

Abstract
Delayed lung repair leads to alveolopleural fistulae, which are a major cause of morbidity after lung resections. We have reported that intrapleural hypercapnia is associated with delayed lung repair after lung resection. Here, we provide new evidence that hypercapnia delays wound closure of both large airway and alveolar epithelial cell monolayers because of inhibition of epithelial cell migration. Cell migration and airway epithelial wound closure were dependent on Rac1-GTPase activation, which was suppressed by hypercapnia directly through the upregulation of AMP kinase and indirectly through inhibition of injury-induced NF-κB–mediated CXCL12 (pleural CXC motif chemokine 12) release, respectively. Both these pathways were independently suppressed, because dominant negative AMP kinase rescued the effects of hypercapnia on Rac1-GTPase in uninjured resting cells, whereas proteasomal inhibition reversed the NF-κB–mediated CXCL12 release during injury. Constitutive overexpression of Rac1-GTPase rescued the effects of hypercapnia on both pathways as well as on wound healing. Similarly, exogenous recombinant CXCL12 reversed the effects of hypercapnia through Rac1-GTPase activation by its receptor, CXCR4. Moreover, CXCL12 transgenic murine recipients of orthotopic tracheal transplantation were protected from hypercapnia-induced inhibition of tracheal epithelial cell migration and wound repair. In patients undergoing lobectomy, we found inverse correlation between intrapleural carbon dioxide and pleural CXCL12 levels as well as between CXCL12 levels and alveolopleural leak. Accordingly, we provide first evidence that high carbon dioxide levels impair lung repair by inhibiting epithelial cell migration through two distinct pathways, which can be restored by recombinant CXCL12.
Keywords: hypercapnia, Rac1, CXCL12, wound healing
Clinical Relevance
Recent literature is beginning to understand the harmful effects of hypercapnia on injury repair. This article provides mechanistic evidence that elevated CO2 delays cell migration and lung wound healing, which contributes to the development of alveolopleural fistula, a major cause of morbidity after lung resection.
Delayed lung healing after surgical lung resection leads to alveolopleural fistulae, which manifest as prolonged air leak (PAL) from the lung surface (1, 2). Development of PAL remains the major cause of morbidity and mortality after lung resection for both nonmalignant and malignant conditions (1, 3, 4). The National Emphysema Treatment Trial reported that 50% of patients undergoing lung resection as part of lung volume reduction surgery developed PAL (5). In patients undergoing lung cancer resection, incidence of PAL can similarly exceed 50% (1). Several clinical risk factors have been shown to be associated with PAL, which include reduced postresection forced expiratory volume in 1 second (1, 6), incomplete parenchymal fissures (7), advanced emphysema (8), low diffusion capacity, or presence of pleural adhesions encountered during lung surgery (5). However, the cellular mechanisms that contribute to the persistence of PAL are not fully understood.
An increase in CO2 levels is often a consequence of diseases with impaired gas exchange, including the acute respiratory distress syndrome and chronic obstructive pulmonary disease (9, 10). Many investigators have suggested the effects of hypercapnia are inconsequential or even beneficial (9, 11). As a result, the clinical practice of tolerating or encouraging “permissive hypercapnia” is the prevalent paradigm in the care for patients with lung injury (11, 12). Recent studies, including our own, have challenged this paradigm by demonstrating that elevations in CO2 activate specific signaling pathways leading to adverse consequences for lung and organismal physiology (13–20). The alveolar epithelial type 2 cells participate in the repair of alveolopleural fistulae, and the airway epithelial cells repair proximal airway injuries, including bronchopleural fistulae (21, 22). Using a recently developed technique of analysis of pleural gas, we reported that pleural hypercapnia predisposed to PAL (6, 23). Furthermore, reduction in pleural CO2 levels was associated with shorter time of air leakage from the residual lung tissue after resection (23). We have also recently reported that reduced pleural CXCL12 (CXC motif chemokine 12) (also known as stromal cell–derived factor 1), is associated with PAL. CXCL12 promotes alveolar epithelial cell migration by regulating Rac1-GTPase and cofilin activation (24). Also, we have shown that high CO2 impairs cell proliferation because of microRNA-183–mediated inhibition of IDH2 (isocytrate dehydrogenase 2) (25). Hence, we set out to test the effects of hypercapnia on lung wound healing. Here, we provide evidence that hypercapnia delays wound closure in both alveolar and large airway epithelial cells by impairing cell migration via decreased Rac1 activity in resting cells through upregulation of AMP kinase (AMPK) and inhibition of injury-associated autocrine NF-κB–mediated CXCL12 production. Importantly, patients undergoing surgical lobectomy had an inverse correlation between pleural carbon dioxide levels and pleural CXCL12 levels, direct correlation between pleural carbon dioxide and duration of air leak (alveolopleural fistula), and inverse correlation between CXCL12 levels and duration of air leak. Our data have biologic and clinical implications in the monitoring and modulation of pleural hypercapnia to ameliorate the disease burden associated with alveolo-bronchopleural fistulae in patients.
Methods
Cells, materials, permanent transfection, adenoviral infection, and immunohistochemistry can be found in the data supplement Method section. CO2 medium and CO2 exposure were performed as previously described (19, 20). Scratch wound healing, Transwell migration, and Rac1 pull-down assays can be found in the data supplement. Cell lysis and Western blot analysis were performed as previously described (16, 25). F/G actin ratio was calculated using a commercially available kit following the manufacturer’s instructions (Cytoskeleton).
Functional Gene Network Analysis
Gene network analysis was performed from our previously published microarray data from mice housed in room air or in normoxic hypercapnic conditions (10% CO2, 21% O2) for 7 consecutive days (16). Detailed description can be found in data supplement.
Luciferase assay, mRNA isolation, and quantitative PCR can be found in the data supplement.
Mice
Adult (9–11 wk old) male C57BL/6J and BALB/cJ mice were obtained from the Jackson Laboratories. CXCL12 transgenic (CXCL12-Tg) mice were a gift from Dr. Hal Broxmeyer, Indiana University (26). These CXCL12-Tg mice endogenously and globally express CXCL12 under a Rous sarcoma virus promoter. All animals were provided with food and water ad libitum, maintained on a 14-hour light/10-hour dark cycle, and handled according to National Institutes of Health guidelines. All of the procedures involving animals were approved by the Northwestern University Institutional Animal Care and Use Committee (IS00001153). For high CO2 exposure (10% CO2, 21% O2), animals were maintained in a Biospherix C-Shuttle Glove Box (BioSpherix) for up to 13 days, as previously described (16). None of the animals developed appreciable distress. At selected time points, animals were killed with Euthasol (pentobarbital sodium–phenytoin sodium) and trachea was excised. Then either airway cells were collected for RNA extraction or tissue was embedded in Tissue Plus O.C.T (Fisher Healthcare) and frozen in dry ice for immunohistochemistry.
Orthotopic Tracheal Transplantation
Orthotopic tracheal transplantation was performed as previously described (27–30). Detailed protocol can be found in the data supplement.
Human Subjects
The study was approved by the institutional review board at Northwestern University (STU00088120). Consecutive patients undergoing lobectomy for stage I lung cancer were included in the study. Informed consent was obtained from all patients in the study. For pleural CXCL12 analysis, 5 ml of fluid was collected on postoperative Day 1 and analyzed using standardized ELISA (R&D Systems Inc.). The gas analysis was performed by connecting a Datex analyzer (GE Healthcare, Inc.) to the sampling port of the chest draining system (Atrium, Inc.). Measurements were performed while the tubes were on water seal drainage. Patients were breathing room air with arterial oxygen saturation >92%. The analyzer was first connected to the chest drainage system, and CO2 values were recorded.
Statistical Analysis
Data are expressed as mean ± SD. When comparing two groups versus time, statistical analysis was performed by two-way ANOVA with Sidak post hoc test. For comparisons between two groups, significance was evaluated by unpaired t test. When more than two groups were compared, one-way ANOVA was used followed by the Tukey post hoc test. Patient cohort data were analyzed by Fisher exact test. Data were considered significant when P < 0.05.
Results
Hypercapnia Delays Monolayer Wound Closure by Inhibiting Epithelial Cell Migration
We assessed the effects of hypercapnia on respiratory epithelial cell wound closure in primary respiratory epithelial cells and epithelial cell lines in vitro, using well-established scratch wound assays (31). As shown in Figure 1 and Figure E1 in the data supplement, we found that hypercapnia delayed wound closure in mouse and human alveolar epithelial cell lines (A549 and MLE12), primary alveolar epithelial cells, and bronchial epithelial cell line (BEAS-2B). To assess whether the effects of hypercapnia were due to changes in cell migration, we analyzed the migration of A549 and MLE12 cells using Boyden-Transwell chambers and found that A549 (Figure 1D) and MLE12 (Figure E1C) cells migrated more slowly across the chambers in hypercapnic conditions. We have previously reported that hypercapnia inhibited the proliferation rate of alveolar epithelial cells, which was rescued by overexpression of IDH2 (25). To determine whether proliferation played a role in the delayed wound closure, we performed scratch wound assays in cells overexpressing IDH2 (Figure 2A) and in cells exposed to hypercapnia in the presence of mitomycin C (inhibitor of proliferation) (Figure 2B) and found no reversal of delayed epithelial wound closure.
Figure 1.
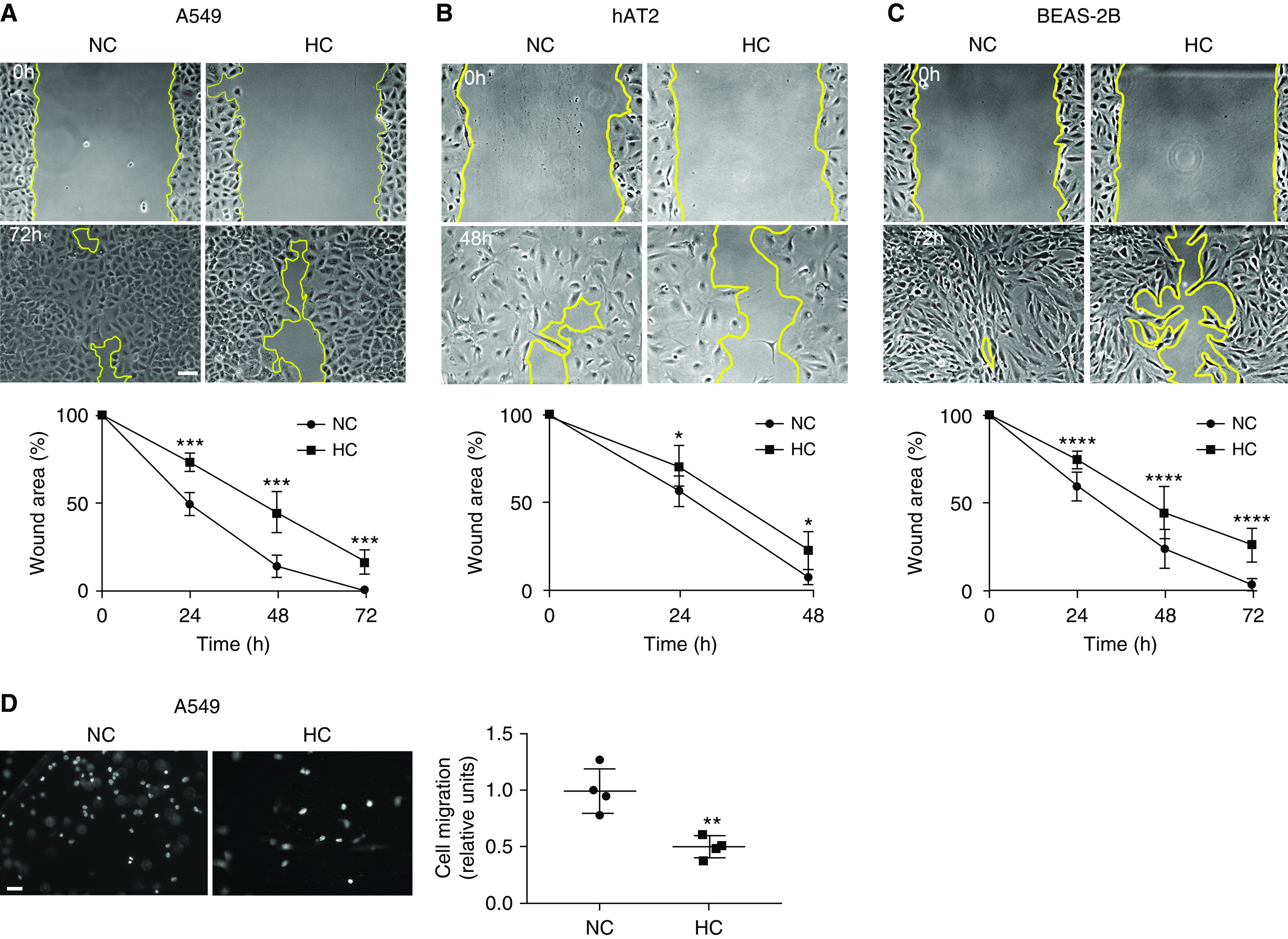
Hypercapnia delays wound closure by suppressing epithelial cell migration. (A) A549 (n = 7), (B) hAT2 (n = 5), and (C) BEAS-2B (n = 10) cells were placed for 24 hours in normocapnia (NC) or hypercapnia (HC) before a scratch wound was made with a sterile pipette tip. Cells were maintained in NC or HC for the indicated time, and photographs were acquired. Scale bar, 100 μm. Graphs show wound area versus time. Data were analyzed by two-way ANOVA followed by Sidak multiple comparison test. *P < 0.05, ***P < 0.001, and ****P < 0.0001. (D) A549 cells were placed for 24 hours in NC or HC, trypsinized, and seeded in Boyden chambers. Migration was determined after 4 hours by staining with Hoechst 33342 and counting nuclei (n = 4). Scale bar, 50 μm. Graph shows quantification of migration. Data were analyzed by unpaired t test. **P < 0.01.
Figure 2.

Hypercapnia delays wound closure by suppressing epithelial cell migration independently of proliferation. (A) A549 cells overexpressing IDH2 (isocytrate dehydrogenase 2) (A549-IDH2) (n = 7), and (B) A549 cells in the presence of 10 μM mitomycin (n = 4) were placed for 24 hours in NC or HC before a scratch wound was made with a sterile pipette tip. Cells were maintained in NC or HC for the indicated time, and photographs were acquired. Scale bar, 100 μm. Graphs show wound area versus time. Data were analyzed by two-way ANOVA followed by Sidak multiple comparison test. **P < 0.01, ***P < 0.001, and ****P 0.0001.
Hypercapnia Inhibits Rac1/Cofilin Pathway in Uninjured Epithelial Cells via AMPK
Cell migration requires remodeling of the actin cytoskeleton via the small GTPase Rac1 (32). Activated Rac1 participates in the formation of lamellipodia, the thin cytoplasmic sheets extended at the front of migrating cells during tissue repair, by phosphorylating cofilin, which promotes F-actin treadmilling driving directional movements (33, 34). Cofilin activation in lung epithelial cells is dependent on Rac1, because W56, a Rac1 inhibitor, prevents cofilin phosphorylation (35). We observed that hypercapnia reduced Rac1 activity and cofilin phosphorylation (Figures 3A and 3B), with an expected decreased F/G actin ratio (Figure 3C) (36) in uninjured resting cells. When we overexpressed an activated form of Rac1 (Rac1-QL) (37), it restored cofilin phosphorylation as well as the delayed wound healing (Figures 3B and 3D) under hypercapnia. To determine the mechanism by which high CO2 decreased Rac1 activity, we first focused on AMPK, a metabolic sensor we have found to be activated in alveolar epithelial cells (19, 38) and a known inhibitor of Rac1 (39). As depicted in Figure 3E, we found that uninjured resting cells infected with an adenovirus expressing dominant negative AMPK (DN-AMPK) (19) did not have decreased cofilin phosphorylation when exposed to high CO2 (37). This suggested that hypercapnia suppressed Rac1-GTPase in the resting cells through the upregulation of AMPK.
Figure 3.
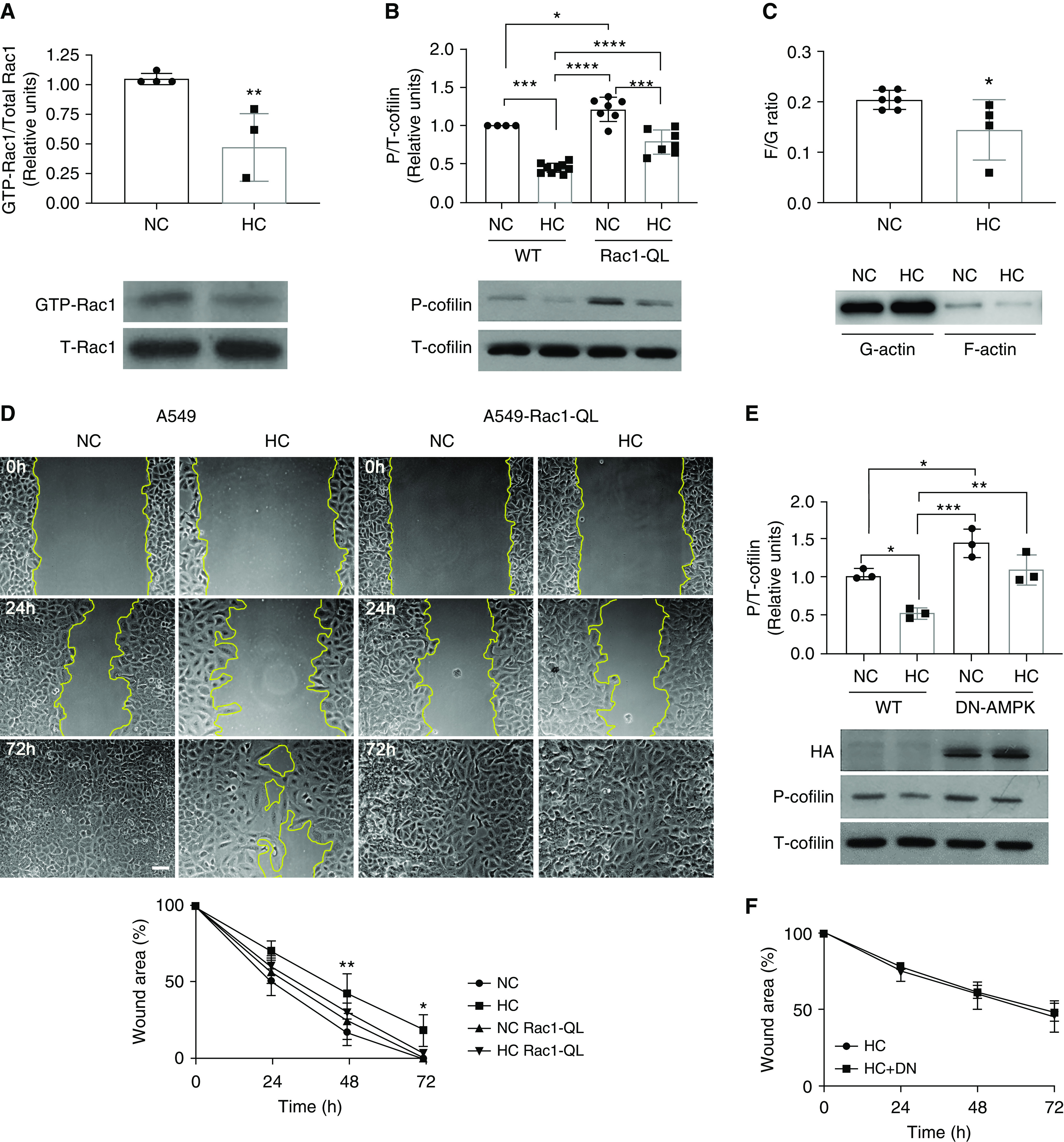
Hypercapnia impairs epithelial cell migration by inhibiting the Rac1/cofilin pathway via AMPK (AMP kinase). (A) Rac1 pull-down assay was performed in cell lysates from A549 cells exposed for 24 hours to NC or HC. Graph shows composite from different experiments (n = 4). Lower panel depicts a representative Western blot. Data were analyzed by unpaired t test. **P < 0.01. (B) A549-wild-type (WT) cells and A549-Rac1-QL cells were exposed for 24 hours to NC or HC, and Western blot against phospho-cofilin (P-cofilin) and total-cofilin (T-cofilin) was performed. Graph shows quantification of different experiments (n = 7–9). Lower panel depicts a representative Western blot. Data were analyzed by one-way ANOVA followed by Tukey multiple comparison test. *P < 0.05, ***P < 0.001, and ****P < 0.0001. (C) F- and G-actin were isolated using a commercially available kit from A549 cells exposed to NC or HC for 24 hours. Graph shows composite from different experiments (n = 4–6) of the F- to G-actin ratio. Lower panel depicts a representative Western blot. Data were analyzed by unpaired t test. *P < 0.05. (D) A549-WT and A549-Rac1-QL cells were placed for 24 hours in NC or HC before a scratch wound was made with a sterile pipette tip. Cells were maintained in NC or HC for the indicated times, and photographs were acquired. Graph shows wound area versus time (n = 4). Data were analyzed by two-way ANOVA followed by Sidak multiple comparison test. *P < 0.05 and **P < 0.01 compared with NC. Scale bar, 100 μm. (E) A549 cells were infected with adenovirus expressing DN-AMPK (dominant negative AMPK), exposed for 24 hours to NC or HC, and Western blot against P- and T-cofilin was performed. Graph shows quantification of different experiments (n = 3). Lower panel depicts a representative Western blot. HA (hemagglutinin) shows expression of the DN-AMPK. Data were analyzed by one-way ANOVA followed by Tukey multiple comparison test. *P < 0.05, **P < 0.01, and ***P < 0.001. (F) A549-WT and A549 cells infected with adenovirus expressing DN-AMPK were placed for 24 hours in NC or HC before a scratch wound was made with a sterile pipette tip. Cells were maintained in NC or HC for the indicated times, and photographs were acquired. Graph shows wound area versus time (n = 3). Data were analyzed by two-way ANOVA followed by Sidak multiple comparison test.
Hypercapnia Inhibits CXCL12 through the Suppression of the NF-κB Pathway after Scratch Wounding of Epithelial Cell Monolayers
Because DN-AMPK rescued Rac1-GTPase and cofilin phosphorylation in the resting cells, we next hypothesized that this should rescue scratch wound healing under hypercapnic conditions. Intriguingly, DN-AMPK did not rescue wound healing when the cells were injured (Figure 3F). These data suggested that hypercapnia might affect a second pathway necessary for injury repair. To explore plausible injury repair pathways affected by hypercapnia, we performed an unbiased functional network analysis of genes altered during exposure to hypercapnia using our previously published murine lung gene array (16). The functional gene network was generated grouping genes on the basis of known interactions and functional similarity within hypercapnia signature (Figure 4A). Using this approach, we found that CXCL12 was significantly downregulated in mouse lungs exposed to hypercapnia. CXCL12 is known to be secreted after scratch wound by lung epithelial cells (35), and we found that this increased transcription was downregulated in hypercapnic conditions (Figure 4B). Addition of recombinant CXCL12 restored the hypercapnia-induced inhibition of Rac1-GTPase (Figure 5A), decreased cofilin phosphorylation (Figure 5B), impaired wound closure (Figures 5C, E2C, and E2D), and cell migration (Figures 5D and E2A) to normocapnic levels. Moreover, inhibition of CXCL12 receptor CXCR4, a known activator of Rac1-GTPase, with AMD-3100 prevented the beneficial effects of CXCL12 in hypercapnia (Figures 5C, 5D, and E2). Taken together, this suggested that hypercapnia suppresses Rac1-GTPase through upregulation of AMPK as well as inhibiting CXCL12 release on injury. However, sufficient amounts of exogenous CXCL12 can overcome the suppression of both pathways by hypercapnia.
Figure 4.

Hypercapnia decreases CXCL12 (pleural CXC motif chemokine 12) production after scratch wound in epithelial cell monolayers. (A) Functional gene network of murine lung genes downregulated during 7-day exposure to HC. (B) A549, MLE-12, hAT2, and mAT2 cells were placed for 24 hours in NC or HC before a scratch wound was made with a sterile comb with 15 teeth. Cells were maintained in NC or HC for 24 hours, mRNA isolated, and quantitative PCR (qPCR) for CXCL12 performed. Graph shows fold change in CXCL12 expression compared with nonscratched monolayers. Data were analyzed by unpaired t test comparing NC versus HC. **P < 0.01 and ***P < 0.001. FC = fold change.
Figure 5.
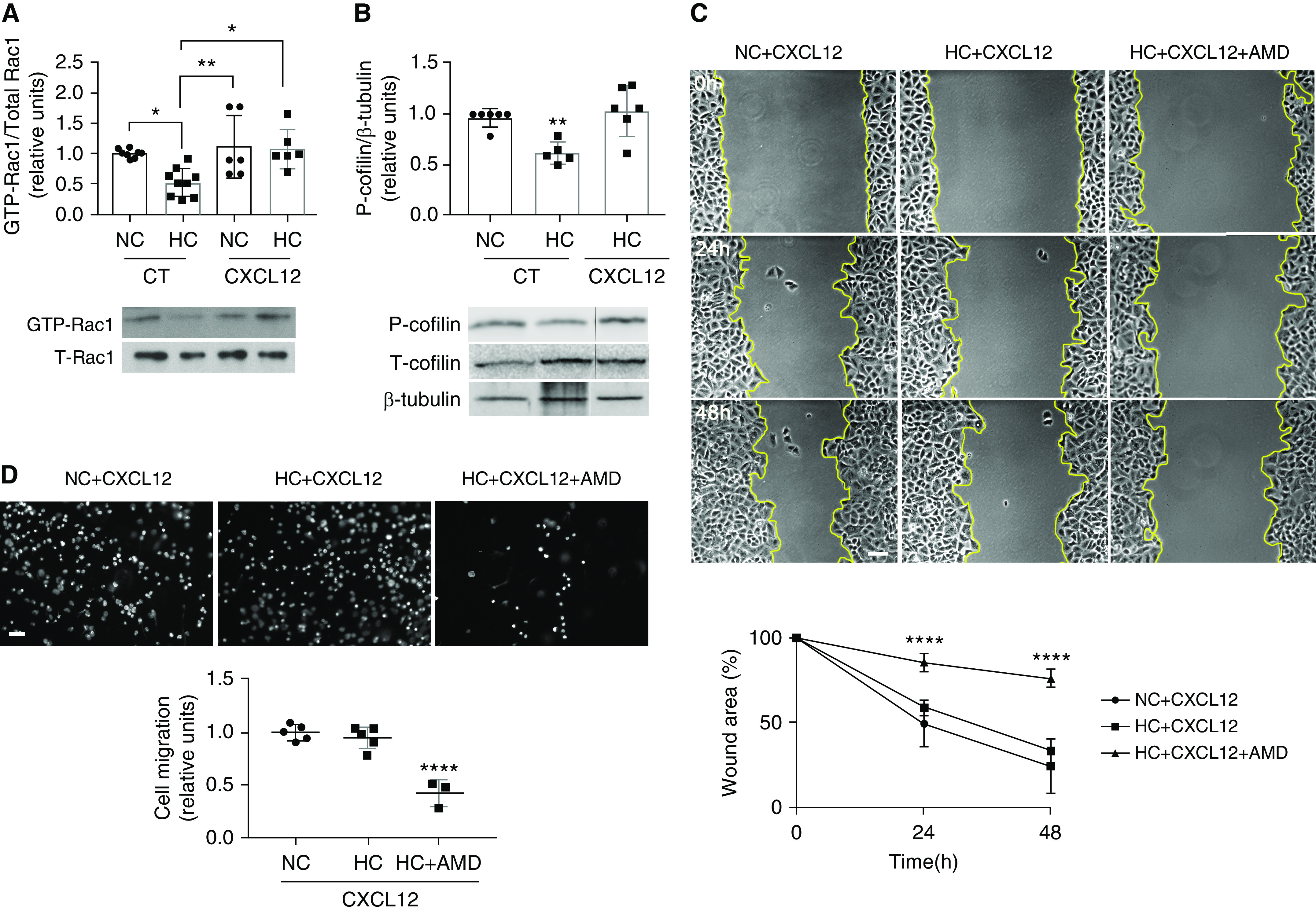
CXCL12 rescues the hypercapnia-inhibited Rac1/cofilin pathway as well as the delayed wound closure and migration. (A) Rac1 pull-down assay was performed in cell lysates from A549 cells exposed for 24 hours to NC or HC and incubated in the presence or absence of vehicle (control [CT]) or 100 ng/ml CXCL12 for 15 minutes. Graph shows composite from different experiments (n = 6–9). Lower panel depicts a representative Western blot. *P < 0.05 and **P < 0.01. (B) A549 cells were exposed for 24 hours to NC or HC and incubated in the presence or absence of vehicle (CT) or 100 ng/ml CXCL12 for 15 minutes. Western blot against P- and T-cofilin and β-tubulin as loading control was performed. Graph shows quantification of different experiments (n = 5–6). Lower panel depicts a representative Western blot. **P < 0.01. (C) A549 cells were placed for 24 hours in NC or HC before a scratch wound was made with a sterile pipette tip. Cells were incubated with 100 ng/ml CXCL12 in the presence or absence of 6 μg/ml AMD-3100 (AMD) for the indicated times, and photographs were acquired. Scale bar, 100 μm. Graph shows wound area versus time (n = 6). Data were analyzed by two-way ANOVA followed by Sidak multiple comparison test. ****P < 0.001 HC + CXCL12 + AMD compared with the other two groups. (D) A549 cells were placed for 24 hours in NC or HC, trypsinized, and seeded in Boyden chambers. Cells were incubated with 100 ng/ml CXCL12 for 15 minutes in the presence or absence of 6 μg/ml AMD. Migration was determined after 4 hours by staining with Hoechst 33342 and counting nuclei (n = 3–5). Scale bar, 50 μm. Graph shows quantification of migration. Data from A, B, and D were analyzed by one-way ANOVA followed by Tukey multiple comparison test.
Hypercapnia is known to inhibit the NF-κB pathway (40, 41), which is essential for the production of CXCL12 (42). High CO2 has been shown to impair NF-κB activity by cleaving the NF-κB member RelB through a process sensitive to the proteasome inhibitor MG-132 (41). Accordingly, we found that preincubation with MG-132 prevented the hypercapnia-induced suppression of NF-κB activation after scratch wounding of the alveolar epithelial cells monolayer (Figure 6A), the decreased CXCL12 production (Figure 6B), and the consequent delayed wound closure (Figure 6C).
Figure 6.

Inhibition of the NF-κB pathway by hypercapnia is responsible for the decreased CXCL12 secretion after wound scratch. (A) A549 cells were exposed to NC or HC for 24 hours in the presence or absence of MG-132 and NF-κB activity after wound scratch was measured with a luciferase assay. Graph shows composite from different experiments (n = 6–7). Data were analyzed by unpaired t test. *P < 0.05. n.s. = not significant. (B) A549 cells were placed in NC or HC in the presence or absence of MG-132 before a scratch wound was made with a sterile comb with 15 teeth. Cells were maintained in NC or HC for 24 hours, mRNA isolated, and qPCR for CXCL12 performed. Graph shows fold change in CXCL12 expression compared with nonscratched monolayers (n = 3). Data were analyzed by one-way ANOVA followed by Tukey multiple comparison test. *P < 0.05. (C) A549 cells were placed for 24 hours in NC or HC in the presence or absence of MG-132 before a scratch wound was made with a sterile pipette tip. Cells were kept in NC or HC for the indicated times and photographs were acquired. Scale bar, 100 μm. Graph shows wound area versus time (n = 6). Data were analyzed by two-way ANOVA followed by Sidak multiple comparison test. **P < 0.05 and ***P < 0.01.
CXCL12 Rescues Hypercapnia-mediated Wound Healing Delay in a Murine Orthotopic Tracheal Transplantation Model
Because of the potential clinical relevance, we developed a murine model to investigate whether hypercapnia-induced suppression of CXCL12 is causally linked to hypercapnia-mediated impaired wound healing. We used a murine orthotopic tracheal transplantation model in which a six-ringed native tracheal segment is replaced with a donor graft (28–30). The recipient epithelium migrates into the transplanted donor graft, from both proximal and distal ends, repopulating it entirely within 11 days (28–30). As shown in Figure 7A, after tracheal transplantation, wild-type allograft recipients had increased CXCL12 mRNA expression in the tracheal epithelial cells. In contrast, murine tracheal allograft recipients had decreased CXCL12 production when kept in hypercapnic conditions (Figure 7A). The wild-type mice kept at room air conditions had complete epithelial repopulation into the donor segment by Day 11 (Figure 7B). In contrast, wild-type transplant recipients exposed to hypercapnic conditions had a delay of 2 days in epithelial repopulation, which was completed by Day 13. When we performed wild-type orthotopic tracheal transplantation into CXCL12-Tg recipient mice in which the CXCL12 was not suppressed during hypercapnia (Figure 7A), transplanted donor tracheas were repopulated by Day 12 under hypercapnic conditions, thus representing a partial rescue (Figure 7B).
Figure 7.
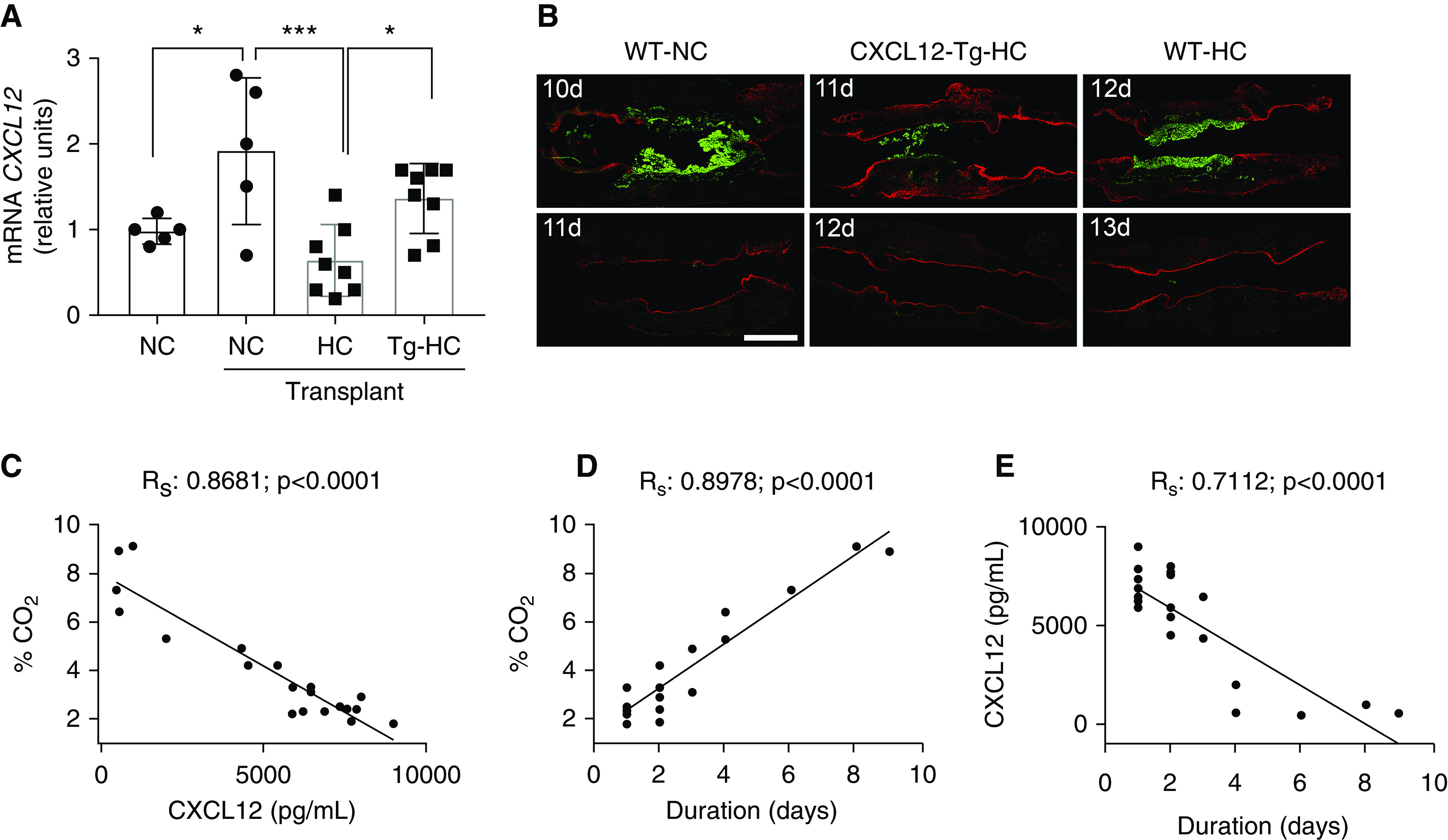
Hypercapnia and CXCL12 in a murine orthotopic tracheal transplantation model and after lung resection in humans. (A) mRNA was isolated from airways from tracheas from WT and CXCL12-transgenic (CXCL12-Tg) mice exposed to NC or HC for 10 days, and qPCR for CXCL12 was performed. Data were analyzed by one-way ANOVA followed by Tukey multiple comparison test. *P < 0.05 and ***P < 0.001 (n = 5–8). (B) Representative immunofluorescent images from postorthotopic tracheal transplantation in the indicated times showing donor tracheal segments (BALB/cJ mice, green) and recipient trachea (C57BL/6J mice, red). Scale bar, 1,000 μm (n = 5–8). (C) Graph represents CO2 versus CXCL12 levels at a given time. (D) Graph represents CO2 levels versus time after lung resection. (E) Graph represents CXCL12 levels versus time after lung resection. Patient cohort data were analyzed by Fisher exact test. Rs = R-squared.
Pleural Hypercapnia Is Associated With Decreased CXCL12 and Delayed Lung Healing after Lung Resection
We have previously reported that pleural hypercapnia is associated with alveolopleural fistula, which manifests as ongoing air leak from the lung surface (23), and that lower levels of CXCL12 are associated with delayed healing after lung resection (24). On the basis of our studies described above, we further tested whether pleural hypercapnia causes delayed lung healing by inhibiting pleural CXCL12 levels in patients undergoing lung resection. Toward this, patients undergoing video-assisted thoracoscopic lobectomy for stage I lung cancer were assessed for pleural CO2, pleural CXCL12, and duration of air leak from the lung. Our hypothesis was that higher levels of CO2 in the pleural space would be associated with reduced local CXCL12 production and increased duration of air leak from the resected lung, reflective of delayed lung wound healing. The demographic profile of the patients is depicted in Table 1. Pleural CO2 levels were inversely related to the CXCL12 levels (Figure 7C) in the pleural fluid, consistent with our observations that hypercapnia can suppress production of CXCL12 in response to injury (Figure 4). Higher levels of CO2 were associated with increased duration of air leak from the resected lung (Figure 7D), indicating that hypercapnia suppresses lung wound closure, which is necessary for the resolution of air leak. Finally, pleural CXCL12 levels inversely correlated with duration of air leak, supporting our findings from the cell culture and murine models (Figure 7E). Taken together, these clinical observations indicate that hypercapnia suppresses CXCL12 production and delays lung wound healing.
Table 1.
Characteristics of Study Patients
| Variable | Overall (n = 20) |
|---|---|
| Age, yr | 59.8 ± 13.6 |
| Female | 9 (45) |
| BMI, kg/m2 | 28.9 ± 9.6 |
| Smoking history | 13 (65) |
| CKD | 1 (5) |
| Pulmonary function | |
| FEV1% | 73.1 ± 21.0 |
| VC% | 67.4 ± 15.6 |
| DlCO% | 67.1 ± 19.8 |
| Laboratory | |
| Hemoglobin, g/dl | 13.2 ± 2.1 |
| WBC, 1,000/μl | 8.7 ± 6.5 |
| Platelets, 1,000/μl | 291.2 ± 104.0 |
| Sodium, mEq/L | 138.6 ± 3.8 |
| Creatinine, mg/dl | 0.8 ± 0.4 |
| BUN, mg/dl | 17.3 ± 6.7 |
| Albumin, g/dl | 3.7 ± 0.7 |
| INR | 1.2 ± 1.1 |
Definition of abbreviations: BMI = body mass index; BUN = blood urea nitrogen; CKD = chronic kidney disease; FEV1 = forced expiratory volume in 1 second; INR = international normalized ratio; VC = vital capacity; WBC = white blood cell.
Data are shown as mean ± SD or n (%).
Discussion
Delayed lung healing leading to alveolopleural or bronchopleural fistulae is a major cause of morbidity and mortality after lung resection (1, 3, 4). Cell migration is a fundamental biological process necessary for wound healing. In this study, we uncover a pathway by which hypercapnia impairs airway as well as alveolar epithelial cell migration, which results in delayed wound healing after injury. Hypercapnia inhibited both the production of CXCL12 and Rac1 activity, which contribute to delayed cell migration and wound closure. As such, our data provide a mechanistic link into the findings that pleural hypercapnia is associated with prolonged alveolopleural fistula in patients undergoing lung resection (6, 23).
We have previously reported that hypercapnia inhibits alveolar epithelial cell as well as fibroblast proliferation by impairing ATP production via the inhibition of IDH2 (25). However, in the present study we found that the hypercapnia-mediated inhibition of alveolar epithelial cell migration and wound healing was not rescued by overexpression of a constitutively active form of IDH2, suggesting that alternative pathways regulate the impaired wound healing. We found that the migration of alveolar epithelial cells was dependent on Rac1-dependent cofilin phosphorylation, which hypercapnia inhibited, whereas overexpression of the constitutively active isoform of Rac1 rescued the effects of hypercapnia on wound healing. Furthermore, suppression of Rac1 in resting cells under hypercapnia was mediated by AMPK, a metabolic sensor that our group has found activated in hypercapnic conditions in multiple cell types (19, 20). However, although DN-AMPK restored the Rac1-GTPase and phospho-cofilin levels to normal levels in resting cells, it did not rescue scratch wound healing, suggesting that an alternate injury repair response mechanism in epithelial cells was suppressed by hypercapnia.
CXCL12 is stimulated during injury and has been shown to promote cell migration in different cell lines (43, 44). Indeed, CXCL12 null mice die in utero, suggesting a critical role in mouse development (45). Here, we report that both alveolar and airway epithelial cells produce CXCL12 in response to injury, which contributes to cell migration and wound healing through its receptor CXCR4; this is consistent with prior findings about CXCR4 (35). We have previously reported that CXCL12 levels are decreased in delayed lung healing after surgery (24), and in the present report we found that hypercapnia inhibits CXCL12, as suggested by unbiased analysis of lung mRNA and quantitative analysis of epithelial cells. The NF-κB pathway has been shown to be highly sensitive to high CO2 (40, 46), in particular the noncanonical pathway involving IKKα and RelB (41, 47). Interestingly, expression of CXCL12 is dependent on the NF-κB noncanonical pathway, which explains the decreased CXCL12 observed in the present study (42, 48). RelB, a member of the NF-κB family, confers transcriptional activity by associating primarily with p52. However, the precursor protein for p52 is p100, which has predominantly inhibitory functions. Controlled degradation of the C-terminal inhibitory domain of the p100 precursor protein within p100:RelB complex leads to the formation of the transcriptionally active p52:RelB heterodimer and secretion of CXCL12 (49). Intriguingly, hypercapnia increases the cleavage and nuclear localization of RelB. However, the RelB translocated to the nucleus under hypercapnia is cleaved in a different position, leading to abnormal transcriptional activity (41). Furthermore, cleavage of RelB to the abnormally short protein, as well as p100 translocation under hypercapnia, is mediated by activation of proteasome and could be reversed by the proteasomal inhibitor MG-132 (41). Our findings indicating that MG-132 reverses hypercapnia-mediated suppression of CXCL12 and wound closure are, therefore, consistent with previously published reports. Together, we hypothesize that hypercapnia suppresses Rac-1GTPase through upregulation of AMPK as well as inhibition of CXCL12 after injury.
We also found that CXCL12 improved wound healing in vivo in a murine orthotopic tracheal transplant. In this transplant model, there is reepithelialization of the transplanted donor segment with the recipient epithelium (28, 29, 50). The tracheal epithelial cells isolated from the recipients after the tracheal transplant demonstrated increase in CXCL12, similar to the epithelial cells in vitro, which was inhibited in hypercapnic conditions. Furthermore, the CXCL12-Tg mice, in which CXCL12 is constitutively expressed (26), did not have a decrease in CXCL12 and improved reepithelialization despite being in hypercapnic conditions (24). The CXCL12-Tg mice did not achieve a complete reversal of wound healing compared with mice kept at room air, which is likely due to the inhibitory effects of hypercapnia on epithelial cell proliferation or other effects that were not rescue in these mice, because CXCL12 does not affect epithelial cell proliferation (24). Alternatively, it may be possible that the constitutive production of CXCL12 in this model is not sufficient to overcome the suppressive effect of AMPK on Rac1-GTPase under hypercapnia. Our patient data demonstrating a strong reverse association between pleural hypercapnia and pleural CXCL12 levels as well as lung healing further provide clinical relevance for our findings.
In summary, we demonstrate a mechanistic link between hypercapnia, cell migration, and alveolopleural and bronchopleural leak resolution after lung resection. We provide evidence of a novel pathway of hypercapnia-mediated inhibition of lung wound healing. Our data are supportive of therapies to decrease pleural hypercapnia to improve lung wound healing after lung resection and reduce the time course of alveolopleural/bronchopleural leak.
Acknowledgments
Acknowledgment
Histology services were provided by the Northwestern University Mouse Histology and Phenotyping Laboratory, which is supported by National Cancer Institute grant P30 CA060553, awarded to the Robert H. Lurie Comprehensive Cancer Center. Imaging work was performed at the Northwestern University Center for Advanced Microscopy, which is generously supported by National Cancer Institute Cancer Center support grant P30 CA060553, awarded to the Robert H. Lurie Comprehensive Cancer Center.
Footnotes
Supported in part by the U.S. National Institutes of Health grants HL125940, HL147575, HL147290, HL145478, HL147070, HL71643, and AG049665.
Author Contributions: A.B., G.R.S.B., E.L., and J.I.S. contributed to all study design and data interpretation and wrote the manuscript. M. Angulo, H.S., A.A., Y.C., L.C.W., J.A.K., and E.L. performed in vitro experiments. M. Angulo, M.S., and S.B. performed and analyzed microarray experiments. A.B. and M. Akbarpour designed and analyzed the human cohort study. H.S. and M. Akbarpour conducted the murine orthotopic tracheal transplantation model. A.B. and J.I.S. provided funding and are guarantors of the paper.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0354OC on April 10, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Okereke I, Murthy SC, Alster JM, Blackstone EH, Rice TW. Characterization and importance of air leak after lobectomy. Ann Thorac Surg. 2005;79:1167–1173. doi: 10.1016/j.athoracsur.2004.08.069. [DOI] [PubMed] [Google Scholar]
- 2.Cornwell LD, Panchal R, Bakaeen FG, Omer S, Preventza O, Lazarus DR, et al. Bronchoscopic management of prolonged air leaks with endobronchial valves in a veteran population. JAMA Surg. 2017;152:207–209. doi: 10.1001/jamasurg.2016.3195. [DOI] [PubMed] [Google Scholar]
- 3.Singhal S, Ferraris VA, Bridges CR, Clough ER, Mitchell JD, Fernando HC, et al. Management of alveolar air leaks after pulmonary resection. Ann Thorac Surg. 2010;89:1327–1335. doi: 10.1016/j.athoracsur.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 4.Quero-Valenzuela F, Piedra-Fernández I, Martínez-Ceres M, Romero-Palacios PJ, Sánchez-Palencia A, De Guevara AC, et al. Predictors for 30-day readmission after pulmonary resection for lung cancer. J Surg Oncol. 2018;117:1239–1245. doi: 10.1002/jso.24973. [DOI] [PubMed] [Google Scholar]
- 5.DeCamp MM, Blackstone EH, Naunheim KS, Krasna MJ, Wood DE, Meli YM, et al. NETT Research Group Patient and surgical factors influencing air leak after lung volume reduction surgery: lessons learned from the National Emphysema Treatment Trial Ann Thorac Surg 200682197–206.[Discussion, pp. 206–207]. [DOI] [PubMed] [Google Scholar]
- 6.Bharat A, Graf N, Cassidy E, Smith S, Gillespie C, Meyerson S, et al. Pleural gas analysis for detection of alveolopleural fistulae. Ann Thorac Surg. 2015;99:2179–2182. doi: 10.1016/j.athoracsur.2014.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez-Caro A, Calvo MJ, Lanzas JT, Chau R, Cascales P, Parrilla P. The approach of fused fissures with fissureless technique decreases the incidence of persistent air leak after lobectomy. Eur J Cardiothorac Surg. 2007;31:203–208. doi: 10.1016/j.ejcts.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 8.Keller CA. Lasers, staples, bovine pericardium, talc, glue and...suction cylinders? Tools of the trade to avoid air leaks in lung volume reduction surgery. Chest. 2004;125:361–363. doi: 10.1378/chest.125.2.361. [DOI] [PubMed] [Google Scholar]
- 9.Feihl F, Perret C. Permissive hypercapnia. How permissive should we be? Am J Respir Crit Care Med. 1994;150:1722–1737. doi: 10.1164/ajrccm.150.6.7952641. [DOI] [PubMed] [Google Scholar]
- 10.Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. LUNG SAFE Investigators; ESICM Trials Group. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315:788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 11.Laffey JG, Kavanagh BP. Carbon dioxide and the critically ill: too little of a good thing? Lancet. 1999;354:1283–1286. doi: 10.1016/S0140-6736(99)02388-0. [DOI] [PubMed] [Google Scholar]
- 12.Hodgson C, Cooper DJ, Arabi Y, Bennett V, Bersten A, Brickell K, et al. PHARLAP Study Investigators and the Australian and New Zealand Intensive Care Society Clinical Trials Group. Permissive Hypercapnia, Alveolar Recruitment and Low Airway Pressure (PHARLAP): a protocol for a phase 2 trial in patients with acute respiratory distress syndrome. Crit Care Resusc. 2018;20:139–149. [PubMed] [Google Scholar]
- 13.Helenius IT, Krupinski T, Turnbull DW, Gruenbaum Y, Silverman N, Johnson EA, et al. Elevated CO2 suppresses specific Drosophila innate immune responses and resistance to bacterial infection. Proc Natl Acad Sci USA. 2009;106:18710–18715. doi: 10.1073/pnas.0905925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nin N, Muriel A, Peñuelas O, Brochard L, Lorente JA, Ferguson ND, et al. VENTILA Group. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017;43:200–208. doi: 10.1007/s00134-016-4611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharabi K, Charar C, Friedman N, Mizrahi I, Zaslaver A, Sznajder JI, et al. The response to high CO2 levels requires the neuropeptide secretion component HID-1 to promote pumping inhibition. PLoS Genet. 2014;10:e1004529. doi: 10.1371/journal.pgen.1004529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shigemura M, Lecuona E, Angulo M, Homma T, Rodríguez DA, Gonzalez-Gonzalez FJ, et al. Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling. Sci Transl Med. 2018;10:eaat1662. doi: 10.1126/scitranslmed.aat1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiruvoipati R, Pilcher D, Buscher H, Botha J, Bailey M. Effects of hypercapnia and hypercapnic acidosis on hospital mortality in mechanically ventilated patients. Crit Care Med. 2017;45:e649–e656. doi: 10.1097/CCM.0000000000002332. [DOI] [PubMed] [Google Scholar]
- 18.Vadász I, Dada LA, Briva A, Helenius IT, Sharabi K, Welch LC, et al. Evolutionary conserved role of c-Jun-N-terminal kinase in CO2-induced epithelial dysfunction. PLoS One. 2012;7:e46696. doi: 10.1371/journal.pone.0046696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vadász I, Dada LA, Briva A, Trejo HE, Welch LC, Chen J, et al. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J Clin Invest. 2008;118:752–762. doi: 10.1172/JCI29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaitovich A, Angulo M, Lecuona E, Dada LA, Welch LC, Cheng Y, et al. High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1) J Biol Chem. 2015;290:9183–9194. doi: 10.1074/jbc.M114.625715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oczypok EA, Perkins TN, Oury TD. Alveolar epithelial cell-derived mediators: potential direct regulators of large airway and vascular responses. Am J Respir Cell Mol Biol. 2017;56:694–699. doi: 10.1165/rcmb.2016-0151PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chambers RC, Mercer PF. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac Soc. 2015;12:S16–S20. doi: 10.1513/AnnalsATS.201410-448MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharat A, Graf N, Mullen A, Kanter J, Andrei AC, Sporn PH, et al. Pleural hypercarbia after lung surgery is associated with persistent alveolopleural fistulae. Chest. 2016;149:220–227. doi: 10.1378/chest.15-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanter JA, Sun H, Chiu S, DeCamp MM, Sporn PH, Sznajder JI, et al. Decreased CXCL12 is associated with impaired alveolar epithelial cell migration and poor lung healing after lung resection. Surgery. 2015;158:1073–1080. doi: 10.1016/j.surg.2015.04.051. [Discussion, pp. 1080–1082.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vohwinkel CU, Lecuona E, Sun H, Sommer N, Vadász I, Chandel NS, et al. Elevated CO(2) levels cause mitochondrial dysfunction and impair cell proliferation. J Biol Chem. 2011;286:37067–37076. doi: 10.1074/jbc.M111.290056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Broxmeyer HE, Cooper S, Kohli L, Hangoc G, Lee Y, Mantel C, et al. Transgenic expression of stromal cell-derived factor-1/CXC chemokine ligand 12 enhances myeloid progenitor cell survival/antiapoptosis in vitro in response to growth factor withdrawal and enhances myelopoiesis in vivo. J Immunol. 2003;170:421–429. doi: 10.4049/jimmunol.170.1.421. [DOI] [PubMed] [Google Scholar]
- 27.Genden EM, Boros P, Liu J, Bromberg JS, Mayer L. Orthotopic tracheal transplantation in the murine model. Transplantation. 2002;73:1420–1425. doi: 10.1097/00007890-200205150-00010. [DOI] [PubMed] [Google Scholar]
- 28.Bharat A, Kuo E, Saini D, Steward N, Hachem R, Trulock EP, et al. Respiratory virus-induced dysregulation of T-regulatory cells leads to chronic rejection Ann Thorac Surg 2010901637–1644.[Discussion, p. 1644.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo E, Bharat A, Goers T, Chapman W, Yan L, Street T, et al. Respiratory viral infection in obliterative airway disease after orthotopic tracheal transplantation. Ann Thorac Surg. 2006;82:1043–1050. doi: 10.1016/j.athoracsur.2006.03.120. [DOI] [PubMed] [Google Scholar]
- 30.Kuo E, Bharat A, Dharmarajan S, Fernandez F, Patterson GA, Mohanakumar T. Animal models for bronchiolitis obliterans syndrome following human lung transplantation. Immunol Res. 2005;33:69–81. doi: 10.1385/IR:33:1:069. [DOI] [PubMed] [Google Scholar]
- 31.McClendon J, Jansing NL, Redente EF, Gandjeva A, Ito Y, Colgan SP, et al. Hypoxia-inducible factor 1alpha signaling promotes repair of the alveolar epithelium after acute lung injury. Am J Pathol. 2017;187:1772–1786. doi: 10.1016/j.ajpath.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steffen A, Ladwein M, Dimchev GA, Hein A, Schwenkmezger L, Arens S, et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 2013;126:4572–4588. doi: 10.1242/jcs.118232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bugyi B, Carlier MF. Control of actin filament treadmilling in cell motility. Annu Rev Biophys. 2010;39:449–470. doi: 10.1146/annurev-biophys-051309-103849. [DOI] [PubMed] [Google Scholar]
- 34.Oleinik NV, Helke KL, Kistner-Griffin E, Krupenko NI, Krupenko SA. Rho GTPases RhoA and Rac1 mediate effects of dietary folate on metastatic potential of A549 cancer cells through the control of cofilin phosphorylation. J Biol Chem. 2014;289:26383–26394. doi: 10.1074/jbc.M114.569657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh MC, Makena PS, Gorantla V, Sinclair SE, Waters CM. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol. 2012;302:L846–L856. doi: 10.1152/ajplung.00321.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song H, Zhang J, He W, Wang P, Wang F. Activation of cofilin increases intestinal permeability via depolymerization of f-actin during hypoxia in vitro. Front Physiol. 2019;10:1455. doi: 10.3389/fphys.2019.01455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teramoto H, Malek RL, Behbahani B, Castellone MD, Lee NH, Gutkind JS. Identification of h-ras, rhoa, rac1 and cdc42 responsive genes. Oncogene. 2003;22:2689–2697. doi: 10.1038/sj.onc.1206364. [DOI] [PubMed] [Google Scholar]
- 38.Welch LC, Lecuona E, Briva A, Trejo HE, Dada LA, Sznajder JI. Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na,K-ATPase downregulation. FEBS Lett. 2010;584:3985–3989. doi: 10.1016/j.febslet.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan Y, Tsukamoto O, Nakano A, Kato H, Kioka H, Ito N, et al. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat Commun. 2015;6:6137. doi: 10.1038/ncomms7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Toole D, Hassett P, Contreras M, Higgins BD, McKeown ST, McAuley DF, et al. Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-κB dependent mechanism. Thorax. 2009;64:976–982. doi: 10.1136/thx.2008.110304. [DOI] [PubMed] [Google Scholar]
- 41.Oliver KM, Lenihan CR, Bruning U, Cheong A, Laffey JG, McLoughlin P, et al. Hypercapnia induces cleavage and nuclear localization of RelB protein, giving insight into CO2 sensing and signaling. J Biol Chem. 2012;287:14004–14011. doi: 10.1074/jbc.M112.347971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madge LA, May MJ. The NFκB paradox: RelB induces and inhibits gene expression. Cell Cycle. 2011;10:6–7. doi: 10.4161/cc.10.1.14291. [DOI] [PubMed] [Google Scholar]
- 43.He W, Yang T, Gong XH, Qin RZ, Zhang XD, Liu WD. Targeting CXC motif chemokine receptor 4 inhibits the proliferation, migration and angiogenesis of lung cancer cells. Oncol Lett. 2018;16:3976–3982. doi: 10.3892/ol.2018.9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng Y, Qu J, Che X, Xu L, Song N, Ma Y, et al. CXCL12/SDF-1α induces migration via SRC-mediated CXCR4-EGFR cross-talk in gastric cancer cells. Oncol Lett. 2017;14:2103–2110. doi: 10.3892/ol.2017.6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Azab AK, Azab F, Blotta S, Pitsillides CM, Thompson B, Runnels JM, et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood. 2009;114:619–629. doi: 10.1182/blood-2009-01-199281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeshita K, Suzuki Y, Nishio K, Takeuchi O, Toda K, Kudo H, et al. Hypercapnic acidosis attenuates endotoxin-induced nuclear factor-κB activation. Am J Respir Cell Mol Biol. 2003;29:124–132. doi: 10.1165/rcmb.2002-0126OC. [DOI] [PubMed] [Google Scholar]
- 47.Keogh CE, Scholz CC, Rodriguez J, Selfridge AC, von Kriegsheim A, Cummins EP. Carbon dioxide-dependent regulation of NF-κB family members RelB and p100 gives molecular insight into CO2-dependent immune regulation. J Biol Chem. 2017;292:11561–11571. doi: 10.1074/jbc.M116.755090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madge LA, May MJ. Classical NF-kappaB activation negatively regulates noncanonical NF-κB-dependent CXCL12 expression. J Biol Chem. 2010;285:38069–38077. doi: 10.1074/jbc.M110.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kew RR, Penzo M, Habiel DM, Marcu KB. The IKKα-dependent NF-κB p52/RelB noncanonical pathway is essential to sustain a CXCL12 autocrine loop in cells migrating in response to HMGB1. J Immunol. 2012;188:2380–2386. doi: 10.4049/jimmunol.1102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernández FG, Jaramillo A, Chen C, Liu DZ, Tung T, Patterson GA, et al. Airway epithelium is the primary target of allograft rejection in murine obliterative airway disease. Am J Transplant. 2004;4:319–325. doi: 10.1111/j.1600-6143.2004.00333.x. [DOI] [PubMed] [Google Scholar]