

Abstract
Chromatin structure is determined by nucleosome positioning, histone modifications, and DNA methylation. How chromatin modifications are coordinately altered under pathological conditions remains elusive. Here we describe a stress-activated mechanism of concerted chromatin modification in the heart. In mice, pathological stress activates cardiomyocytes to express Brg1 (nucleosome-remodeling factor), G9a/Glp (histone methyltransferase), and Dnmt3 (DNA methyltransferase). Once activated, Brg1 recruits G9a and then Dnmt3 to sequentially assemble repressive chromatin—marked by H3K9 and CpG methylation—on a key molecular motor gene (Myh6), thereby silencing Myh6 and impairing cardiac contraction. Disruption of Brg1, G9a or Dnmt3 erases repressive chromatin marks and de-represses Myh6, reducing stress-induced cardiac dysfunction. In human hypertrophic hearts, BRG1–G9a/GLP–DNMT3 complex is also activated; its level correlates with H3K9/CpG methylation, Myh6 repression, and cardiomyopathy. Our studies demonstrate a new mechanism of chromatin assembly in stressed hearts and novel therapeutic targets for restoring Myh6 and ventricular function. The stress-induced Brg1–G9a–Dnmt3 interactions and sequence of repressive chromatin assembly on Myh6 illustrates a molecular mechanism by which the heart epigenetically responds to environmental signals. This article is part of a Special Issue entitled: Cardiomyocyte Biology: Integration of Developmental and Environmental Cues in the Heart edited by Marcus Schaub and Hughes Abriel.
Keywords: Histone methylation, DNA methylation, Chromatin remodeling, Gene silencing, Myosin heavy chain, G9a, Dnmt, Brg1, H3K9me2, Cardiac hypertrophy, Cardiomyopathy, Heart failure
1. Introduction
Genes are tightly packed in chromatin, which acts as a dynamic DNA scaffold responsive to external cues to coordinate gene expression programs. Pathological stress induces transcriptional gene reprogramming in the heart muscle, leading to myopathy and heart failure. However, the chromatin mechanism that underlies such pathological cardiac gene reprogramming remains largely undefined. Chromatin could adopt covalent modifications that impact on the transcriptional regulation in response to various biological stimuli [4–6,24]. It remains elusive how chromatin modifiers act in concert with each other to establish individual chromatin marks, particularly in mammalian organs under stress conditions. Of special interest are the mechanistic interactions between enzymes that covalently modify chromatin and the ATP-dependent chromatin remodelers that position the nucleosomes.
Here we describe a new mechanism that controls the chromatin scaffold of a key molecular motor gene in the heart. The molecular motor genes encode myosin heavy chain (MHC) that hydrolyzes ATP and drive heart muscle contraction. Two isoforms of MHC—Myh6 (α-MHC) and Myh7 (β-MHC)—exist in the mammalian heart. Myh6 hydrolyzes ATP 3 times as fast as Myh7 and generates more contractile power. MHC isoform amount changes under different pathophysiological conditions. MHC of a normal human left ventricle comprises ~10% Myh6 and 90% Myh7 [22]; however, when human hearts are diseased, Myh6 is virtually eliminated [22], causing contractile dysfunction. Transfer of Myh6 gene into failing human cardiomyocytes enhances muscle contraction by 3–4-fold [10]. Similarly, in other large mammals such as the pig, the increase of Myh6 from 0 to 10% augments cardiac contraction by 90% [19]. A small amount of Myh6 thus has a large impact on cardiac contractility. In smaller mammals, Myh6 is also reduced in stressed hearts [9], and like in humans, increase of Myh6 enhances cardiac contractility and resistance to pathological stress [10,11,14]. Heart function is thus sensitive to Myh6 expression, and cardiac contractile force increases within the full range of Myh6 expression from 0 to 100% [19,20].
Given Myh6’s evolutionary conservation and importance in cardiac contractility, we focused on determining the epigenetic regulatory mechanism of Myh6 in pathologically stressed hearts and how the dynamic changes of Myh chromatin scaffold from embryos to adult hearts can provide a molecular avenue for identifying new pathogenic factors of hypertrophy and heart failure. Certain essential regulators of cardiac hypertrophy, such as miR208 [29,30], Brg1 [9] and Myheart [8], are embedded within or capable of controlling the Myh loci, suggesting that Myh loci can provide a useful molecular tool for identifying new regulators of hypertrophy. Therefore, instead of using a genomic approach, we demonstrated this specific use of Myh promoters by (1) determining the chromatin changes of Myh6 promoter in embryonic, normal adult, and stressed adult hearts, (2) identifying enzymes that catalyze such chromatin changes (G9a and Dnmt3), (3) demonstrating the in vivo requirements of G9a and Dnmt3 for pathological hypertrophy, and finally (4) validating the thesis by defining the genetic and molecular interactions of Brg1, G9a, and Dnmt3 on the Myh promoter and associated chromatin changes. Our focus on interactions of the three critical epigenetic regulators on Myh6 established a novel Brg1-G9a-Dnmt3-mediated mechanism of repressive chromatin assembly on an essential gene promoter in stressed hearts. Future genome-wide approaches will be necessary for defining broader effects of Brg1-G9a-Dnmt3 complex on other cardiac target genes.
2. Results
2.1. H3K9 methylation is associated with Myh6 repression
Myh6 expression changes dynamically under different pathophysiological conditions: it has low expression in fetal hearts, is up-regulated in adult hearts, but repressed in hypertrophic hearts. To test how the chromatin of Myh6 promoter was regulated during pathophysiological changes, we surveyed by chromatin immunoprecipitation (ChIP) major repressive histone methylation marks, including methylation of histone 3 lysine 9 (H3K9) [15,28,31] and histone 3 lysine 27 (H3K27) on the Myh6 promoter in fetal, adult, and hypertrophic mouse hearts. We focused on the evolutionarily conserved Myh6 proximal promoter (−673 to −73), the activity of which is cardiac-specific and is required for Myh6 transcription regulation [8,9]. Among these histone marks, only dimethylated H3K9 (H3K9me2), but not H3K27me3, showed dynamic changes that correlated with Myh6 repression (Supplemental Text, Fig. 1A, and S1). This promoter region was highly enriched with H3K9me2 in fetal hearts at embryonic day 12.5 (E12.5), whereas H3K9me2 decreased 4.4-fold on Myh6 promoter in healthy adult hearts (Fig. 1A). When the adult heart was stressed by pressure overload through surgical constriction of the transverse aorta (transverse aortic constriction, TAC) [9], H3K9me2 on Myh6 promoter was reformed and increased by 3.6-fold on the promoter, comparable to the fetal level of H3K9me2 (Fig. 1A). Conversely, very low H3K9me2 was present on the proximal Myh7 promoter (−322 to +278 that is critical for Myh7 transcription regulation [9]), and H3K9me2 showed no significant changes on Myh7 promoter during heart development and hypertrophy (Fig. 1B). These findings suggest that H3K9 methylation underlies Myh6 repression under different pathophysiological conditions.
Fig. 1.
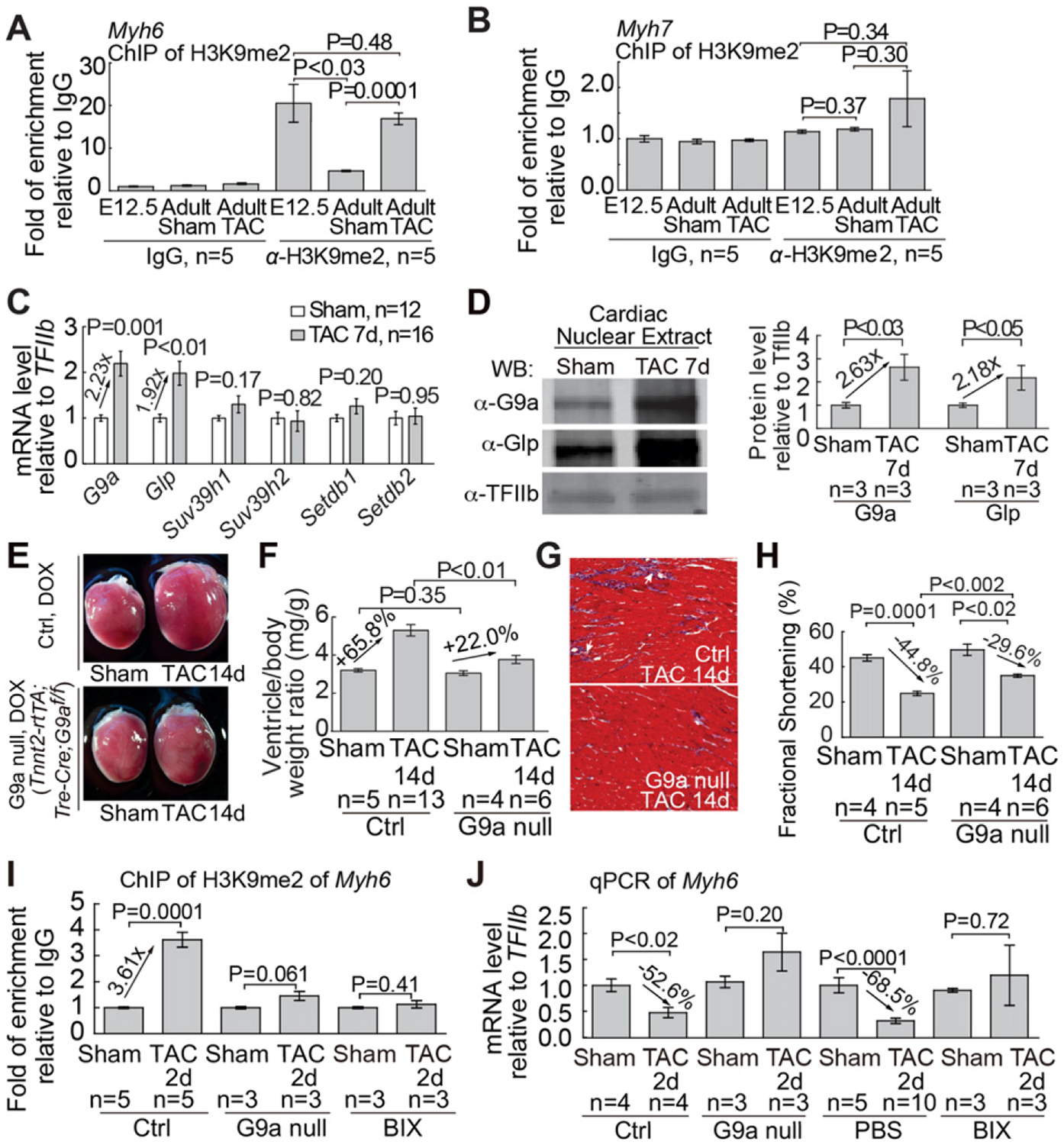
G9a/Glp is essential for cardiac hypertrophy and dysfunction. (A and B) Quantitation of H3K9me2 ChIP of the proximal promoters of Myh6 (A) and Myh7. (B) in fetal hearts (E12.5), sham-operated adult hearts, and TAC-operated adult hearts. Data are presented as H3K9me2 enrichment relative to IgG control. (C) mRNA expression of H3K9 methyltransferases in adult hearts 7 days after TAC. (D) Western blot and quantitation of G9a, Glp proteins 7 days after TAC. TFIIb proteins were used as the internal control. (E–H) Gross morphology of ventricle (E), quantitation of ventricle/body weight ratio (F), trichrome staining of left ventricles (G), and echocardiographic measurement of left ventricular fractional shortening (H) in littermate control and mutant mice lacking myocardial G9a 14 days after sham or TAC operation. Ctrl: control mice. G9a null: Tnnt2-rtTA;Tre-Cre;G9af/f mice. (I) Quantitation of H3K9me2 on the Myh6 proximal promoter (I) and Myh6 mRNA (J) 2 days after sham or TAC operation. BIX: BIX-treated heart. H3K9me2 ChIP data are presented as the enrichment normalized to the sham-operated hearts in control mice. Myh6 mRNA data are presented as mRNA levels normalized to the sham-operated hearts in control mice or PBS-treated mice, respectively. P-value: Student’s t-test. Error bar: SEM, standard error of the mean.
2.2. G9a/Glp is the primary enzyme catalyzing H3K9 methylation to silence Myh6
Given H3K9me2 is critical for silencing euchromatin within generich areas [16,28,31] (Supplemental Text), we focused on enzymes that could catalyze H3K9me2 on Myh6 promoter. By RT–qPCR, we examined the expression of known histone H3K9 methyltransferases (H3K9MTs) [32]—G9a/Ehmt2, Glp/Ehmt1, Suv39h1, Suv39h2, Setdb1, and Setdb2 (Supplemental Text)—in mouse heart ventricles with or without TAC by normalization to TFIIb [25]. Among these enzymes, only G9a and Glp mRNAs, which encode proteins to form a heteromeric enzyme complex [26], were induced 1.9–2.2-fold by TAC; the other enzymes had no significant changes (Fig. 1C). G9a and Glp activation by TAC was confirmed by western blot analysis (Fig. 1D) and by immunostaining that showed abundant G9a and Glp proteins in the fetal myocardium (Figure S2A and S2B), minimal G9a/Glp in healthy adult myocardium (Figure S2C and S2E), and reactivation of G9a/Glp in TAC-stressed adult myocardium (Figure S2D and S2F). Such G9a/Glp dynamics corresponded to H3K9me2 dynamics on Myh6 promoter and correlated with Myh6 repression, suggesting an essential role of G9a/Glp in cardiac hypertrophy.
To test G9a/Glp requirement for Myh6 silencing and cardiac hypertrophy, we used the doxycycline-inducible Tnnt2-rtTA;Tre-Cre mouse line [9] to effect deletion of G9a floxed alleles (G9af/f) in adult cardiomyocytes to abolish G9a/Glp methyltransferase complex. After a 7-day doxycycline treatment to activate myocardial deletion of G9a, the mice appeared grossly normal, indicating that short-term G9a disruption has no significant effect on baseline heart function, consistent with low G9a expression in unstressed hearts. We then used 27-gauge needle–perform TAC to exert high pressure load on the hearts of littermate control (G9af/+, G9af/f, or Tnnt2-rtta;Tre-Cre;G9af/+) and mutant (Tnnt2-rtTA;Tre-Cre;G9af/f) mice to induce hypertrophy. Immunostaining of TAC-stressed mutant hearts showed 81% disruption of G9a in cardiomyocytes without changes of G9a in endothelial cells (Figure S2G and S2H), confirmed by western blot analysis (Supplemental Fig. 2 l). Within 2 weeks after TAC, control mice fed with doxycycline diet developed cardiac hypertrophy with grossly enlarged hearts (Fig. 1E), increased ventricle/body-weight ratio (Fig. 1F), enlarged cardiomyocytes (Figure S2J–S2N), interstitial fibrosis (Fig. 1G and S2O), reduced left ventricular fractional shortening (FS), and increase of end-systolic left ventricular size (Fig. 1H and S2P). Conversely, doxycycline-treated mutant mice (Tnnt2-rtTA;Tre-Cre;G9af/f) exhibited mild cardiac hypertrophy (Fig. 1E, F, and S2J–S2N), minimal fibrosis (Fig. 1G and S2O), and less cardiac dysfunction (Fig. 1H and S2P). To examine the longer-term effect of G9a disruption on cardiac function, we used a less stringent 26-gauge needle to perform TAC, with less pressure load and slower hypertrophy progression. We found that in the absence of myocardial G9a, heart function began to improve 4 weeks after TAC, and the beneficial effects lasted for at least 8 weeks with 52% reduction of cardiac dysfunction (P = 0.002) and 41% reduction of hypertrophy (P = 0.025) (Figure S2Q–S2S). Therefore, stress-induced G9a activation in cardiomyocytes is essential for pathological hypertrophy to develop. Moreover, pharmacological inhibition of G9a/Glp with the nanomolar inhibitor BIX-01,294 (BIX) [16], which did not have significant effect on baseline heart function, blocked TAC-induced cardiac hypertrophy, fibrosis, and contractile dysfunction (Figure S3A–S3J). Collectively, without G9a/Glp activity, cardiac hypertrophy was reduced by 41–67%, FS decline lessened by 33–52%, and LV dilation reduced by 60–100%. Therefore, myocardial G9a/Glp is essential for stress-induced cardiomyopathy.
To test G9a requirement for H3K9 methylation, we used ChIP to examine H3K9me2 on Myh6 promoter with/without G9a. To avoid measuring secondary effects from cardiac hypertrophy, we conducted the experiments within 2 days of TAC when there was no/minimal cardiac hypertrophy (Figure S3K and S3L). Within 2 days of TAC, G9a disruption by gene deletion or BIX treatment abolished TAC-induced H3K9me2 of Myh6 (Fig. 1I) and reversed TAC-induced Myh6 repression (Fig. 1J). Conversely, G9a disruption had no significant influence on stress-induced Myh7 changes (Figure S3M and S3N). The data indicate that stress-induced G9a/Glp activation is required for H3K9 methylation, Myh6 repression, and cardiac hypertrophy.
2.3. CpG methylation by Dnmt3 is essential for Myh6 repression and hypertrophy
We examined another potent epigenetic marker for gene silencing: DNA methylation at CpG dinucleotides. We identified five CpG sites on Myh6 proximal promoter and eight CpG sites on the proximal promoter/5′-untranslated region of Myh7 (Fig. 2A and B). Bisulfite genomic sequencing showed that Myh6 CpG sites were highly methylated in E12.5 fetal heart ventricles, and the methylation decreased by 30.1% in healthy adult heart ventricles (from 45.9% to 32.1%, Fig. 2A and C). Interestingly, in adult heart ventricles stressed by TAC for 2, 7 and 14 days, the degree of Myh6 CpG methylation increased by 1.7–1.9 fold (from 32.1% to 59.9%, 54.5% and 53.8%, Fig. 2A and C). Because bisulfite sequencing does not distinguish between methylation (5mC) and hydroxymethylation (5hmC) of DNA [12], we performed Methylated-DNA Immunoprecipitation (MeDIP) analysis of Myh promoters using specific antibodies against methylated or hydroxymethylated DNA [7]. MeDIP studies showed dramatic increase of methylated DNA on Myh6 promoter, but not Myh7, and no significant changes of hydroxymethylated DNA on Myh promoters (Fig. 2D and E), indicating TAC induces CpG methylation but not hydroxymethylation on Myh6 promoter. Such TAC-induced CpG methylation was a rapid process with near saturation of CpG methylation within 2 days of TAC. Conversely, on the Myh7 locus, the methylation of CpG sites was absent in fetal hearts, modest in healthy adult hearts, and sparse in TAC-stressed hearts (Fig. 2B and C). Such differential CpG methylation correlates inversely with Myh expression at various pathophysiological conditions, suggesting a functional role of CpG methylation in Myh gene regulation.
Fig. 2.
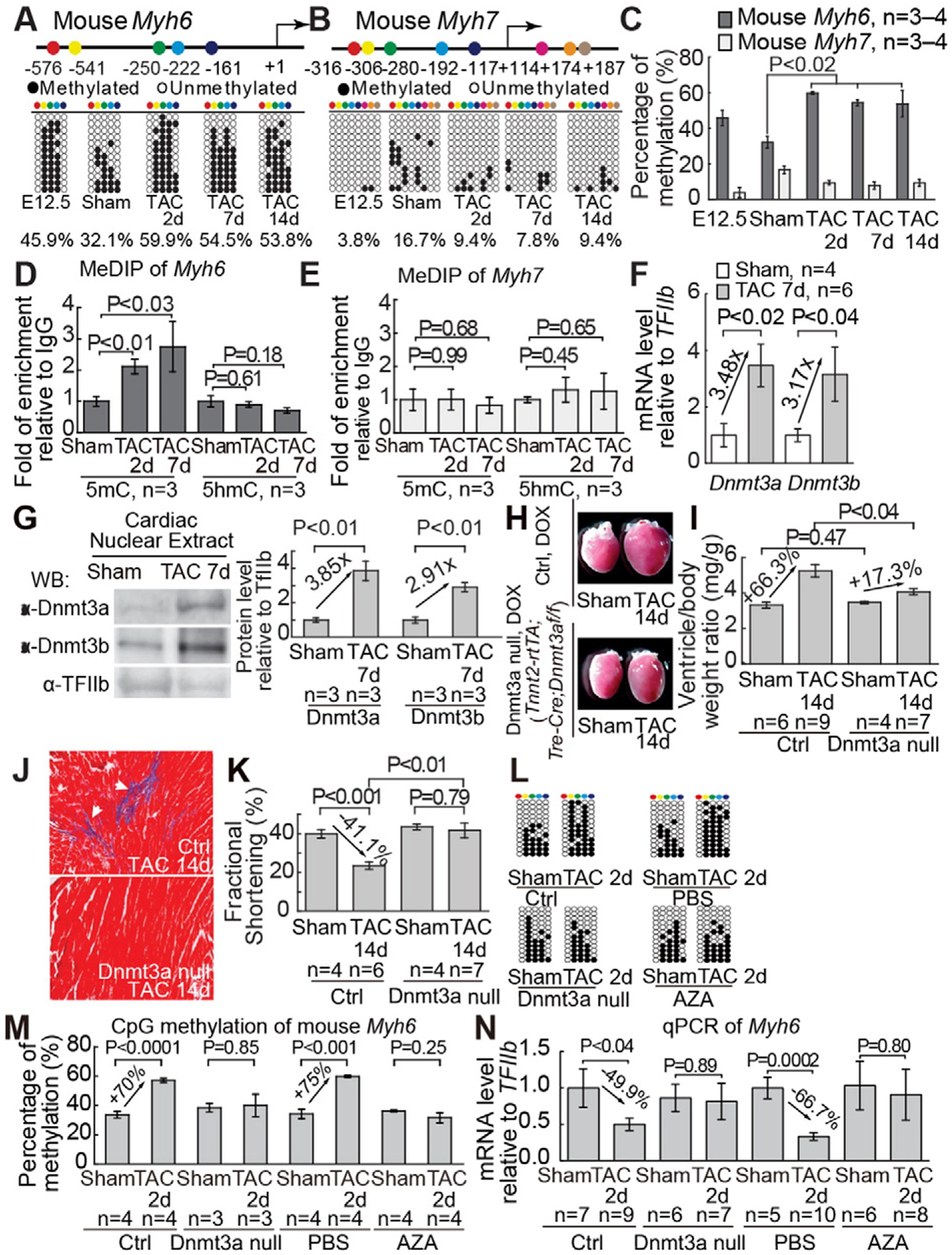
Dnmt3a is essential for cardiac hypertrophy and dysfunction. (A and B) Distribution of CpG sites across proximal promoters or 5′-untranslated regions of mouse Myh6 (A) and Myh7 (B), as well as the methylation of CpG sites in fetal heart ventricles (E12.5) and adult heart ventricles after 2–14 days of sham/TAC operation. The numbers denote CpG sites relative to the transcriptional start site (+1). CpG sites are color coded. Open and closed circles represent unmethylated and methylated CpG sites, respectively. (C) Quantification of the percentage of methylated CpG sites on mouse Myh6 and Myh67 indicated in Fig. 2A, B. “n” represents the number of different hearts used for analysis, with each heart having 12 randomly selected clones sequenced. (D and E) Quantitation of MeDIP of 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC) on the proximal promoters of Myh6 (D) and Myh7 (E) in adult heart ventricles after 2 days and 7 days of sham/TAC operation. Data are presented as enrichment relative to IgG control. (F) Quantitation of mRNA of Dnmt3a and Dnmt3b in adult hearts 7 days after sham/TAC operation. (G) Western blot and quantitation of Dnmt3a, Dnmt3b proteins 7 days after TAC. Internal control: TFIIb. (H–K) Gross morphology of ventricle (H), quantitation of ventricle/body weight ratio (I), trichrome staining of left ventricles (J), and echocardiographic measurement of left ventricular fractional shortening (K) in littermate control and mutant mice lacking myocardial Dnmt3a 14 days after sham/TAC operation. Ctrl: control mice. Dnmt3a null: Tnnt2-rtTA;Tre-Cre;Dnmt3af/f mice. (L and M) Methylation of CpG sites on the proximal promoter of Myh6 in hearts 2 days after sham or TAC operation. Representative sequencing results (L) and quantitation (M) of CpG methylation of individual hearts are shown. “n” represents the number of different hearts used for analysis, with each heart having 12 randomly selected clones sequenced. (N) Quantitation of Myh6 mRNA 2 days after sham or TAC operation. Data are presented as mRNA levels normalized to the sham-operated hearts in control mice or PBS-treated mice, respectively. Ctrl: control heart. P-value: Student’s t-test. Error bar: SEM, standard error of the mean.
To identify enzymes responsible for TAC-induced DNA methylation of Myh6, we examined the expression of two DNA methyltransferases (Dnmt 3a and 3b) required for de novo CpG methylation (Supplemental Text) [2,23]. Dnmt3a and Dnmt3b mRNAs in healthy adult hearts were low but up-regulated 3–4-fold by TAC (Fig. 2F). This was confirmed by western blot analysis (Fig. 2G) and by immunostaining: abundant Dnmt3a/3b proteins in the fetal myocardium (Figure S4A and S4B), minimal Dnmt3a/3b in healthy adult myocardium (Figure S4C and S4D), and Dnmt3a/3b reactivation in TAC-stressed adult myocardium (Figure S4C and S4D). Such Dnmt3 dynamics corresponded to that of CpG methylation, G9a/Glp, and H3K9me2, suggesting that Dnmt3 reactivation is necessary for CpG methylation, cardiac hypertrophy, and dysfunction.
Given that the induction of Dnmt3a protein by TAC was higher than that of Dnmt3b (Fig. 2G) and that Dnmt3a has higher enzymatic activity than Dnmt3b [27], we tested the necessity of Dnmt3a for TAC-induced hypertrophy. We generated Tnnt2-rtTA;Tre-Cre;Dnmt3af/f mice to enable doxycycline-inducible deletion of Dnmt3a in cardiomyocytes. After a 7-day doxycycline treatment to activate myocardial deletion of Dnmt3a, the mice appeared grossly normal, indicating that short-term Dnmt3a disruption in the heart has no significant effect at the baseline, consistent with minimal Dnmt3 expression in unstressed hearts. We then used 27-gauge needle to perform TAC on littermate control (Dnmt3af/+, Dnmt3af/f, or Tnnt2-rtTA;Tre-Cre;Dnmt3af/+) and mutant (Tnnt2-rtTA;Tre-Cre;Dnmt3af/f) mice. Dnmt3a immunostaining showed 80% disruption of myocardial Dnmt3a activation without changes of Dnmt3a in endothelial cells (Figure S4E and S4F), confirmed by western blot analysis (Figure S4G). Within 2 weeks after TAC, control mice on doxycycline diet developed cardiac hypertrophy with grossly enlarged hearts (Fig. 2H), increased ventricle/body-weight ratio (Fig. 2I), enlarged cardiomyocytes (Figure S4H–S4L), cardiac fibrosis (Fig. 2J), contractile dysfunction with FS reduction (Fig. 2K), and increased chamber size (Figure S4M). In contrast, doxycycline-treated mutant mice (Tnnt2-rtTA;Tre-Cre;Dnmt3af/f) exhibited mild cardiac hypertrophy, minimal fibrosis, and no cardiac dysfunction or chamber dilatation (Fig. 2H–K, and S4H–S4M). To examine the longer-term effect of Dnmt3a on cardiac function, we used a less stringent 26-gauge needle to perform TAC, with less pressure load and slower disease progression. In the absence of myocardial Dnmt3a, FS began to improve 4 weeks after TAC, and the improvement lasted for at least 8 weeks with 71% reduction of cardiac dysfunction (P = 0.002) and 45% reduction of hypertrophy (P = 0.041) (Figure S4N–S4P). Furthermore, pharmacological inhibition of DNA methylation with the nanomolar inhibitor 5-Azacytidine (AZA) [13,33] abolished TAC-induced cardiac hypertrophy, fibrosis, and dysfunction (Figure S5A–S5J). Overall, without myocardial Dnmt activity, there was a 45–100% reduction of hypertrophy and 69–100% reduction of cardiac dysfunction and chamber dilatation, indicating Dnmt3a plays a major role in stress-induced cardiac pathology. To test the requirement of Dnmt3a for CpG methylation, we used bisulfite sequencing to examine CpG methylation on Myh6 promoter. Within two days of TAC, Dnmt3 disruption by myocardial deletion or AZA treatment abolished TAC-induced CpG methylation of Myh6 (Fig. 2L and M) and reversed TAC-induced Myh6 repression (Fig. 2N), without significant influence on Myh7 (Figure S5K). Therefore, stress-induced Dnmt3a is essential for CpG methylation, Myh6 repression, and cardiac hypertrophy.
2.4. G9a recruits Dnmt3 to methylate CpG sites on Myh6
ChIP analyses of mouse left ventricles showed that TAC triggered the binding of G9a and Dnmt3a to Myh6, but not Myh7 promoter (Fig. 3A and B). Co-immunoprecipitation showed that G9a and Dnmt3a formed a physical complex in stressed left ventricles (Fig. 3C), suggesting that G9a and Dnmt3a cooperate to silence Myh6. Interestingly, disruption of Dnmt3a in cardiomyocytes didn’t change G9a expression (Figure S6A–S6C) and had no effects on G9a binding or H3K9 methylation of Myh6 promoter (Fig. 3D). Conversely, in stressed hearts that lacked G9a/Glp in cardiomyocytes, the binding of Dnmt3a to Myh6, in the absence of Dnmt3a expression changes (Figure S6D–S6F), was eliminated (Fig. 3E), accompanied by a complete loss of TAC-induced Myh6 CpG methylation (Fig. 3F). Therefore, G9a doesn’t require Dnmt3a to target Myh6 or catalyze H3K9 methylation, whereas Dnmt3a requires G9a to bind to and methylate CpG sites of Myh6.
Fig. 3.
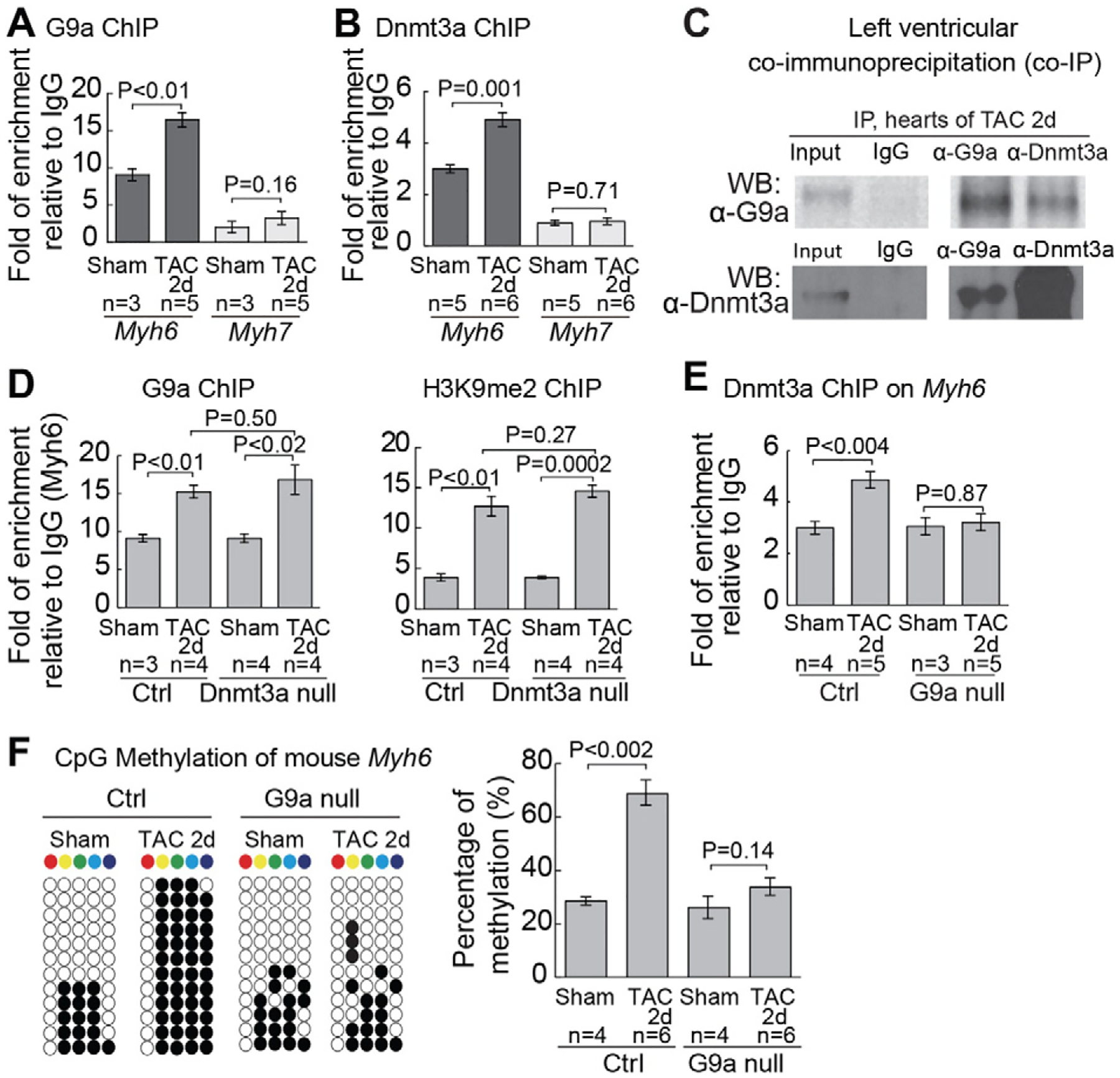
G9a recruits Dnmt3a to Myh6 promoter for CpG methylation (A) Quantitation of G9a ChIP on proximal promoters of Myh6 and Myh7 two days after sham/TAC operation. Data are presented as G9a enrichment relative to IgG control. (B) Quantitation of Dnmt3a ChIP on proximal promoters of Myh6 and Myh7 2 days after sham/TAC operation. Data are presented as Dnmt3a enrichment relative to IgG control. (C) Co-immunoprecipitation of G9a and Dnmt3a in left ventricles 2 days after TAC. (D) Quantitation of G9a and H3K9me2 ChIP on the proximal promoter of Myh6 2 days after sham/TAC operation in control and Dnmt3a null hearts. Data are presented as G9a or H3K9me2 enrichment normalized to sham-operated hearts in control mice. (E) Quantitation of Dnmt3a ChIP on the proximal promoter of Myh6 in control and G9a null hearts 2 days after sham/TAC operation. G9a null: Tnnt2-rtTA;Tre-Cre;G9af/f heart. (F) Quantitation of CpG methylation of Myh6 in control and G9a null hearts. Representative sequencing results and quantitation analysis of CpG methylation of individual hearts are shown. “n” represents the number of different hearts used for analysis, with each heart having 12 randomly selected clones sequenced.
2.5. Chromatin methylation by G9a and Dnmt requires Brg1
Given that Brg1 is a stress-activated chromatin-remodeling factor that controls Myh and cardiac hypertrophy [9], we tested if G9a and Dnmt3a required Brg1 to silence Myh6. Co-IP studies showed that Brg1 complexed with G9a and Dnmt3a in TAC-stressed hearts (Fig. 4A). However, Brg1 did not require G9a or Dnmt3a to target Myh6 promoter, as evidenced by the comparable ChIP quantitation of TAC-induced Brg1 enrichment on Myh6 with or without G9a or Dnmt3a in cardiomyocytes (Fig. 4B). Conversely, G9a and Dnmt3a required Brg1 to bind to Myh6 promoter. In TAC-stressed mice that lacked Brg1 in cardiomyocytes (dox-treated Tnnt2-rtTA;Tre-Cre;Brg1f/f mice) but had normal G9a and Dnmt3a expression (Figure S6G–S6K), G9a and Dnmt3 were absent or greatly reduced from Myh6 promoter with a consequent loss of H3K9me2 and CpG methylation (Fig. 4C–F). The need of Brg1 for G9a and Dnmt3a to bind to Myh6 promoter occurred within 2 days of TAC before there’s apparent hypertrophy (Figure S3K and S3L). Re-ChIP (sequential ChIP) analyses confirmed the presence of G9a and Dnmt3a proteins on Brg1-immunoprecipited chromatin of Myh6 (but not Myh7) promoter (Fig. 4G). Given G9a was essential for recruiting Dnmt3a to Myh6, the data suggest a stress-activated sequence of recruitment from Brg1, G9a to Dnmt3a to methylate and establish repressive chromatin for Myh6 silencing.
Fig. 4.
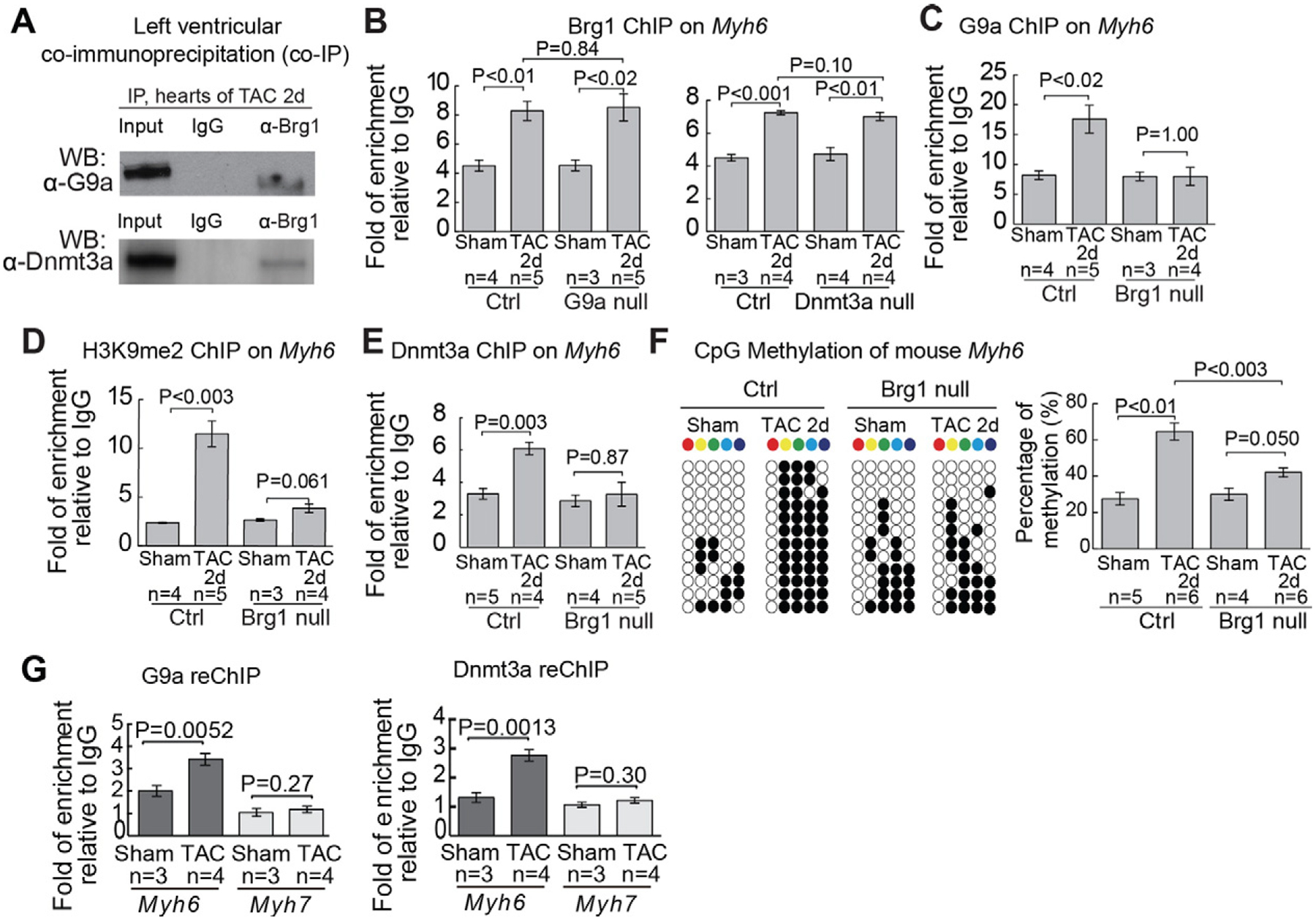
Brg1 recruits G9a and Dnmt3a to Myh6 promoter for H3K9 and CpG methylation (A) Co-immunoprecipitation of Brg1 with G9a and Dnmt3a in left ventricles 2 days after TAC. (B) Quantitation of Brg1 ChIP on the proximal promoter of Myh6 in control, G9a null, and Dnmt3a null hearts 2 days after sham/TAC operation. (C and D) Quantitation of G9a (C) and H3K9me2 (D) ChIP on the proximal promoter of Myh6 in control and Brg1 null hearts 2 days after sham/TAC operation. Brg1 null: Tnnt2-rtTA;Tre-Cre;Brg1f/f heart. (E) Quantitation of Dnmt3a ChIP on the proximal promoter of Myh6 in control and Brg1 null hearts 2 days after sham/TAC operation. (F) Quantitation of CpG methylation of Myh6 in control and Brg1 null hearts. Representative sequencing results and quantitation analysis of CpG methylation of individual hearts are shown. “n” represents the number of different hearts used for analysis, with each heart having 12 randomly selected clones sequenced. (G) Quantitation of G9a and Dnmt3a on Brg1-immunoprecipitated proximal promoters of Myh6 and Myh7 after 7 days of sham/TAC operation. Data are presented as G9a or Dnmt3a enrichment relative to IgG control. P-value: Student’s t-test. Error bar: SEM, standard error of the mean.
2.6. G9a/GLP, DNMT3, and chromatin methylation in human hypertrophic hearts
We examined G9a/GLP and DNMT3 expression, as well as H3K9me2 and CpG methylation of MYH loci in human hearts. Human left ventricular tissues were sampled from donor hearts that were not transplanted due to left ventricular hypertrophy (LVH) or the lack of matched recipients within 4–6 h after harvest of the heart. The donors did not have known family history of inheritable heart disease (Figure S7), and LVH (diagnosed by echocardiography) was linked to chronic hypertension and/or morbid obesity. We validated the normal/disease status of the donor hearts by analysis of MYH expression [1,21,22]. The hypertrophic left ventricles had down-regulated MYH6 mRNA (by 82.4%) and up-regulated MYH7 (by 4–5 fold) (Fig. 5A), consistent with the clinical diagnosis of LVH. ChIP analysis revealed that H3K9me2 was enriched 6.7-fold on MYH6 promoter without significant enrichment on MYH7 promoter of hypertrophic left ventricles (Fig. 5B). Bisulfite sequencing showed a 1.9-fold increase of CpG methylation on MYH6 locus (Fig. 5, C and D) and a converse 55% decrease of CpG methylation on MYH7 locus (Fig. 5E and F). These changes of H3K9me2 and CpG methylation of MYH promoters correlated inversely with MYH mRNA levels (Fig. 5A) and resembled changes observed in mouse hearts. The results suggest that H3K9 and CpG methylation provides a conserved mechanism for MYH6 repression in human hypertrophic hearts.
Fig. 5.
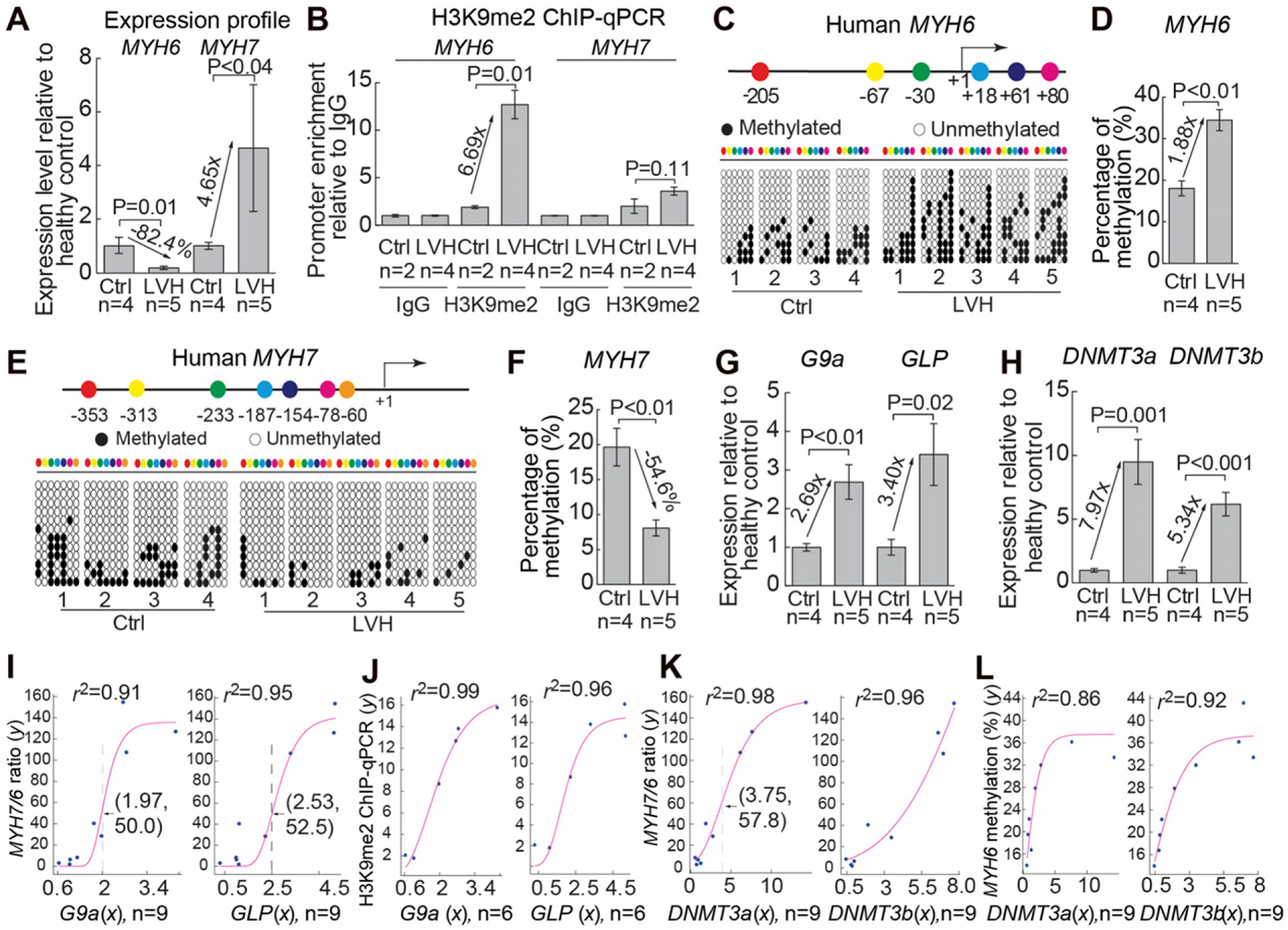
G9a/GLP, DNMT3, and chromatin methylation in human hypertrophic hearts. (A) Quantitation of Myh6 and Myh7 mRNAs in normal and hypertrophic left ventricles of human hearts. Ctrl: control. LVH: left ventricular hypertrophy. (B) Quantitation of H3K9me2 ChIP on proximal promoters of human MYH6 and MYH7 in normal and hypertrophic left ventricles of human hearts. (C and D) Distribution of the CpG sites across the proximal promoter of human MYH6 (C) and quantitation of CpG methylation on MYH6 (D). The numbers denote CpG sites relative to the transcriptional start site (+1). The CpG sites are color coded. Open and closed circles represent unmethylated and methylated CpG sites, respectively. “n” represents the number of different hearts, with each heart having 12 randomly selected clones sequenced. (E and F) Distribution of the CpG sites across the proximal promoter of human MYH7 (E) and quantitation of CpG methylation on MYH7 (F). “n” represents the number of different hearts used for analysis. (G) Quantitation of human G9a and GLP mRNAs. (H) Quantitation of human DNMT3a and DNMT3b mRNA levels. (I and J) Correlation of G9a and GLP mRNA level (x axis) with Myh7/Myh6 mRNA ratio (y axis) (I) and with H3K9 methylation of Myh6 (y axis) (J). Red: regression curve. e, the base of natural logarithm (~2.718). Arrow and dashed line, the inflection point. (K and L) Correlation of DNMT3a and DNMT3b mRNA level (x axis) with Myh7/Myh6 mRNA ratio (y axis) (K) and with CpG methylation of Myh6 (y axis) (L). Red: regression curve. e, the base of natural logarithm (~2.718).
We then examined G9a/GLP and DNMT3 expression in human hypertrophic left ventricles. RT–qPCR showed that G9a and GLP expression was elevated in the hypertrophic hearts by 2.7–3.4-fold (Fig. 5G), whereas DNMT3a and DNMT3b elevated by 5.3–8.0-fold (Fig. 5H), resembling G9a/Glp and Dnmt3 increase in TAC-stressed mouse hearts and suggesting that G9a and DNMT3 may play essential roles in human cardiac hypertrophy. Non-linear regression analysis revealed a strong correlation between G9a/GLP mRNA and the disease severity as defined by MYH7/MYH6 ratio [1,21,22] (regression r2 = 0.91 and 0.95, Fig. 5I). Also, G9a and GLP level correlated strongly with H3K9me2 of human MYH6 promoter (regression r2 = 0.99 and 0.96, Fig. 5J), consistent with the necessity of G9a for hypertrophy and H3K9 methylation on MYH6 in mice. DNMT3a and 3b level also correlated strongly with the disease severity (regression r2 = 0.98 and 0.96, Fig. 5K) and with CpG methylation of MYH6 (regression r2 = 0.86 and 0.92, Fig. 5L), consistent with the necessity of Dnmt3a for hypertrophy and Myh6 CpG methylation in mice. Although the human heart tissue studies are correlative and limited in sample number, the human and mouse studies, collectively, suggest that the activation of G9a/GLP, DNMT3a, and H3K9/CpG methylation contributes to human cardiac hypertrophy.
3. Discussion
We demonstrated a new mechanistic link from chromatin remodeling to histone (H3K9) and DNA (CpG) methylation that contributes to cardiomyopathy (Fig. 6). Activated by stress, Brg1 governs this epigenetic event by recruiting G9a and then Dnmt3 proteins to catalyze chromatin methylation on Myh6 promoter. The current studies, together with our previous work [9], show that five classes of chromatin-regulating factors—Brg1, G9a/GLP, Dnmt3, Hdac2, and Parp1—converge on Myh6 loci to create repressive chromatin for gene control in hypertrophic hearts. Interestingly, among these factors that regulate cardiac hypertrophy, only the expression of Brg1, G9a/Glp, and Dnmt3—but not Hdac (Hdac1, 2, 3) or Parp1—is stress-induced in the mouse and human hearts (Figure S8). Because all these five factors are essential for cardiomyopathy, it will be crucial to elucidate the biochemical basis of how stress-induced factors integrate with constitutive ones to modify chromatin substrates for gene control. Identification of other target genes of this epigenetic complex will require genome-wide studies to determine how the Brg1–G9a/Glp–Dnmt3a complex modifies the epigenome and reprograms genetic pathways in stressed hearts.
Fig. 6.
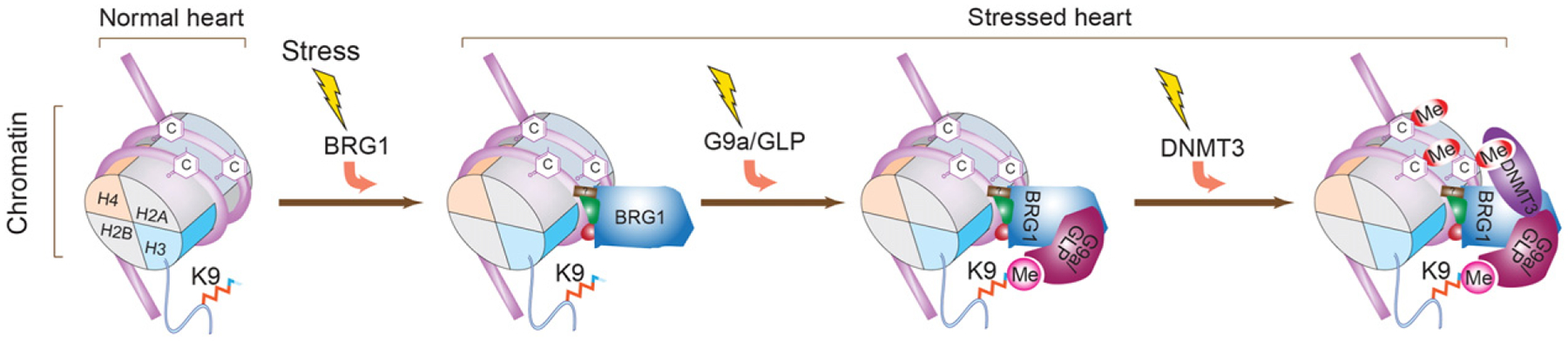
A model of how chromatin remodeling is mechanistically linked to histone (H3K9) and DNA (CpG) methylation on Myh6 promoter Working model showing that cardiac stress triggers sequential recruitment of chromatin regulators on the Myh6 locus to establish a repressive chromatin scaffold. H: histone. K9: the ninth lysine residue of histone H3 N-terminal tail. C: cytosine at the CpG site. Me: methyl group on H3K9 or CpG sites.
A recent study shows that the proximal promoter DNA sequence—which is mostly unperturbed by environmental fluctuations—is both necessary and sufficient to determine its own DNA methylation states [17]. This brings up an intriguing question [3]: whether the widespread differences in DNA methylation of individual human epigenomes [18] reflect variations of the underlying DNA sequence or differences in the experience of external environment. Because DNMTs are actively recruited to effect DNA methylation of α-MHC only upon external cardiac stress, our studies suggest that external environment plays a role in defining the DNA methylation state. Future investigations will be needed to illuminate how environmental factors interact with specific DNA sequence to determine DNA methylation of the epigenome.
Chromatin analyses in mouse and human hearts provide new insights into the mechanism of heart failure. Chemically stable DNA methylation marks on crucial genes may contribute to the clinical deterioration of failing hearts. These marks are difficult to erase without altering key factors involved in their formation. Disrupting the Brg1–G9a–Dnmt3 sequence that leads to the long-lasting methylation may halt disease progression through restoration of Myh6 and likely other essential genes (Supplemental text). We showed that small molecules that inhibit G9a/GLP or DNMT are capable of erasing H3K9/DNA methylation marks on the Myh6 promoter and reducing cardiac dysfunction, revealing a new therapeutic avenue.
4. Experimental Procedures
4.1. Mice
The mouse embryonic date was determined by the conventional method, in which the date of observing a vaginal plug was set as embryonic day E0.5. The use of mice for studies is in compliance with the regulations of Indiana University, Stanford University, and National Institute of Health.
4.2. Immunostaining, trichrome Staining, and quantification
The following primary antibodies were used for immunostaining: anti-G9a (Cat# PP-A8620A-00, R&D Systems), anti-GLP (Cat# PP-B0422–00, R&D Systems), anti-DNMT3a (H-295, Cat# sc20703, Santa Cruz Biotechnology), anti-DNMT3b (Cat# ab16049, Abcam). The immunofluorescence staining of G9a or Dnmt3a by measuring the intensity of nuclear fluorescence signals and background using Image J. The corrected intensity (intensity — background) of staining was used. At least four measurements were made for each heart. N represents the number of animals used within each group. To quantitate cardiac interstitial fibrosis, the perivascular fibrotic areas were excluded. The blue channel (fibrotic) and red channel (non-fibrotic) were separated by Photoshop and quantitated by Image J. The ratios of (area of blue)/(area of blue and red) were used to evaluate the fibrosis. At least four measurements were made for each heart. N represents the animals used within a group.
4.3. Transaortic constriction (TAC)
Mice were fed with doxycycline food 7 days before the operation to induce deletion of G9a, Dnmt3a, or Brg1. TAC was performed on littermate mice of 8–10 weeks of age and approximately between 20 and 25 g of weight. Mice were fed with doxycycline food pellets (6 mg doxycycline/kg of food, Bioserv, Frenchtown, NJ) 7 days before the operation. Mice were anesthetized with isoflurane (2–3%, inhalation) in an induction chamber and then intubated with a 20-gauge intravenous catheter and ventilated with a mouse ventilator (Minivent, Harvard Apparatus, Inc). Anesthesia was maintained with inhaled isoflurane (1–2%). A longitudinal 5-mm incision of the skin was made with scissors at midline of sternum. The chest cavity was opened by a small incision at the level of the second intercostal space 2–3 mm from the left sternal border. While opening the chest wall, the chest retractor was gently inserted to spread the wound 4–5 mm in width. The transverse portion of the aorta was bluntly dissected with curved forceps. Then, 6–0 silk was brought underneath the transverse aorta between the left common carotid artery and the brachiocephalic trunk. 27 or 26-gauge needle was placed directly above and parallel to the aorta. The loop was then tied around the aorta and needle, and secured with a second knot. The needle was immediately removed to create a lumen with a fixed stenotic diameter. The chest cavity was closed by 6–0 silk suture. Sham-operated mice underwent similar surgical procedures, including isolation of the aorta, looping of aorta, but without tying of the suture. The pressure load caused by TAC was verified by the pressure gradient across the aortic constriction measured by echocardiography. The mean pressure gradients of mice with TAC were 20–30 mm Hg (peak gradient 30–50 mm Hg).
4.4. Echocardiography
The echocardiographer was blinded to the genotypes, surgical, or pharmacological treatment of the mice. Transthoracic ultrasonography with a GE Vivid 7 ultrasound platform (GE Health Care, Milwaukee, WI) and a 13 MHz transducer was used to measure aortic pressure gradient and left ventricular function. Echocardiography was performed on control, Tnnt2-rtTA;Tre-Cre;G9af/f, and Tnnt2-rtTA;Tre-Cre;Dnmt3af/f mice, as well as on mice treated with PBS (phosphate buffered saline), BIX, and AZA at 8 to 12 weeks of age. To minimize the confounding influence of different heart rates on aortic pressure gradient and left ventricular function, the flow of isoflurane (inhalational) was adjusted to anesthetize the mice while maintaining their heart rates at 450–550 beats per minute. The peak aortic pressure gradient was measured by continuous wave Doppler across the aortic constriction. The left ventricular function was assessed by the M-mode scanning of the left ventricular chamber, standardized by two-dimensional, short-axis views of the left ventricle at the mid papillary muscle level. The fractional shortening (FS) of the left ventricle was defined as 100% × (1 − endsystolic/enddiastolic diameter), representing the relative change of left ventricular diameters during the cardiac cycle. The mean FS of the left ventricle was determined by the average of FS measurement of the left ventricular contraction over 5 beats. P-values were calculated by the Student-t test. Error bars indicate standard error of mean.
4.5. Chromatin immunoprecipitation–quantitative PCR (ChIP–qPCR)
Hearts from E12.5 mouse embryos, adult mice, and human subjects were used for ChIP assay as described previously [9]. Chromatin was sonicated to generate average fragment sizes of 200–600 bp, and immunoprecipitated using anti-BRG1 J1 antibody [9], anti-G9a antibody (Cat# G6919, Sigma), anti-H3K9Me2 antibody (Cat# 17–648, Millipore), anti-DNMT3a antibody (H-295, Cat# sc20703, Santa Cruz Biotechnology), or normal control IgG. Isolation and purification of immunoprecipitated and input DNA were done according to the manufacturer’s protocol (Magna ChIP Protein G Magnetic Beads, Cat# 16–662, Millipore), and qPCR analysis of immunoprecipitated DNA was performed. ChIP–qPCR signals of individual ChIP reaction was standardized to its own input qPCR signals and normalized to IgG ChIP signals. PCR primers (listed below) were designed to amplify the proximal promoter regions of mouse Myh6 (−426, −320), mouse Myh7 (−102, +58), human MYH6 (−169, −5) and human MYH7 (−343, −189). The DNA positions are denoted relative to the transcriptional start site (+1).
PCR primers for ChIP–qPCR:
Mouse ChIP-Myh6-F (GCAGATAGCCAGGGTTGAAA),
Mouse ChIP-Myh6-R (TGGGTAAGGGTCACCTTCTC),
Mouse ChIP-Myh7-F (GTGACAACAGCCCTTTCTAAAT),
Mouse ChIP-Myh7-R (CTCCAGCTCCCACTCCTACC),
Human ChIP-MYH6-F (AAATCAGGGGGCCCTGCTG),
Human ChIP-MYH6-R (GTCCTCAAAGCTCCAGTTCCT),
Human ChIP-MYH7-F (GGACATTGGCTGCCTGTGGT),
Human ChIP-MYH7-R (TCATTGTTATGGCATGGACTGT).
4.6. Bisulfite genomic sequencing
For each reaction, genomic DNA was extracted from 25 mg of the left ventricular tissues of mouse or human hearts by ZR Tissue & Insect DNA MiniPrep kit (Cat# D6016, Zymo Research). Bisulfite treatment of 2 μg DNA per reaction was performed using the EZ DNA Methylation-Gold Kit (Cat# D5005, Zymo Research). PCR primers are listed below. Amplified products by ZymoTaq PreMix (Cat# E2003, Zymo Research) were cloned into pDrive Cloning Vector (Qiagen). For each gene promoter, twelve randomly selected clones were sequenced with the T7 primer. N number in the figures represents the number of different hearts used for analysis, with each heart having 12 randomly selected clones sequenced.
4.7. Methylated-DNA immunoprecipitation–quantitative PCR (MeDIP–qPCR)
Left ventricles from adult mice were used for MeDIP assay 2 days and 7 days after sham or TAC operations. Genomic DNA was extracted by ZR Tissue & Insect DNA MiniPrep kit (Cat# D6016, Zymo Research) and sonicated to generate average fragment sizes of 200–600 bp, and immunoprecipitated using anti-5mC (5-methylcytosine, Cat# 39,649, Active Motif), anti-5hmC (5-hydroxymethylcytosine, Cat#39,791, Active Motif), or normal control IgG combined with Magna ChIP Protein G Magnetic Beads (Cat# 16–662, Millipore). Immunoprecipitated and input DNA were isolated and purified, followed by qPCR analysis of immunoprecipitated DNA. MeDIP–qPCR signals of individual ChIP reaction was standardized to its own input qPCR signals and normalized to IgG ChIP signals.
4.8. Statistical analysis
N numbers were denoted in each test in the figures. We used mouse littermates to control and perform our experiments. The assignment to each experimental subgroups was based on genotypes. Littermate mice with same genotypes were randomly picked from the cage and assigned to control and experimental subgroups. Data, presented as mean ± standard error of mean, were evaluated by Student’s t-test. P values of less than 0.05 were considered significant.
4.9. Construction of regression curves and derivation of equations
The regression curves and r-square (r2) for Fig. 5I to L were derived using the Levenburg–Marquardt non-linear regression method and a statistics/curve modeling software (XLfit, 2005). Different curve models ranging from linear, polynomial, exponential/log, power series, hyperbolic and sigmoidal curves were tested. Curves that fitted best with the data points based on r2 are shown in Fig. 5.
Supplementary Material
Acknowledgements
We thank Yoichi Shinkai and Eugene Oltz for providing G9af/f mice.C-P.C. is the Charles Fisch Scholar of Cardiology and was supported by American Heart Association (AHA, Established Investigator Award). National Institutes of Health (NIH, R01HL118087, R01HL121197), March of Dimes Foundation (MOD), California Institute of Regenerative Medicine (CIRM), Indiana University (IU) School of Medicine-IU Health Strategic Research Initiative, and the IU Physician-Scientist Initiative, endowed by Lilly Endowment, Inc. W.L. was supported by Oak Foundation; Y.X. by Oak Foundation, AHA, and Lucile Packard Children’s Foundation; C.S. by MOD and NIH fellowship; J.W. and H.-S.V.C by the CIRM and NIH.
Footnotes
This article is part of a Special Issue entitled: Cardiomyocyte Biology: Integration of Developmental and Environmental Cues in the Heart edited by Marcus Schaub and Hughes Abriel.
Clarification: Biological Sciences-Medical Sciences.
Conflict of interest
The authors have not conflict of interest to report at this time.
Transparency document
The Transparency document associated with this article can be found, in the online version.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbamcr.2016.03.002.
References
- [1].Abraham WT, Gilbert EM, Lowes BD, Minobe WA, Larrabee P, Roden RL, Dutcher D, Sederberg J, Lindenfeld JA, Wolfel EE, et al. , Coordinate changes in Myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype, Mol. Med 8 (2002) 750–760. [PMC free article] [PubMed] [Google Scholar]
- [2].Bestor TH, The DNA methyltransferases of mammals, Hum. Mol. Genet 9 (2000) 2395–2402. [DOI] [PubMed] [Google Scholar]
- [3].Bird A, Putting the DNA back into DNA methylation, Nat. Genet 43 (2011) 1050–1051. [DOI] [PubMed] [Google Scholar]
- [4].Cedar H, Bergman Y, Linking DNA methylation and histone modification: patterns and paradigms, Nat. Rev. Genet 10 (2009) 295–304. [DOI] [PubMed] [Google Scholar]
- [5].Clapier CR, Cairns BR, The biology of chromatin remodeling complexes, Annu. Rev. Biochem 78 (2009) 273–304. [DOI] [PubMed] [Google Scholar]
- [6].Greer EL, Shi Y, Histone methylation: a dynamic mark in health, disease and inheritance, Nat. Rev. Genet 13 (2012) 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gupta R, Nagarajan A, Wajapeyee N, Advances in genome-wide DNA methylation analysis, Biotechniques 49 (2010) iii–ixi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST, Jin KK, Xu W, Lin CY, Lin CJ, et al. , A long noncoding RNA protects the heart from pathological hypertrophy, 2014. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, Zhou B, Chang CP, Chromatin regulation by Brg1 underlies heart muscle development and disease, Nature 466 (2010) 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Herron TJ, Devaney E, Mundada L, Arden E, Day S, Guerrero-Serna G, Turner I, Westfall M, Metzger JM, Ca2+-independent positive molecular inotropy for failing rabbit and human cardiac muscle by alpha-myosin motor gene transfer, FASEB J. 24 (2010) 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Herron TJ, McDonald KS, Small amounts of alpha-myosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments, Circ. Res 90 (2002) 1150–1152. [DOI] [PubMed] [Google Scholar]
- [12].Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A, The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing, PLoS One 5 (2010), e8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Issa JP, Kantarjian H, Azacitidine, Nat. Rev. Drug Discov. Suppl (2005) S6–S7. [DOI] [PubMed] [Google Scholar]
- [14].James J, Martin L, Krenz M, Quatman C, Jones F, Klevitsky R, Gulick J, Robbins J, Forced expression of alpha-myosin heavy chain in the rabbit ventricle results in cardioprotection under cardiomyopathic conditions, Circulation 111 (2005) 2339–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jenuwein T, The epigenetic magic of histone lysine methylation, FEBS J. 273 (2006) 3121–3135. [DOI] [PubMed] [Google Scholar]
- [16].Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro Miguel L., Rea S, Mechtler K, Kowalski JA, Homon CA, et al. , Reversal of H3K9me2 by a Small-Molecule Inhibitor for the G9a Histone Methyltransferase, Mol. Cell 25 (2007) 473–481. [DOI] [PubMed] [Google Scholar]
- [17].Lienert F, Wirbelauer C, Som I, Dean A, Mohn F, Schubeler D, Identification of genetic elements that autonomously determine DNA methylation states, Nat. Genet 43 (2011) 1091–1097. [DOI] [PubMed] [Google Scholar]
- [18].Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR,Lee L, Ye Z, Ngo QM, et al. , Human DNA methylomes at base resolution show widespread epigenomic differences, Nature 462 (2009) 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Locher MR, Razumova MV, Stelzer JE, Norman HS, Moss RL, Effects of low-level α-myosin heavy chain expression on contractile kinetics in porcine myocardium, Am. J. Physiol. Heart Circ. Physiol 300 (2011) H869–H878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Locher MR, Razumova MV, Stelzer JE, Norman HS, Patel JR, Moss RL, Determination of rate constants for turnover of myosin isoforms in rat myocardium: implications for in vivo contractile kinetics, Am. J. Physiol. Heart Circ. Physiol 297 (2009) H247–H256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, et al. , Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium, J. Clin. Invest 100 (1997) 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Miyata S, Minobe W, Bristow MR, Leinwand LA, Myosin heavy chain isoform expression in the failing and nonfailing human heart, Circ. Res 86 (2000) 386–390. [DOI] [PubMed] [Google Scholar]
- [23].Okano M, Bell DW, Haber DA, Li E, DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development, Cell 99 (1999) 247–257. [DOI] [PubMed] [Google Scholar]
- [24].Saha A, Wittmeyer J, Cairns BR, Chromatin remodelling: the industrial revolution of DNA around histones, Nat. Rev. Mol. Cell Biol 7 (2006) 437–447. [DOI] [PubMed] [Google Scholar]
- [25].Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K,Kayama Y, Harada M, et al. , p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload, Nature 446 (2007) 444–448. [DOI] [PubMed] [Google Scholar]
- [26].Tachibana M, Ueda J, Fukuda M, Takeda N, Ohta T, Iwanari H, Sakihama T, Kodama T, Hamakubo T, Shinkai Y, Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9, Genes Dev. 19 (2005) 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Takeshima H, Suetake I, Shimahara H, Ura K, Tate S, Tajima S, Distinct DNA methylation activity of Dnmt3a and Dnmt3b towards naked and nucleosomal DNA, J. Biochem 139 (2006) 503–515. [DOI] [PubMed] [Google Scholar]
- [28].Tarakhovsky A, Tools and landscapes of epigenetics, Nat. Immunol 11 (2010) 565–568. [DOI] [PubMed] [Google Scholar]
- [29].van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ Jr., E.N. Olson, A family of microRNAs encoded by MYOSIN GENES governs myosin expression and muscle performance, Dev. Cell 17 (2009) 662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN, Control of stress-dependent cardiac growth and gene expression by a microRNA, Science 316 (2007) 575–579. [DOI] [PubMed] [Google Scholar]
- [31].Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP, Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells, Nat. Genet 41 (2009) 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, et al. , Structural biology of human H3K9 methyltransferases, PLoS One 5 (2010), e8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xiao D, Dasgupta C, Chen M, Zhang K, Buchholz J, Xu Z, Zhang L, Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats, Cardiovasc. Res 101 (2014) 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.