

Abstract
The airway epithelium plays a critical role in innate responses to airborne allergens by secreting IL-1 family cytokines such as IL-1α and IL-33 as alarmins that subsequently orchestrate appropriate immune responses. Previous studies revealed that epithelial IL-33 secretion by allergens such as Alternaria alternata or house dust mite involves Ca2+-dependent signaling, via initial activation of ATP-stimulated P2YR2 (type 2 purinoceptor) and subsequent activation of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase DUOX1. We sought to identify proximal mechanisms by which epithelial cells sense these allergens and here highlight the importance of PAR2 (protease-activated receptor 2) and TRP (transient receptor potential) Ca2+ channels such as TRPV1 (TRP vanilloid 1) in these responses. Combined studies of primary human nasal and mouse tracheal epithelial cells, as well as immortalized human bronchial epithelial cells, indicated the importance of both PAR2 and TRPV1 in IL-33 secretion by both Alternaria alternata and house dust mite, based on both pharmacological and genetic approaches. TRPV1 was also critically involved in allergen-induced ATP release, activation of DUOX1, and redox-dependent activation of EGFR (epidermal growth factor receptor). Moreover, genetic deletion of TRPV1 dramatically attenuated allergen-induced IL-33 secretion and subsequent type 2 responses in mice in vivo. TRPV1 not only contributed to ATP release and P2YR2 signaling but also was critical in downstream innate responses to ATP, indicating potentiating effects of P2YR2 on TRPV1 activation. In aggregate, our studies illustrate a complex relationship between various receptor types, including PAR2 and P2YR2, in epithelial responses to asthma-relevant airborne allergens and highlight the central importance of TRPV1 in such responses.
Keywords: lung, DUOX1, ATP, IL-33, type 2 immune responses
Clinical Relevance
The findings presented here highlight the importance of the nonselective ion channel TRPV1 (transient receptor potential vanilloid 1) in airway epithelial secretion of IL-33 in response to airborne protease allergens. In light of the proposed importance of IL-33 secretion in asthma exacerbations, selective strategies to inhibit TRPV1 may be beneficial in such cases.
The respiratory epithelium forms a protective barrier in the lung and plays a critical role in innate airway responses to environmental pathogens and airborne allergens (1, 2). An important component of innate epithelial responses to various environmental triggers is the rapid release of the IL-1 family member IL-33, normally expressed within the nucleus of epithelia, as an alarmin to induce appropriate immune responses by stimulating IL-33–specific receptors (IL1RL1/ST2) on various effector cells, such as T lymphocytes, innate lymphoid cells (ILC2), and mast cells (3–5). Such IL-33–dependent type 2 immune responses contribute to tissue regenerative responses and maintenance of homeostasis, but excessive IL-33 release and activation of type 2 inflammation has also been implicated in chronic lung pathologies, such as allergic inflammation and pulmonary fibrosis (6–8).
The mechanisms by which different external triggers promote epithelial IL-33 release are still incompletely understood. Although this is typically believed to involve passive IL-33 release during epithelial necrosis, recent studies indicated the importance of signaling events independent of necrosis in epithelial IL-33 release by asthma-relevant allergens such as Alternaria alternata (ALT) or Dermatophagoides pteronyssinus (house dust mite, HDM), which involve initial release of ATP as a cellular damage signal and Ca2+ signaling due to activation of purinergic receptors on the epithelial surface (3, 9). Previous studies by our group demonstrated that allergen-induced IL-33 release requires the Ca2+-sensitive NADPH oxidase DUOX1, which is primarily expressed within the respiratory epithelium and generates H2O2 to activate the tyrosine kinases Src and EGFR (epidermal growth factor receptor), thereby promoting processing and nonclassical secretion of IL-33 (9).
The proximal mechanisms by which allergens such as ALT or HDM are sensed by airway epithelial cells or other innate immune cells are not well established, but serine or cysteine proteinases associated with these allergens, such as Der p 1, are believed to play a major role (10–12). Allergen-derived proteases can act on epithelial cells in different ways, including direct disruption of junctional proteins, production of TLR4 (toll-like receptor 4) ligands, and activation of cell surface PARs (protease-activated receptors), G protein-coupled receptors that comprise four members (PAR1, PAR2, PAR3, and PAR4). Among these, PAR2 has been most strongly implicated in epithelial responses to protease allergens, including HDM and ALT (13–15). PAR2 is also the main isoform that is enhanced in asthmatic airways (16) and is involved in allergen-induced loss of epithelial integrity, allergen sensitization, and induction of Th2-polarizing cytokines (17, 18). Notwithstanding these various findings, the direct importance of PAR receptors in allergen-induced epithelial IL-33 secretion has not yet been established.
Although PAR2 has been implicated in allergen-induced epithelial responses, it is not fully responsible for allergen-induced ATP release and Ca2+ signaling, and other Ca2+ channels have been implicated (15, 19). An important class of proteins that mediates intracellular Ca2+ increase in response to diverse stimuli comprises the TRP (transient receptor potential) Ca2+ channels, which are Ca2+-permeable, nonselective cation channels that sense a variety of chemical and physical stimuli. Diverse TRP channels have been implicated in airway responses to various environmental triggers (e.g., tobacco smoke) and in various lung pathologies (20, 21). Although typically believed to be confined to nociceptive neurons, several TRP channels, particularly members of the TRPV (TRP vanilloid) subfamily such as TRPV1 and TRPV4, are also found within the bronchial epithelium (20), and nonneuronal TRPV1 has been shown to be significantly upregulated in the airway epithelium of patients with asthma (22) and in nasal polyps from patients with chronic rhinosinusitis (23). Population-based epidemiological studies revealed the involvement of TRPV1 in symptoms typically associated with asthma (24), and TRPV1 was found to contribute to airway hyperresponsiveness and airway inflammation in mouse models of asthma (25, 26). Intriguingly, G-protein–coupled receptors such as PAR2 can dramatically sensitize TRPV1 responses during pain responses (27, 28), and TRPV1 was found to contribute to PAR2-induced hypersensitivity of dorsal root ganglion (DRG) neurons (29). However, the involvement of TRP channels in innate airway epithelial responses to allergens, and IL-33 secretion in particular, has to date not been addressed.
The present studies were undertaken to investigate the importance of PAR receptors and TRP channels in innate responses to asthma-relevant airborne allergens HDM and ALT, in studies with primary human nasal epithelial cells and murine tracheal epithelial cells. Our findings demonstrate that PAR2 and TRPV1 both critically contribute to allergen-induced ATP release as an initial damage signal that promotes activation of DUOX1 and subsequent redox-dependent EGFR activation and IL-33 secretion. Moreover, TRPV1 was also critical for IL-33 secretion and subsequent type 2 inflammation after in vivo allergen challenge and contributed to ATP-mediated IL-33 secretion, indicating potentiating effects of type 2 purinoceptor (P2YR2) on TRPV1 activation. Collectively, our findings illustrate a central role of TRPV1 in innate epithelial responses to protease allergens, which acts in concert with PAR2 and P2YR2 to promote epithelial IL-33 secretion and subsequent activation of type 2 responses.
Methods
Cell Culture and Treatments
Primary human nasal epithelial (HNE) cells were collected from healthy volunteers and patients with allergic rhinitis and cultured as described previously (9). Immortalized human bronchial epithelial (HBE1) cells were cultured as described previously (9). Mouse tracheal epithelial (MTE) cells were isolated from wild-type (WT) C57BL6/J mice, BALB/c mice, or TRPV1-deficient mice (B6.129X1-Trpv1tm1Jul/J) and cultured as described previously (9). Before treatment with indicated stimuli or inhibitors, cells were incubated overnight in EGF-free complete media to suppress basal EGFR activity. Cells were pre-treated with various inhibitors for 30 minutes and stimulated with extracts of HDM (D. pteronyssinus; Lot. #213051; Greer Laboratories) or A. alternata (Lot. #217252; Greer Laboratories) or other reagents of interest for either 10 minutes or 2 hours at 37°C and 5% CO2. Alternatively, cells were transfected with human siRNA (Dharmacon; see Table E1 in the data supplement) using DharmaFECT1 transfection reagent (Thermo Scientific), before treatment. Conditioned culture media were collected for cytokine analysis, and cell extracts were prepared for mRNA or protein extraction for RT-PCR or Western blot analysis, respectively. Further experimental details are described in the data supplement.
In vivo Animal Studies
C57BL6/J or TRPV1−/− mice (B6.129X1-Trpv1tm1Jul/J; originally obtained from Jackson Laboratories and kindly provided by George Wellman, Department of Pharmacology, University of Vermont) aged 8 to 12 weeks were subjected to brief isoflurane anesthesia and administration of 50 μl of 1 μg/ml HDM extract (Lot #218862, 1.27 endotoxin U/mg; Greer Laboratories) or 50 μl PBS vehicle control, via the oropharyngeal route, and BAL and lung tissues were collected 1 or 6 hours after challenge. In separate experiments, WT C57BL6/NJ mice and DUOX1−/− mice (originally provided by Geiszt [9] and backcrossed to C57BL6/NJ background) were subjected to oropharyngeal instillation of capsaicin (30 μg/kg body weight in 50 μl PBS/0.5% EtOH; M2028; Sigma) or vehicle control under isoflurane anesthesia. After instillation, mice were maintained under 1% isoflurane anesthesia for a total duration of 1 hour, after which mice were killed, and BAL and lung tissues were collected. All animal procedures were reviewed and approved by the Animal Care and Use Committee of the University of Vermont.
Biochemical Assays
Cell culture supernatants or BAL fluids were analyzed for IL-33, KC/IL-8, IL-13, and IL-1α using DuoSet ELISAs (R&D Systems) according to the manufacturer’s instructions. Extracellular H2O2 production was determined by lactoperoxidase-catalyzed crosslinking of o-tyrosine, which was analyzed by HPLC and fluorescence detection (9). ATP release into the medium was examined using a luciferase/luciferin bioluminescence ATP determination assay, according to the manufacturer’s instructions (Molecular Probes). Activation of EGFR signaling was assessed by Western blot analysis of phospho-EGFR.
Statistical Analysis
All quantitative data are presented as the mean ± SE. Statistical differences between groups were analyzed using one- or two-way ANOVA with Bonferroni post hoc analysis in GraphPad Prism (version 7.0; GraphPad Software). P values < 0.05 were considered significant.
Results
Allergen-induced IL-33 Secretion from HNE Cells Involves Protease Activity and PAR2 Activation
We used primary HNE cells from four healthy volunteers to assess mechanisms of innate responses to two common asthma-relevant allergens, ALT or HDM, which both possessed detectable protease activity (Figure 1A). Apical stimulation of HNE cells in Transwell inserts with either ALT or HDM resulted in rapid apical IL-33 release, consistent with previous studies (9), but this was not observed in the presence of either PMSF or antipain (Figure 1B), two inhibitors of serine proteases. Because proteases can induce epithelial responses by activating PAR receptors (13), we evaluated mRNA expression of various PAR isoforms in HNE cells and observed that HNE cells primarily express PAR2 and to a lesser extent PAR1 (Figure E1). Accordingly, allergen-induced IL-33 secretion was not observed in the presence of the PAR2 inhibitor FSLLRY-NH2 (Figure 1B). Moreover, IL-33 secretion could also be evoked by direct stimulation with PAR2 agonist peptides (SLIGLR-NH2 and f-LIGRLO-NH2) but not by agonists of PAR1 (thrombin and TFLLR-NH2) (Figure 1C). Similarly, direct activation of PAR2, but not PAR1, promoted HNE cell production of extracellular H2O2, reflecting DUOX1 activation, which was recently implicated in allergen-induced IL-33 secretion (9). Hence, protease allergens evoke epithelial IL-33 secretion largely by stimulating PAR2 and subsequent activation of DUOX1.
Figure 1.

Protease allergens induce IL-33 release from nasal epithelial (HNE) cells via PAR2 (protease-activated receptor 2) and TRPV1 (transient receptor potential vanilloid 1). (A) Assessment of protease activity in allergen extracts, relative to papain (μg/ml; mean ± SE; n = 2–6) after exposure to 0, 3, 10, and 30 μg/ml allergen (Alternaria alternata [ALT] or house dust mite [HDM]). *P < 0.05 versus no allergen by one-way ANOVA. (B) Inhibition of allergen-induced IL-33 release from HNE cells by the serine protease inhibitors PMSF (100 μM) or antipain (50 μg/ml) or by the PAR2 inhibitor FSLLRY-NH2 (10 μM). (C) IL-33 release and extracellular H2O2 production induced by agonists of PAR2 (SLIGRL-NH2, 50 μM; f-LIGRLO-NH2, 5 μM), but not by agonists of PAR1 (thrombin, 30 nM; TFLLR-NH2, 50 μM). *P < 0.05 versus PBS control by one-way ANOVA. (D) Inhibition of allergen-induced IL-33 release by the TRPV1 inhibitors JNJ17203212 (1 μM) or capsazepine (10 μM), but not by the TRPV4 inhibitor RN1734 (10 μM). (E) Stimulation of IL-33 release or H2O2 production by TRP channel agonists, capsaicin (10 μM), allyl isothiocyanate (AITC, 100 μM), GSK1016790A (10 nM), or acrolein (ACR; 30 μM). *P < 0.05 versus control by one-way ANOVA. Data represent mean ± SE from independent studies from two to four different donors. *P < 0.05 versus control treatment. #H2O2 production by capsaicin could not be assessed because of its interference with the H2O2 assay. CTL = control; GSK = GSK1016790A; PMSF = phenylmethyl sulfonyl fluoride.
Allergen-induced Epithelial IL-33 Secretion Involves TRPV1
Airway epithelial cells may also express various TRP Ca2+ channels, which could be involved in allergen-induced Ca2+ signaling and DUOX1 activation. Therefore, we sought to address the mRNA expression of TRP channel isoforms in HNE cells, which revealed expression of both TRPV1 and TRPV4 (Figure E1). We used various TRP channel inhibitors to address the potential involvement of TRPV1/4 in allergen-induced responses. Studies with HNE cells from at least three subjects indicated increased IL-33 release by ALT or HDM, which appeared to be suppressed by two TRPV1 inhibitors, JNJ17203212 and capsazepine, but not by the TRPV4 inhibitor RN1734, although neither result was statistically significant (Figure 1D). Moreover, direct stimulation of HNE cells with agonists of TRPV1 (capsaicin or allyl isothiocyanate [AITC], although the latter also activates TRP ankyrin 1 [TRPA1]) could evoke IL-33 secretion, as well as H2O2 production (reflecting DUOX1 activation), whereas agonists of TRPV4 (GSK1016790A) or TRPA1 (acrolein) were much less effective (Figure 1E). It should be noted that H2O2 production by capsaicin could not be assessed because it was found to interfere with the H2O2 assay. Collectively, these findings indicate that allergen-induced H2O2 production and IL-33 secretion by HNE cells involves the contribution of TRP channels and primarily TRPV1.
Activation of PAR2 and TRPV1 Promotes ATP Release and P2YR2-Dependent Signaling
Previous studies implicated epithelial ATP release and P2YR2 activation in allergen-induced IL-33 secretion (3, 9). We therefore examined whether activation of PAR2 or TRPV1 could evoke apical ATP release from HNE cells and act through P2YR2-dependent mechanisms. Indeed, direct activation of PAR2 (but not PAR1) or TRPV1 was capable of evoking rapid ATP release, comparable to stimulation with HDM or ALT (Figure 2A). Although measured extracellular ATP levels in cell culture media were in the nanomolar range, concentrations near the cell surface are likely higher and sufficient to activate purinoceptors such as P2YR2 (30). The importance of P2YR2 in allergen-induced responses in HNE cells was confirmed by inhibition of both H2O2 production and IL-33 secretion by the P2YR inhibitor suramin (Figure 2B), consistent with previous findings (3, 9). These findings suggest that protease allergens induce epithelial ATP release by a concerted mechanism involving initial activation of both PAR2 and TRPV1 (20, 27, 28), subsequently resulting in activation of P2YR2, DUOX1-dependent H2O2 production (9), and IL-33 secretion. Follow-up studies were performed in HBE1 cells to determine the importance of P2YR2 in TRPV1/4-mediated responses, using siRNA silencing of P2YR2. As expected (3, 9), IL-33 secretion in response to allergens or to exogenous ATP was attenuated after silencing of P2YR2 (Figure 2C). Similarly, P2YR2 silencing also attenuated IL-33 secretion in response to the TRPV1 agonist capsaicin or the TRPV4 agonist GSK1016790A, confirming that TRPV1/4-induced IL-33 secretion requires P2YR2-dependent DUOX1 activation.
Figure 2.
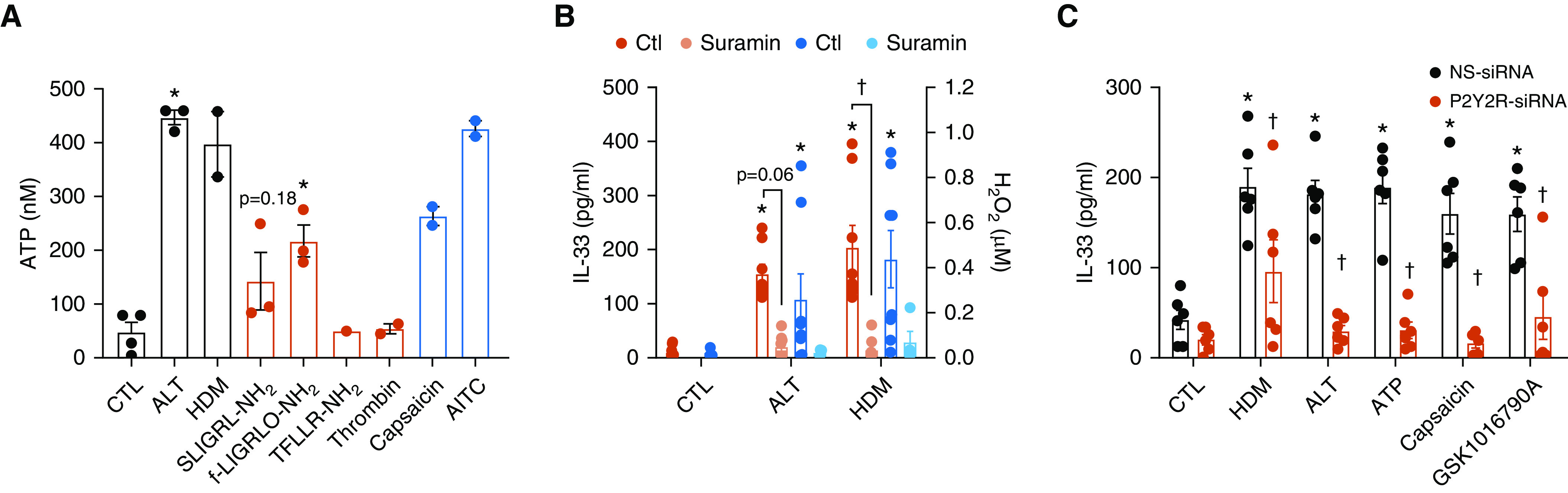
Importance of type 2 purinoceptor (P2YR2) and DUOX1 in innate allergen responses. (A) Effects of protease allergens (black), PAR agonists (red), or TRPV1 agonists (blue) on extracellular ATP release from HNE cells after 15 minutes of stimulation. *P < 0.05 versus control by one-way ANOVA. (B) Inhibition of allergen-induced H2O2 production and IL-33 secretion from HNE cells by the P2YR2 inhibitor suramin (100 μM). Data represent mean ± SE from independent studies from three to four different donors. *P < 0.05 versus control by one-way ANOVA. †P < 0.05 versus corresponding treatment without inhibitor by one-way ANOVA. (C) Measurement of IL-33 release from HBE1 cells (mean ± SE; n = 5–6) after 2 hours of stimulation with HDM (50 μg/ml), ALT (50 μg/ml), ATP (100 μM), the TRPV1 agonist capsaicin (10 μM), or the TRPV4 agonist GSK1016790 (10 nM), after siRNA silencing of P2YR2. *P < 0.05 versus control treatment. †P < 0.05 versus corresponding non-specific (NS)-siRNA treatment. DUOX1 = dual oxidase 1.
TRPV1 Activation Is Crucial for Allergen-induced DUOX1 Activation and EGFR Signaling
To further elucidate the importance of TRPV1 in allergen-induced responses, we performed complementary studies with HBE1 cells and MTE cells. Western blot analysis indicated the presence of TRPV1 protein in immortalized human bronchial epithelial HBE1 cells (Figure E1). Consistent with our findings in HNE cells (Figure 1), HDM-induced IL-33 secretion from HBE1 cells as well as MTE cells from either C57BL6/J mice or BALB/c mice could in each case be attenuated by inhibitors of TRPV1 (JNJ17203212 or capsazepine) or mimicked by direct stimulation of TRPV1 (capsaicin, AITC) as well as TRPV4 (GSK1016790A) (Figures 3A and 3B and E2). On the basis of our previous studies indicating that innate epithelial responses to protease allergens involve redox-dependent activation of the tyrosine kinases Src and EGFR (9), we addressed whether TRPV1 was also involved in allergen-induced activation of these signaling pathways. Indeed, pharmacological inhibition of TRPV1 strongly attenuated allergen- or ATP-induced activation of EGFR in HBE1 cells (Figures 3C and 3D) as well as MTE cells (Figure E3), and agonists of TRPV1 and TRPV4 were capable of inducing EGFR activation (Figures 3C–3E and E3). Because of the potential lack of specificity of pharmacological TRPV inhibitors, we also used siRNA silencing to assess the specific involvement of TRPV1 in allergen responses (Figures E1 and E5B). Indeed, IL-33 release induced by allergens, ATP, and the TRPV1 activator capsaicin was markedly inhibited after TRPV1 siRNA (Figure 3F). Interestingly, IL-33 secretion by GSK1016790A was not affected by TRPV1 siRNA (Figure 3F), illustrating the specificity of this agonist for TRPV4 and not TRPV1. Similar inhibitory effects of TRPV1 siRNA were also apparent with respect to H2O2 production by these triggers (Figure 3G). Collectively, these findings indicate that allergen-induced DUOX1 activation, EGFR activation, and IL-33 secretion primarily involve TRPV1.
Figure 3.

Involvement of TRPV1 in allergen-induced innate epithelial responses. (A) Human bronchial epithelial (HBE1) cells were pretreated with either the TRPV1 inhibitor JNJ17203212 (1 μM) or capsazepine (10 μM) and stimulated for 2 hours with HDM (50 μg/ml) or ATP (100 μM) for analysis of IL-33 secretion in the medium (mean ± SE; n = 4). (B) Activation of IL-33 release from HBE1 cells by 2-hour stimulation with TRPV1 agonists capsaicin (10 μM) or AITC (100 μM) or the TRPV4 agonist GSK1016790 (10 nM) (mean ± SE; n = 4). (C) Representative Western blot analysis of HBE1 cell lysates for EGFR (epidermal growth factor receptor) phosphorylation after 10-minute stimulation with HDM (50 μg/ml) or ATP (100 μM), in the absence or presence of TRPV1 inhibitors JNJ17203212 (1 μM) or capsazepine (10 μM), or after stimulation with the TRPV1 agonist capsaicin (10 μM) or TRPV4 agonist GSK1016790 (10 nM). (D and E) Semi-quantitative analysis (mean ± SE; n = 4) of pEGFR/tEGFR ratios, calculated using ImageJ software and normalized to either HDM or AITC groups. Effect of TRPV1-siRNA on (F) IL-33 secretion (mean ± SE; n = 6) or (G) extracellular H2O2 production (mean ± SE; n = 2) on stimulation with HDM (50 μg/ml), ALT (50 μg/ml), ATP (100 μM), the TRPV1 agonist capsaicin (10 μM), or the TRPV4 agonist GSK1016790 (10 nM). Note that H2O2 production by capsaicin stimulation could not be assessed because of its interference with H2O2 assay. *P < 0.05 versus control treatment. †P < 0.05 versus corresponding treatment without inhibitor or NS-siRNA treatment. capz = capsazepine; JNJ = JNJ17203212; pEGFR = phosphorylated EGFR on Y1068; tEGFR = total EGFR.
TRPV1 Potentiates P2YR2-mediated Responses in Airway Epithelial Cells
One unanticipated finding in our studies was the fact that TRPV1 inhibition and siRNA silencing also dramatically suppressed IL-33 release, H2O2 production, and EGFR activation in response to exogenous ATP (Figures 3, E2, and E3). Although ATP is a ligand for P2YR2, high doses of exogenous ATP could potentially activate cells by P2YR-independent mechanisms, and we therefore evaluated IL-33 secretion by lower concentrations of ATP and demonstrated that ATP induced IL-33 secretion at concentrations as low as 10 μM (which is relatively selective for P2YR), prevented by TRPV1 silencing in each case (Figure E4). Of note, TRPV1 siRNA also modestly reduced P2YR2 mRNA expression (Figure E5). In aggregate, these findings indicate that TRPV1 is not only involved in allergen-induced ATP release and downstream P2YR2-dependent signaling but also critically contributes to ATP-dependent signaling in cooperation with P2YR2.
TRPV1 Is Critical for Innate Airway Epithelial Responses to HDM
To more firmly address the importance of TRPV1 in allergen-induced innate responses, we performed studies with TRPV1-deficient mice. We verified that TRPV1 deficiency did not affect expression levels of Duox1, P2yr2, or Trpv4 (Figure E6). Studies using isolated MTE cells from either WT or TRPV1-deficient mice confirmed that TRPV1 was critical for acute ATP- or HDM-induced production of IL-33 as well other innate cytokines, such as IL-1⍺, or the murine IL-8 homolog KC (although ATP stimulation did not significantly increase KC production in this case) (Figures 4A–4C). Next, WT and TRPV1−/− mice were subjected to acute in vivo airway challenge with HDM (50 μg/ml), and lung lavage fluids were collected at various time points for analyses of IL-33 and related type 2 immune responses. As expected, HDM challenge resulted in rapid and transient production of IL-33 (Figure 5A), IL-1⍺ (Figure 5B), amphiregulin (Areg) (Figure 5C), and KC (Figure 5D), which peaked at 1 hour after challenge and in most cases declined after 6 hours. Each of these responses was significantly attenuated in TRPV1-deficient mice (Figure 5A–5D). In addition, HDM challenge induced delayed airway production of IL-5 and IL-13 (Figures 5E and 5F), most likely as a result of initial IL-33 secretion, which was also attenuated in TRPV1-deficient mice. Finally, to address the involvement of DUOX1 in these TRPV1-mediated airway responses, WT or DUOX1−/− mice were subjected to acute airway challenge with the TRPV1 agonist capsaicin, which was found to similarly evoke significant acute airway production of IL-33 and KC and tended to increase IL-1α, responses that were largely attenuated in DUOX1−/− mice (Figures 6A–6C). Combined with earlier studies indicating the importance of epithelial DUOX1 in production of these various cytokines by allergens (9), these results suggest a critical role for epithelial TRPV1 in DUOX1-mediated innate airway responses to airborne allergens such as HDM, particularly in IL-33 secretion and subsequent activation of type 2 immune responses.
Figure 4.

TRPV1 deficiency attenuates innate HDM responses. Mouse tracheal epithelial cells (MTECs) isolated from wild-type (WT) or TRPV1−/− mice were stimulated with HDM (50 μg/ml) or ATP (100 μM) for 2 hours, and production of (A) IL-33, (B) IL-1⍺, or (C) KC was measured in the medium by ELISA (mean ± SE; n = 8). *P < 0.05 compared with control treatment. †P < 0.05 compared with corresponding treatment in WT MTECs. KC = keratinocyte chemoattractant (also known as neutrophil chemokine CXCL1).
Figure 5.
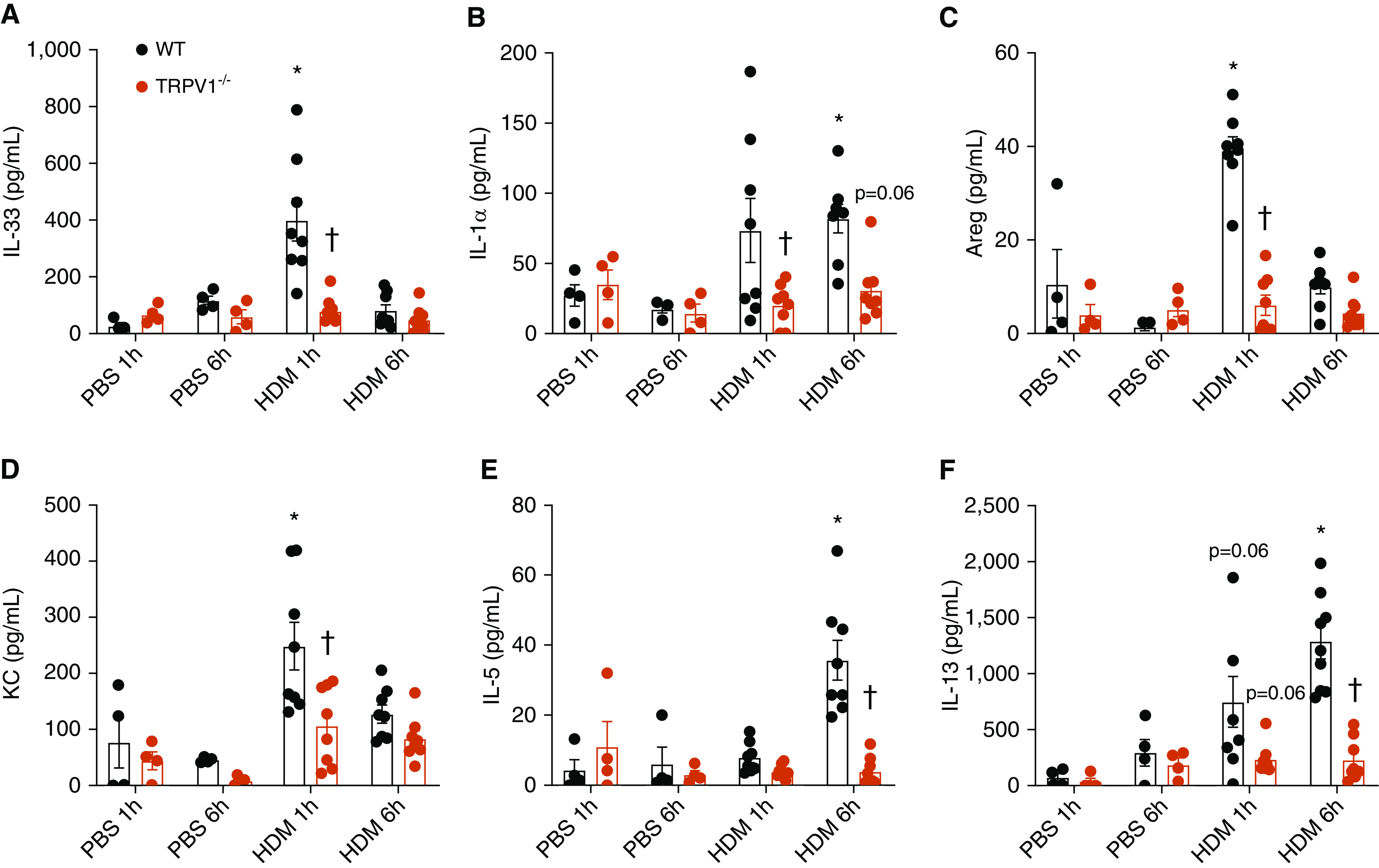
TRPV1 deficiency attenuates innate HDM responses and type 2 inflammation in vivo. WT C57Bl6/J and TRPV1−/− mice were challenged with HDM extract or vehicle control, and lung lavage was collected after 1 or 6 hours, for ELISA analysis of (A) IL-33, (B) IL-1⍺, (C) Areg (amphiregulin), (D) KC, (E) IL-5, or (F) IL-13 (mean ± SE; n = 4–8). *P < 0.05 compared with corresponding PBS control. †P < 0.05 compared with corresponding treatment in WT mice.
Figure 6.

DUOX1 deficiency attenuates innate cytokine responses in response to capsaicin. WT C57BL6/NJ and DUOX1−/− mice were challenged with capsaicin (30 μg/kg), and BAL was collected after 1 hour for ELISA analysis of (A) IL-33, (B) IL-1⍺, or (C) KC (mean ± SE; n = 5–6). *P < 0.05 compared with corresponding PBS control. †P < 0.05 compared with corresponding treatment in WT mice.
TRPV1 and P2YR2 Expression Are Increased in HNE Cells from Subjects with Asthma
We obtained HNE cells from both healthy control subjects and subjects with allergic asthma and rhinitis (9) and addressed expression of various genes addressed in this study. Notably, mRNA expression of both P2YR2 and TRPV1 were significantly enhanced in HNE cells from subjects with asthma compared with control subjects without asthma, whereas expression of PAR1 and PAR2 also tended to be increased (Figure 7). This indicates that enhanced expression of these receptors may contribute to amplified type 2 immune responses to allergens or other exacerbating triggers in these subjects.
Figure 7.

Enhanced mRNA expression of P2YR2 and TRPV1 in HNE cells isolated from patients with asthma compared with control subjects. Relative mRNA expression of P2YR2, PAR1, PAR2, TRPV1, and TRPV4 in HNEs isolated from control subjects (black) or patients with asthma (red). Data points represent the averages of duplicate analysis (mean ± SE) in cells isolated from 10 control donors and 9 donors with asthma.
Discussion
IL-33, a member of the IL-1 family, has received extensive recent interest as an epithelial-derived alarmin that mediates type 2 immune responses to various allergens and because of its important role in type 2 asthma (31, 32). The mechanisms by which IL-33 is released from the epithelium during, for example, allergen challenge or exacerbations has remained somewhat enigmatic (4). Although IL-33 can be released passively from epithelial cells as an active cytokine because of necrosis, emerging studies indicate that various asthma-relevant allergens can promote active IL-33 secretion from airway epithelia by initial release of ATP and activation of purinoceptor-dependent signaling pathways, which involve the NADPH oxidase DUOX1, redox-dependent activation of tyrosine kinases such as Src and EGFR, and activation of calpains (3, 9). The upstream mechanisms by which allergens initiate these responses have remained largely unclear. However, airway epithelial cells have been shown to express many diverse receptor types (e.g., TLRs, dectin-1) that may recognize different components of common allergens such as HDM or ALT (33). Extending previous findings implicating allergen-associated proteases in airway responses (10, 12), our present studies highlight the critical importance of protease-activated receptors, particularly PAR2, in epithelial IL-33 secretion in response to either ALT or HDM, which is associated with initial ATP release and activation of DUOX1. Moreover, although previous studies have implicated additional Ca2+ channels in airway epithelial responses to these allergens, such as P2X7 receptors (19) or CRAC (Ca2+ release-activated Ca2+) channels (15), our findings also demonstrate the critical importance of the transient receptor potential channel TRPV1 as a common mediator of allergen-induced activation of DUOX1 and IL-33 secretion.
Although PAR2 receptors can be directly activated by allergen-derived proteases, it is not clear how these allergens activate TRPV1. In addition to capsaicin, several other chemical ligands from exogenous (e.g., resiniferatoxin) or endogenous sources (so-called “endovanilloids,” including lipid-derived products such as anandamide), are believed to directly activate TRPV1 (34, 35). Interestingly, segmental allergen challenge of patients with allergic asthma was found to enhance local production of anandamide (36). TRPV1 can also be activated by physical stimuli such as heat (>42°C), alterations in membrane potential, low pH (<6), and various inflammatory mediators, although in these latter cases it is not always clear to what extent TRPV1 activation is due to direct stimulation or sensitization by additional cellular pathways. Our attempts to unravel the relative role(s) of the various receptor types in innate allergen responses yielded the surprising observation that, although direct activation of both PAR2 and TRPV1 evoked ATP release and subsequent purinoceptor activation, TRPV1 was also critical for IL-33 release in response to extracellular ATP. This latter finding is consistent with previous studies indicating that ATP stimulation of neuronal cells activates Ca2+ responses and nociception through combined activation of TRPV1 and P2YR2 (37) and with the general concept that GPCRs such as P2YR2 as well as PAR2 can potentiate or sensitize TRPV1 activation (29, 38). Hence, our findings imply that concerted or combined activation of PAR2, P2YR2, and TRPV1 by protease allergens such as HDM and ALT is necessary for optimal activation of Ca2+ signaling and DUOX1 activation, as a central mechanism of IL-33 secretion and production of related cytokines. The precise sequence of molecular events between these different receptor pathways is difficult to decipher, because they are subject to various reciprocal interactions. For example, Ca2+ signaling mechanisms can enhance ATP release (39), but ATP can also activate Ca2+ signaling by both P2YR2 and TRPV1. Moreover, because TRP channels including TRPV1 are also subject to redox alterations (40), it is possible that DUOX1 activation may also contribute to TRPV1 activation in a positive feedback mechanism. Nevertheless, the main novel aspect of our present studies is the apparent central role of TRPV1 in these innate airway epithelial responses by allergens or by ATP, as illustrated in Figure E7. Our findings may not be unique to TRPV1, as TRPV4 is also expressed within the nasal or airway epithelium (e.g., Figure 7) and may contribute to pulmonary inflammatory disease (41). Moreover, TRPV4 can also be sensitized by PAR2 receptor activation in, for example, DRG neurons, intensifying neurogenic inflammation and responses to various stimuli (42). Indeed, our studies suggest that TRPV4 activation can also induce DUOX1 activation and IL-33 secretion from airway epithelial cells, although TRPV1 appears to be the primary isoform involved in similar responses to allergens.
We did not specifically address what form(s) of IL-33 are being released from airway epithelial cells on exposure to allergen proteases, although our previous studies indicate that IL-33 is secreted primarily in its cleaved 18-kD form (9). Recent studies indicated that exogenous allergen cysteine proteases (in, e.g., A. alternata), as well as endogenous calpains from damaged airway epithelial cells, can process full-length IL-33 (33 kD) to its cleaved form (18 kD), to increase its cytokine activity (12, 43). Conversely, IL-33 is also subject to inactivation by oxidation of critical cysteine residues (44), which would suggest that, although DUOX1-dependent oxidative mechanisms are critical for initiating IL-33 secretion in response to allergen challenge, DUOX1 might also contribute to IL-33 inactivation by oxidation. Nevertheless, our findings indicate that PAR2-P2YR2-TRPV1-DUOX1 is critical for initial IL-33 alarmin activation, as indicated by subsequent type 2 responses (Reference 9 and present study).
Various TRP channels, including TRPV1, have been implicated in the pathology of asthma or related allergic diseases (21, 23, 45), and a loss-of-function SNP within TRPV1 (I585V) has been associated with lower risk of childhood asthma (24). Some studies in animal models of allergic asthma have suggested a contribution of TRPV1 to type 2 inflammation and airway hyperresponsiveness (25, 26), but other reports indicate that TRPV1 deficiency does not affect allergic asthma (46) or may even enhance Th2-biased inflammation (47). The contribution of TRPV1 to asthma has been attributed primarily to its role in nociceptor responses in sensory neurons, which are critical in development of cough and bronchospasm or airways hyperactivity, and also in allergic inflammation through neuroimmune crosstalk (45, 48). In addition to its presence in sensory neurons, TRPV1 has also been detected in the bronchial epithelium of patients with asthma (22), and our studies highlight a specific role of epithelial TRPV1 in allergen-induced IL-33 secretion as a major factor in activation of type 2 inflammation. Interestingly, the IL-33 receptor ST2 is also expressed in DRG neurons and was found to mediate itch responses in a model of poison ivy–induced allergic contact dermatitis (49). Thus, the contribution of TRPV1 in allergen-induced cough or bronchospasm in subjects with asthma could conceivably also involve initial IL-33 secretion through TRPV1 activation within the airway epithelium. Future studies with epithelia-specific deletion of TRPV1 would be needed to address this possibility.
In conclusion, our present findings highlight the importance of PAR2-P2YR2-TRPV1-DUOX1–mediated responses in innate epithelial responses to protease allergens, particularly with respect to IL-33 secretion and activation of type 2 inflammation. We recognize that our studies were based on acute challenge with relatively high doses of either HDM or ALT that may not necessarily reflect realistic exposure conditions. Although it is well established that exposure to HDM in dust has been associated with asthma development (e.g., Reference 50), the actual levels of airway HDM exposure are likely much lower than those used in the present study. Similarly, although fungal infection can contribute to asthma severity or complications (51), local concentrations of fungal allergens are difficult to estimate. However, the observed levels of IL-33 secretion in our present studies are quite similar to those observed in nasal secretions of subjects with asthma during exacerbations by respiratory syncytial virus (52) or in BAL of children with severe asthma with fungal sensitization (53). Therefore, we believe that our findings are informative with respect to proximal mechanisms that may contribute to such responses in subjects with asthma during, for example, exacerbations and offer mechanistic insight into the molecular pathways involved in exacerbated IL-33 responses observed in these settings. Observations from previous studies (9) and our present findings (Figure 7) indicating that P2YR2, PAR2, TRPV1, and DUOX1 are all elevated in basal nasal epithelial cells from patients with allergic asthma or chronic rhinosinusitis, combined with recent observations that basal epithelial cells from patients with allergic chronic rhinosinusitis display features of allergic inflammatory “memory” (54), would suggest that such elevations in PAR2-P2YR2-TRPV1-DUOX1 in the airways of these subjects may underlie increased risk for IL-33 activation and exacerbations (53, 55). In light of the current interest in developing biologics against IL-33 and related epithelial cytokines in treating allergic disease (5), our study indicates that selective targeting of upstream factors in this pathway, such as TRPV1, may also deserve consideration.
Footnotes
Supported by National Institutes of Health grants HL085646 and HL138708 (A.v.d.V.) and an unrestricted grant from Chiesi Pharmaceuticals (E.F.M.W.).
Author Contributions: C.S., M.H., and A.H. performed and analyzed the experiments; C.M.D. and K.D. assisted with interpretation of data; N.L.R. and E.F.M.W. assisted with experimental design and interpretation of data; C.S. and A.v.d.V. were responsible for planning, discussion and interpretation of experiments, and for writing the manuscript; A.v.d.V. was responsible for the conception and overall supervision of the project. All authors read and approved the final manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0170OC on March 17, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Holtzman MJ, Byers DE, Alexander-Brett J, Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol. 2014;14:686–698. doi: 10.1038/nri3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambrecht BN, Hammad H. Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol. 2014;134:499–507. doi: 10.1016/j.jaci.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 3.Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186:4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cayrol C, Girard JP. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev. 2018;281:154–168. doi: 10.1111/imr.12619. [DOI] [PubMed] [Google Scholar]
- 5.Roan F, Obata-Ninomiya K, Ziegler SF. Epithelial cell-derived cytokines: more than just signaling the alarm. J Clin Invest. 2019;129:1441–1451. doi: 10.1172/JCI124606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med. 1999;190:895–902. doi: 10.1084/jem.190.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–1432, e11. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Hristova M, Habibovic A, Veith C, Janssen-Heininger YM, Dixon AE, Geiszt M, et al. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol. 2016;137:1545–1556, e11. doi: 10.1016/j.jaci.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter PC, Yang T, Luong A, Delclos GL, Abramson SL, Kheradmand F, et al. Proteinases as molecular adjuvants in allergic airway disease. Biochim Biophys Acta. 2011;1810:1059–1065. doi: 10.1016/j.bbagen.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snelgrove RJ, Gregory LG, Peiro T, Akthar S, Campbell GA, Walker SA, et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134:583–592, e6. doi: 10.1016/j.jaci.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott IC, Majithiya JB, Sanden C, Thornton P, Sanders PN, Moore T, et al. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep. 2018;8:3363. doi: 10.1038/s41598-018-21589-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asokananthan N, Graham PT, Stewart DJ, Bakker AJ, Eidne KA, Thompson PJ, et al. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immunol. 2002;169:4572–4578. doi: 10.4049/jimmunol.169.8.4572. [DOI] [PubMed] [Google Scholar]
- 14.Yee MC, Nichols HL, Polley D, Saifeddine M, Pal K, Lee K, et al. Protease-activated receptor-2 signaling through β-arrestin-2 mediates Alternaria alkaline serine protease-induced airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2018;315:L1042–L1057. doi: 10.1152/ajplung.00196.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jairaman A, Maguire CH, Schleimer RP, Prakriya M. Allergens stimulate store-operated calcium entry and cytokine production in airway epithelial cells. Sci Rep. 2016;6:32311. doi: 10.1038/srep32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knight DA, Lim S, Scaffidi AK, Roche N, Chung KF, Stewart GA, et al. Protease-activated receptors in human airways: upregulation of PAR-2 in respiratory epithelium from patients with asthma. J Allergy Clin Immunol. 2001;108:797–803. doi: 10.1067/mai.2001.119025. [DOI] [PubMed] [Google Scholar]
- 17.Kauffman HF, Tomee JF, van de Riet MA, Timmerman AJ, Borger P. Protease-dependent activation of epithelial cells by fungal allergens leads to morphologic changes and cytokine production. J Allergy Clin Immunol. 2000;105:1185–1193. doi: 10.1067/mai.2000.106210. [DOI] [PubMed] [Google Scholar]
- 18.Page K, Ledford JR, Zhou P, Dienger K, Wills-Karp M. Mucosal sensitization to German cockroach involves protease-activated receptor-2. Respir Res. 2010;11:62. doi: 10.1186/1465-9921-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Grady SM, Patil N, Melkamu T, Maniak PJ, Lancto C, Kita H. ATP release and Ca2+ signalling by human bronchial epithelial cells following Alternaria aeroallergen exposure. J Physiol. 2013;591:4595–4609. doi: 10.1113/jphysiol.2013.254649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grace MS, Baxter M, Dubuis E, Birrell MA, Belvisi MG. Transient receptor potential (TRP) channels in the airway: role in airway disease. Br J Pharmacol. 2014;171:2593–2607. doi: 10.1111/bph.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belvisi MG, Birrell MA. The emerging role of transient receptor potential channels in chronic lung disease. Eur Respir J. 2017;50:1601357. doi: 10.1183/13993003.01357-2016. [DOI] [PubMed] [Google Scholar]
- 22.McGarvey LP, Butler CA, Stokesberry S, Polley L, McQuaid S, Abdullah H, et al. Increased expression of bronchial epithelial transient receptor potential vanilloid 1 channels in patients with severe asthma. J Allergy Clin Immunol. 2014;133:704–712, e4. doi: 10.1016/j.jaci.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 23.Tóth E, Tornóczky T, Kneif J, Perkecz A, Katona K, Piski Z, et al. Upregulation of extraneuronal TRPV1 expression in chronic rhinosinusitis with nasal polyps. Rhinology. 2018;56:245–254. doi: 10.4193/Rhin17.108. [DOI] [PubMed] [Google Scholar]
- 24.Cantero-Recasens G, Gonzalez JR, Fandos C, Duran-Tauleria E, Smit LA, Kauffmann F, et al. Loss of function of transient receptor potential vanilloid 1 (TRPV1) genetic variant is associated with lower risk of active childhood asthma. J Biol Chem. 2010;285:27532–27535. doi: 10.1074/jbc.C110.159491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi JY, Lee HY, Hur J, Kim KH, Kang JY, Rhee CK, et al. TRPV1 blocking alleviates airway inflammation and remodeling in a chronic asthma murine model. Allergy Asthma Immunol Res. 2018;10:216–224. doi: 10.4168/aair.2018.10.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rehman R, Bhat YA, Panda L, Mabalirajan U. TRPV1 inhibition attenuates IL-13 mediated asthma features in mice by reducing airway epithelial injury. Int Immunopharmacol. 2013;15:597–605. doi: 10.1016/j.intimp.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, et al. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol. 2006;575:555–571. doi: 10.1113/jphysiol.2006.111534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Yang C, Wang ZJ. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. 2011;193:440–451. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- 29.Mrozkova P, Spicarova D, Palecek J. Hypersensitivity induced by activation of spinal cord PAR2 receptors is partially mediated by TRPV1 receptors. PLoS One. 2016;11:e0163991. doi: 10.1371/journal.pone.0163991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC. Physiological regulation of ATP release at the apical surface of human airway epithelia. J Biol Chem. 2006;281:22992–23002. doi: 10.1074/jbc.M603019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Préfontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125:752–754. doi: 10.1016/j.jaci.2009.12.935. [DOI] [PubMed] [Google Scholar]
- 32.Fahy JV. Type 2 inflammation in asthma: present in most, absent in many. Nat Rev Immunol. 2015;15:57–65. doi: 10.1038/nri3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salazar F, Ghaemmaghami AM. Allergen recognition by innate immune cells: critical role of dendritic and epithelial cells. Front Immunol. 2013;4:356. doi: 10.3389/fimmu.2013.00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geron M, Hazan A, Priel A. Animal toxins providing insights into TRPV1 activation mechanism. Toxins (Basel) 2017;9:E326. doi: 10.3390/toxins9100326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunthorpe MJ, Chizh BA. Clinical development of TRPV1 antagonists: targeting a pivotal point in the pain pathway. Drug Discov Today. 2009;14:56–67. doi: 10.1016/j.drudis.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Zoerner AA, Stichtenoth DO, Engeli S, Batkai S, Winkler C, Schaumann F, et al. Allergen challenge increases anandamide in bronchoalveolar fluid of patients with allergic asthma. Clin Pharmacol Ther. 2011;90:388–391. doi: 10.1038/clpt.2011.94. [DOI] [PubMed] [Google Scholar]
- 37.Malin SA, Davis BM, Koerber HR, Reynolds IJ, Albers KM, Molliver DC. Thermal nociception and TRPV1 function are attenuated in mice lacking the nucleotide receptor P2Y2. Pain. 2008;138:484–496. doi: 10.1016/j.pain.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veldhuis NA, Poole DP, Grace M, McIntyre P, Bunnett NW. The G protein-coupled receptor-transient receptor potential channel axis: molecular insights for targeting disorders of sensation and inflammation. Pharmacol Rev. 2015;67:36–73. doi: 10.1124/pr.114.009555. [DOI] [PubMed] [Google Scholar]
- 39.Cordeiro JV, Jacinto A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nat Rev Mol Cell Biol. 2013;14:249–262. doi: 10.1038/nrm3541. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa N, Kurokawa T, Mori Y. Sensing of redox status by TRP channels. Cell Calcium. 2016;60:115–122. doi: 10.1016/j.ceca.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 41.Scheraga RG, Southern BD, Grove LM, Olman MA. The role of transient receptor potential vanilloid 4 in pulmonary inflammatory diseases. Front Immunol. 2017;8:503. doi: 10.3389/fimmu.2017.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W, et al. Protease-activated receptor 2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) protein coupling is required for sustained inflammatory signaling. J Biol Chem. 2013;288:5790–5802. doi: 10.1074/jbc.M112.438184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. 2018;19:375–385. doi: 10.1038/s41590-018-0067-5. [DOI] [PubMed] [Google Scholar]
- 44.Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. 2015;6:8327. doi: 10.1038/ncomms9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serhan N, Basso L, Sibilano R, Petitfils C, Meixiong J, Bonnart C, et al. House dust mites activate nociceptor-mast cell clusters to drive type 2 skin inflammation. Nat Immunol. 2019;20:1435–1443. doi: 10.1038/s41590-019-0493-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tränkner D, Hahne N, Sugino K, Hoon MA, Zuker C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc Natl Acad Sci USA. 2014;111:11515–11520. doi: 10.1073/pnas.1411032111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mori T, Saito K, Ohki Y, Arakawa H, Tominaga M, Tokuyama K. Lack of transient receptor potential vanilloid-1 enhances Th2-biased immune response of the airways in mice receiving intranasal, but not intraperitoneal, sensitization. Int Arch Allergy Immunol. 2011;156:305–312. doi: 10.1159/000323889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabata H, Artis D. Neuro-immune crosstalk and allergic inflammation. J Clin Invest. 2019;130:1475–1482. doi: 10.1172/JCI124609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu B, Tai Y, Achanta S, Kaelberer MM, Caceres AI, Shao X, et al. IL-33/ST2 signaling excites sensory neurons and mediates itch response in a mouse model of poison ivy contact allergy. Proc Natl Acad Sci USA. 2016;113:E7572–E7579. doi: 10.1073/pnas.1606608113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sporik R, Holgate ST, Platts-Mills TA, Cogswell JJ. Exposure to house-dust mite allergen (Der p I) and the development of asthma in childhood: a prospective study. N Engl J Med. 1990;323:502–507. doi: 10.1056/NEJM199008233230802. [DOI] [PubMed] [Google Scholar]
- 51.Denning DW, Pashley C, Hartl D, Wardlaw A, Godet C, Del Giacco S, et al. Fungal allergy in asthma-state of the art and research needs. Clin Transl Allergy. 2014;4:14. doi: 10.1186/2045-7022-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190:1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Castanhinha S, Sherburn R, Walker S, Gupta A, Bossley CJ, Buckley J, et al. Pediatric severe asthma with fungal sensitization is mediated by steroid-resistant IL-33. J Allergy Clin Immunol. 2015;136:312–322, e7. doi: 10.1016/j.jaci.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018;560:649–654. doi: 10.1038/s41586-018-0449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ravanetti L, Dijkhuis A, Dekker T, Sabogal Pineros YS, Ravi A, Dierdorp BS, et al. IL-33 drives influenza-induced asthma exacerbations by halting innate and adaptive antiviral immunity. J Allergy Clin Immunol. 2019;143:1355–1370, e16. doi: 10.1016/j.jaci.2018.08.051. [DOI] [PubMed] [Google Scholar]