

Abstract
Air pollution particulate matter <2.5 μm (PM2.5) exposure is associated with poor respiratory outcomes. Mechanisms underlying PM2.5-induced lung pathobiology are poorly understood but likely involve cellular and molecular changes to the airway epithelium. We extracted and chemically characterized the organic and water-soluble components of air pollution PM2.5 samples, then determined the whole transcriptome response of human nasal mucociliary airway epithelial cultures to a dose series of PM2.5 extracts. We found that PM2.5 organic extract (OE), but not water-soluble extract, elicited a potent, dose-dependent transcriptomic response from the mucociliary epithelium. Exposure to a moderate OE dose modified the expression of 424 genes, including activation of aryl hydrocarbon receptor signaling and an IL-1 inflammatory program. We generated an OE-response gene network defined by eight functional enrichment groups, which exhibited high connectivity through CYP1A1, IL1A, and IL1B. This OE exposure also robustly activated a mucus secretory expression program (>100 genes), which included transcriptional drivers of mucus metaplasia (SPDEF and FOXA3). Exposure to a higher OE dose modified the expression of 1,240 genes and further exacerbated expression responses observed at the moderate dose, including the mucus secretory program. Moreover, the higher OE dose significantly increased the MUC5AC/MUC5B gel-forming mucin expression ratio and strongly downregulated ciliated cell expression programs, including key ciliating cell transcription factors (e.g., FOXJ1 and MCIDAS). Chronic OE stimulation induced mucus metaplasia–like remodeling characterized by increases in MUC5AC+ secretory cells and MUC5AC mucus secretions. This epithelial remodeling may underlie poor respiratory outcomes associated with high PM2.5 exposure.
Keywords: air pollution particulate matter 2.5, whole transcriptome RNA-seq, human nasal airway epithelium, mucus metaplasia, airway remodeling
Clinical Relevance
This transcriptomic study details the molecular mechanisms that are dysregulated in the airway epithelium during exposure to increasing doses of air pollution particulate matter <2.5 μm (PM2.5) organic constituents, and connects deleterious reprogramming of gene expression programs to disease-relevant tissue remodeling processes at the cellular and protein level. We highlight potential mechanisms by which PM2.5 exposure may initiate disease-related airway pathobiology, thereby providing targetable pathways for future study and clinical intervention in air pollution–mediated/exacerbated airway disease. Furthermore, our PM2.5 response profile can be leveraged in future nasal gene expression studies by aiding in identification of subjects with prior PM2.5 exposure and contributing to the identification of a PM2.5 molecular endotype of airway disease.
In 2014, the World Health Organization cited air pollution as the world’s most prominent single environmental health risk (1). Exposure to indoor and outdoor pollution is a major causal factor for mortality due to cardiovascular and respiratory conditions, including stroke, ischemic heart disease, lung cancer, chronic respiratory diseases, and acute respiratory infections. Furthermore, chronic exposure to air pollution components has been linked to impaired lung function development in children, and even short-term exposure has been shown to be associated with poor health outcomes in subjects with childhood asthma, including increased hospitalization and mortality (2–4).
Air pollution is a heterogeneous mixture, primarily composed of gaseous pollutants (e.g., carbon monoxide, nitrogen oxides, ozone, and sulfur dioxide) and suspended solid or liquid droplet particulate matter (PM) derived from both anthropogenic and geogenic sources. Fine air pollution particle matter with an aerodynamic diameter <2.5 μm (PM2.5) has proven particularly detrimental to human health, as exposure to ambient PM2.5 was associated with 4.2 million deaths and 103.1 million disability-adjusted life years globally in 2015 (5). These respirable particles are usually generated as combustion by-products and serve as a vehicle for transport of potentially noxious heavy metals, hydrocarbons, and reactive ions deep into the respiratory tract to the sites of gas exchange.
On inhalation, PM2.5 first encounters the mucociliary barrier formed by highly specialized secretory and ciliated cells of the airway epithelium. However, most studies investigating molecular/cellular responses of the airway epithelium to PM2.5 have used transformed lung epithelial cells that lack the cellular diversity and functional capability of the in vivo airway epithelium (6, 7). In addition, cellular responses to PM2.5 have typically been determined by using PM standard reference material from the National Institute of Standards and Technology (NIST) in place of real-world air pollution–derived PM2.5. Unfortunately, these NIST standard reference materials were collected in bulk from air filtration systems in Europe and baghouses in Washington D.C. over 40 years ago; therefore, the composition and particle size of these samples are poorly representative of ambient air pollution PM2.5 generated and inhaled in modern U.S. cities (8). Only a few studies have evaluated the effects of air pollution–derived PM2.5 extracts in primary mucociliary airway epithelial cultures (9, 10). In general, these studies determined responses of a limited number of specific candidate genes, pathways, or mechanisms, generating only an isolated piece of the full epithelial molecular response to PM2.5. These studies have reported effects on metabolic and inflammatory pathways, mucus production, and perturbation of defense mechanisms, reinforcing the significant influence of PM2.5 on epithelial functions (11). However, no study has evaluated genome-wide responses to contemporary urban air pollution PM2.5 constituents in primary mucociliary epithelial cultures generated from a large number of donors.
Here, we obtained air pollution PM2.5 from California urban areas in 2011, and fractionated the PM2.5 into chemically characterized, water-soluble (WE) and organic-soluble (OE) extracts. We used these PM2.5 extracts in dose–response studies of mucociliary airway epithelial cell (AEC) cultures generated from donors in the GALA II (Genes-Environments and Admixture in Latino Americans II) childhood asthma study. To comprehensively determine epithelial PM2.5 responses, we performed gene and network analysis of whole transcriptome RNA sequencing (RNA-seq) data from these cultures. We then harnessed powerful single-cell RNA-seq (scRNA-seq) data sets to infer how PM2.5 affects epithelial cell type composition and function and confirmed many of these effects in a model of chronic PM2.5 exposure. These analyses reveal linked networks of PM2.5-induced inflammation coinciding with and likely driving pathologic mucociliary remodeling of the airway epithelium. Some of the results of this study have been previously reported in the form of an abstract (12).
Methods
PM2.5 Collection and Characterization
PM2.5 samples collected on 47-mm polytetrafluoroethylene filters from three cities in California in 2011 (Bakersfield/Fresno, Sacramento, and Yuba City) were provided by the California Air Resources Board. Material collected on filters was pooled by region and fractionated into WE and OE. Chemical species were identified and quantified using inductively coupled plasma mass spectrometry and ion chromatography for WE and gas chromatography-mass spectrometry for OE. Blank filters were also extracted to produce mock extract controls for stimulations.
Human Subject Information
Nasal AECs used for culture came from subjects recruited as part of the GALA II childhood asthma study, which was approved by local institutional review boards (University of California San Francisco, institutional review board number 10-00889, reference number 153543, National Jewish Health HS-2627). All subjects and their parents provided written informed assent and consent, respectively (13, 14).
Mucociliary Air–Liquid Interface Culture and Stimulation
Nasal AECs from five healthy subjects were selected randomly for California extract dose–response experiments, whereas NIST 2786 stimulations were conducted on four donors. Full characterization of moderate OE dose–response was conducted with six healthy donors and six donors with asthma (n = 12). Expansion and differentiation at air–liquid interface (ALI) in vitro was conducted according to established protocols (15). On mucociliary differentiation (Day 28–32 of ALI culture), cultures were washed with PBS and then apically treated with mock extract or Bakersfield/Fresno PM2.5 extracts in a final volume of 20 μl culture media. PBS wash and stimulation were repeated at 24 hours, followed by on-membrane lysis using RNA lysis buffer (Zymo Research) containing 40 mM DTT for total RNA collection after an additional 24 hours of culture incubation.
For chronic stimulation experiments, cultures were washed with PBS and then apically treated with media control or high OE dose from the Yuba City PM2.5 sample (because of limited Bakersfield/Fresno sample) in a final volume of 20 μl media. Alternatively, cultures were treated with 10 ng/ml IL-13 in media on apical (20 μl) and basolateral sides (500 μl). PBS washes and stimulations were repeated every 24 hours for a total of five stimulations before cultures were analyzed. Details for quantitative PCR, immunofluorescence labeling, Western blot, and mucin secretion assays can be found in the data supplement.
Whole Transcriptome RNA-seq
KAPA mRNA HyperPrep (Roche) whole transcriptome libraries were constructed with 250 ng RNA per sample and 12 cycles of library amplification. Libraries were sequenced using the Illumina NovaSeq 6000 system. Details for RNA-seq data processing and analysis can be found in the data supplement.
Gene Enrichment Analysis
Enrichment of differentially expressed genes (DEGs) was performed with Enrichr (16), separately for upregulated and downregulated genes, using the most current gene ontology (GO) databases (GO Biological Processes 2018, GO Molecular Function 2018, GO Cellular Component 2018, KEGG 2016, and Reactome 2016). We also tested for DEG enrichment within cell type marker lists derived from scRNA-seq data as described in the online supplement. Enrichment P values were corrected for multiple testing.
Gene Network Analysis
FGNet and GeneTerm Linker databases were used to construct gene networks on the basis of GO- and KEGG-enriched terms for groups of genes significantly upregulated by moderate OE stimulation (17–19).
Results
Characterization of California Urban PM2.5 Reveals Diverse Organic and Water-Soluble Chemical Components
PM2.5 collected on filters from three California air pollution monitoring sites (Bakersfield/Fresno, Sacramento, and Yuba City) in 2011 was extracted and fractionated into WE and OE as described (see Figure E1 in the data supplement). Chemical characterization of WE revealed nitrate (NO3−), ammonium (NH4+), and sulfate (SO42−) (listed in order of abundance; Figure 1A). In contrast to the other two sites, phosphate (PO43−) was not present at measurable levels in the Yuba City sample. Among the inorganic elements, metals that are characteristic of traffic-related pollution, including copper (Cu), zinc (Zn), barium (Ba), and strontium (Sr), were measured at a greater mass fraction in the Sacramento site sample than in the other site samples (Figure 1B). Chemical characterization of OE revealed an array of carcinogenic and immunotoxic polycyclic aromatic hydrocarbon (PAH) species, including benzo[a]pyrene, indeno[1,2,3-c,d]pyrene, and dibenz[a,h]anthracene (Figure 1C). The Bakersfield/Fresno OE consistently contained the highest level for individual as well as combined PAH species measured per microgram of extracted material (Figures 1C and 1D).
Figure 1.
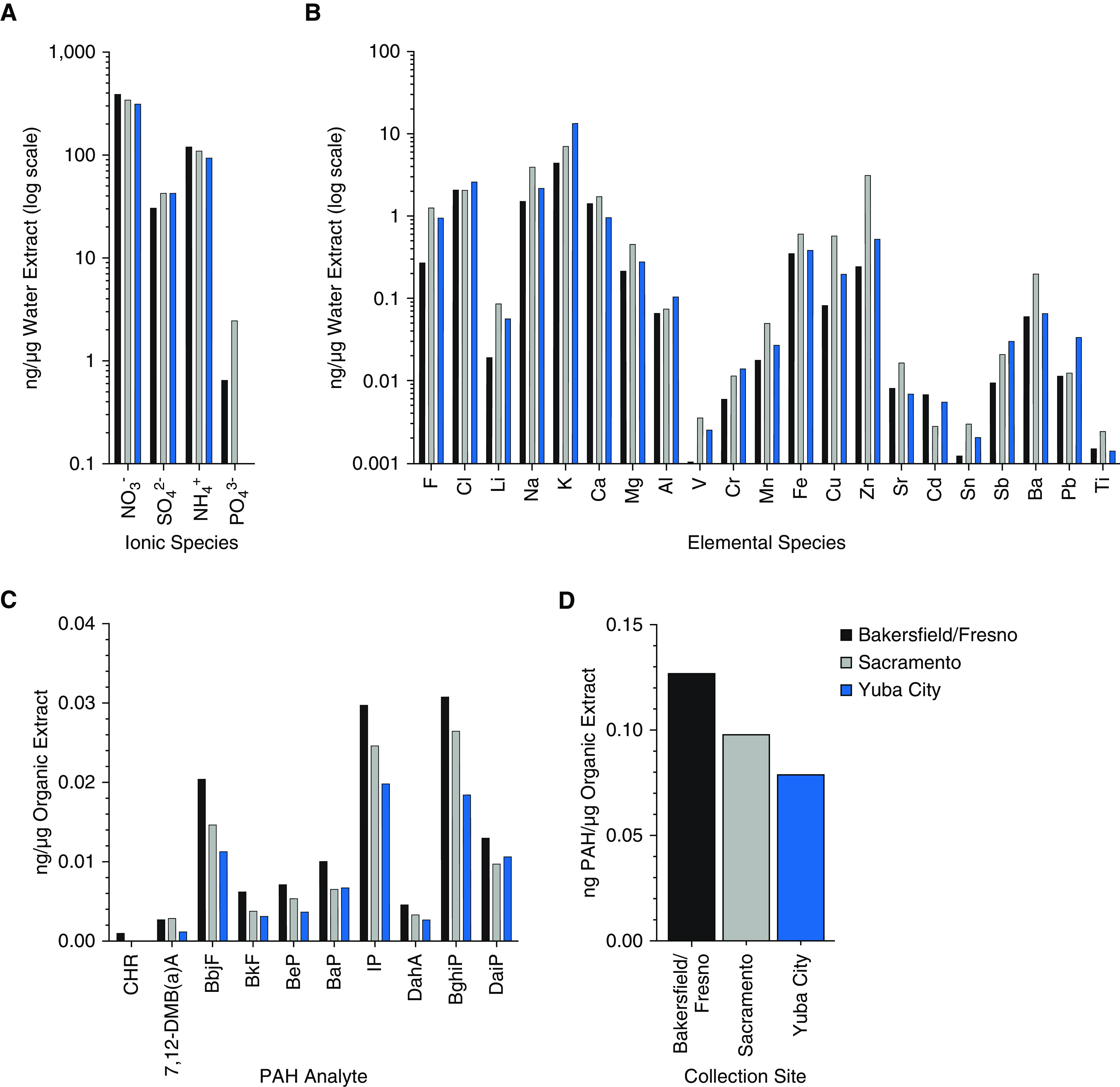
Chemical analysis of urban air pollution particulate matter <2.5 μm (PM2.5) extracts. (A) Relative abundance of ionic chemical species per microgram of water-soluble extract (WE) detected by ion chromatography. (B) Relative abundance of inorganic elemental chemical species per microgram of WE detected by inductively coupled plasma mass spectrometry. (C) Relative abundance of polycyclic aromatic hydrocarbon (PAH) species per microgram of organic extract (OE) detected by gas chromatography–mass spectrometry. (D) Combined concentration of all assayed PAH species for each pool. BaP = benzo[a]pyrene; BbjF = benzo[bj]fluoranthene; BeP = benzo[e]pyrene; BghiP = benzo[g,h,i]perylene; BkF = benzo[k]fluoranthene; CHR = chrysene; DahA = dibenz[a,j]anthracene; DaiP = dibenz[ai]pyrene; IP = indeno[1,2,3-c,d]pyrene.
PM2.5 Modifies Airway Epithelial Gene Expression through Organic Components
To model the acute transcriptional effects of PM2.5 components on the human airway epithelium, we leveraged mature mucociliary ALI cultures derived from primary human nasal AECs (Figure 2A). To determine biologically active, yet nontoxic concentrations of WE and OE, we performed stimulations of five independent donor cultures using a range of extract concentrations (WE: 3 and 30 μg/cm2 culture area; OE: 0.045, 0.45, and 4.5 μg/cm2 culture area). To benchmark the effects of our unique PM2.5 extracts, we also stimulated cultures from these donors with an unseparated fine PM standard reference material, NIST 2786 (<4 μm diameter; stimulation concentration 30 μg/cm2 culture area) (8). Experiments were conducted by performing two consecutive stimulations at the indicated concentrations 24 hours apart, with culture harvest 24 hours after the second stimulation. We did not observe significant toxicity (<1% by lactate dehydrogenase assay) with either PM2.5 extract or NIST 2786 at the tested concentrations (data not shown). To evaluate whether these PM types mediated significant transcriptional changes in our differentiated mucociliary model of the airway epithelium, we generated whole transcriptome RNA-seq data using total RNA isolated from all stimulated as well as mock-stimulated cultures. We performed paired differential gene expression analyses between the mock-stimulated cultures and cultures treated with each of the PM2.5 WE, OE, and NIST dosages to identify all genes with significantly modified expression. No genes exhibited significantly modified expression with either WE stimulation concentration tested. In contrast, OE stimulations resulted in DEGs with a dose-dependent increase in the number of DEGs detected (Figure 2B and Table E2). Specifically, each 10-fold increase in OE concentration resulted in an ∼10-fold increase in the number of DEGs detected, from 11 (low dose) to 124 (moderate dose) to 1,296 (high dose). The genes detected across these OE exposures appeared to be highly conserved, as 9 of the 11 low dose DEGs were also DEGs for the moderate dose, and 71% of the moderate dose DEGs were also DEGs for the high dose (Figure 2C). We also detected 111 DEGs induced by NIST 2786 stimulation, a response comparable in size to the moderate OE dose stimulation (Table E2). Moreover, 35% of the moderate OE DEGs were also DEGs for NIST, and the log2 fold changes (LFCs) of all DEGs resulting from these two stimulants were highly correlated (Pearson correlation coefficient, 0.75; P = 1.68 × 10−34; Figures 2C and 2D). Together, these results indicate that PM2.5 OE exposure can modify airway epithelial gene expression in a dose-dependent manner and that WE does not induce significant transcriptomic changes in our model, even at greater than six times higher concentration than the highest OE dose tested.
Figure 2.
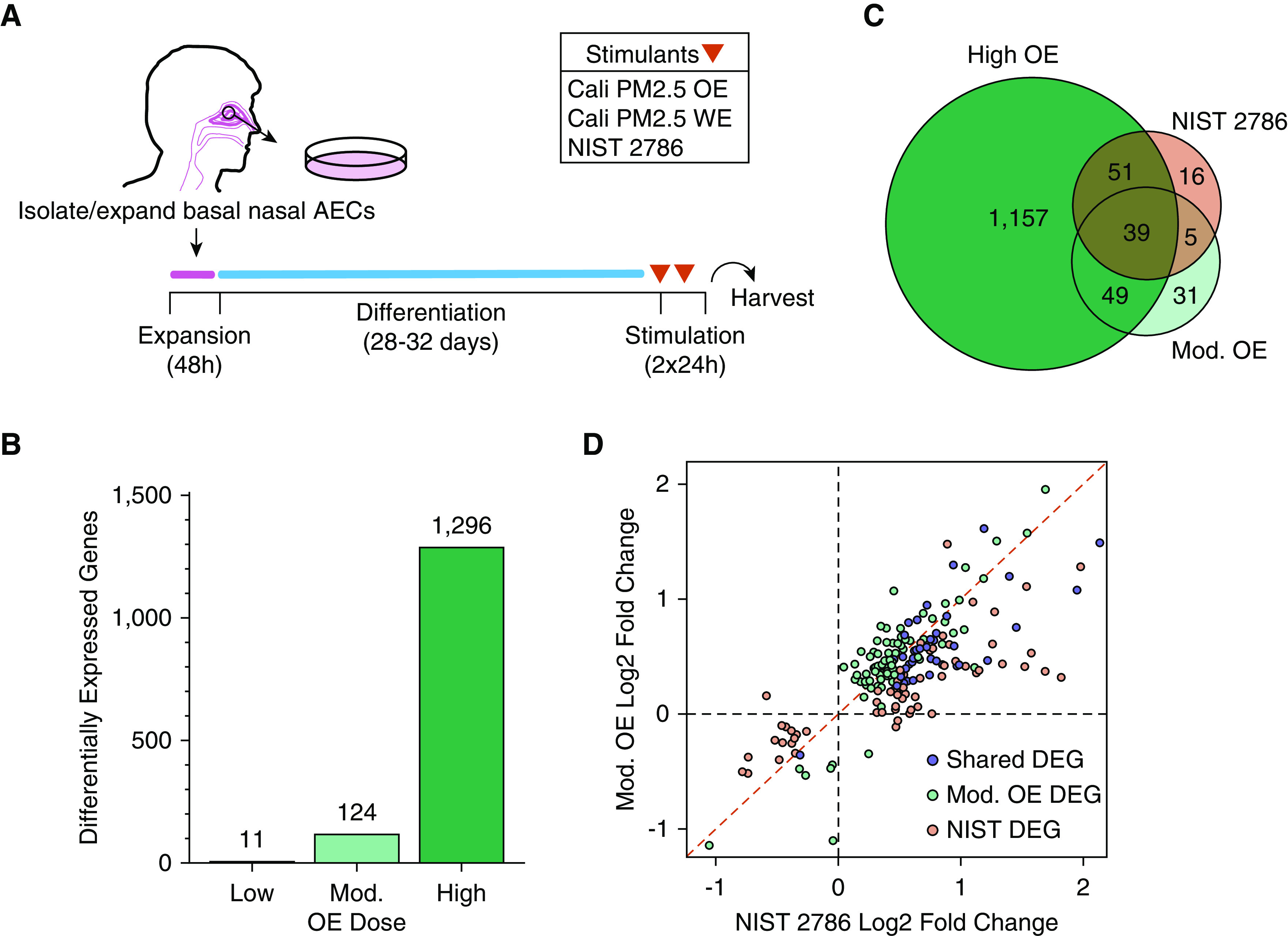
OE drives the airway epithelial response to PM2.5 stimulation. (A) Schematic of experimental design starting from isolation of nasal airway epithelial cells (AECs) to mucociliary air–liquid interface culture differentiation and acute (48 h) stimulation with PM2.5 extracts (Cali PM2.5) or National Institute of Standards and Technology (NIST) 2786. (B) Number of differentially expressed genes (DEGs) after stimulation with three separate PM2.5 OE doses. (C) Venn diagram showing shared and unique DEGs across moderate (Mod.) OE dose, high OE dose, and NIST 2786. (D) Comparison of effect sizes between the union of DEGs from moderate OE and NIST 2786 stimulations.
Moderate Levels of PM2.5 Organic Extract Trigger Transcriptional Upregulation of Inflammatory and Mucus Secretory Cell Gene Expression
On the basis of our dose–response experiment, we selected our moderate OE dose (0.45 μg/cm2) for further stimulation experiments to elucidate the biological effects of OE exposure on the airway epithelium. We performed this moderate OE dose stimulation on mature airway epithelial cultures from an additional 7 donors (for 12 donors total), followed by whole transcriptome RNA-seq, increasing our power to characterize the genome-wide response. Paired differential gene expression analysis of these donors identified 424 DEGs consisting of 281 upregulated and 143 downregulated genes (Table E2). Principal component analysis of these DEGs verified consistency in this response across donors (Figure 3A). Examining this gene set we found that the most highly OE-induced gene was the xenobiotic metabolizing cytochrome P450 enzyme, CYP1A1 (LFC, 6.21; Figure 3B). CYP1A1 is known to be induced by PAHs, a confirmed component of our OE extract, through aryl hydrocarbon receptor (AHR)-mediated transcriptional activation (20). Consistent with OE activation of the AHR receptor, we observed significant induction of other AHR-inducible xenobiotic metabolizing enzymes, including CYP1B1 and genes encoding the AHR battery enzymes NQO1 (NAD(P)H quinone dehydrogenase 1) and ALDH3A1 (aldehyde dehydrogenase 3 family member A1) (21).
Figure 3.
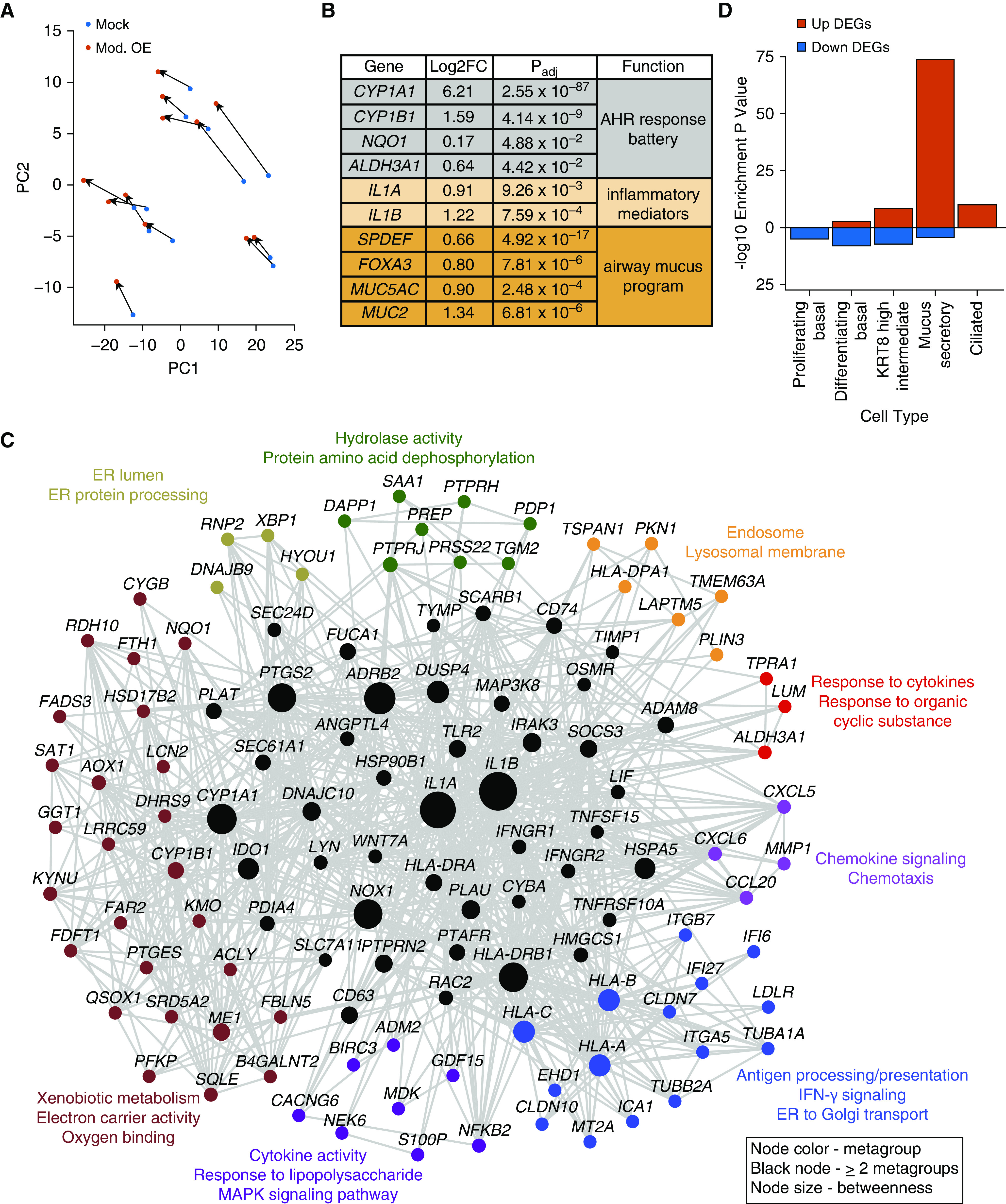
PM2.5 OE stimulates an inflammatory network that induces a mucus secretory cell program. (A) Principal component (PC) analysis on 424 DEGs from moderate OE and mock comparison. (B) Table of select DEGs induced by moderate OE stimulation. (C) Functional gene network of moderate OE-upregulated DEGs. Node colors refer to metagroup membership, and black nodes represent genes shared between two or more metagroups. Node size represents betweenness across entire network. (D) Enrichment of moderate OE DEGs for cell type–specific marker genes. AHR = aryl hydrocarbon receptor; ER = endoplasmic reticulum.
Interestingly, we found that IL1A and IL1B (IL-1α and -1β) inflammatory cytokines were both among the most highly upregulated genes with OE stimulation (LFC, 0.91 and 1.22, respectively; Figure 3B), indicating that OE stimulation triggered epithelial inflammation. To interrogate this and other biological processes underlying OE-upregulated genes, we performed gene set enrichment analyses using FGNet (17). Genes were filtered and organized based on the relationship between, and similarity of, significant enrichment terms, thereby constructing an OE functional gene response network (Figure 3C and Table E3). This OE response network was composed of eight significant clusters (metagroups). These metagroups included several defined by functional terms relating to an inflammatory response, including cytokine-mediated signaling, IFN-γ signaling, antigen processing and presentation, response to liposaccharide, and chemotaxis. The CYP1A1, IL1A, and IL1B genes were shared by multiple metagroups and exhibited especially high betweenness centrality, indicating the likely critical nature of these genes in the transcriptional response to OE stimulation.
We also found significant enrichments for Golgi subcompartment and endoplasmic reticulum (ER) protein processing terms (Figure 3C and Table E3), both of which contained genes involved in the production of mucin proteins. Furthermore, we found that one of the two primary gel-forming mucin genes in the airway, MUC5AC (mucin 5AC), was strongly induced by OE stimulation, as was another gel-forming mucin, MUC2 (mucin 2), which is normally expressed in the gut and lowly expressed in the airway (LFC, 0.90 and 1.34, respectively; Table E2) (22). Together, these results suggest that the number and/or function of mucus secretory cells within the epithelium is increasing with OE exposure. To more comprehensively investigate whether OE exposure DEGs are enriched in genes expressed by mucus secretory or other epithelial cell types, we tested all moderate OE DEGs for enrichment among a catalog of epithelial cell type marker genes generated from scRNA-seq of the human airway epithelium (Figure 3D and Table E4). Although several cell type enrichments were observed, the most significant enrichment was for mucus secretory cell marker genes among OE-upregulated DEGs (Padj = 1.19 × 10−74).
To confirm a functional relationship among mucus secretory genes that were upregulated by moderate OE, we tested whether any of these genes were coexpressed in the data set (Figure E2). Interestingly, this analysis resulted in a mucus secretory coexpression module containing MUC5AC as well as two of the BPI-fold containing genes (BPIFA1 and BPIFB1), which encode antimicrobial defense proteins produced by secretory epithelial cells (23). This cluster also contained XBP1 (X-box binding protein 1), a cellular stress response transcription factor (TF) that initiates ER programs to accommodate accumulation of secretory proteins (24). Most interestingly and perhaps most central to this cluster was SPDEF (SAM pointed domain containing ETS transcription factor), a canonical TF reported to drive mucus production and metaplasia in response to the type 2 cytokine, IL-13 (25). Although found in a different cluster, we also observed upregulation of FOXA3 (forkhead box A3), an additional mucus metaplasia TF functionally related to SPDEF (26). Together, these results suggest that moderate OE exposure triggers AHR signaling and IL-1–mediated inflammation, which in turn initiates a mucus secretory program in the airway epithelium.
High Levels of PM2.5 Organic Extract Cause Dysregulation of Mucociliary Transcriptional Programs
On the basis of the number of DEGs induced, our OE dose–response analysis suggested that the highest OE dose (4.5 μg/cm2) induced a more extensive epithelial gene expression response than the moderate dose. Using Enrichr (16), we performed gene set enrichment analysis of upregulated and downregulated DEGs across moderate and high OE dose to identify shared terms as well as those unique to the high dose (Table E5). Most of the significant enrichments for the moderate OE dose DEGs (from n = 12 data set) were also enrichments for the high OE dose DEGs (from n = 5 data set). In addition, we found that of the 169 upregulated DEGs that were shared between doses, 164 were further upregulated with the high dose, suggesting that a stronger stimulus exacerbates dysregulated expression of these genes (Figure 4A and Table E6). In particular, we noticed that genes central to the moderate OE dose–response (IL1A, IL1B, and CYP1A1) were further upregulated with the high OE compared with moderate OE dose (Figure 4B and Table E6). In addition, although the enrichment of high dose DEGs within mucus secretory cell markers was significant (Padj = 8.27 × 10−100; Figure 4C and Table E4), as was observed with the moderate OE dose, the high dose exhibited even higher average levels of upregulation for master mucus secretory TFs (SPDEF, FOXA3, and XBP1) (Figure 4B and Table E6). Moreover, this activation of mucus secretory genes with high OE dose stimulation was matched by repression of SCGB1A1 (secretoglobin family 1A member 1) (LFC, −0.92; Padj = 1.13 × 10−3; Figure 4B and Table E2), the marker gene of club secretory cells. This is consistent with mucus metaplasia of club secretory cells, as has been reported for IL-13–induced mucus metaplasia (27). In line with this mucus metaplastic signal, MUC5AC expression was increased with high OE dose, similar to its induction with moderate OE dose (Figure 4D and Table E2). However, we observed a trend toward decreased expression of the other major gel-forming mucin, MUC5B (mucin 5B), only with the high OE dose (LFC, −0.79; Padj = 1.0 × 10−1). The viscoelastic properties of airway mucus are largely determined by the ratio of levels of the “healthy” homeostatic mucin MUC5B and the inflammation-induced MUC5AC (28–32). It is notable that a significant increase in the MUC5AC/MUC5B ratio with moderate OE dose (P = 3.19 × 10−2) was increased further with the high OE dose (P = 2.68 × 10−2; Figure 4D), suggesting an important relationship between OE stimulation and alteration of mucus properties. Accompanying this change in the gel-forming mucin ratio was a high OE dose–specific induction of genes involved in the O-linked glycosylation of mucins, including five members of the GalNac-T family of glycosyltransferase enzymes (GALNT3, 5, 6, 7, and 12) and N-acetylglucosaminyltransferase enzyme GCNT3 (Table E2), which likely contribute to modifications in the viscoelastic properties of mucus induced by the OE-stimulated airway epithelium (31).
Figure 4.
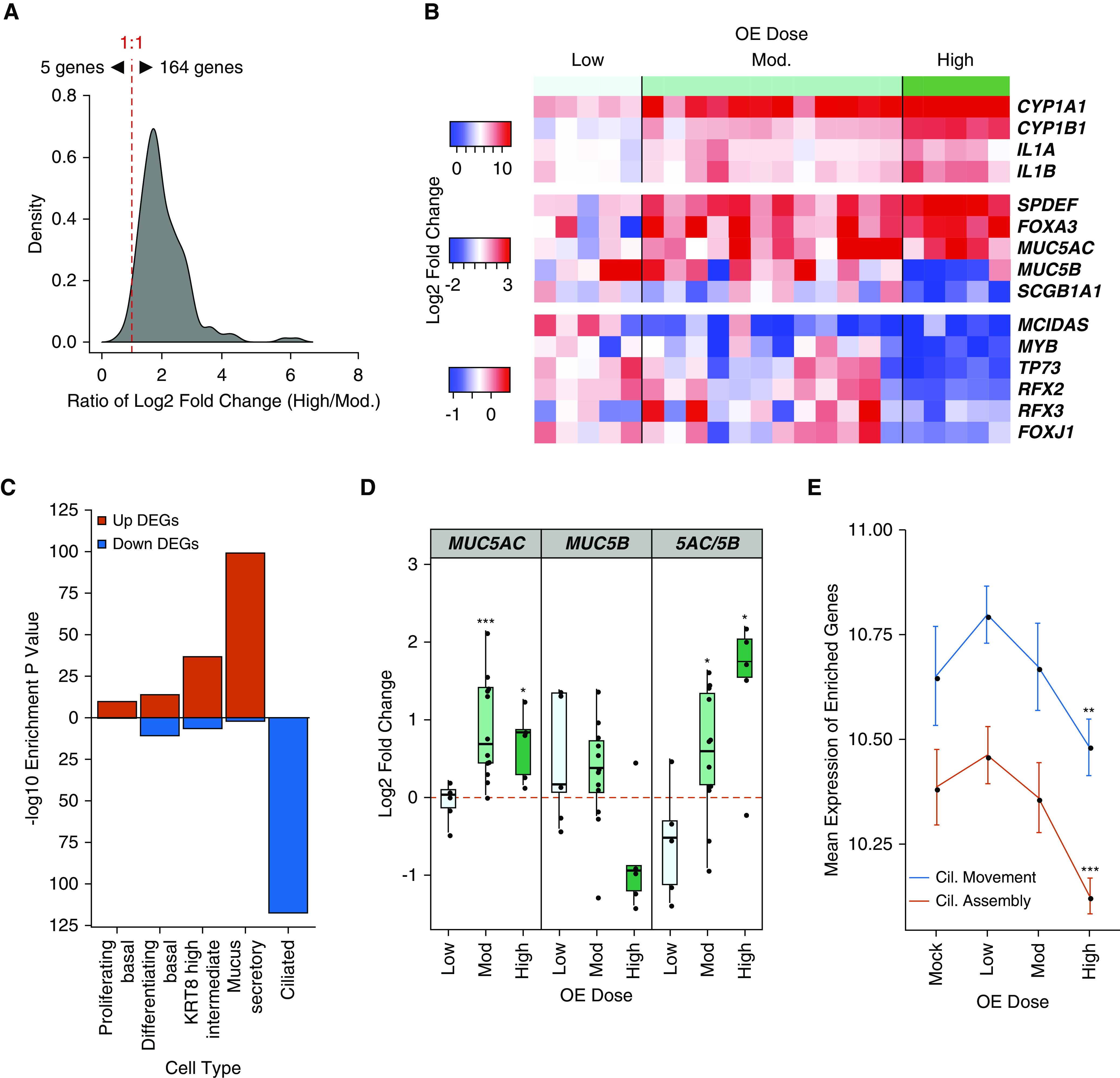
High OE dose induces transcriptional remodeling of the airway epithelium. (A) Histogram of the ratio of log2 fold change (High/Mod.) for shared upregulated DEGs between moderate and high OE dose stimulation. (B) Heat map of the log2 fold change in expression between mock and OE dose-stimulated donor culture pairs (columns) for key response genes across OE dose. (C) Strength of enrichment for high OE dose DEGs within groups of cell type–specific marker genes. (D) First and second panels: log2 fold change of MUC5AC (mucin 5AC) and MUC5B (mucin 5B) across OE dose (*Padj < 0.05, and ***Padj < 0.001); third panel: ratio of log2 fold changes (MUC5AC/MUC5B). Statistically significant differences in ratio compared with mock-treated cultures (*P < 0.05) were determined by two-tailed, paired t test. (E) Mean expression of cilia (Cil.) assembly and cilia movement genes across mock and OE dose. Statistically significant differences in module gene expression between mock and OE dose (**P < 0.01 and ***P < 0.001) were determined by two-tailed, paired t test. For all plots, mock and moderate OE, n = 12 independent donors; low and high OE, n = 5 independent donors.
Enrichment analysis of downregulated genes yielded multiple terms specific to the high OE dose, the majority of which were related to the development and function of cilia (Table E5). Furthermore, in contrast to moderate OE DEGs, we found highly significant enrichment of high OE dose–downregulated DEGs within ciliated cell marker genes (Padj = 3.57 × 10−118; Figure 4C and Table E4). Mean expression of genes within the ciliary movement (P = 3.05 × 10−3) and ciliary assembly (P = 4.20 × 10−4) GO terms exhibited significant downregulation in expression only with high OE dose (Figure 4E). Supporting this loss in ciliated cell gene expression, we found canonical TFs controlling ciliated cell differentiation (MCIDAS, MYB, TP73, RFX2, RFX3, and FOXJ1) were consistently decreased with high OE dose (Figure 4B and Table E2) (33–38). Together, these data show that high OE dose mediates the same mucus secretory changes observed with moderate OE dose but also downregulates ciliary expression programs.
Chronic OE Stimulation Results in Mucus Secretory Remodeling of the Airway Epithelium
To investigate whether transcriptional reprogramming induced by high OE dose culminates in demonstrative changes to protein expression, cellular composition, and in the mucus secretions of the airway epithelium, we chronically stimulated mature airway epithelial cultures with a high dose of OE (5 days of repeat exposure). As a benchmark for metaplastic expansion of mucus secretory cells, we also performed chronic stimulations of additional cultures from the same donors with the cytokine IL-13. We found that both chronic OE and IL-13 stimulation resulted in upregulation of MUC5AC (OE LFC, 1.72; P = 1.27 × 10−2; IL-13 LFC, 3.71; P = 8.86 × 10−3) paired with an increase in SPDEF expression (OE LFC, 0.84; P = 3.19 × 10−2; IL-13 LFC, 2.91; P = 2.54 × 10−2), characteristic of mucus metaplasia (Figure E3A). Moreover, these stimuli resulted in decreased expression of SCGB1A1 (OE LFC, −1.51; P = 2.94 × 10−2; IL-13 LFC, −1.74; P = 4.09 × 10−2) and a trend toward decreased MUC5B expression (OE LFC, −0.72; P = 9.74 × 10−2; IL-13 LFC, −5.24; P = 2.55 × 10−2), as well as downregulation of FOXJ1 (forkhead box J1) (OE LFC, −0.36; P = 1.92 × 10−2; IL-13 LFC, −1.15; P = 6.34 × 10−3). These results support that OE induces a mucus metaplasia process similar to IL-13 (Figure E3A).
To confirm these results histologically, we examined cultures with alcian blue–periodic acid-Schiff staining to identify mucin-positive cells and found higher numbers of mucin-positive cells in chronic OE-stimulated versus mock-stimulated cultures for three of the four donor pairs examined (average fold change [FC] = 2.01; P = 6.52 × 10−2; Figure 5A). Using immunofluorescent labeling for MUC5AC and MUC5B, we found an increase in MUC5AC+ cells with chronic OE stimulation for all four donors (FC = 2.88; P = 3.19 × 10−2; Figure 5B), suggesting that this mucin was the primary source of increased alcian blue–periodic acid-Schiff staining with chronic OE stimulation. In contrast, cultures for all four donors exhibited a decrease in MUC5B+ cells with chronic OE exposure (FC, 0.74; P = 8.84 × 10−2; Figure 5B). Consistent with these changes in mucin expression being mediated by the action of SPDEF, we found that chronic OE stimulation resulted in a 1.5-fold increase in SPDEF protein levels (P = 1.07 × 10−2; Figure 5C).
Figure 5.
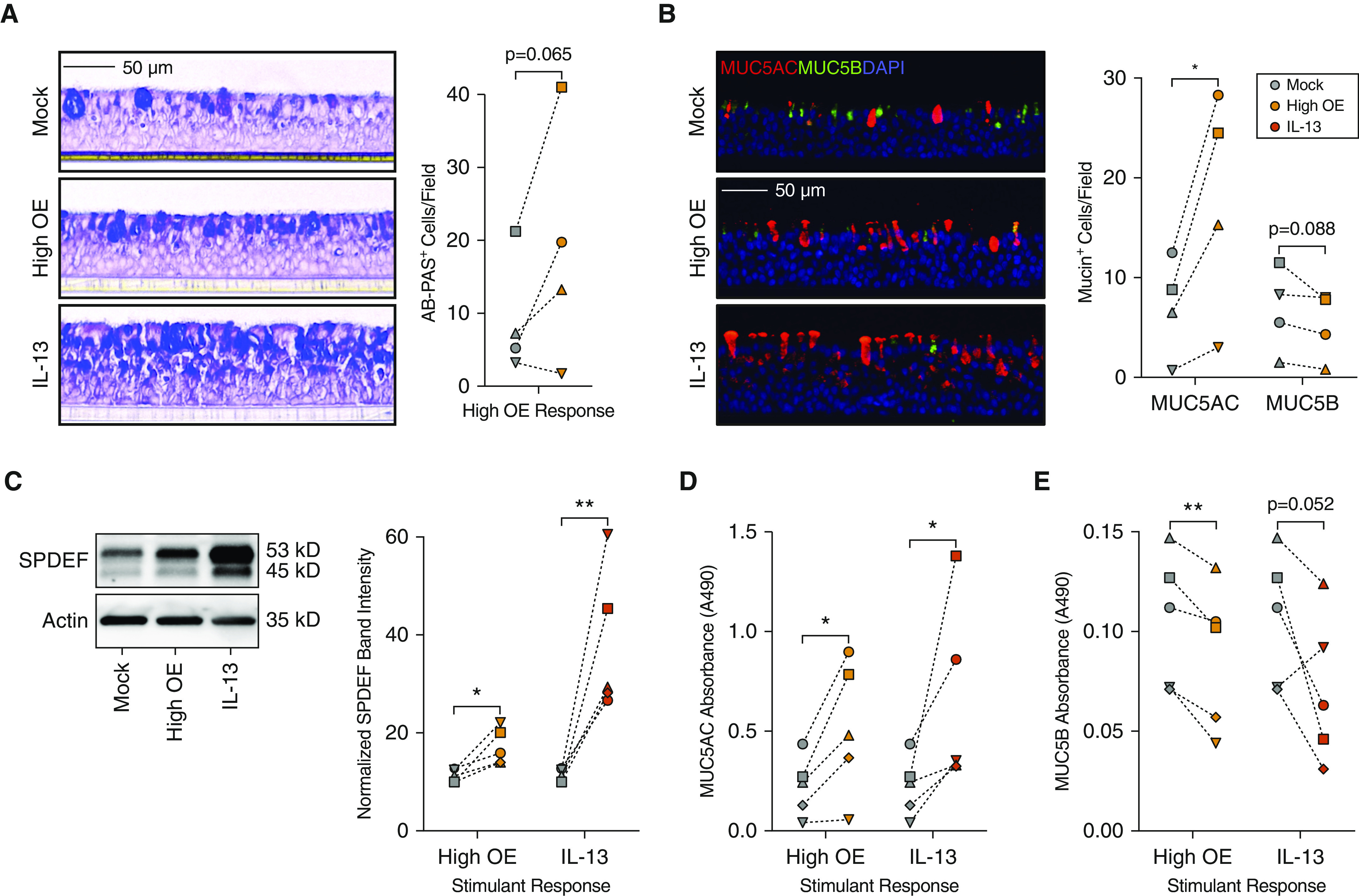
Chronic OE stimulation increases the number of MUC5AC+ cells and MUC5AC secretion. (A) Left panel: alcian blue–periodic acid-Schiff (AB-PAS) staining of culture histological sections for mucopolysaccharides/mucins. Right panel: quantification of AB-PAS+ cells. Scale bar, 50 μm. (B) Left panel: immunofluorescence labeling of culture histological sections for gel-forming mucins MUC5AC (red) and MUC5B (green). Nuclei were stained with DAPI (blue). Right panel: quantification of MUC5AC+ and MUC5B+ cells. Scale bar, 50 μm. Images for A and B are representative of labeling performed in cultures from four donors. (C) Left panel: Western blot analysis of total protein for SPDEF (SAM pointed domain containing ETS transcription factor) expression. Images are representative of data from five independent donors. Right panel: quantification of SPDEF Western blot band intensity normalized to actin expression as a loading control. (D and E) Colorimetric ELISA absorbance readings at 490 nm (A490) for MUC5AC (D) and MUC5B (E) detection in ATP-induced secretions of cultures from five donors. Statistically significant differences (*P < 0.05 and **P < 0.01) were determined by paired, one-tailed t test (on the basis of direction suggested by transcriptomic analyses). Independent donors are represented by data point shape, and connecting lines indicate paired mock- and OE- or IL-13–treated cultures.
To examine the effect of these mucus secretory cell changes on mucin secretion, we used an ATP-induced mucus secretion assay (39), followed by examination of mucin content in these secretions with ELISA assays. Chronic OE induced a 2.2-fold increase in total secreted MUC5AC (P = 1.54 × 10−2), comparable with the robust induction in MUC5AC secretion stimulated by IL-13 (FC, 3.87; P = 3.85 × 10−2; Figure 5D). Moreover, we observed a significant decrease in the secretion of MUC5B with chronic OE (FC, 0.81; P = 3.59 × 10−3), again similar to the effect mediated by IL-13 stimulation (FC, 0.70; P = 5.20 × 10−2; Figure 5E). These data demonstrate that chronic OE stimulation induces simultaneous expansion and reduction of MUC5AC+ and MUC5B+ mucus secretory cells, respectively, resulting in corresponding changes in secretion of these mucins. Together, these effects are consistent with the observed increase in SPDEF protein levels with chronic OE stimulation.
We next investigated the consequences of chronic OE stimulation on ciliated cells by immunofluorescence labeling of ciliated cells for the transcription factor FOXJ1 and for cilia with acetylated α-tubulin. Confocal microscopy was used to produce maximum projections of labeled cultures and revealed that ciliary structure and abundance were relatively consistent across all conditions (Figure 6). We did note that the acetylated α-tubulin staining (cilia) appeared weaker in OE-stimulated cultures than in either mock or IL-13–stimulated cultures. However, OE-stimulated cultures displayed a 75% average decrease in FOXJ1+ nuclei compared with mock-treated cultures (P = 1.20 × 10−2; Figures 6 and E3B). Interestingly, this effect was more pronounced than the nonsignificant 40% average decrease in FOXJ1+ nuclei observed for IL-13–treated cultures (P = 1.02 × 10−1). The lack of nuclear FOXJ1 in ciliated cells from chronic OE-stimulated cultures is consistent with the decrease in ciliated cell gene expression observed in our transcriptome analysis.
Figure 6.
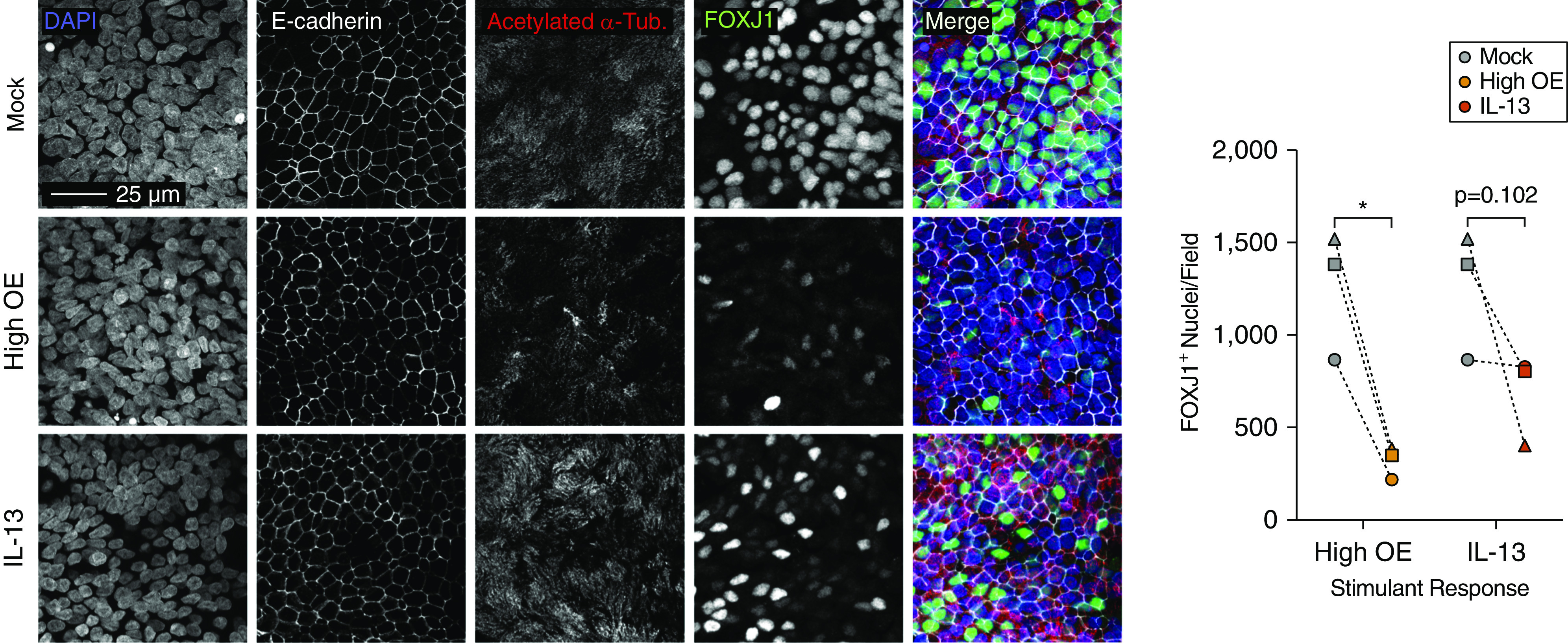
Chronic OE impairs FOXJ1 (forkhead box J1) expression in ciliated cells. Left panel: confocal fluorescence microscopy maximum projections of intact epithelial cultures labeled for E-cadherin (white), acetylated α-tubulin (red), and FOXJ1 (green). Nuclei were stained with DAPI (blue). Images are representative of labeling performed in cultures from three donors. Scale bar, 25 μm. Right panel: quantification of FOXJ1+ nuclei. Statistically significant differences (*P < 0.05) were determined by paired, one-tailed t test (on the basis of direction suggested by transcriptomic analyses). Independent donors are represented by data point shape and connecting lines indicate paired mock-treated and OE- or IL-13–treated cultures.
Discussion
Decades of epidemiologic research have confirmed the detrimental influence of PM2.5 exposure on the development and exacerbation of lung diseases. Yet, the cellular and molecular mechanisms, as well as the chemical components through which PM2.5 drives these poor outcomes, are incompletely understood. As the first and primary interface through which the respiratory tract interacts with PM2.5 and other inhaled matter, the nasal airway epithelium acts as a sentinel, capable of directing responses by other structural and immune cells of the lower airways and lung. Building on prior studies investigating candidate mechanisms involved in airway epithelial responses to PM2.5, we agnostically and comprehensively defined airway epithelial responses to the organic components of PM2.5 using whole transcriptome analysis and further examined whether chronic OE exposure modulates cellular composition and function of the epithelium.
Our study represents one of the first to examine molecular responses of the nasal epithelium to the organic and water-soluble components of PM2.5, independently. Although it is difficult to determine how closely the concentrations we tested relate to in vivo exposures, we attempted to use concentrations that were not toxic and did not have any significant effect on epithelial integrity, yet that influenced the molecular function of the epithelium. The limited quantities of the sample prevented us from identifying a concentration of PM2.5 WE which altered molecular function of the epithelium. Despite this, considering heavy metals present in PM2.5 WE are known to trigger epithelial responses, we anticipate higher WE concentrations would mediate an epithelial response of sorts. In contrast, all tested concentrations of PM2.5 OE altered epithelial gene expression in a dose-dependent manner, providing dosage guidelines for future primary epithelial exposure studies. Our results in primary airway epithelial cells are in line with and extend previous studies identifying OEs and PAHs as the most harmful components of PM2.5 for transformed lung epithelial cells (40, 41).
Rather than just a simple perturbation of xenobiotic metabolism pathways, we find that the organic components of PM2.5 modulate the expression of many genes and networks, leading to changes in the cellular composition of the airway epithelium. The high network interconnectedness of CYP1A1 suggests that activation of AHR signaling by PM2.5 PAHs is an early and central event underlying the larger epithelial response to PM2.5 observed. In addition, our network analysis also identified IL1A and IL1B as hub genes in the PM2.5 response. These potent cytokines were part of, and the likely drivers of, an intense inflammatory expression profile we found to be activated by PM2.5 OE. Prior studies have established IL-1β, and to a lesser extent IL-1α, as major drivers in chronic inflammatory lung diseases, including asthma and chronic obstructive pulmonary disease (COPD) (42). For example, IL-1β secretion has been found to be increased in the sputum of patients with COPD and asthma at baseline and further increased during acute exacerbations (43–46). In addition, expression of IL1B in sputum and serum is positively correlated with systemic inflammation and negatively correlated with pulmonary function (forced expiratory volume in 1 second percent predicted) in patients with COPD (46, 47). In asthma, IL-1α and IL-1β are reported to independently regulate immune subtypes of disease characterized by neutrophilic and eosinophilic inflammation, respectively (48). Together, these data make clear that the IL-1 inflammatory axis is involved in respiratory inflammation and disease, and our work suggests that extensive exposure to air pollution PM2.5 may underlie activation of these mechanisms in many patients.
Perhaps most interestingly, our study of primary mucociliary epithelial cultures allowed us to observe dramatic PM2.5 effects on mucus secretory cells. Importantly, we found the inflammation-related mucin, MUC5AC, was strongly upregulated by PM2.5 OE. Moreover, the MUC5AC/MUC5B gel-forming mucin ratio, which is known to be critical for the physical properties of airway mucus, was strongly upregulated in OE-stimulated cultures. In addition, the activation of an array of enzymes involved in O-glycosylation of mucins further supported the likely highly modified nature of mucus produced by the PM2.5-stimulated airway. We also found that PM2.5 induced the XBP1 transcription factor and target ER chaperone genes, which are activated by cellular stress caused by increased mucin production. Enrichment analysis of upregulated DEGs within mucus secretory cell marker genes revealed strong evidence that this increase in the expression of mucin production and secretion pathways resulted from an increase in the proportion of mucus secretory cells within the epithelium. The strong activation of both SPDEF and FOXA3 expression, coupled with a loss in SCGB1A1 expression, suggests that the generation of new mucus secretory cells may occur through mucus metaplasia of club secretory cells, not dissimilar to the mucus metaplasia process stimulated by the type 2 cytokine IL-13 (27). We note recent work by Chen and colleagues finding that either IL-1α or IL-1β stimulation can induce mucus cell metaplasia in mice and that SPDEF is a critical downstream effector in this process (49). We speculate that the IL-1 cytokine-driven OE inflammatory response network triggers SPDEF, FOXA3, and XBP1 production, which form a regulatory axis that drives mucus metaplasia of the airway epithelium.
Our dose–response analysis strongly supports worldwide public health efforts to mitigate ambient air pollution levels. In fact, not only do we find the magnitude of gene expression alteration increases with higher PM2.5 OE doses, but the number of epithelial genes whose expression is affected also scales with the PM2.5 OE dose. Importantly, we found the highest OE dose tested was sufficient to strongly downregulate ciliated cell gene expression, suggesting OE-mediated dysfunction of ciliated cells. Supporting this, we observed a dramatic decrease in the expression of transcriptional regulators of ciliated cell specification and ciliogenesis (e.g., MCIDAS, FOXJ1, and MYB). Moreover, our chronic OE exposure model provides evidence for the epithelial remodeling suggested by our whole transcriptome analysis. Epithelial remodeling included an increase in MUC5AC+ cells and a decrease in MUC5B+ cells, matched by a corresponding change in secretion of these mucins. These changes also occur with IL-13 stimulation, revealing similarities in the epithelial response to these two stimuli. Although we did not observe a decrease in ciliated cell frequency, we did find that OE induces a decrease in nuclear FOXJ1 labeling among ciliated cells. Loss of nuclear FOXJ1 may underlie the downregulation in ciliated cell gene expression we observed in our whole transcriptome analysis. Although these ciliated cells appeared structurally normal, continued OE exposure may eventually impede the maintenance of ciliated cells and result in loss of this critical cell type or block ciliated cell regeneration in response to injury. On the basis of these results, we hypothesize that long-term exposure to PM2.5 could lead to decreased mucociliary clearance and increased mucostasis/mucus plugging in vivo. In fact, the mechanisms we outline here could partially explain the strong inverse correlation between PM2.5 and forced expiratory volume in 1 second (2), as well as the increased susceptibility to respiratory infections among subjects with high exposures to PM2.5 (50).
The PM2.5-induced changes to the airway epithelium we observed approach the level of an airway endotype (pathobiological mechanism), not dissimilar to that exhibited by individuals with type 2–high asthma (51, 52). These similarities include the induction of SPDEF and accompanying MUC5AC+ mucus metaplasia, loss of SCGB1A1 expression, as well as downregulation in ciliated cell gene expression. However, type 2 epithelial disease is driven through IL-13, whereas it appears that IL-1 cytokines drive PM2.5-mediated epithelial dysfunction. Moreover, differences in the secretory proteins upregulated and downregulated by PM2.5 versus IL-13 (e.g., BPIFA1, BPIFA2, and MMP2) and many other genes suggest the nature of the metaplastic mucus secretory cells and other cell states differs between these stimuli as well. Importantly, these differences suggest a highly plastic airway epithelium capable of remodeling to multiple cell compositions and containing different cell states, likely underlying the complexity in mechanisms driving common airway disease. In the future, our PM2.5 response profile could be used as a biomarker in nasal gene expression studies to identify subjects with excessive PM2.5 exposure and possibly a PM2.5 airway endotype. Identification of these subjects in expression studies would allow direct in vivo comparison of endotype mechanisms as well as precise determination of the clinical consequences of PM2.5 exposure.
There are several limitations to the present study and multiple areas of investigation that should be pursued based on our findings. Although our results suggest that PM2.5 OE activates cellular remodeling of the epithelium, these changes will need to be replicated and investigated in depth by methods such as lineage tracing and scRNA-seq. Moreover, longer-term chronic PM2.5 stimulation experiments will need to be conducted to model effects on the epithelium occurring in subjects with years of PM2.5 exposure. Finally, although our study involved more human primary epithelial donors than any in vitro PM2.5 study to date, in the future, larger studies will be needed to determine if PM2.5 response mechanisms are enhanced or different among subjects with respect to variables such as race/ethnicity, age, sex, and disease status.
In summary, we found that the organic components of PM2.5 are sufficient to drive broad-scale transcriptomic, cellular, and mucin secretory changes to the human airway epithelium. These changes appear to emanate from PM2.5 induction of AHR signaling and IL-1–driven inflammation of the epithelium, resulting in mucus secretory changes and ciliated cell dysfunction. These effects on the epithelium appear to occur through alteration of core transcription factors in the specification of these cell types. These results provide potential mechanisms underlying PM2.5 exposure effects on respiratory health and provide targetable pathways for further study and clinical intervention in air pollution–mediated or -exacerbated lung and airway diseases.
Acknowledgments
Acknowledgment
The authors thank the Genomics and Microarray Core Facility at the University of Colorado Anschutz Medical Campus for sequencing services, the NJH Regenerative Medicine and Genome Editing Program for access to scRNA-seq data sets, and Dave Heinz (NJH Pathology Laboratory) for assistance with tissue processing and staining. The authors also thank Alicia Adams and the California Air Resources Board for supplying air pollution filters that served as the source of PM2.5 samples used in this study. The authors thank the patients, families, recruiters, health care providers, and community clinics for their participation. In particular, the authors thank Sandra Salazar for her support as the GALAII/SAGE II study coordinator.
Footnotes
Supported by U.S. National Institutes of Health (NIH) grants R01MD010443, R01HL128439, R01HL135156, and P01HL132821 and Department of Defense grant W81WH-16-2-0018 (M.A.S.). Also supported by the Sandler Family Foundation; the American Asthma Foundation; the Robert Wood Johnson Foundation Amos Medical Faculty Development Program; Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II; NIH grants R01HL117004, R01HL128439, R01HL135156, 1X01HL134589, R01HL141992, and R01HL141845; NIH and Environmental Health Sciences grants R01ES015794 and R21ES24844; the National Institute on Minority Health and Health Disparities grants P60MD006902, RL5GM118984, R01MD010443, and R56MD013312; the Tobacco-Related Disease Research Program under Award Numbers 24RT-0025 and 27IR-0030; and the National Human Genome Research Institute U01HG009080 (E.G.B.).
Author Contributions: Conception and experimental design: M.T.M., S.P.S., S.-H.C., J.L.E., C.L.R., S.S.O., and M.A.S. Acquisition of data: M.T.M., S.-H.C., J.L.E., C.L.R., M.C., C.E., V.M., J.R.E., S.S.O., J.R.-S., E.K.V., E.G.B., and M.A.S. Analysis of data: M.T.M., S.P.S., S.-H.C., N.D.J., B.S., J.R.E., and S.S.O. Interpretation of work: M.T.M., S.P.S., S.-H.C., J.L.E., K.C.G., N.D.J., S.S.O., E.K.V., E.G.B., and M.A.S. Manuscript preparation for intellectual content: M.T.M., S.P.S., S.-H.C., J.L.E., K.C.G., N.D.J., S.S.O., J.R.-S., E.G.B., and M.A.S.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0454OC on April 10, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.7 million deaths annually linked to air pollution. Cent Eur J Public Health. 2014;22:53, 59. [PubMed] [Google Scholar]
- 2.Gauderman WJ, Urman R, Avol E, Berhane K, McConnell R, Rappaport E, et al. Association of improved air quality with lung development in children. N Engl J Med. 2015;372:905–913. doi: 10.1056/NEJMoa1414123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Pan J, Zhang H, Shi C, Li G, Peng Z, et al. Short-term exposure to ambient air pollution and asthma mortality. Am J Respir Crit Care Med. 2019;200:24–32. doi: 10.1164/rccm.201810-1823OC. [DOI] [PubMed] [Google Scholar]
- 4.Iskandar A, Andersen ZJ, Bønnelykke K, Ellermann T, Andersen KK, Bisgaard H. Coarse and fine particles but not ultrafine particles in urban air trigger hospital admission for asthma in children. Thorax. 2012;67:252–257. doi: 10.1136/thoraxjnl-2011-200324. [DOI] [PubMed] [Google Scholar]
- 5.Cohen AJ, Brauer M, Burnett R, Anderson HR, Frostad J, Estep K, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet. 2017;389:1907–1918. doi: 10.1016/S0140-6736(17)30505-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu CW, Lee TL, Chen YC, Liang CJ, Wang SH, Lue JH, et al. PM2.5-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-κB-dependent pathway. Part Fibre Toxicol. 2018;15:4. doi: 10.1186/s12989-018-0240-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X, Liu J, Zhou J, Wang J, Chen C, Song Y, et al. Urban particulate matter (PM) suppresses airway antibacterial defence. Respir Res. 2018;19:5. doi: 10.1186/s12931-017-0700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schantz MM, Cleveland D, Heckert NA, Kucklick JR, Leigh SD, Long SE, et al. Development of two fine particulate matter standard reference materials (<4 μm and <10 μm) for the determination of organic and inorganic constituents. Anal Bioanal Chem. 2016;408:4257–4266. doi: 10.1007/s00216-016-9519-7. [DOI] [PubMed] [Google Scholar]
- 9.Leclercq B, Platel A, Antherieu S, Alleman LY, Hardy EM, Perdrix E, et al. Genetic and epigenetic alterations in normal and sensitive COPD-diseased human bronchial epithelial cells repeatedly exposed to air pollution-derived PM2.5. Environ Pollut. 2017;230:163–177. doi: 10.1016/j.envpol.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Boublil L, Assémat E, Borot MC, Boland S, Martinon L, Sciare J, et al. Development of a repeated exposure protocol of human bronchial epithelium in vitro to study the long-term effects of atmospheric particles. Toxicol In Vitro. 2013;27:533–542. doi: 10.1016/j.tiv.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 11.Huff RD, Carlsten C, Hirota JA. An update on immunologic mechanisms in the respiratory mucosa in response to air pollutants. J Allergy Clin Immunol. 2019;143:1989–2001. doi: 10.1016/j.jaci.2019.04.012. [DOI] [PubMed] [Google Scholar]
- 12.Cromie M, Saef B, Montgomery MT, Rios C, Cho S-H, Everman JL, et al. Organic components of air pollution particulate matter 2.5 (PM2.5) elicit changes in the airway epithelial transcriptome of asthmatic children [abstract] Am J Respir Crit Care Med. 2019;199:A2142. [Google Scholar]
- 13.Neophytou AM, White MJ, Oh SS, Thakur N, Galanter JM, Nishimura KK, et al. Air pollution and lung function in minority youth with asthma in the GALA II (Genes-Environments and admixture in Latino Americans) and SAGE II (study of African Americans, asthma, genes, and environments) studies. Am J Respir Crit Care Med. 2016;193:1271–1280. doi: 10.1164/rccm.201508-1706OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, et al. Early-life air pollution and asthma risk in minority children: the GALA II and SAGE II studies. Am J Respir Crit Care Med. 2013;188:309–318. doi: 10.1164/rccm.201302-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everman JL, Rios C, Seibold MA. Utilization of air-liquid interface cultures as an in vitro model to assess primary airway epithelial cell responses to the type 2 cytokine i-13. Methods Mol Biol. 2018;1799:419–432. doi: 10.1007/978-1-4939-7896-0_30. [DOI] [PubMed] [Google Scholar]
- 16.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aibar S, Fontanillo C, Droste C, De Las Rivas J. Functional Gene Networks: R/Bioc package to generate and analyse gene networks derived from functional enrichment and clustering. Bioinformatics. 2015;31:1686–1688. doi: 10.1093/bioinformatics/btu864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontanillo C, Nogales-Cadenas R, Pascual-Montano A, De las Rivas J. Functional analysis beyond enrichment: non-redundant reciprocal linkage of genes and biological terms. PLoS One. 2011;6:e24289. doi: 10.1371/journal.pone.0024289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47:D330–D338. doi: 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Q. Induction of CYP1A1: the AhR/DRE paradigm: transcription, receptor regulation, and expanding biological roles. Curr Drug Metab. 2001;2:149–164. doi: 10.2174/1389200013338603. [DOI] [PubMed] [Google Scholar]
- 21.Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- 22.Thai P, Loukoianov A, Wachi S, Wu R. Regulation of airway mucin gene expression. Annu Rev Physiol. 2008;70:405–429. doi: 10.1146/annurev.physiol.70.113006.100441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canny G, Levy O, Furuta GT, Narravula-Alipati S, Sisson RB, Serhan CN, et al. Lipid mediator-induced expression of bactericidal/ permeability-increasing protein (BPI) in human mucosal epithelia. Proc Natl Acad Sci USA. 2002;99:3902–3907. doi: 10.1073/pnas.052533799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh KD, Siddiqui S, Cheng D, Bonser LR, Sun DI, Zlock LT, et al. Efficient RNP-directed human gene targeting reveals SPDEF is required for IL-13-induced mucostasis. Am J Respir Cell Mol Biol. 2020;62:373–381. doi: 10.1165/rcmb.2019-0266OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, et al. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest. 2009;119:2914–2924. doi: 10.1172/JCI39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans CM, Williams OW, Tuvim MJ, Nigam R, Mixides GP, Blackburn MR, et al. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am J Respir Cell Mol Biol. 2004;31:382–394. doi: 10.1165/rcmb.2004-0060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8:e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T, et al. Localization of secretory mucins MUC5AC and MUC5B in normal/healthy human airways. Am J Respir Crit Care Med. 2019;199:715–727. doi: 10.1164/rccm.201804-0734OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. 2014;505:412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol. 2008;70:459–486. doi: 10.1146/annurev.physiol.70.113006.100702. [DOI] [PubMed] [Google Scholar]
- 32.Bonser LR, Zlock L, Finkbeiner W, Erle DJ. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J Clin Invest. 2016;126:2367–2371. doi: 10.1172/JCI84910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan FE, Vladar EK, Ma L, Fuentealba LC, Hoh R, Espinoza FH, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140:4277–4286. doi: 10.1242/dev.094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma L, Quigley I, Omran H, Kintner C. Multicilin drives centriole biogenesis via E2f proteins. Genes Dev. 2014;28:1461–1471. doi: 10.1101/gad.243832.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.You Y, Huang T, Richer EJ, Schmidt JE, Zabner J, Borok Z, et al. Role of f-box factor foxj1 in differentiation of ciliated airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L650–L657. doi: 10.1152/ajplung.00170.2003. [DOI] [PubMed] [Google Scholar]
- 36.Nemajerova A, Kramer D, Siller SS, Herr C, Shomroni O, Pena T, et al. TAp73 is a central transcriptional regulator of airway multiciliogenesis. Genes Dev. 2016;30:1300–1312. doi: 10.1101/gad.279836.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El Zein L, Ait-Lounis A, Morlé L, Thomas J, Chhin B, Spassky N, et al. RFX3 governs growth and beating efficiency of motile cilia in mouse and controls the expression of genes involved in human ciliopathies. J Cell Sci. 2009;122:3180–3189. doi: 10.1242/jcs.048348. [DOI] [PubMed] [Google Scholar]
- 38.Chung MI, Peyrot SM, LeBoeuf S, Park TJ, McGary KL, Marcotte EM, et al. RFX2 is broadly required for ciliogenesis during vertebrate development. Dev Biol. 2012;363:155–165. doi: 10.1016/j.ydbio.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdullah LH, Wolber C, Kesimer M, Sheehan JK, Davis CW. Studying mucin secretion from human bronchial epithelial cell primary cultures. Methods Mol Biol. 2012;842:259–277. doi: 10.1007/978-1-61779-513-8_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh SM, Kim HR, Park YJ, Lee SY, Chung KH. Organic extracts of urban air pollution particulate matter (PM2.5)-induced genotoxicity and oxidative stress in human lung bronchial epithelial cells (BEAS-2B cells) Mutat Res. 2011;723:142–151. doi: 10.1016/j.mrgentox.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Yang L, Wang WC, Lung SC, Sun Z, Chen C, Chen JK, et al. Polycyclic aromatic hydrocarbons are associated with increased risk of chronic obstructive pulmonary disease during haze events in China. Sci Total Environ. 2017;574:1649–1658. doi: 10.1016/j.scitotenv.2016.08.211. [DOI] [PubMed] [Google Scholar]
- 42.Osei ET, Brandsma CA, Timens W, Heijink IH, Hackett TL. Current perspectives on the role of Interleukin-1 signaling in the pathogenesis of asthma and COPD. Eur Respir J. 2020;55:1900563. doi: 10.1183/13993003.00563-2019. [DOI] [PubMed] [Google Scholar]
- 43.Baines KJ, Simpson JL, Wood LG, Scott RJ, Gibson PG. Transcriptional phenotypes of asthma defined by gene expression profiling of induced sputum samples. J Allergy Clin Immunol. 2011;127:153–160, 160.e1–9. doi: 10.1016/j.jaci.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 44.Pauwels NS, Bracke KR, Dupont LL, Van Pottelberge GR, Provoost S, Vanden Berghe T, et al. Role of IL-1α and the Nlrp3/caspase-1/IL-1β axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38:1019–1028. doi: 10.1183/09031936.00158110. [DOI] [PubMed] [Google Scholar]
- 45.Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184:662–671. doi: 10.1164/rccm.201104-0597OC. [DOI] [PubMed] [Google Scholar]
- 46.Fu JJ, McDonald VM, Baines KJ, Gibson PG. Airway IL-1β and systemic inflammation as predictors of future exacerbation risk in asthma and COPD. Chest. 2015;148:618–629. doi: 10.1378/chest.14-2337. [DOI] [PubMed] [Google Scholar]
- 47.Zou Y, Chen X, Liu J, Zhou DB, Kuang X, Xiao J, et al. Serum IL-1β and IL-17 levels in patients with COPD: associations with clinical parameters. Int J Chron Obstruct Pulmon Dis. 2017;12:1247–1254. doi: 10.2147/COPD.S131877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W, Liu S, Verma M, Zafar I, Good JT, Rollins D, et al. Mechanism of TH2/TH17-predominant and neutrophilic TH2/TH17-low subtypes of asthma. J Allergy Clin Immunol. 2017;139:1548–1558, e4. doi: 10.1016/j.jaci.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen G, Sun L, Kato T, Okuda K, Martino MB, Abzhanova A, et al. IL-1β dominates the promucin secretory cytokine profile in cystic fibrosis. J Clin Invest. 2019;129:4433–4450. doi: 10.1172/JCI125669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Croft DP, Zhang W, Lin S, Thurston SW, Hopke PK, Masiol M, et al. The association between respiratory infection and air pollution in the setting of air quality policy and economic change. Ann Am Thorac Soc. 2019;16:321–330. doi: 10.1513/AnnalsATS.201810-691OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poole A, Urbanek C, Eng C, Schageman J, Jacobson S, O’Connor BP, et al. Dissecting childhood asthma with nasal transcriptomics distinguishes subphenotypes of disease. J Allergy Clin Immunol. 2014;133:670–678, e12. doi: 10.1016/j.jaci.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]