

Abstract
Introduction:
Acquired cancer therapy resistance evolves under selection pressure of immune surveillance and favors mechanisms that promote drug resistance through cell survival and immune evasion. AXL receptor tyrosine kinase is a mediator of cancer cell phenotypic plasticity and suppression of tumor immunity, and AXL expression is associated with drug resistance and diminished long-term survival in a wide range of malignancies, including NSCLC.
Methods:
We aimed to investigate the mechanisms underlying AXL-mediated acquired resistance to first- and third-generation small molecule EGFR tyrosine kinase inhibitors (EGFRi) in NSCLC.
Results:
We found that EGFRi resistance was mediated by up-regulation of AXL, and targeting AXL reduced reactivation of the MAPK pathway and blocked onset of acquired resistance to long-term EGFRi treatment in vivo. AXL-expressing EGFRi-resistant cells revealed phenotypic and cell signaling heterogeneity incompatible with a simple bypass signaling mechanism, and were characterized by an increased autophagic flux. AXL kinase inhibition by the small molecule inhibitor bemcentinib or siRNA mediated AXL gene silencing was reported to inhibit the autophagic flux in vitro, bemcentinib treatment blocked clonogenicity and induced immunogenic cell death in drug-resistant NSCLC in vitro, and abrogated the transcription of autophagy-associated genes in vivo. Furthermore, we found a positive correlation between AXL expression and autophagy-associated gene signatures in a large cohort of human NSCLC (n = 1018).
Conclusion:
Our results indicate that AXL signaling supports a drug-resistant persister cell phenotype through a novel autophagy-dependent mechanism and reveals a unique immunogenic effect of AXL inhibition on drug-resistant NSCLC cells.
Keywords: NSCLC, AXL receptor tyrosine kinase, Acquired EGFR TKI resistance, Phenotypic plasticity, Tumor immune microenvironment, Autophagic flux
Introduction
Acquired therapy resistance to oncogene-targeted cancer therapeutics invariably diminishes long-term clinical outcome, and the lack of efficacy of immune-based therapy in oncogene-driven tumors limits treatment options for patients with recurrent tumors. Molecular analysis reveals that relapsing tumors utilize both genetic and nongenetic resistance mechanisms, including additional gene mutations that could abrogate or bypass drug-target interactions and affect epigenetic reprogramming mechanisms that would alter the expression of various drug targets involved in cell survival.1 In lung adenocarcinomas with activating EGFR mutations, secondary mutations in the EGFR tyrosine kinase domain (T790M) are detected in 50% to 60% of tumors displaying acquired resistance to first-generation EGFR tyrosine kinase inhibitors (TKIs).2–5 In addition, bypass signaling through MET gene amplification or epistatic gene activating mutations in downstream signaling proteins (e.g., MAPK/PI3K) can also restore the oncogenic driver signaling, and these two phenomena are considered to be the main acquired resistance mechanisms to EGFR TKIs. Nonmutational acquired resistance through induced cellular phenotypic plasticity, including epithelial-to-mesenchymal transition (EMT) and small cell trans-differentiation, represents an alternative mode of drug resistance that relies on epigenetically regulated phenotypic transition to an EGFR-independent cellular state.6 Enhanced stem cell–like features allowing adaptation to dynamic tumor microenvironments frequently accompany this cellular transition. However, the early events leading to acquired resistance are less thoroughly understood, and an outstanding question remains as to whether an epigenetically regulated drug tolerant state, triggered by prolonged drug exposure, precedes the emergence of permanent drug resistance through the manifestation of genetic resistance mechanisms.7 This drug tolerant state is suggested to entail drug-induced epigenetic and transcriptional reprogramming mechanisms.8 A post-treatment tumor microenvironment that selects for a drug-resistant phenotype comprises cell debris from dying tumor cells and pro-inflammatory mediators derived from macrophages.9 The importance of reversible transcriptional reprogramming in the development of acquired drug resistance was recently reported by Shaffer and colleagues,10 who reported that rare melanoma cells in culture are transiently poised to undergo drug-induced epigenetic reprogramming and thus may represent the source of subsequent drug-resistant colonies.10 Of note, these cells were characterized by high levels of AXL and other genes with known associations to drug resistance and are further referred to as “AXL jackpot cells.”10 Thus, accumulating evidence indicates that AXL signaling may uniquely contribute to an early pro-survival state that seems to be a prerequisite for the subsequent development of acquired resistance. Correspondingly, elevated expression of AXL has been reported in a wide range of cancers, including NSCLCs, and has been associated with aggressive clinical behavior and drug resistance.11–15
Autophagy, a conserved cellular process by which cytoplasmic vacuoles are shuttled to lysosomal compartments for bulk degradation, is increasingly recognized as a pro-survival mechanism for cells in response to intrinsic and extrinsic stress, and autophagy is frequently exploited by neoplastic cells during cancer progression.16 EMT has been associated with increased autophagy and tumor progression and may allow cancer cells to overcome microenvironmental stress, and also to escape immune surveillance by cytotoxic T-lymphocytes.17–20 We have recently shown that EMT, and in particular expression of AXL, in NSCLC cells is correlated with increased cancer cell–intrinsic resistance to both natural killer (NK)- and cytotoxic T-lymphocyte (CTL)–mediated killing.21 We hypothesized that a small molecule drug targeting AXL could sensitize mesenchymal lung cancer cells to cytotoxic lymphocyte-mediated killing, and we subsequently reported that targeting AXL overcomes NSCLC resistance to NK- and CTL–mediated cytotoxicity.21
The antitumor efficacy of widely used cancer treatment strategies, including common chemotherapies, radiation, and more selective targeted approaches has been attributed, in part, to the induction of immunogenic cell death (ICD).22 ICD inducer screening supports the contention that Food and Drug Administration–approved anticancer agents are more likely to promote ICD than approved agents from remaining non-oncology pharmacologic specialties.23 Cancer cells undergoing ICD elicit immunostimulatory capacity owing to the spatio-temporally defined release of potent danger signals, referred to as danger- or damage-associated molecular patterns (DAMPs), also known as alarmins.22 Of note, three DAMPs, in particular, have been reported to play a crucial role in the immunogenic potential of nearly all ICD inducers identified thus far22: (1) the translocation of calreticulin (CALR) from the endoplasmic reticulum to the outer leaflet of the plasma membrane, (2) premortem autophagy-dependent release of the “find me” signal adenosine triphosphate (ATP), and (3) the passive release of the high-mobility group box 1 (HMGB1) nuclear protein. When successfully induced, ICD could provide the adjuvant immunostimulatory signals necessary for long-term protection against recurrence and metastasis of carcinoma cells caused by the activation of protective immunity. Premortem stress-related autophagy can drive an immunogenic form of cell death, indicating that an increased autophagic flux can be exploited to bridge the innate and the adaptive immune system, and may be a critical determinant of immune-related therapy-induced cancer cell death.24 A high premortem autophagic flux is a prerequisite for the accumulation of ATP, one of the alarmins critical for induction of ICD.25
AXL signaling mediates acquired resistance to EGFR tyrosine kinase inhibition by driving a pro-survival pathway in NSCLC cells. In the current study, we report that targeting AXL with the small molecule inhibitor bemcentinib delayed the onset of resistance to EGFR inhibition and affected the transcription of autophagy-associated gene signatures in vivo, whereas in vitro the apoptosis resistance of NSCLC cells that were resistant to the EGFR TKI erlotinib was attributed to their high autophagic flux. Furthermore, in samples from a large human cohort of NSCLC (n = 1018) we found a positive correlation between AXL expression and autophagy-associated gene signatures, which supports the clinical relevance of our findings. We hypothesized that the high premortem autophagic flux of the AXL-expressing cells could be exploited in a therapeutic setting for EGFRi–resistant NSCLC tumors because the release of the alarmin ATP from dying autophagic cells would be expected to kick-start the cancer immunity cycle. Here, we report for the first time that targeted inhibition of AXL signaling by bemcentinib abrogates this high premortem autophagic flux in NSCLC cells resistant to first- and third-generation EGFR inhibitors and thus enhances tumor immunogenicity through the release of DAMPs while undergoing an immunogenic form of cell death. Thus, our data reveal a novel link between AXL, autophagy, and tumor immunogenicity.
Materials and Methods
Drugs
Erlotinib hydrochloride salt (E-4007, LC Laboratories, Woburn, Massachusetts), bemcentinib (BGB324/R428, BerGenBio ASA, Bergen, Norway), rociletinib (CO-1686, Clovis Oncology, Boulder, CO), staurosporine (ab120056, Abcam, Cambridge, United Kingdom), bafilomycin A (B1793, Sigma-Aldrich, St. Louis, Missouri), and rapamycin (R8781, Sigma-Aldrich) were prepared in DMSO for in vitro studies. Erlotinib and bemcentinib were prepared in 0.5% (w/w) hydroxypropyl methyl-cellulose/0.1% (w/w) Tween-80 for in vivo administration. Chloroquine diphosphate (ab142116, Abcam) was dissolved in water.
Cell Culture
The human NSCLC epithelial cell line HCC827 (CRL-2868, ATCC, Manassas, Virginia) has an activating EGFR mutation (E746–A750 deletion) that engenders sensitivity to erlotinib. HCC827 cells were maintained in RPMI-1640 medium (R8758, Sigma-Aldrich) containing 5% heat-inactivated fetal bovine serum (FBS) (Gibco), 20 U/mL penicillin, 20 μg/mL streptomycin (penicillin-streptomycin, P-0781, Sigma-Aldrich) and 2 mM L-glutamine (G-0781, Sigma-Aldrich) at 37°C, 5% CO2, 5% O2, and 95% relative humidity in a multigas incubator (MCO-5M-PE, Panasonic, Osaka Prefecture, Japan). Erlotinib-resistant clones (ER3 and ER10) derived from HCC827 parental cells were established in vitro by culture in increasing concentrations of erlotinib, as described previously.14 The ER3 cells were established in Professor Trever G. Bivona’s laboratory (UCSF, California), whereas the ER10 cells were established in Professor Henrik J. Ditzel’s laboratory (University of Southern Denmark, Odense, Denmark). Erlotinib-resistant cells were maintained in culture in the presence of 1 μM erlotinib. The human NSCLC cell line H1975 and two rociletinib (CO-1686)-resistant (COR) cell clones (COR1–1 and COR10–1) were provided by Clovis Oncology.26 H1975 cells were cultured in the same way as HCC827 cells and rociletinib-resistant clones were maintained in culture with 1 μM rociletinib. Cell lines were routinely tested for Mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (LT07–218, Lonza, Basel, Switzerland). All cell lines used in these studies were authenticated by short tandem repeat (STR) profiling using the LGC service Promega’s PowerPlex 18D System. The AACR STR database (135-XV-5, ATCC) was applied as a reference for authentication of STR profiles.
Animal Studies
Animal experiments were approved by the Institutional Animal Care Research Authority and in accordance with the European convention for the protection of vertebrates used for scientific purposes. Animals were housed in a germ-free environment in filter top cages. Environmental parameters were monitored by the Laboratory Animal Facility at the University of Bergen following institutional standard operating procedures. Animals were provided certified laboratory feed and sterile drinking water ad libitum. Athymic Nude-Foxn1nu mice 6 to 8 weeks old were obtained from Harlan Laboratories (Horst, The Netherlands). For xenograft studies, 8 million HCC827 cells (at least 95% viable as determined by Tryphan Blue exclusion test) in 1:1 nonsupplemented RPMI1640 medium and Matrigel (Matrigel Basement Membrane Matrix Growth Factor Reduced, BD Biosciences, San Jose, CA) were implanted subcutaneously into the right and left flank of female nude mice. Randomization was conducted using the Latin square method on the basis of total tumor volume of each animal. Randomization of animals was performed so that the average tumor volume for all treatment groups (eight animals per group) was approximately 110 mm3. Bemcentinib was administered twice a day at a concentration of either 50 mg/kg or 100 mg/kg by oral gavage. Erlotinib was administered once a day at 50 mg/kg by oral gavage. Tumor growth was measured two to three times weekly with digital hand-held calipers, and tumor volumes were calculated using the formula (L × W × W)/2. For the 50 mg/kg bemcentinib + 50 mg/kg erlotinib group, one tumor was identified by the Grubbs test (description under Methods section of Statistics) as a statistically significant outlier (p < 0.01). This tumor was eliminated from further analysis. Clinical observations of animal appearance and body weight measurements were recorded daily, and tumor measurements were performed two to three times weekly. In general, a decrease in body weight greater than 20% would indicate toxicity and should lead to euthanization of mice. None of the treatment groups had reduction in body weight which would be indicative of toxicity. At study termination, the animals were anesthetized by sevoflurane and euthanized by cervical dislocation following institutional standard operating procedures.
Gene Expression Analysis
On the day of study termination (after 25 d of treatment), animals were euthanized, and harvested tumors were snap-frozen in liquid nitrogen and stored in a nitrogen tank. Total RNA from HCC827 tumors were isolated with RNeasy Mini columns (Qiagen, Hilden, Germany) and RNA integrity was evaluated using a Bio-analyzer instrument (Agilent Technologies, Santa Clara, CA). Gene expression arrays were performed at the genomics core facility at the University of Bergen. Gene expression levels were measured by hybridization to Illumina Human HT-12 v4 Expression BeadChip arrays and analyzed in GeneStudio and J-Express Pro (http://jexpress.bioinfo.no). Four samples were excluded on the basis of poor quality control data (one vehicle, two erlotinib-treated, one combination-treated). Significance analysis of microarray data (J-Express Pro) was used to generate a list of up- and down-regulated genes in erlotinib-treated versus combination-treated tumors with associated fold change and significance calculations. Significance analysis of microarray whole-ranked lists were used for gene ontology analyses by employing GOrilla software (http://cbl-gorilla.cs.technion.ac.il). A volcano plot was generated to display changes in gene expression versus significance (log2[fold change] versus -log10[p value]) between HCC827 tumors treated for 25 days either with erlotinib alone (50 mg/kg, once a day) or a combination of erlotinib (50 mg/kg, once a day) and bemcentinib (50 mg/kg, twice a day), n = 4. KM-plotter was used to assess the effect of sprouty genes (SPRY1, SPRY2, and SPRY4) on NSCLC survival (http://kmplot.com/analysis/).
Cell Viability Assays
For 96-well plate viability assays, 1000 cells per well were seeded and allowed to attach overnight. The next day, treatment was added in fresh cell medium containing varying concentrations of erlotinib (5083S, Cell Signaling Technology) or rociletinib (Clovis Oncology) alone, or in combination with bemcentinib (BerGenBio). After 5 days of treatment, the cell viability was measured by incubating with resazurin (25 μg/mL, R-7017, Sigma-Aldrich) for 3 hours at 37°C, 5% O2, 5% CO2, and fluorescence was measured for 1 second at 560Ex/590Em in a 1420 Multilabel Counter VICTOR3 (Perkin Elmer, Waltham, Massachusetts) using the Wallac 1420 software. Background fluorescent signal was removed by subtracting the mean value of fluorescence from blank wells containing only cell culture medium and resazurin, and the cell viability was normalized against vehicle (DMSO) control.
Cell viability for the assessment of drug synergism was calculated using the Bliss independence model.27 Delta Bliss values were calculated as expected cell death (fold change) from the combined therapy minus observed cell death (fold change) at the same concentrations. The Bliss sum is then calculated as the sum of all delta Bliss values for all the different drug combinations in one experiment. Negative delta Bliss values indicate synergy; positive delta Bliss values indicate that the drugs are working as antagonists, whereas delta Bliss values close to zero indicate that the drugs are acting independently. Cell death was calculated as (one-cell viability [fold change]), and a mean value of six parallels was used for further analysis. Assuming that the drug response from erlotinib and bemcentinib are mutually independent, the expected cell death from treatment with a combination of erlotinib and bemcentinib at concentration i and was calculated using the formula:; where is the observation cell death for erlotinib treatment at concentration i and is the observed cell death at bemcentinib concentration j, and i and j are all drug concentrations used in the experiment.27
Clonogenic Assay
Clonogenic assays were performed according to a protocol adapted from Franken et al.28 Briefly, cells were detached and counted with a Nucleocounter NC-100 (Chemometec, Allerod, Denmark) and the number of viable cells per mL was calculated. Subsequently, 1000 viable single cells were plated per well in six-well cell culture plates. Cells were allowed to attach for 16 hours before initiation of treatment and cells in three replicate wells were treated per condition. The cells were treated as indicated for 14 days before fixation and staining by incubation in a mixture of 25% methanol and 0.01% crystal violet for 30 minutes at room temperature. Crystal violet binds to DNA in the nuclei of mammalian cells, staining them a deep purple and helping to visualize colonies. On removal of the fix and stain solution, the cells were rinsed carefully and immersed in running tap water. The colonies were allowed to dry in normal air at room temperature overnight before imaging by an EPSON Perfection V850 Pro scanner (Epson, Suwa, Japan). Clonogenic assays were quantified using the “ColonyArea” plugin29 for ImageJ.30 Each experiment was independently repeated three times and the results revealed the mean colony area (%) ± SD from a representative experiment.
Caspase 3/7 Luminescence Assay
For determination of induced apoptosis in treated cells, Caspase-Glo 3/7 luminescence-based assay (G8092, Promega, Madison, Wisconsin) was used according to the manufacturer’s instructions. Briefly, 5000 cells per well were seeded in a 96-well plate and incubated overnight before treatment, as indicated, in 100 μL culture medium. For the assay, 50 μL cell medium was removed and 50 μL Caspase-Glo reagent was added per replicate. The content was mixed by gently shaking the plate for 30 seconds, and the plate was incubated at room temperature for 1 hour before measuring luminescence for 0.1 second in a Perkin Elmer 1420 Multilabel Counter VICTOR3 (Perkin Elmer) using the Wallac1420 software. Two-way analysis of variance (ANOVA) was followed by Dunnett’s t test comparing the treated cells against the vehicle control for each cell line.
Western Blotting
Tumor material harvested after 51 days of treatment was lysed in radioimmunoprecipitation assay (RIPA) buffer (phosphate buffered saline [PBS] with 1% [vol/vol] NP-40, 0.5% [wt/vol] sodium deoxycholate, and 0.1% [wt/vol] sodium dodecyl sulfate [SDS]) supplemented with protease and phosphatase inhibitor tablets (Roche, Basel, Switzerland) and 0.2 mM phenylmethylsulfonyl fluoride. In vitro cell cultures were treated as indicated and washed with ice-cold PBS, scraped in PBS on ice, collected by centrifugation, and lysed in RIPA buffer (sc-24948A, Santa Cruz Biotechnology, Dallas, Texas). Protein concentration was measured using a Pierce BCA Protein Assay Kit in accordance with manufacturer’s instructions (23225/23227, Thermo Fisher Scientific, Waltham, Massachusetts). Lysates were dissolved in water with BIO-RADxT sample buffer (1610791, Bio-Rad, Hercules, California) and incubated for 5 minutes at 90°C. The samples were then centrifuged and 10 to 20 μg protein loaded per well on 4% to 20% Mini-PROTEAN TGX Stain-Free Gels (4561096, Bio-Rad). The proteins were separated by electrophoresis at 90 V for 10 minutes, followed by 130 V for 1 hour. The stain-free gels were then activated by 2.5 minute-exposure to ultraviolet light (ChemiDoc XRS+, Bio-Rad) to allow total protein quantification. Semidry blotting to a transblot turbo mini-size low fluorescence poly-vinylidene fluoride membrane (1704156, Bio-Rad) was performed using the Bio-Rad Trans-Blot system (2.5 A, 25 V, 7 min). Tris-glycine-SDS buffer (1610772, Bio-Rad) was used for the transfer. Images of the total protein amount of the membrane were obtained directly after the transfer and these images were later used for normalization against total protein and further quantifications of Western blots as described for stain-free gels by the manufacturer (ChemiDoc XRS+, Bio-Rad). Membranes were blocked with 5% bovine serum albumin (BSA) (A2058, Sigma-Aldrich) or 5% nonfat dry milk (170–6404, Bio-Rad) in tris-buffered saline with 0.1% polysorbate 20, then incubated with primary antibody overnight at 4°C (see Supplementary Table 2A). Membranes were washed three times for 5 minutes with tris-buffered saline and 0.1% polysorbate 20, and incubated for 1 hour at room temperature with one-to-10,000 ratio of goat anti-mouse horseradish peroxidase labeled (170–6516, Bio-Rad) or goat anti-rabbit horseradish peroxidase labeled (170–6515, Bio-Rad) secondary antibodies. Chemiluminescent substrate was added (Super Signal West Femto Maximum Sensitivity Substrate, 34095, Thermo Fisher Scientific) and chemiluminescence was measured by a Molecular Imager ChemiDoc XRS+ (Bio-Rad). ImageLab v5.0 (Bio-Rad) software was used for analysis and quantification of the results. Quantification of Western blot results are presented as chemiluminescent signal normalized against total protein, as described by Gürtler et al.31 Although not used for quantification per se, a housekeeping protein (actin, β-actin, or glyceraldehyde 3-phosphate dehydrogenase) are shown as loading controls in all figures to improve instant interpretability for readers.
Mass Cytometry
Antibody Panel.
An antibody panel was designed to include antibodies against some of the major signaling proteins downstream of the AXL-EGFR pathways. Available data on heavy metal signal overlap were used to minimize spillover using the Fluidigm MaxPar Panel Designer. All antibodies were previously verified and described by Pelissier Vatter et al.,32 or commercially available and verified by Fluidigm. Phospho-antibodies were titrated using lambda-phosphatase–treated cells as a negative control (data not shown). An overview of the antibodies and antibody concentrations is given in Supplementary Table 2B.
Sample Preparation and Data Acquisition.
Cells were detached using 2 mL TrypLE Express Enzyme (12604021, ThermoFisher Scientific) per T75 flask. Cells were exposed to TrypLE at 37°C until the cells had completely detached (approximately 5 minutes), and then TrypLE was inactivated by adding 8 mL complete cell medium. Approximately, 2 million cells per sample were resuspended in 1 mL Cell-ID Cisplatin 194-Pt (1:2000 dilution in PBS, stock concentration; 1 mM, 201194, Fluidigm, South San Fransisco, CA) and incubated for 1 minute at room temperature; 9 mL serum-free cell medium was then added, and the cells were collected by centrifugation. Cells were resuspended in 1 mL 1.6% paraformaldehyde (PFA) (15710, ElectronMicroscopySciences) in serum-free medium, and incubated for 10 minutes at room temperature. The cells were then spun down and the samples was stored at −80°C until use (< 2 mo). Each sample was washed with 200 μL Maxpar Barcode Perm buffer (201057, Fluidigm), collected by centrifugation and then resuspended in 195 μL Maxpar Barcode Perm buffer. Five microliters of barcoding solution (Cell-ID 20-Plex Pd Barcoding kit, 201060, Fluidigm) was added and the cells were incubated in this solution for 30 minutes at room temperature. Then, the samples were washed two times with 200 μL Maxpar Barcode Perm buffer, followed by one wash with 1 mL cell staining buffer (CSB) (201068, Fluidigm) and one wash with 1 mL 1% BSA (A9647, Sigma-Aldrich) containing 1× DNAse (DN25, Sigma-Aldrich) in Dulbecco’s PBS (14040–153, ThermoFisher Scientific). Samples were pooled into one sample tube and permeabilized with 1 mL ice-cold methanol for 20 minutes at −20°C. The cells were then washed two times with 1 mL CSB and stained with antibody mixture (Supplementary Table 2B) dissolved in 300 μL CSB for 1 hour at room temperature. The sample was then washed three times in 1 mL CSB, one time in 1 mL PBS (201058, Fluidigm), and then incubated with 1 mL 1.6% PFA in PBS with 0.3 μL Cell-ID Intercalator Iridium (201192B, Fluidigm) overnight at 4°C. The next day, the sample was washed once with PBS, incubated 10 minutes at room temperature in 1 mL 1% BSA containing 1× DNAse in Dulbecco’s phosphate buffered saline, washed twice with 1 mL PBS, twice with 1 mL MilliQ water, and dissolved in MilliQ water with EQ Four Element Calibration Beads (201078, Fluidigm) at a concentration of approximately 1 million cells per mL. The sample was then analyzed using a Helios mass cytometer (Fluidigm).
Data Analysis.
Data was normalized and debarcoded using the CyTOF software (Fluidigm). For the debar-coding, the minimum barcode separation was set to 0.14 and the maximum Mahalanobis distance was set to three; the rest of the settings were kept as default. Cells were gated as iridium positive events, and cisplatin staining was used to discriminate dead cells. Iridium plotted against event length was used to gate ion clouds from single cells. After gating, signal from approximately 100,000 viable and single cells per sample were included in the histograms (Supplementary Fig. 4). ViSNE, a visualization tool for high-dimensional single-cell data, which is based on the t-Distributed Stochastic Neighbor Embedding (tSNE) algorithm,33 was then applied using the Cytobank software with the following settings: downsampling; equal sampling; 10,000 cells per sample, 5000 iterations, perplexity: 30, theta: 0.5, seed: random (1730472643), channels used in tSNE: E-cadherin (CDH1), vimentin (VIM), CD44, EGFR, AXL, and programmed death-ligand 1.
Immunofluorescence and Confocal Imaging
Immunocytochemistry for the Characterization of EMT Markers.
For immunocytochemistry, 20,000 cells per well was plated on crystal-clear German glass coverslips with Poly-D-lysine coating (Neuvito Corporation, Vancouver, Washington) in a 24-well cell culture plate and allowed to attach overnight. Cells were washed in PBS and fixed with 3.7% formalin for 15 minutes at room temperature. The cells were then washed again with phosphate buffered saline containing 0.1% Tween-20 (PBS-T), permeabilized with 0.3% Triton X-100 for 20 minutes, and blocked with 5% goat serum (G9023, Sigma-Aldrich) for 1 hour at room temperature. Incubation with primary antibody against E-cadherin (CDH1, 14472S, Cell Signaling Technology, 1:100) and vimentin (VIM, ab92547, Abcam, 1:100) overnight at 4°C was done, followed by washing and 1 hour of incubation with secondary antibodies (goat anti-mouse AF488, 1:200, A11029, Invitrogen and goat anti-rabbit AF546, 1:200, A11035, Invitrogen, Carlsbad, California) at room temperature. Cells were washed again and mounted with ProLong Diamond Antifade Mountant with 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain (P36962, Molecular Probes, Eugene, Oregon). Images were obtained on a Leica TCS SP5 confocal microscope (Leica, Wetzlar, Germany) using a 63× objective lens magnification (HCX PL Apo Cs, oil, NA = 1.4, WD = 0.1 mm).
Immunocytochemistry for the Co-Localization of LC3 Puncta and LysoTracker.
Cells were treated with various drug combinations as indicated for 7 days, whereas 50 μM chloroquine was added for the final 16 hours. Live cells were stained with 50 nM LysoTracker Red DND-99 (LY528, Invitrogen) in prewarmed medium for 15 minutes at 37°C. Cells were plated, fixed, permeabilized, and blocked using the same method as the E-cadherin (CDH1) and vimentin (VIM) staining. Cells were then incubated in primary Ab LC3 A/B (DU4C) (12741, Cell Signaling Technology) at one-to-100 ratio overnight at 4°C, carefully rinsed three times with PBS-T, and incubated with secondary antibody Alexa Fluor-488 labeled goat anti-rabbit (A11008, Invitrogen, 1:200) for 1 hour at room temperature; it was then subsequently washed four times with PBS-T before mounting with ProLong Diamond Antifade Mountant with DAPI (P36962, Molecular Probes). This protocol was adapted from Kang et al.34 Images were obtained on a Leica TCS SP8 confocal microscope using 100× objective lens magnification (HC PL Apo STED white, oil, NA = 1.4, WD = 0.13 mm). The co-localization intensity spatial profile was obtained by drawing a one pixel-wide square across the images, and the same area was selected for the green and red channels for each image. The plot profile function in ImageJ was then used to find the intensity values that were plotted in GraphPad Prism.
Autophagic Flux Experiments by Tandem Sensor LC3 Constructs and Confocal Imaging
The Premo Autophagy Tandem Sensor RFP-GFP-LC3B Kit (P36239, Thermo Fisher Scientific) was used according to the manufacturer’s instructions. Ten thousand cells were seeded on Poly-D-lysine coated coverslips (12 mm, 1.5 thickness, crystal-clear German glass coverslips with Poly-D-lysine coating, Neuvito Corporation) in 24-well plates for 48 hours before incubation with 10 μL of BacMam reagent (insect baculovirus with a mammalian promoter containing tandem RFP-GFP-LC3B DNA) for 24 hours. Cells were then treated with DMSO (vehicle control), bemcentinib (0.8 μM), chloroquine (50 μM), or a combination of bemcentinib and chloroquine for 24 hours. Rapamycin (200 nM) was applied as an additional positive control. Cells were rinsed once with PBS and fixed with 4% PFA in PBS for 15 minutes at room temperature before mounting with ProLong Diamond Antifade Mountant with DAPI (P36962, Thermo Fisher Scientific). Images were obtained on a Leica TCS SP8 confocal microscope using 100× objective lens magnification (HC PL Apo STED white, oil, NA = 1.4, WD = 0.13 mm).
Lentiviral Expression Plasmids
Lentiviral expression plasmids encoding mCherry-EGFP-LC3B were produced in the pLVX-Tight-Puro backbone (Clontech Laboratories, Mountain View, CA) using a Gibson Assembly of the individual PCR products. Triple transfection of HEK293 packaging cells was performed with either the p-LVX-Tight-Puro-mCherry-EGFP-LC3B expression plasmid or with pLVX-Tet-On Advanced transactivator expression plasmid, both accompanied by the pMD2.G packaging plasmid and pVSV-G envelope plasmid (Tronolab), as described previously.35 Target cells were subsequently simultaneously transduced with transactivator and mCherry-EGFP-LC3B encoding viral particles in the presence of 4 μg/mL protamine sulfate (P4020, Sigma-Aldrich). Transgene expression was induced by doxycycline (1 μg/mL, 631311, Clontech), and cell populations were sorted twice by fluorescence-activated cell sorting (FACS) (Sony SH800) to obtain a cell population containing a high percentage of stably transduced cells with medium to high transgene expression. Stably transduced cells were subsequently used for the live cell confocal imaging described below.
Live Cell Confocal Imaging
We seeded 10,000 cells per well in four-well plates (1μ-Slide four-well IbidiTreat, Ibidi GmbH, Martinsried, Germany) and allowed them to attach overnight before treatment with DMSO (vehicle control), bemcentinib (0.8 μM), or chloroquine (50 μM). All treatments were done in the presence of doxycycline (1 μg/mL, 631311, Clontech) to induce transgene expression. After 24 hours of treatment, cell culture medium was exchanged with phenol red–free cell culture medium (D5030, Sigma-Aldrich) supplemented with 5% FBS, 1 μg/mL doxycycline, and Draq5 (62254, Thermo Fisher Scientific 1:1000), and the indicated concentrations of vehicle (DMSO), bemcentinib, or chloroquine. Live cell imaging was performed with a Leica TCS SP8 confocal microscope equipped with a TokaiHit (Shizuoka, Japan) tabletop incubator and CO2 control using 40× objective lens magnification (HC PL Apo motCORR CS2, water, NA = 1.1, WD = 0.65 mm). Images were obtained every 10 minutes with the following settings: sequence 1: 488 nm laser, PMT detector (495–556 nm), gain 833 and 640 laser, HyD detector (650–750 nm); sequence 2: 561 laser, HyD detector (570–625nm). Autofocus control was used to hold current position, and a line averaging of two was applied for all images.
Long-Lived Protein Degradation Assay by Flow Cytometry
For the quantification of autophagic long-lived protein degradation, we performed a flow cytometry–based protocol developed by Zhang et al.,36 with minor modifications. In principle, L-azidohomoalanine (AHA) was used as a surrogate for the essential amino acid L-methionine. A click reaction between the incorporated azide and fluorescently labeled alkyne renders long-lived proteins fluorescently labeled for FACS quantification. Cells were seeded in six-well plates at a density of 2.5 × 105 cells per well. The following day, the cells were washed with prewarmed PBS and cultured in L-methionine–free RPMI1640 (R7513, Sigma-Aldrich) for 30 minutes to deplete the methionine reserves. The cells were then pulsed with the Click-IT AHA (C10102, Sigma-Aldrich) for 24 hours at a final concentration of 25 μM in L-methionine–free medium supplemented with 5% FBS and 2 mM L-glutamine (Sigma-Aldrich). After the pulse, the AHA-containing medium was removed and the cells were washed with 2 mL PBS with 2.5% FBS to remove unincorporated AHA, and cultured in regular complete RPMI1640 with 5% FBS and L-glutamine containing 10 times L-methionine (final concentration 2 mM) to prevent reincorporation of AHA surrogate for 2 hours to chase out the short-lived proteins. The cells were then pretreated (as indicated in the figure legend, Fig. 5D) for 16 hours in complete RPMI1640 with 5% FBS and L-glutamine (Sigma-Aldrich) containing 10 times L-methionine (Sigma-Aldrich). Then, the cells were washed with either 2 mL complete RPMI1640 with 5% FBS or Earle’s balanced salt solution (EBSS) with 0.1% BSA two times, and both media were supplemented with 10 times L-methionine. The cells were then incubated with the complete media or starvation media with various treatments for 3 hours. After treatment, cells were harvested with trypsin and fixed in 4% formaldehyde in PBS for 15 minutes at room temperature. After fixation, cells were washed twice with PBS containing 3% BSA and permeabilized with 0.5% Triton X-100 in PBS for 20 minutes at room temperature. Finally, the cells were washed in PBS and incubated in the click reaction mixture for 2 hours at room temperature. A final concentration of 5 μM AF-488–labeled alkyne (A10267, Invitrogen) in the copper-containing Click-IT reaction buffer (C10269, Invitrogen) was used. After tagging and washing, single-cell signal intensities were detected in the fluorescence intensity-1 channel, and the ratio of fluorescence intensity in treated cells to unlabeled control cells was calculated. Relative fluorescence intensity was calculated as follows: (%) = ([treated group MFI – unstained control MFI) / (vehicle control group MFI – unstained control MFI]) × 100. This represents the rate of degradation of long-lived proteins at given time points.
Figure 5.
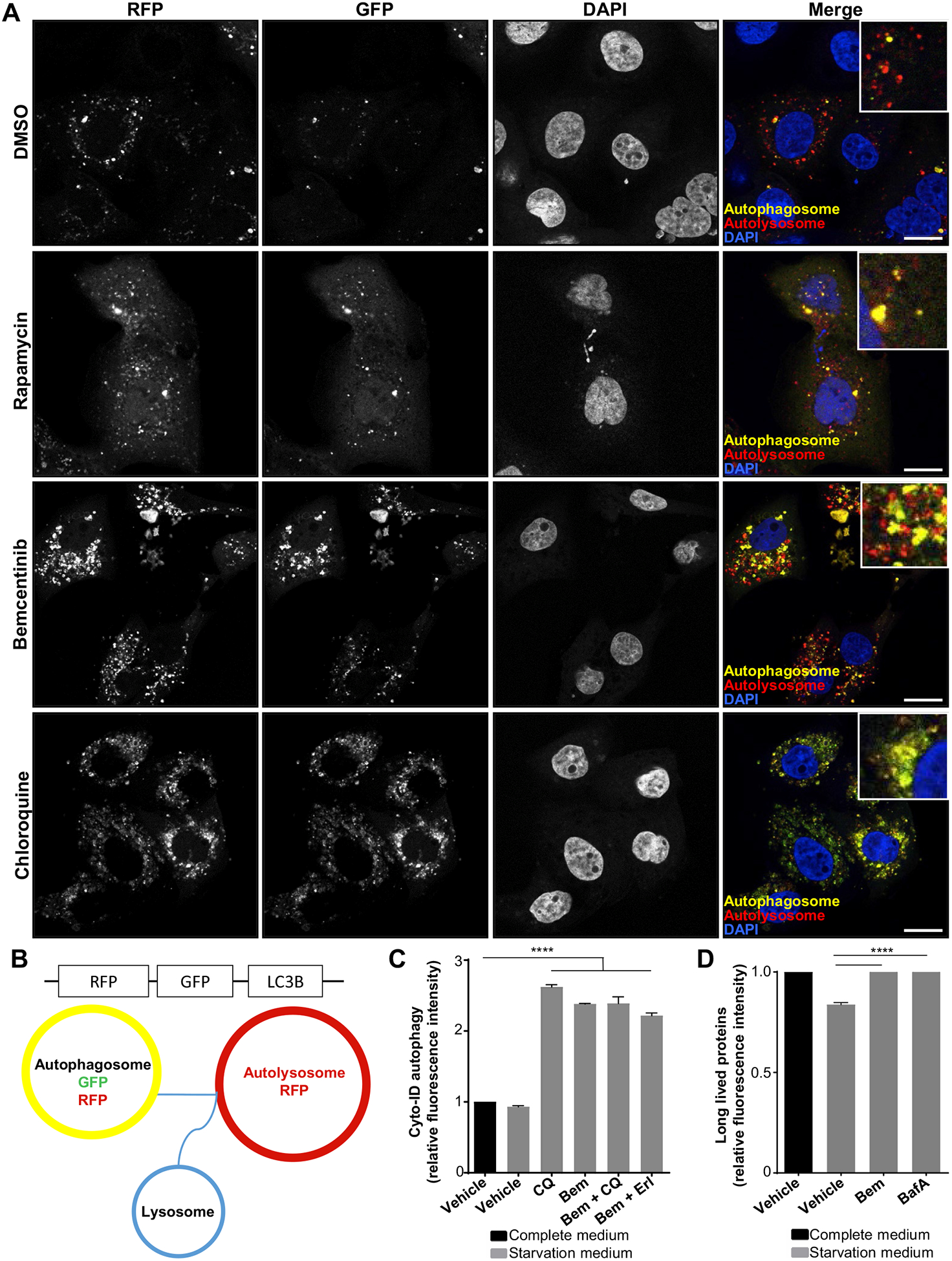
The specific AXL inhibitor bemcentinib inhibits the high premortem autophagic flux of the erlotinib-resistant cells. (A) Dynamics of autophagosome formation and subsequent fusion with lysosomes to form autolysosomes was assessed by Premo Autophagy Tandem Sensor RFP-GFP-LC3B experiment. ER10 cells transfected with RFP-GFP-LC3B tandem sensor were treated as indicated for 24 hours with vehicle (DMSO), rapamycin (200 nM), bemcentinib (0.8 μM), or chloroquine (50 μM). Representative confocal images are shown. Scale bar: 10 μm. (B) Schematics for the assay described above. The LC3-associated autophagosomes seem yellow because of the simultaneous expression of both RFP and EGFP under neutral pH, (B) as on the fusion of the LC3-associated vesicles with the lysosome, the resultant autolysosomes express RFP only because of the loss of fluorescence from the acid–sensitive EGFP fluorochrome under acidic conditions. (C) Autophagic flux was assessed in ER3 cells by measuring the balance of autophagosome formation and clearance by pretreating the cells 16 hours (Vehicle [DMSO], 50 μM chloroquine [CQ], 1.2 μM bemcentinib, 10 μM erlotinib). Autophagy was induced through starvation in EBSS with 0.1% BSA for 3 hours (in the presence of treatment). Relative green fluorescence intensity from the Cyto-ID probe compared with the vehicle-treated cells in complete cell culture medium is given for a representative experiment ± SD (n = 3). Unpaired t test (****p < 0.0001). (D) Flow cytometry Click-IT AHA chemistry–based long-lived protein degradation assay of ER3 cells pretreated as indicated for 16 hours with bemcentinib 1.2 μM or Bafilomycin A 50 nM, before starvation in EBSS with 0.1% BSA for 3 hours (in the presence of treatment). The alkyne-AF488 fluorescence intensity ± SD (n = 3) was given for the starved samples relative to the vehicle-treated (DMSO) cells in complete RPMI-1640 cell culture medium. Unpaired t test (****p < 0.0001). BSA, bovine serum albumin; EBSS, Earle’s Balanced Salt Solution.
Detection of Autophagic Flux by CYTO-ID Flow Cytometry Assay
A quantitative assay for monitoring the autophagic activity at the single-cell level (CYTO-ID Autophagy detection kit, ENZ-51031, Enzo Life Sciences, Farmingdale, New York) was performed according to the manufacturer’s instructions in combination with autophagy induction and inhibition to measure the autophagosome generation and clearance by live cell flow cytometry by BD Accuri C6 flow cytometer (BD Biosciences). In this assay, following 16 hours pretreatment, starvation for 3 hours in EBSS supplemented with 0.1% BSA was used for autophagy induction, and chloroquine at a concentration of 50 μM for 16 hours treatment and a final concentration of 300 μM for 3 hours was used to inhibit the successful fusion between autophagosomes and lysosomes. Induction and inhibition of autophagy were performed in combination with the treatments, as indicated in the figure legend (Fig. 5C).
Gene Expression Analysis of Human Lung Adenocarcinoma (LUAD) and Lung Squamous Cell Carcinoma (LUSC) Samples
We investigated the association between AXL expression and autophagy score in The Cancer Genome Atlas (TCGA) LUAD cohort (n = 517) and the combined LUAD and LUSC cohort (n = 1018).37 Gene expression values in fragments per kilobase of transcripts per million mapped reads were downloaded from Broad firehose, version 2016_01_28.38 Autophagy signatures were downloaded from Gene Set Enrichment Analysis Molecular Signatures Database v6.1,39,40 the Autophagy database,41 and Human Autophagy Database (last accessed: 2019 Apr 22; http://autophagy.lu/index.html). Enrichment scores were subsequently estimated using R v3.5.1, Bioconductor v3.8 GSVA v1.28.0.42
Transmission Electron Microscopy
For the preparation of treated cells for transmission electron microscopy, cells were seeded in six-well plates and treated with drugs, as indicated (7 days of treatment with erlotinib and bemcentinib, then chloroquine was added for the final 16 hours), washed twice with PBS, and then fixed for 1 hour at room temperature in 2.5% ultrapure glutaraldehyde in PBS. Postfixation was done by 1% osmium tetroxide for 1 hour on ice, before washing and dehydrating with ascending concentrations of alcohol. Infiltration was carried out using ethanol with propylene oxide (1:1 ratio) for 1 hour. Three individual specimens per sample were collected and placed in fresh resin and polymerized at 60°C for 48 hours. Thick sections were cut at 1 μm and stained with 0.6% toluidine blue in 0.3% sodium bicarbonate; 70 nm sections were cut and stained with saturated uranyl acetate in 50% ethanol followed by Reynolds lead citrate. Electron microscopy images were obtained using a Jeol JEM-1230 transmission electron microscope (Jeol, Tokyo, Japan).
Real-Time Growth Curve Measurement by IncuCyte Confluence Measurement
The IncuCyte ZOOM microscope and software (Essen Bioscience) were used to measure % confluence in real time. Cells were seeded and treated with bemcentinib as described, for the resazurin cell viability assays. Two images were taken per hour per well of the 96-well plate by IncuCyte microscope using Nikon 10× objective lens magnification. Percent confluence was quantified using the IncuCyte software, and the settings for the quantification of confluence measurements by the IncuCyte software were optimized individually for the different cell lines of the experiment and were given to the various cell lines as per the following: HCC827 (segmentation adjustment: 1.2; Hole fill: 100 μm2; adjust size: −6 pixels; Filters: Area min: 100 μm2), ER3 (segmentation adjustment: 1.5; Hole fill: 200 μm2; adjust size: −1 pixels; Filters: Area min: 200 μm2), and ER10 (segmentation adjustment: 1.2; Hole fill: 100 μm2; adjust size: −1 pixels; Filters: Area min: 200 μm2). Time-lapse cytotoxicity assay to determine the cytotoxic effect of staurosporine treatment in HCC827, ER3, and ER10 cells was established using the IncuCyte CytoTox Green reagent 200 μM to stain the dead cells. Green area relative to the percent confluence for each well was plotted ± SE. Calculations where n equals three wells per condition and four images per well per time point was done using the IncuCyte software in accordance with the manufacturer’s instructions.
AXL Silencing by siRNA Transfection
For siRNA transfection, HCC827 and ER3 cells were seeded 2 × 105 cells per well in six-well plates and allowed to adhere overnight. Cells were transfected with Individual ON-TARGETplus AXL-siRNA#1, #2 (Dharmacon, Lafayette, CO, J-003104–11 and J-003104–12, respectively), or ON-TARGETplus Non-targeting siRNA (Dharmacon, D-001810–01) at a final concentration of 10 nM in 2 mL culture medium per well, using HiPerFect transfection reagent (Qiagen, Cat# 301704) in accordance with the manufacturer’s protocol. After 24 hours, the cells were treated with vehicle (DMSO), erlotinib (1 μM), or chloroquine (10 μM or 100 μM, as indicated) for another 16 hours. After treatment, cells were harvested and lysed with RIPA buffer with protease and phosphatase inhibitors, and knockdown was confirmed by SDS-polyacrylamide gel electrophoresis and Western blot analysis. The effect of AXL knockdown on the regulation of autophagy was analyzed by Western blotting as described for drug treatment studies.
ATP Release Assay
Release of extracellular ATP was measured by the luciferin-based ENLITEN ATP Assay System Bioluminescence Detection Kit for ATP Measurement in excess of luciferin and luciferase, as indicated by the manufacturer (FF2000, Promega). Beforehand, 100,000 cells per well were seeded in 12-well plates in the serum-free RPMI1640 medium containing 1% BSA. Cells were allowed to attach overnight and then treated with the drugs as indicated for 24 hours in the presence of 50 μM ecto-ATPase inhibitor (ARL67156, A265, Sigma-Aldrich). Harvested samples were transferred to ice-cold Eppendorf tubes and spun down at 3200 g for 5 minutes at 4°C. The supernatant was transferred to a new ice-cold Eppendorf tube and stored at −80°C until assayed. Cell medium was used for background subtraction. ATP-driven chemiluminescence was recorded at 0.1 s with a 1420 Multilabel Counter VICTOR3 (Perkin Elmer) with the Wallac1420 software. Three biological replicates were given per condition, and the biological replicate was represented by the mean value of two technical duplicates.
HMGB1 Immunoassay
To evaluate the HMGB1 release from treated cells in culture, 100,000 cells were allowed to adhere to 12-well culture dishes overnight and then treated with the indicated drugs for 72 hours. Serum-free cell culture media containing 1% BSA was used for this experiment to avoid the detection of HMGB1 from the serum. Supernatants were collected after treatment, cleared from dying cancer cells by centrifugation (800 g, 5 min), and frozen at −80°C or immediately analyzed for HMGB1 abundance. The HMGB1 quantification was performed using an HMBG1 enzyme immunoassay kit (ST51011, IBL International GmbH, Hamburg, Germany), according to the manufacturer’s instructions, for high-sensitivity range detection.
Flow Cytometry
For the detection of AXL by flow cytometry, cells were harvested by trypsin, washed with PBS containing 1% BSA, and blocked in 5% goat serum in 1% BSA with PBS for 20 minutes. The cells were then stained with mouse monoclonal anti-AXL antibody (MAB154, R&D Systems, Minneapolis, Minnesota) for 30 minutes at room temperature, washed as above, and incubated with AF488-conjugated secondary goat anti-mouse antibody (A11029, Invitrogen) for 30 minutes at room temperature. The cells were washed and analyzed using a FACS Accuri C6 flow cytometer (BD Biosciences). For the detection of surface-exposed CALR, trypsinized cells were fixed with 0.25% paraformaldehyde in PBS for 5 minutes on ice, washed in PBS, and subjected to indirect immunofluorescence with a rabbit anti-CALR antibody (mouse monoclonal anti-CALR antibody, PE-conjugated, ab83220, Abcam). Ab91357 (Mouse IgG1, PE-conjugated, Abcam) was used as an isotype control for these experiments. Samples were run on an Accuri Flow Cytometer (BD Biosciences), and data were analyzed by Cytobank (Santa Clara, California).
Statistics
Data were presented as mean values (± SD) or as fold changes from a representative experiment, if not otherwise indicated in the figure legends. In general, all statistical analyses were performed using the GraphPad Prism 7 software (San Diego, California). For in vivo experiments, comparison of tumor volumes between treatment groups were performed by one-way ANOVA with Bonferroni’s Multiple Comparison Test using the Graph-Pad Prism 7 software. Survival was analyzed by Kaplan-Meier estimation. Outliers were determined using Grubbs’ test, also called the extreme studentized deviate, with a significance level 0.01 (two-sided). The Spearman correlation coefficient rank test was performed using Matlab R2016b version 9.1.0.441655 (MathWorks; Natick, Massachusetts). The following symbols are given to report statistical significance: (1) NS = p greater than 0.05; (2) *p smaller than or equal to 0.05; (3) **p smaller than or equal to 0.01; (4) ***p smaller than or equal to 0.001; and (5) ****p smaller than or equal to 0.0001.
Results
AXL Signaling is Required for Acquired Resistance to Erlotinib In Vivo
Acquired resistance to BRAF–targeting agents in melanoma is shown to proceed through a two-step process in which rare, transient, AXL-expressing drug-resistant (AXL jackpot) cells persist on drug treatment and lead to stable drug resistance without new mutations.10 The prevalence of AXL expression in EGFRi-resistant NSCLC indicates that this mechanism may be an important step in acquired resistance to targeted therapies in lung cancer.13,14,43 To address the role of AXL in acquired resistance to erlotinib, we designed three independent in vivo experiments. In the first in vivo experiment, we aimed to evaluate whether inhibition of the AXL–mediated signaling by the oral small molecule AXL tyrosine kinase inhibitor bemcentinib (BGB324) could delay the onset of erlotinib resistance.44,45 In this experiment, animals harboring tumors established from HCC827 (EGFRDelE746-A750) NSCLC cells were randomly assigned to six treatment groups and treated with one of the following: vehicle, erlotinib monotherapy (50 mg/kg, once a day), bemcentinib monotherapy (50 mg/kg or 100 mg/kg, twice a day), or a combination of erlotinib (50 mg/kg) and bemcentinib (50 mg/kg or 100 mg/kg) for 5 months (138 d). Animals in the vehicle and bemcentinib monotherapy groups were killed within 75 days owing to their large tumor volumes. As expected, established HCC827 tumors responded to erlotinib treatment, showing an initial rapid decrease in tumor burden—68% decrease within 10 days of treatment initiation (Fig. 1A). The maximal antitumor response was maintained for 3 months (Fig. 1A). However, after 100 days on erlotinib therapy, tumor relapse was observed in 70% of the mice belonging to the erlotinib treatment group (50 mg/kg, once a day), confirming that the model closely mimics the transient duration of response observed for the vast majority of patients with NSCLC treated with erlotinib in the clinic. Upon termination of experiment (138 d), average recurrent tumor volumes had exceeded the tumor volumes recorded at the start of therapy (Fig. 1A and B). Although bemcentinib treatment alone exhibited no effect on the HCC827 tumor growth in the nude mice, statistically significant tumor growth delay (p < 0.01) was observed in the bemcentinib-erlotinib combination treatment groups (erlotinib 50 mg/kg once a day and bemcentinib 50 mg/kg or 100 mg/kg twice a day). More importantly, at the time of termination of the experiment, maximal antitumor response comprising non-palpable residual nodules was maintained (Fig. 1B and D). The prominent effect of the AXL inhibitor bemcentinib in this tumor model suggests that AXL signaling is required for acquired erlotinib resistance in vivo in the HCC827 model.
Figure 1.

AXL inhibition delays emergence of acquired resistance to the EGFR tyrosine kinase inhibitor erlotinib in vivo, and AXL signaling is associated with a novel gene signature of post-treatment cancer cell persistence that correlates with decreased patient survival. (A) The growth of human NSCLC HCC827 xenograft tumors in nude mice displays the expansive growth of vehicle-treated and bemcentinib-treated tumors (terminated at d 75). The tumors were initially sensitive to erlotinib treatment but tumors treated with erlotinib alone eventually regrew. Group mean tumor volumes and SE are shown. (B) Zoomed segment from (A) showing mean tumor volumes of groups treated with erlotinib (50 mg/kg, once a day) or erlotinib in combination with bemcentinib (erlotinib 50 mg/kg, once a day and bemcentinib 50 or 100 mg/kg, twice a day, respectively). The graph displays the final phase of the experiment from day 100 until study termination of experiment at day 138, with 14 to 16 tumors per group. (C) Representative images of tumor–bearing mice treated with erlotinib (50 mg/kg, once a day) or erlotinib in combination with bemcentinib (50 mg/kg, once a day, and 100 mg/kg, twice a day, respectively). (D) Final tumor volumes at termination of study (d 138) of individual HCC827 tumors for different treatment groups are shown. Differences between treatment groups (n = 14–16 tumors per group) were calculated using one-way ANOVA followed by Tukey’s multiple comparisons test (**p ≤ 0.01). (E) HCC827 tumors from a separate in vivo experiment were harvested after 51 days of treatment with vehicle or erlotinib (50 mg/kg, once a day) to perform Western blot analysis for the expression of AXL (140 kDa), pAXL (Tyr779, 140 kDa), pAKT (Ser473, 60 kDa), and vimentin (VIM, 57 kDa). GAPDH (37 kDa) is shown as a loading control. (F) GO analysis of the whole-ranked list of genes differentially expressed in erlotinib and bemcentinib–treated versus erlotinib-treated tumors harvested after 25 days of treatment. The list is computed using the GOrilla software taking the whole-ranked list of deregulated genes into account. REViGO visualization of the GO terms and with redundant GO terms removed is displayed. The table is generated on the basis of the GO terms and associated p values from GOrilla, and the “frequency” represents the proportion of this GO term in the underlying database; that is, higher frequency represents more general terms, whereas the lower represents more specific ones. The “uniqueness” measures whether the term is an outlier when compared semantically with the whole list. More unique terms tend to be less dispensable. The list is ranked based on “dispensability,” which is the semantic similarity threshold at which the term was removed from the list and assigned to a cluster. (G) Volcano plot displaying differentially expressed genes (cutoff: 1.5-fold change and p < 0.01) between HCC827 tumors treated for 25 days with erlotinib (50 mg/kg, once a day) or combination of erlotinib and bemcentinib (50 mg/kg, once a day, and 50 mg/kg, twice a day, respectively) (n = 6 tumors). The x-axis specifies the gene expression log2 (fold change) values and the y-axis specifies significance -log10 (p value) for each gene. The position of selected down- and up-regulated genes are indicated. (H) Bemcentinib-induced transcriptional changes in mesenchymal markers involved in negative regulation of MAPK activity (SPRY1, SPRY2, and SPRY4) correlate with overall survival in patients with lung adenocarcinoma. Kaplan-Meier survival analysis with survival time (mo) on the x-axis and survival probability on the y-axis. Hazard ratio = 0.38 (0.29–0.48). Logrank p = 1.9E-15. ANOVA, analysis of variance; GO, gene ontology.
AXL Signaling Is Associated With a Novel Gene Signature of Post-Treatment NSCLC Cell Persistence That Correlates With Decreased Patient Survival
To evaluate whether AXL-mediated signaling was activated by erlotinib treatment in vivo, we designed a second in vivo experiment in which tumors established from HCC827 (EGFRDelE746-A750) NSCLC cells were treated with erlotinib (50 mg/kg, once a day) and harvested 51 days after the initiation of erlotinib treatment. To decipher the molecular basis for the role of AXL in acquired resistance to erlotinib, we analyzed the expression and activation of AXL, and a panel of markers for EMT. Residual erlotinib-treated tumors exhibited increased AXL expression compared with the vehicle controls and increased phosphorylation of AXL and AKT indicated that persistent HCC827 cells up-regulated AXL and relied on AXL-mediated signaling (Fig. 1E). To gain a better understanding of the early changes in erlotiniband bemcentinib-erlotinib–treated tumors, we designed a third in vivo experiment in which residual tumor tissues were harvested at day 25 and processed for transcriptomic analyses. Gene ontology (GO) analysis was computed on the basis of the whole-ranked list of genes (Fig. 1F) using the GOrilla software.46,47 The GO term autophagy was reported as one of the top biological processes affected by bemcentinib-erlotinib co-treatment versus erlotinib alone in these tumors (Fig. 1F). Bemcentinib and erlotinib co-treatment resulted in down-regulation of several autophagy-associated genes, including KLH24, PDK4 FBXO32, and CDKN1B in the list significantly altered genes (cutoff: 1.5-fold change and p < 0.01) (Fig. 1G, Supplementary Table 1). Analysis of differentially expressed genes in bemcentinib-erlotinib–treated tumors versus erlotinib-treated tumors identified GO biological processes, which included neutrophil chemotaxis, negative regulation of the MAPK activity and positive regulation of inflammatory response, and apoptotic processes (Fig. 1G; Supplementary Tables 1A and B). Bemcentinib treatment induced transcriptional changes in negative regulators of RAS/MAPK activity, including the sprouty family.48 Three sprouty family members (SPRY1, SPRY2, SPRY4) were up-regulated upon treatment with bemcentinib in combination with erlotinib versus erlotinib alone. A higher expression of SPRY1, SPRY2, and SPRY4, as observed on bemcentinib treatment, correlated with better overall survival in patients with LUAD (Fig. 1H). SPRY2 expression has been shown to be consistently lower in NSCLC tissue when compared with the corresponding normal lung epithelium,49 whereas SPRY4 was recently reported to correlate with AXL-mediated osimertinib resistance.50 The role of the complex MAPK signaling pathway in the regulation of autophagy is context-dependent and has not yet been fully described.51 Elucidation of the signaling cascades that regulate autophagy and its mechanisms will therefore be highly beneficial to disease treatment and prevention.
Erlotinib-resistant NSCLC Cells Display Epithelial Plasticity and Broad Resistance to Apoptosis
To better understand the basis for AXL-mediated drug resistance and survival, we examined the pro-survival effects of AXL signaling in independent erlotinib-resistant HCC827 cell clones (ER3, ER10) that do not have secondary mutations in EGFR associated with erlotinib resistance (e.g., T790M).13,14,52,53 As expected, HCC827 parental cells were extremely sensitive to erlotinib (IC50 ≈ 6 nM), whereas ER3 and ER10 were unaffected by erlotinib at concentrations up to 10 μM (Fig. 2A). We observed a morphologic switch in the epithelial HCC827 cells from a cobblestone-like shape in the parental HCC827 cells to a spindle-like shape in the ER cells. We then determined that the morphologic switch was accompanied by induction of EMT hallmarks, including down-regulation of E-cadherin (CDH1), up-regulation of N-cadherin (CDH2) and vimentin (VIM), and profound up-regulation of AXL in the ER cells compared with the parental cells (Fig. 2B and C, Supplementary Figs. 1, 2A, and B).13,14,52,53 Immunofluorescence single channel displays are shown in Supplementary Figure 1. The combination of apoptosis assays and time-lapse experiment established that the parental cells were more sensitive to induction of apoptosis than the erlotinib-resistant clones under the conditions and time points tested. Luminescence-based caspase 3/7 assay (Fig. 2D), time-lapse evaluation of cell death on staurosporine treatment (Fig. 2E), and Western blot for cleaved caspase 3, cleaved caspase 8, and cleaved poly (ADP-ribose) polymerase (Fig. 2F) are shown.
Figure 2.

Erlotinib-resistant subclones underwent epithelial-to-mesenchymal transition and up-regulation of AXL and are characterized by resistance to apoptosis. (A) Erlotinib sensitivity of HCC827 parental cells and the erlotinib-resistant subclones (ER3 and ER10) was measured by resazurin assay after 5 days of erlotinib treatment. The mean IC50 value for HCC827 cells was 5.6 nM, and greater than 10 μM for the two erlotinib-resistant subclones ER3 and ER10 (n = 11–13, independent experiments). (B) Representative confocal images of HCC827, ER3, and ER10 cells immunofluorescently labeled for epithelial cell adhesion molecule E-cadherin (CDH1, green) and mesenchymal intermediate filament vimentin (VIM, red); DAPI nuclear stain (blue); scale bar: 50 μm. Single channel displays are shown in Supplementary Figure 1. (C) Western blot of lysates from HCC827 parental and ER cells using the indicated antibodies for epithelial marker E-cadherin (CDH1, 135 kDa), and mesenchymal markers N-cadherin (CDH2, 135 kDa), vimentin (VIM, 54 kDa), and AXL (140 kDa). β-Actin (42 kDa) is shown as a loading control. (D) Relative resistance to apoptosis was measured by induction of caspase activity of the HCC827 parental cells and ER10 cells upon 4 hours treatment with increasing doses of staurosporine by caspase 3/7 Glo luciferase assay. Y-axis represents bioluminescence as photon flux (photons/s). One-way analysis of variance was followed by Dunnett’s multiple comparisons test comparing every treatment against vehicle (DMSO) control (*** p < 0.001, **** p < 0.0001). (E) Time-lapse cytotoxicity assay to determine the cytotoxic effect of staurosporine treatment in HCC827, ER3, and ER10 cells. Green area (stained with IncuCyte CytoTox Green reagent 200 μM) representing dead cells is plotted relative to the percent confluence for each well ± SE. Three wells per condition and four images per well per time point were used for the calculations using the IncuCyte software. (F) Assessment of apoptosis induction in HCC827, ER3, and ER10 cells upon 24-hour treatment with 1 μM staurosporine by Western blot; cleaved PARP (89 kDa), procaspase 3 (32 kDa), cleaved caspase 3 (19, 17 kDa) and cleaved caspase 8 (CC8; 43, 41, and 18 kDa) expression is shown. HeLa cells were included as a positive control in this experiment. Actin (42 kDa) serves as a loading control. (G) High-dimensional analysis of the HCC827 parental cell line and the erlotinib-resistant clone ER3 using a 22-parameter time of flight mass cytometry (CyTOF) panel. Nonlinear dimensionality reduction, tSNE33 created a 2D projection of the three cell lines on the basis of their E-cadherin (CDH1), vimentin (VIM), CD44, EGFR, AXL, and PD-L1 expression. Each point in the Cytobank visual representation of data (viSNE plot) represents an ion-cloud representing a single cell (equal downsampling at 10,000 cells per sample, 5000 iterations were applied). Distribution of cells within the viSNE plot is shown as viSNE contour plots colored by density of ion clouds representing single cells. (H) The color range represents the intensity of markers included in the CyTOF panel and the distribution is shown for each for the cell lines in separate viSNE plots. DAPI, 4′,6-diamidino-2-phenylindole PARP, poly (ADP-ribose) polymerase; PD-L1, programmed death-ligand 1; tSNE, t-distributed stochastic neighbor embedding.
AXL Is a Unifying Marker for Drug Resistance and Plasticity in the Resistant NSCLC Clones
To better understand the phenotypic and signaling heterogeneity within the HCC827 and ER cell populations, we performed single-cell mass cytometry profiling with a 22-marker antibody panel (Supplementary Table 2). Nonlinear dimensionality reduction by the tSNE algorithm was performed and 2D viSNE projections of the three cell lines were created on the basis of their E-cadherin (CDH1), vimentin (VIM), CD44, EGFR, AXL, and programmed death-ligand 1 expression using Cytobank software (Fig. 2G). The viSNE distribution revealed that the epithelial HCC827 cells form a distinct cluster from the mesenchymal (E-cadherin low, vimentin high) erlotinib-resistant cells (Fig. 2G and H, Supplementary Fig. 4A–C). Cell signaling differed dramatically between the parental HCC827 and ER populations (Fig. 2H, Supplementary Fig. 4A and B). More importantly, the erlotinib-resistant cells exhibited loss of pPLCy, pSTAT5, and EGFR/pEGFR expression and strong AXL expression consistent with erlotinib resistance (Fig. 2H, Supplementary Fig. 4A and C). AXL signaling supports a unique pro-survival mode in drug-resistant cells. We conducted viability assay to evaluate the dose-response of erlotinib and bemcentinib, and the Bliss independence analysis was applied to determine potential synergy between the drugs in HCC827 and ER cells upon 5 days treatment.27 Interestingly, this analysis reported that erlotinib and bemcentinib action is independent and exert an additive effect on ER cell viability (Supplementary Fig. 3A–D). Clonogenic assays revealed that bemcentinib treatment was sufficient to block ER cell colony formation, revealing AXL dependence under these conditions (Fig. 3A and B). Real-time growth curve measurements for HCC827 parental cells and ER3 and ER10 erlotinib-resistant clones in response to increasing doses of bemcentinib were performed over a time course of 120 hours (Fig. 3C), and revealed that the effect of bemcentinib under these conditions is caused at least in part by growth arrest (Fig. 3C). Bemcentinib treatment did not induce caspase 3/7 activity at 24 hours in ER3 cells (Fig. 3D). Caspase 3/7 assays (Fig. 3D) and Western blot analysis for detection of induced apoptosis (Supplementary Fig. 3E) of HCC827 parental cells and ER cells treated for 24 hours with combinations of erlotinib and bemcentinib verified that the treatment combination did not induce prominent apoptosis at the earliest time points, and only at the highest doses in ER10 cells. Taken together, these data suggest that AXL signaling supports a unique pro-survival mode in drug-resistant cells, that AXL inhibition induces growth arrest, and that cell death is induced at increased doses and time point. Representative images of HCC827 and ER3 on various concentrations of bemcentinib 48 hours after treatment initiation is shown in Supplementary Figure 2C. Timelapse videos from IncuCyte monitoration of HCC827 and ER3 cells upon treatment with bemcentinib are available through this link (web-link with time-lapse video will be made available to the readers),
Figure 3.
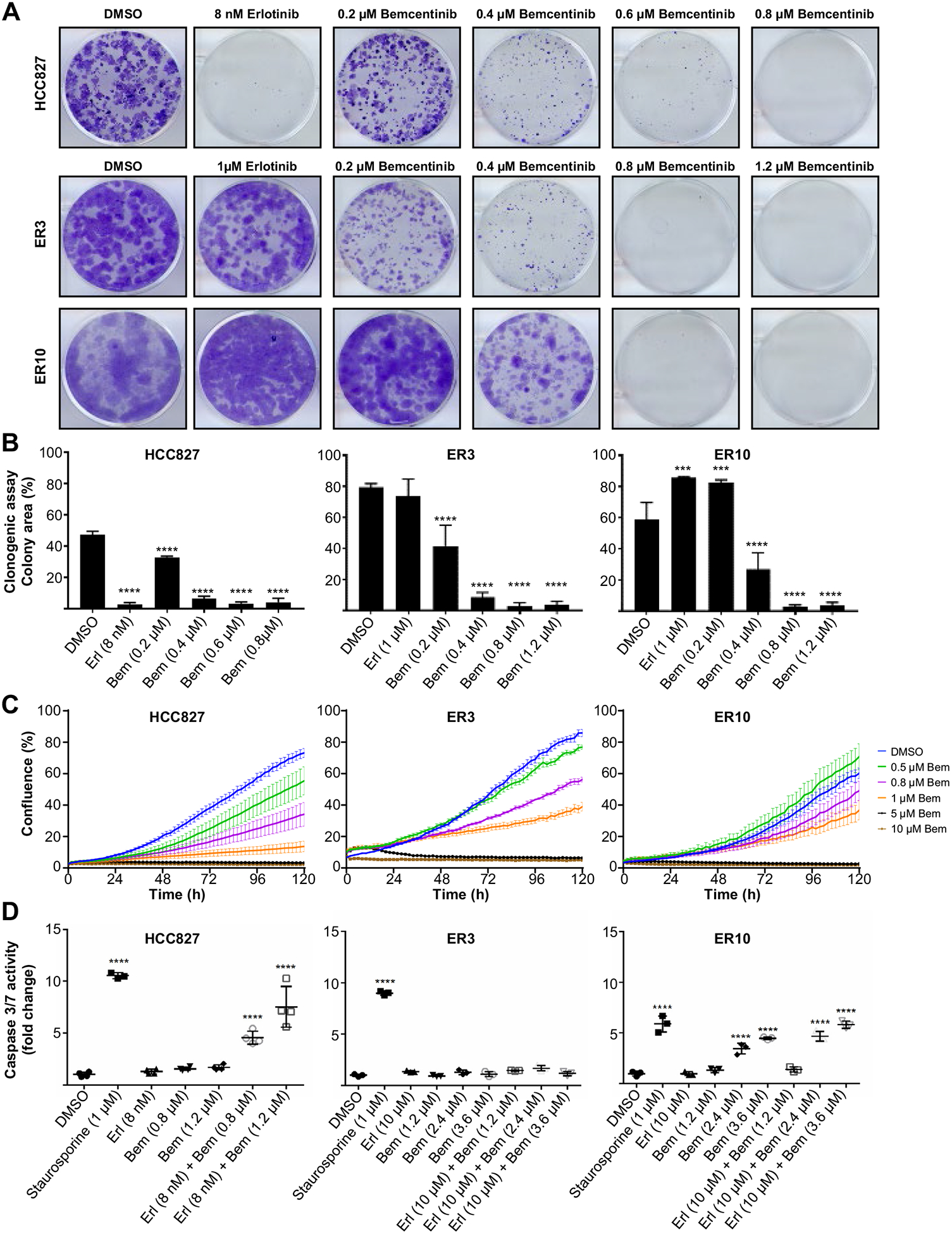
Bemcentinib treatment reduces clonogenic survival of ER cells. (A) Representative images of crystal violet-stained colonies from clonogenic survival assay of HCC827, ER3, and ER10 cells treated for 14 days with vehicle (DMSO), bemcentinib, or erlotinib as monotherapy. (B) Survival fraction of HCC827, ER3, and ER10 cells after treatment as indicated in (A). Each experiment was performed in triplicate (n = 2, independent experiments). Data represent survival fraction ± SD. Colony area was quantified using the “ColonyArea” plugin in ImageJ and one-way ANOVA was followed by Dunnett’s multiple comparisons test comparing every treatment against vehicle (DMSO) control (***p < 0.001, ****p < 0.0001). (C) Growth curves for HCC827 parental cells and ER3 and ER10 erlotinib-resistant clones in response to increasing doses of bemcentinib as indicated. Real-time measurements were performed over a time course of 120 hours, and percent confluence measurements calculated by the IncuCyte software are shown on the y-axis. The mean value of six replicate wells per condition with SE is shown for each experimental condition. (D) Relative resistance to apoptosis was measured by caspase 3/7 Glo luciferase activity assay in HCC827 parental cells, ER3, and ER10 cells on treatment as indicated for 24 hours. Bioluminescence as photon flux (photons/s) was measured, and fold change relative to vehicle control (DMSO) is given (y-axis). One-way ANOVA was followed by Dunnett’s multiple comparisons test comparing every treatment against vehicle (DMSO) control (****p < 0.0001). ANOVA, analysis of variance.
Erlotinib-Resistant NSCLC Cells Display an Increased Autophagic Flux
As the ER cells were more susceptible to undergo apoptosis at the earliest time points compared with parental cells (Fig. 2D), we next aimed to assess whether the less sensitive AXL-expressing ER cells were also characterized by an increased autophagic flux. Lipidation of microtubule–associated proteins 1A/1B light chain 3 A/B (MAPLC3A/B, hereafter referred to as LC3A/B, and in the figures LC3 for short) is frequently used as a surrogate marker for disrupted autophagic flux54). On induction of autophagy, the cytosolic form of the ubiquitin-like protein LC3A/B (LC3-I) is conjugated to phosphatidylethanolamine in the autophagic membrane (LC3-II), and remains bound to autophagic vesicles throughout the pathway, therefore the ratio between LC3-II and LC3-I is frequently used to quantify the level of autophagy.55 We found that compared with HCC827 cells, both ER3 and ER10 cells exhibited increased expression of LC3A/B, and, in particular, an increase in the membrane-bound form LC3-II (Fig. 4A). A shift in LC3A/B from the cytosolic form LC3-I to the lipid-bound form LC3-II is usually associated with increased autophagosome formation but could also indicate a block in fusion of autophagosomes with lysosomes. Thus, in order to draw conclusions regarding the autophagic flux, the formation of autophagic compartments in HCC827 cells and erlotinib-resistant ER3 cells was further quantified by a flow cytometry–based selective detection of autolysosomes and earlier autophagic compartments using the Cyto-ID detection probe to measure autophagic activity. The Cyto-ID assay allow efficient measurement of the dynamic balance between autophagosome generation and clearance. Chloroquine inhibits autophagy by preventing lysosomal acidification, decreasing autophagosome-lysosome fusion, and consequently reduces lysosomal protein degradation.56 Interestingly, the starvation–induced autophagic flux was significantly (p ≤ 0.0001) increased in ER cells when compared to HCC827 parental cells (Fig. 4B). Morphometric transmission electron microscopy confirmed increased vacuolization in ER cells upon chloroquine treatment (Fig. 4C), further strengthening the notion that the autophagic flux is increased in the erlotinib-resistant cells.
Figure 4.
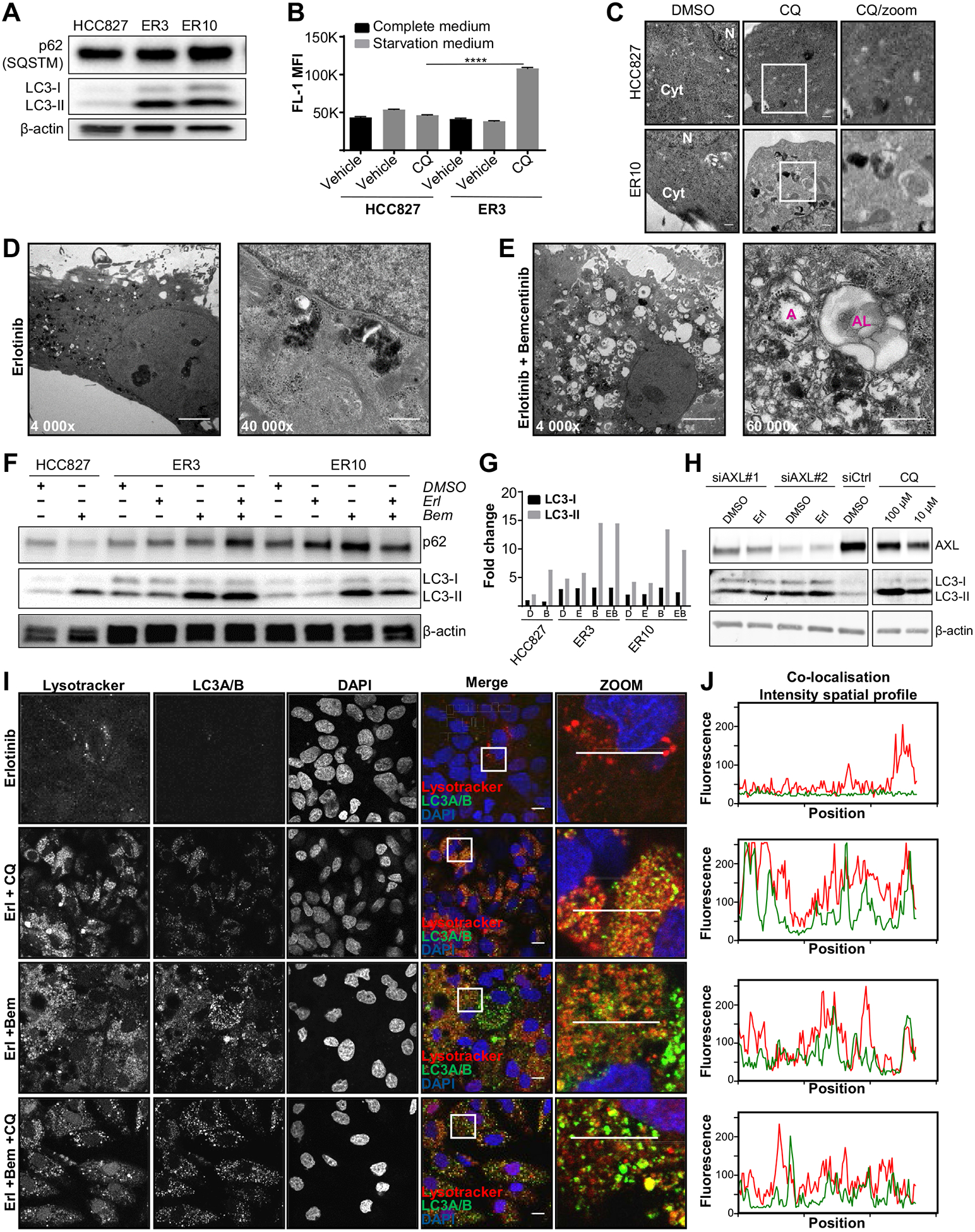
AXL expressing erlotinib-resistant NSCLC cells are characterized by an increased cytoprotective autophagic flux that is abrogated by inhibition with the selective AXL inhibitor bemcentinib and gene silencing by AXL siRNA. (A) Relative protein levels of p62 (SQSTM, 62 kDa) and LC3A/B (LC3-I 17 kDa, LC3-II 19 kDa) were assessed in lysates of HCC827, ER3, and ER10 cells by Western blot. β-Actin (42 kDa) is shown as a loading control. (B) HCC827 parental cells and ER3 cells grown in complete medium or starved in EBSS (with 0.1% BSA in the absence or presence of chloroquine) were stained by the Cyto-ID probe and analyzed by flow cytometry. MFI ± SD is shown. (C) Transmission electron micrographs of HCC827 and ER10 cells with or without the autophagy inhibitor chloroquine (50 μM, 16 h); scale bar: 500 nm. (D, E) Representative transmission electron microscopy images of erlotinib-resistant ER3 cells treated with erlotinib (D; 1 μM) alone or in combination with the small molecule AXL inhibitor bemcentinib (E; 0.8 μM) for 7 days. On combination treatment, massive vacuolization and the presence of autophagosomes (A) and autolysosomes (AL) are shown; scale bar; 5 μm for 4000× magnification images, 500 nm for 40,000× magnification, and 60,000× magnification images. (F) Western blot analysis of protein levels of p62 (SQSTM, 62 kDa) and the nonlipidated (LC3-I, 17 kDa) and the lipidated (LC3-II, 19 kDa) forms of LC3A/B in the HCC827, ER3, and ER10 cells treated for 24 hours under various treatment conditions as indicated (1 μM erlotinib, 0.8 μM bemcentinib). β-Actin (42 kDa) is shown as a vehicle control. (G) Quantification of LC3A/B levels shown in (F). Normalization was performed against total protein using Bio-Rad stain-free imaging technology. Data are presented as fold change normalized against LC3-I expression level of the HCC827 vehicle (DMSO)-treated sample. (H) Alterations in LC3A/B lipidation on specific knockdown of AXL by siRNA in erlotinib-resistant ER3 cells. Western blot analysis to verify the reduced protein levels of AXL (144 kDa) on specific knockdown of AXL by two siRNA constructs (siAXL #1 and siAXL #2) versus control siRNA (siCtrl). Combination of siRNA knockdown with vehicle (DMSO) or erlotinib (1 μM) treatment is also shown. The nonlipidated (LC3-I, 17 kDa) and the lipidated (LC3-II, 19 kDa) forms of LC3A/B in the ER3 chloroquine treated samples are shown to indicate expected LC3A/B alterations upon interrupted autophagic flux. Housekeeping protein β-Actin (42 kDa) is shown as a control for loading accuracy in this experiment. (I) Staining of the erlotinib-resistant ER3 cells by LysoTracker (red DN-99) in combination with anti-LC3A/B antibody reveals the presence of lysosomes (red puncta), autophagosomes (green puncta), and autolysosomes (yellow puncta) in cells treated with erlotinib (1 μM), bemcentinib (0.8 mM), or both for 7 days. Chloroquine (50 μM) was added as indicated 16 hours before fixation and staining. Scale bar; 10 μm. (J) Co-localization intensity profiles from the zoomed images is shown. Marker in zoomed insert in (I) indicates the selected area analyzed and the marker used for all experimental conditions is 1 pixel high and 130 pixels wide. Fluorescence intensity (y-axis) along the x-position (x-axis) of this selected area is presented. BSA, bovine serum albumin; EBSS, Earle’s Balanced Salt Solution; MFI, Median fluorescence intensity.
Moreover, we observed a massive vacuolization and accumulation of autophagic vesicles (autophagosomes and autolysosomes) in ER cells after 7 days treatment with erlotinib and bemcentinib, compared with erlotinib alone (Fig. 4D and E). Next, we evaluated the expression of autophagy markers in erlotinib–sensitive versus erlotinib-resistant cells in response to AXL inhibition. We observed a shift from cytosolic LC3-I to lipidated LC3-II in both HCC827 parental cells and ER cells after bemcentinib treatment, suggesting that AXL inhibition interferes with autophagy independently of erlotinib (Fig. 4F and G). In order to evaluate whether bemcentinib mediated effect on autophagy was specific to inhibition of AXL, we performed siRNA mediated genetic silencing of AXL. Upon successful knockdown of AXL by siRNA, the levels of lipidated LC3 (LC3-II) were markedly increased compared to siCtrl samples and comparable to the chloroquine treated positive control samples (Fig. 4H).
Furthermore, co-localization of lysosomes and LC3-positive vesicles confirmed the massive accumulation of LC3-positive autophagosomes, lysosomes, and corresponding fused autolysosomes in the erlotinib- and bemcentinib-treated cells (Fig. 4I). Although inhibition of autophagic flux with chloroquine 16 hours before fixation and staining caused a substantial accumulation of LC3 and LysoTracker-positive vesicles in the erlotinib-treated cells, no further accumulation of LC3 and LysoTracker double-positive vesicles was observed on addition of chloroquine to cells co-treated with erlotinib and bemcentinib, indicating that bemcentinib treatment abrogated the autophagic degradation. Co-localization intensity spatial profiles support these observations (Fig. 4I and J).
AXL Inhibition Abrogates the Elevated Premortem Autophagic Cell Flux in Erlotinib-Resistant Cells
To dynamically monitor the formation of autophagosomes versus autolysosomes over time in the absence or presence of chloroquine, we utilized the Autophagy Tandem Sensor RFP-GFP-LC3B probe (Fig. 5A and B). When autophagic flux is normal, the fusion of autophagosomes with acidic lysosomes resulted in the formation and acidification of the fused autolysosomes. Owing to the nature of the fluorescent probes of the tandem sensor, autophagosomes will seem yellow (GFP + RFP) whereas autolysosomes seem red (RFP) owing to the quenching of pH-sensitive GFP fluorochrome in the acidic autolysosome (Fig. 5B). As shown in Figure 5A, bemcentinib treatment of ER cells increased the numbers of both yellow and red RFP-GFP-LC3B puncta, in contrast with chloroquine treatment, which completely inhibited the formation of red puncta (Fig. 5A). The mTOR inhibitor rapamycin induces autophagic flux and was included as a control. To further elucidate how bemcentinib affects autophagic flux, we generated ER10 cells stably expressing a lentiviral doxycycline-inducible mCherry-EGFP-LC3B construct. This enabled live cell imaging and time-lapse studies visualizing real-time dynamics of autophagic vesicles on pretreatment, as indicated (Supplementary Fig. 5). The vehicle (DMSO) treated ER10-mCherry-EGFP-LC3B cells displayed a moderate to low level of LC3B–positive vesicles, and during the 120-minute course of the time lapse, the color of individual puncta did not change from yellow to red at a constant rate. Furthermore, newly formed yellow puncta were seen, indicating that the autophagic flux was retained (Supplementary Fig. 5, lower panels). ER10-mCherry-EGFP-LC3B cells treated with bemcentinib (Supplementary Figure 5, middle panels) did, however, seem similar to cells treated with chloroquine, showing increased frequency of yellow autophagosomes and a prominent decrease in the movement and transition from yellow to red LC3-associated puncta, indicating that fusion between autophagosomes and lysosomes is compromised on bemcentinib or chloroquine treatment (Supplementary Fig. 5).
The Cyto-ID assay allowed efficient measurement of the dynamic balance between autophagosome generation, and clearance in ER3 cells treated with bemcentinib in the presence or absence of autophagy inducers or inhibitors (starvation and chloroquine, respectively). As expected, no significant alteration was detected in ER3 cells cultured in starvation medium (EBSS with 0.1% BSA) for 3 hours relative to culture in complete medium, consistent with an uninterrupted flux in both conditions. A significant increase in the relative fluorescence intensity was detected in the starved cells when autophagic degradation was inhibited by chloroquine, indicating that the autophagic flux is intact in the ER3 cells (Fig. 5C). The relative fluorescence intensity was also significantly increased on bemcentinib treatment of the starved ER3 cells compared with the vehicle-treated controls (Fig. 5C), confirming that bemcentinib abrogates the balance between autophagosome generation and clearance.
To further corroborate our findings, we took advantage of a flow cytometry–based assay to quantify the degradation of long-lived proteins, known to occur predominantly through autophagy. The methionine analogue AHA was incorporated into newly synthesized proteins during a long pulse, followed by a chase to degrade short-lived proteins. The long-lived protein degradation was then evaluated at the single-cell level upon autophagy stimulation by starvation. Specific labeling of the incorporated AHA with a fluorescently labeled alkyne, was followed by flow cytometry analysis. Using this assay, we reported that bemcentinib treatment significantly reduced the starvation–induced degradation of long-lived proteins in ER3 cells when compared with the vehicle-treated starvation controls (Fig. 5D). Bafilomycin A, an inhibitor of the lysosomal proton pump, inhibits fusion between autophagosomes and lysosomes, and was included as a control in these experiments.
The H1975 cell line harbors the T790M gatekeeper mutation predictive of erlotinib resistance. NSCLC cells harboring the T790M mutation can be targeted by third-generation EGFR TKIs, e.g. rociletinib (CO-1686). AXL has recently been shown to confer intrinsic resistance to osimertinib and promote the emergence of osimertinib tolerant cells.50 Thus, we aimed to evaluate if autophagy was deregulated in the rociletinib-resistant (COR) clones (COR1–1 and COR10–1) versus the H1975 parental cells (Supplementary Fig. 6). Similar to what we observed in the HCC827 model, development of acquired resistance against rociletinib (Supplementary Fig. 6A) was accompanied with increased AXL expression (Supplementary Fig. 6C and D) as well as a switch against a more mesenchymal phenotype (Supplementary Fig. 6D and E). Furthermore, we observed an increased expression of LC3A/B in the drug-resistant COR1–1 and COR10–1 clones compared with the parental H1975 cells (Supplementary Fig. 6D). When these cells were treated with the AXL TKI bemcentinib (Supplementary Fig. 6B), we observed an increase in LC3A/B by Western blot analysis, and prominent vacuolization was observed by lysotracker and LC3A/B co-staining (Supplementary Fig. 6F and G), consistent with an abrogation of the autophagic flux by bemcentinib treatment. These studies indicate that an increased flux is a mechanism broadly associated with AXL up-regulation in NSCLC, and reports that bemcentinib treatment abrogates the autophagic flux in NSCLC cells resistant to first- and third-generation EGFR TKIs.
AXL Expression Is Widely Associated With Autophagy in LUAD and LUSC
To determine whether AXL expression is broadly correlated with positive regulation of autophagy in human adenocarcinomas, we evaluated correlation between AXL expression and positive regulation of autophagy in human NSCLC. For this purpose, we first analyzed RNAseq data from the TCGA LUAD cohort (n = 517),37 and these analyses established that AXL expression is significantly associated with positive regulation of autophagy gene expression signatures from the Human Autophagy Database (http://autophagy.lu/index.html), (Rho = 0.3765, p < 1E-200, Fig. 6A). The significant correlation between AXL expression and autophagy was further validated by three additional autophagy gene signatures generated from independent datasets (Fig. 6B–D). These include autophagy signatures downloaded from GSEA Molecular signature database v6.139 (Rho = +0.5071, p < 1E-200, Fig. 6B), Kumar Autophagy Network enrichment score40 (Rho = +0.3987, p < 1E-200, Fig. 6C), and the Autophagy database41 (Rho = +0.3396, p = 2.7E-15, Fig. 6D). Furthermore, the strong positive correlation between AXL expression and positive regulation of autophagy was also confirmed in a combined group of LUAD and LUSC from the TCGA cohort (LUAD + LUSC, n = 1018)37 (Spearman correlation Rho = +0.5017, p < 1E-200).
Figure 6.

AXL is widely associated with positive regulation of autophagy signatures in human NSCLC transcriptome samples, and bemcentinib treatment induces programmed cell death displaying hallmarks of immunogenic cell death with the release of DAMPs (A–D) The correlation between AXL mRNA (fragments per kilobase of transcripts per million mapped reads; log2, × axis) and four individually generated autophagy gene signatures (enrichment score, y axis) in human patient samples from TCGA lung adenocarcinoma cohort (n = 517). Correlation Rho and p value was computed by Spearman correlation coefficient rank test. Correlation between AXL (FKPM log2) (x-axis) and enrichment score (y-axis). (A) Positive regulation of autophagy gene expression signatures from the Human Autophagy Database (http://autophagy.lu/index.html), (B) MSigDB v6.1.39 (C) Kumar Autophagy Network enrichment score,40 (D) the Autophagy database.41 (E) Release of extracellular ATP after 24 hours treatment as indicated was measured by ATP luciferase assay for HCC827 parental cells and erlotinib-resistant clone ER3. ER10 cells responded similar to ER3 cells (data not shown). Fold change values of extracellular ATP normalized against vehicle control (DMSO) is shown for a representative experiment (n = 4). One-way analysis of variance was followed by Dunnett’s multiple comparisons test comparing every treatment against vehicle (DMSO) control (* p < 0.05, ****p < 0.0001). (F) Release of HMGB1 after 72 hours treatment as indicated was measured by ELISA for HCC827 parental and ER3 cells. Parental cells were treated with erlotinib (8 nM) with or without bemcentinib (0.8 μM), and ER3 cells were treated with erlotinib (1 μM) ± bemcentinib (0.8 μM). ER10 cells responded similar to ER3 cells (data not shown). Samples were normalized against the corresponding resazurin measurement for each well, and further normalized against the vehicle control (DMSO) for each cell line. Data are shown for a representative experiment (n = 4). One-way analysis of variance was followed by Dunnett’s multiple comparisons test comparing every treatment against vehicle (DMSO) control (****p < 0.0001). (G) Flow cytometric assessment of extracellular calreticulin (CALR). Histograms of ER10 cells treated for 7 days as indicated are shown, and MFI for all cell lines from a representative experiment is shown in the corresponding table (n = 4). HCC827 cells were treated with 0.8 μM bemcentinib, and ER3 and ER10 cells with 1.2 μM bemcentinib. (H) Alteration in the expression of major histocompatibility complex 1 assessed by flow cytometry after 7 days treatment with erlotinib (10 μM) with or without bemcentinib (0.8 μM for HCC827 parental cells, 1.2 μM for ER-cells). Histogram shown for ER3 cells. Median fluorescence intensity values for all cell lines are given in the corresponding table. ATP, adenosine triphosphate; CALR, calreticulin; HMGB1, high-mobility group box 1; MFI, median fluorescence intensity; MSigDB, Gene Set Enrichment Analysis Molecular Signatures Database; TCGA, The Cancer Genome Atlas.
Bemcentinib Induces Immunogenic Cell Death in Erlotinib-Resistant Cells
ICD is characterized by active secretion of ATP, exposure of CALR on the outer leaflet of the plasma membrane, and the release of the high-mobility group box 1 (HMGB1) protein. Premortem autophagy is an essential determinant of the immunogenicity of drug-induced cancer cell death by affecting the secretion of ATP. On the basis of our findings above, we evaluated whether bemcentinib treatment resulted in ICD in the ER cells by measuring the release of ATP in addition to the two other DAMPs; HMGB1 and surface-exposed CALR (Fig. 6E–G). Using a luciferase–based assay, we evaluated the release of ATP from ER cells treated with erlotinib and bemcentinib for 24 hours, either as single agents or in combination. We found that ATP secretion was significantly (p < 0.0001) increased in the erlotinib-resistant clones treated with the combination of erlotinib and bemcentinib (Fig. 6E). The release of the ATP damage signal into the extracellular space increased in a dose-dependent manner (Fig. 6E).
HMGB1 secreted from dying cancer cells functions as a potent DAMP, with the potential to attract antigen-presenting cells to the tumor bed. ELISA was used to assess the release of HMGB1 from cancer cells on treatment with erlotinib and bemcentinib administered as single agents or in combination for 72 hours. A significant (p < 0.0001) increase in extracellular HMGB1 after bemcentinib treatment alone or in combination with erlotinib was observed in the ER3 cells. The HMGB1 increase was not significant on erlotinib-bemcentinib combination treatment in parental HCC827 cells (Fig. 6F).
CALR is externalized at the cell surface on cells during ICD, and this potent “eat me” signal promotes efficient phagocytosis by dendritic cells, thereby facilitating tumor antigen presentation and incitement of tumor-associated antigen-specific cytotoxic T-cells. Externalized CALR was monitored at the single-cell level by flow cytometry, which exhibited an increase in the extracellular exposure of CALR after bemcentinib treatment in erlotinib-resistant ER10 cells (Fig. 6G). In addition, bemcentinib treatment resulted in a moderate increase in major histocompatibility complex-1 (MHC-I) (Fig. 6H). Antigen presentation by MHC-I is essential for adaptive immunity. Thus, three potent regulators of immunogenicity were up-regulated in the erlotinib-resistant tumor cells on treatment with bemcentinib, indicating an alteration of the tumor immune microenvironment beneficial for tumor cell recognition and eradication by the immune system.
Discussion
The sequence of events that lead to acquired resistance after an initial antitumor response to targeted agents is complex and remain poorly understood. Current models envision that acquired drug resistance requires both robustness, the ability to withstand environmental and genetic variability, and evolvability or the ability to acquire selectable and heritable phenotypic changes.57 Adaptive intrinsic phenotypic plasticity of cancer cells facilitates a robust drug tolerant, slow cycling state, which eventually transitions to permanent mutational resistance.7,57,58 We report that AXL kinase activity is necessary to acquire resistance to long-term erlotinib therapy in an in vivo model of mutant EGFR NSCLC. This supports the notion that AXL signaling underpins a critical pro-survival function necessary for long-term tumor cell persistence that is prerequisite to attain durable drug resistance. This was recently and independently confirmed using a different AXL kinase inhibitor (DS-1205), in which combinational treatment with DS-1205 and the third-generation EGFR inhibitor osimertinib significantly delayed the onset of resistance compared with osimertinib treatment alone in an HCC827 xenograft model.59 This is consistent with a recent study showing that acquired resistance to BRAFi in melanoma cells in vitro is derived from transient AXL-expressing drug-resistant (AXL jackpot) cells.10 In addition, in NSCLC, a recent report suggests AXL as an important contributor to overcome the initial drug response against osimertinib.50 A current clinical trial (NCT02424617) is evaluating bemcentinib in combination with erlotinib in EGFR-mutant NSCLC.
Several assays may be used to assess the autophagic flux, and great efforts have been made to standardize the nomenclature and the guidelines for monitoring autophagy in different model systems.60 In this context, it is important to note that monitoring autophagic elements may be used as a surrogate marker for autophagy, although that is not equivalent to monitoring the autophagic flux per se.60 In multicellular eukaryotes, monitoring autophagic flux is particularly challenging, and it is difficult to discriminate a block in the autophagic flux from an increased flux based solely on available surrogate markers. Thus, care is needed when interpreting these results, and these challenges have likely contributed to some of the seemingly contradictory findings in the field.61 We report that AXL signaling supports enhanced autophagic flux that prevents caspase-mediated apoptosis.17 This observation is aligned with previous reports linking EMT and erlotinib resistance to an increased cytoprotective autophagic flux.17,62,63 Exactly how the increased autophagic flux interferes with the cells ability to undergo apoptosis is still not known. However, based on our single-cell high-dimensional mass cytometry analysis, we hypothesize that this cytoprotective property of AXL-expressing cells may function independent of EMT. Indeed, AXL-expressing erlotinib-resistant NSCLC cells report substantial phenotypic and cell signaling heterogeneity that is incongruent with the prevailing bypass signaling hypothesis of acquired EGFRi resistance.2 The close correlation between AXL and CD44, and the anti-clonogenic effect of bemcentinib on ER cells suggests that AXL signaling mediates survival in the context of stem cell–like plasticity that includes epithelial, mesenchymal, and intermediate epithelial-mesenchymal states.6 Overall, these findings support the hypothesis that these features are critical for the survival of rare cancer cells under extreme stress induced by initial drug exposure.18,19,62,64–66
We found that treatment with the selective AXL inhibitor bemcentinib abrogated the cytoprotective autophagic flux and significantly inhibited the degradation of long-lived proteins upon autophagy induced by starvation, resulting in cell death with accumulated autophagic vacuoles in the cytoplasm. Taken together, our results support a model in which AXL-mediated signaling sustains a high autophagic flux, which is disrupted when AXL signaling is inhibited, either by the small molecule AXL inhibitor bemcentinib or siRNA mediated knockdown. AXL has previously been linked to autophagy in a study of acute liver injury in mice, in which AXL was reported to induce autophagy in macrophages after interaction with its ligand GAS6, mediated by MAPK (mitogen-activated protein kinase) 14 activity.67 In the immune system, AXL acts as an inhibitor of the inflammatory response to pathogens, and AXL together with the other TAM receptors are important for efficient efferocytosis of apoptotic cells.68 Because of the similarities between the mechanisms of efferocytosis and autophagy, one can speculate that the cancer cells exploit the physiological role of AXL in the regulation of efferocytosis to induce an increased cytoprotective autophagic flux to overcome acquired drug resistance against anticancer therapeutics.
The exact mechanism underlying how high premortem autophagy contributes to ATP release remains elusive, although it has been speculated that it might preserve a bioenergetic status in which, despite massive cellular damage and energy-consuming repair attempts, high intracellular ATP levels are maintained.24 Targeting AXL in ER cells induced a massive premortem autophagic vacuolization in the erlotinib-resistant carcinoma cells accompanied by ICD. One of the three hallmarks of ICD is the active release of ATP by cells that are committed to undergo, but have not yet succumbed to, apoptosis. Intriguingly, our findings suggest that induction of ICD can be a potential benefit of AXL targeting of drug-resistant EGFR-mutant NSCLC.
Recently, gene signatures enriched for wound healing and EMT, including AXL up-regulation, were found to be associated with innate resistance to programmed cell death protein-1 blockade.21 In a recent study, we have shown that the expression of AXL in the mesenchymal NSCLC cells was correlated with an increased cancer cell–intrinsic resistance to killing by both NK cells and autologous CTLs. Notably, small molecule AXL targeting potently sensitized mesenchymal lung cancer cells to cytotoxic lymphocyte–mediated killing.21 Furthermore, recent publications have suggested that AXL also suppresses proper antigen presentation by MHC-I, and that targeting AXL could therefore mediate a favorable reprogramming of the immune suppressive tumor microenvironment.69,70 In this context, increased immuno-adjuvanticity—the recruitment of professional antigen-presenting cells to educate the naive CTL population—is critical to maximize the potential therapeutic benefit of immune checkpoint blockade. Our novel finding that bemcentinib is a potent inducer of ICD is of particular interest as it implicates a favorable adjuvant effect and alteration of the tumor immune microenvironment upon AXL inhibition. AXL has previously been shown to suppress antigen presentation by MHC-I, whereas AXL inhibition enhances cytokine release and increases T-cell response in syngeneic models, resulting in an elevated cytotoxic T-cell–dependent antitumor immune response after radiation.69 Indeed, ongoing clinical trials are evaluating AXL inhibition in combination with immune checkpoint inhibitors.70 To the best of our knowledge, this is the first report to establish an abrogated autophagic flux by AXL inhibition through bemcentinib treatment. Monitoring autophagic flux in vivo represents a challenge, and thus, uncertainty on the role of autophagic flux as a regulator of DAMP secretion still remains. Much remains to be learned about the role of autophagy as a mediator of adjuvanticity and of polarization of cancer cell phenotypes and immune cell compartments in complex tumor immune microenvironments.61 Despite the challenges in monitoring DAMPs in vivo, and the obvious limitations of the nude mouse model used in this study, particularly with respect to the inability to examine immune effector cell infiltration, it is tempting to speculate that the ICD observed in our study may explain the previously reported favorable alteration of the tumor immune microenvironment observed in syngeneic lung carcinoma models on bemcentinib treatment.69,71,72 In one of these studies, reduced tumor growth and significant reduction of myeloid–derived suppressor cells were observed in the tumors of the animals treated with a combination of programmed cell death protein-1 signaling inhibitors and an AXL inhibitor,69,70 reinforcing the concept that immune checkpoint therapies are not effective unless an antitumor immune response is generated. Together with the immunogenicity of cancers, it is evident that proper adjuvanticity is required for a robust initiation of the cancer immunity cycle, and several clinical trial protocols are currently exploring the potential clinical benefit of increasing adjuvanticity in the context of immune checkpoint inhibitor treatment regimens. The release of DAMPs, including cell-surface translocation of CALR and the release of HMGB1 and ATP represent hallmarks of immunomodulation. This immunomodulation, in turn, promotes an immune response toward the cancer cells through the recruitment of antigen-presenting dendritic cells and immune effector cells to the tumor bed. Thus, the immunogenic form of cell death activates both the innate and adaptive arms of the immune system by initiating the cancer immunity cycle.73
In conclusion, we report that AXL signaling is necessary for acquired resistance to erlotinib in vivo in a model of mutant EGFR–driven NSCLC. Our results also reveal a positive correlation between AXL expression and autophagy in a cohort of human LUAD and LUSC samples and implicate a cytoprotective, AXL–dependent autophagy-related mechanism that supports a robust drug tolerant state from which permanent resistance can emerge. It has been shown experimentally that surprisingly few combinations of drugs exert a true synergistic effect; however, some drug cocktails may exert their superior efficacy by affecting heterogenous cellular subpopulations.74,75 In the case of AXL inhibition as a means of preventing emergence of EGFR inhibitor–resistant cells, we cannot rule out that the inhibition of resistance could be attributed to more effective initial antiproliferative, pro-apoptotic effects. Nevertheless, the prevention of emergent AXL-positive cell populations with increased autophagy pro-survival signaling deserves further study. As recently reported by Schaffer and colleagues, acquired resistance to BRAF–targeting agents in melanoma were shown to proceed through a two-step process in which rare, transient, AXL-expressing drug-resistant (AXL jackpot) cells persisted on drug treatment and led to emergence of stable drug resistance without acquisition of new mutations.10 With regard to the case in which the drugs affect the same cell, the convergence of downstream signaling pathways related to cell death might limit the efficiency of the combined treatment.74,75 In these cases, the exploitation of alternative modes of cell death, particularly cell death with increased potential to elicit an immune response, represents a promising therapeutic possibility.76,77 A growing body of evidence demonstrates a central role for autophagy in modulating adjuvanticity, which is an essential component for increasing both natural and therapy-induced immunosurveillance.61 Of note, abrogation of this cytoprotective autophagic flux by AXL targeting resulted in ICD, which is expected in turn to enhance antitumor immunity and potentiate anticancer effects. This suggests that AXL-expressing EGFR inhibitor–resistant NSCLC may be sensitive to AXL targeting in combination with immunotherapy.78 Further experiments are warranted to evaluate the contribution of the alterations in the various immune cell compartments to the clinical efficacy of AXL inhibition.
Supplementary Material
Acknowledgments
This work was partly supported by the Research Council of Norway through its Centres of Excellence funding scheme, project number 223250 (CCBIO affiliates). Dr. Lotsberg was supported by a PhD fellowship grant from Helse Vest RHF (the Western Norway Regional Health Authority, grant number 911934). Dr. Minna was supported by National Cancer Institute grant Lung Cancer SPORE (P50CA070907), Cancer Prevention Research Institute of Texas (CPRIT), and Margot Johnson Foundation grants. Dr. Chouaib was supported by la Ligue Contre le Cancer (EL2015.LNCC/SaC). Dr. Lorens was supported by grants from the Norwegian Research Council (grant number 204868) and Norwegian Cancer Society (grant number 190330). Dr. Engelsen was supported by the FRIPRO Mobility Grant Fellowship from the Research Council of Norway co-funded by the EU’s Seventh Framework Programme’s Marie Skłodowska Curie Actions (MSCA COFUND, grant agreement number 608695), Legat for Forskning av Kreftsykdommer fund at University of Bergen (UiB), and Familien Blix fund for this project. Flow cytometry, cell sorting analysis, and mass cytometry were performed at the Flow Cytometry Core Facility, Department of Clinical Science, at UiB. Flow cytometry was also performed at the Imaging and Cytometry Platform (PFIC) at Gustave Roussy Cancer Campus Grand Paris. Gene expression analysis was performed at the genomics core facility (GCF) at UiB. Imaging was performed at the Molecular Imaging Center (MIC) at UiB. The results published here are in part based upon data generated by the TCGA Research Network (https://www.cancer.goc/tcga). We acknowledge the TCGA network and the lung cancer patients that consented to donate tumor tissue for application in cancer research. The authors also thank Sissel Vik Berge, Ingrid Sandven Gavlen, Eline Milde Nævdal, and Anna Boniecka; Endy Spriet, Hege Avsnes Dale and Anne Karin Nyhaug at MIC; Marianne Enger, Brith Bergum and Jørn Skavland at the Flow Cytometry Core Facility/UiB; and Bendik Nordanger at the Department of Pathology, University of Bergen and Haukeland University Hospital for their skillful technical assistance.
Footnotes
Disclosure: Drs. Lorens and Micklem are founders, shareholders, and employees of BerGenBio ASA. Dr. Gausdal is employed by and stock option holder of BerGenBio ASA. Drs. Lorens, Micklem, Gausdal, Lotsberg, and Engelsen are co-inventors of patent(s) pending or issued to BerGenBio ASA. Drs. Wnuk-Lipinska and Hellesøy are former employees of BerGenBio ASA. Drs. Chouaib and Brekken signed Sponsored Research Agreements with BerGenBio ASA related to separate research projects. Dr. Bivona reports grants from National Institutes of Health during the conduct of the study; grants and other from Novartis, Revolution Medicines, personal fees from AstraZeneca, Takeda, Strategia, Springworks, Array, Pfizer, and Rain outside the submitted work. Dr. Minna reports grants from National Cancer Institute, Margot Johnson Foundation, and CPRIT during the conduct of the study, and personal fees from National Cancer Institute and University of Texas Southwestern Medical Center outside the submitted work. Dr. Thiery is the scientific founder of Biocheetah Pte. Ltd., Singapore and advisor of Biosyngen Pte. Ltd., Singapore. The remaining authors declare no conflict of interest.
Supplementary Data
Note: To access the supplementary material accompanying this article, visit the online version of the Journal of Thoracic Oncology at www.jto.org and at https://doi.org/10.1016/j.jtho.2020.01.015.
References
- 1.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. [DOI] [PubMed] [Google Scholar]
- 2.Neel DS, Bivona TG. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis Oncol. 2017;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19:2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. [DOI] [PubMed] [Google Scholar]
- 7.Hammerlindl H, Schaider H. Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance. J Cell Commun Signal. 2018;12:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell sub-populations. Cell. 2010;141:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sulciner ML, Serhan CN, Gilligan MM, et al. Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med. 2018;215:115–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaffer SM, Dunagin MC, Torborg SR, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davidsen KT, Haaland GS, Lie MK, Lorens JB, Engelsen AST. The role of AXL receptor tyrosine kinase in tumor cell plasticity and therapy resistance In: Watnick RS, AAkslen LA, eds. Biomarkers of the Tumor Microenvironment. Springer; 2017:351–376. [Google Scholar]
- 12.Gjerdrum C, Tiron C, Hoiby T, et al. AXL is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107:1124–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies AXL as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin PA, Brekken RA, Byers LA, Heymach JV, Gerber DE. Axl receptor axis: a new therapeutic target in lung cancer. J Thorac Oncol. 2016;11:1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gugnoni M, Sancisi V, Manzotti G, Gandolfi G, Ciarrocchi A. Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 2016;7:e2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akalay I, Janji B, Hasmim M, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013;73:2418–2427. [DOI] [PubMed] [Google Scholar]
- 19.Akalay I, Janji B, Hasmim M, et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy. 2013;9:1104–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terry S, Buart S, Tan TZ, et al. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology. 2017;6: e1271858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terry S, Abdou A, Engelsen AST, et al. AXL targeting overcomes human lung cancer cell resistance to NK- and CTL-mediated cytotoxicity. Cancer Immunol Res. 2019;7:1789–1802. [DOI] [PubMed] [Google Scholar]
- 22.Garg AD, Galluzzi L, Apetoh L, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kepp O, Senovilla L, Kroemer G. Immunogenic cell death inducers as anticancer agents. Oncotarget. 2014;5:5190–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martins I, Michaud M, Sukkurwala AQ, et al. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy. 2012;8:413–415. [DOI] [PubMed] [Google Scholar]
- 25.Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–734. [DOI] [PubMed] [Google Scholar]
- 26.Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3:1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bliss CI. The toxicity of poisons applied JOINTLY1. Ann Appl Biol. 1939;26:585–615. [Google Scholar]
- 28.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. [DOI] [PubMed] [Google Scholar]
- 29.Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9: e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurtler A, Kunz N, Gomolka M, et al. Stain-free technology as a normalization tool in Western blot analysis. Anal Biochem. 2013;433:105–111. [DOI] [PubMed] [Google Scholar]
- 32.Pelissier Vatter FA, Schapiro D, Chang H, et al. High-dimensional phenotyping identifies age-emergent cells in human mammary epithelia. Cell Rep. 2018;23:1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amir el AD, Davis KL, Tadmor MD, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang M, Jeong CW, Ku JH, Kwak C, Kim HH. Inhibition of autophagy potentiates atorvastatin-induced apoptotic cell death in human bladder cancer cells in vitro. Int J Mol Sci. 2014;15:8106–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Svendsen A, Kmiecik J, et al. Targeting the NG2/CSPG4 proteoglycan retards tumour growth and angiogenesis in preclinical models of GBM and melanoma. PLoS One. 2011;6:e23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Wang J, Ng S, Lin Q, Shen HM. Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy. 2014;10:901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Broad Institute TCGA Genome Data Analysis Center. Firehose VERSION run stddata_2016_01_28. Broad Institute of MIT and Harvard; 2016, 10.7908/C11G0KM9. [DOI] [Google Scholar]
- 39.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar D, Nath L, Kamal MA, et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 2010;140:731–743. [DOI] [PubMed] [Google Scholar]
- 41.Homma K, Suzuki K, Sugawara H. The autophagy database: an all-inclusive information resource on autophagy that provides nourishment for research. Nucleic Acids Res. 2011;39:D986–D990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanzelmann S, Castelo RGJ. GSV: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu F, Li J, Jang C, Wang J, Xiong J. The role of AXL in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int J Clin Exp Pathol. 2014;7:6653–6661. [PMC free article] [PubMed] [Google Scholar]
- 44.Holland SJ, Pan A, Franci C, et al. R428, a selective small molecule inhibitor of AXL kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544–1554. [DOI] [PubMed] [Google Scholar]
- 45.Kimani SG, Kumar S, Davra V, et al. Normalization of TAM post-receptor signaling reveals a cell invasive signature for AXL tyrosine kinase. Cell Commun Signal. 2016;14:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007;3:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of Ras signaling. Cell. 1999;96:655–665. [DOI] [PubMed] [Google Scholar]
- 49.Sutterluty H, Mayer CE, Setinek U, et al. Down-regulation of Sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol Cancer Res. 2007;5:509–520. [DOI] [PubMed] [Google Scholar]
- 50.Taniguchi H, Yamada T, Wang R, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barutcu SA, Girnius N, Vernia S, Davis RJ. Role of the MAPK/cJun NH2-terminal kinase signaling pathway in starvation-induced autophagy. Autophagy. 2018;14: 1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bae SY, Hong JY, Lee HJ, Park HJ, Lee SK. Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget. 2015;6:10146–10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacobsen K, Bertran-Alamillo J, Molina MA, et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat Commun. 2017;8:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19: 5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kucharavy A, Rubinstein B, Zhu J, Li R. Robustness and evolvability of heterogeneous cell populations. Mol Biol Cell. 2018;29:1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jimbo T, Hatanaka M, Komatsu T, et al. DS-1205b, a novel selective inhibitor of AXL kinase, blocks resistance to EGFR-tyrosine kinase inhibitors in a non-small cell lung cancer xenograft model. Oncotarget. 2019;10:5152–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lotsberg ML, Chen S, Dhakal S, et al. Autophagy mediated danger signaling regulates tumor immunosurveillance and may potentiate the effects of anticancer immunotherapy through increased adjuvanticity. In: Chouaib S, Bonavida B, eds. Autophagy in Immune Response: Impact on Cancer Immunotherapy. Elsevier Academic Press; 2020. [Google Scholar]
- 62.Zou Y, Ling YH, Sironi J, Schwartz EL, Perez-Soler R, Piperdi B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J Thorac Oncol. 2013;8:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu X, Shi S, Wang H, et al. Blocking autophagy improves the anti-tumor activity of afatinib in lung adenocarcinoma with activating EGFR mutations in vitro and in vivo. Sci Rep. 2017;7:4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu D, Yang Y, Zhao S. Autophagy facilitates the EGFR-TKI acquired resistance of non-small-cell lung cancer cells. J Formos Med Assoc. 2014;113:141–142. [DOI] [PubMed] [Google Scholar]
- 65.Han W, Pan H, Chen Y, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011;6: e18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li YY, Lam SK, Mak JC, Zheng CY, Ho JC. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer. 2013;81:354–361. [DOI] [PubMed] [Google Scholar]
- 67.Han J, Bae J, Choi CY, et al. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy. 2016;12:2326–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grabiec AM, Goenka A, Fife ME, Fujimori T, Hussell T. Axl and MerTK receptor tyrosine kinases maintain human macrophage efferocytic capacity in the presence of viral triggers. Eur J Immunol. 2018;48:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aguilera TA, Rafat M, Castellini L, et al. Reprogramming the immunological microenvironment through radiation and targeting AXL. Nat Commun. 2016;7:13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aguilera TA, Giaccia AJ. Molecular pathways: oncologic pathways and their role in T-cell exclusion and immune evasion-a new role for the AXL receptor tyrosine kinase. Clin Cancer Res. 2017;23:2928–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wnuk-Lipinska K, Davidsen K, Blø M, et al. BGB324, a selective small molecule inhibitor of receptor tyrosine kinase AXL, abrogates tumor intrinsic and microenvironmental immune suppression and enhances immune checkpoint inhibitor efficacy in lung and mammary adenocarcinoma models. Cancer Res. 2017, abstract 626;77(13 suppl). [Google Scholar]
- 72.Davidsen K, Wnuk-Lipinska K, Du W, et al. Bemcentinib (BGB324)-a selective small molecule inhibitor of receptor tyrosine kinase AXL, targets tumor immune suppression and enhances immune checkpoint inhibitor efficacy. Poster presented at: AACR Annual meeting 2018. Chicago, IL; April 14–18, 2018. [Google Scholar]
- 73.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 74.Pritchard JR, Bruno PM, Hemann MT, Lauffenburger DA. Predicting cancer drug mechanisms of action using molecular network signatures. Mol Biosyst. 2013;9:1604–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pritchard JR, Bruno PM, Gilbert LA, Capron KL, Lauffenburger DA, Hemann MT. Defining principles of combination drug mechanisms of action. Proc Natl Acad Sci U S A. 2013;110:E170–E179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Galluzzi L, Bravo-San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bezu L, Gomes-de-Silva LC, Dewitte H, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bloy N, Garcia P, Laumont CM, et al. Immunogenic stress and death of cancer cells: contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev. 2017;280:165–174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.