

Abstract
Homologous recombination/end joining (HR/HEJ)-deficient cancers with BRCA mutations utilize alternative DNA double-strand break repair pathways, particularly alternative non-homologous end joining or microhomology-mediated end joining (alt-EJ/MMEJ) during S and G2 cell cycle phases. Depletion of alt-EJ factors, including XRCC1, PARP1 and POLQ, is synthetically lethal with BRCA2 deficiency; yet, XRCC1 roles in HR-deficient cancers and replication stress are enigmatic. Here, we show that after replication stress, XRCC1 forms an active repair complex with POLQ and MRE11 that supports alt-EJ activity in vitro. BRCA2 limits XRCC1 recruitment and repair complex formation to suppress alt-EJ at stalled forks. Without BRCA2 fork protection, XRCC1 enables cells to complete DNA replication at the expense of increased genome instability by promoting MRE11-dependent fork resection and restart. High XRCC1 and MRE11 gene expression negatively impacts Kaplan–Meier survival curves and hazard ratios for HR-deficient breast cancer patients in The Cancer Genome Atlas. The additive effects of depleting both BRCA2 and XRCC1 indicate distinct pathways for replication restart. Our collective data show that XRCC1-mediated processing contributes to replication fork degradation, replication restart and chromosome aberrations in BRCA2-deficient cells, uncovering new roles of XRCC1 and microhomology-mediated repair mechanisms in HR-deficient cancers, with implications for chemotherapeutic strategies targeting POLQ and PARP activities.
INTRODUCTION
Mutations can confer selective growth advantages on cancer cells, leading to their clonal selection and expansion. Analysis of tumor genome sequences led to the identification of several different patterns of mutations or mutational signatures (1). Many such mutational signatures are linked to underlying external causes such as exposures to sunlight or cigarette smoke. Additionally, specific defects in DNA repair mechanisms also generate unique mutational signatures, linking DNA repair abnormalities to mutation accumulation and genomic instability in cancer cells (1,2).
The discovery that hereditary forms of breast cancer are caused by a defect in homologous recombination (HR), a repair pathway that utilizes the undamaged sister chromatid as a template to repair DNA double-strand breaks (DSBs), led to the development of PARP inhibitors as therapeutics to selectively target cancers with HR defects (3,4). To date, key HR factors, including BRCA1, BRCA2, PALB2 and RAD51, have been identified as cancer susceptibility genes. Mutations in these genes are associated with a specific mutational signature, namely signature 3 (5). Furthermore, PARP inhibitors are effective clinically for breast and ovarian cancers with this signature (6).
Importantly, in HR-deficient tumors the repair of DSBs that arise during the S/G2 phases of the cell cycle occurs via less understood alternative end joining pathways, which are often error-prone (7–11). These pathways, initially discovered as backup pathways for non-homologous end joining (NHEJ), frequently utilize microhomologies to bridge the broken ends. They typically involve limited resection at DSBs by MRE11 and CtIP, gap-filling synthesis by DNA polymerase θ (POLQ), scaffolding by XRCC1 and ligation by DNA ligase 1 or 3 (LIG1/LIG3) (12). Although microhomology-mediated end joining (MMEJ) has a minor role in NHEJ- and HR-proficient cells, it contributes to chromosomal rearrangements and genomic instability in aneuploid cancer cells (13–15), and is upregulated in HR-deficient tumors, where POLQ is critical for genome maintenance and tumor survival (7–11,16). Additional MMEJ factors, PARP1 and XRCC1, were also identified as being synthetically lethal with BRCA2 in a recent CRISPR screen (17).
Error-prone DSB repair (DSBR) in sister chromatids by MMEJ contributes to deletions and translocations in HR-deficient cancers (5,11). In addition, there is emerging evidence that degradation of stalled replication forks followed by replication restart also contributes to genome instability in these tumors (18–22). Interestingly, while BRCA1, BRCA2 and RAD51 have HR-independent roles in fork protection (21,23,24), MRE11 and PARP1 are involved in fork degradation (25,26). Since PARP1 and MRE11 are also MMEJ factors (27), we reasoned that MMEJ likely contributes to fork degradation and subsequent replication restart at the degraded forks. This hypothesis is consistent with recent studies showing that POLQ is critical for replication-associated DSBR (28,29).
While XRCC1 engages with MRE11 and CtIP in response to ionizing radiation to form a MMEJ-competent complex (27,30) in addition to its canonical role as a scaffolding protein that coordinates base excision repair (BER) and single-strand break repair (SSBR) (31), the role of XRCC1 at stalled replication forks is less well defined. Here, we examined whether XRCC1-dependent MMEJ contributes to the repair of single-ended DSBs (seDSBs) that arise at collapsed replication forks. We find that replication fork collapse induces formation of a MMEJ-competent XRCC1 complex containing end resection factors and DNA polymerases in BRCA2-deficient cells. Furthermore, XRCC1 promotes stalled fork degradation and replication restart in BRCA2-deficient cells. Thus, in the absence of fork protection by BRCA2, XRCC1 enables cells to complete DNA replication at the expense of increased genome instability by promoting fork resection followed by replication restart.
MATERIALS AND METHODS
Cell culture
All in-cell and in vitro repair assays were performed with U2OS cells. Stable inducible shRNA-expressing cell lines (scrambled control, scr; and BRCA2, B2) were a generous gift from Ryan Jensen (Yale University). U2OS-EJ2 cells were a kind gift from Jeremy Stark (City of Hope) (32). All cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM, high-glucose, Gibco-BRL) supplemented with 10% fetal calf serum (Sigma) and 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco-BRL). Inducible cell lines were cultured in 2 μg/ml puromycin (InvivoGen), and shRNA expression was induced with 10 μg/ml doxycycline for 72 h. Cells were pretreated with 100 μM mirin (Sigma) for 1 h to inhibit MRE11 exonuclease activity or 10 μM rucaparib (Selleck) to inhibit PARP activity for EJ2-U2OS assays. Ataxia telangiectasia and Rad3-related kinase inhibitor (ATRi) VE-821 (Sigma) and hydroxyurea (HU; Sigma) were used to induce replication stress as indicated in the figure legends.
Plasmid and siRNA transfection
The XRCC1WT cDNA sequences were subcloned from respective 6X-His-tag-containing pCDE2 vectors into the p3XFLAG-CMV14 vector. myc-hPolQ-Flag vector was purchased from Addgene (Plasmid #73132) (deposited by Agnel Sfeir) (8). Treatment with 100 nM siRNA oligonucleotides (Sigma, TX) for XRCC1, CtIP, FEN1, LIG3, POLQ, BRCA1, BRCA2 and LIG1 (Supplementary Table S1) depleted the respective proteins (Supplementary Figure S1A). Exponentially growing cells were transfected with plasmids or siRNA at the indicated concentrations with Lipofectamine 2000 in OptiMEM media (Gibco-BRL) following the manufacturer’s protocol. The media were changed after 4 h, and the cells were incubated for 48 h for XRCC1 and POLQ expression and 72 h for siRNA treatment.
Clonogenic cell survival assay
U2OS cells were transfected with 100 nM control or XRCC1 siRNA, and after 72 h incubation the cells were treated with HU and/or the ATRi, VE-821, for the indicated times. The cells were then detached with trypsin and 500 cells from each sample were plated in triplicate in six-well dishes. After 10 days, the cells were fixed and stained with 0.5% crystal violet solution in 50% methanol and colonies were counted.
MTT assay
U2OS cells pretreated with siRNA and doxycycline were seeded in triplicate at a density of 3–5 × 103 cells/well in a 96-well plate, incubated overnight and exposed to rucaparib (Selleck) at indicated concentrations for 120 h. The cells were then exposed to a tetrazolium compound (TACS MTT Reagent, Trevigen) for 4 h, followed by solubilization for 2 h. Absorbance at 570 nm was measured using an Infinite M1000 microplate reader (TECAN).
Comet assay
Neutral comet assay was performed using the Trevigen Comet Assay Kit (4250-050-K) according to manufacturer’s protocol. At least 50 random comets for each sample were analyzed using CaspLab (33).
Antibodies
Primary antibodies used were mouse monoclonal ANTI-FLAG® M2-peroxidase (HRP) antibody (A8592, Sigma), mouse monoclonal ANTI-FLAG® M2 antibody (F1804, Sigma), rabbit polyclonal anti-DYKDDDDK tag antibody (#2368, Cell Signaling Technology), mouse monoclonal anti-XRCC1 antibody (#MS-434-P0, Thermo Scientific), rabbit monoclonal anti-XRCC1 antibody (ab134056, Abcam), rabbit polyclonal anti-PARP-1 antibody (H-300) (sc-25780, Santa Cruz Biotechnology), mouse monoclonal anti-DNA ligase 3 antibody (E-7) (sc-390922, Santa Cruz Biotechnology), mouse monoclonal anti-DNA ligase 1 antibody (ab615, Abcam), rabbit polyclonal anti-DNA polymerase beta antibody (18003-1-AP, Proteintech), rabbit polyclonal anti-MRE11 antibody (#4895, Cell Signaling Technology), rabbit monoclonal anti-phospho-histone H2A.X antibody (Ser139) (20E3) (#9718, Cell Signaling Technology), mouse monoclonal anti-phospho-histone H2A.X (Ser139) antibody (#05-636, EMD Millipore), mouse monoclonal anti-BRCA1 antibody (D-9) (sc-6954, Santa Cruz Biotechnology), mouse monoclonal anti-BRCA2 antibody (2B) (OP95, EMD Millipore), mouse monoclonal anti-FEN1 antibody (B4) (sc-28355, Santa Cruz Biotechnology), mouse monoclonal anti-BrdU antibody (IIB5) (ab8152, Abcam) and mouse monoclonal anti-β-Actin antibody (A5316, Sigma). Secondary antibodies for western blotting were from GE Healthcare (anti-mouse, NA9310V; anti-rabbit, NA934V). Secondary antibodies for immunofluorescence were from Invitrogen (Alexa Fluor, anti-rabbit 594, A11037; anti-mouse 594, A11005; anti-rabbit 488, A11008; anti-mouse 488, A11001).
γH2AX and XRCC1 foci analysis
U2OS cells were transfected with 100 nM scrambled control or XRCC1 siRNA; after 48 h, the cells were plated in eight-chamber slides, incubated overnight and then treated with HU and/or ATRi as indicated. The cells were then fixed with 4% paraformaldehyde for 15 min, followed by permeabilization with 0.5% Triton X solution in phosphate-buffered saline (PBS) for 30 min. Subsequently, after blocking with 3% bovine serum albumin (BSA) solution in PBS for 1 h, the cells were incubated with anti-phosphoserine H2A.X antibody (Cell Signaling Technology, #9718) or anti-XRCC1 antibody (#MS-434-P0, Thermo Scientific) diluted 1:500 in PBS for 2 h at room temperature. After washing with PBS, the cells were incubated with Alexa Fluor secondary antibody (1:500). After the final wash, the slides were dried for 5–10 min at 37°C and mounted with mounting media with DAPI (Invitrogen, P36941) and coverslips. Samples were observed under 60× oil immersion lens, and images were captured from at least 10 random fields for each sample. Cells were marked positive if they contained >10 foci.
Proximity ligation assay and immunostaining
For proximity ligation assay (PLA) and immunostaining, U2OS cells grown in an eight-chamber slide were treated as described in the figure legends and then fixed with 4% paraformaldehyde in PBS followed by permeabilization with 0.5% Triton X in PBS. For each PLA experiment, the respective primary antibodies raised in different species were used. Assays were performed with the Duolink kit (Olink Bioscience, Uppsala, Sweden) according to the manufacturer’s instructions. PLA foci/immunostaining were visualized using a 60× oil immersion lens in an inverted Zeiss bright-field/fluorescent microscope. For XRCC1-BrdU PLA, asynchronous U2OS cells were pulsed with 10 μM BrdU for 15 min before treatment with replication stalling agents for various times. Genomic DNA was denatured after fixation and permeabilization. Images were merged and analyzed with ImageJ software. For the PLA to detect co-localization of XRCC1 and POLQ, U2OS cells were transfected with the construct expressing POLQ-FLAG 48 h before treatment.
Chromosomal MMEJ assay
Incubation of U2OS-EJ2 cells with HU and/or ATRi, depletion with siRNA, DSB induction and cell harvesting were carried out as described (32). Flow cytometry was performed using a BD FACS LSRII and data were analyzed using Flowing Software (Perttu Terho, Turku Centre for Biotechnology).
In-cell plasmid circularization assay
U2OS cells grown in 60 mm plates (50% confluent) were treated with HU and/or ATRi as indicated, and then transfected with 100 ng pNS (the linearized DSBR substrate, Figure 3B) (30) using Lipofectamine 2000 and incubated overnight (15 h). Plasmids (5 μl) were isolated using the Qiagen plasmid miniprep kit prior to transformation of XL10-gold ultracompetent Escherichia coli cells (Agilent) following the manufacturer’s protocol. Forty colonies were randomly selected for plasmid sequencing using the CMV-F primer by Genewiz, Inc. Sequences with insertions/deletions of 1–4 nt at the DSB site were scored as NHEJ products, whereas the plasmids with deletion of one of the 5 nt microhomology sequences were scored as products of MMEJ/alt-EJ products, as described (30). Nonspecific extended deletions (>10 nt) at the 3′ or 5′ end of the DSB were not considered when plotting MMEJ versus NHEJ.
Figure 3.

Replication stress activates MMEJ. (A) Upper panel: scheme for repair of I-SceI-induced DSBs via MMEJ in the EJ2-U2OS cell line; lower panel: repair of I-SceI-induced DSBs after replication stress. EJ2-U2OS cells were treated with the indicated doses of HU and VE-821 (ATRi) for 8 h before DSB induction. (B) Upper panel: scheme for repair of pNS plasmid in mammalian cells after transfection; lower panel: repair of linearized plasmid substrate pNS after replication stress. U2OS cells were treated with the indicated doses of HU and VE-821 for 8 h before transfection of pNS; repaired sequences were analyzed according to (30). (C) Upper panel: in vitro MMEJ repair activity assay scheme; lower panel: MMEJ repair activity of XRCC1-FLAG IP. U2OS cells were transfected with XRCC1-FLAG and immunoprecipitated after treatment. Immunoprecipitated XRCC1-FLAG complexes were incubated with linearized plasmid substrate and competent E. coli cells were transformed with repaired product according to (30). Total colony number reflects individual repair events. Western blot of IP bead eluate after incubation is shown.
Preparation of whole cell and nuclear extracts
U2OS cells were harvested, washed with Dulbecco’s phosphate-buffered saline solution (Cellgro, Corning) and then pelleted by centrifugation at 800 rpm for 5 min. For preparing whole cell lysates, the cell pellet was resuspended in whole cell lysis buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl and 1% Triton X) with protease inhibitor cocktail (Thermo Fisher Scientific), vortexed at 4°C for 15 min and then clarified by centrifugation at 14 000 rpm. For preparation of nuclear extracts, the cell pellet was resuspended in cytoplasmic extraction buffer (10 mM Tris–HCl, pH 7.9, 0.34 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT and 0.1% Nonidet P-40) with protease inhibitor cocktail. The cell suspension was vortexed briefly, centrifuged at 3500 × g for 15 min at 4°C. The pellet of nuclei was resuspended in whole cell lysis buffer and a soluble extract prepared as described above. For co-immunoprecipitation assays, the nuclear extracts were incubated with 0.15 U/μl benzonase (EMD Millipore) to avoid DNA-mediated immunoprecipitation of protein complexes.
In vitro MMEJ assay
Exponentially growing U2OS cells transiently expressing XRCC1-FLAG were treated with HU and/or ATRi as indicated. After treatment, the irradiated and control cells were harvested for preparation of nuclear extracts. XRCC1-FLAG IP was isolated by incubating the nuclear extract with FLAG-M2 agarose beads for 2 h at 4°C. The beads were directly incubated for 30 min with 5 ng pNS in a reaction buffer containing 2 mM MgCl2, 60 mM NaCl, 50 mM HEPES, 2 mM DTT, 1 mM ATP, 1 mM dNTPs and 50 μg/ml BSA, with gentle shaking at 30°C. This was followed by addition of 14 ng XRCC1/LIG3 recombinant protein complex (as the ligation step is rate limiting) to the reaction mix for further incubation for 15 h at 16°C. After removal of the beads by low-speed centrifugation, 5 μl supernatant was used for transformation of XL10-gold ultracompetent E. coli cells (Agilent Technologies). The separated beads were also eluted with 4× LDS loading buffer for western blot analysis. The colonies in each plate were counted and submitted for sequence analysis using the CMV-F primer (Genewiz Inc.) similar to that used in the in-cell assays. Details on pNS synthesis are given in (30).
SSBR assay
The in vitro ligation activity assay was performed as previously described (34). Briefly, annealed oligomers labeled with Cy3 fluorescent dye were mixed with IP complexes in 1× T4 ligation buffer, and the mixture was incubated in a water bath at 30°C for 20 min, followed by incubating with 2× TBE sample buffer at 100°C for 3 min and on ice for another 3 min. Oligomers were separated by denaturing urea polyacrylamide gel electrophoresis, and Cy3 fluorescence was detected by a Typhoon FLA 7000 system.
DNA fiber analysis
Cells were labeled with 50 μM CldU (Sigma), exposed to HU (4 mM) and labeled with 50 μM IdU (Sigma) as indicated in the figures. DNA fibers were spread as previously described (35), and fiber tracts were detected using anti-IdU (BD Biosciences, 347580) and anti-CldU (Novus Biologicals, NB500-169) primary antibodies and Alexa Fluor 488 and 555 secondary antibodies (Invitrogen). Fibers were imaged at 60× magnification with oil immersion using a Zeiss microscope and analyzed with ImageJ.
Statistical and bioinformatic analyses
Most statistics were performed using Prism software. Fiber assay distributions were analyzed using the Mann–Whitney U test. PLA foci distributions, comet assay distributions, MMEJ in vitro assay, EJ2 repair events and clonogenic survival assay were analyzed using Student’s t-test. Foci formation and in-cell MMEJ assay results were analyzed using Fisher’s exact test. Mutational signature data were from COSMIC (https://cancer.sanger.ac.uk/cosmic/signatures); heat plots were generated in R using the ‘gplots’ package. TCGA data for gene expression were obtained through the TCGA Assembler utility and analyzed using custom scripts. P-values were obtained from a Welch’s t-test with unequal variance. Survival curves and hazard ratios were obtained from the ‘survminer’, ‘survival’ and ‘dplyr’ R packages. ns: P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001 in all figures.
RESULTS
XRCC1 has minor impact on cell survival and DSBR after replication stress in HR-proficient cells
Because XRCC1-depleted cells are sensitive to inhibition of ATR, the key proximal kinase activated by replication stress (36,37), we sought to examine XRCC1’s role in the replication stress response. XRCC1-depleted U2OS cells (Figure 1A) were modestly sensitive to the ATRi VE-821 (Figure 1B). HU, a ribonucleotide reductase inhibitor, induces replication stress by depleting cell deoxyribonucleotides, causing replication fork stalling. Similar to the ATRi results, XRCC1-depleted cells were modestly sensitive to HU (Figure 1C). HU induced a larger but not significant increase in DSBs in XRCC1-depleted cells compared to control cells as measured by the comet assay (Figure 1D). There was also a modest but not significant reduction in the rate of repair of HU-induced DSBs in XRCC1-depleted cells (Figure 1E). These results demonstrate a less robust effect of ATR inhibition on XRCC1-deficient cell survival than previously reported (36), likely in part due to the usage of U2OS cells rather than CHO cells in our experiments. Taken together, these results indicate that XRCC1 has a minor role in the response to replication stress in cells that are NHEJ and HR proficient.
Figure 1.

Effect of XRCC1 depletion and replication stress in WT cells. (A) Western blot of XRCC1 depletion by siRNA. (B) Clonogenic survival assay of XRCC1-depleted cells to ATRi. (C) Clonogenic survival assay of XRCC1-depleted cells to HU. (D) Neutral comet assay in ctrl and XRCC1 siRNA-treated cells after HU. Cells were treated with 3 mM HU for 8 h. At least 50 cells were analyzed for each experiment. (E) Relative fraction of U2OS cells positive for the DSB marker γH2AX by immunofluorescence. Cells were treated with 3 mM HU for 3 h and then allowed to recover for the indicated times. Cells were marked positive if they contained >10 foci. The number of cells analyzed in each experiment varied from 55 to 72. P-values obtained from z-tests at each time point varied from 0.88 (time 0) to 0.35 (2 h). The fractions of positive cells at time 0 were 0.49 (control siRNA, 55 cells) and 0.47 (XRCC1 siRNA, 65 cells).
XRCC1 is recruited to replication stress sites and co-localizes with DNA damage response factors
Since most forks stalled in response to HU treatment are stable without prolonged incubation, we examined the effect of treating cells with HU and simultaneously inhibiting ATR to induce the collapse of stalled forks (36). Cortez and colleagues have demonstrated the formation of DSBs with dissociation of replisome components both after prolonged treatment of cells with HU and more quickly after combined treatment of HU and ATRi (37). As expected, distinct XRCC1 foci, which co-localized with phosphorylated histone H2AX (γH2AX) in the proximity of DSBs, formed in response to exposure to HU and to a greater extent after combined exposure to HU and ATRi (Figure 2A and B). To determine whether XRCC1 is recruited to DSBs at stalled and collapsed replication forks, we used the PLA, which produces a fluorescent focus when two distinct antibodies are in close proximity (∼40 nm; Duolink). Asynchronous U2OS cells were pulse-labeled with BrdU to mark sites of DNA replication and then incubated with either HU or HU together with ATRi. The co-localization of XRCC1 with BrdU increased in a time-dependent manner both in the HU-treated cells and, to a greater extent, in cells co-incubated with HU and ATRi (Figure 2C and E).
Figure 2.

XRCC1 is recruited to sites of replication stress-induced DNA damage. (A) XRCC1 foci formation after treatment of U2OS cells with 3 mM HU and/or 3 μM ATRi for 8 h where indicated. Cells were marked positive if they contained >10 foci. At least 50 cells were analyzed for each experiment. (B) Co-localization of XRCC1 with γH2AX after ATRi + HU. (C) Representative images of the PLA data related to (E). (D) Representative images of the PLA data related to (F). (E) XRCC1 localization to sites of replication stress, as measured by BrdU-XRCC1 PLA. Asynchronous cells were pulsed with 10 μM BrdU for 15 min before treatment with 3 mM HU and/or 3 μM ATRi for the indicated times. At least 50 cells were analyzed for each experiment. (F) PLA between XRCC1 and the indicated factors in control cells and after treatment for 8 h with 3 mM HU and 3 μM ATRi. At least 50 cells were analyzed for each experiment.
We then utilized PLA to quantify changes in association between XRCC1 and other DNA damage response proteins (Figure 2D and F). Significant increases in the co-localization of XRCC1 with the MMEJ factors PARP1, MRE11 and POLQ, as well as γH2AX were observed, supporting a role for XRCC1 at DSBs formed in response to replication stress. In contrast, the association between XRCC1 and POLβ, the canonical gap-filling polymerase in BER and SSBR, did not change after replication stress.
Replication stress stimulates microhomology-mediated end joining by forming an XRCC1 repair complex
Based on the increased association between XRCC1 and other MMEJ factors in response to replication stress and collapsed replication forks, we measured MMEJ activity utilizing a chromosomally integrated MMEJ reporter system (EJ2) (Figure 3A, upper panel) (32). This measures MMEJ by restoration of GFP after DSB induction by I-SceI. In parallel, we used a linearized plasmid reporter system (Figure 3B, upper panel) that measures both MMEJ and NHEJ based on sequencing DSB joints in plasmids recovered from transfected cells (30). While neither of these assays directly measures events at stalled/collapsed replication forks, they do detect increased MMEJ activity that includes resection, annealing and subsequent ligation. We suggest that these MMEJ assays serve as surrogates for the activity of MMEJ factors at stalled/collapsed replication forks. Treatment of U2OS-EJ2 cells with HU or a combination of HU and ATRi led to an increase in the total number of MMEJ events, although the increase with HU alone did not reach statistical significance (Figure 3A, lower panel). In the linearized plasmid reporter system, pretreatment of U2OS cells with HU or a combination of HU and ATRi led to an increase in the number of MMEJ events relative to NHEJ events (Figure 3B, lower panel). Repair sequences are listed in Supplementary Figure S2. Thus, replication stress promotes cellular MMEJ repair activity, supporting and extending previous observations (38). As expected, this enhancement was dependent on XRCC1 and CtIP levels, in addition to PARP1 and MRE11 activity (Supplementary Figure S1B).
To further test whether replication stress and fork collapse enhance formation of active MMEJ complexes, we measured the ability of XRCC1 immunocomplexes to repair linearized pNS plasmid via MMEJ (30) (Figure 3C, upper panel). Pre-incubation of WT U2OS cells expressing FLAG-tagged XRCC1 with ATRi and HU significantly increased the repair of plasmid DNA by the XRCC1 immunocomplex (Figure 3C, lower panel). The sequences of the repaired plasmids confirmed that, as expected (30), all the repair events occurred by MMEJ rather than increased ligation activity (Supplementary Figure S3).
BRCA2- and XRCC1-depleted cells are sensitive to replication stress and repair DSBs inefficiently
Given the synthetic lethal relationship between BRCA2 and XRCC1 (17,39), we examined the impact of depleting either BRCA2 or BRCA1 on replication stress-induced MMEJ. While BRCA2 depletion led to a marked increase in chromosomal MMEJ events, BRCA1 depletion led to a decrease (Figure 4A). These results are consistent with published studies on the effect of BRCA1 and BRCA2 deficiency on MMEJ frequency (10,40) although there is a conflicting report (41). To further test the relationship between XRCC1 and BRCA2, we utilized a set of isogenic U2OS cells that allow reversible knockdown of BRCA2 in the presence of doxycycline (Figure 4B). Depletion of XRCC1 in BRCA2-deficient cells significantly increased the cytotoxicity of the combination of ATRi and HU (Figure 4C). These cells both accumulated more DSBs (Figure 4D) and repaired DSBs less efficiently (Figure 4E) following HU treatment. We therefore investigated whether XRCC1 depletion affects the sensitivity of BRCA2-deficient cells to PARP inhibition using the MTT assay, a colorimetric method of assessing metabolic activity. As expected, XRCC1-deficient cells were moderately sensitive to rucaparib, and BRCA2-deficient cells were highly sensitive (Figure 4F). Surprisingly, co-depleted cells were less sensitive than BRCA2-depleted cells, raising the possibly that XRCC1 promotes formation of toxic, DNA-trapped PARP1. Collectively, these data indicate that XRCC1 plays a more prominent role in the repair of DSBs arising from replication stress in BRCA2-deficient cells.
Figure 4.

XRCC1 plays prominent roles in replication stress responses and DSBR in BRCA2-deficient cells. (A) Dependence of EJ2-U2OS MMEJ events on BRCA1 and BRCA2. Cells were treated with siRNA according to (32). Data are plotted relative to ctrl siRNA-treated samples and scaled according to their fold depletion as measured via western blot. (B) Western blot of inducible knockdown of BRCA2 in scr-U2OS (scr) and BRCA2-U2OS (BRCA2) cells. scr-U2OS and BRCA2-U2OS (also referred to as scr and BRCA2 or B2) cells can be induced with doxycycline to express scrambled shRNA and BRCA2 shRNA, respectively. (C) Clonogenic survival assay of scr-U2OS and BRCA2-U2OS cells treated with ctrl or XRCC1 siRNA and ATRi + HU. (D) Neutral comet assay in scr-U2OS and BRCA2-U2OS cells treated with ctrl and XRCC1 siRNA and/or HU. Cells were treated with 3 mM HU for 8 h where indicated. At least 50 cells were analyzed for each experiment. (E) Relative fraction of scr-U2OS and BRCA2-U2OS cells positive for the DSB marker γH2AX by immunofluorescence. XRCC1 or ctrl siRNA-treated cells were treated with 3 mM HU for 3 h and then allowed to recover for the indicated times. Cells were marked positive if they contained >10 foci. The number of cells analyzed in each experiment ranged from 87 to 158. P-values were obtained from pairwise z-tests between control and each of the other conditions. *P-value 0.034, P(α)0.05 0.565; **P-value 0.006, P(α)0.05 0.793. (F) MTT assay of scr-U2OS and BRCA2-U2OS treated with either ctrl siRNA or XRCC1 siRNA and exposed to varying concentrations of the PARP inhibitor rucaparib for 120 h.
BRCA2 suppresses MMEJ by preventing XRCC1 recruitment and repair complex formation
To better define the relationship between XRCC1 and BRCA2, we examined XRCC1 localization and complex formation in BRCA2-deficient cells. More XRCC1 foci formed in response to replication stress in BRCA2-deficient cells (Figure 5A), although the result was not statistically significant. This correlated with increased co-localization of XRCC1 with BrdU (Figure 5B and E) and with γH2AX, PARP1, POLQ and MRE11 (Figure 5C and F). To further test whether replication stress causes increased association of MMEJ factors in BRCA2-deficient cells, we analyzed co-immunoprecipitation of MRE11 and XRCC1 with FLAG-tagged POLQ expressed in BRCA2-proficient and BRCA2-deficient U2OS cell lines. In accord with the increased co-localization (Figure 5F), higher levels of XRCC1 (∼2.7-fold) and MRE11 (∼4-fold) were found in POLQ-FLAG IP from BRCA2-deficient cells, showing that BRCA2 suppressed the association of POLQ with both XRCC1 and MRE11 (Figure 5D, upper panel). Similarly, there was less POLQ-FLAG and MRE11 in endogenous XRCC1 immunoprecipitates from BRCA2-proficent cells compared with BRCA2-deficient cells (∼1.5-fold, POLQ-FLAG; ∼2-fold, MRE11) after replication stress (Figure 5D, lower panel).
Figure 5.

BRCA2 suppresses XRCC1–POLQ–MRE11 complex formation and MMEJ. (A) XRCC1 foci formation after treatment of scr-U2OS or BRCA2-U2OS cells with 3 mM HU and/or 3 μM ATRi for 8 h where indicated. Cells were marked positive if they contained >10 foci. At least 50 cells were analyzed for each experiment. (B) Representative images of the PLA data related to (E). (C) Representative images of the PLA data related to (F). (D) Upper panel: western blot of POLQ-FLAG IP from scr-U2OS and BRCA2-U2OS cells with and without ATRi + HU treatment. Lower panel: western blot of endogenous XRCC1 IP from scr-U2OS and BRCA2-U2OS cells expressing POLQ-FLAG with and without ATRi + HU treatment. (E) XRCC1 localization to sites of replication stress in scr-U2OS or BRCA2-U2OS cells, as measured by BrdU-XRCC1 PLA. Asynchronous cells were pulsed with 10 μM BrdU for 15 min before treatment with 3 mM HU and/or 3 μM ATRi for the indicated times. At least 50 cells were analyzed for each experiment. (F) PLA between XRCC1 and the indicated factors in scr-U2OS and BRCA2-U2OS cells treated for 8 h with 3 mM HU and 3 μM ATRi. At least 50 cells were analyzed for each experiment. (G) MMEJ repair activity of XRCC1-FLAG IP. scr-U2OS and BRCA2-U2OS cells were transfected with XRCC1-FLAG and treated with 3 mM HU and 3 μM ATRi where indicated. Immunoprecipitated XRCC1-FLAG complexes were incubated with linearized plasmid substrate and competent bacterial cells were transformed with repaired product according to (30). Western blot of IP bead eluate after incubation is shown.
To test whether replication stress causes increased activity of the MMEJ complex in BRCA2-deficient cells, we examined the effect of BRCA2 depletion on the MMEJ activity of XRCC1-FLAG immunocomplexes. The XRCC1-FLAG IP from BRCA2-depleted cells performed MMEJ at a significantly higher level than control cells (Figure 5G), whereas SSBR was carried out at a moderately higher level in BRCA2-depleted cells relative to control cells (Supplementary Figure S3). Together these data indicate that the presence of BRCA2 suppresses the localization of XRCC1 to sites of replication stress, formation of the XRCC1–MMEJ repair complex and the repair activity of this complex.
BRCA2 deficiency changes the effect of XRCC1 depletion on replication fork dynamics and chromosome aberrations
Since XRCC1 is recruited to sites of replication stress, we considered the possibility that it has functions at the stalled replication fork in addition to MMEJ. Using DNA fiber analysis (35), we found that XRCC1 depletion did not affect DNA replication progression or fork protection in WT U2OS cells (Supplementary Figure S4), whereas it did significantly limit the extent of replication fork restart (Figure 6A), extending previous observations (42).
Figure 6.

XRCC1 depletion limits fork degradation, fork restart and chromosome aberration accumulation in BRCA2-deficient cells. (A) Effect of XRCC1 depletion on replication restart in U2OS cells. (B) Effect of XRCC1 depletion on replication restart in scr-U2OS and BRCA2-U2OS cells. (C) Effect of XRCC1 depletion and MRE11 inhibition (100 μM Mirin) on replication fork protection in scr-U2OS and BRCA2-U2OS cells. At least 200 individual fibers were analyzed for each experiment. (D) Quantification of chromosome aberrations from 20 separate metaphases in scr- and BRCA2-U2OS cells with or without XRCC1 siRNA treatment. (E) Representative metaphase chromosome spread of BRCA2-U2OS with HU treatment. (F) Western blot of BRCA2 and XRCC1 co-depletion.
Based on the degradation of nascent DNA at stalled forks by nucleases, including MRE11, that leads to chromosomal aberrations and genomic instability in BRCA2-deficient cells (21), together with our data showing increased association of MRE11 and XRCC1 at replication forks in response to replication stress, we examined the effect of depleting XRCC1 in BRCA2-depleted and control U2OS cells (Figure 6F) on replication fork dynamics. Depletion of BRCA2 further inhibited replication restart caused by XRCC1 knockdown (Figure 6B), suggesting that BRCA2 and XRCC1 participate in distinct pathways that promote replication restart. Interestingly, XRCC1 depletion alleviated the degradation of stalled forks in BRCA2-deficient cells (Figure 6C) and the increase in chromosome aberrations induced by replication stress (Figure 6D and E). XRCC1 knockdown in BRCA2-depleted cells has a similar effect to inhibiting MRE11 nuclease activity with mirin on fork degradation (Figure 6C), as well as a similar effect to mirin treatment in BRCA2-deficient cells on chromosome aberration number (Figure 6D) (21). Since MRE11 inhibition fully restores replication fork protection in BRCA2-deficient cells, MRE11 is likely the primary nuclease responsible for fork degradation in these cells. The observed effect of XRCC1 in fork protection, combined with observations indicating co-localization (Figure 5D) and complex formation (Figure 5F), leads us to reason that MRE11 and XRCC1 function in the same pathway responsible for resecting stalled forks in BRCA2-deficient cells. Collectively, our results reveal specific roles for XRCC1 in BRCA2-deficient cells that balance fork degradation and fork restart to promote chromosome aberrations and cell survival.
XRCC1 and MRE11 deficiency are more strongly correlated with survival in BRCA-deficient breast cancers than BRCA-proficient breast cancers
Based on our observation that XRCC1 and MRE11 are critical not only for DSBR by MMEJ but also for replication fork restart in BRCA2-deficient cancer cells, we analyzed XRCC1 and MRE11 expression in breast cancers compiled in The Cancer Genome Atlas (TCGA). We first partitioned all TCGA breast cancers into BRCA-proficient and BRCA-deficient groups using their signature 3 score (a pattern of single-base substitutions strongly associated with large insertions and deletions with overlapping microhomology at chromosome breakpoint junctions; signature 3+ cancers are strongly associated with homologous recombination deficiency) from COSMIC (1) (sig3+: sig3score > 0; sig3−: sig3score = 0) (Figure 7A). Then, we partitioned breast cancers within these groups into high and low gene expression groups (top and bottom ∼20% of tumors) (Figure 7D and G), and calculated the survival curves for those groups. We found that in all breast cancers (regardless of HR status), low XRCC1 expression is associated with poor survival (Figure 7B) (P-value = 0.031). This association is stronger in HR-proficient cells (Figure 7E) (P-value = 0.0008), and is absent in HR-deficient cells (Figure 7F) (P-value = 0.52), consistent with a distinct role of XRCC1 in the promotion of cancer fitness in HR-deficient breast cancers. Similarly, MRE11 expression is significantly associated with poor survival only in the HR-deficient subset of breast cancers (Figure 7C, F and I) (P-value = 0.016), consistent with promotion of cancer fitness by MRE11 specifically in this subset of breast cancers. In contrast, levels of the SSBR/BER polymerase POLβ and the NHEJ factor Ku70 (XRCC6) did not show association with survival in either BRCA-proficient or BRCA-deficient breast cancers (Supplementary Figure S5). These data collectively suggest that in HR-deficient breast cancers, high expression of XRCC1 and MRE11 leads to poor survival as a result of either increased genome instability or therapy resistance due to increased replication fork restart and DNA repair (Figure 8).
Figure 7.

XRCC1 and MRE11 gene expression is correlated with poor survival in HR-deficient breast cancer patients. (A) Distribution of signature 3 score across all breast cancers in TCGA. (B) Kaplan–Meier survival curve and hazard ratio for all TCGA breast cancer patients with high (top 20%) and low (bottom 20%) gene expression levels (panel D) of XRCC1. (C) Kaplan–Meier survival curve and hazard ratio for all TCGA breast cancer patients with high (top 20%) and low (bottom 20%) gene expression levels (panel G) of MRE11. Kaplan–Meier survival curves and hazard ratios for Sig3-negative (HR-positive, E) and Sig3-positive (HR-negative, F) breast cancer patients with high and low expression of XRCC1. Kaplan–Meier survival curves and hazard ratios for Sig3-negative (HR-positive, H) and Sig3-positive (HR-negative, I) breast cancer patients with high and low expression of MRE11.
Figure 8.
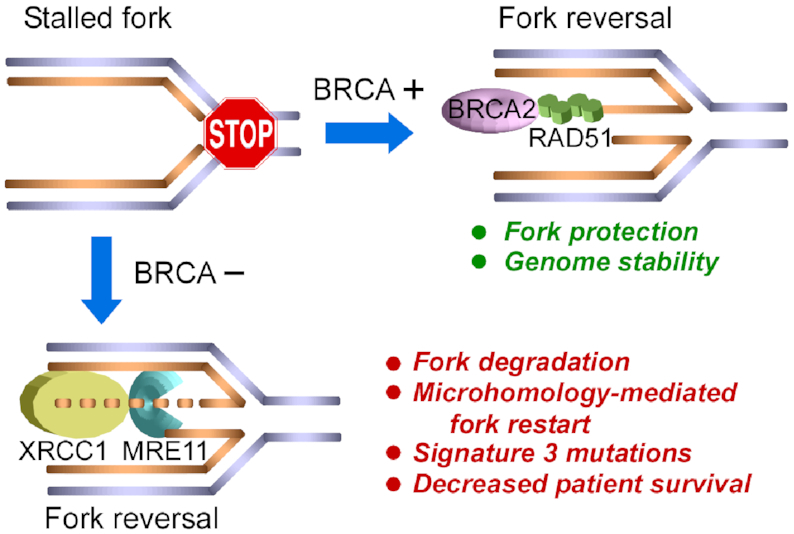
Graphical scheme summarizing results. In BRCA2-proficient cells, BRCA2 loads RAD51 to protect reversed replication forks, leading to replication restart by homologous recombination and genome stability. In BRCA2-deficient cells, MRE11 and XRCC1 degrade reversed forks, leading to replication restart using microhomology-based mechanisms, the accumulation of signature 3 mutations and poor patient survival.
DISCUSSION
XRCC1 has key scaffolding roles in coordinating repair of base damage and single-strand breaks by housekeeping DNA repair pathways (31). Also, it was proposed to have an analogous role in backup error-prone DSBR pathways, including MMEJ, that can become critical for the viability of HR-deficient tumor cells (12). Previously, we found that ionizing radiation exposure enhances formation of functional XRCC1-containing MMEJ complexes and increases MMEJ activity (30). Here, we support and extend those results to show that XRCC1-mediated MMEJ is also increased in response to replication stress and fork collapse. Furthermore, we find that XRCC1 and other MMEJ proteins are recruited to stalled and/or damaged replication forks. While this study is limited by the usage of only U2OS cells for isogenic BRCA2 depletion, we suggest that the reported results on replication, repair substrates and mutational signature are likely to be generalizable to other cell types in light of the fundamentally conserved roles of XRCC1 in DNA damage responses. Analyses of XRCC1-deficient rodent and human cells have revealed similarities but also some differences in the contribution of XRCC1 to the cellular response to DNA damage and replication stress, such as the modest sensitivity of XRCC1-depleted human U20S cells to HU compared with CHO EM9 cells (43). Additional studies in other cancer and non-malignant human cell lines will explore the role of XRCC1 in dealing with replicative stress, such as that induced by oncogenes, in the absence or presence of functional recombinational repair.
A role of XRCC1 in PARP-dependent replication-associated DNA repair was identified previously (44), but was attributed to the canonical role of XRCC1 SSBR/BER without examination of possible roles of XRCC1 in replication and microhomology-mediated repair processes. We have extended that work here to demonstrate a clear role for XRCC1 in DNA replication processes beyond its SSBR/BER function. Recently, low levels of poly(ADP-ribose) (pAR) synthesis were detected at the replication fork during unperturbed DNA replication (45). As there are higher levels of pAR synthesis in DNA Ligase 1 (Lig1)-deficient cells but Lig1-null cells are viable because of the presence of the XRCC1 partner protein, Lig3, it is likely that Lig3 and XRCC1 participate in a PARP-dependent backup pathway that joins Okazaki fragments at the replication fork (45). Since depletion of XRCC1 had only a small effect on cellular sensitivity to replication stress and the level of replication stress-induced DSBs, it appears that, despite enhanced recruitment to replication forks and increased MMEJ activity, XRCC1 also has only a minor role at stalled or collapsed replication forks in HR-proficient cells.
In contrast, in HR-deficient cells, it is evident that MMEJ plays a critical role in the mutagenic repair of DSBs in sister chromatids (5,11). There is, however, emerging evidence that HR factors contribute to genome stability not only through error-free repair of DSBs but also by protecting stalled replication forks. In BRCA2-mutant cancer cells, stalled and collapsed forks, which are not substrates for the NHEJ machinery, are degraded by the MRE11 nuclease (21). Since this increased fork degradation was dependent upon XRCC1, we suggest that complex formation with XRCC1 at the replication fork is likely to be required for fork degradation. Given the increased association of XRCC1, MRE11 and other MMEJ factors in response to replicative stress, it appears likely that this complex carries out resection at stalled forks that is limited by the exposure and annealing of microhomologies. This type of resection is compatible with a RAD51-independent pathway of break-induced replication (BIR) termed microhomology-mediated BIR (MMBIR), which utilizes microhomology to facilitate multiple template switching events and is a significant source of rearrangements observed in human cancers and other genetic diseases (46–48). Notably, replication-associated seDSBs are repaired by MMBIR in HR-deficient cells that are unable to utilize RAD51-dependent, homology-dependent BIR (49,50). While we are currently working to establish the cell cycle dependence of these processes, the clear effect on replication fork dynamics and its suppression by BRCA2 demonstrated here provide strong evidence that these repair processes are highly active in S/G2 phases of the cell cycle.
As depletion of both BRCA2 and XRCC1 had an additive effect on replication restart, these proteins likely act in two distinct pathways for replication restart. Thus, it appears that BRCA2 may protect stalled forks from degradation and promotes replication restart, possibly via homology-dependent BIR (21). The increased formation of MMEJ complexes and the increased co-localization of MMEJ factors at stalled forks in HR-proficient tumors may reflect replication stress-induced signaling and/or recruitment to a subset of stalled forks that are not protected by BRCA2. Our assay results imply that XRCC1 promotes microhomology-mediated repair at stalled or collapsed forks. Moreover, the increased formation of MMEJ complexes at sites of stalled replication, their increased resection and annealing activities, plus promotion of fork degradation, restart and chromosome aberrations argue that the MMEJ repair complex acts at replication sites in HR-deficient cancer cells. We posit that this replication-associated activity may contribute to observed genome rearrangements in HR-deficient cancers and thus merits further investigation due to its implications for chemotherapeutic strategies targeting POLQ and PARP activities.
Our collective results suggest that, in the absence of BRCA2 and homology-dependent pathways for fork restart, MMEJ directs fork degradation in order to expose microhomologies that mediate template switching and replication restart via a pathway involving XRCC1 as well as the MMEJ factors POLQ, MRE11 and PARP1 (7,16,26). While this pathway is mutagenic and presumably contributes to genomic instability by generating deletions with microhomologies at the breakpoints to enable replication restart at microhomologies, it would enable HR-deficient cancer cells to complete replication (22,51). The enrichment of such breakpoints and the biological importance of MMEJ factors in HR-deficient cancers indicate a connection between replication restart, fork protection and MMBIR/MMEJ that warrants further exploration. For example, defining the mechanism of POLQ and PARP activity more precisely during fork degradation and replication restart will inform the applications of PARP and POLQ inhibitors to BRCA-deficient cancer therapy (6). Furthermore, specific inhibitors of the poly(ADP-ribose) glycohydrolase (PARG) cause replication fork stalling and cancer cell death (52). In concert with the results reported here, including the finding that the sensitivity of BRCA2-deficient cells to PARP inhibitors is partially dependent on XRCC1, it appears that the XRCC1–MRE11 axis connects PARP, PARG and POLQ at stalled forks as well as at DSBs. These findings are significant in the context of PARP inhibition in HR-deficient tumors, which extend beyond BRCA deficiency itself, particularly in platinum-sensitive ovarian cancer patients who demonstrate improved progression-free survival following maintenance therapy with rucaparib, olaparib, veriparib or niraparib (53–55).
Since increased activity of this XRCC1–MRE11-dependent pathway of fork degradation and restart will lead to genome instability, our finding that expression levels of XRCC1 and MRE11 correlate with survival outcomes for patients with HR-deficient breast cancers, in contrast to NHEJ and SSBR/BER factors, supports our model in which XRCC1 and MRE11 have fundamentally different roles in HR-deficient and HR-proficient breast cancers. Notably, high expression of XRCC1 and MRE11 correlates with decreased overall survival (relative to HR-proficient survival curves) specifically in HR-deficient breast cancers. While there are multiple possible explanations for this observation, we suggest that, given the high burden of MH-containing genome rearrangements in HR-deficient cancers, the ability of HR-deficient cancers to complete replication via XRCC1 and MRE11 correlates with tumor fitness, and thus decreased overall survival.
In sum, we herein have identified a novel role for an MMEJ-competent protein complex that is enhanced by replication stress at stalled replication forks in BRCA2-deficient cells. Notably, XRCC1 and MRE11 promote degradation of forks in BRCA2-deficient cells, presumably exposing microhomologies that are utilized to prime replication restart. Thus, in HR-deficient tumors, resection and annealing mediated by the MMEJ complex may be important for replication fork restart and cell survival. Overall, the identification here of an XRCC1–MRE11 axis connecting PARP, PARG and POLQ at stalled forks as well as at DSBs appears relevant to therapeutic strategies targeting POLQ and PARP activities.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Katharina Schlacher for discussions. This research used computing resources at the Texas Advanced Computing Center, which is supported by National Science Foundation.
Contributor Information
Bradley J Eckelmann, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA; Texas A&M Health Science Center, College of Medicine, Bryan, TX 77807, USA.
Albino Bacolla, Departments of Cancer Biology and Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Haibo Wang, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA.
Zu Ye, Departments of Cancer Biology and Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Erika N Guerrero, Gorgas Memorial Institute for Health Studies, Panama City, Panama.
Wei Jiang, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital & Shenzhen Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Shenzhen, 518036, China.
Randa El-Zein, Department of Radiology, Houston Methodist Research Institute, Houston, TX 77030, USA.
Muralidhar L Hegde, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA; Weill Cornell Medical College, Cornell University, New York, NY 10065, USA.
Alan E Tomkinson, Departments of Internal Medicine and Molecular Genetics & Microbiology, and the University of New Mexico Cancer Center, University of New Mexico, Albuquerque, NM 87131, USA.
John A Tainer, Departments of Cancer Biology and Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Sankar Mitra, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA; Weill Cornell Medical College, Cornell University, New York, NY 10065, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health [CA158910 and GM105090 to S.M., ES12512 to A.E.T., NS088645 to M.L.H., CA092548 to J.A.T., A.E.T. and S.M., CA220430 to J.A.T.]; National Space Biomedical Research Institute [NCC 9-58 to B.J.E.]; Cancer Prevention and Research Institute of Texas (CPRIT) [RP180813 and RP130397 to J.A.T.]; Robert A. Welch Chemistry Chair [to J.A.T.]; National Science Foundation [ACI-1134872].
Conflict of interest statement. None declared.
REFERENCES
- 1. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L.et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bacolla A., Ye Z., Ahmed Z., Tainer J.A. Cancer mutational burden is shaped by G4 DNA, replication stress and mitochondrial dysfunction. Prog. Biophys. Mol. Biol. 2019; 147:47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917. [DOI] [PubMed] [Google Scholar]
- 4. Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C.et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921. [DOI] [PubMed] [Google Scholar]
- 5. Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C.et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pilie P.G., Tang C., Mills G.B., Yap T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019; 16:81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kent T., Chandramouly G., McDevitt S.M., Ozdemir A.Y., Pomerantz R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015; 22:230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mateos-Gomez P.A., Gong F., Nair N., Miller K.M., Lazzerini-Denchi E., Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature. 2015; 518:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mateos-Gomez P.A., Kent T., Deng S.K., McDevitt S., Kashkina E., Hoang T.M., Pomerantz R.T., Sfeir A. The helicase domain of Polθ counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 2017; 24:1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Han J., Ruan C., Huen M.S.Y., Wang J., Xie A., Fu C., Liu T., Huang J. BRCA2 antagonizes classical and alternative nonhomologous end-joining to prevent gross genomic instability. Nat. Commun. 2017; 8:1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ceccaldi R., Liu J.C., Amunugama R., Hajdu I., Primack B., Petalcorin M.I., O’Connor K.W., Konstantinopoulos P.A., Elledge S.J., Boulton S.J.et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015; 518:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sallmyr A., Tomkinson A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018; 293:10536–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mattarucchi E., Guerini V., Rambaldi A., Campiotti L., Venco A., Pasquali F., Lo Curto F., Porta G. Microhomologies and interspersed repeat elements at genomic breakpoints in chronic myeloid leukemia. Genes Chromosomes Cancer. 2008; 47:625–632. [DOI] [PubMed] [Google Scholar]
- 14. Nussenzweig A., Nussenzweig M.C. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010; 141:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sallmyr A., Tomkinson A.E., Rassool F.V. Up-regulation of WRN and DNA ligase IIIα in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008; 112:1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roy S., Tomaszowski K.H., Luzwick J.W., Park S., Li J., Murphy M., Schlacher K. p53 orchestrates DNA replication restart homeostasis by suppressing mutagenic RAD52 and POLθ pathways. eLife. 2018; 7:e31723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mengwasser K.E., Adeyemi R.O., Leng Y., Choi M.Y., Clairmont C., D’Andrea A.D., Elledge S.J. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol. Cell. 2019; 73:885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding X., Ray Chaudhuri A., Callen E., Pang Y., Biswas K., Klarmann K.D., Martin B.K., Burkett S., Cleveland L., Stauffer S.et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 2016; 7:12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ray Chaudhuri A., Callen E., Ding X., Gogola E., Duarte A.A., Lee J.E., Wong N., Lafarga V., Calvo J.A., Panzarino N.J.et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016; 535:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., Ray Chaudhuri A., Nussenzweig A., Janscak P.et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017; 8:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011; 145:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willis N.A., Frock R.L., Menghi F., Duffey E.E., Panday A., Camacho V., Hasty E.P., Liu E.T., Alt F.W., Scully R. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature. 2017; 551:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schlacher K., Wu H., Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51–BRCA1/2. Cancer Cell. 2012; 22:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daza-Martin M., Starowicz K., Jamshad M., Tye S., Ronson G.E., MacKay H.L., Chauhan A.S., Walker A.K., Stone H.R., Beesley J.F.J.et al. Isomerization of BRCA1–BARD1 promotes replication fork protection. Nature. 2019; 571:521–527. [DOI] [PubMed] [Google Scholar]
- 25. Kolinjivadi A.M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Techer H., Baldi G., Shen R., Ciccia A., Pellegrini L.et al. Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell. 2017; 67:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bryant H.E., Petermann E., Schultz N., Jemth A.S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009; 28:2601–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Syed A., Tainer J.A. The MRE11–RAD50–NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu. Rev. Biochem. 2018; 87:263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z., Song Y., Li S., Kurian S., Xiang R., Chiba T., Wu X. DNA polymerase theta (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J. Biol. Chem. 2019; 294:3909–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feng W., Simpson D.A., Carvajal-Garcia J., Price B.A., Kumar R.J., Mose L.E., Wood R.D., Rashid N., Purvis J.E., Parker J.S.et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019; 10:4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dutta A., Eckelmann B., Adhikari S., Ahmed K.M., Sengupta S., Pandey A., Hegde P.M., Tsai M.S., Tainer J.A., Weinfeld M.et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017; 45:2585–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanssen-Bauer A., Solvang-Garten K., Akbari M., Otterlei M. X-ray repair cross complementing protein 1 in base excision repair. Int. J. Mol. Sci. 2012; 13:17210–17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gunn A., Stark J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 2012; 920:379–391. [DOI] [PubMed] [Google Scholar]
- 33. Konca K., Lankoff A., Banasik A., Lisowska H., Kuszewski T., Gozdz S., Koza Z., Wojcik A. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. 2003; 534:15–20. [DOI] [PubMed] [Google Scholar]
- 34. Wang H., Guo W., Mitra J., Hegde P.M., Vandoorne T., Eckelmann B.J., Mitra S., Tomkinson A.E., Van Den Bosch L., Hegde M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat. Commun. 2018; 9:3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jackson D.A., Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell Biol. 1998; 140:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sultana R., Abdel-Fatah T., Perry C., Moseley P., Albarakti N., Mohan V., Seedhouse C., Chan S., Madhusudan S. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS One. 2013; 8:e57098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dungrawala H., Rose K.L., Bhat K.P., Mohni K.N., Glick G.G., Couch F.B., Cortez D. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell. 2015; 59:998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Truong L.N., Li Y., Shi L.Z., Hwang P.Y., He J., Wang H., Razavian N., Berns M.W., Wu X. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl Acad. Sci. U.S.A. 2013; 110:7720–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fan J., Wilson P.F., Wong H.K., Urbin S.S., Thompson L.H., Wilson D.M. 3rd XRCC1 down-regulation in human cells leads to DNA-damaging agent hypersensitivity, elevated sister chromatid exchange, and reduced survival of BRCA2 mutant cells. Environ. Mol. Mutagen. 2007; 48:491–500. [DOI] [PubMed] [Google Scholar]
- 40. Tutt A., Bertwistle D., Valentine J., Gabriel A., Swift S., Ross G., Griffin C., Thacker J., Ashworth A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001; 20:4704–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Badie S., Carlos A.R., Folio C., Okamoto K., Bouwman P., Jonkers J., Tarsounas M. BRCA1 and CtIP promote alternative non-homologous end-joining at uncapped telomeres. EMBO J. 2015; 34:410–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ying S., Chen Z., Medhurst A.L., Neal J.A., Bao Z., Mortusewicz O., McGouran J., Song X., Shen H., Hamdy F.C.et al. DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016; 76:1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brem R., Hall J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005; 33:2512–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor R.M., Moore D.J., Whitehouse J., Johnson P., Caldecott K.W. A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Mol. Cell. Biol. 2000; 20:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hanzlikova H., Kalasova I., Demin A.A., Pennicott L.E., Cihlarova Z., Caldecott K.W. The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol. Cell. 2018; 71:319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carvalho C.M., Lupski J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016; 17:224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei P.C., Chang A.N., Kao J., Du Z., Meyers R.M., Alt F.W., Schwer B. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell. 2016; 164:644–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seo S.H., Bacolla A., Yoo D., Koo Y.J., Cho S.I., Kim M.J., Seong M.W., Kim H.J., Kim J.M., Tainer J.A.et al. Replication-based rearrangements are a common mechanism for SNCA duplication in Parkinson’s disease. Mov. Disord. 2020; 35:868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sotiriou S.K., Kamileri I., Lugli N., Evangelou K., Da-Re C., Huber F., Padayachy L., Tardy S., Nicati N.L., Barriot S.et al. Mammalian RAD52 functions in break-induced replication repair of collapsed DNA replication forks. Mol. Cell. 2016; 64:1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kwon M.S., Lee J.J., Min J., Hwang K., Park S.G., Kim E.H., Kim B.C., Bhak J., Lee H. Brca2 abrogation engages with the alternative lengthening of telomeres via break-induced replication. FEBS J. 2019; 286:1841–1858. [DOI] [PubMed] [Google Scholar]
- 51. Hartlerode A.J., Willis N.A., Rajendran A., Manis J.P., Scully R. Complex breakpoints and template switching associated with non-canonical termination of homologous recombination in mammalian cells. PLos Genet. 2016; 12:e1006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Houl J.H., Ye Z., Brosey C.A., Balapiti-Modarage L.P.F., Namjoshi S., Bacolla A., Laverty D., Walker B.L., Pourfarjam Y., Warden L.S.et al. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 2019; 10:5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Swisher E.M., Lin K.K., Oza A.M., Scott C.L., Giordano H., Sun J., Konecny G.E., Coleman R.L., Tinker A.V., O’Malley D.M.et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017; 18:75–87. [DOI] [PubMed] [Google Scholar]
- 54. Gonzalez-Martin A., Pothuri B., Vergote I., DePont Christensen R., Graybill W., Mirza M.R., McCormick C., Lorusso D., Hoskins P., Freyer G.et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2019; 381:2391–2402. [DOI] [PubMed] [Google Scholar]
- 55. Mirza M.R., Coleman R.L., Gonzalez-Martin A., Moore K.N., Colombo N., Ray-Coquard I., Pignata S. The forefront of ovarian cancer therapy: update on PARP inhibitors. Ann. Oncol. 2020; doi:10.1016/j.annonc.2020.06.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.