

Summary
Chimeric antigen receptor (CAR) T cell costimulation mediated by CD28 and 4–1BB is essential for CAR-T cell-induced tumor regression. However, CD28 and 4–1BB differentially modulate kinetics, metabolism and persistence of CAR-T cells, and the mechanisms governing these differences are not fully understood. We found that LCK recruited into the synapse of CD28-encoding CAR by co-receptors causes antigen-independent CAR-CD3ζ phosphorylation and increased antigen-dependent T cell activation. In contrast, the synapse formed by 4–1BB-encoding CAR recruits the THEMIS-SHP1 phosphatase complex that attenuates CAR-CD3ζ phosphorylation. We further demonstrated that the CAR synapse can be engineered to recruit either LCK to enhance the kinetics of tumor killing of 4–1BB CAR-T cells or SHP1 to tune down cytokine release of CD28 CAR-T cells.
Graphical Abstract
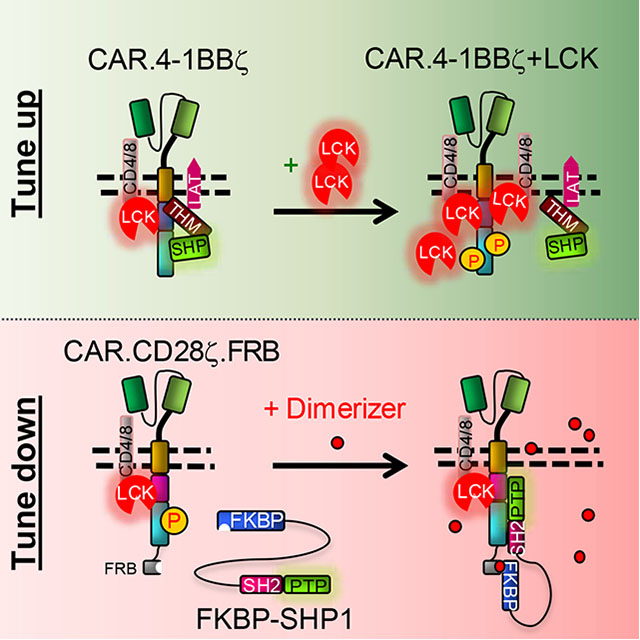
In Brief
Sun et al. uncover the mechanisms by which CD28 and 4–1BB differentially regulate the equilibrium of phosphorylation and dephosphorylation of CAR-CD3ζ to impact the magnitude of CAR-T cell activation. LCK kinase and SHP1 phosphatase can be engineered in CAR-T cells to tune their activity.
Introduction
Chimeric antigen receptors (CARs) are synthetic molecules composed of a single chain variable fragment (scFv), co-stimulatory moieties (either CD28 or 4–1BB) and a CD3ζ signaling domain that when expressed by T lymphocytes trigger their lytic machinery and costimulation upon antigen engagement (Dotti et al. 2014;Finney et al. 1998;Gross et al. 1989;Imai et al. 2004;Sadelain et al. 2013). In clinical studies, CAR co-stimulation plays an essential role in promoting the expansion of CAR-redirected T cells, and both CD28 and 4–1BB lead to equally significant clinical responses in B cell malignancies (Brentjens et al. 2013;Maude et al. 2014;Savoldo et al. 2011). However, CD28- and 4–1BB-mediated costimulation in CAR-T cells has been associated with distinct antitumor kinetics as CD28 endodomain promotes faster antitumor activity as compared to 4–1BB endodomain (Zhao et al. 2015). This phenomenon correlates with the observed pronounced glycolytic metabolism and higher susceptibility to exhaustion of the CD28-mediated costimulation as opposed to the predominantly oxidative metabolism and lower susceptibility to exhaustion of the 4–1BB-mediated co-stimulation (Kawalekar et al. 2016;Long et al. 2015).
Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and tumor necrosis factor receptor associated factors (TRAFs) are known downstream signaling molecules recruited by CD28 and 4–1BB, respectively (Arch and Thompson 1998;Frauwirth et al. 2002). However, PI3K and TRAF signaling do not explain the observed functional differences between CD28 and 4–1BB co-stimulation and it remains elusive whether key signaling events occur within the CAR synapse causing the observed kinetics of antitumor activity. Here, we investigated the molecular mechanisms underlying the functional differences between CD28 and 4–1BB co-stimulation to identify strategies for generating CAR-T cells with more predictable activity and safer clinical profiles.
Results
CD28 costimulation promotes higher activation of CAR-T cells via LCK-mediated constitutive phosphorylation of the CAR-CD3ζ domain.
To stringently compare the CD28 and 4–1BB proximal signaling in CAR-T cells, we generated two CARs that encode the same CD19-specific scFv and CD8α stalk, and either the CD28 or the 4–1BB intracytoplasmic co-stimulatory domain followed by the intracytoplasmic tail of the CD3ζ chain (CAR19.28ζ and CAR19.BBζ) (Fig.S1A). Upon activation, transduction and expansion of CAR-T cells for 10 – 14 days following clinically validated standard operating procedures (Ramos et al. 2016), the magnitude of CAR19.28ζ-T and CAR19.BBζ-T cell activation was measured by stimulating them with titrated doses of an anti-idiotype Ab (α-CAR19 Ab) that crosslinks the CAR (Diaconu et al. 2017). CAR19.28ζ -T cells showed a significantly higher magnitude of activation than CAR19.BBζ -T cells as measured by Ca2+ influx (Fig.1A) and higher expression of the early T cell activation marker CD69 in both CD4+ and CD8+ T cells (Fig.1B). Accordingly, CAR19.28ζ -T cells released more IFNγ than CAR19.BBζ-T cells (Fig.1C). In contrast, no significant differences in the expression of activation markers and cytokine release were observed when CAR19.28ζ -T and CAR19.BBζ -T cells were stimulated via T-cell-receptor (TCR) crosslinking (Fig.S1B), indicating that the costimulation associated with the CAR determines the magnitude of activation upon CAR engagement. Similar results were obtained when CAR19.28ζ -T and CAR19.BBζ -T cells were simulated via titration of tumor cells expressing the target antigen (Fig.1D and Fig.S1C). To confirm our results in vivo, CAR19.28ζ -T and CAR19.BBζ -T cells were differentially labeled and infused simultaneously in NSG (NOD-scid IL2Rgnull) mice bearing CD19+ tumor cells. T cells were then harvested 6 hr after infusion. Tumor cells (CD45+CD3−) and T cells (CD45+CD3+) were detected in the peripheral blood, bone marrow, lung, and spleen by flow cytometry (Fig.1E). Gating on CD45+CD3+ T cells and differentially labeled cells, CAR19.28ζ-T cells showed higher expression of CD69 than CAR19.BBζ -T cells in organs that contained less tumor cells, such as blood, lung and spleen (Fig.1F and Fig.S1D–F). The stronger activation of CAR19.28ζ -T cells translated into a more pronounced short-term antitumor activity as compared to CAR19.BBζ -T cells when low doses of CAR-T cells were used (Fig.S1G).
Fig.1. 28ζ-T cells show higher magnitude of activation than CAR19.BBζ-T cells after CAR crosslinking.
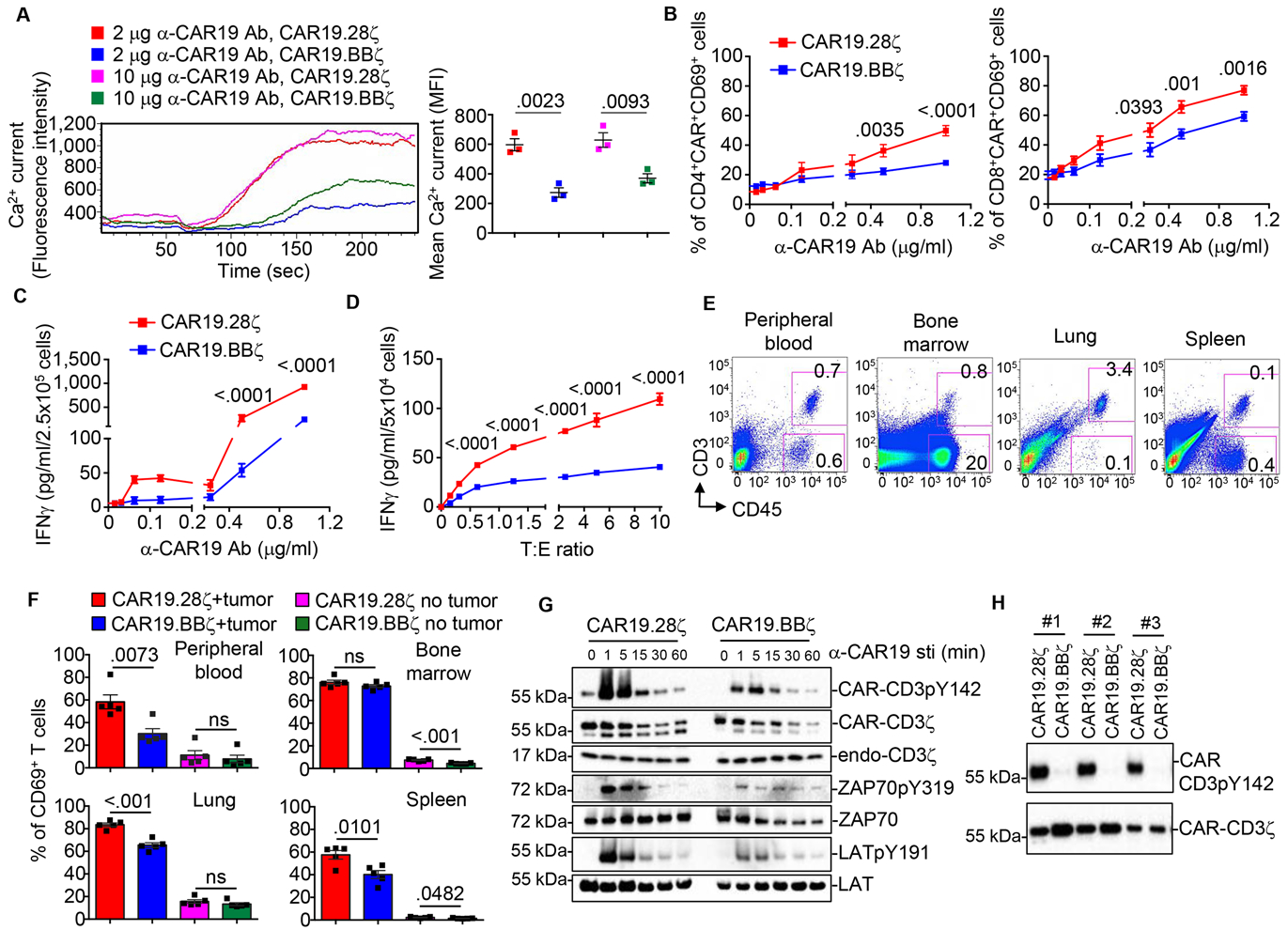
(A-C) CAR19.28ζ-T and CAR19.BBζ-T cells were stimulated with different concentrations of α-CAR19 Ab and Ca2+ influx (A), CD69 expression in CD4+ and CD8+ T cells (B), and IFNγ release (C) were measured (n = 3 in (A), one-way ANOVA; n = 17 in (B), n = 3 in (C), two-way ANOVA). (D) CAR19.28ζ-T and CAR19.BBζ-T cells were stimulated with the CD19+ BV173 cell line at different tumor to T cell ratios (T:E). IFNγ levels in the culture supernatants were measured (n = 4, two-way ANOVA). (E and F) NSG mice engrafted with the CD19+ Daudi cell line were infused with differentially labeled CAR19.28ζ-T and CAR19.BBζ-T cells mixed at a 1:1 ratio. Samples were collected 6 hr after infusion. Non-tumor bearing NSG mice, infused with mixed CAR19.28ζ-T and CAR19.BBζ-T cells, were used as a negative control. Representative flow cytometry plots showing T cells (CD45+CD3+) and Daudi tumor cells (CD45+CD3−) identified in the peripheral blood, bone marrow, lung, and spleen (E). Summary of CD69 expression in T cells (n = 5, two-tailed unpaired t-test) (F). (G) Phosphorylation of CAR-CD3ζ, ZAP70, and LAT in CAR19.28ζ-T and CAR19.BBζ-T cells after stimulation with the α-CAR19 Ab at 10 μg/ml. Cells were incubated with the α-CAR19 Ab followed by incubation with a goat anti-mouse IgG secondary antibody on ice. CAR-T cells were then transferred to 37°C for indicated time to be activa ted (n = 3). (H) CAR-CD3ζ phosphorylation of CAR19.28ζ-T and CAR.19.BBζ-T cells in the absence of CAR crosslinking. Total CAR detected by the CD3ζ chain Ab was used as equal loading. Results of 3 representative donors were shown. All data were presented as mean ± SEM. See also Figure S1 and Table S1.
Proximal signaling molecules are rapidly phosphorylated upon TCR activation in T cells (Smith-Garvin et al. 2009). We observed that CAR19.28ζ -T cells exhibited higher phosphorylation of the downstream proximal signaling molecules CAR-CD3ζ, ZAP70 and LAT when stimulated with the α-CAR19 Ab as compared to CAR19.BBζ -T cells (Fig.1G), and CAR19.28ζ -T cells consistently showed higher antigen-independent/basal phosphorylation of the CAR-CD3ζ (Fig.1H, Fig.S1H, and Table S1). The latter effect was observed with two other CARs encoding the CD28 endodomain (Fig.S1I,J), and regardless of the type of hinge or transmembrane domain used within the CAR19 constructs (Fig.S1K–N). Furthermore, both CD4+ and CD8+ T cells (Fig.S1O,P) growing with different cytokine(s) (Fig.S1Q,R) showed higher basal phosphorylation of CAR-CD3ζ in CAR encoding the CD28 endodomain.
LCK kinase confers more profound phosphorylation events in CAR19.28ζ-T cells as compared with CAR19.BBζ-T cells (Salter et al. 2018). We observed that the CAR-CD3ζ basal phosphorylation of CAR19.28ζ-T cells was abrogated by the addition of Src family kinase (PP2) or LCK inhibitor (Inh-II) (Awale and Mohan 2008) (Fig.2A). Furthermore, pretreatment of CAR19–28ζ-T cells with PP2, which significantly decreases CAR-CD3ζ basal phosphorylation, reduced their responsiveness to CAR crosslinking with low doses of α-CAR19 Ab to similar levels of CAR19.BBζ-T cells, as shown by the expression of CD69 and release of IFNγ and IL-2 (Fig.2B,C and Fig.S2A). In contrast, high doses of α-CAR19 Ab partially overcame the PP2-mediated inhibition in CAR19.28ζ-T cells (Fig.2B,C and Fig.S2B). This suggests that LCK is rapidly recruited in the CAR19.28ζ synapse and that a strong CAR aggregation mediated by high doses of α-CAR19 Ab rapidly overcomes the inhibitory effects of PP2 pretreatment. Collectively, these data indicate that higher CAR-CD3ζ basal phosphorylation primes CAR19.28ζ-T cells to a higher magnitude of activity in response to low antigen stimulation.
Fig.2. CAR-CD3ζ basal phosphorylation causes higher magnitude of activation of CAR19.28ζ-T cells than CAR19.BBζ-T cells after CAR crosslinking.
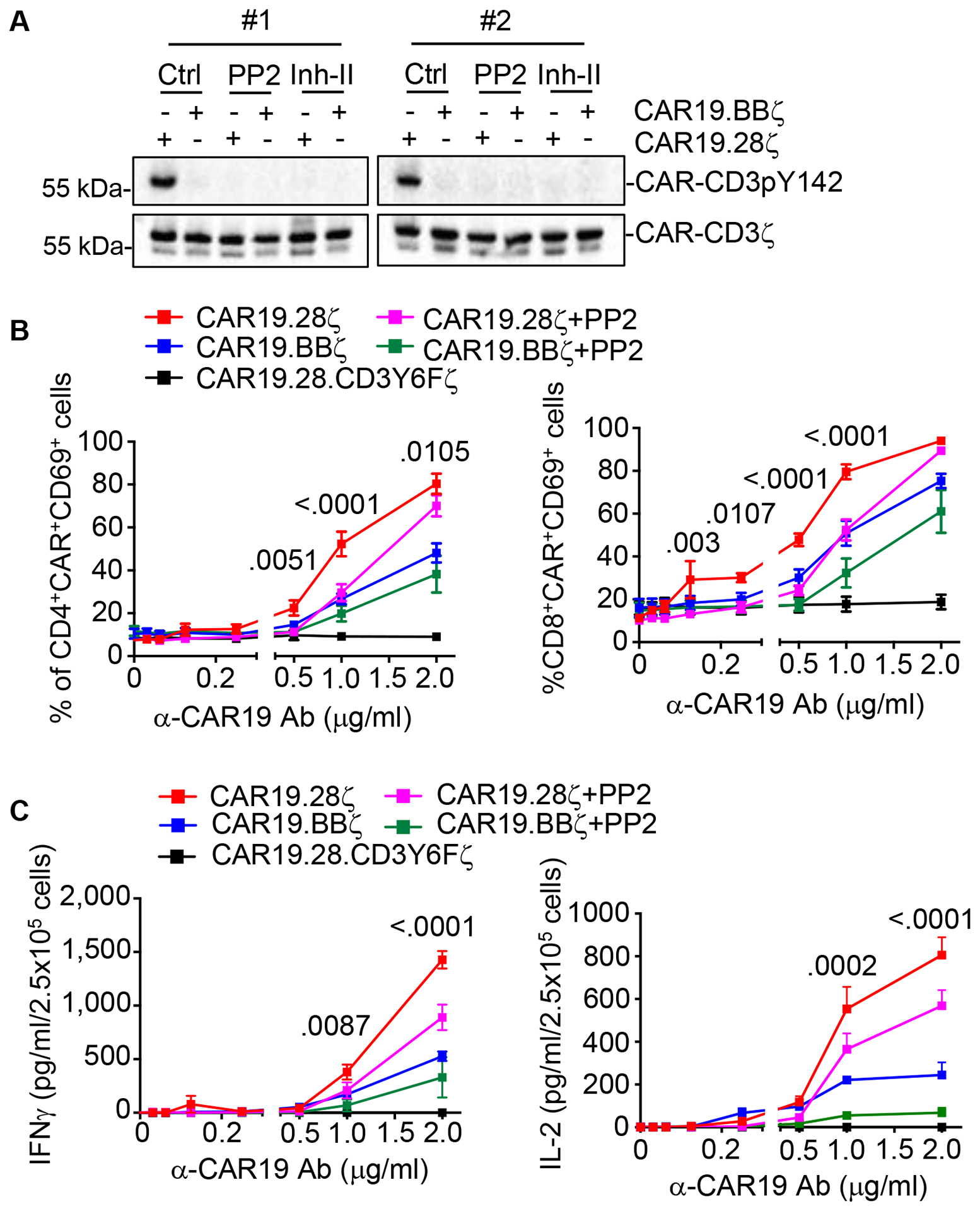
(A) Phosphorylation of CAR-CD3ζ in CAR19.28ζ-T and CAR19.BBζ-T cells treated with DMSO (Ctrl), Src family kinase (PP2) or LCK (Inh-II) inhibitors at 10 μM for 16 hr. Results of 2 representative donors were shown. (B) CD69 expression in CAR19.28ζ-T and CAR.19.BBζ-T cells pretreated with 10 μM PP2 for 16 hr and then stimulated with α-CAR19 Ab at various concentrations for 6 hr. CAR19.28ζ-T and CAR19.BBζ-T cells not exposed to PP2 were used as control. An additional negative control is represented by CAR19.CD3Y6Fζ-T cells, which are T cells expressing the CAR19 in which all six tyrosine (Y) residues of the three ITAMs of CAR-CD3ζ were mutated to phenylalanine (F) to completely abrogate tyrosine phosphorylation of the CAR-CD3ζ. (C) CAR19.28ζ-T and CAR19.BBζ-T cells were treated as in (B). Cytokine release were quantified (n = 3 for cytokine release, n = 5 for CD69 expression, two-way ANOVA; p value between CAR19.28ζ-T and CAR19.28ζ-T cells + PP2 groups). All data were presented as mean ± SEM. See also Figure S2.
Co-receptors recruits LCK within the CAR synapse.
LCK binds to the PYAPP motif of CD28 (Dobbins et al. 2016;Hofinger and Sticht 2005;Holdorf et al. 1999). However, we observed that introduction of loss-of-function mutations to the PYAPP motif of CAR19.28ζ (CAR19.28AAAζ, CAR19.28YFζ, CAR19.28AFAAζ) (Fig.3A,B) did not completely abolish the association of LCK to CAR19.28ζ (Fig.S3A). Similarly, basal CAR-CD3ζ phosphorylation was only modestly reduced (Fig.S3B,C), and the cytokine production of CAR19.28ζ-T cells remained unaffected (Fig.3C). Since no significant differences were observed between CD28 mutants and CAR19.28ζ in both phosphorylation of the CAR-CD3ζ and cytokine production, we chose one representative mutant to monitor CD69 expression after α-CAR19 activation. There were no significant differences on CD69 level between CAR19.28ζ and CAR19.28AAAζ mutation either upon CAR or TCR crosslinking (Fig.S3D,E). Furthermore, in CAR molecules pulled down from CAR.19.BBζ-T cells we observed the presence of LCK (Fig.3D) even if the human 4–1BB endodomain does not contain any known LCK binding motifs (Kim et al. 1993;Wen et al. 2002). These observations suggest that LCK is recruited to the CAR synapse by co-receptors rather than by the CAR-associated CD28 moiety (Rudd et al. 1988;Veillette et al. 1988). By co-immunoprecipitation-mass spectrometry (Co-IP-MS), we detected unique CD8α peptides spanning regions absent in the CAR-CD8α stalk in CARs pulled down from both CAR19.28ζ-T and CAR.19.BBζ-T cells (Table S2). Furthermore, overexpression of a CD8α mutant that cannot bind to LCK (CD8α-SKS, CKCP mutated to SKSP) (Turner et al. 1990) greatly reduced the basal CAR-CD3ζ phosphorylation in CD4+ T cells expressing the CAR19.28ζ, while CD4+ T cells co-expressing CAR.19.BBζ and the wild-type CD8α (CD8α-WT) showed increased basal CAR-CD3ζ phosphorylation (Fig.3E and Fig.S3F). We did not find any peptide of the CD4 co-receptor in the CAR19.28ζ pull-down product in Co-IP-Ms even if CD4+ T cells were still present at day 10 – 14 of culture. However, overexpression of the wild-type CD4 in both CD4+ and CD8+ CAR19.BBζ-T cells increased the basal phosphorylation of CAR-CD3ζ (Fig.3F). As observed for TCR signaling (Wiest et al. 1996), our data support the conclusion that LCK recruited to the CAR synapse by either CD8 or CD4 co-receptors plays a major role in triggering the basal CAR-CD3ζ phosphorylation.
Fig.3. Co-receptors bring LCK into the CAR synapse.
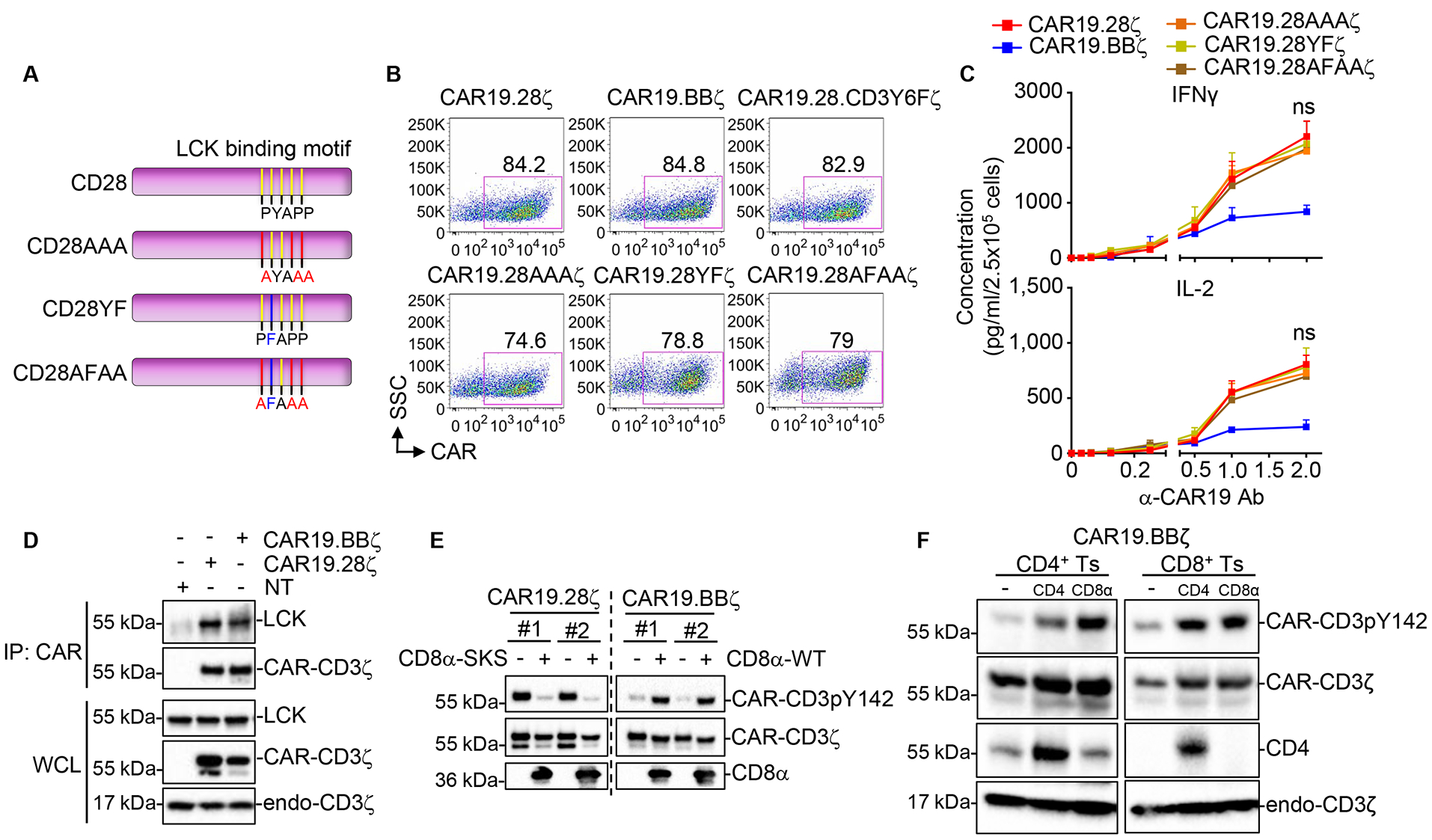
(A) Schema of the CAR19.28ζ constructs in which specific mutations were included to generate CD28AAA, CD28YF, and CD28AFAA. (CD28AAA, mutation of PYAPP to AYAAA; CD28YF, mutation of PYAPP to PFAPP; CD28AFAA, mutation of PYAPP to AFAAA). (B) Representative flow plots showing the expression of CAR19 in transduced T cells at day 6 of culture. (C) Cytokine release of CAR-T cells stimulated with the α-CAR19 Ab at different concentrations for 6 hr. Cells were collected at day 14 of culture (n = 3, two-way ANOVA; ns, not significant between CAR19.28ζ-T and CAR19.28AAAζ-T, CAR19.28YFζ-T, or CAR19.28AFAAζ-T cells). (D) LCK detection by IP in CAR molecules pulled down from CAR19.28ζ-T and CAR19.BBζ-T cells collected at day 14 of culture. (E) Phosphorylation of CAR-CD3ζ Y142 in CD4+ T cells expressing either CAR19.28ζ or CAR19.BBζ and co-transduced with the CD8α mutant (CD8α-SKS) or wild-type CD8α (CD8α-WT). Results of 2 representative donors are shown. (F) CAR-CD3ζ pY142 phosphorylation in CD4+ or CD8+ CAR19.BBζ-T cells overexpressing either CD4 or CD8α. All data were presented as mean ± SEM. See also Figure S3 and Table S2.
THEMIS-SHP1 counteracts the effect of LCK in the CAR synapse of CAR-T cells encoding 4–1BB.
Although co-receptors bring LCK into the CAR19.BBζ synapse (Fig.3D–F, Fig.S3F, and Table S2), we observed very low basal phosphorylation of the CAR-CD3ζ, suggesting that 4–1BB may recruit phosphatases to the CAR synapse that counter the LCK-mediated phosphorylation. Indeed, the tyrosine phosphatase inhibitor Na3VO4 promoted antigen-independent phosphorylation of CAR-CD3ζ in CAR19.BBζ-T cells (Fig.4A). Accordingly, Co-IP-MS demonstrated that THEMIS is more abundantly associated with CAR19.BBζ than CAR19.28ζ (Table S2). While THEMIS does not have direct phosphatase activity, it binds to the phosphatase SHP1, and then the THEMIS-SHP1 complex is recruited by LAT to the TCR synapse to regulate T cell activation (Paster et al. 2015). THEMIS pull-down in T cells and Jurkat cells co-expressing THEMIS and CARs confirmed a stronger interaction between THEMIS and CAR19.BBζ than THEMIS and CAR19.28ζ (Fig.S4A,B). Knockdown of THEMIS or SHP1 in CAR19.BBζ-T cells using shRNA increased their CAR-CD3ζ basal phosphorylation (Fig.4B,C and Fig.S4C,D), indicating that the THEMIS-SHP1 complex negatively regulates CAR-CD3ζ phosphorylation of CAR19.BBζ. To map the domain of 4–1BB interacting with THEMIS, we generated 4–1BB mutants and found that the COOH-terminal deletion of 10 amino acids abolishes 4–1BB interaction with THEMIS (Fig.4D,E and S4E), leading to increased CAR-CD3ζ phosphorylation (Fig.4F), Ca2+ influx (Fig.4G and Fig.S4F), CD69 expression, and IFNγ release (Fig.4H and Fig.S4G,H) by the activated CAR19.BBζ-T cells. These data support the conclusion that the THEMIS-SHP1 complex selectively attenuates CAR-CD3ζ phosphorylation in CAR-T cells expressing 4–1BB.
Fig.4. THEMIS-SHP1 complex attenuates CAR-CD3ζ basal phosphorylation of CAR19.BBζ-T cells.
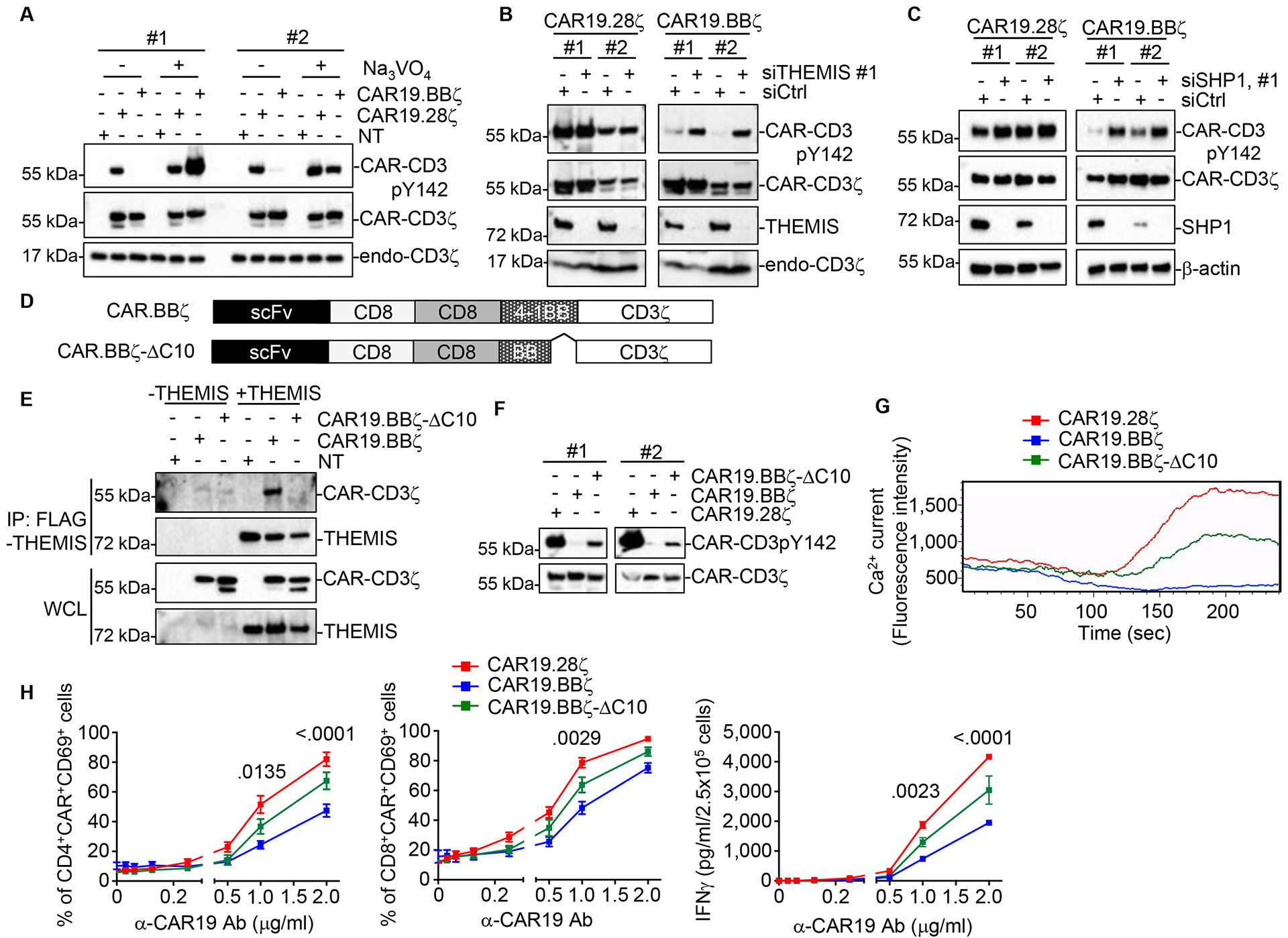
(A) Phosphorylation of CAR-CD3ζ Y142 in CAR19.28ζ-T and CAR19.BBζ-T cells treated with 200 μM phosphatase inhibitor Na3VO4. NT, non-transduced control. Results of 2 representative donors are shown. (B) THEMIS expression and CAR-CD3ζ Y142 phosphorylation in CAR19.28ζ-T and CAR19.BBζ-T cells co-transduced with vectors encoding shRNA specific for THEMIS and selected by puromycin. Results of 2 representative donors are shown. (C) CAR19.28ζ-T and CAR19.BBζ-T cells were co-transduced with vectors encoding shRNA specific for SHP1 and selected by puromycin. CAR-CD3ζ Y142 phosphorylation and SHP1 were measured by western blot. Results of 2 representative donors were shown. (D) Structures of CAR19.BBζ and its mutant CAR19.BBζ-ΔC10 (deletion of 10 AA at the COOH of 4–1BB). (E) CAR19.BBζ-T or CAR19.BBζ-ΔC10-T cells were co-transduced with a vector encoding FLAG-tagged THEMIS. At day 14 of culture cells were collected and THEMIS was immunoprecipitated by FLAG IP. CAR in the IP product was detected by western blot using the α-CD3ζ Ab. (F) Phosphorylation of CAR-CD3ζ Y142 in CAR19.BBζ-T and CAR19.BBζ-ΔC10-T cells. Results of 2 representative donors are shown. (G) Ca2+ influx in CAR19.28ζ-T cells, CAR19.BBζ-T cells and CAR19.BBζ-ΔC10-T cells following stimulation with the α-CAR19 Ab. Representative of 3 donors. (H) CD69 expression and IFNγ release in CAR19.28ζ-T cells, CAR19.BBζ-T cells, and CAR19.BBζ-ΔC10-T cells upon α-CAR19 Ab stimulation (n = 3 for IFNγ release, n = 5 for CD69 expression, two-way ANOVA; p value between CAR19.BBζ-T cells and CAR19.BBζ-ΔC10-T cells groups). All data were presented as mean ± SEM. See also Figure S4.
Engineering LCK kinase fine-tunes the antitumor activity of CAR-T cells encoding 4–1BB.
LCK has access to the CAR19.BBζ synapse, but its kinase activity is limited by the presence of the THEMIS-SHP1 complex. While the COOH-terminal deletion of 10 amino acids abolishing the 4–1BB interaction with THEMIS promotes the rapid activation of CAR19.BBζ-T cells, this deletion also abrogates the binding to TRAF2, which would compromise 4–1BB signaling (Jang et al. 1998). In contrast, LCK overexpression in CAR19.BBζ-T cells may break the balance of kinases and phosphatases within the CAR synapse and promote basal CAR-CD3ζ phosphorylation (Fig.5A). We found that overexpressed LCK retains its native N-terminal myristoylation and palmitoylation (Resh 1994), and thus accumulates in the cell membrane of CAR19.BBζ-T cells (Fig.5B) increasing the basal phosphorylation of CAR-CD3ζ (Fig.5C) and Ca2+ influx upon antigen stimulation (Fig.5D and Fig.S5A). CAR19.BBζ-T cells co-expressing LCK expanded numerically in vivo in a lymphoma tumor model (Fig.5E), and showed better control of tumor growth at a suboptimal cell dose as compared to CAR19.BBζ-T cells (Fig.5F and Fig.S5B). Moreover, CAR19.BBζ-T cells co-expressing LCK better controlled tumor growth after tumor re-challenge as compared to CAR19.28ζ-T cells (Fig.5F). The beneficial effect of LCK overexpression in CAR-T cells encoding 4–1BB was also observed in a neuroblastoma model targeting the GD2 antigen (Fig.5G,H and Fig.S5C), without causing any increase in the expression of PD-1 and TIM3 in CAR-T cells (Fig.5I). These data support the conclusion that LCK overexpression in CAR-T cells expressing 4–1BB promotes faster antitumor activity without compromising their intrinsic enhanced persistence.
Fig. 5. Engineering LCK kinase in CAR-T cells encoding 4–1BB enhances their antitumor activity in vitro and in vivo.
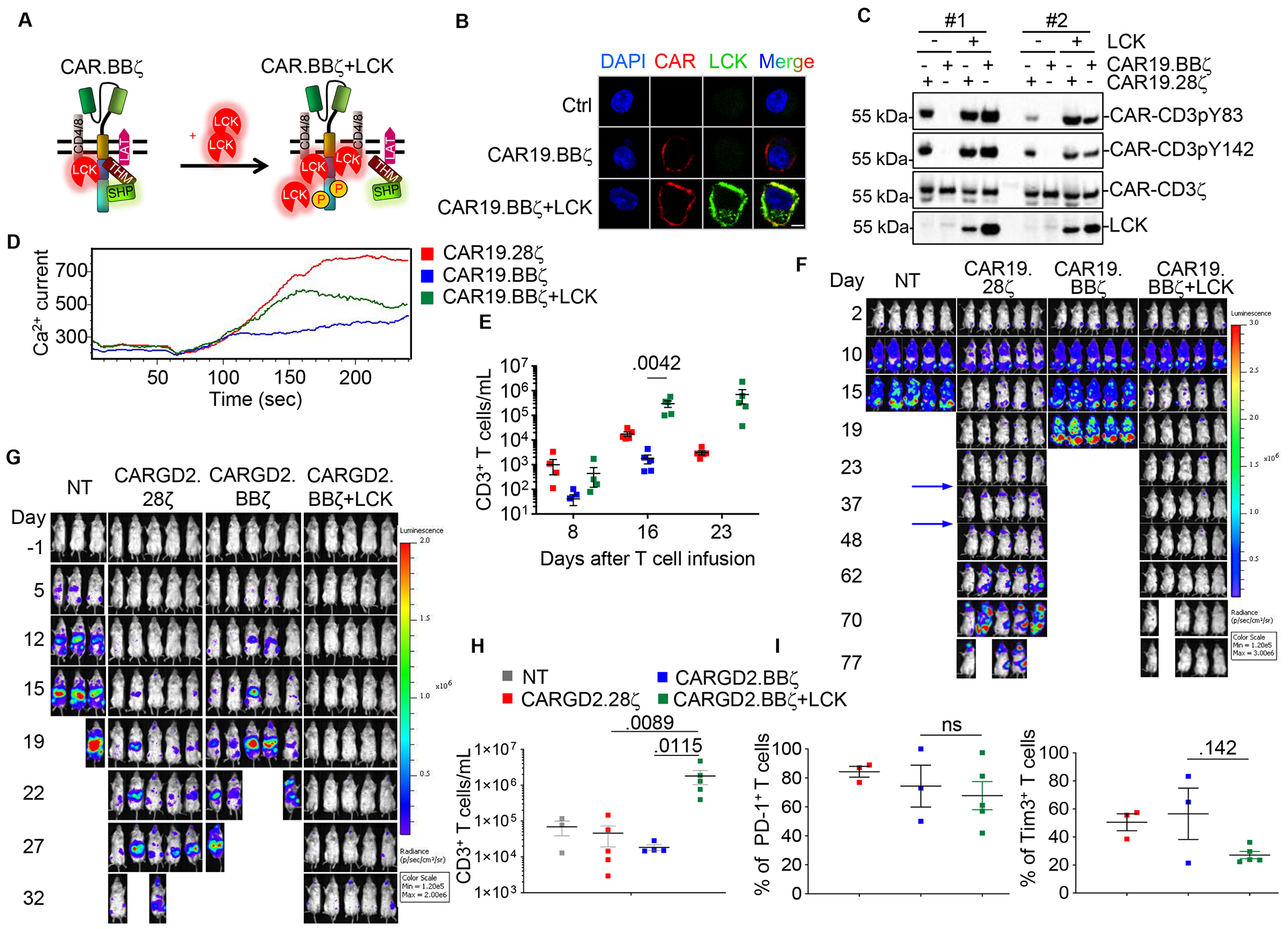
(A) Schema illustrating LCK engineering to counter the phosphatase activity of the THEMIS-SHP1 complex in CAR19.BBζ-T cells. (B) Confocal microscopy showing the LCK accumulation in the membrane in CAR19.BBζ-T cells co-transduced with the FLAG-tagged LCK. Scale bar represents 5 μm. The experiment was replicated in 3 donors. (C) Phosphorylation of the CAR-CD3ζ Y83 and Y142 in CAR19.28ζ-T and CAR19.BBζ-T cells expressing LCK. Results of 2 representative donors were shown. (D) Representative of Ca2+ influx in CAR19.28ζ-T cells and CAR19.BBζ-T cells with or without co-expression of LCK. (E and F) Quantification of T cells in the peripheral blood (E) and tumor growth monitored by BLI (F) in NSG mice engrafted with the CD19+ Daudi cells and infused with CAR19.28ζ-T cells or CAR19.BBζ-T cells with or without LCK. Re-challenging with Daudi cells was performed at day 25 and 40 after initial CAR-T cell infusion (indicated by arrows) (n = 5, one-way ANOVA). (G) Tumor growth monitored by BLI in NSG mice engrafted with the neuroblastoma tumor cell line CHLA-255 and infused with either CARGD2.28ζ -T or CARGD2.BBζ -T cells with or without LCK. (H and I) T cell number at day 18 (H), and expression of PD-1 and TIM3 at day 22 (I) in circulating CAR-T cells in mice engrafted with the neuroblastoma tumor cells and treated as described in (G) (n = 3 in NT group, n = 5 in other groups, one-way ANOVA). All data were presented as mean ± SEM. See also Figure S5.
Engineering SHP1 phosphatase fine-tunes the effector function of CAR-T cells encoding CD28.
CAR-T cells infused in patients with significant leukemia or lymphoma tumor burden cause cytokine release syndrome (CRS) (Lee et al. 2014). While the administration of a monoclonal antibody blocking the IL-6 receptor has been demonstrated effective in attenuating the CRS, the implementation of a precise pharmacologic control of CAR-T cells remains highly appealing (Diaconu et al. 2017;Foster et al. 2017;Wu et al. 2015). We modified SHP1 allowing it to be pharmacologically recruited to the CAR synapse via the FKBP-FRB heterodimerization process (Fig.6A and Fig.S6A). In response to the heterodimerization small molecule AP21967, FKBP-SHP1 forms heterodimers with CAR19.28ζ.FRB, tunes down CAR-CD3ζ phosphorylation upon antigen binding (Fig.6B) and reduces IFNγ release by CAR19.28ζ-T cells in a reversible manner without compromising their antitumor activity in vitro (Fig.6C and Fig.S6B,C). Similarly, in a xenograft lymphoma model (Fig.6D), the administration of the AP21967 in vivo reduced IFNγ release by CAR19.28ζ-T cells co-expressing SHP1, without impairing their antitumor effects (Fig.6E,F).
Fig. 6. Engineering SHP1 phosphatase in CAR-T cells encoding CD28 ameliorates cytokine release syndrome.
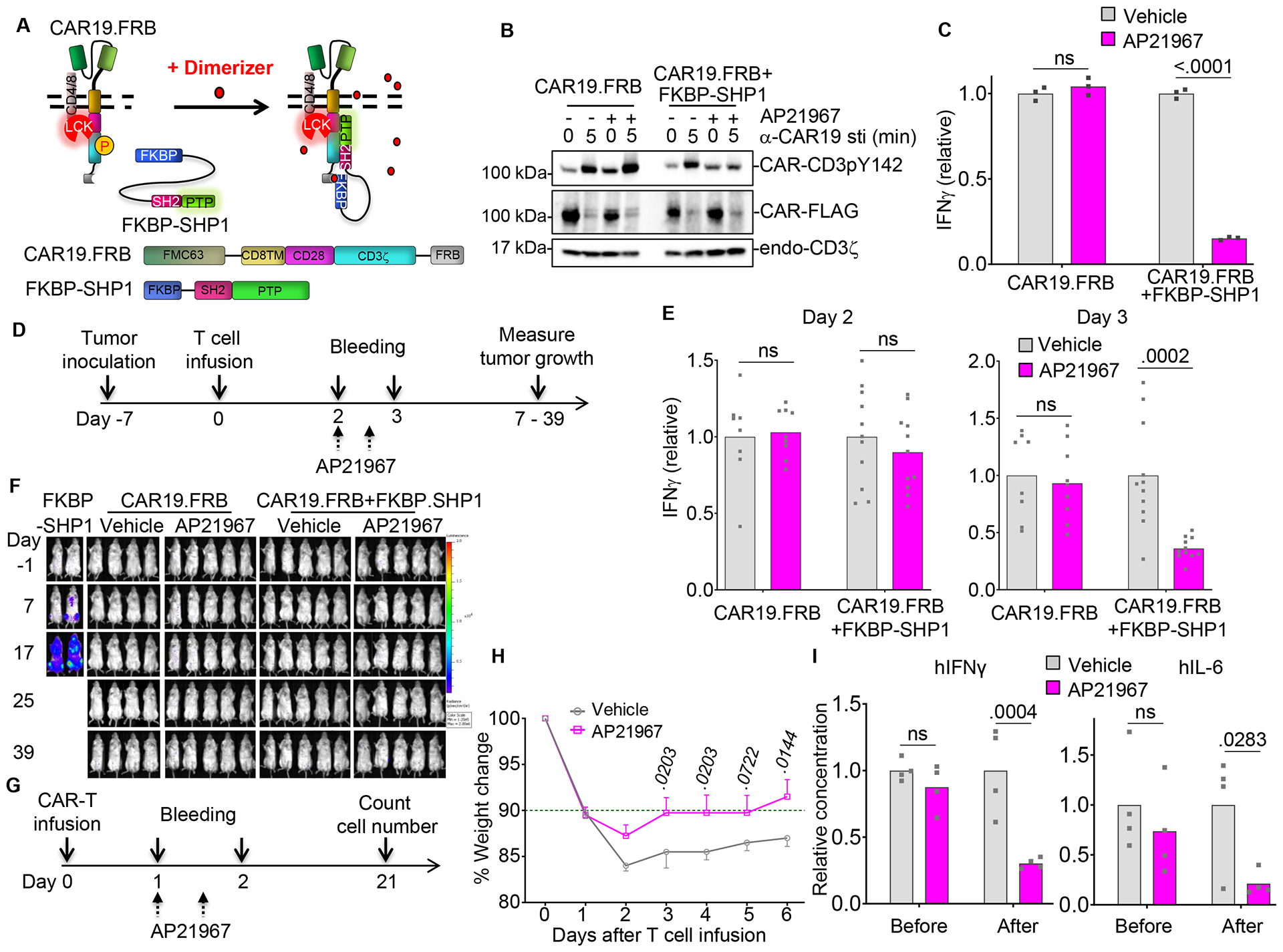
(A) Schema of the FRB and FKBP domain engineering of SHP1 and CARs to pharmacologically control SHP1 recruitment to the CAR19.28ζ synapse. (B) Phosphorylation of CAR-CD3ζ pY142 in CAR19.28ζ.FRB-T cells alone or co-transduced with FKBP-SHP1 after the stimulation with αCAR19 Ab in the presence of vehicle (ethanol) or 1 μM AP21967. The experiment was replicated in 3 donors. (C) IFNγ release in the supernatant by CAR19.28ζ.FRB-T cells alone or co-transduced with FKBP-SHP1 and incubated with the CD19+ BV173 tumor cell line at a 1:5 ratio in the presence of vehicle or AP21967 (n = 3, values were normalized to average values in vehicle groups, two-way ANOVA; representative of 3 donors). (D) Schema of the NSG mouse model to evaluate the effects of SHP1 heterodimerization in CAR-T cells in vivo. Plasma was collected at day 2 and 3 after T cell infusion. AP21967 was administrated twice at day 2 every 12 hr. (E) IFNγ level in the plasma of mice illustrated in (D) before (Day 2) and after (Day 3) the administration of vehicle or AP21967 (n = 9 – 11, values were normalized to average values in vehicle groups, two-way ANOVA; pooled data from 2 independent experiments). (F) Representative tumor growth monitored by BLI in NSG mice described as in (D) (n = 4 – 5, representative of 2 independent experiments). (G) Schema of the humanized mouse model to evaluate the effects of SHP1 heterodimerization in CAR-T cells in vivo. Plasma was collected at days 1 and 2 after the infusion of CAR-T cells targeting CD19 and co-expressing SHP1. AP21967 was administrated twice at day 1 every 12 hr. (H and I) Weight change (H), and cytokine detection (I) in humanized mice illustrated in (G) after CAR-T cell infusion. Weight was normalized to the starting weight before CAR-T cell infusion. Human IFNγ and IL-6 were detected before (Day 1) and after (Day 2) the administration of vehicle or AP21967 (n = 4, two-way ANOVA). All data were presented as mean ± SEM. See also Figure S6.
Since NSG immunodeficient mice are not appropriate models for CRS we used a humanized mouse model in which CAR-T cell inoculation causes the release of human IL-6 in the plasma of the treated mice (Diaconu et al. 2017;Norelli et al. 2018). This model reconstitutes high amount of normal human B lymphocytes expressing CD19 that can be targeted by CD19-specific CAR-T cells (Diaconu et al. 2017). In these humanized mice (Fig.6G), SHP1 recruitment induced by the transient administration of AP21967 alleviated their weight loss (Fig.6H), and significantly reduced the release in the plasma of human IFNγ, IL-6, GM-CSF, and TNFα (Fig.6I and Fig.S6D). Importantly, the transient use of AP21967 did not impair the therapeutic effect of CAR19.28ζ-T cells in vivo (Fig.S6E). The effect of SHP1 heterodimerization in vivo on cytokine release was further validated in another CRS model, using SCID-beige mice (Giavridis et al. 2018) (Fig.S6F,G). Taken together, these data demonstrate that the transient recruitment of SHP1 within the CAR19.28ζ synapse by the heterodimerization small molecule AP21967 can tune down the severity of CRS without impairing the antitumor effects of CAR-T cells.
Discussion
Costimulation plays a fundamental role in promoting the antitumor effects and persistence of CAR-redirected T cells. Here, we demonstrated that CD28 and 4–1BB endodomains incorporated into CAR molecules differentially regulate the equilibrium of phosphorylation and dephosphorylation of the CAR-CD3ζ endodomain and this in turn regulates the magnitude of CAR-T cell activation. Furthermore, we provide data supporting the notion that the equilibrium of LCK/THEMIS-SHP1 can be genetically manipulated to modulated efficacy and safety of CAR-T cells.
It has been recognized in clinical trials that CD19-specific CAR-T cells encoding either CD28 or 4–1BB costimulatory endodomains promote equal antitumor activity in patients with B cell malignancies (Brentjens et al. 2013;Maude et al. 2014). However, preclinical models showed that CD28 and 4–1BB have fundamental biological differences that may be critical in inducing antitumor effects and in promoting persistence of CAR-T cells. CD28 costimulation is generally associated with a very rapid tumor clearance as compared to 4–1BB, but also more pronounced propensity to exhaustion or less formation of memory T cells as indicated by their short term persistence (Long et al. 2015;Zhao et al. 2015). Here we mechanistically link the rapid kinetics of CD28 expressing CAR-T cells with the basal phosphorylation of the CAR-CD3ζ endodomain. The basal phosphorylation is caused by LCK recruited within the CAR synapse, which is largely mediated by co-receptors rather than the CD28 incorporated within the CAR, and imprints CAR-T cells to higher magnitude of response immediately upon encountering the antigen. In sharp contrast, the synapse formed by CAR molecules containing the 4–1BB endodomain selectively recruits the THEMIS-SHP1 complex that dephosphorylates the CAR-CD3ζ endodomain and attenuates T cell activation. We demonstrated a direct interaction between THEMIS and 4–1BB encoded within the CAR by overexpressing a tagged THEMIS, while we were not able to show the direct interaction with the endogenous THEMIS. We attribute this weakness to the lack of appropriate reagents to immune precipitate the endogenous THEMIS. However, all our performed experiments highlight the biological relevance of the discovered interaction between THEMIS and 4–1BB in CAR molecules.
Our mechanistic discoveries have immediate translational implications because kinases and phosphatases can be engineered in CAR-T cells to either enhance or tune down their activation. We demonstrated that LCK when overexpressed in CAR19.BBζ-T cells breaks the balance of kinases and phosphatases within the CAR synapse increasing the basal phosphorylation of CAR-CD3ζ and the speed of their antitumor effects. However, while CD28 costimulation prone CAR-T cells to rapid contraction and possible exhaustion, LCK overexpression in 4–1BB costimulated CAR-T cells does not affect the intrinsic property of 4–1BB signaling to promote T cell long persistence since the antitumor response is maintained after tumor re-challenge. Enhancing the kinetic of antitumor activity of 4–1BB, while preserving their longevity, may be critical in controlling rapidly progressive solid tumors.
Toxicities associated with CAR-T cells such as CRS remain of concern (Lichtman and Dotti 2017). Pharmacologic and remote control of CAR-T cells has been explored to modulate CAR-T cell expansion in vivo and mitigate side effects (Diaconu et al. 2017;Foster et al. 2017;Wu et al. 2015). While the remote control of T cell proliferation remains challenging in the clinical setting, our mechanistic observation suggests that a temporary break of T cell activation can be achieved via phosphatase engineering. Phosphatases play a critical role in attenuating CAR signaling and we demonstrated that SHP1 can be pharmacologically recruited to the CAR synapse and temporarily attenuate T cell function without eliminating CAR-T cells.
In summary, CD28 and 4–1BB differentially regulate the equilibrium of phosphorylation and dephosphorylation of the CAR-CD3ζ, which in turn regulates the magnitude of CAR-T cell activation. Engineering kinases and phosphatases can be used to tune CAR-T cell function for adoptive immunotherapy.
STAR ★★★ METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Plasmids generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gianpietro Dotti (gdotti@med.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and cell culture.
293T cells were cultured in IMDM (Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS, HyClone, Thermo Scientific), 2 mM GlutaMax, 100 I.U./mL penicillin and 100 μg/mL streptomycin (Invitrogen). BV173, Jurkat, Daudi-FFLuc, Raji-FFluc, and CHLA-255-FFluc cell lines were cultured in RPMI-1640 (Gibco, Invitrogen) supplemented with 10% FBS (HyClone), 2 mM GlutaMax, 100 I.U./mL Penicillin and 100 μg/mL Streptomycin (Invitrogen). All cell lines were routinely tested for mycoplasma.
Human samples.
For the humanized mouse construction, human fetal liver tissues were obtained from elective or medically indicated termination of pregnancy through a non-profit intermediary working with outpatient clinics (Advanced Bioscience Resources, Alameda, CA). The use of the tissue in research had no influence on the decision regarding termination of the pregnancy. Informed consent of the maternal donor is obtained in all cases, under regulation governing the clinic. We were provided with no information regarding the identity of the patients, nor is this information traceable. The project was reviewed by Office of Human Research Ethics at the University of North Carolina at Chapel Hill, which has determined that this submission does not constitute human subjects research as defined under federal regulations [45 CFR 46.102 (d or f) and 21 CFR 56.102(c)(e)(l)] and does not require IRB approval.
Mouse studies.
Male and female NSG (NOD-scid IL2Rgnull) mice were purchased from the Animal Core Facility at UNC. Female SCID-beige (C.B-Igh-1b/GbmsTac-Prkdcscid-LystbgN7) mice were purchased from Taconic Biosciences. All the mice were housed in the Animal Core Facility at UNC. All mouse experiments were performed in accordance with UNC Animal Husbandry and Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by UNC IACUC. For the humanized mouse model to evaluate the effects of SHP1 heterodimerization in CAR-T cells in vivo, humanized NSG mice (NSG-hu HSC) were generated as previously reported (Li et al. 2014). Briefly, human fetal liver tissues were obtained from elective or medically indicated termination of pregnancy through a non-profit intermediary working with outpatient clinics (Advanced Bioscience Resources). CD34+ hematopoietic stem cells (HSC) were transplanted in newborn NSG mice through intra-liver injection of 2 × 105 purified HSC. Human immune cell engraftment was detected by flow cytometry 12 weeks after transplantation. This model reconstitutes high amount of normal human B cells that can be targeted by CD19-specific CAR-T cells, which causes the release of human IL-6 in the plasma of the treated mice.
METHOD DETAILS
Plasmids.
CD19-specific CARs were constructed using the scFv from the FMC63 monoclonal antibody (Ab) the CD8α stalk including hinge and transmembrane domain and CD3ζ chain intracytoplasmic domain (Diaconu et al. 2017). CAR19.28ζ and CAR19.BBζ contained CD28 and 4–1BB endoplasmic domains, respectively. CAR19.28AAAζ (mutation of PYAPP to AYAAA), CAR19.28YFζ (mutation of PYAPP to PFAPP), CAR19.28AFAAζ (mutation of PYAPP to AFAAA) and CAR19.BBζ-ΔC10 were generated by overlapping PCR. CARGD2 (targeting the GD2 antigen) and CAR138 (targeting the CD138 antigen) were generated by replacing the scFv of CAR19. CARs with the IgG1 hinge or the CD28 transmembrane domain were cloned by overlapping PCR to replace CD8α hinge or transmembrane domain. CAR19.28.CD3Y6Fζ and CAR19.BB.CD3Y6Fζ were generated by gene synthesis (GeneArt, Thermo Scientific) and cloned into the original CAR19.28ζ construct. The full-length human THEMIS (accession_NM_001164685.1) and LCK (accession_NM_001042771.2) were PCR amplified from a cDNA library of activated T cells, and cloned into the SFG retroviral vector after the addition of the HA or FLAG tags. Gene expression was verified in both 293T and T cells by western blot. FRB and FKBP domains were cloned by PCR from plasmid PM-FRB-mRFP-T2A-FKBP-5-ptase (Addgene #40896). SHP1 full length (accession_NM_002831.5) was PCR amplified from a T cell cDNA library, and cloned into the SFG vector with FKBP. CAR19.28ζ.FRB.FLAG was generated by overlapping PCR. Lentiviral constructs encoding shRNAs were obtained from UNC shRNA Core Lab and tested for knockdown efficiency in primary T cells. Two functional shRNAs were selected for functional assays.
Retrovirus and lentivirus production.
Retroviral supernatants were prepared as previously described (Vera et al. 2006). Briefly, 293T cells were transfected with 3 plasmids (retroviral transfer vector, Peg-Pam-e encoding gag-pol, and RDF encoding the RD114 envelope), using GeneJuice transfection reagent (Novagen). Supernatants were collected at 48 and 72 hr. Lentiviral supernatants were produced in 293T cells with 3 different plasmids (lentiviral transfer vector, ps.pAX2 for lentiviral gag-pol and pMD.2G for VSV-G envelope). Supernatant was collected at 48 hr for transduction of T cells.
Transduction and expansion of human T cells.
Buffy coats from healthy donors were obtained through the Gulf Coast Regional Blood Center, Houston, TX. Peripheral blood mononuclear cells (PBMCs) were isolated with Lymphoprep density separation (Fresenius Kabi Norge) and activated using 1μg/mL α-CD3 (Miltenyi Biotec) and 1 μg/mL α-CD28 (BD Biosciences) mAb coated plates. Forty eight hr later, T lymphocytes were transduced with retroviral or lentiviral supernatants using retronectin-coated plates (Takara Bio), and expanded in complete medium (45% RPMI-1640 and 45% Click’s medium (Irvine Scientific), 10% FBS (Hyclone), 2mM GlutaMAX, 100 I.U./mL of Penicillin and 100 μg/mL of Streptomycin) with IL-7 (10 ng/mL; PeproTech) and IL-15 (5 ng/mL; PeproTech) or IL-2 (50 U/ml; R&D) (Zhou et al. 2014). Four to seven days later, cells were collected for in vitro or in vivo experiments. Lentiviral transduced cells were selected in 1 μg/ml puromycin (Sigma) for 3 – 5 days before T cells were used in functional assays.
Flow cytometry and cell phenotyping.
CAR19 expression on T cells was detected with CAR19 α-idiotype antibody followed by a goat α-mouse APC secondary Ab (BD bioscience) (Diaconu et al. 2017). Murine α-human CD3, CD4, CD8, CD45, CD69 and CD19 Abs were obtained from BD Bioscience. Samples were acquired on a Canto II or Fortessa flow cytometer from BD and data were analyzed using the FlowJo software (Tree star).
Activation of CD19-specific CAR-T cells with the anti-idiotype Ab or CD19+ tumor cells.
α-CAR19 and α-CD3 Abs were serially diluted 2-fold, and coated on non-tissue culture treated 96-well plates for 16 hr. Plates were washed twice before plating T cells (2.5 × 105 cells/well). Plates were centrifuged at 1,000g for 5 min, and incubated at 37°C for 6 hr. Plates were then briefly spun, and 200 ml of the supernatant was collected for ELISA. Cells were collected and stained with α-CAR19, CD4-PE, CD69-FITC Abs and Zombie Aqua™ dye (Biolegend) at 4°C for 30 min. Samples were fixed and stored for flow cytometry analysis. For tumor cell-mediated activation, 5 × 104 BV173 tumor cells were seeded in each well of 96-well plates, and T cells were serially diluted and seeded. Six hr later, supernatant was collected for ELISA.
Cytokine measurements.
Cytokines in culture supernatants and plasma were measured using enzyme-linked immunosorbent assay (ELISA) or magnetic luminex assay following manufacturer’s instructions (R&D Systems). Data were collected and analyzed using the Lumina-200 System and the Bio-Plex Manager 6.1 software (Bio-Rad).
Ca2+ influx assay.
T cells were incubated with Ca2+ indicator as per manufacturer’s instructions (Ca2+ influx assay, BD Bioscience). Cells were incubated with α-CAR19 Ab followed by goat α-mouse secondary Ab on ice. Ca2+ current was measured by flow cytometry for time-lapsed fluorescence change. Cells were first collected on ice as the baseline of the Ca2+ current, and then activated at 37°C for the Ca2+ current during T cell activation.
Immunoprecipitation (IP).
Proteins from T cells were extracted in RIPA lysis buffer (Thermo Scientific) supplemented with 1 × protease/phosphatase Inhibitors (Thermo Scientific). Equal amount of total proteins was used for IP. Rabbit/mouse IgG (Thermo Scientific) with protein G magnetic beads (Bio-Rad) was used to pre-clear the lysate and α-CAR19, α-HA or α-FLAG Abs were incubated with lysate for 16 hr, and protein G beads were then used for the pull-down. IP products were dissolved in 2 × SDS Laemmli buffer for Western blot analysis. For mass spectrometry, >5 × 108 T cells were lysed and α-CAR19 Ab was first crosslinked on protein G beads with Dimethyl pimelimidate (DMP, Thermo Scientific). 2 × SDS Laemmli buffer without β-mercaptoethanol was used to dissolve the IP product, and β-mercaptoethanol was supplemented before western blot.
Western blot.
Protein lysate was normalized according to the amount of CAR expression and resolved on 4% - 15% SDS polyacrylamide gel electrophoresis gels (SDS-PAGE, Bio-Rad). After protein transfer onto Polyvinylidene fluoride membranes (Bio-Rad), membranes were blocked in 5% non-fat milk in TBS-T and incubated with primary and secondary Abs in TBS-T with 1% milk. The following Abs were used: α-CD3pY83 and α-CD3pY142 (Abcam), α-ZAP70pY319, α-ZAP70, α-LATpY191, α-LAT, α-LCK, α-THEMIS, α-SHP1 and α-HA tag (Cell Signaling Technology), α-CD3ζ, α-FKBP, α -β-actin (Santa Cruz), α-FLAG (M2) (Sigma) and horseradish peroxidase conjugated secondary Abs (Goat-α-mouse, Goat-α-Rabbit from Thermo Scientific). Membranes were developed with with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) on a Gel station (Bio-Rad).
Mass spectrometry.
Immunoprecipitated samples (3 biological replicates) were subjected to SDS-PAGE and stained with coomassie. Lanes for each sample were excised and the proteins were reduced, alkylated, and in-gel digested with trypsin overnight at 37°C. Peptides were extracted, desalted with C18 spin columns (Pierce) and dried via vacuum centrifugation. Peptide samples were stored at −80°C until further analysis. The peptide samples were analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) in 3 separated experiments using a Thermo Easy nLC 1000 coupled to a QExactive HF or a Waters nanoAcquity coupled to a Thermo LTQ-Orbitrap Velos. Samples were injected onto a PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 90 or 120 min gradient where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The LTQ-Orbitrap Velos was operated in data-dependent mode where the 10 most abundant precursors were selected for CID fragmentation (35% CE). The QExactive HF was operated in data-dependent mode where the 15 most intense precursors were selected for subsequent HCD fragmentation (27 NCE). For the second replicate, a targeted analysis of THEMIS peptides was conducted. The QExactive HF was operated in PRM mode, and an inclusion list was used to target previously identified THEMIS peptides. Raw data files were processed using Proteome Discoverer version 2.1 (Thermo Scientific). Peak lists were searched against a reviewed Uniprot human database and appended with the CAR-T sequences using Sequest. The following parameters were used to identify tryptic peptides for protein identification: 10 ppm precursor ion mass tolerance; 0.02 Da product ion mass tolerance for QE HF data and 0.6 Da for Velos data; up to two missed trypsin cleavage sites; carbamidomethylation of Cys was set as a fixed modification; oxidation of Met, acetylation of N-terminus and phosphorylation of Ser, Thr and Tyr were set as variable modifications. The ptmRS node was used to localize the sites of phosphorylation. Peptide false discovery rates (FDR) were calculated by the Percolator node using a decoy database search and data were filtered using a 5% FDR cutoff.
Xenograft mouse models.
For long-term in vivo cytotoxicity, male or female NSG (NOD-scid IL2Rgnull) mice were injected intravenously (i.v.) with 2 × 106 CD19+ Daudi tumor cell line labeled with the Firefly luciferase gene (Daudi-FFLuc). Four to seven days later, mice received T cells control or expressing CARs intravenously (i.v.). For tumor re-challenging experiments, 2 × 106 CD19+ Daudi-FFLuc cells were i.v. injected at indicated time point. Tumor growth was monitored every 2 – 3 days by injecting mice intraperitoneally (i.p.) with D-luciferin (150 mg/kg, Xenolight, PerkinElmer). Photon emission was analyzed using the Xenogen-IVIS Imaging System as previously validated (Diaconu et al. 2017). For short-term in vivo T cell activation, NSG mice were injected with Daudi-FFLuc cells i.v. Two weeks later, CAR19.28ζ-Ts were labeled with Cell-trace Violet (Thermo Scientific) and CAR19.BBζ-Ts were labeled with CFSE (Thermo Scientific) as per manufacturer’s instructions. Cells were mixed 1:1, and a total 1 × 107 cells were i.v. injected in each mouse. Peripheral blood, bone marrow, lung and spleen were harvested 6 hr after T cell injection, dissociated into single cells and stained with α-human CD45-PE, CD3-APC and CD69-PE-Cy7 Abs. For the neuroblastoma metastatic model, 6 – 8-week-old male or female NSG mice were injected i.v. with CHLA-255-FFluc tumor cell line (2 × 106 cells/mouse). Fourteen days after tumor inoculation, CAR-T cells were infused i.v. (Chen et al. 2019). For the NSG mouse model to evaluate the effects of SHP1 heterodimerization in CAR-T cells in vivo, mice were injected i.v. with 2 × 106 Daudi-FFLuc cells. Seven days later, 2 × 106 T cells were infused i.v.. AP21967 was administrated intraperitoneally twice at 10 mg/kg dose. Tumor growth was monitored twice a week.
Cytokine release syndrome models.
Evaluation of CRS in humanized NSG mice was performed as previously described (Diaconu et al. 2017). Mice were infused with 5 × 106 CAR-T cells at day 0, and plasma was collected at days 1 and 2. AP21967 was administrated intraperitoneally at 10 mg/kg dose (Rivera et al. 2012). Weight of each mouse was normalized to starting weight before CAR-T cell infusion. In a second CRS murine model, 7 – 10-week-old female SCID-beige (C.B-Igh-1b/GbmsTac-Prkdcscid-LystbgN7, Taconic Biosciences) mice were injected i.p. with Raji-FFluc cells (Giavridis et al. 2018). After 21 days, mice were grouped based on the BLI (total flux). Mice were infused i.p. with 30 × 106 CAR-T cells. Weight of each mouse was normalized to starting weight before CAR-T cell infusion.
Co-culture assays.
CD19+ BV173 tumor cells were seeded into 24-well plates with 5 × 105 cells/well and T cells were added at different effector to target (E:T) ratios (E:T=1:1 or 1:5). After 24 hr, supernatant was collected for ELISA. Three days later, cells were collected and stained with α-human CD3 and CD19 Abs for flow cytometry analysis.
Immunofluorescence and confocal microscopy.
Ctrl and CAR19.BBζ-T cells with or without FLAG-tagged LCK at day 6 of culture were first stained with CAR19 α-idiotype Ab followed by the goat anti-mouse IgG conjugated with AlexaFluor 647 (Invitrogen) secondary Ab to detect CAR19 expression. Cells were then fixed and permeabilized with Cytofix/Cytoperm solution (BD Bioscience) according to manufacturer’s instructions. Intracellular FLAG-tagged LCK was detected with Rabbit α-FLAG (Cell Signaling Technology) and goat anti-Rabbit IgG conjugated with AlexaFluor 488 (Invitrogen). Cells were then loaded on slides by Cytospin Cytocentrifuge (Thermo Scientific) and mounted with ProLong Diamond Antifade Mountant with DAPI (Thermo Scientific). Slides were imaged using confocal microscopy (Zeiss LSM710) and images were analyzed with Fiji software (ImageJ).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were presented as mean ± SEM unless indicated otherwise. Statistical analyses were performed using GraphPad Prism software. Two-tailed unpaired t-test, one-way ANOVA, and two-way ANOVA were used. Bonferroni’s correction for multiple comparisons was used to calculate adjusted p value when appropriate. The exact p values were shown in figures; ns, not significant. Specific statistical test used for each figure was described in the corresponding figure legend.
DATA AND CODE AVAILABILITY
This study did not generate datasets.
ADDITIONAL RESOURCES
N/A
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CAR1219 (clone 233–4A) | Gift from Dr. Lawrence Cooper, MD Anderson | N/A |
| Anti-CD3 (clone OKT3) | Miltenyi Biotec | Cat# 130-093-387 |
| Anti-CD28 (clone CD28.2) | BD Biosciences | Cat# 555725 |
| Goat anti-mouse Ig-APC | BD Biosciences | Cat# 550826 |
| hCD3-PE (clone SK7) | BD Biosciences | Cat# 347347 |
| hCD3-APC (clone SK7) | BD Biosciences | Cat# 340440 |
| hCD4-PE (clone SK3) | BD Biosciences | Cat# 347327 |
| hCD8-FITC (clone SK1) | BD Biosciences | Cat# 347313 |
| hCD19-PE (clone SJ25C1) | BD Biosciences | Cat# 340364 |
| hCD45-FITC (clone 2D1) | BD Biosciences | Cat# 347463 |
| hCD45-PE (clone HI30) | BD Biosciences | Cat# 555483 |
| hCD69-FITC (clone L78) | BD Biosciences | Cat# 347823 |
| hCD69-PE-Cy7 (clone FN50) | BD Biosciences | Cat# 557745 |
| hPD-1-PE-Cy7 (clone EH12.1) | BD Biosciences | Cat# 561272 |
| hTIM3-BV711 (clone 7D3) | BD Biosciences | Cat# 565566 |
| Anti-CD3pY83 (clone EP7769(2)Y) | Abcam | Cat# ab68236 |
| Anti-CD3pY142 (clone EP265(2)Y) | Abcam | Cat# ab68235 |
| Anti-HA (clone C29F4) | Cell Signaling Tech | Cat# 3724 |
| Anti-FLAG (clone D6W5B) | Cell Signaling Tech | Cat# 14793 |
| Anti-ZAP70pY319 | Cell Signaling Tech | Cat# 2701 |
| Anti-ZAP70 (clone D1C10E) | Cell Signaling Tech | Cat# 3165 |
| Anti-LATpY191 | Cell Signaling Tech | Cat# 3584 |
| Anti-LAT | Cell Signaling Tech | Cat# 9166 |
| Anti-LCK (clone L22B1) | Cell Signaling Tech | Cat# 2657 |
| Anti-LCK (clone V49, for IP) | Cell Signaling Tech | Cat# 2714 |
| Anti-THEMIS | Cell Signaling Tech | Cat# 4482 |
| Anti-SHP1 (clone C14H6) | Cell Signaling Tech | Cat# 3759 |
| Anti-CD3z (clone F-3) | Santa Cruz | Cat# SC-166275 |
| Anti-FKBP (clone H-5) | Santa Cruz | Cat# SC-133067 |
| Anti-β-actin (clone C4) | Santa Cruz | Cat# SC-47778 |
| Anti-CD3ζ-HRP (clone F-3) | Santa Cruz | Cat# SC-166275 |
| Anti-FLAG (clone M2) | Sigma | Cat# F1084 |
| Anti-CD4-HRP (clone EPR6855) | Abcam | Cat# ab195842 |
| Anti-CD8α (clone 144B) | Abcam | Cat# ab17147 |
| Biological Samples | ||
| Buffy coats from healthy donors | Gulf Coast Regional Blood Center, Houston, TX | N/A |
| Fetal liver tissues | Advanced Bioscience Resources, Alameda, CA | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AP21967 (A/C Heterodimerizer) | Takara/Clontech | Cat# 635055 |
| RetroNectin | Takara/Clontech | Cat# T100B |
| Calcium Assay Kit | BD | Cat# 640176 |
| PP2 | Sigma | Cat# P0042 |
| Lck Inhibitor II (CAS 918870-43-6) | Millipore | Cat# 428206 |
| Human IL-7 | PeproTech | Cat# 200–07 |
| Human IL-15 | PeproTech | Cat# 200–15 |
| GeneJuice | Merck Millipore | Cat# 70967 |
| Phusion PCR master mix | Thermo | Cat# F-531 |
| XenoLight D-Luciferin | Perkin Elmer | Cat# 122799 |
| Critical Commercial Assays kit | ||
| Human IFN-gamma ELISA kit | R&D systems | Cat# DY285B |
| Human IL-2 ELISA kit | R&D systems | Cat# DY202 |
| Mouse IL-6 DuoSet ELISA kit | R&D systems | Cat# DY406–05 |
| ELISA substrate reagent pack | R&D systems | Cat# DY999 |
| Luminex Human Magnetic Assay | R&D systems | Cat# LXSAHM |
| Luminex Mouse Magnetic Assay | R&D systems | Cat# LXSAMSM |
| Experimental Models: Cell Lines | ||
| BV173 | German Cell Culture Collection | Cat# ACC 20 |
| Jurkat | ATCC | Cat# TIB-152 |
| Daudi-FFLuc | This lab (Vera et al. 2006) | PMID: 16926291 |
| Raji-FFluc | This lab (Hoyos et al. 2010) | PMID: 20428207 |
| CHLA-255-FFluc | Gift from Dr. Metelitsa, Baylor College of Medicine, Houston TX | N/A |
| Recombinant DNA | ||
| CAR19.28ζ | This lab (Savoldo et al. 2011) | PMID: 21540550 |
| CAR19.BBζ | This lab (Diaconu et al. 2017) | PMID: 28187946 |
| CAR19.28AAAζ | This manuscript | N/A |
| CAR19.28YFζ | This manuscript | N/A |
| CAR19.28AFAAζ | This manuscript | N/A |
| CAR19.BBζ-ΔC10 | This manuscript | N/A |
| CAR19.28.CD3Y6Fζ | This manuscript | N/A |
| CAR19.BB.CD3Y6Fζ | This manuscript | N/A |
| CAR.CD8h.CD28TM.28ζ | This manuscript | N/A |
| CAR.CD8h.CD28TM.BBζ | This manuscript | N/A |
| CAR.IgG1h.CD8TM.28ζ | This manuscript | N/A |
| CAR.IgG1h.CD8TM.BBζ | This manuscript | N/A |
| CARGD2 | This lab (Chen et al. 2019) | PMID: 30617136 |
| CAR138 | This lab (Sun et al. 2019) | PMID: 31040928 |
| PM-FRB-mRFP-T2A-FKBP-5-ptase | Addgene | Cat# 40896 |
| FKBP-SHP1 | This manuscript | N/A |
| CAR19.28ζ.FRB.FLAG | This manuscript | N/A |
| SFG.THEMIS | This manuscript | N/A |
| SFG.LCK | This manuscript | N/A |
| SFG.CD8α-SKS | This manuscript | N/A |
| SFG.CD8α-WT | This manuscript | N/A |
| SFG.CD4 | This manuscript | N/A |
| Oligonucleotides | ||
| THEMIS shRNA #1: 5’-GAGATCACTGAAGAGCAATAT-3’ | UNC shRNA Core Lab | TRCN0000128476 |
| THEMIS shRNA #2: 5’-CCCATAGTGACTGAAGTCATA-3’ | UNC shRNA Core Lab | TRCN0000130264 |
| SHP1 shRNA #1: 5’-GCATGACACAACCGAATACAA-3’ | UNC shRNA Core Lab | TRCN0000006886 |
| SHP1 shRNA #2: 5’-CGACATGCTCATGGAGAACAT-3’ | UNC shRNA Core Lab | TRCN0000006887 |
| Software and Algorithms | ||
| Flowjo v10 | FlowJo, LLC | N/A |
| GraphPad Prism | GraphPad Software Inc. | N/A |
| Living Image v4.5.2 (IVIS imaging) | Perkin Elmer | N/A |
| Other | ||
| Counting beads | Invitrogen | Cat# C36950 |
Attached in a separate Word document (Chen et al. 2019;Diaconu et al. 2017;Hoyos et al. 2010;Savoldo et al. 2011;Sun et al. 2019;Vera et al. 2006).
Highlights.
LCK promotes basal CAR-CD3ζ phosphorylation in the synapse of CAR.CD28ζ
THEMIS-SHP1 counteracts the effect of LCK in the synapse of CAR.4–1BBζ
Engineering LCK kinase tunes up the antitumor activity of CAR.4–1BBζ-T cells
Engineering druggable SHP1 phosphatase tunes down the function of CAR.CD28ζ-T cells
Statement of significance.
Costimulation mediated by CD28 and 4–1BB endodomains integrated into CAR molecules is critical for their antitumor activity. In this study, we have identified that LCK and THEMIS-SHP1 complex are differentially recruited by CD28 and 4–1BB, respectively. Based on these mechanistic findings, we have developed strategies to engineer either LCK or SHP1 in CAR-T cells to specifically tune their functions.
Acknowledgement
The UNC Small Animal Imaging Facility at the Biomedical Imaging Research Center, Microscopy Services Laboratory, Flow Cytometry Core Facility, and Michael Hooker Proteomics Core Facility are supported in part by an NCI Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center (P30-CA016086). This work was supported in part by R01-CA193140-03 (G.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The engineering processes described in the manuscript have been included in a filed patent application. Dr. Dotti serves in the SAB of Bellicum Pharmaceutical s.p.a. and MolMed s.p.a. No potential conflicts of interest were disclosed by the other authors.
References
- Arch RH & Thompson CB 1998. 4–1BB and 0×40 are members of a tumor necrosis factor (TNF) -nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol. Cell Biol, 18, (1) 558–565 available from: PM:9418902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awale M & Mohan CG 2008. Molecular docking guided 3D-QSAR CoMFA analysis of N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of leukocyte-specific protein tyrosine kinase. J MoiModel, 14, (10) 937–947 available from: PM: 18626671 [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T,He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, & Sadelain M 2013. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. TransI.Med,5, (177) 177ra38 available from: PM:23515080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, & Savoldo B 2019. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin Cancer Res. available from: PM:30617136 [DOI] [PubMed] [Google Scholar]
- Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, & Savoldo B 2017. Inducible Caspase-9 Selectively Modulates the Toxicities of CD 19-Specific Chimeric Antigen Receptor-Modified T Cells. Moi. Ther, 25, (3 ) 580–592 available from: PM:28187946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbins J, Gagnon E, Godec J, Pyrdol J, Vignali DA, Sharpe AH, & Wucherpfennig KW 2016. Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling. Sci.Signal, 9, (438) ra75 available from: PM:27460989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti G, Gottschalk S, Savoldo B, & Brenner MK 2014. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol.Rev, 257, (1) 107–126 available from: PM:24329793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney HM, Lawson AD, Bebbington CR, & Weir AN 1998. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J.Immunol 61, (6) 2791–2797 available from: PM:9743337 [PubMed] [Google Scholar]
- Foster AE, Mahendravada A, Shinners NP, Chang WC, Crisostomo J, Lu A, Khalil M, Morschl E, Shaw JL, Saha S, Duong MT, Collinson-Pautz MR, Torres DL, Rodriguez T, Pentcheva-Hoang T, Bayle JH, Slawin KM, & Spencer DM 2017. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol. Ther, 25, (9) 2176–2188 available from: PM:28697888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, & Thompson CB 2002. The CD28 signaling pathway regulates glucose metabolism. Immunity, 16, (6) 769–777 available from: PM:12121659 [DOI] [PubMed] [Google Scholar]
- Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, & Sadelain M 2018. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat.Med, 24, (6) 731–738 available from: PM:29808005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G, Gorochov G, Waks T, & Eshhar Z 1989. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant.Proc, 21, (1 Pt 1) 127–130 available from: PM:2784887 [PubMed] [Google Scholar]
- Hofinger E & Sticht H 2005. Multiple modes of interaction between Lck and CD28. J Immunol, 174, (7) 3839–3840 available from: PM:15778335 [DOI] [PubMed] [Google Scholar]
- Holdorf AD, Green JM, Levin SD, Denny MF, Straus DB, Link V, Changelian PS, Allen PM, & Shaw AS 1999. Proline residues in CD28 and the Src homology (SH) 3 domain of Lck are required for T cell costimulation. J Exp.Med, 190, (3) 375–384 available from: PM:10430626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, & Dotti G 2010. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia,24, (6) 1160–1170 available from: PM:20428207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, & Campana D 2004. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia, 18, (4) 676–684 available from: PM:14961035 [DOI] [PubMed] [Google Scholar]
- Jang IK, Lee ZH, Kim YJ, Kim SH, & Kwon BS 1998. Human 4–1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-kappa B. Biochem.Biophys.Res.Commun, 242, (3) 613–620 available from: PM:9464265 [DOI] [PubMed] [Google Scholar]
- Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., Patel PR, Guedan S, Scholler J, Keith B, Snyder NW, Blair IA, Milone MC, & June CH 2016. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity., 44, (2) 380–390 available from: PM:26885860 [DOI] [PubMed] [Google Scholar]
- Kim YJ, Pollok KE, Zhou Z, Shaw A, Bohlen JB, Fraser M, & Kwon BS 1993. Novel T cell antigen 4–1 BB associates with the protein tyrosine kinase p56lck1. J Immunol, 151, (3) 1255–1262 available from: PM:8335927 [PubMed] [Google Scholar]
- Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, & Mackall CL 2014. Current concepts in the diagnosis and management of cytokine release syndrome. Blood, 124, (2) 188–195 available from: PM:24876563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Cheng M, Nunoya J, Cheng L, Guo H, Yu H, Liu YJ, Su L, & Zhang L 2014. Plasmacytoid dendritic cells suppress HIV-1 replication but contribute to HIV-1 induced immunopathogenesis in humanized mice. PLoS.Pathog, 10, (7) e1004291 available from: PM:25077616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman E.l. & Dotti G 2017. Chimeric antigen receptor T-cells for B-cell malignancies. Trans/.Res, 187, 59–82 available from: PM:28719798 [DOI] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, & Mackall CL 2015. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat.Med, 21, (6) 581–590 available from: PM:25939063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, & Grupp SA 2014. Chimeric antigen receptor T cells for sustained remissions in leukemia. N.Engl.J.Med, 371, (16) 1507–1517 available from: PM:25317870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, Traversari C, Bordignon C, Ciceri F, Ostuni R, Bonini C, Casucci M, & Bondanza A 2018. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat.Med, 24, (6) 739–748 available from: PM:29808007 [DOI] [PubMed] [Google Scholar]
- Paster W, Bruger AM, Katsch K, Gregoire C, Roncagalli R, Fu G, Gascoigne NR, Nika K, Cohnen A, Feller SM, Simister PC, Molder KC, Cordoba SP, Dushek O, Malissen B, & Acuto O 2015. A THEMIS:SHP1 complex promotes T-cell survival. EMBO J, 34, (3) 393–409 available from: PM:25535246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, Liu E, Carrum G, Kamble RT, Gee AP, Mei Z, Wu MF, Liu H, Grilley B, Rooney CM, Brenner MK, Heslop HE, & Dotti G 2016. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J.Clin.Invest, 126, (7) 2588–2596 available from: PM:27270177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh MD 1994. Myristylation and palmitylation of Src family members: the fats of the matter. Cell, 76, (3) 411–413 available from: PM:8313462 [DOI] [PubMed] [Google Scholar]
- Rivera VM, Berk L, & Clackson T 2012. Dimerizer-mediated regulation of gene expression in vivo. Cold Spring Harb.Protoc, 2012, (7) 821–824 available from: PM:22753599 [DOI] [PubMed] [Google Scholar]
- Rudd CE, Trevillyan JM, Dasgupta JD, Wong LL, & Schlossman SF 1988. The CD4 receptor is complexed in detergent lysates to a protein-tyrosine kinase (pp58) from human T lymphocytes. Proc.Nat/.Acad.Sci.U.S.A, 85, (14) 5190–5194 available from: PM:2455897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R, & Riviere I 2013. The basic principles of chimeric antigen receptor design. Cancer Discov, 3, (4) 388–398 available from: PM:23550147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, Voytovich UJ, Lin C, Sommermeyer D, Liu L, Whiteaker JR, Gottardo R, Paulovich AG, & Riddell SR 2018. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Scl.Signal, 11, (544) available from: PM:30131370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, Liu H, Grilley B, Rooney CM, Heslop HE, Brenner MK, & Dotti G 2011. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin.Invest, 121, (5) 1822–1826 available from: PM:21540550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, & Jordan MS 2009. T cell activation. Annu.Rev.Immunol, 27, 591–619 available from: PM:19132916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Mahendravada A, Ballard B, Kale B, Ramos C, West J, Maguire T, McKay K, Lichtman E, Tuchman S, Dotti G, & Savoldo B 2019. Safety and efficacy of targeting CD 138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget, 10, (24) 2369–2383 available from: PM:31040928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JM, Brodsky MH, Irving BA, Levin SD, Perlmutter RM, & Littman DR 1990. Interaction of the unique N-terminal region of tyrosine kinase p56lckwith cytoplasmic domains of CD4 and CD8 is mediated by cysteine motifs. Cell, 60, (5) 755–765 available from: PM:2107025 [DOI] [PubMed] [Google Scholar]
- Veillette A, Bookman MA, Horak EM, & Bolen JB 1988. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell, 55, (2) 301–308 available from: PM:3262426 [DOI] [PubMed] [Google Scholar]
- Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C, Wu J, Heslop HE, Rooney CM, Brenner MK, & Dotti G 2006. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood, 108, (12) 3890–3897 available from: PM: 16926291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen T, Bukczynski J, & Watts TH 2002. 4–1BB ligand-mediated costimulation of human T cells induces CD4 and CD8 T cell expansion, cytokine production, and the development of cytolytic effector function. J Immunol, 168, (10) 4897–4906 available from: PM: 11994439 [DOI] [PubMed] [Google Scholar]
- Wiest DL, Ashe JM, Abe R, Bolen JB, & Singer A 1996. TCR activation of ZAP70 is impaired in CD4+CD8+ thymocytes as a consequence of intrathymic interactions that diminish available p56lck. Immunity, 4, (5) 495–504 available from: PM:8630734 [DOI] [PubMed] [Google Scholar]
- Wu CY, Roybal KT, Puchner EM, Onuffer J, & Lim WA 2015. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science, 350, (6258) aab4077 available from: PM:26405231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Condomines M, van derStegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, & Sadelain M 2015. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell, 28, (4) 415–428 available from: PM:26461090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Di SA, Tey SK, Krance RA, Martinez C, Leung KS, Durett AG, Wu MF, Liu H, Leen AM, Savoldo B, Lin YF, Grilley BJ, Gee AP, Spencer DM, Rooney CM, Heslop HE, Brenner MK, & Dotti G 2014. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood, 123, (25) 3895–3905 available from: PM:24753538 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets.