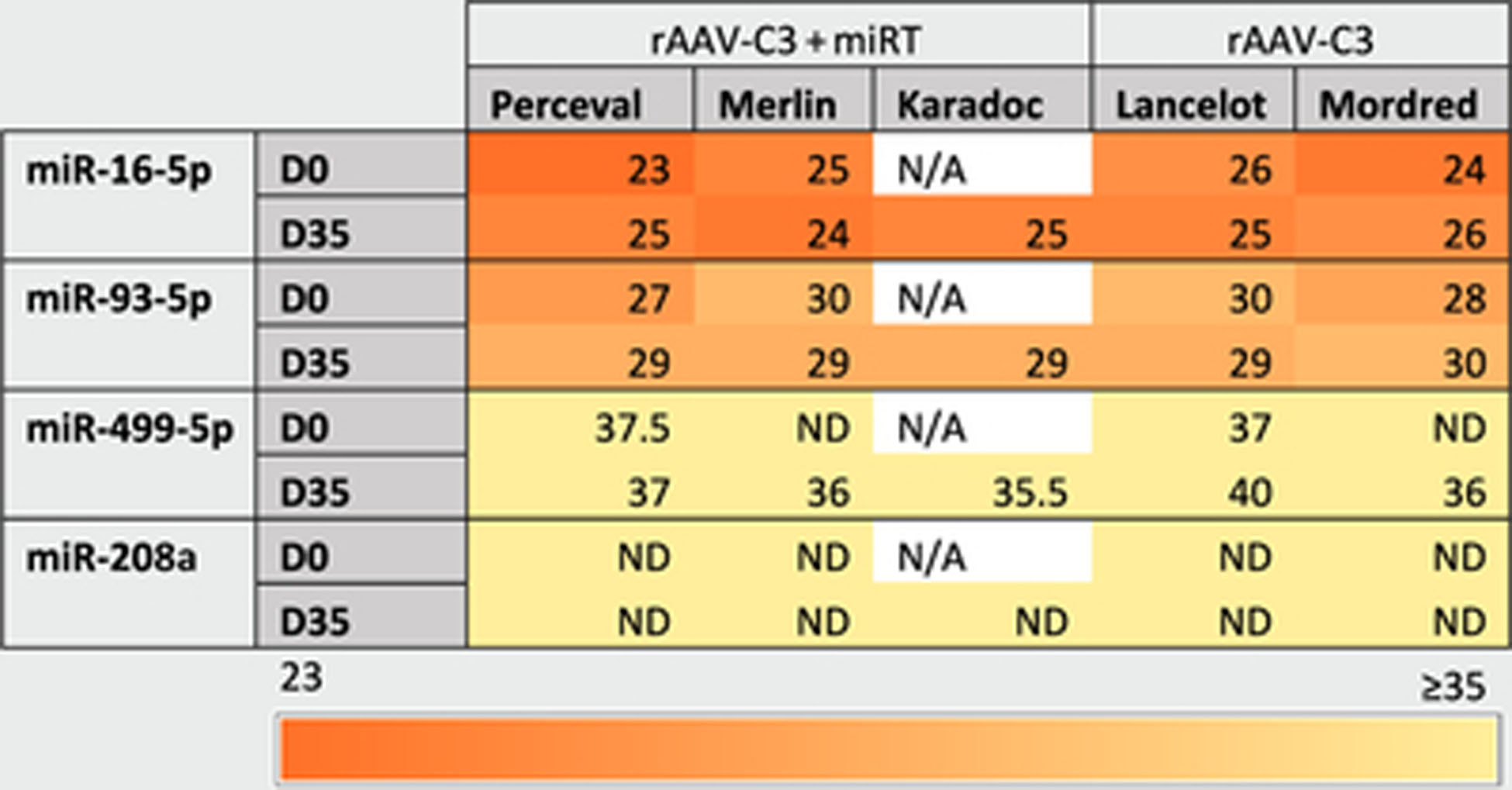
Table 2. Measurements of cardiac miRNA expression.
Cardiac miRs (miR-208a and miR-499) and control miR (miR-16 and miR-93) were measured in the serum of injected primates. CT values were averaged over two technical replicates. N/A: not available; ND, not detected.
|
One calpain 3 binding site on titin shows species-specific differences
Because of the lack of toxicity in primates and the known toxicity when calpain 3 is expressed in the mouse heart, we analyzed calpain 3 binding sites on titin in these two species. Four binding sites of calpain 3 on titin have been previously reported: three in the N2A/PEVK region, corresponding to the Ig80-IS domain, Ig82-Ig83 Ig repeats, and an N-terminal region of PEVK coded by exons 116 to 135 (25), and one unique insertion in the M band—IS7 coded by the Mex5 exon (Fig. 5A). We performed RT-PCR of these regions in murine, NHP, and human heart and skeletal muscle samples (Fig. 5B). Although no size difference was observed in the first two binding sites in N2A and the M-line site, we observed bands of larger sizes in Macaca, human, and mouse skeletal muscles compared to cardiac tissues for the third I-band binding site (Fig. 5). To confirm these isoform data, we performed RNA-sequencing analysis on mouse, primate, and human heart samples and defined the percent spliced-in index of the exons containing calpain 3 binding sites. We observed a differential inclusion of exons and species- and genotype-dependent isoform expression (Fig. 5C). This observation suggested that the third binding site of the N2A/PEVK region might explain the difference between calpain 3 proteolytic activity regulation in skeletal muscle compared to that in the heart. Titin transcripts in primate and human heart samples incorporate more exons in the N2A/PEVK region compared to mouse, suggesting that human sensitivity toward the cardiac toxicity induced by calpain 3 ectopic expression may be more similar to that of primate than mouse.
Fig. 5. CAPN3 binding sites on titin.
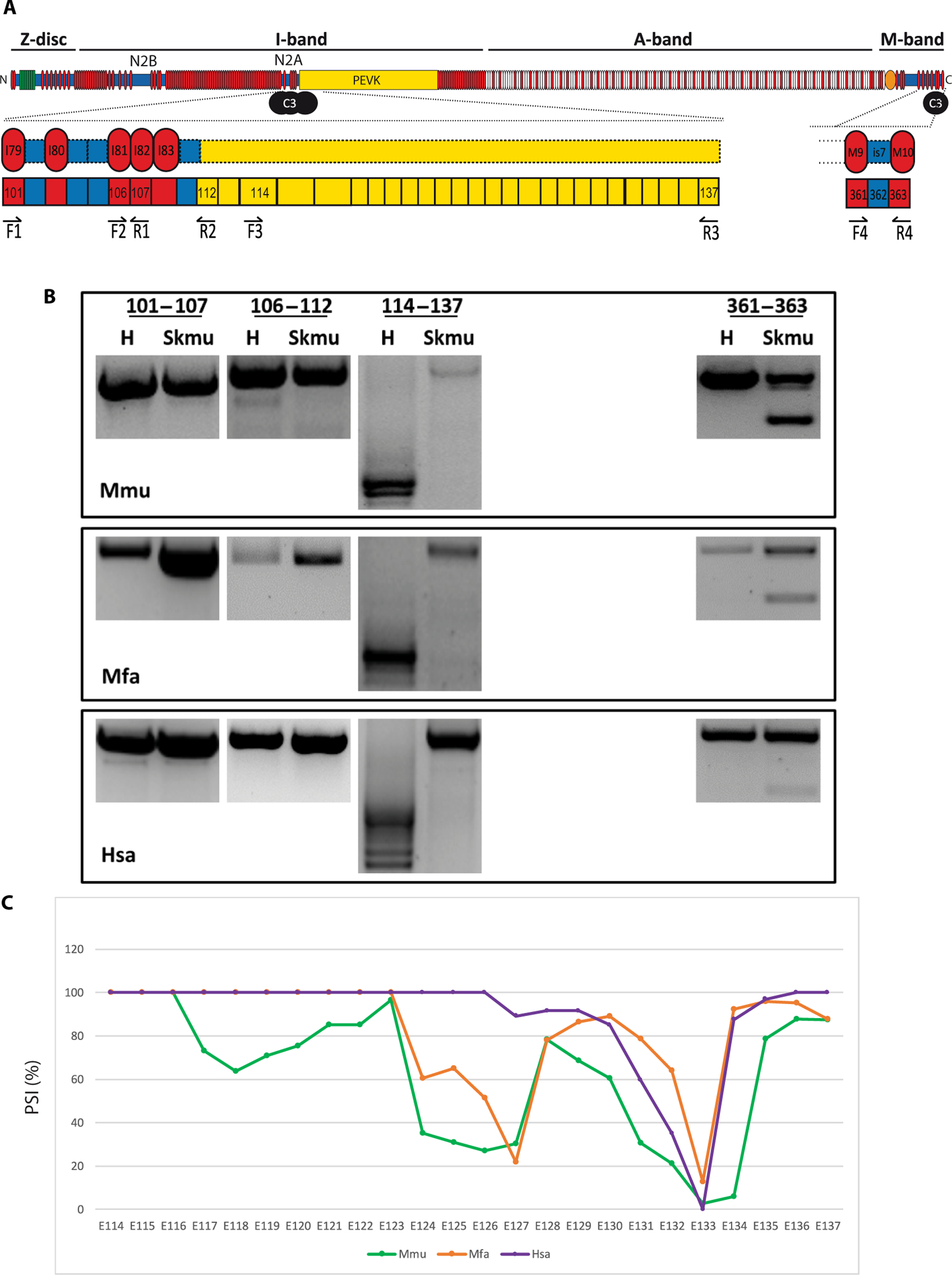
(A) Known binding sites of calpain 3 on the titin protein. Three sites are located in N2A/PEVK region and one in the M-line at the C-terminal part. Position of the primers used for each amplicon is drawn below the corresponding exons. (B) RT-PCR of the CAPN3 binding sites on mouse, primate, and human cardiac and skeletal muscles (exons 101 to 107 and 106 to 112 in the N2A domain, exons 114 to 137 for the domain in the upstream region of the PEVK domain, and exons 362 to 364 in the M-line). The experiments were performed in triplicate. (C) Percent spliced-in index (PSI) between mouse (Mmu), primate (Mfa), and human (Hsa) titin in the PEVK region (exons 114 to 137).
A specific PEVK muscle domain regulates calpain 3 proteolytic activity in vivo
To define whether the presence or absence of the third binding site of the N2A/PEVK region plays a role in the different outcomes of AAV-mediated expression of calpain 3 in cardiac and skeletal tissues, we took advantage of a mouse model KO for RNA binding protein, RBM20, that was previously shown to be a transcriptional modulator of titin alternative splicing (26, 27). The deletion of RBM20 in this murine model leads to an inclusion of all RBM20-regulated exons in the cardiac isoforms of titin. We first validated the inclusion of the alternative binding domain in heart and skeletal muscle of the RBM20 KO model by RT-PCR. Our analysis indicated that the exons corresponding to the third binding site in the N2A/PEVK region were expressed, whereas the other binding sites were not affected (Fig. 6A), consistent with the literature (28). This analysis confirms that this mouse model expresses a titin isoform in the heart that is more similar to that of skeletal muscle, as far as calpain 3 binding sites are concerned. The model is therefore relevant for investigating the mechanism of cardiac toxicity related to calpain 3 activity.
Fig. 6. Effect of AAV-mediated expression of calpain 3 in RBM20 KO mice.
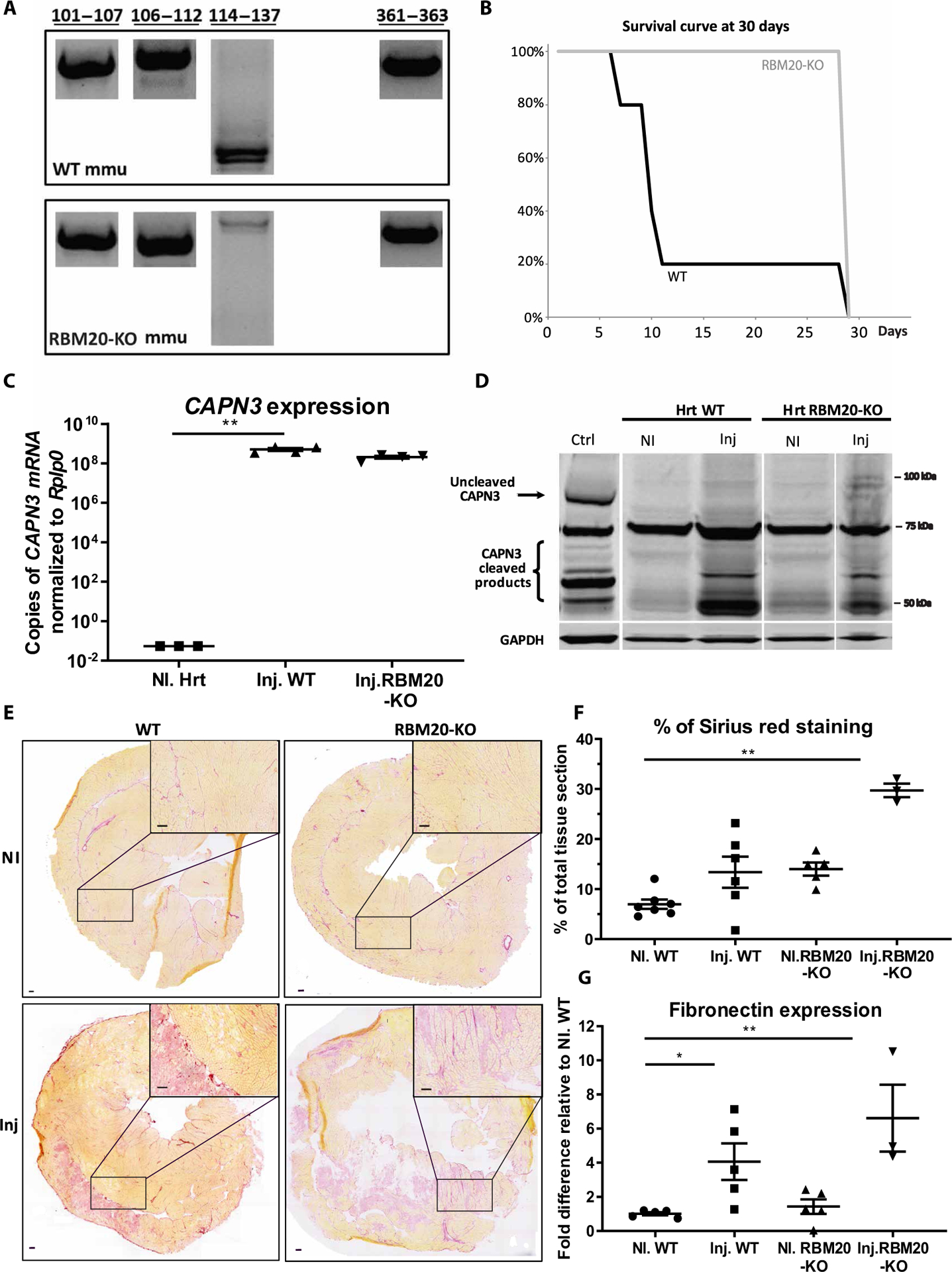
(A) RT-PCR performed on cardiac samples. The amplified regions corresponding to the four known binding sites are shown. (B) Survival curve of CAPN3- injected animals (WT and RBM20 KO) over a 30-day test period (n = 3 to 7). At the last time point (D30), all animals were sacrificed. (C) Transgenic CAPN expression in cardiac tissue was quantified by RT-qPCR (copies per microgram of RNA). Data were normalized to Rplp0. rAAV-injected mice (Inj.) versus phosphate-buffered saline (PBS) (NI). Statistical analyses were performed using nonparametric Kruskal-Wallis test and post hoc multiple comparisons Dunn’s test (n = 3 to 4). **P < 0.01. (D) Western blot of calpain 3 in cardiac tissue. Whole calpain 3 is expected at 94 kDa (black arrow), and its cleaved products are expected at ~55 to 65 kDa (bracket) according to the antibody used. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize the amount of protein loaded. rAAV-injected mice versus PBS. WT skeletal muscle (TA) was used as the positive control. (E) Heart sections were stained with Sirius red to highlight fibrotic areas. Representative examples of the entire sections as well as ×2 magnification of specific areas are shown. rAAV-injected mice versus PBS. Scale bars, 100 μm. (F) Sirius red quantification. Amount of staining is expressed in percentage of the total tissue section. rAAV-injected mice versus PBS is shown. Statistical analyses were performed using nonparametric Kruskal-Wallis test and post hoc multiple comparisons Dunn’s test (n = 3 to 7). **P < 0.01. (G) Fibronectin was quantified by RT-qPCR. Results were normalized to Rplp0 and compared to a PBS-injected WT condition. rAAV-injected mice (Inj.) versus PBS (NI). Statistical analyses were performed using nonparametric Kruskal-Wallis test and post hoc multiple comparisons Dunn’s test (n = 3 to 5). *P < 0.05 and **P < 0.01.
To investigate the role of this titin domain in the control of calpain 3 activity, we injected RBM20 KO mice with the rAAV9-C3 vector at a dose (2.5 × 1013 vg/kg) we previously demonstrated to be lethal in WT mice (19) and monitored the mice for a period of 30 days. The WT mice did not survive more than 10 days (as expected), whereas the injected RBM20 KO mice were still alive at the end of the protocol (Fig. 6B). To confirm the correct transfer of the calpain 3 vector, we injected another group of mice and compared WT and KO animals 10 days after injection. Transgene expression was analyzed by qRT-PCR, demonstrating high expression of calpain 3 in heart samples transduced with rAAV9-C3 with an efficient and comparable gene transfer in both mouse strains (Fig. 6C). Western blot analysis of heart lysates showed no calpain 3 in noninjected animals (as expected) but expression of cleavage products of calpain 3 (~60 to 65 kDa) in RBM20 KO mice, suggesting the presence of active calpain 3 in heart tissue (Fig. 6D). Histological and mRNA quantification of fibrosis and fibronectin expression showed that RBM20 KO hearts were fibrotic because of the absence of RBM20 (Fig. 6, E to G), a consequence that was previously reported in a RBM20 KO rat model (26). The calpain 3 injection increased this condition (Fig. 6, E to G). In conclusion, even if calpain 3 expression led to an increase of fibrosis in the tissue of the RBM20 KO mouse, the presence of the calpain 3 binding domain of interest resulted in a protective effect as seen by the absence of lethality. This suggests that titin exons 116 to 135 play a role in controlling the proteolytic activity of calpain 3.
Is7 muscle domain plays a major role in calpain 3 stabilization
Because calpain 3 expression led to increased fibrosis in the heart of RBM20 KO mice, it is possible that other molecular players are involved in calpain 3 regulation. Therefore, we investigated the role of another binding site of calpain 3 on titin at the M-line: the titin Is7 domain encoded by the penultimate Mex5 exon. We used a mouse model lacking this domain, the titinMex5−/Mex5− (hereafter referred to as “delMex5”) model that was previously characterized (29). We validated that the absence of this domain did not affect the other calpain 3 binding sites (Fig. 7A).
Fig. 7. Effect of AAV-mediated expression of calpain 3 in DelMex5 mice.
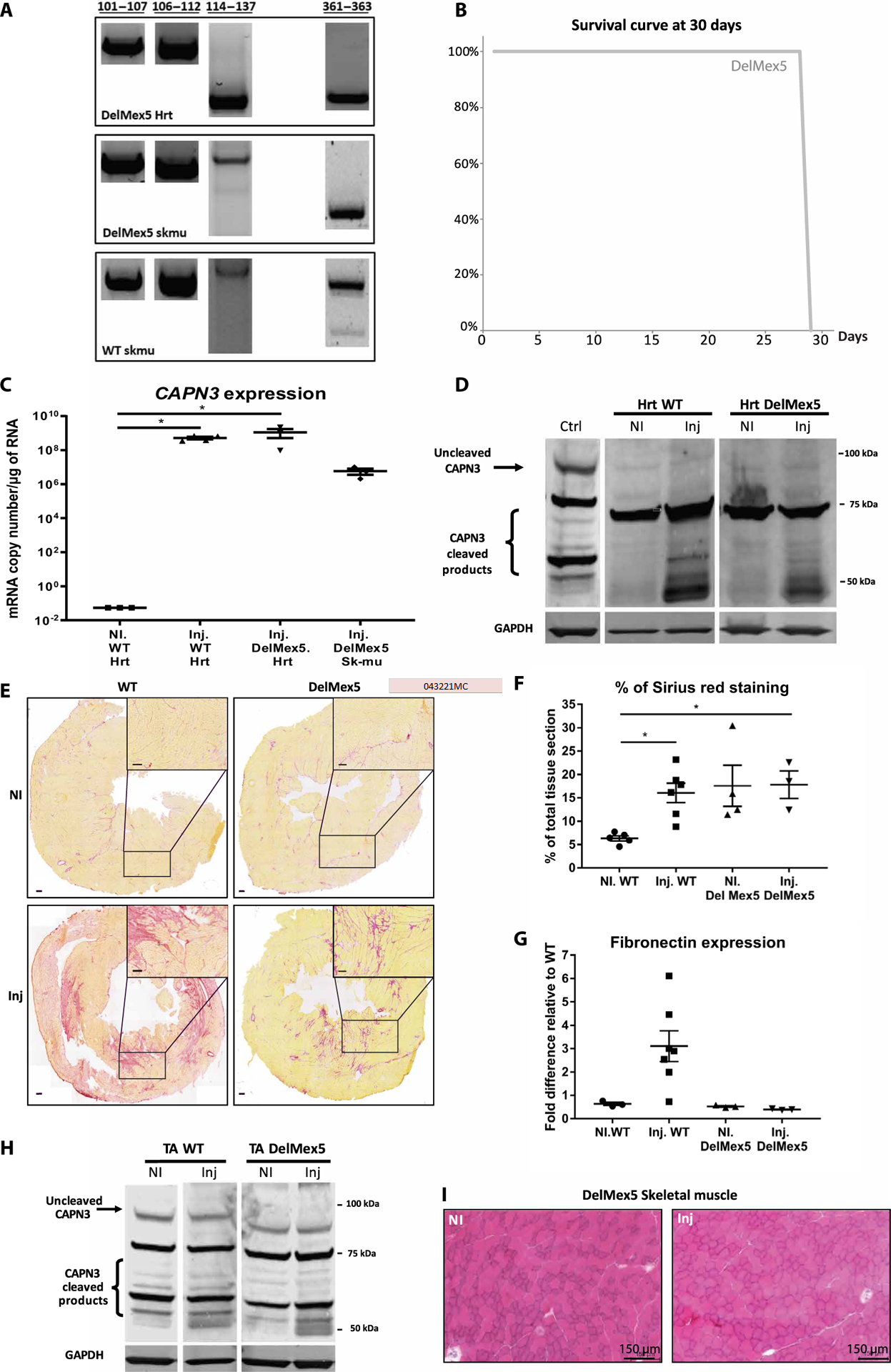
(A) RT-PCR was performed using primers located in exons flanking each known C3 binding site on titin using extracts from heart of DelMex5 mice and skeletal muscle of DelMex5 and WT mice. (B) Survival curve of DelMex5 mice over a 30-day period after CAPN3 injection (n = 3 to 6). All mice were sacrificed at day 30. Control mice for this experiment are shown in Fig. 6B. (C) Expression of CAPN3 mRNA after systemic injection, quantified by RT-qPCR (copies per microgram of RNA), normalized to Rplp0 (rAAV-injected mice versus PBS). Statistical analyses were performed using nonparametric Kruskal-Wallis test and the post hoc multiple comparisons Dunn’s test (n = 3 to 4). *P < 0.05. (D) Western blot of calpain 3 in cardiac tissue. rAAV-injected mice versus PBS. (E) Heart sections were stained with Sirius red to highlight areas of fibrosis. Representative images of entire sections as well as ×2 magnification of one area are shown. rAAV-injected mice versus PBS. Scale bars, 100 μm. (F) Sirius red quantification. The amount of staining is expressed in percentage of the total tissue section. rAAV-injected mice versus PBS. Statistical analyses were performed using nonparametric Kruskal-Wallis test and post hoc multiple comparisons Dunn’s test (n = 3 to 6). *P < 0.05. (G) Fibronectin was quantified by RT- qPCR. Results were normalized to Rplp0 and compared to a PBS-injected WT condition. rAAV-injected mice versus PBS. Statistical analyses were performed using nonparametric Kruskal-Wallis test and post hoc multiple comparisons Dunn’s test (n = 3 to 7). (H) Western blot of calpain 3 in muscle tissue. Whole calpain 3 is expected at 94 kDa (black arrow), and cleavage products are expected at ~55 to 65 kDa (bracket). GAPDH was used to normalize the amount of protein loaded. rAAV-injected mice versus PBS. WT skeletal muscle (TA) was used as a positive control. (I) Stained HPS muscle sections. rAAV-injected mice versus PBS.
To evaluate the consequences of calpain 3 overexpression in the absence of the Is7 domain, we systemically delivered a dose of calpain 3 vector similar to the above-discussed experiment with the RBM20 KO mice (2.5 × 1013 vg/kg). The delMex5 mice survived the full 30-day period after the injection despite the complete absence of the Is7 domain (Fig. 7B), and contrary to the WT mice, many of which died. In another group of animals, samples were collected 10 days after injection. We confirmed by qRT-PCR that the transgene expression corresponded to efficient gene transfer in cardiac and skeletal muscles (Fig. 7C). However, it was not possible to detect calpain 3 on Western blot for the delMex5 samples, whereas it was detected in the WT condition, suggesting protein instability in the absence of Is7 (Fig. 7D). Fibrosis quantification and fibronectin expression analysis on heart sections showed that calpain 3 expression after vector injection did not worsen the fibrotic state naturally present in this delMex5 model (Fig. 7, E to G) (29). There were no deleterious consequences of the transfer of calpain 3 in skeletal muscle: no abnormalities in the stability or activity of calpain 3 protein compared to the WT TA muscle (Fig. 7, H and I), whereas we previously showed that gene transfer was deleterious in the absence of the whole M-line (30). In conclusion, our results indicate that calpain 3 overexpression in the absence of Is7 is not deleterious, likely because of instability of the protease that prevents the cardiotoxicity related to the absence of the third N2A/PEVK calpain 3 binding site. This contrasts with the toxicity seen when the PEVK binding site is absent, as is the case in the normal mouse heart. Together, we show that the presence of Is7 is required to stabilize calpain 3, and PEVK exons 116 to 134 are a prerequisite for modulating calpain 3 activity.
DISCUSSION
The main purpose of this study was to perform preclinical assessments of the use of AAV-mediated transfer of the calpain 3 gene, as a potential therapeutic for patients with LGMD2A. We used a severe dKO mouse model, deficient in both calpain 3 and dysferlin, validating this model for dose assessment. We also assessed the cardiac safety profile of rAAV expressing CAPN3 after systemic injection in primates. Last, we investigated the underlying causes of the cardiac toxicity observed after gene transfer in mice and the role of titin in this phenomenon. Such studies are essential before applying gene therapy in patients with LGMD2A.
We investigated the amount of calpain 3 expression required in skeletal muscle to reach a therapeutic effect using a dKO mouse model, whose phenotype was more severe than the mild CAPN3 mouse model. This study parallels previous work done in the dystrophin field in which the most widely used model, the mdx mouse, does not reproduce the same disease impairment as that seen in humans. This observation has led to the use of different dKO mouse models, of which the most frequently used is the dKO deficient in utrophin and dystrophin (31, 32). Here, we selected dysferlin, a known partner of calpain 3, which, when mutated, can also cause muscular dystrophy. A dKO deficient in dystrophin and dysferlin demonstrated a more severe muscular dystrophy than the mdx model (33), possibly because dysferlin and dystrophin play different roles in maintaining muscle cell integrity. We previously showed that calpain 3 KO muscle does not present a defect in membrane repair; therefore, it is conceivable that calpain 3 and dysferlin function via different pathways and, consequently, that a cumulative effect of both deficiencies is present in the dKO. Here, we validated this hypothesis: The dKO presented a higher disease severity than either single KO model, raising interesting questions regarding the functional relationship between the two proteins and presenting a useful model for our efficacy study. This dKO model appears adequate for precise evaluation of the rAAV dose because of the marked difference in phenotype between mice with double deficiencies and the sole dysferlin deficiency. We observed that a considerable therapeutic effect was achieved in the dKO with a dose leading to similar calpain 3 expression as attained in primates. It is optimal to use the lowest dose that achieves efficacy, considering not only virus production requirements but also possible toxic effects that may occur in patients when a high dose of vector is administered in one treatment.
Our previous observations indicated that the ectopic expression of calpain 3 after systemic administration of rAAV-C3 leads to cardiotoxicity due to unregulated calpain 3 proteolytic activity. Even if it was possible to circumvent this toxicity by preventing expression in cardiac tissue using a miR-regulated vector, we considered it vital to investigate the safety of the vectors in a translational model similar to humans. Here, we performed systemic injections of both regular and miR-regulated AAV vector in primates. One month after delivery of 3 × 1013 vg/kg by single intravenous infusion, correct distribution of the vectors in the heart was observed in the absence of adverse effects or detectable toxicity in NHP. The AAV9 serotype was selected for this study to correspond to the worst-case scenario with respect to the evaluation of the cardiotoxicity of our vectors. This serotype is well known for driving transduction in the heart tissue of mice after systemic delivery (34). Evidence of AAV9 transduction in the heart of primates has also been reported, albeit in few studies (35–37). Our study demonstrated roughly the same biodistribution as these previous studies but with a higher efficiency, corroborating the choice of this serotype for this evaluation.
The AAV transfer of calpain 3 demonstrated an absence of cardiac toxicity in primates, as evaluated by serum markers, histology, and echocardiography, even without the vector being miR regulated. In mice, lethality can be observed as early as 10 days after injection of the vector without miR; in the absence of lethality, fibrosis was observed with doses as low as 4 × 1012 vg/kg using the less efficient AAV1 serotype (19). Considering the biodistribution and expression data, the differences in outcomes between the murine and Macaca species are unlikely to be due to a difference in biodistribution, efficacy of transfer of AAV9, or transgene expression. An alternative hypothesis is that the different outcomes are due to a disparity in the regulation of calpain 3 activity. Because it has been previously shown that titin can regulate the activity of calpain 3 (12, 25) and considering that skeletal and cardiac muscles express distinct titin isoforms (38), we investigated the alternative splicing of titin’s calpain 3 binding sites using RT-PCR. We observed a difference in one of the N2A/PEVK binding sites in the hearts of macacas compared to those of mice. This region (in I81 to I83 domains of the N2A region) has previously been shown to regulate calpain 3 activity (12, 25). Thanks to the use of two animal models presenting different deletions of calpain 3 binding sites on titin, we were able to demonstrate that the absence of the third N2A/PEVK calpain 3 binding site participates in the generation of the cardiac toxicity, that the absence of Is7 leads to an important instability of calpain 3, and that this instability is sufficient to prevent toxicity in the absence of the third N2A/PEVK calpain 3 binding site. This complex interplay is interesting to consider in the view of previous reports that demonstrate that calpain 3 is redistributed from the M-line to the N2A region as sarcomeres lengthen and that the redistribution is dependent on calpain’s proteolytic activity (17, 39). These reports suggested that the conformational change resulting from the autoproteolytic processing of calpain 3 played a role in its dynamics within the sarcomere. It is therefore possible to propose a model in which Is7 binding is mandatory for calpain 3 to attain an active form (fig. S3). It should be noted that the mode of binding in the two regions differs: The binding in the M-line requires the full-length calpain 3, whereas in the N2A region, only the Is2 region of calpain 3 is required (11). This difference suggests that the mode of activation of calpain 3 within the two regions could also differ. We propose that once activated, calpain 3 translocates into the N2A region where it activates calpain 3 present in this region. However, this only occurs if the condition of activation is fulfilled: The binding at the third PEVK/ N2A region has to be unlocked, possibly through a mechanical signal.
Potential limitations of our study relate to animal models. We used a limited number of animals in the primate study for ethical and cost reasons; the models are also imperfect, considering, for example, the existing cardiac phenotype in the RBM20 KO mice. The results obtained with the WT primates using only one dose of rAAV may not accurately predict results in human patients. It is not known whether the loss of function of calpain 3 or the presence of the dystrophic process will affect the efficacy of gene transfer in patients. Second, defining potential adverse effects would require additional doses. Furthermore, we have only assessed the acute toxicity of the vector in NHP and not the long-term effect of the vector administration. Some of these limitations can be addressed by regulatory biodistribution/safety studies performed during further preclinical development of the gene therapy approach.
In summary, we have developed a mouse model in which the effect of calpain 3 can be monitored with high sensitivity. We also demonstrated that a single systemic delivery of rAAV vectors at a dose of 3 × 1013 vg/kg induced the expression of the calpain 3 transgene in NHP without eliciting detectable toxicity or adverse effects. These data further support the gene delivery strategy as a safe approach for treating patients with LGMD2A. Our studies into the cause of the cardiac toxicity observed in murine models elucidated the regulation of calpain 3 activation, supporting a species-specific, two-hit mechanism involving binding sites at different locations on titin.
MATERIALS AND METHODS
Study design
This study was designed with the primary aim to investigate the safety of rAAV-mediated transfer of calpain 3 in primates with an emphasis on cardiac evaluation. A secondary objective was to investigate the mechanism of the cardiac toxicity that we previously observed in mice. A therapeutic dose was defined in a newly developed, severe dystrophic mouse model deficient in both calpain 3 and dysferlin. The NHP study used randomly selected primates after screening for AAV9 seronegativity. Investigation of the involvement of titin in the cardiotoxicity in mice was performed using two mouse models with littermates as controls and random assignment of the animals between groups. Animal experimentation was approved by the Ethical Committee for Animal Experimentation no. 6 of Nantes and C2AE-51 of Evry under numbers 04810.02 and 2015–003-A, respectively, for the primate and mouse studies. The rAAV injection and dissection experiments were conducted in a nonblinded fashion. Blinding approaches were used during histology validation, immunostaining analysis, and biomarker analysis. Details on experimental procedures are presented in Supplementary Materials and Methods. Individual subject-level data are presented in data file S1.
Statistical analysis
Results are presented as scatter plots with the mean (thick line) and SEM (thin lines). Statistical significance between groups was calculated by the nonparametric Kruskal-Wallis test (α = 0.05) and the post hoc multiple comparisons Dunn’s test using the Prism package (GraphPad Software). Values of P < 0.05 were considered as statistically significant.
Supplementary Material
Fig. S1. Supplementary data for the dKO experiments.
Fig. S2. Supplementary data for the primate experiments.
Fig. S3. Proposed model of calpain 3 activation.
Table S1. Screening of primates for AAV9 seronegativity.
Table S2. Antibody response against the AAV9 serotype on sera (preimmune and sacrifice).
Table S3. Blood parameters of primates.
Table S4. Summary of echocardiogram analysis before and after systemic injection in primates.
Data file S1. Individual subject-level data.
References (40-43)
Acknowledgments:
We would like to thank the Vector Core of Atlantic Gene Therapy at the University Hospital of Nantes for producing the rAAV used in this study and the Center of Boisbonne, Nantes, especially V. Guilloux and S. Moullec, for the helpful interaction during the primate study. We acknowledge the help of the team of F. Mingozzi (Généthon, France) for performing serology screening and providing primate control samples and for helpful discussion. We are thankful to A. Bemelmans and C. Jan from MIRCen (Molecular Imaging Research Center, CEA, Fontenay-aux-Roses) for providing primate samples. We are grateful to the Généthon support teams for in vivo experimentation and histology and the “Imaging and Cytometry Core Facility” of Généthon for technical support and the Ile-de-France Region, GIP Genopole, Evry, and INSERM for the purchase of the equipment.
Funding: This work was supported by the “Association Française contre les Myopathies” (AFM), the Fondation de France, the “Coalition Against Calpain 3,” the Leducq Foundation (13CVD04), and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR073179).
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: The authors declare that they have no competing financial interests. A patent related to the vectors used in this study has been filed with I.R. as an inventor under the number EP 14721468.8 and is held by Généthon and INSERM.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, Hillaire D, Passos-Bueno MR, Zatz M, Tischfield JA, Fardeau M, Jackson CE, Cohen D, Beckmann JS, Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 81, 27–40 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Urtasun M, Sáenz A, Roudaut C, Poza JJ, Urtizberea JA, Cobo AM, Richard I, García Bragado F, Leturcq F, Kaplan JC, Marti Massó JF, Beckmann JS, López de Munain A, Limb-girdle muscular dystrophy in Guipúzcoa (Basque Country, Spain). Brain 121 (Pt 9), 1735–1747 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Fanin M, Nascimbeni AC, Fulizio L, Angelini C, The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscul. Disord 15, 218–224 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Fardeau M, Hillaire D, Mignard C, Feingold N, Feingold J, Mignard D, de Ubeda B, Collin H, Tome FM, Richard I, Beckmann J, Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain 119 (Pt 1), 295–308 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Richard I, Hogrel JY, Stockholm D, Payan CA, Fougerousse F, Eymard B, Mignard C, Lopez de Munain A, Fardeau M, Urtizberea JA, Natural history of LGMD2A for delineating outcome measures in clinical trials. Ann. Clin. Transl. Neurol 3, 248–265 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quick S, Schaefer J, Waessnig N, Schultheiss T, Reuner U, Schoen S, Reichmann H, Strasser R, Speiser U, Evaluation of heart involvement in calpainopathy (LGMD2A) using cardiovascular magnetic resonance. Muscle Nerve 52, 661–663 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, Suzuki K, Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem 264, 20106–20111 (1989). [PubMed] [Google Scholar]
- 8.Herasse M, Ono Y, Fougerousse F, Kimura E, Stockholm D, Beley C, Montarras D, Pinset C, Sorimachi H, Suzuki K, Beckmann JS, Richard I, Expression and functional characteristics of calpain 3 isoforms generated through tissue-specific transcriptional and posttranscriptional events. Mol. Cell. Biol 19, 4047–4055 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fougerousse F, Anderson LV, Delezoide AL, Suel L, Durand M, Beckmann JS, Calpain3 expression during human cardiogenesis. Neuromuscul. Disord 10, 251–256 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Sorimachi H, Kinbara K, Kimura S, Takahashi M, Ishiura S, Sasagawa N, Sorimachi N, Shimada H, Tagawa K, Maruyama K, Suzuki K, Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J. Biol. Chem 270, 31158–31162 (1995). [DOI] [PubMed] [Google Scholar]
- 11.Kinbara K, Sorimachi H, Ishiura S, Suzuki K, Muscle-specific calpain, p94, interacts with the extreme C-terminal region of connectin, a unique region flanked by two immunoglobulin C2 motifs. Arch. Biochem. Biophys 342, 99–107 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Ono Y, Torii F, Ojima K, Doi N, Yoshioka K, Kawabata Y, Labeit D, Labeit S, Suzuki K, Abe K, Maeda T, Sorimachi H, Suppressed disassembly of autolyzing p94/CAPN3 by N2A connectin/titin in a genetic reporter system. J. Biol. Chem 281, 18519–18531 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Taveau M, Bourg N, Sillon G, Roudaut C, Bartoli M, Richard I, Calpain 3 is activated through autolysis within the active site and lyses sarcomeric and sarcolemmal components. Mol. Cell. Biol 23, 9127–9135 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia Diaz BE, Gauthier S, Davies PL, Ca2+ dependency of calpain 3 (p94) activation. Biochemistry 45, 3714–3722 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Duguez S, Bartoli M, Richard I, Calpain 3: A key regulator of the sarcomere? FEBS J 273, 3427–3436 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Kramerova I, Kudryashova E, Venkatraman G, Spencer MJ, Calpain 3 participates in sarcomere remodeling by acting upstream of the ubiquitin-proteasome pathway. Hum. Mol. Genet 14, 2125–2134 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Ojima K, Kawabata Y, Nakao H, Nakao K, Doi N, Kitamura F, Ono Y, Hata S, Suzuki H, Kawahara H, Bogomolovas J, Witt C, Ottenheijm C, Labeit S, Granzier H, Toyama-Sorimachi N, Sorimachi M, Suzuki K, Maeda T, Abe K, Aiba A, Sorimachi H, Dynamic distribution of muscle-specific calpain in mice has a key role in physical-stress adaptation and is impaired in muscular dystrophy. J. Clin. Invest 120, 2672–2683 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartoli M, Roudaut C, Martin S, Fougerousse F, Suel L, Poupiot J, Gicquel E, Noulet F, Danos O, Richard I, Safety and efficacy of AAV-mediated calpain 3 gene transfer in a mouse model of limb-girdle muscular dystrophy type 2A. Mol. Ther 13, 250–259 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Roudaut C, Le Roy F, Suel L, Poupiot J, Charton K, Bartoli M, Richard I, Restriction of calpain3 expression to the skeletal muscle prevents cardiac toxicity and corrects pathology in a murine model of limb-girdle muscular dystrophy. Circulation 128, 1094–1104 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP, Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Mellgren RL, Miyake K, Kramerova I, Spencer MJ, Bourg N, Bartoli M, Richard I, Greer PA, McNeil PL, Calcium-dependent plasma membrane repair requires m- or mu-calpain, but not calpain-3, the proteasome, or caspases. Biochim. Biophys. Acta 1793, 1886–1893 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laure L, Suel L, Roudaut C, Bourg N, Ouali A, Bartoli M, Richard I, Daniele N, Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling. FEBS J 276, 669–684 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Lostal W, Bartoli M, Bourg N, Roudaut C, Bentaib A, Miyake K, Guerchet N, Fougerousse F, McNeil P, Richard I, Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum. Mol. Genet 19, 1897–1907 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Millay DP, O’Rourke JR, Sutherland LB, Bezprozvannaya S, Shelton JM, Bassel-Duby R, Olson EN, Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499, 301–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi C, Ono Y, Doi N, Kitamura F, Tagami M, Mineki R, Arai T, Taguchi H, Yanagida M, Hirner S, Labeit D, Labeit S, Sorimachi H, Multiple molecular interactions implicate the connectin/titin N2A region as a modulating scaffold for p94/calpain 3 activity in skeletal muscle. J. Biol. Chem 283, 14801–14814 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Ozcelik C, Saar K, Hubner N, Gotthardt M, RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med 18, 766–773 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Methawasin M, Hutchinson KR, Lee EJ, Smith JE III, Saripalli C, Hidalgo CG, Ottenheijm CA, Granzier H, Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 129, 1924–1936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Guo W, Dewey CN, Greaser ML, Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res 41, 2659–2672 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charton K, Suel L, Henriques SF, Moussu JP, Bovolenta M, Taillepierre M, Becker C, Lipson K, Richard I, Exploiting the CRISPR/Cas9 system to study alternative splicing in vivo: Application to titin. Hum. Mol. Genet 25, 4518–4532 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Charton K, Sarparanta J, Vihola A, Milic A, Jonson PH, Suel L, Luque H, Boumela I, Richard I, Udd B, CAPN3-mediated processing of C-terminal titin replaced by pathological cleavage in titinopathy. Hum. Mol. Genet 24, 3718–3731 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE, Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR, Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 90, 729–738 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Han R, Rader EP, Levy JR, Bansal D, Campbell KP, Dystrophin deficiency exacerbates skeletal muscle pathology in dysferlin-null mice. Skelet. Muscle 1, 35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE, Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther 16, 1073–1080 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Murrey DA, Naughton BJ, Duncan FJ, Meadows AS, Ware TA, Campbell KJ, Bremer WG, Walker CM, Goodchild L, Bolon B, La Perle K, Flanigan KM, McBride KL, McCarty DM, Fu H, Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: Toxicology, biodistribution, and immunological assessments in primates. Hum. Gene. Ther. Clin. Dev 25, 72–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, Zolotukhin I, Tarantal AF, Byrne BJ, Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res 99, e3–e9 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, Wilson JM, Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther 29, 285–298 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S, Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ. Res 86, 1114–1121 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Ojima K, Ono Y, Doi N, Yoshioka K, Kawabata Y, Labeit S, Sorimachi H, Myogenic stage, sarcomere length, and protease activity modulate localization of muscle-specific calpain. J. Biol. Chem 282, 14493–14504 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Ayuso E, Blouin V, Lock M, McGorray S, Leon X, Alvira MR, Auricchio A, Bucher S, Chtarto A, Clark KR, Darmon C, Doria M, Fountain W, Gao G, Gao K, Giacca M, Kleinschmidt J, Leuchs B, Melas C, Mizukami H, Muller M, Noordman Y, Bockstael O, Ozawa K, Pythoud C, Sumaroka M, Surosky R, Tenenbaum L, van der Linden I, Weins B, Wright JF, Zhang X, Zentilin L, Bosch F, Snyder RO, Moullier P, Manufacturing and characterization of a recombinant adeno-associated virus type 8 reference standard material. Hum. Gene Ther 25, 977–987 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohr UP, Wulf MA, Stahn S, Steidl U, Haas R, Kronenwett R, Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time PCR. J. Virol. Methods 106, 81–88 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Dobin A, Gingeras TR, Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics 51, 11, 14.1–11.14.19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C, Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther 21, 704–712 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Supplementary data for the dKO experiments.
Fig. S2. Supplementary data for the primate experiments.
Fig. S3. Proposed model of calpain 3 activation.
Table S1. Screening of primates for AAV9 seronegativity.
Table S2. Antibody response against the AAV9 serotype on sera (preimmune and sacrifice).
Table S3. Blood parameters of primates.
Table S4. Summary of echocardiogram analysis before and after systemic injection in primates.
Data file S1. Individual subject-level data.
References (40-43)