

Abstract
Adverse Outcome Pathway Bayesian Networks (AOPBNs) are a promising avenue for developing predictive toxicology and risk assessment tools based on Adverse Outcome Pathways (AOPs). Here we describe a process for developing AOPBNs. AOPBNs use causal networks and Bayesian statistics to integrate evidence across key events. In this paper, we use our AOPBN to predict the occurrence of steatosis under different chemical exposures. Since it is an expert-driven model, we use external data (i.e., data not used for modeling) from the literature to validate predictions of the AOPBN model. The AOPBN accurately predicts steatosis for the chemicals from our external data. In addition, we demonstrate how end users can utilize the model to simulate the confidence (based on posterior probability) associated with predicting steatosis. We demonstrate how the network topology impacts predictions across the AOPBN, and how the AOPBN helps us identify the most informative key events that should be monitored for predicting steatosis. We close with a discussion of how the model can be used to predict potential effects of mixtures and how to model susceptible populations (e.g., where a mutation or stressor may change the conditional probability tables in the AOPBN). Using this approach for developing expert AOPBNs will facilitate the prediction of chemical toxicity, facilitate the identification of assay batteries, and greatly improve chemical hazard screening strategies.
Keywords: toxicology, risk assessment, adverse outcome pathway, computational toxicology
SUMMARY:
Bayesian Networks + Adverse Outcome Pathways = improved predictive toxicology and causal understanding for single chemicals, mixtures, and susceptible populations.
1. INTRODUCTION
The integration of data from high content and high throughput assays remains a key challenge in chemical safety prediction, especially with respect to understanding the biology underlying pathogenesis. The challenge in data integration is balancing the net benefits of reduced overall costs (including decreased animal use) against the disadvantages associated with having sufficient biological knowledge to make the causal arguments required for hazard characterization, especially when an apical endpoint may not be observed. At this point, AOPs are a good starting point for leveraging these data.
Adverse outcome pathways (AOPs) describe pathogenesis from the molecular initiating event (MIE) through a series of key events (KEs) causally linked by key-event relationships (KERs) and to the apical adverse outcome. KEs generally occur at the molecular, cellular and tissue levels but also, in some cases at the organ, organism and population levels as well. (Ankley et al., 2010; Villeneuve et al., 2014). Thus, AOPs describe the causal chain moving from the chemical-receptor interaction to the adverse outcome. Recently, AOPs have been used to make predictions about whether or not a chemical causes steatosis or skin sensitization (Burgoon, Druwe, Painter, & Yost, 2016; Strickland et al., 2016). In fact, the skin sensitization AOP has changed how chemicals are tested under the REACH annexes (European Chemicals Agency, 2019). By combining multiple AOPs together, we can build AOP Networks, which are more complete descriptions of the routes that lead to a particular adverse outcome (Villeneuve et al., 2014).
In this study, we focused on the AOP network describing hepatic steatosis, or fatty liver. Steatosis has an estimated human prevalence of 21.4% in the United States (Lazo et al., 2013). Drug-induced liver injury, which includes drug-induced steatosis, may hamper drug development or approval (Kullak-Ublick et al., 2017). In addition, the US EPA has cited steatosis as a serious liver injury that is considered when developing toxicity reference values in the Integrated Risk Information System, the US EPA program that evaluates toxicity information independent of the regulatory program offices (e.g., IRIS assessments of dichloromethane).
AOP networks can be modeled using expert-driven Adverse Outcome Pathway Bayesian Networks (AOPBNs) as a way to make probabilistic predictions (i.e., predictions based on a probability of occurrence that communicates the uncertainty of the prediction, like the probability of rain at a certain time) of adverse outcomes and probabilistic weight of evidence data integration to support hazard identification and risk assessments. AOPBNs combine multiple AOPs together into a network and add tables that represent the probability of moving from one key event to another (this includes moving from MIE to the first KE through intermediary KEs to the last KEs and ultimately to the AO).
By using causal networks we can identify the minimally sufficient set of key events (MinSSKE) that need to be measured to predict an adverse outcome (Burgoon et al., 2016). We can also look at the network topology and devise scenarios to identify other less statistically certain sets of key events than the MinSSKEs that may, after taking costs into account, become more optimal to run as assay batteries. This in turn allows us to design efficient predictive toxicity assay batteries that only test the key events necessary to make a prediction. It is important to note that given a particular AOPBN this may be one or more nodes/key events.
The AOPBN for steatosis that we describe here was developed for both predictive toxicology and mechanistic investigation in support of hazard identification. We outline our process of developing the AOPBN, starting with existing AOPs and metabolic and disease pathways. We then demonstrate its ability to accurately predict chemical effects using in vitro assay data as inputs to the model.
2. MATERIALS AND METHODS
2.1. Model construction
An AOPBN describing the biological events in the human liver leading to steatosis was constructed from pathways described in Reactome (Fabregat et al., 2018) and the peer-reviewed literature (see Results section for full information). The AOPBN for steatosis consists of a series of conditional probabilities for each node. See the supplemental table for more details.
More specifically, we started with the AOP described by Burgoon et al. (2016) and combined it with information from Reactome, and the literature. We used literature information, especially knock-out, knock-in, and knock-down information to facilitate the expert-driven development of the conditional probability tables – see the supplemental information for more details and references. Note that these conditional probabilities are generated using expert judgment to ascertain the uncertainty and thus the probability, of the conditional events. This is common when developing expert models to be used as Bayesian Networks.
The model has been implemented in the Bayesian Inference for Substance and Chemical Toxicity tool (BISCT). BISCT is freely available and open source software that can be downloaded from GitHub (https://github.com/DataSciBurgoon/bisct/releases/tag/1.1.4) and cited with a DOI (DOI: 10.5281/zenodo.3349914).
By using the literature and existing expert-driven AOPs, our AOPBN is classified as an expert model, as opposed to a computationally driven reversed engineered model. The model is based on expert judgment and analysis of literature and available data. We evaluated model performance using previously published external data in Angrish, et al. (2017).
2.2. Data for Model Validation
Data were taken without reanalysis from Angrish, et al. (2017). We converted threshold data from continuous to binary TRUE/FALSE values using the same threshold values as the original author (Table I). For instance, fatty acid beta oxidation (as measured using the Seahorse Mito Stress Test with exogenous palmitate) was detected following exposure to all chemicals, so they all scored TRUE. Cytosolic fatty acids (measured using fluorescence intensities and the dyes Nile Red and Hoechst 33342, see Angrish, et al. (2017) for details) were decreased for only certain chemicals therefore those were scored as FALSE. We also only used the results for the highest concentrations as this is a hazard identification case study where we desired to detect any potential ability to cause steatosis.
Table I.
Assay results from Angrish et al (2017) Converted to True/False
| Chemical | FABO | PPARG | SREBP1 | Cytosolic Fatty Acids | Lipogenesis |
|---|---|---|---|---|---|
| 22(R)-hydroxycholesterol | TRUE | TRUE | TRUE | FALSE | FALSE |
| amiodarone | TRUE | TRUE | TRUE | FALSE | FALSE |
| cyclosporin A | TRUE | TRUE | TRUE | TRUE | TRUE |
| T0901317 | TRUE | TRUE | TRUE | TRUE | TRUE |
| Troglitazone | TRUE | TRUE | TRUE | FALSE | FALSE |
| Wyeth-14,643 | TRUE | TRUE | TRUE | FALSE | FALSE |
Note that the efforts described here and those in Angrish, et al. (2017) were completely separate; none of the information from the Angrish, et al. (2017) study was used to develop the AOPBN presented here, making that data external to this validation study.
2.3. Testing Mathematical Sufficiency
For predictive toxicology and hazard assessment, we can use causal networks to identify the minimal set of nodes within the AOPBN that are sufficient to infer that the adverse outcome will occur. We call this the Minimally Sufficient Set of Key Events (MinSSKE). We used Pearl’s backdoor algorithm (Pearl, 2010) to identify the minimal set of sufficient key events. We verified it by testing whether activation/deactivation of those nodes is sufficient to predict the adverse outcome.
2.3.1. Backdoor Algorithm:
Create an empty set, called SufficientNodes, to collect the sufficient nodes
Place new undirected edges connecting parent nodes that share a child node (parent nodes are those nodes that are upstream and feed into a child node).
Find shortest path from adverse outcome to molecular initiating event
With adverse outcome as the first node in the shortest path, the second node is a sufficient node
Add sufficient node to set SufficientNodes, and remove this sufficient node
Repeat from Step 2 until all paths have been explored from the adverse outcome to the molecular initiating event
Note that the AOPBN contains many MIEs. This means that there are several potential entry points for chemical-receptor interactions to jump-start the AOPBN.
3. RESULTS
3.1. Description of the AOPBN for Steatosis
Steatosis is when there is too much fat in the cytoplasm of hepatocytes. This occurs either because lipids are not being metabolized fast enough, lipids are not being exported fast enough, or both. Thus, lipids are either being shuttled into the cell too quickly (influx), being created too fast (lipogenesis), not being shuttled out fast enough (problems of efflux), not being metabolized fast enough (problems with fatty acid beta oxidation; FABO), or a combination of all of these.
The AOPBN for steatosis has a number of possible MIEs, including farsenoid X receptor (FXR), peroxisome proliferator-activated receptor (PPAR) alpha (PPARA), PPAR-gamma (PPARG), liver X receptor (LXR), and insulin receptor (IR). The adverse outcome is steatosis. The conditional probability tables are listed in the Supplemental Materials. The structure of the AOPBN is given in Figure 1.
Fig. 1.
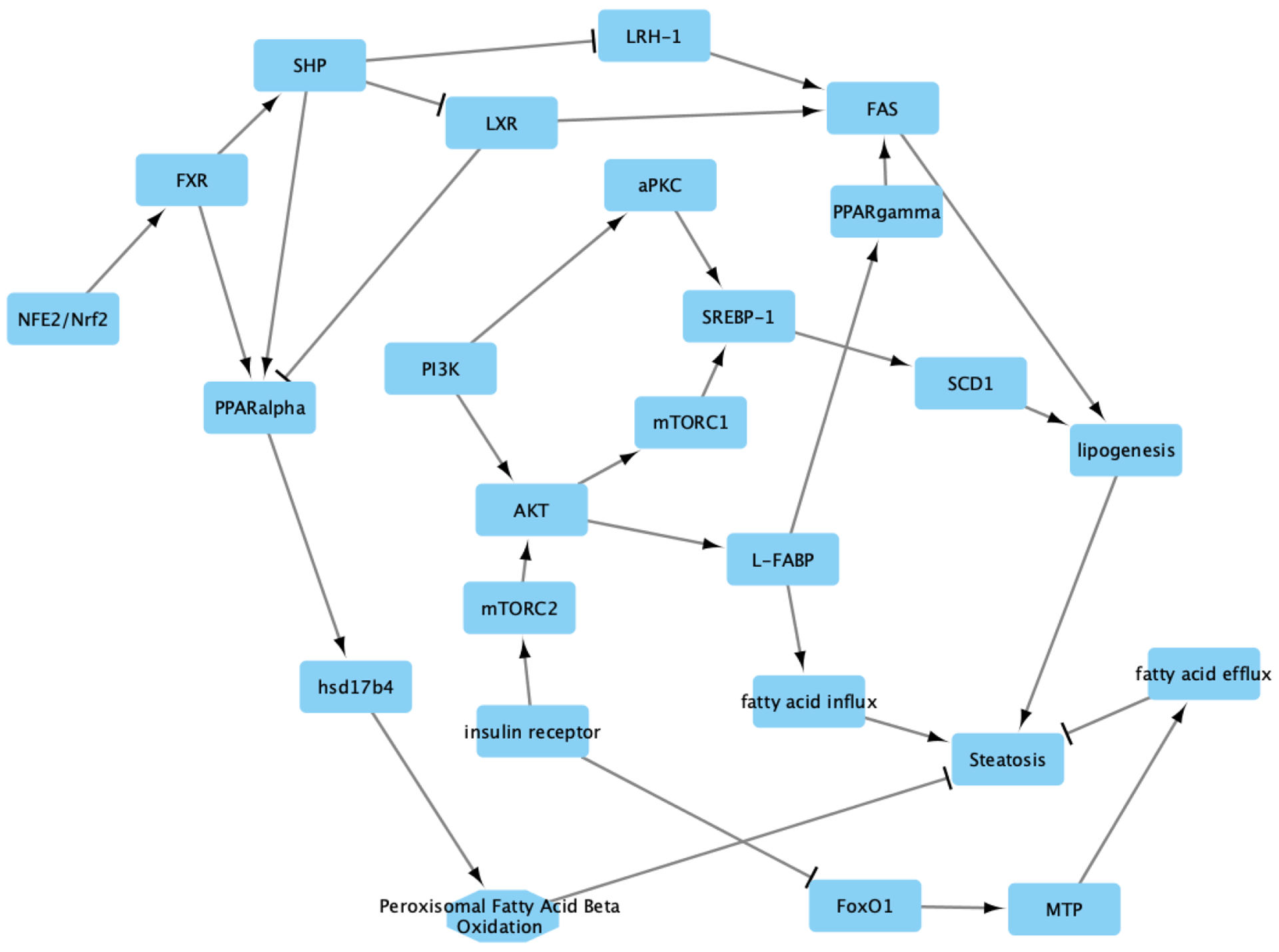
The AOPBN for steatosis is a combination of several different adverse outcome pathways and disease pathways. The molecular initiating events include FXR, PPAR-alpha, LXR, PPAR-gamma, and insulin receptor. The adverse outcome is steatosis. Each node/vertex in the graph represents a protein (e.g., PI3K), metabolite (e.g., cytosolic fatty acids), or action (e.g., fatty acid influx). Edges with arrow heads represent activation, while the T-shaped edges represent inhibition. Each edge represents a conditional probability that the child node is activated/deactivated given the activation of the parent node. The conditional probability tables are given in the Supplemental Materials.
Nuclear factor erythroid 2-related factor (Nrf2) mediates protection from developing steatosis in one branch of our AOPBN. Here, Nrf2 activates FXR dependent induction of small heterodimer partner (SHP). SHP likely outcompetes LXR for binding to retinoid X receptor (RXR) which in turn represses LXR-mediated gene expression (Kay et al., 2011; Kim et al., 2009). In addition, SHP inhibits liver receptor homolog-1 (LRH1/LRH-1) (Goodwin et al., 2000). LRH1 serves as a cofactor that increases the ability for LXR to drive fatty acid synthase (FAS) transcription (Matsukuma, Wang, Bennett, & Osborne, 2007). Thus, SHP activation would lead to less FAS expression and activity, leading to a decrease in lipogenesis, thus avoiding steatosis (Kay et al., 2011; Matsukuma et al., 2007). We also note that PPAR-gamma knockout mice (Cre/flox mice) had decreased expression of FAS (Matsusue et al., 2003), and that knockout of PPARG protected mice from developing steatosis when treated with rosiglitazone.
Mutation analysis identified a functional FXR-binding site in the PPAR-alpha gene regulatory region (Pineda Torra et al., 2003. SHP has been noted to augment the transcriptional activity of PPAR-alpha by itself (Kassam, Capone, & Rachubinski, 2001). There is some evidence that LXR-PPAR-alpha heterodimers can form, thus resulting in the overall decrease in PPAR-alpha activity (Ide et al., 2003).
Hydroxysteroid 17-beta dehydrogenase (HSD17B4) is a PPAR-alpha target gene (Corton et al., 1996; Fan, Cattley, & Corton, 1998; Rakhshandehroo, Knoch, Muller, & Kersten, 2010). HSD17B4 performs beta-oxidation of various fatty acids (Möller, van Grunsven, Wanders, & Adamski, 2001). It is known that inhibition of beta-oxidation leads to steatosis and steatohepatitis (Rao & Reddy, 2001; Reddy, 2001), and that loss of HSD17B4 activity, a key component of the beta-oxidation machinery, is sufficient to infer steatosis (Burgoon et al., 2016).
Our AOPBN contains another branch that starts at IR. Here, IR activation leads to mammalian target of rapamycin (mTOR) complex 2 (mTORC2) activation (Laplante & Sabatini, 2009). Activated mTORC2 activates protein kinase B (AKT) (Laplante & Sabatini, 2009), which activates cascades that drive a pro-steatotic environment. This includes AKT activation of liver-type fatty acid binding protein (L-FABP) which drives fatty acid influx (Chattopadhyay, Selinger, Ballou, & Lin, 2011), and AKT activation of mTOR complex 1 (mTORC1) which mediates sterol regulatory element-binding protein 1 (SREBP-1) activation (Laplante & Sabatini, 2009), which upregulates the expression of and thus likely activates stearoyl-coA desaturase 1 SCD1 (Ruiz et al., 2014) and leads to increased lipogenesis (Strable & Ntambi, 2010). Phosphoinositide 3-kinase (PI3K) also plays a role, where it can activate atypical protein kinase C (aPKC) to lead to SREBP-1 activation (Matsumoto et al., 2003; Schmitz-Peiffer & Biden, 2008, p.), as well as activate AKT (Laplante & Sabatini, 2009) and L-FABP (Chattopadhyay et al., 2011)
3.2. Validating the Expert-Driven AOPBN Using Existing Data
We validated the model differently than when we use machine learning or data-driven approaches to develop the model as our AOPBN is expert-driven, and not model driven. Specifically, we used existing information (see previous section) to build our model. Next, we ran real-world data through the model to assess the performance of the model. In this case we are using data from Angrish, et al (2017). Note that the data used for validation was not used in model development.
This dataset consists of 6 chemicals, where 2 are known to cause steatosis. The investigators in that study sought to develop assays that represent key events within a steatosis AOP – which fits well with our purposes for validating our model. Table I lists the chemicals that were tested as well as the activation/inactivation of specific key events (TRUE or FALSE, respectively). Angrish, et al (2017) confirmed that T0901317 and cyclosporin A both caused steatosis; the rest of the chemicals did not.
Predictions from the AOPBN for steatosis agreed with the results published in Angrish, et al (2017) (Table II). It is important to recall that the AOPBN is completely independent of the data from Angrish, et al (2017) therefore this test represents a true model validation.
Table II.
Comparison of Results from AOPBN Model to Angrish et al (2017)
| Chemical | Angrish et al (2017) Results for High Concentration | AOPBN Model for Steatosis for High Concentration |
|---|---|---|
| 22(R)-hydroxycholesterol | No | No (99% certain) |
| amiodarone | No | No (99% certain) |
| cyclosporin A | Yes | Yes (99% certain) |
| Wyeth-14,643 | No | No (99% certain |
| Troglitazone | No | No (99% certain) |
| T0901317 | Yes | Yes (99% certain) |
In addition, we also wanted to demonstrate how one could use information about chemical actions to see what the model would predict. This gave us another opportunity to validate the model. Kawano and Cohen (2013) noted that microsomal triglyceride transfer protein (MTP) inhibitors shut down fatty acid efflux from hepatocytes, leading to steatosis. When we ran a simulation where we inhibited MTP in our model we noticed that the model predicted that steatosis would occur (probability of 74.2%). This is consistent with the literature (Ksander et al., 2001; Liao, Hui, Young, & Davis, 2003; Welty, 2014).
3.3. Identifying Key Events as Sufficient for Predicting Steatosis
We identified fatty acid influx, lipogenesis, fatty acid beta oxidation, and fatty acid efflux as one MinSSKE using the backdoor algorithm. Here, sufficient nodes means that these are the only nodes that need to be measured to make predictions about whether or not steatosis will occur.
To test the sufficiency, we created nine scenarios:
Fatty acid beta oxidation = FALSE; fatty acid influx = TRUE; lipogenesis = TRUE; fatty acid efflux = FALSE – this is what we expect will lead to steatosis
FXR = FALSE; PPAR-alpha = FALSE; PPAR-gamma = TRUE; LXR = TRUE; insulin receptor = TRUE – this should also lead to steatosis
FXR = FALSE; PPAR-alpha = FALSE; PPAR-gamma = TRUE; LXR = TRUE; insulin receptor = TRUE; fatty acid efflux = FALSE
Fatty acid beta oxidation = FALSE
Fatty acid beta oxidation = FALSE; fatty acid efflux = FALSE
Fatty acid beta oxidation = FALSE; PPAR-gamma = TRUE
Fatty acid beta oxidation = FALSE; lipogenesis = TRUE
PPAR-gamma = TRUE, insulin receptor = TRUE, LXR = TRUE
FXR = FALSE, PPAR-alpha = FALSE
We hypothesized that the posterior probability from using the activation/inactivation of the sufficient set of key events leading to steatosis should be higher than the posterior probability of using just information about the MIEs alone. Thus, activation/inactivation of the minimal set of sufficient key events leading to steatosis, should result in a high posterior probability, near 99%. In the case of the molecular initiating events only scenarios, the posterior probability should be considerably lower. If the molecular initiating events only scenario still results in a near 99% posterior probability, then it suggests the molecular initiating events are also sufficient to infer steatosis.
The results are shown in Table III. The first scenario, where the minimal set of sufficient key events are the only ones used, yields a 99% probability of steatosis. If we use only the most distant MIEs, FXR and PPAR-alpha (Scenario 9), we see the posterior probability of steatosis drop to 67.3%. If we only use the molecular initiating events that are closest to the adverse outcome (Scenario 8), we see a posterior probability of 97.1%. This demonstrates the key point of the Markov property – we have the most certainty about whether or not there will be steatosis if we measure assays that are closest to steatosis. The further the key events are from the adverse outcome, the greater the uncertainty.
Table III.
Various Scenarios to Test Node Sensitivity and Demonstrate AOPBN Capability
| Scenario | Parameters | Posterior Probability of Steatosis |
|---|---|---|
| 1. Sufficient Key Events | Fatty acid beta oxidation = FALSE Fatty acid influx = TRUE Lipogenesis = TRUE Fatty acid efflux = FALSE |
99.0% |
| 2. Molecular Initiating Events | FXR = FALSE PPAR-alpha = FALSE PPAR-gamma = TRUE LXR = TRUE Insulin receptor = TRUE |
97.3% |
| 3. Molecular Initiating Events + Fatty Acid Efflux | FXR = FALSE PPAR-alpha = FALSE PPAR-gamma = TRUE LXR = TRUE Insulin receptor = TRUE Fatty acid efflux = FALSE |
98.8% |
| 4. Sufficient Key Event Subset 1 | Fatty acid beta oxidation = FALSE | 66.0% |
| 5. Sufficient Key Event Subset 2 | Fatty acid beta oxidation = FALSE Fatty acid efflux = FALSE |
96.8% |
| 6. Sufficient Key Event Subset 3 | Fatty acid beta oxidation = FALSE PPAR-gamma = TRUE |
90.5% |
| 7. Sufficient Key Event Subset 4 | Fatty acid beta oxidation = FALSE Lipogenesis = TRUE |
76.8% |
| 8. Molecular Initiating Events Closest to Adverse Outcome | PPAR-gamma = TRUE Insulin receptor = TRUE LXR = TRUE |
97.1% |
| 9. Molecular Initiating Events Furthest from Adverse Outcome | FXR = FALSE PPAR-alpha = FALSE |
67.3% |
| 10. Molecular Initiating Events Closest to Adverse Outcome + Fatty Acid Efflux | PPAR-gamma = TRUE Insulin receptor = TRUE LXR = TRUE Fatty acid efflux = FALSE |
98.5% |
Another key lesson of the Markov property: the value of information from an assay to predict an adverse outcome decreases as the distance from the adverse outcome increases. In other words, value of information is inversely proportional to the distance between the key event and the adverse outcome. Consider, the information gain we obtain going from Scenario 8 (molecular initiating events closest to steatosis) to Scenario 2 (all molecular initiating events; Table III). We go from a posterior probability of 97.1% in Scenario 8 to a posterior probability of 97.3% in Scenario 2 – adding in the assays furthest from the adverse outcome only increased our certainty of predicting steatosis by 0.2% in our model.
Scenario 4 tests if we need to use the sufficient key events as a set, or if we can just use one of the sufficient key events alone. In this case, just having knowledge of fatty acid beta oxidation being inhibited results in a posterior probability of having steatosis of just 66%. Adding on fatty acid efflux inhibition in Scenario 5 raises the posterior probability to 96.8%. In Scenario 6, we take Scenario 4 and add on PPAR-gamma activation. This results in a posterior probability of 90.5%. Adding on lipogenesis activation to fatty acid beta oxidation (Scenario 7) also only raises the posterior probability modestly, to 76.8%.
The Bayesian Network approach also allows us to see the probabilities that any node/key event is active or inactive. In Table IV we show the probability of the node being active for Scenario 1. This is helpful in tracking down potential modes of action, or building hypotheses for additional testing.
Table IV.
Probability That a Node/Protein Is Active in Scenario 1
| Node Name | Probability Active |
|---|---|
| nrf2 | 0.16 |
| ir | 1.0 |
| pi3k | 0.50 |
| fxr | 0.12 |
| shp | 0.08 |
| lxr | 0.92 |
| ppara | 0.04 |
| hsd17b4 | 0.01 |
| fatty_acid_beta_oxidation | 0.0 |
| lrh1 | 0.89 |
| mtorc2 | 1.0 |
| foxo1 | 0.003 |
| mtp | 0.001 |
| efflux | 0.0 |
| akt | 0.99 |
| lfabp | 1.0 |
| influx | 1.0 |
| pparg | 0.96 |
| fas | 0.99 |
| mtorc1 | 0.94 |
| apkc | 0.50 |
| srebp1 | 0.96 |
| scd1 | 0.95 |
| lipogenesis | 1.0 |
| steatosis | 0.99 |
4. DISCUSSION
AOPBNs drive the AOP concept toward utility as predictive toxicology and mechanistic investigation tools. As they stand, AOPs are a knowledge source that summarizes mechanistic pathways leading from chemical-receptor interaction through regulatory adverse outcomes. However, AOPs themselves have limited utility for predictive toxicology. The most obvious reason is that AOPs are focused, on purpose, to make them more manageable. Thus, any individual AOP is likely to ignore other AOPs that share key events, or that may intersect in some other way to impact each other (e.g., transrepression mechanisms). AOP Networks represent the best way to integrate AOPs together, to better reflect the cross-talk and integration of signals from several AOPs that may modulate the same adverse outcome.
AOPBNs are AOP Networks with a probability twist – because they are causal networks, they can be represented as Bayesian Networks, with conditional probabilities at each edge between the key event nodes.
Generally speaking, the minimal set of sufficient key events is unlikely to include the MIEs. This can become a source of confusion for some, who believe that one must understand the MIEs to predict hazards. However, knowledge of which MIEs are activated is not necessarily relevant for risk assessment or hazard identification – knowledge of the potential adverse outcome is. Also biology is complicated – signals can intervene within a network to interfere with the signaling that starts at an MIE. This means that in a network context, a chemical may activate the MIE, but if another signal from, say another chemical within a mixture, or from an ongoing biological process, shunts the signal from the MIE, then the key events just prior to the adverse outcome may not be perturbed. This would result in a lack of AO activation.
Ultimately, the most informative key events in the AOPBN for predicting adverse outcomes will be those closest to the adverse outcome. This is a result of the Markov property which states that the probability that a node will be active or inactive is solely determined by its parents. In the case of AOPBNs, that means that the nodes that feed into the adverse outcome are those that determine if the adverse outcome will occur or not. The Markov property is true for all causal networks (Pearl, 2010). Thus, the MinSSKE will always by definition include those nodes which are the parents of the adverse outcome. We can see the Markov property at work, when we compare the information gain in probabilistic certainty (posterior probability) comparing various scenarios in Table III.
For instance, the key events that are the furthest from the adverse outcome have the least amount of information – they change the overall certainty in the results the least. This was demonstrated when comparing the information gain (measured as change in posterior probability, which here is a measure of certainty) between Scenario 2 (all MIEs) and Scenario 8 (only the MIEs closest to the adverse outcome; Table III). We go from a posterior probability of 97.1% in Scenario 8 to a posterior probability of 97.3% in Scenario 2 – adding in the assays for the MIEs furthest from the adverse outcome (FXR and PPAR-alpha) only increased our certainty of predicting steatosis by 0.2% in our model. This is also demonstrated when comparing Scenario 8 to Scenario 9 (MIEs furthest from the adverse outcome) where we see a difference in information gain of 29.8% or comparing Scenario 1 (the MinSSKE) to Scenario 9 where we see a difference in information gain of 31.7%.
This makes it clear that when you want to predict the adverse outcome, knowledge of the most upstream events within the network are not as important as knowledge of the downstream events most proximal to the adverse outcome; this is a demonstration of the Markov property.
If we wish to consider the information content of nodes other than those in the MinSSKE (e.g., perhaps there are no assays for the MinSSKEs), then network topology matters (and for clarity, topology here means the arrangement of nodes with respect to each other, and the conditional probabilities at each edge connecting nodes). For instance, we can see that measuring all of the MIEs and nothing else results in a steatosis prediction of 97.3% (Table III). However, in previous versions of the model that lacked FoxO1 and MTP, the performance of just the MIEs was approximately 75%. By adding in FoxO1 and MTP, we have shortened the path from insulin receptor to steatosis considerably, increasing the value of the information we obtain from insulin receptor.
What are the real world implications of all this causal network modeling, Markov properties, MinSSKEs, and using the model to identify alternative sets of potential key events to predict the adverse outcome? We can exploit these properties of causal networks when designing assay batteries based on AOPBNs to predict the adverse outcome. Specifically, an ideal assay battery should be efficient – it should balance financial constraints and predictive performance. We demonstrated here that concentrating on the sufficient key events results in the highest predictive performance for an assay battery (Table III). However, the scenario results also demonstrate that we can achieve less than optimal, yet still meaningful and high certainty information, if we use assays that are closer to the adverse outcome, rather than those that are further away. In addition, our network approach demonstrates that sometimes the molecular initiating events exist within the middle of biological networks – a point that can be missed when only considering linear adverse outcome pathways.
Thus, we recommend that end-users consider the availability of technologies and assays to measure the key events, especially the MinSSKEs, costs and benefits when designing ideal assay batteries for their adverse outcomes of interest with predictive performance being only one consideration. For instance, after considering cost, the most optimal approach may no longer be the use of the MinSSKEs if there are no assays that exist. Rather, in that case, the most optimal from a cost and certainty (where posterior probability of adverse outcome is the measure of certainty) standpoint may actually be a combination of assays that yield less certainty, but where the assays actually exist, or they may be of such significantly lower cost that the decrease in certainty is worth the cost savings. Approaches for considering utility, combining cost and predictive performance, are beyond the scope of this paper, but that type of operations research optimization problem is something we may work on in the future.
We are confident that our AOPBN’s posterior probabilities for developing steatosis can be used for relative ranking of chemicals. For instance, given a list of otherwise equally efficacious chemicals (i.e., they meet some performance criteria), the posterior probabilities could be used to rank and prioritize the chemicals for further development. For instance, chemicals with lower posterior probabilities across a suite of AOs may be better candidates for development than those with higher posterior probabilities across the same suite of AOs. In this case in particular, we might recommend pushing forward chemicals that have the lowest posterior probability of causing the AO at the dose/concentration that will likely be used.
What about those cases where we want to identify potential modes of action? End users can use the full model output from the AOPBN to look at the posterior probabilities of each of the nodes in the AOPBN being active. Using that information, end users can make predictions about what may have happened upstream and downstream. For instance, in the case of Scenario 1 (Table III) where we model that fatty acid influx and lipogenesis are occurring, while fatty acid efflux and fatty acid beta oxidation are not occurring, we can look at the model posterior probabilities for each node in the model in Table IV. Here we can see that there is a low likelihood that FXR is activated (12%). Likewise, PPAR-gamma is most likely activated (96%), as is FAS (99%). We also see that LXR and LRH1 are both likely active (92% and 89%, respectively). In this scenario, we also anticipate that insulin receptor is activated (100%) while PPAR-alpha is likely inactivated (4%). Thus, the most likely hypothesized mode of action is activation of insulin receptor, LXR, and PPAR-gamma, which can be tested more directly in other systems.
AOPBNs will also help us make hypotheses and predictions about what may occur following exposure to complex mixtures and stressor mixtures. One way to make predictions using AOPBNs is to do a completely theoretical exposure. We could start with protein docking models for all of the proteins in our KEs. For instance, we might find that benzo[k]flouranthene (BkF) is a direct or allosteric inhibitor of HSD17b4. In our mixture, we might also find that benzo[a]pyrene (BaP) has much lower affinity for binding to PPARa, but benz[a]anthracene (BaA) has very high affinity for PPARa. If we are dealing with a contaminated site that is primarily BaA, with less BkF and BaP then we would anticipate that steatosis is not likely with a probability of 43.9%.
If, on the other hand, the site was evenly split between BaA, BaP and BkF, then our query of the model would probably look more like PPARa being activated (since BaA has more affinity for PPARa than BaP it is unlikely that BaP will outcompete BaA for binding to PPARa) and BkF would likely inhibit the hsd17b4 activity. In this case, the probability of steatosis is only 53.8%, which is still practically equivocal.
Using similar types of logic, we can assign evidence as to what may be happening at a KE based on genetic and other factors. For instance, there may be a mutation that causes hsd17b4 to have lower affinity for BkF. Or there may be a mutation that causes PPARa to bind more strongly to its DNA binding sites, thus increasing the likelihood of transcription following activation by an agonist. We can model these types of situations by turning the evidence on or off more often.
We could also model entire populations. In that case rather than have simple probability tables, we would use probability distributions. We would then perform Monte Carlo analysis to generate a population of individuals and look at how the chemicals are impacting each member of the population. These types of analyses are more computationally intensive to implement than these simple Bayesian networks.
One note to consider is that our AOPBN works well under most circumstances based on the literature we reviewed. However, an important caveat with respect to Nrf2 is that there is evidence that in some instances Nrf2 activity will lead to steatosis. It appears that these situations are mostly in aging rodents (Chambel, Santos-Gonçalves, & Duarte, 2015). The applicability of this dual nature of Nrf2 to humans remains unclear.
In addition, there may be concerns about converting high throughput screening (HTS) data into strictly binary (1 vs 0) data to draw conclusions. Becker et al (2017) rightly claim that IARC’s approach (IARC, 2018) to using HTS data led to conclusions that weren’t much better than random chance. IARC’s approach discretized the HTS data to 1s when a chemical was “active” in an assay and 0 when it was “inactive”, and then they summed those values up in a type of evidence integration. We want to make clear that IARC’s use of binaries is not the same as our use of binaries. Our use of binary responses is consistent with the idea that there are thresholds below which chemicals are not likely to have a biological response. That is why we focused on the high dose phenomena from the Angrish et al (2017) paper where they quite clearly did or did not see a biological response in their assays. This is akin to the idea of tipping points (Shah et al., 2016), or concentrations/doses at which cells are not likely to return to a normal state, which is itself consistent with the idea of toxicity thresholds. At issue in the IARC monograph is that IARC uses the fact that a chemical activates an HTS assay as evidence to state that a phenotype characteristic of cancer is present.
We do not agree with using this type of binary approach for evidence integration. Rather, for evidence integration we would recommend authors use a Bayesian approach to integrate the information, such as a bootstrap meta-regression used by Burgoon et al (Burgoon et al., 2016) to generate a reasonable distribution of concentration-response curves for identifying the threshold. Any concentrations above the threshold would be assigned a 1, and concentrations below would be assigned a value of 0. Those would then form the basis of the inputs to the AOPBN. The AOPBN would then output the posterior probability, of steatosis in this case, on a per-concentration basis. For the adventurous, formal Bayesian approaches would be superior for evidence integration compared to our AOPBN; however, the AOPBN would be a decent approximation.
5. CONCLUSION
We have discussed an approach for developing AOPBNs and ways in which AOPBNs can be used to predict the probability of an adverse outcome based on assay data. The AOPBNs can be used to develop and simulate assay battery results. That information combined with cost information could provide a measure of utility, which would help decision-makers in applying predictive toxicology to screen and prioritization of chemicals, and to predict hazards.
Supplementary Material
6. ACKNOWLEDGEMENTS
This work is based on discussions held at the Pellston Workshop on “Advancing the Adverse Outcome Pathway Concept, Cornwall, ON, 2017”. This work was funded in part by the US Army Environmental Quality Program. Permission for publishing this information has been granted by the Chief of Engineers. Mention of specific products or trade names does not indicate endorsement by the US Army Corps of Engineers, US Environmental Protection Agency, or the federal government. The AOPXplorer source for the steatosis Bayesian Network model can be found at https://github.com/DataSciBurgoon/aop_bayes_nets/tree/master/steatosis
8. REFERENCES
- Angrish MM, McQueen CA, Cohen-Hubal E, Bruno M, Ge Y, & Chorley BN (2017). Editor’s Highlight: Mechanistic Toxicity Tests Based on an Adverse Outcome Pathway Network for Hepatic Steatosis. Toxicological Sciences, 159(1), 159–169. 10.1093/toxsci/kfx121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, … Villeneuve DL (2010). Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environmental Toxicology and Chemistry, 29(3), 730–741. 10.1002/etc.34 [DOI] [PubMed] [Google Scholar]
- Becker RA, Dreier DA, Manibusan MK, Cox L. A. (Tony), Simon TW, & Bus JS (2017). How well can carcinogenicity be predicted by high throughput “characteristics of carcinogens” mechanistic data? Regulatory Toxicology and Pharmacology, 90, 185–196. 10.1016/j.yrtph.2017.08.021 [DOI] [PubMed] [Google Scholar]
- Burgoon LD, Druwe IL, Painter K, & Yost EE (2016). Using In Vitro High-Throughput Screening Data for Predicting Benzo[k]Fluoranthene Human Health Hazards. Risk Analysis. 10.1111/risa.12613 [DOI] [PubMed] [Google Scholar]
- Chambel SS, Santos-Gonçalves A, & Duarte TL (2015). The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism [Research article]. 10.1155/2015/597134 [DOI] [PMC free article] [PubMed]
- Chattopadhyay M, Selinger ES, Ballou LM, & Lin RZ (2011). Ablation of PI3K p110-α Prevents High-Fat Diet–Induced Liver Steatosis. Diabetes, 60(5), 1483–1492. 10.2337/db10-0869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, Bocos C, Moreno ES, Merritt A, Marsman DS, Sausen PJ, … Gustafsson JA (1996). Rat 17 beta-hydroxysteroid dehydrogenase type IV is a novel peroxisome proliferator-inducible gene. Molecular Pharmacology, 50(5), 1157–1166. [PubMed] [Google Scholar]
- European Chemicals Agency. (2019). News—ECHA. ECHA News, https://echa.europa.eu/view-article/-/journal_content/title/registrants-to-use-alternative-test-methods-for-skin-sensitisation, accessed 7-23-2019. [Google Scholar]
- Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, … D’Eustachio P (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Research, 46(D1), D649–D655. 10.1093/nar/gkx1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LQ, Cattley RC, & Corton JC (1998). Tissue-specific induction of 17 beta-hydroxysteroid dehydrogenase type IV by peroxisome proliferator chemicals is dependent on the peroxisome proliferator-activated receptor alpha. The Journal of Endocrinology, 158(2), 237–246. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, … Kliewer SA (2000). A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Molecular Cell, 6(3), 517–526. 10.1016/S1097-2765(00)00051-4 [DOI] [PubMed] [Google Scholar]
- IARC. (2018). IARC Monograph on the Evaluation of Carcinogenic Risks to Humans: DDT, Lindane, and 2,4-D (Vol. 113). Retrieved from https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/DDT-Lindane-And-2-4-D-2016 [Google Scholar]
- Ide T, Shimano H, Yoshikawa T, Yahagi N, Amemiya-Kudo M, Matsuzaka T, … Yamada N (2003). Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. II. LXRs suppress lipid degradation gene promoters through inhibition of PPAR signaling. Molecular Endocrinology (Baltimore, Md.), 17(7), 1255–1267. 10.1210/me.2002-0191 [DOI] [PubMed] [Google Scholar]
- Kassam A, Capone JP, & Rachubinski RA (2001). The short heterodimer partner receptor differentially modulates peroxisome proliferator-activated receptor α-mediated transcription from the peroxisome proliferator-response elements of the genes encoding the peroxisomal β-oxidation enzymes acyl-CoA oxidase and hydratasedehydrogenase. Molecular and Cellular Endocrinology, 176(1), 49–56. 10.1016/S0303-7207(01)00475-0 [DOI] [PubMed] [Google Scholar]
- Kawano Y, & Cohen DE (2013). Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. Journal of Gastroenterology, 48(4), 434–441. 10.1007/s00535-013-0758-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay HY, Kim WD, Hwang SJ, Choi H-S, Gilroy RK, Wan Y-JY, & Kim SG (2011). Nrf2 Inhibits LXRα-Dependent Hepatic Lipogenesis by Competing with FXR for Acetylase Binding. Antioxidants & Redox Signaling, 15(8), 2135–2146. 10.1089/ars.2010.3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T-H, Kim H, Park J-M, Im S-S, Bae J-S, Kim M-Y, … Ahn Y-H (2009). Interrelationship between Liver X Receptor α, Sterol Regulatory Element-binding Protein-1c, Peroxisome Proliferator-activated Receptor γ, and Small Heterodimer Partner in the Transcriptional Regulation of Glucokinase Gene Expression in Liver. Journal of Biological Chemistry, 284(22), 15071–15083. 10.1074/jbc.M109.006742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksander GM, deJesus R, Yuan A, Fink C, Moskal M, Carlson E, … Hospattankar A (2001). Diaminoindanes as Microsomal Triglyceride Transfer Protein Inhibitors. Journal of Medicinal Chemistry, 44(26), 4677–4687. 10.1021/jm010294e [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Andrade RJ, Merz M, End P, Benesic A, Gerbes AL, & Aithal GP (2017). Drug-induced liver injury: Recent advances in diagnosis and risk assessment. Gut, gutjnl-2016–313369. 10.1136/gutjnl-2016-313369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, & Sabatini DM (2009). MTOR signaling at a glance. Journal of Cell Science, 122(20), 3589–3594. 10.1242/jcs.051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, … Clark JM (2013). Prevalence of Nonalcoholic Fatty Liver Disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. American Journal of Epidemiology, 178(1), 38–45. 10.1093/aje/kws448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Hui TY, Young SG, & Davis RA (2003). Blocking microsomal triglyceride transfer protein interferes with apoB secretion without causing retention or stress in the ER. Journal of Lipid Research, 44(5), 978–985. 10.1194/jlr.M300020-JLR200 [DOI] [PubMed] [Google Scholar]
- Matsukuma KE, Wang L, Bennett MK, & Osborne TF (2007). A Key Role for Orphan Nuclear Receptor Liver Receptor Homologue-1 in Activation of Fatty Acid Synthase Promoter by Liver X Receptor. Journal of Biological Chemistry, 282(28), 20164–20171. 10.1074/jbc.M702895200 [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Ogawa W, Akimoto K, Inoue H, Miyake K, Furukawa K, … Noda T (2003). PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. The Journal of Clinical Investigation, 112(6), 935–944. 10.1172/JCI18816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsusue K, Haluzik M, Lambert G, Yim S-H, Gavrilova O, Ward JM, … Gonzalez FJ (2003). Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. Journal of Clinical Investigation, 111(5), 737–747. 10.1172/JCI200317223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller G, van Grunsven EG, Wanders RJA, & Adamski J (2001). Molecular basis of d-bifunctional protein deficiency. Molecular and Cellular Endocrinology, 171(1), 61–70. 10.1016/S0303-7207(00)00388-9 [DOI] [PubMed] [Google Scholar]
- Pearl J (2010). An Introduction to Causal Inference. The International Journal of Biostatistics, 6(2). 10.2202/1557-4679.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart J-C, & Staels B (2003). Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Molecular Endocrinology (Baltimore, Md.), 17(2), 259–272. 10.1210/me.2002-0120 [DOI] [PubMed] [Google Scholar]
- Rakhshandehroo M, Knoch B, Muller M, & Kersten S (2010). Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Research, 2010 10.1155/2010/612089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, & Reddy JK (2001). Peroxisomal beta-oxidation and steatohepatitis. Seminars in Liver Disease, 21(1), 43–55. [DOI] [PubMed] [Google Scholar]
- Reddy JK (2001). III. Peroxisomal β-oxidation, PPARα, and steatohepatitis. American Journal of Physiology - Gastrointestinal and Liver Physiology, 281(6), G1333–G1339. [DOI] [PubMed] [Google Scholar]
- Ruiz R, Jideonwo V, Ahn M, Surendran S, Tagliabracci VS, Hou Y, … Morral N (2014). Sterol Regulatory Element-binding Protein-1 (SREBP-1) Is Required to Regulate Glycogen Synthesis and Gluconeogenic Gene Expression in Mouse Liver. Journal of Biological Chemistry, 289(9), 5510–5517. 10.1074/jbc.M113.541110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz-Peiffer C, & Biden TJ (2008). Protein Kinase C Function in Muscle, Liver, and β-Cells and Its Therapeutic Implications for Type 2 Diabetes. Diabetes, 57(7), 1774–1783. 10.2337/db07-1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah I, Setzer RW, Jack J, Houck KA, Judson RS, Knudsen TB, … Kavlock RJ (2016). Using ToxCast™ Data to Reconstruct Dynamic Cell State Trajectories and Estimate Toxicological Points of Departure. Environmental Health Perspectives, 124(7), 910–919. 10.1289/ehp.1409029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, & Espenshade PJ (2012). Expanding roles for SREBP in metabolism. Cell Metabolism, 16(4), 414–419. 10.1016/j.cmet.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strable MS, & Ntambi JM (2010). Genetic control of de novo lipogenesis: Role in diet-induced obesity. Critical Reviews in Biochemistry and Molecular Biology, 45(3), 199–214. 10.3109/10409231003667500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland J, Zang Q, Kleinstreuer N, Paris M, Lehmann DM, Choksi N, … Casey W (2016). Integrated decision strategies for skin sensitization hazard. Journal of Applied Toxicology, 36(9), 1150–1162. 10.1002/jat.3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, … Whelan M (2014). Adverse Outcome Pathway (AOP) Development I: Strategies and Principles. Toxicological Sciences, 142(2), 312–320. 10.1093/toxsci/kfu199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welty FK (2014). Hypobetalipoproteinemia and Abetalipoproteinemia. Current Opinion in Lipidology, 25(3), 161–168. 10.1097/MOL.0000000000000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Borrmann CM, Borchers T, & Spener F (2001). Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: A signaling path to the nucleus. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2323–2328. 10.1073/pnas.051619898 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.