

Abstract
The gene therapy field has been galvanized by two technologies that have revolutionized treating genetic diseases: vectors based on adeno-associated viruses (AAVs), and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas gene-editing tools. When combined into one platform, these safe and broadly tropic biotherapies can be engineered to target any region in the human genome to correct genetic flaws. Unfortunately, few investigations into the design compatibility of CRISPR components in AAV vectors exist. Using AAV-genome population sequencing (AAV-GPseq), we previously found that self-complementary AAV vector designs with strong DNA secondary structures can cause a high degree of truncation events, impacting production and vector efficacy. We hypothesized that the single-guide RNA (sgRNA) scaffold, which contains several loop regions, may also compromise vector integrity. We have therefore advanced the AAV-GPseq method to also interrogate single-strand AAV vectors to investigate whether vector genomes carrying Cas9-sgRNA cassettes can cause truncation events. We found that on their own, sgRNA sequences do not produce a high degree of truncation events. However, we demonstrate that vector genome designs that carry dual sgRNA expression cassettes in tail-to-tail configurations lead to truncations. In addition, we revealed that heterogeneity in inverted terminal repeat sequences in the form of regional deletions inherent to certain AAV vector plasmids can be interrogated.
Keywords: adeno-associated virus, AAV-genome population sequencing, CRISPR-Cas, single molecule, real-time sequencing, gene therapy vectors, ssAAV, scAAV, sgRNA, ITR
Graphical Abstract
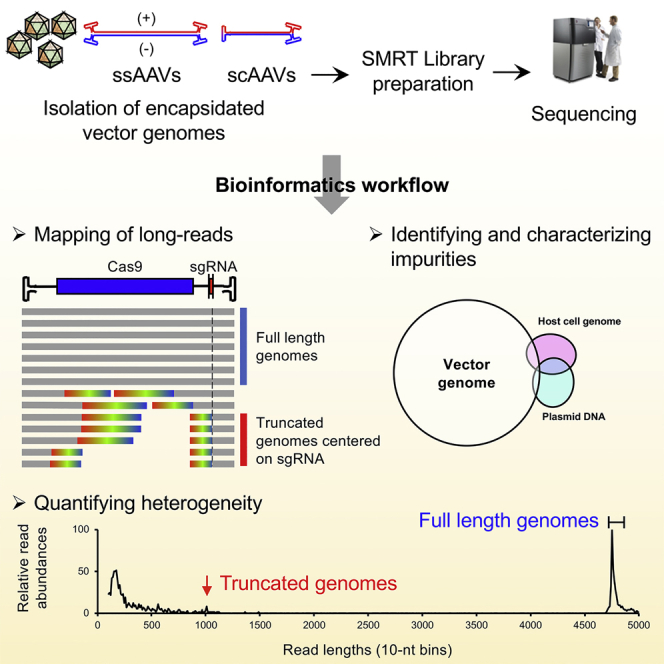
AAV-GPseq can quantify vector genome heterogeneity in AAV preparations from ITR to ITR. Using AAV-GPseq, Tran and colleagues show that CRISPR-Cas cassettes in vectors do not yield a high degree of truncation events, but certain dual-guide designs predicted to have strong secondary structure predominantly produce truncated genomes.
Introduction
Vectors based on adeno-associated viruses (AAVs) have in recent years been at the center of the gene therapy revolution. Due to their favorable safety profiles in countless pre-clinical studies and more than 120 clinical trials worldwide, recombinant AAVs (rAAVs) are now considered the leading vector platform for gene replacement, gene knockdown, and gene addition therapies.1 The AAV capsid is a 60-mer icosahedral virion consisting of three capsids proteins named VP1, VP2, and VP3 that are respectively expressed at an approximate 1:1:10 ratio and are encoded by a single open reading frame.1 Typically, rAAV vectors consist of a transgene that is under the control of a ubiquitous or tissue/cell-specific promoter, as well as auxiliary elements required for strong transgene expression and/or stabilization, such as a polyadenylation sequence or a synthetic intron.1 These components are flanked by inverted terminal repeats (ITRs), which are essential for viral genome replication as self-priming structures and harbor sequence motifs that are required for genome packaging into the pre-assembled capsid.
The merger of gene-editing methods and rAAV-mediated therapeutics has further expanded the utility of rAAVs toward an unlimited potential to treat genetic diseases. The leading gene-editing tools in this regard are those derived from the class of bacterial genes called clustered regularly interspaced short palindromic repeats (CRISPR) and their associated Cas proteins.2 These revolutionary tools have changed the face of genomics and medicine by giving researchers the ability to alter the genome through relatively straightforward molecular biology approaches. Genomic specificity is defined by the guide RNA (gRNA) sequence, which can be designed to be complementary to the target sequence. Provided that the gene-editing target is within proximity to a protospacer-adjacent motif (PAM), on-target editing is relatively specific and efficient.2 Cas proteins are by convention expressed from RNA polymerase II (RNA Pol II) promoters, lending to the potential for spatial and temporal control of gene-editing events. Cas-mediated gene editing has proven to be highly efficient in cell culture and can be achieved by standard transient-transfection procedures or by sustained transduction using viral vectors. The generation of transgenic animals that carry CRISPR-Cas components has also produced very powerful tools for creating relevant animal models for research.3 Continuing work to make these tools more specific and amendable to expanded protospacer rules have allowed researchers to achieve higher precision in gene editing. Additionally, recombinant engineering of the CRISPR-Cas system has now led to the development of gene promoter activity modulation, precision base editing, prime editing, and RNA-editing strategies.2 Unfortunately, translation of these tools into in vivo biotherapies is less straightforward. Among the wide-range of strategies to deliver CRISPR-Cas components into patients, rAAVs have been shown to be efficacious vehicles for conferring high-level Cas and single-guide RNA (sgRNA) transgene expression in vivo.4, 5, 6, 7, 8 Despite the potential compatibility of Cas9-mediated gene editing and AAV vectors, their relative safety is still poorly understood.
Previously, we reported that certain self-complementary (sc)AAV designs carrying small interfering RNA (siRNA) cassettes to knock down gene expression via RNA interference (RNAi) led to the formation of truncated genomes.9 Our results revealed that DNA sequences with high secondary structure can cause template switching events that lead to truncations. Interestingly, truncations were also found to center on non-siRNA cassette sequences such as promoters and even within the EGFP transgene,9,10 which is commonly used in pre-clinical AAV studies. In order to quantify the heterogeneity of AAV genomes in vector preparations, we developed AAV-genome population sequencing (AAV-GPseq), a method based on single-molecule, real-time (SMRT) sequencing as a universal pipeline to profile and characterize the integrity of scAAV vector genomes.10 Owing to the strand-displacement polymerase that is used during rolling-circle replication of the template strand, full resolution of individual vector genomes are obtained from ITR to ITR as an intact read without sequence reconstruction. With AAV-GPseq, we were also able to identify contaminating DNAs originating from the vector packaging cell line and packaging plasmid sequences. In addition, we were able to provide evidence that these contaminating genomes were chimeric with vector sequences, providing a mechanism for unwanted genomes to be actively packaged into AAV virions via Rep-mediated action. Importantly, these results reveal a means for these problematic species to persist in non-replicating cells following ITR-driven episome formation.11,12 Unfortunately, applying AAV-GPseq for analyzing single-stranded (ss)AAV vectors remained challenging, since the vector genomes are ostensibly incompatible with the SMRT sequencing approach, which requires adaptering of double-stranded DNA fragments.
In this study, we aimed to address whether sgRNA sequences, which are characterized by short hairpin loops,13 may cause truncations during the packaging of AAV. To achieve this goal, we further developed the AAV-GPseq approach to profile ssAAVs, since these are the typical platforms for housing the Cas9 cDNA, which exceeds the packaging capacity of scAAVs. Part of this advancement was our demonstration that ssAAVs can be successfully adaptered for SMRT sequencing. Since ssAAVs are on their own not readily adapterable as solitary genomes such as their scAAV counterparts, we have now established the capacity to adapter annealed plus and minus stranded genomes. In doing so, we are able to obtain high sequence resolution from ITR to ITR of ssAAV and scAAV vectors carrying sgRNA cassettes. Based on our analyses, we found that sgRNA expression cassettes on their own do not cause a high degree of truncation events. However, construct designs that house dual sgRNA expression cassettes, which are often used in AAV vectors for large deletion strategies in gene editing, can cause a high frequency of truncation and yield very poor abundance of functional vector during production if orientated in a tail-to-tail fashion. Such designs evidently would form long palindromic sequences with strong secondary structure. In addition, we found that similar to other vectors, there was a high percentage of contaminating genomes that were chimeras of cellular DNAs and an ITR-bearing vector sequence, once again posing concerns over whether AAV vectors may have long-term effects for patients receiving these therapies. Furthermore, the capacity for AAV-GPseq to query ITR composition in packaged ssAAV genomes provides an additional benefit to our pipeline development to profile the integrity of ITRs, essential elements in vector genome potency. These new findings further guide improvements to vector design for increasing the efficacy and safety of these and related rAAV approaches.
Results
Singe-Unit sgRNA Sequences Engineered into Vector Genomes Are Not Inherent Hotspots for Truncations during rAAV Vector Replication and Packaging
To assess whether vectors that carry CRISPR-Cas9 components exhibit a high degree of truncation events and subsequent genome heterogeneity, we examined an all-in-one vector that packages SaCas9 and a sgRNA (Figure S1A, sample 4635). Vector was generated by the standard triple transfection method in HEK293 cells with the cis plasmid containing the transgene cassette flanked by ITR elements, the trans plasmid containing the rep and cap genes, and an adenovirus (Ad) helper plasmid containing essential adenoviral genes.14 Vectors were purified by cesium chloride purification, and vector genomic DNA was isolated by phenol/chloroform extraction.10,14 Agarose gel electrophoresis of DNA extracted from this vector demonstrated that the predominant and only visible species was the expected 4.7-kb full-length genome (Figures S1B and S1C).
We aimed to determine whether undesirable genomes do in fact persist in preparations by subjecting the isolated DNA to SMRT sequencing15 (Figure 1). A prerequisite of this approach is that DNA molecules need to have double-stranded free 5′ and 3′ ends to be adaptered by the SMRTbell adapter.10,15 We previously reported the use of SMRT sequencing to profile scAAV genomes,10 which are typically double-stranded structures sealed at one end with the mutant ITR (mITR) and with a single adapterable double-stranded free end as the other end. With these molecules, the generation of circular consensus sequences needed to consider single adaptering and, therefore, inserts were treated as “linear consensus sequences.”10 In contrast, ssAAVs are not inherently double-stranded. Therefore, genomes need to be converted to a double-stranded configuration before they can be properly ligated to the SMRTbell adapters. We note that when we subject vector genomes to agarose gel electrophoresis, ssAAV genomes migrate at the expected sizes, as if they were double-stranded molecules (Figure S1C, sample 4635). It is known that wild-type (WT) AAVs package plus and minus strand genomes at a roughly 50:50 ratio.16 It has also been theorized that plus- and minus-stranded genomes can undergo strand annealing following uncoating of the genome in the cell nucleus to form double-stranded species, crucial for the expression of viral genes.17 The same is true for rAAV vectors with intact ITRs at both ends of the genome. Therefore, our interpretation of how the SaCas9-sgRNA genome migrates by gel electrophoresis is that following isolation, the majority of plus- and minus-stranded genomes base-pair with each other. This presumably circumvents the need to convert the single-stranded genomes into a double-stranded configuration via in vitro second-strand synthesis. We note that certain ssAAV vector genomes, under neutral conditions, have been observed to run as “doublets.”18,19 These species have been suggested to be the annealed plus and minus strands that migrate at the predicted size, and non-annealed single-stranded genomes that migrate faster and as a smear. The non-annealed species, we speculate, may reflect an imbalance in packaged plus- and minus-stranded genomes that is typically resolved when run under alkaline conditions.18
Figure 1.

Schematic of SMRT Sequencing-Based AAV-GPseq Workflow
Single-stranded or self-complementary (ssAAV and scAAV) genomes are purified from virions. The plus (+) and minus (−) strands of the ssAAV genomes undergo strand annealing in solution to form adapterable ends. ssAAVs are adaptered on both ends of the genome by ligation to SMRTbell adapters (green loops). scAAVs are adaptered only on one end. Libraries are subjected to SMRT sequencing to produce long reads that can be processed by strand-specific consensus reconstruction to separate plus- and minus-stranded genomes as independent reads. Since scAAVs are only adaptered on one end, a single pass encompasses both the forward and reverse strand of the full-length genome. Hence, strand-specific consensus does not impact representation.
Material presumed to be predominantly annealed and double-stranded were subjected to SMRT sequencing library builds (Figure 1). Adaptered libraries were then sequenced. Since each adaptered full-length ssAAV molecule consists of annealed plus- and minus-strand genomes, the forward- and reverse-stranded SMRT subreads must be considered separate and unique genomes. To generate separate consensus reads for forward and reverse strands, consensus sequences were established using the -byStrand option. Reads were then mapped to the reference genomes accordingly. As expected, we observed a diversity of reads, the majority of which mapped to sequences residing between the ITRs (Figures 2A and 2B). There was a minor population that mapped to the plasmid backbone despite purification with Benzonase treatment and an additional DNase I treatment preceding vector genome isolation to remove any plasmid DNA carryover. Although gel electrophoresis suggested that full-length genomes predominantly exist in this preparation (Figure S1C), our SMRT sequencing results revealed that truncated forms were indeed packaged into particles. As observed before, we detected a relatively large abundance of reads smaller than 500 nt in length, peaking at 150–200 nt10 (Figure 2C). As mentioned above, these reads may be overrepresented as a consequence of preferential loading of shorter molecules. Although the abundance of read lengths was corrected for using the λDNA spike-in, these reads may be artifacts of library preparation, since they are not observed by gel electrophoresis. Excluding reads under 500 nt in size, approximately 52% of the reads are ∼4.7 kb in length (Figure 2C). This lower-than-anticipated percentage is due to the sequencing strategy’s ability to capture all low-abundance reads that are distributed throughout all observed genome lengths. In other words, although peak values at ∼4.7 kb stand out in both the read-length traces and by DNA gels, their sums only make up a fraction of all read lengths. Importantly, a small population of truncated reads centered on the sgRNA cassette was indeed revealed by SMRT sequencing (Figure 2A, red arrow). However, these accounted for 2.48% of the population (Figure 2C, red arrow). Overall, the read alignment confirmed that the sgRNA sequence does not substantially cause a high degree of truncation events.
Figure 2.

SMRT Sequencing Summary of the ssAAV-SaCas9-sgRNA Vector
(A) IGV display of reads mapping to the ssAAV-SaCas9-sgRNA cis plasmid reference. The top track displays the alignment summary in log10 scale. The bottom track displays individual reads mapping to the reference, squished down to visualize all reads. A linear diagram of the cis plasmid construct is displayed above the alignment tracks. Sequences matching the reference are displayed in gray, and those not matching the reference are colored as individual bases. Gaps are displayed as black dashes. In this squished view, they appear as speckles in the alignment. The display also shows soft-clipped bases to highlight truncated genomes that are in most of the cases in a self-complementary conformation. The blue arrow indicates a portion of reads with full-length genomes. The red arrow demarcates a population of truncated, self-complementary reads that are centered on the sgRNA cassette. The colored portion of the alignment summary track reflects sequence variation at the ITR regions that mark the distribution of flip- and flop-orientated ITRs. The ITR orientations within the reference are flop at both 5′ and 3′ ITRs. (B) Non-squished zoom-in displays of full-length genomes and truncated genomes from (A) (blue and red arrows). Each row is a continuous single SMRT read circular consensus. Mismatches at the 5′ and 3′ flanks indicate flip-orientated ITRs. Truncated genomes are self-complementary in structure, as revealed by their partial alignments, which spans half of the read. A color legend of the IGV-displayed matches, mismatches, gaps, and inserts is shown. (C) Lengths of all reads mapping to the vector reference were determined and their distributions are plotted. The relative abundances before normalization (blue trace) and after normalizing to the DNA (red trace) are shown. The relative read abundances of major peaks (brackets) are displayed as percentages of all mapped reads greater than 500 bp in length. Since the abundance of reads with lengths under 500 bp cannot be formally confirmed, they are discounted from the analysis.
High-Confidence Detection of Chimeric Genomes in ssAAV Vectors
In our previous study on profiling scAAV genomes by AAV-GPseq, we described the identification of chimeric genomes where vector genomic sequences were recombined with non-vector sequences, such as host-cell genomic sequences and plasmid DNA.10 In that study, blunt-end ligation to the SMRTbell adapters was employed and, therefore, the identification of chimeric genomes may have been partially skewed by fragment-to-fragment ligation (https://www.biorxiv.org/content/10.1101/245241v1.full). In consideration of this shortcoming, we have taken the opportunity in this study to validate chimeric vector genomes as genuine reads that do not result from the library build artifacts. In order to be fully confident in the detection of chimeric genomes explored in this study, we performed A-tailed-overhang ligation for adaptering the vector genomes to the SMRTbells. This procedure uses the addition of an overhanging adenosine to the 3′ ends of the target DNA strands to ensure that fragment-to-fragment ligations are eliminated. To demonstrate the effectiveness of overhang ligation to abolish fragment-to-fragment ligation, we spiked in with the vector genomes a non-specific plasmid DNA (pMax-GFP) that was independently digested with three four-base, blunt-end cutters and mixed at equimolar ratios.
Sequencing of the ssAAV-SaCas9-sgRNA vector with or without the spike-in produced equivalent read depths (Table S1). Reads that mapped to the vector genomes for either library also did not yield substantial differences in count representation. To observe the abundance of chimeric genomes in this study, we selected to observe reads that mapped specifically to the hg38 genome. Excluding reads related to spike-in material, we detected about 2.76% (182 total) and 2.53% (250 total) reads mapping to the hg38 genome among the vector libraries with and without spike-ins, respectively (Table S1; Figure 3A). As demonstrated by us and others,10,19, 20, 21 we also obtained reads that also mapped to the trans plasmid, the pAd helper plasmid, and elements of the vector backbones (Figure S2; Table S2). As demonstrated by the alignment display and the varied lengths of reads mapping to the vector backbone, these impurities are highly heterogeneous (Figure S2B). In agreement with our previous observations,10 we did not find any enrichment of specific genomic regions that favored packaging into virions (Figure 3B). Reads that mapped to hg38 were then remapped to the vector genome to assess the subset of reads sharing both the vector genome and hg38 sequences. We observed that 58%–63% of hg38-aligned reads were chimeric with the ssAAV-SaCas9-sgRNA vector genomes (Figure 3A). Interestingly, chimeric reads revealed that they predominantly share the 3′ ITR in common (Figure 3C). When displayed as aligned reads by the Integrative Genomics Viewer (IGV), we observed that the majority have host-cell genome sequences that are continuous with vector genome sequences, specifically the 3′ ITR. This reveals that the chimeric species are a product of non-random recombination events during vector production. To assess whether chimeras are perhaps due to fragment-to-fragment ligation, we analyzed reads that mapped to the non-related plasmid DNA spike-in. We generated a reference spanning a region of the plasmid that does not overlap with any sequence associated with the trans or Ad helper plasmids (Figure S3). Reads mapping to the non-related reference accounted for 3,360 out of 26,351 total reads (12.75%, Table S1) (Figure 3A, right Venn diagram). Importantly, none of the reads mapping to the non-related spike-in co-mapped to the vector reference. This finding suggests that the spike-in DNA did not form chimeric reads with vector genomes, directly demonstrating that chimeric genomes originate from the vector preparation and do not arise as a consequence of artifactual ligation during the library preparation.
Figure 3.

Evaluation of Host-Cell DNAs Encapsidated within ssAAV-SaCas9-sgRNA Vectors
(A) Venn diagrams of mapped read abundances related to the vector genome (white circles), the host-cell genome (gray circles), or a non-related DNA spike-in (red circle). Non-overlapping portions represent reads that map exclusively to either reference. Regions of overlap represent the counts of reads that co-mapped to both the host-cell genome and the vector genome. The percentages of co-mapped reads are displayed. (B) Histograms summarizing the number of unique regions throughout the host-cell genome (hg38) to which sequencing reads are mapped. Left graphs, without non-related DNA spike-in; right graphs, with spike-in. (C) Squished IGV display of chimeric reads mapped to the vector genome. Sequence regions that align to the vector reference are in gray, while those that do not are colored as their respective bases. A unifying feature of chimeric reads is that they are anchored at the 3′ ITR region. The lower left schematic illustrates the hypothesized self-complementary chimeric structures for reference.
SMRT Sequencing Reveals ITR Heterogeneity of Intact Genomes
As mentioned above, the ITRs are essential structures for replication and packaging and are involved in episomal formation once the vector genome is delivered into the nucleus. The ITRs also contain a terminal resolution sequence (TRS) that is nicked by Rep to initiate another round of replication.22 A hallmark of SMRT sequencing is that processivity through strong secondary structures within the template DNA is relatively high, owing to the isothermal strand-displacing polymerase used during sequencing.15 We previously demonstrated that the resolution of SMRT sequencing across scAAV ITRs is deep enough to reveal the distributions of flip and flop configurations among full-length vector genomes.10 Heterogeneity of AAV ITRs has recently raised concerns over whether mutations and/or recombined ITRs may impact packaging, vector titers, and transgene efficacy and safety.23,24 Due to the inherent structure of the ITR, it is highly recombinogenic within bacterial plasmids. Therefore, ITR stability during plasmid DNA manipulation and what is ultimately packaged into rAAVs is difficult to predict and assess. We therefore asked whether read processivity through ITRs by SMRT sequencing can help to reveal the heterogeneity of ITRs in preparations. Importantly, a positive demonstration that vector genomes carrying gene-editing components can be fully resolved from ITR to ITR with AAV-GPseq strengthens the method as the ideal approach for profiling CRISPR-Cas-AAV vectors.
We first gauged the representation of flip and flop configurations of the single-sgRNA design. As stated above, each ssAAV genome is defined by plus or minus strand; each ssAAV has two ITRs, at the 5′ and 3′ ends of the genome; and ITRs can have two orientations, flip and flop.25 Therefore, for every intact ssAAV genome, there are eight possible configurations. Unfortunately, since each template consists of annealed plus- and minus-stranded genomes, we cannot know the true percentage of plus and minus strands in an ssAAV population. As the WT AAV dependoparvovirus has four ITR-defined genome forms that are distributed equally, we observed that the single-sgRNA design yielded a near equal distribution of ITR-defined forms, that is, flip:flip, flip:flop, flop:flip, and flop:flop (Figure 4A). Although we were able to obtain the predicted distribution of ITR configurations in vector genome populations, all full-length reads are reliant on strand annealing of the plus- and minus-stranded genomes. We therefore speculated whether allowing the strands to anneal following DNA extraction was efficient enough to produce adapterable ends that are free of bias. This is problematic, since there are four ITR-defined forms. For example, flip- and flop-oriented ITRs on complementary strands will not fully undergo Watson-Crick base pairing. To determine this, we quantified the distribution of ITR annealing events between all read pairs of ssAAV-SaCas9-sgRNA vector genomes. Read pairs here are defined by the plus and minus strands that are separated following circular consensus using the -byStrand option. We observed that genome annealing with homologous ITRs (flip annealed to flip, or flop annealed to flop) on both ends are as abundantly represented as genomes with heterologous ITRs annealed on just one end (Figure S4). Unfortunately, genomes with heterologous annealing on both ends are very poorly represented. Although these heterologous-annealed ITRs can be adaptered, these results do demonstrate that there may be some bias in representation.
Figure 4.

Quantification of ITR Configuration and Heterogeneity with SMRT Sequencing
(A) Stacked histogram of flip and flop configuration percentage among all full-length genomes. (B and C) Tabulation of read starts and ends to reveal frequency of partial ITRs. (B) Traces representing absolute counts of read alignment starts (red) and alignment ends (blue) for vector DNA. The vector genome from ITR to ITR is displayed above to indicate where start and stops are mapped. (C) Heatmap display (log10 scale) of read alignment starts and stops to indicate vector genome termini throughout the 145-nt ITR structure. All data reflect samples with or without treatment to heating and slow cooling (HT). Percentages of read termini among reads ending within the 145-nt ITR are displayed. (D–F) Quantification of ITR damage/repair of an 11-nt deletion within the C-domain found in certain AAV cis vectors in circulation. (D) mfold DNA structures of an intact ITR (left) and mutant ITR with 11-nt deletion within the C-domain. (E) IGV display of aligned SMRT reads zoomed into the ITR domain. All bases are displayed with their respective color scheme. Gaps are shown as dashed lines. Red arrows indicate reads containing the C-domain deletion. (F) Pie chart summarizing the absolute counts of ITRs carrying the C-domain deletion in the flip or flop orientations.
The underrepresentation of genomes with heterologous ITR annealing at both genome termini may be a result of improper base-pairing. We therefore tested whether heat treatment followed by slow cooling to allow for proper annealing of the ITR ends could improve read representation. Interestingly, we found that heating of the same sample DNA preparation did not change the distribution of ITR configurations (Figure 4A). Read representation was however notably increased (Table S1), suggesting that heat treatment resulted in improved adaptering. Additionally, we observed a decrease in the abundance of reads that were less than 500 nt in length (Figure S5). These observations suggest that read representation for longer species was improved following heating and slow cooling of samples. However, read pairs with heterologous-annealed ITRs on both ends were still underrepresented (Figure S4). This finding suggests that ITR structures impact adaptering of molecules and may potentially skew representation.
One interesting aspect of being able to obtain resolution at the single-genome scale is that the heterogeneity of mutated ITRs can be gauged. Several reports have demonstrated that vectors can package partial ITRs or ITRs with deleted BB′ or CC′ arms of the ITR structure.26, 27, 28 It has been long reported that through plasmid recombination, ITRs can gain deletions.26 Presently, the extent to which these mutations can impact titers and vector performance is unclear. Due to the imprecision of standard sequencing techniques to accurately span the ITR region, gauging the heterogeneity of these structures in packaged genomes was never precisely assessed. This limitation is heavily influenced by the strong secondary structure of the ITR that prohibits efficient processivity by standard polymerases. Since the phi29-derived enzyme that the SMRT sequencing technology is based on has strand displacement activity,15 read coverage across ITR sequences of adaptered genomes is not significantly lost. We found that most of the ssAAV-SaCas9-sgRNA vector genomes with read ends within the 145-nt ITR region had their terminal positions at the defined TRS, accounting for more than 77% of reads ending within the ITR (Figures 4B and 4C). We did observe multiple reads that did not terminate at the TRS, but most were within the distal A sequence. One particular position, 16 nt from either TRS, comprised 2.85% of the left ITR terminal position and 2.55% of the right ITR terminus. Overall, there were very few reads that terminated at the BB′ or CC′ arms.
Unbeknownst to some in the field, mutations within the ITR sequence in plasmid constructs can propagate through subclones unintentionally and are inherently problematic.23 Fortunately, mutations have been shown to be corrected through replication,26,27 but the extent of correction was previously not accurately quantifiable. Incidentally, the ssAAV-SaCas9-sgRNA vector construct we profiled harbors an 11-nt deletion within the 5′ ITR (left ITR) of the plasmid (Figure 4D). We therefore gauged the degree of ITR “repair”; namely, the prevalence of WT-ITR sequences in packaged virions versus those that remained mutated. Interestingly, among all full-length, ITR-to-ITR spanning reads (910 total), we observed that the extent of packaged genomes with repaired left ITRs was quite dramatic (>99%) (Figures 4E and 4F). Less than 1% of genomes (9 reads out of 910 full-length genomes) were detected to have the 11-nt C-domain deletion. We note that the packaged genomes with repaired genomes were likely formed by intramolecular ITR replication (Figure S6) as previously proposed.26,29 Interestingly, we also detected 11-nt C-domain deletions at the 3′ ITR (right ITR). This occurred at even lower frequencies (3 reads out of 910 genomes), one in the flop orientation and two in the flip orientation (Figure 4F). Detection of the 11-nt C-domain deletion in both flip and flop orientations suggests that these mutated ITRs can undergo replication. However, they may be less efficient or the resulting genomes are packaged at much lower frequencies.
Configuration of Dual sgRNA Cassettes in Vector Genomes Impacts Packaging and Heterogeneity of Genomes
We were able to demonstrate that sgRNA sequences on their own do not result in severe truncation events. However, we also wanted to gauge the extent that AAV vectors carrying dual sgRNA cassettes (i.e., those used in large DNA deletions) could promote truncation events. We designed a vector construct containing two sgRNA cassettes oriented in a tail-to-tail configuration (ssAAV-2xT2T[sgRNA]), creating a long, inverted, repeat sequence (Figure S1A). We predicted that this design would produce a vector genome with strong secondary structure. As expected, gel electrophoresis analysis of this vector revealed a high degree of genome heterogeneity (Figure S1C). Bands that migrate close to the expected 2.8-kb full-length genomes were relatively faint. Instead, predominant bands migrating at ∼1.3 and ∼0.7 kb were observed. Additionally, heating and slow cooling did not significantly change the distribution of gel bands. Profiling of vector genomes by SMRT sequencing confirmed that there was a predominance of truncated genomes, with five main populations. Alignments of reads indicate that these genomes are self-complementary in structure, as previously observed.10 We hypothesized that the truncation events are mainly centered at the junction of the two sgRNA sequences (Figure 5A). Reads longer than 500 nt with approximate lengths of 710 nt (20.15%), 1,380 nt (45.31%), and 2,580 nt (2.46%) define the majority of packaged genomes by this vector design (Figure 5B). Read lengths that were approximately 2.8 kb in size, the expected full-length size, made up only 1.05% of all reads. More accurately, only 12 legitimate full-length genomes (0.21% of all reads mapping to the vector genome) were detected (Figures 5A and 5B). Notably, dominant species observed by gel electrophoresis migrating at ∼1.3 and ∼0.7 kb likely correlate with the self-complementary 2,580-nt and 1,380-nt reads, respectively.
Figure 5.

SMRT Sequencing of AAV Vectors Carrying Dual sgRNA Reveals Tail-to-Tail-Oriented Designs Yield Truncated Genomes
(A and C) Squished IGV displays of SMRT sequencing reads of a single-strand vector carrying dual sgRNAs in a tail-to-tail orientation (ssAAV-2sgRNA-T2T, A) and a self-complementary vector carrying dual sgRNAs in a tail-to-head orientation (scAAV-2sgRNA-T2H, C) mapping to their respective references. Aligned sequences that match the reference are in gray, while mismatches are colored as individual bases. Gaps are displayed as dashes. Soft-clipped bases are shown to reveal complement strand misaligning to the reference. The squished view results in speckled appearance of gaps. Each alignment is accompanied by an alignment summary track and a diagram of the vector plasmid reference. (C) Box image displays the predicted structure of self-complementary (truncated) vector genomes. The scAAV-2sgRNA-T2H vector packages both scAAV genomes and unexpected ssAAV genomes spanning from the 5′ ITR to the 3′ ITR (reads marked by brackets). Arrows indicate the direction of the U6-driven sgRNAs (forward, magenta; reverse, cyan). (B and D) Traces displaying the relative abundances of mapped reads distributed by length for (A) and (C), respectively. Percentages of read abundances in major peaks (brackets) are shown.
In contrast to the ssAAV-2xT2T[sgRNA] vector, a design where the sgRNAs are oriented in a tail-to-head configuration (scAAV-2xT2H[sgRNA]) (Figure S1A) did not result in a significant abundance of truncated genomes as assessed by gel electrophoresis analysis (Figure S1C). Notably, a minor band is observed under the predominant band. Note that scAAV genomes run as double-stranded DNA. However, their genome size is actually double the size of a single-stranded molecule. Therefore, SMRT sequencing reads of the full-length scAAV-2xT2H[sgRNA] genome is predicted to be approximately 4.5 kb in length (Figures S1B and S1C). The alignment and lengths of reads at ∼4.5 confirm that full-length scAAV genomes are packaged. However, SMRT sequencing of vector DNA unexpectedly revealed an abundance of reads that were shorter than full-length (Figures 5C and 5D). The vector genome population was comprised of two predominant species, one that represents the anticipated full-length self-complementary genome (4.5 kb), making up about 17.5% of all reads greater than 500 bp, and a second population that is half the size (2.3 kb), comprising 10.67% of reads. As expected, the self-complementary genomes aligned half of their read lengths to the reference, while the remaining half did not (Figure 5C). As reported previously, each read is a continuous strand of forward and reverse sequences that are linked by the mITR.10 However, for the second shorter population, reads were found to be single-stranded, encompassing the full expression cassette and spanning from ITR to ITR (Figure 5D). The reason behind this is unclear and warrants further exploration.
We also evaluated read termini positioning among these species (Figure S6). As demonstrated with the SaCas9-sgRNA vector, most reads that terminated at the ITRs end at the defined TRS (Figure S6B). These reads comprise about ∼68% of either the plus or minus strand ITR. Interestingly, for this vector, more reads terminated before the TRS, with approximately 4% of summed reads terminating within the most terminal inverted repeat arm. The most intriguing observation was that some sequences exhibited termination at the 5′ mITR (Figure S7), in agreement with reads that appear to be ssAAV in conformation (Figures 5C and 5D). Of note, there were also reads terminating at the start of the second pU6 promoter (Figure S7A). From the 3′ ITR to this position, the length of the double-stranded molecule would be approximately 1.7 kb in length, possibly correlating with the fainter second band observed by gel electrophoresis (Figure S1C).
We also assessed whether improvement to vector homogeneity reduced the abundance of chimeric genomes. Thus, reads from ssAAV-2xT2T[sgRNA] and scAAV2xT2H[sgRNA] vector libraries were aligned to hg38 to quantify chimeric genomes. Similar to the SaCas9-sgRNA vector, we also observed a degree of host cell genomes packaged into particles (Figures S8C–S8F). Interestingly, the ssAAV-2xT2T[sgRNA] vector resulted in very few reads mapping to the hg38 genome (Figures S8C and S8D). As before, treating extracted vector DNA by heating and slow cooling did not seemingly alter read representation throughout the hg38 genome (Figure S8). Of the heat and slow-cooled samples, approximately 60%–80% of the reads mapping to hg38 were chimeric with vector genomic DNA (Figures S8B, S8D, and S8F), demonstrating that recombination events during vector replication and packaging is not rare and may be common for all preparations. However, the host-cell chromosomal locales where chimeric reads seem to originate are distributed throughout the genome and do not seem to exhibit any preferential representation. These species are definitely a cause for concern since many are 1–2 kb in length and can potentially encompass full genes or promoter sequences.
Discussion
The use of AAV-based vectors to deliver CRISPR gene-editing components has been transformative for treating a range of genetic diseases that cannot be treated by gene replacement or gene addition strategies. However, the compatibility and efficacies of these tools together have only been demonstrated in cell culture and in some pre-clinical animal models. The gene therapy field has increasingly become aware that vector integrity and purity can impact the efficacy and safety of the therapy. Unfortunately, there are still many unknown aspects of the vector genome structure, especially from novel platforms, that may impact the manufacturing of vector and may influence the effective doses for treatment. In recent years, multiple methods of gauging vector genome integrity have been developed to capture the homogeneity of genomes that are packed into AAV capsids,10,19,20,30 each with its own advantages and disadvantages. Based on the structure of the sgRNA cassette, which harbors four loop regions, we predicted that the sgRNA sequence may serve as a scaffold for truncation events during AAV replication, similar to what we previously found for siRNA transgene cassettes.9,10 In turn, this could possibly impact the percentage of cells that undergo genome editing following vector administration. To our knowledge, SMRT sequencing and AAV-GPseq methods we described previously and here are the only means available to sequence the whole AAV vector genome to query truncation events and chimeric species that may arise from vector genomes harboring strong secondary structures. Our only challenge was to advance the AAV-GPseq to also profile ssAAVs, which are unlike scAAVs and need to be converted to a double-stranded configuration. We relied on the observation that AAV vectors package both plus- and minus-stranded genomes that undergo strand-annealing upon isolation.16 This phenomenon was also key for a recently developed short-read sequencing platform.19 By improving this action with heating and slow cooling the extracted vector DNA, we were able to obtain double-stranded genomes that are adapterable by SMRTbell adapters. To demonstrate the reliability of adaptering, we showed heterogeneity of ITR sequences, elements that, aside from being difficult to sequence by conventional means, exhibit flip and flop orientations, display unintended mutations, and can terminate within the ITR structure apart from the conventional TRS at low frequencies. We do recognize the potential for our approach to harbor skewed representation, based on the observation that heterologously annealed ITRs species are drastically underrepresented (Figure S3). Nonetheless, AAV-GPseq is the only method to accurately profile the entire vector genome as an intact molecule without the need for bioinformatics reconstruction, revealing consistency with current models for AAV replication and packaging proven by classical studies. In addition, long-read sequencing methods are the only means by which chimeric reads can be identified. We note that representations of these chimeric genomes were low, and therefore limited us from further exploration of whether these sequences harbored motifs that made them conducive to recombination events with AAV vector genomes. We previously found that chimeric reads did aggregate at transcriptional starts sites,10 but further explorations into these specific populations are undeniably needed.
In this study, we have additionally explored a phenomenon regarding ITR function that remains a puzzle: their inherent heterogeneity. ITRs are the last remaining elements still originating from viral sequence that are retained in the final recombinant AAV. However, they are not fully understood.24 The field has yet to devise and engineer a synthetic ITR sequence that can mimic the multi-functional role of the WT-ITR, whose job during production (genome rescue, replication, and Rep-mediated packaging) and during transduction (second-strand synthesis, episome formation, transcriptional regulation, and integration into the host genome) are still not completely understood. The use of SMRT sequencing and the downstream pipelines we describe herein to specifically profile AAV genomes from ITR to ITR in both ssAAVs and scAAVs will further advance vector designs to improve expression and safety.24 This ability allows for the validation and quantification of ITR repair during vector production, to ensure vector homogeneity and safety, when unstable ITR-bearing rAAV cis plasmids may result in mutated ITRs.
Most importantly, using our approach, we have found that on their own sgRNA cassettes do not cause a high degree of truncation events. This study demonstrates for the first time that at the level of the vector genome, AAV vectors can package CRISPR components without compromising vector integrity. However, we provide evidence that the design of dual guides, which may be used to increase editing efficiencies or induce large deletion events, should not be created in a tail-to-tail (or head-to-head) configuration. We note that such vector designs have not been reported to our knowledge. This lack of use may reflect an overall poor performance of such designs that have gone undescribed. We hope that our findings will inform novice investigators of such outcomes, and will further substantiate the idea that AAV vector platforms must avoid designs with strong secondary structures. All long-palindromic sequences are to be avoided if full-length vector genomes are the goal.
Materials and Methods
Constructs and Vector Production
The ssAAV-TBG-SaCas9-U6-sgRNA (vector lot ID 4635) vector construct was derived from pX602 (https://www.addgene.org/61593/). The sgRNA cassette targets the mouse aspartoacylase (Aspa) gene. Vector was packaged with AAV8 capsids. The ssAAV-2xT2T[U6sgRNA]-CB6-mCherry (vector lot ID 4015) vector was generated starting with the ssAAV-CB-PI-EGFP plasmid construct. The mCherry cDNA was cloned into the vector to replace the EGFP transgene by Gibson assembly. The sgRNAs were cloned into phU6 and pmU6 plasmid vectors, and the resulting U6-sgRNA cassettes were subsequently inserted upstream of the CB promoter by Golden Gate cloning, as previously described.31 The vector was packaged with AAV9 capsids. The scAAV-2xT2H-[U6-sgRNA]-U1a-EGFP vector construct was described previously (vector lot ID 3757).32 The vector was packaged with AAV6 capsids. All vectors were produced by triple transfection in HEK293 cells and purified by cesium chloride density gradient ultracentrifugation.14
Library Generation and SMRT Sequencing
Extraction of vector DNA was performed by phenol/chloroform as described previously.10 Briefly, ∼6E11–3E12 vector genomes were treated with 20 U of DNase I in a 200-μL vol for 15 min at 37°C. Genomes were then treated with Pronase solution (0.1% [w/v] Pronase [Sigma-Aldrich] in 50 mM Tris [pH 7.6] [Invitrogen], 1 mM EDTA [Invitrogen], and 0.5% SDS [Invitrogen]) for 4 h at 37°C. DNA was extracted using equal vol of phenol/chloroform/isoamyl alcohol (25:24:1) (Invitrogen), followed by 2× vol of chloroform/isoamyl alcohol (24:1). Samples were then subjected to standard ethyl alcohol (EtOH) precipitation and resuspended in nuclease-free H2O. Samples designated for heat treatment and slow cooling were heated in annealing buffer (25 mM NaCl, 10 mM Tris-HCl [pH 8.5], 0.5 mM EDTA [pH 8]) at 95°C for 5 min and then cooled to 25°C (1 min for every −1°C) on a thermocycler (Eppendorf Mastercycler). Lambda phage DNA (λDNA) digested with BstEII (NEB, Ipswich, MA, USA) was spiked into all libraries (10% by mass) and used as a normalizer for size loading bias.10 The non-vector-related DNA spike-in used was a pMaxGFP construct (Lonza) that was digested with three four-base, blunt-ended cutters: DpnI, AluI, and HaeIII (NEB, Ipswich, MA, USA). The spike-in was added along with the λDNA, following vector genome isolation. Due to overlapping sequences with plasmids used in the triple transfection method, the reference used for aligned reads was restricted to a 2.8-kb unique region. Libraries for vector DNA along with spike-ins were constructed using the Express Template Prep Kit 2.0 (end-repair/A-tailing) (PN 100-938-900) and ligated to indexed SMRTbell adapters with the barcoded overhang adapter kit (PN 101-628-400/500). Libraries were pooled and purified using 1.8× AMPure beads. Sequencing was performed on a Sequel I instrument following standard procedures defined by the manufacturer and the UMMS Deep Sequencing Core: Pacific Biosciences Core Enterprise. The distribution of reads mapping to vector genomes, human genomes (hg38), and λDNA is summarized in Table S1. All data presented herein are from a single flow cell to ensure accurate inter-library comparisons and to maximize experimental conditions in a cost-effective manner.
Data Analysis
Consensus reads, in fastq format, were generated using the ccs command in SMRT Link (v7.0.1.66975) using the following options: --minSnr=3.75 --minPasses=2 --minZScore=−10 -byStrand. Custom workflows for downstream analyses were processed on the Galaxy web platform (https://usegalaxy.org/),33 unless specified otherwise. Reads were de-multiplexed into their eight respective libraries. Each library was then mapped to its corresponding vector reference genome, the human genome (hg38), the Rep/Cap open reading frames (ORFs) for AAV6, AAV8, and AAV9 for corresponding vectors, and the pΔF6 helper plasmid by using BWA-MEM (https://arxiv.org/abs/1303.3997) using the option -x pacbio. Reads were also mapped to a single reference sequence consisting of bacterial plasmid backbone elements. This was done because the plasmid backbone and housed bacterial elements are shared between the cis and trans plasmids, and the pΔF6 plasmid. Distinction between the three sources is not reliable. Similarly, each library was mapped to the λDNA reference genome to identify reads that were associated with the λDNA spike-in. The lengths of reads mapping to the vector genome references were grouped into 10-nt bins. Relative abundances of reads distributed by length were corrected for size bias by generating polynomial splines of the abundances of reads mapping to the λDNA spike-in at peak summits ±10 nt (predicted from restriction fragment lengths) using the R package smooth.spline. Splines were used to calculate the predicted abundances of reads with observed lengths that mapped to the vector genome. The observed abundances were divided by the predicted abundances to yield the relative representation of reads by length. All read alignments are displayed with the IGV tool (v2.3)34 with soft-clipping on. Alignments displayed in this way summarize the composition of the genomes identified by SMRT sequencing. Alignment summaries are displayed in log10 scale, and variant display thresholds were set at 0.2. To discriminate flip and flop configurations for each vector genome, four references from ITR to ITR representing each configuration were created in a fasta file. In this study, the flip orientation is defined as the B-arm being closest to the open end of the vector genome. The B-arm is defined as 5′-CGGGCGACCTTTGGTCGCCCG-3′ or its reverse complement, and the C-arm is defined as 5′-CGCCCGGGCAAAGCCCGGGCG-3′ or its reverse complement. Full-length reads were mapped to the reference, and reads mapping to the respective ITR configuration were tabulated. Counting of ITRs bearing mutations was accomplished by taking the 145-nt sequences from the 5′ or 3′ ends of reads that fully map to the reference from ITR to ITR. Sequences were then centroid-based clustered by using the cluster_fast option in USEARCH35 with a similarity threshold of 95% (ID = 0.95). The option consout was also used to generate the consensus sequence for each cluster. The 2D structure of WT-ITR or mutated ITRs were displayed by using mfold (http://unafold.rna.albany.edu/?q=mfold). Venn diagrams were generated using eulerAPE_3.0.0.36
Data Availability
The datasets generated and/or analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) under the BioProject accession: PRJNA608034.
Author Contributions
N.T.T. and P.W.L.T. designed, conducted, and interpreted the bioinformatics analysis. P.W.L.T. and G.G. conceived and directed the project. J.X., D.Wang, and A.B. designed the rAAV vectors. C.H., K.W., M.W., D.Wilmot, and M.L.Z. helped to develop the SMRT sequencing strategy and performed the primary quality assessments. S.M. and Q.S. generated the vectors. N.T.T., P.W.L.T., and G.G. wrote the manuscript with contributions from D.W. and A.B.
Conflicts of Interest
G.G. is a scientific co-founder of Voyager Therapeutics and Aspa Therapeutics, and holds equity in these companies. G.G. is an inventor on patents with potential royalties licensed to Voyager Therapeutics, Aspa Therapeutics, and other biopharmaceutical companies. The remaining authors declare no competing interests.
Acknowledgments
G.G. is supported by grants from the University of Massachusetts Medical School (an internal grant) and by the National Institutes of Health (R01NS076991-01, 1P01AI100263-01, 4P01HL131471-02, UG3 HL147367-01, and R01HL097088).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.07.007.
Contributor Information
Guangping Gao, Email: guangping.gao@umassmed.edu.
Phillip W.L. Tai, Email: phillip.tai2@umassmed.edu.
Supplemental Information
References
- 1.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Broeders M., Herrero-Hernandez P., Ernst M.P.T., van der Ploeg A.T., Pijnappel W.W.M.P. Sharpening the molecular scissors: advances in gene-editing technology. iScience. 2020;23:100789. doi: 10.1016/j.isci.2019.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mou H., Kennedy Z., Anderson D.G., Yin H., Xue W. Precision cancer mouse models through genome editing with CRISPR-Cas9. Genome Med. 2015;7:53. doi: 10.1186/s13073-015-0178-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 5.Ibraheim R., Song C.Q., Mir A., Amrani N., Xue W., Sontheimer E.J. All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol. 2018;19:137. doi: 10.1186/s13059-018-1515-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bengtsson N.E., Hall J.K., Odom G.L., Phelps M.P., Andrus C.R., Hawkins R.D., Hauschka S.D., Chamberlain J.R., Chamberlain J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruan G.X., Barry E., Yu D., Lukason M., Cheng S.H., Scaria A. CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital amaurosis 10. Mol. Ther. 2017;25:331–341. doi: 10.1016/j.ymthe.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakim C.H., Wasala N.B., Nelson C.E., Wasala L.P., Yue Y., Louderman J.A., Lessa T.B., Dai A., Zhang K., Jenkins G.J. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3:e124297. doi: 10.1172/jci.insight.124297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie J., Mao Q., Tai P.W.L., He R., Ai J., Su Q., Zhu Y., Ma H., Li J., Gong S. Short DNA hairpins compromise recombinant adeno-associated virus genome homogeneity. Mol. Ther. 2017;25:1363–1374. doi: 10.1016/j.ymthe.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tai P.W.L., Xie J., Fong K., Seetin M., Heiner C., Su Q., Weiand M., Wilmot D., Zapp M.L., Gao G. Adeno-associated virus genome population sequencing achieves full vector genome resolution and reveals human-vector chimeras. Mol. Ther. Methods Clin. Dev. 2018;9:130–141. doi: 10.1016/j.omtm.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penaud-Budloo M., Le Guiner C., Nowrouzi A., Toromanoff A., Chérel Y., Chenuaud P., Schmidt M., von Kalle C., Rolling F., Moullier P., Snyder R.O. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 2008;82:7875–7885. doi: 10.1128/JVI.00649-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan D., Sharma P., Yang J., Yue Y., Dudus L., Zhang Y., Fisher K.J., Engelhardt J.F. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J. Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao G., Sena-Esteves M. Introducing genes into mammalian cells: viral vectors. In: Green M.R., Sambrook J., editors. Molecular Cloning: A Laboratory Manual, Vol. 2. Cold Spring Harbor Laboratory Press; 2012. pp. 1209–1313. [Google Scholar]
- 15.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 16.Mayor H.D., Torikai K., Melnick J.L., Mandel M. Plus and minus single-stranded DNA separately encapsidated in adeno-associated satellite virions. Science. 1969;166:1280–1282. doi: 10.1126/science.166.3910.1280. [DOI] [PubMed] [Google Scholar]
- 17.Nakai H., Storm T.A., Kay M.A. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver in vivo. J. Virol. 2000;74:9451–9463. doi: 10.1128/jvi.74.20.9451-9463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z., Ma H.I., Li J., Sun L., Zhang J., Xiao X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003;10:2105–2111. doi: 10.1038/sj.gt.3302133. [DOI] [PubMed] [Google Scholar]
- 19.Guerin K., Rego M., Bourges D., Ersing I., Haery L., Harten DeMaio K., Sanders E., Tasissa M., Kostman M., Tillgren M. A novel next-generation sequencing and analysis platform to assess the identity of recombinant adeno-associated viral preparations from viral DNA extracts. Hum. Gene Ther. 2020;31:664–678. doi: 10.1089/hum.2019.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maynard L.H., Smith O., Tilmans N.P., Tham E., Hosseinzadeh S., Tan W., Leenay R., May A.P., Paulk N.K. Fast-Seq: a simple method for rapid and inexpensive validation of packaged single-stranded adeno-associated viral genomes in academic settings. Hum. Gene Ther. Methods. 2019;30:195–205. doi: 10.1089/hgtb.2019.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lecomte E., Tournaire B., Cogné B., Dupont J.B., Lindenbaum P., Martin-Fontaine M., Broucque F., Robin C., Hebben M., Merten O.W. Advanced characterization of DNA molecules in rAAV vector preparations by single-stranded virus next-generation sequencing. Mol. Ther. Nucleic Acids. 2015;4:e260. doi: 10.1038/mtna.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snyder R.O., Samulski R.J., Muzyczka N. In vitro resolution of covalently joined AAV chromosome ends. Cell. 1990;60:105–113. doi: 10.1016/0092-8674(90)90720-y. [DOI] [PubMed] [Google Scholar]
- 23.Wilmott P., Lisowski L., Alexander I.E., Logan G.J. A user’s guide to the inverted terminal repeats of adeno-associated virus. Hum. Gene Ther. Methods. 2019;30:206–213. doi: 10.1089/hgtb.2019.276. [DOI] [PubMed] [Google Scholar]
- 24.Berns K.I. The unusual properties of the AAV inverted terminal repeat. Hum. Gene Ther. 2020;31:518–523. doi: 10.1089/hum.2020.017. [DOI] [PubMed] [Google Scholar]
- 25.Lusby E., Fife K.H., Berns K.I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. J. Virol. 1980;34:402–409. doi: 10.1128/jvi.34.2.402-409.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samulski R.J., Srivastava A., Berns K.I., Muzyczka N. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell. 1983;33:135–143. doi: 10.1016/0092-8674(83)90342-2. [DOI] [PubMed] [Google Scholar]
- 27.Wang X.S., Ponnazhagan S., Srivastava A. Rescue and replication of adeno-associated virus type 2 as well as vector DNA sequences from recombinant plasmids containing deletions in the viral inverted terminal repeats: selective encapsidation of viral genomes in progeny virions. J. Virol. 1996;70:1668–1677. doi: 10.1128/jvi.70.3.1668-1677.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Q., Tian W., Liu C., Lian Z., Dong X., Wu X. Deletion of the B-B′ and C-C′ regions of inverted terminal repeats reduces rAAV productivity but increases transgene expression. Sci. Rep. 2017;7:5432. doi: 10.1038/s41598-017-04054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samulski R.J., Berns K.I., Tan M., Muzyczka N. Cloning of adeno-associated virus into pBR322: rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA. 1982;79:2077–2081. doi: 10.1073/pnas.79.6.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penaud-Budloo M., Lecomte E., Guy-Duché A., Saleun S., Roulet A., Lopez-Roques C., Tournaire B., Cogné B., Léger A., Blouin V. Accurate identification and quantification of DNA species by next-generation sequencing in adeno-associated viral vectors produced in insect cells. Hum. Gene Ther. Methods. 2017;28:148–162. doi: 10.1089/hgtb.2016.185. [DOI] [PubMed] [Google Scholar]
- 31.Kabadi A.M., Ousterout D.G., Hilton I.B., Gersbach C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42:e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D., Niu Y., Ren L., Kang Y., Tai P.W.L., Si C., Mendonca C.A., Ma H., Gao G., Ji W. Gene delivery to nonhuman primate preimplantation embryos using recombinant adeno-associated virus. Adv. Sci. (Weinh.) 2019;6:1900440. doi: 10.1002/advs.201900440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 36.Micallef L., Rodgers P. eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS ONE. 2014;9:e101717. doi: 10.1371/journal.pone.0101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the NCBI Sequence Read Archive (SRA) under the BioProject accession: PRJNA608034.