

Abstract
Rationale: Long-term antibiotic use for managing chronic respiratory disease is increasing; however, the role of the airway resistome and its relationship to host microbiomes remains unknown.
Objectives: To evaluate airway resistomes and relate them to host and environmental microbiomes using ultradeep metagenomic shotgun sequencing.
Methods: Airway specimens from 85 individuals with and without chronic respiratory disease (severe asthma, chronic obstructive pulmonary disease, and bronchiectasis) were subjected to metagenomic sequencing to an average depth exceeding 20 million reads. Respiratory and device-associated microbiomes were evaluated on the basis of taxonomical classification and functional annotation including the Comprehensive Antibiotic Resistance Database to determine airway resistomes. Co-occurrence networks of gene–microbe association were constructed to determine potential microbial sources of the airway resistome. Paired patient-inhaler metagenomes were compared (n = 31) to assess for the presence of airway–environment overlap in microbiomes and/or resistomes.
Measurements and Main Results: Airway metagenomes exhibit taxonomic and metabolic diversity and distinct antimicrobial resistance patterns. A “core” airway resistome dominated by macrolide but with high prevalence of β-lactam, fluoroquinolone, and tetracycline resistance genes exists and is independent of disease status or antibiotic exposure. Streptococcus and Actinomyces are key potential microbial reservoirs of macrolide resistance including the ermX, ermF, and msrD genes. Significant patient-inhaler overlap in airway microbiomes and their resistomes is identified where the latter may be a proxy for airway microbiome assessment in chronic respiratory disease.
Conclusions: Metagenomic analysis of the airway reveals a core macrolide resistome harbored by the host microbiome.
Keywords: respiratory disease, metagenomics, antimicrobial resistance, macrolides, resistome
At a Glance Commentary
Scientific Knowledge on the Subject
Long-term and prophylactic antibiotics including macrolides are being increasingly used in the management of frequently exacerbating patients with chronic respiratory disease including severe asthma, chronic obstructive pulmonary disease, and bronchiectasis. The airway resistome, although recognized, is poorly characterized, and its relationship to the host microbiome unknown.
What This Study Adds to the Field
We, for the first time, in the largest and deepest metagenomics assessment of the airway evaluate airway resistomes across chronic respiratory disease states and relate them to host microbiomes. We identify a “core” airway resistome, harbored by the host microbiome and dominated by macrolide resistance genes but with high prevalence of β-lactam, fluoroquinolone, and tetracycline resistance. This core resistome is independent of health status or antibiotic exposure and shares significant overlap with resistomes detected on paired patient-inhaler devices where the latter represents a proxy for the host microbiome. Metagenomic analysis of the airway reveals a core macrolide resistome with implications for potential macrolide antibiotic resistance in the management of respiratory disease.
The development, progression, and associated phenotypes of chronic airways disease has been associated with perturbation of the airway microbiome. Such airway dysbiosis is often characterized by predominance of pathogenic bacteria such as Haemophilus, Streptococcus, and/or Pseudomonas, which coexist with a gamut of other taxa forming the airway “pathobiome” (1, 2). Patients with airways disease and frequent exacerbations are therefore prescribed long-term antibiotic regimes aiming to reduce bacterial burden and inflammation and improve clinical symptoms with variable results (3–5). The selective pressure resulting from such an approach, however, promotes antimicrobial resistance, a key global concern and serious threat to public health (5).
The impact of antimicrobial exposure on lung microbiome architecture and mechanisms promoting antimicrobial resistance remains an intense area of clinical and research interest. In addition, the broader implications of antibiotic exposure on the resident airway resistome, beyond that related to the target bacterial pathogen alone, has lacked study. This is important as interspecies interactions are plentiful in the airway, and the emergence of resistance in nonpathogenic organisms is a key factor potentially influencing therapeutic outcomes (6). Moreover, the environment remains a vast, mobilizable reservoir of resistance determinants with great potential to seed the airway resistome but remains inadequately characterized in the setting of chronic respiratory disease states (7).
As next-generation sequencing becomes cheaper, the era of clinical metagenomics represents an emerging and robust molecular tool allowing characterization of airway microbiomes in tandem to assessment of their functional properties for use in diagnosis, treatment, and/or patient risk stratification (8, 9). Work performed in cystic fibrosis (CF) reveals the cumulative effect of antibiotic exposure on the airway microbiome, and recent data in chronic obstructive pulmonary disease (COPD) confirm the airway as an important reservoir for antimicrobial resistance genes; however, no work has been performed using metagenomics, and no dedicated studies including severe asthma or bronchiectasis (10, 11). Unlike targeted amplicon-sequencing approaches (e.g., 16S rRNA), metagenomic shotgun sequencing remains less susceptible to PCR amplification bias, is not influenced by copy number variation, and, critically, provides scope to probe the functional aspects of the microbiome including its resistome (9, 12, 13).
Here, we report the largest application of deep metagenomic shotgun sequencing to airway samples across a range of chronic respiratory disease states (severe asthma, COPD, and bronchiectasis) and include nondiseased (healthy) individuals to characterize resident airway resistomes. We provide taxonomic and functional insight to airway-based resident antibiotic resistance in health and disease and further assess patient-inhaler devices as a potential environmental reservoir of resistance. Some of the results of these studies have been previously reported in the form of an abstract (14).
Methods
Study Population(s)
Patients with respiratory disease were prospectively recruited during routine attendance at respiratory outpatient clinics at two tertiary hospital sites in Singapore (Table 1): Singapore General Hospital and Tan Tock Seng Hospital. Nondiseased (healthy) individuals were recruited through an established voluntary exercise program at Nanyang Technological University, Singapore. Patients with severe asthma were graded as being on at least step four of the Global Initiative for Asthma guideline treatment ladder and met current criteria for severe asthma. COPD was defined according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria, and bronchiectasis (non-CF) was defined by radiological confirmation of bronchiectasis by either dynamic computed tomography or high-resolution computed tomography thorax in accordance with British Thoracic Society guidelines along with the absence of any other major concurrent chronic respiratory disease state (15–18). Nondiseased (healthy) individuals had no active or past history of any respiratory or other medical disease and normal spirometry measured in accordance with European Respiratory Society/American Thoracic Society criteria. Nondiseased individuals were free from any exposure to inhaled medications or antibiotic use in the preceding 12-month period. Demographics and associated clinical data were collated including age, sex, ethnicity, body mass index, lung function (FEV1% predicted), smoking status, and antibiotic use in the preceding 6-month period. In addition, the number of exacerbations in the year preceding study recruitment was recorded for all patients with respiratory disease (severe asthma, COPD, and bronchiectasis), and these patient groups had their respective disease severity/control scores recorded by the Asthma Control Test, GOLD score, and Bronchiectasis Severity Index, respectively (15, 19, 20).
Table 1.
Patient Demographics
| Demographic | ND | D |
P Value |
|||
|---|---|---|---|---|---|---|
| Severe Asthma | COPD | Bronchiectasis | ND vs. D | D vs. D | ||
| n | 13 | 11 | 15 | 15 | ||
| Age, yr | 34 ± 8 | 70 ± 17 | 70 ± 9 | 64 ± 15 | <0.001 | 0.472 |
| Sex | 0.528 | 0.012 | ||||
| M | 62% (8) | 73% (8) | 100% (15) | 53% (8) | ||
| F | 38% (5) | 27% (3) | 0% (0) | 47% (7) | ||
| BMI, kg/m2 | 23 ± 3 | 27.9 ± 8 | 21.7 ± 8 | 18.4 ± 6 | 0.237 | 0.009 |
| FEV1 % predicted | 109 ± 14 | 72 ± 14 | 45 ± 13 | 65 ± 20 | <0.001 | 0.005 |
| Disease severity* | ||||||
| Mild | — | 0% (0) | 7% (1) | 6% (1) | — | 0.797 |
| Moderate | — | 27% (3) | 40% (6) | 27% (4) | — | — |
| Severe | — | 73% (8) | 53% (8) | 67% (10) | — | — |
| Exacerbations in year preceding study recruitment | 0 ± 0 | 0 ± 1.5 | 3 ± 3 | 2 ± 1 | 0.032 | 0.013 |
| Smoking status | <0.001 | <0.001 | ||||
| Never-smoker | 100% (13) | 91% (10) | 0% (0) | 67% (10) | ||
| Ex-smoker | 0% (0) | 0% (0) | 47% (7) | 27% (4) | ||
| Current smoker | 0% (0) | 9% (1) | 53% (8) | 7% (1) | ||
| Antibiotic use in 6 mo preceding recruitment | 0.050 | <0.001 | ||||
| Yes | 0% (0) | 0% (0) | 67% (10) | 20% (3) | ||
| No | 100% (13) | 100% (11) | 33% (5) | 80% (12) | ||
| Inhaled corticosteroid use | ||||||
| Yes | — | 100% (11) | 53% (8) | 53% (8) | — | 0.064 |
| No | — | 0% (0) | 47% (7) | 47% (7) | — | — |
| Inhaled bronchodilator use | ||||||
| Yes | — | 100% (11) | 100% (15) | 53% (8) | — | 0.009 |
| No | — | 0% (0) | 0% (0) | 47% (7) | — | — |
Definition of abbreviations: ACT = Asthma Control Test; BMI = body mass index; BSI = Bronchiectasis Severity Index; COPD = chronic obstructive pulmonary disease; D = diseased; GOLD =Global Initiative for Chronic Obstructive Lung Disease; ND = nondiseased.
Demographic data is presented as median values ± interquartile range or percentages (patient numbers) as appropriate. Significant P values (P < 0.05) are bolded.
Defined according to disease-specific criteria. Severe asthma: ACT score; ≥20 = mild, 19–15 = moderate, <15 = severe. COPD: GOLD stage; 1 = mild, 2 = moderate, ≥3 = severe. Bronchiectasis: BSI; 0–4 = mild, 5–8 = moderate, ≥9 = severe.
Whole-Genome Shotgun Sequencing of Clinical and Environmental Samples
Representative, spontaneously expectorated sputum samples were obtained from all participants through directed coughing using the huff cough maneuver. An independent and prospective cohort of patients with severe asthma, COPD, or bronchiectasis (as defined above) using regular inhaled bronchodilator and/or corticosteroid therapy were recruited and the inhaler used most frequently sampled (Table E1 in the online supplement). Patients had documented self-reported inhaler adherence over the preceding 6-month period and demonstrated objective evidence of stable and/or improved pulmonary function on spirometry. Paired airway specimen (sputum) and an inhaler swab was obtained and subjected to metagenomic analysis. DNA was extracted from airway and environmental samples according to previously described clinical and environmental sampling methods (21, 22). DNA was used in the preparation of metagenomic shotgun-sequencing libraries as described (23). Resultant sequence data were processed and quality-trimmed before subjecting it to secondary analysis to derive taxonomic and functional genomic profiles including analysis with specific reference to the Comprehensive Antimicrobial Resistance Database (CARD) to assess the metagenomic resistome in all patient and environmental specimens (24).
Full details on DNA extraction, metagenomic sequencing, functional and taxonomic assignment of metagenomic sequence reads, data analysis, visualization, statistical analysis, and details on ethical approvals are provided in the online supplement.
Results
The Airway Metagenome Exhibits Functional Metabolic Dysbiosis and Antibiotic Resistance
Functional classification of microbial gene content based on read assignment to functional categories illustrates variability in patients with COPD and bronchiectasis in contrast to relatively comparable profiles in healthy individuals and patients with severe asthma (Figure 1A). A dysbiotic shift in the abundance of carbohydrate- and amino acid–related pathways toward lipid-associated pathways is evident in COPD and bronchiectasis with the latter exhibiting greatest change (Figure 1A). Classification of metagenomic data with reference to the Kyoto Encyclopedia of Genes and Genomes functional database further reveals alteration in genes associated with antibiotic-associated pathways including degradation, detoxification, and antimicrobial resistance (25). Although drug-related and xenobiotic metabolism is increased in COPD and bronchiectasis, genes involved in streptomycin, butirosin, neomycin, and β-lactamase biosynthesis are abundant in severe asthma and healthy individuals (Figure 1A). To probe specifically for the composition of antibiotic resistance genes within the airway metagenome, reads were classified with reference to the CARD database—a dataset of curated antibiotic resistance genes (24). Derived antibiotic resistance gene profiles exhibit variability across the healthy and diseased states with contrasting abundances of aminoglycoside, bicyclomycin, diaminopyrimidine, multidrug, peptide, phenicol, sulphonamides, sulfones, and triclosan resistance genes (Figure 1B). Critically, recent antibiotic exposure (selective pressure) did not translate to a detectably higher relative abundance of antimicrobial resistance (AMR), and, importantly, AMR profiles of healthy individuals and patients unexposed to antibiotics in the preceding 6-month period exhibit a significant presence of AMR suggesting the presence of a core airway resistome (Figure 1C and Table 2).
Figure 1.
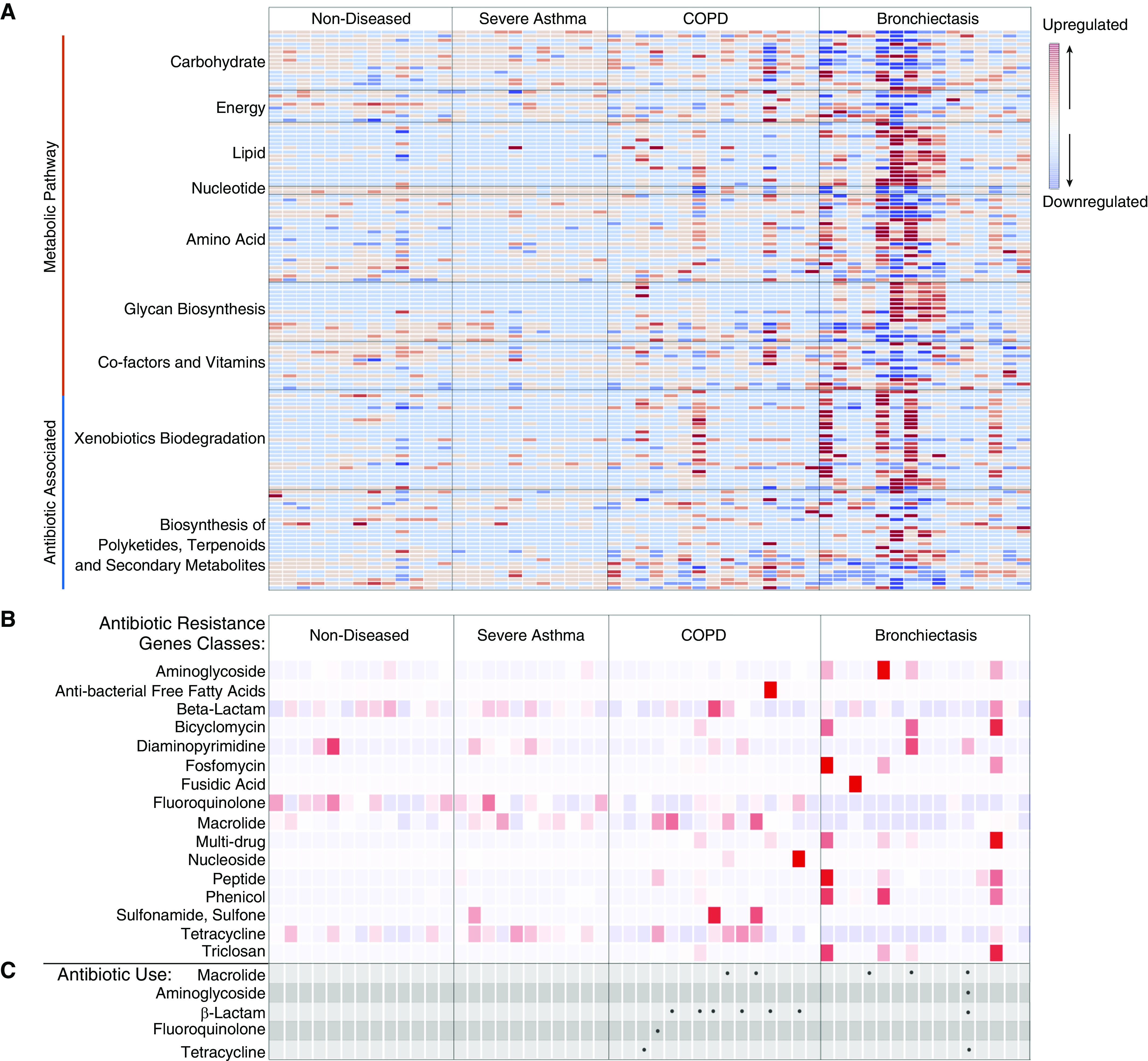
Airway shotgun metagenomics reveals functional metabolic dysbiosis and an increased antibiotic resistance gene abundance across chronic respiratory disease states. (A) Heatmap illustrating the relative abundance of functionally classified sequence reads assigned to functional categories (Kyoto Encyclopedia of Genes and Genomes) in each microbiome profile. Values are expressed as z-scores (calculated on the basis of the deviation from the mean abundance in each group and scaled to the SD). Higher abundance (indicated in red) is associated with specific functional pathways including lipid metabolism, xenobiotic biodegradation, and antibiotic-associated biosynthetic pathways. These are highest in patients with chronic obstructive pulmonary disease (COPD) and bronchiectasis. (B) Heatmap illustrating specific antibiotic resistance gene abundance (by class) based on read alignment to the Comprehensive Antimicrobial Resistance Database. β-Lactam, fluoroquinolone, macrolide, and tetracycline resistance genes are detectable in subjects with and without disease. Patients with COPD and bronchiectasis have the highest load of antibiotic resistance determinants. (C) Patient antibiotic usage and respective class (in the 6 mo preceding airway sampling) is indicated by black dots.
Table 2.
Genes of the Core Resistome (n = 18) Identified across Study Cohorts
| Gene Name | Drug Class | Resistance Mechanism |
|---|---|---|
| aac (3)-VIIa | Aminoglycoside | Antibiotic inactivation |
| aph (3)-IIIa | Aminoglycoside | Antibiotic inactivation |
| oxa-255 | Cephalosporin, penam (β-lactam) | Antibiotic inactivation |
| cfxA2 | Cephamycin (β-lactam) | Antibiotic inactivation |
| dfrA1 | Diaminopyrimidine | Antibiotic target replacement |
| pmrA | Fluoroquinolone | Antibiotic efflux |
| mel (msrD) | Macrolide, lincosamide, streptogramin, tetracycline, phenicol, oxazolidinone, pleuromutilin | Antibiotic target protection |
| ermB | Macrolide, lincosamide, streptogramin | Antibiotic target alteration |
| ermF | Macrolide, lincosamide, streptogramin | Antibiotic target alteration |
| ermX | Macrolide, lincosamide, streptogramin | Antibiotic target alteration |
| lnuC | Lincosamide | Antibiotic inactivation |
| efrB | Rifamycin, macrolide, fluoroquinolone | Antibiotic efflux |
| catS | Phenicol | Antibiotic inactivation |
| tetA (46) | Tetracycline | Antibiotic efflux |
| tetW | Tetracycline | Antibiotic target protection |
| tetB (46) | Tetracycline | Antibiotic efflux |
| tet(D) | Tetracycline | Antibiotic efflux |
| tetO | Tetracycline | Antibiotic target protection |
The Core Airway Resistome
Variable assemblages of antibiotic resistance genes are identified across individual disease cohorts including healthy individuals. Patients with COPD and bronchiectasis have the greatest repertoire of antibiotic resistance determinants, although this was potentially biased by the greater number of patients in these groups (Figure 2A). Patients with COPD harbored the highest diversity of resistance genes (n = 92; 85% shared with other cohorts) distinguished by the presence of specific β-lactam, fluoroquinolone, macrolide, multidrug, phenicol, and tetracycline resistance determinants. Although healthy individuals and severe asthmatics had a slightly lower number of cumulative AMR sequences, analysis revealed a strikingly consistent subset of 18 AMR genes common to all cohorts (including healthy) representing a “core resistome” (Figure 2B and Table 2). Importantly, this core resistome was predominated by AMR genes from the β-lactam, fluoroquinolone, macrolide, and tetracycline classes (Figures 1B and 2B and 2C and Table 2); detectable in every individual recruited into our study irrespective of health or disease status; and did not differ by type of chronic respiratory disease. In aggregate, genes encompassing a core macrolide resistome were most abundant and included msrD (mel), ermB, ermX, and ermF accompanied by genes encoding tetracycline (tetW, tetA [46], tetB [46]), β-lactam (cfxA2), and fluoroquinolone (pmrA) resistance (Figure 2C and Table 2).
Figure 2.
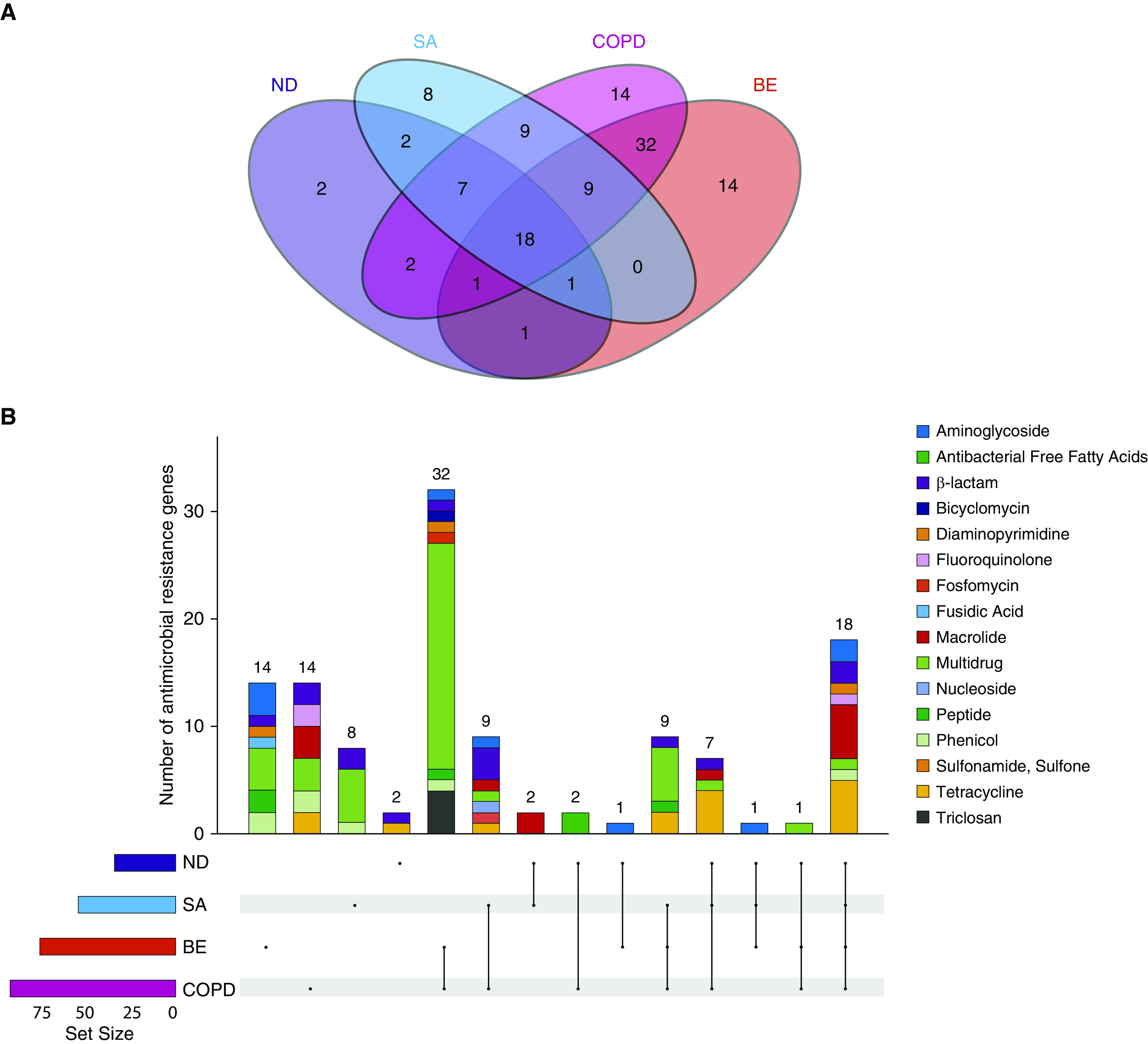
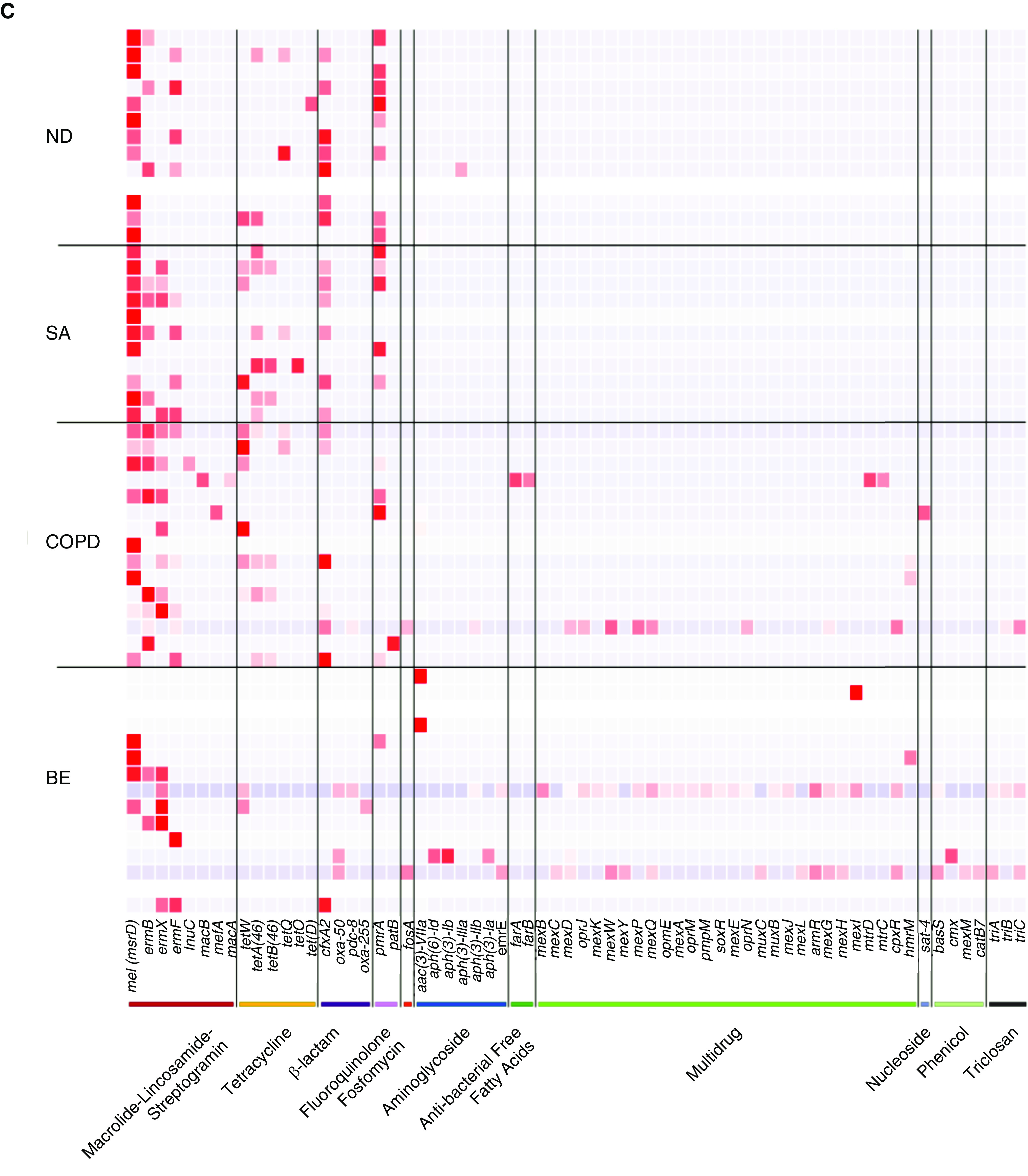
A core airway resistome exists across respiratory disease states including antibiotic-naive and nondiseased (healthy) individuals. (A) Venn diagram illustrating the number of individual antibiotic resistance genes among the study cohorts and their intersections. (B) An upset plot, corresponding to the presented Venn diagram in A, illustrating the antibiotic resistance gene composition across individual cohorts and their intersections. Stacked bar charts reflect the detected antibiotic resistance genes colored according to antibiotic class. Individual groups and their intersections are indicated for each cohort separately (ND, SA, COPD, and BE), followed by their respective intersection by a matrix (located below stacked bars). Set size (i.e., the number of resistance genes detected per group) is indicated by horizontal bars (ND < SA < COPD < BE). Black dots indicate sets, and connecting lines indicate relevant intersections related to each stacked bar chart. An 18-gene core resistome was identified (across all four cohorts) and largely comprises genes conferring macrolide, tetracycline, β-lactam, and aminoglycoside resistance, whereas the 32 genes shared by patients with COPD and patients with bronchiectasis are predominantly multidrug and triclosan resistance classes. (C) Heatmap illustrating specific antibiotic resistance genes by class and individual cohort. Specific antibiotic resistance genes grouped by colored class (x axis) are plotted against individual cohorts (ND, SA, COPD, and BE) (y axis). Genes are presented in order of detected abundance with msrD (mel), ermB, ermF, and ermX macrolide resistance genes most frequently observed across all four cohorts followed by genes encoding tetracycline, β-lactam, and fluoroquinolone resistance. BE = bronchiectasis; COPD = chronic obstructive pulmonary disease; ND = nondiseased; SA = severe asthma.
The Microbial Ecology of Sputum Samples across Respiratory Disease States
Deep sequencing of sputum revealed a microbiome profile dominated by bacteria, with fungi and viruses accounting for <0.01% and <0.25% average relative abundance, respectively (Figure E1). Dysbiosis of the respiratory microbiome was evident across chronic respiratory disease in line with established literature (Figure 3A) (26). Microbiome α-diversity between groups varied with healthy individuals exhibiting highest Shannon diversity compared with disease (Figure E3A). Simpson diversity was comparable across cohorts, whereas patients with severe asthma and bronchiectasis exhibited reduced Chao1 (Figures E3B and E3C). Among the respiratory disease cohorts, microbiome profiles were distinguished by Actinobacteria (Rothia spp.) and proteobacteria (Pseudomonas and Haemophilus spp.), which exhibited increased abundance in diseased cohorts with corresponding reductions in Prevotella spp., Treponema spp. Fusobacteria spp., and diverse Firmicutes compared with healthy individuals on the basis of linear discriminant analysis (Figures E3D and E3E). Among patients with disease, the increased abundance of Rothia mucilaginosa and Pseudomonas aeruginosa were among the most striking species-level differences associated with disease status in our analysis (Figure 3A). Fungi were detected sporadically and at low abundance, and microbiome β-diversity reveals heterogeneity to be highest in bronchiectasis, followed by COPD (Figure 3B). Conversely, healthy subjects and patients with severe asthma have more evenly distributed and diverse bacterial species in their airways further supported by analysis of average centroid distances, which is greatest in COPD and bronchiectasis (Figure 3C).
Figure 3.
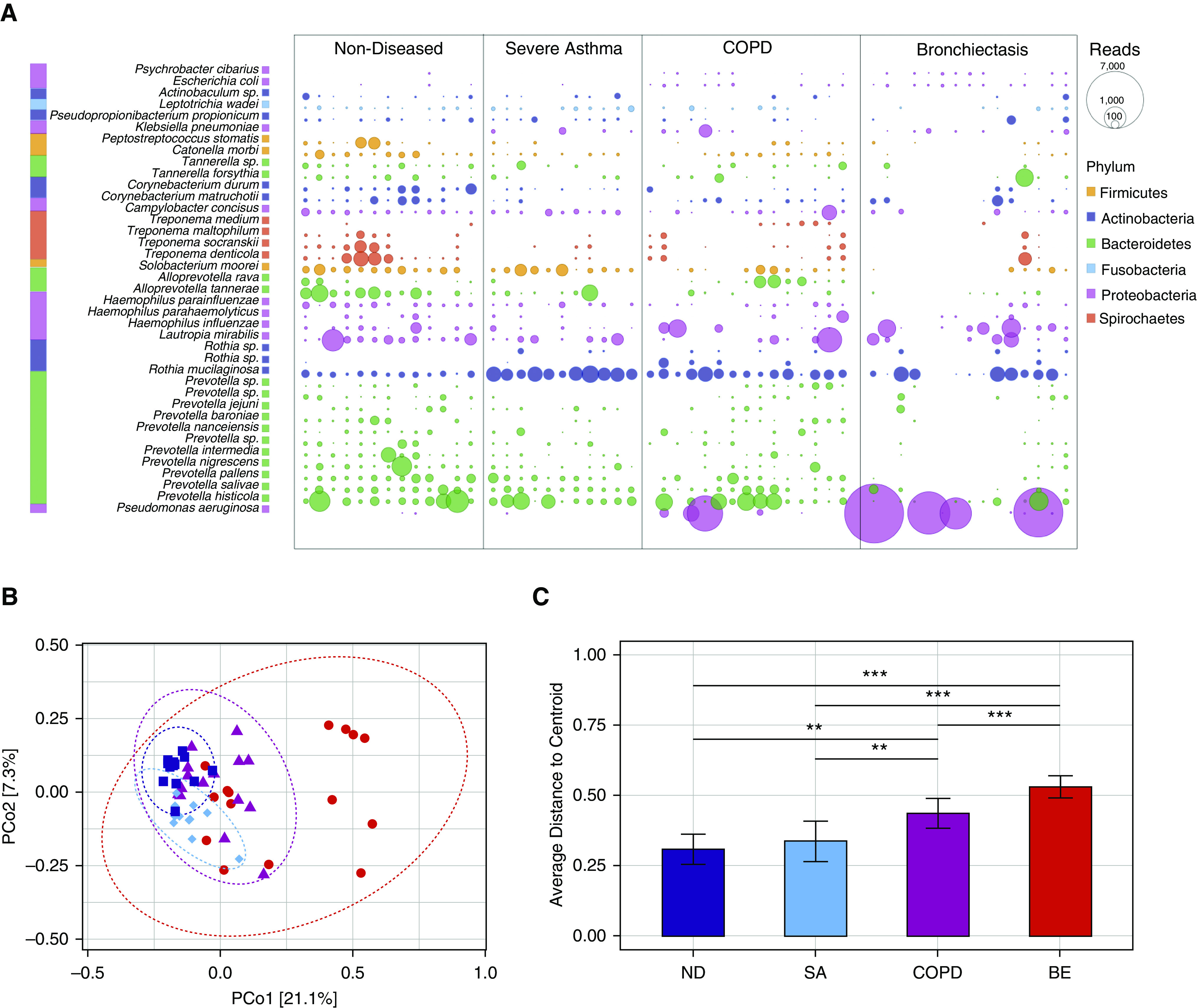
Metagenomic microbiome taxonomic composition exhibits disease-associated signatures with greatest heterogeneity in COPD and bronchiectasis. (A) Bubble chart illustrating microbial abundance of discriminant taxa in nondiseased versus diseased cohorts based on species-level classification. Bubble size corresponds to read count, and phylum membership is color-coded. Rothia mucilaginosa was consistently increased in subjects with disease versus subjects without disease (Dunn’s test, P = 0.01), whereas Pseudomonas aeruginosa was also increased, most notably among patients with bronchiectasis (Dunn’s test, P = 0.02). (B) A principle coordinate analysis plot illustrating β-diversity between the study groups including ND (dark blue), SA (light blue), COPD (purple), and BE (red), each highlighted by a colored ellipse. (C) The average distances to centroid from the principle coordinate analysis was plotted for each respective study group illustrating the heterogeneity of their respective microbiome profiles (error bars reflect SD). Difference in average distance to centroid was formally confirmed by ANOVA and Tukey post hoc analysis; **P < 0.01 and ***P < 0.001. BE = bronchiectasis; COPD = chronic obstructive pulmonary disease; ND = nondiseased; PCo1 = principle coordinate 1; PCo2 = principle coordinate 2; SA = severe asthma.
Gene–Microbe Co-occurrence in the Core Resistome
We next looked at correlation between microbial taxa and the core resistome by constructing a co-occurrence network of microbes and their respective antibiotic resistance genes (Figure 4). This revealed significant relationships (including correlation) between resistance genes (Figure 4A) and airway microbiota (Figure 4B) allowing inference of gene–microbe associations within an integrated holistic network. We focused on the core macrolide resistome because it formed a key component of the core resistome (Figures 2B and 2C) and is an antibiotic class gaining widespread use across chronic respiratory disease states (17, 27, 28). The macrolide resistance gene ermX represents a highly connected node associated with several microbial taxa (Figure 4C). These predominantly consist of upper airway commensals containing only few overtly pathogenic species (Figure 4C). Network inference further identifies a lesser number of microbial associations with ermF (Figure 4D) and msrD (Figure 4E), respectively. Streptococci and Actinomyces were associated with ermX and ermB, whereas the strongest association detected is between ermF and the gut microbe Bacteroidetes thetaiotaomicron. Associations between msrD and the gut pathogen Clostridioidies difficile and the largely unstudied Morococcus cerebrosus were also detected (Figures 4D and 4E).
Figure 4.
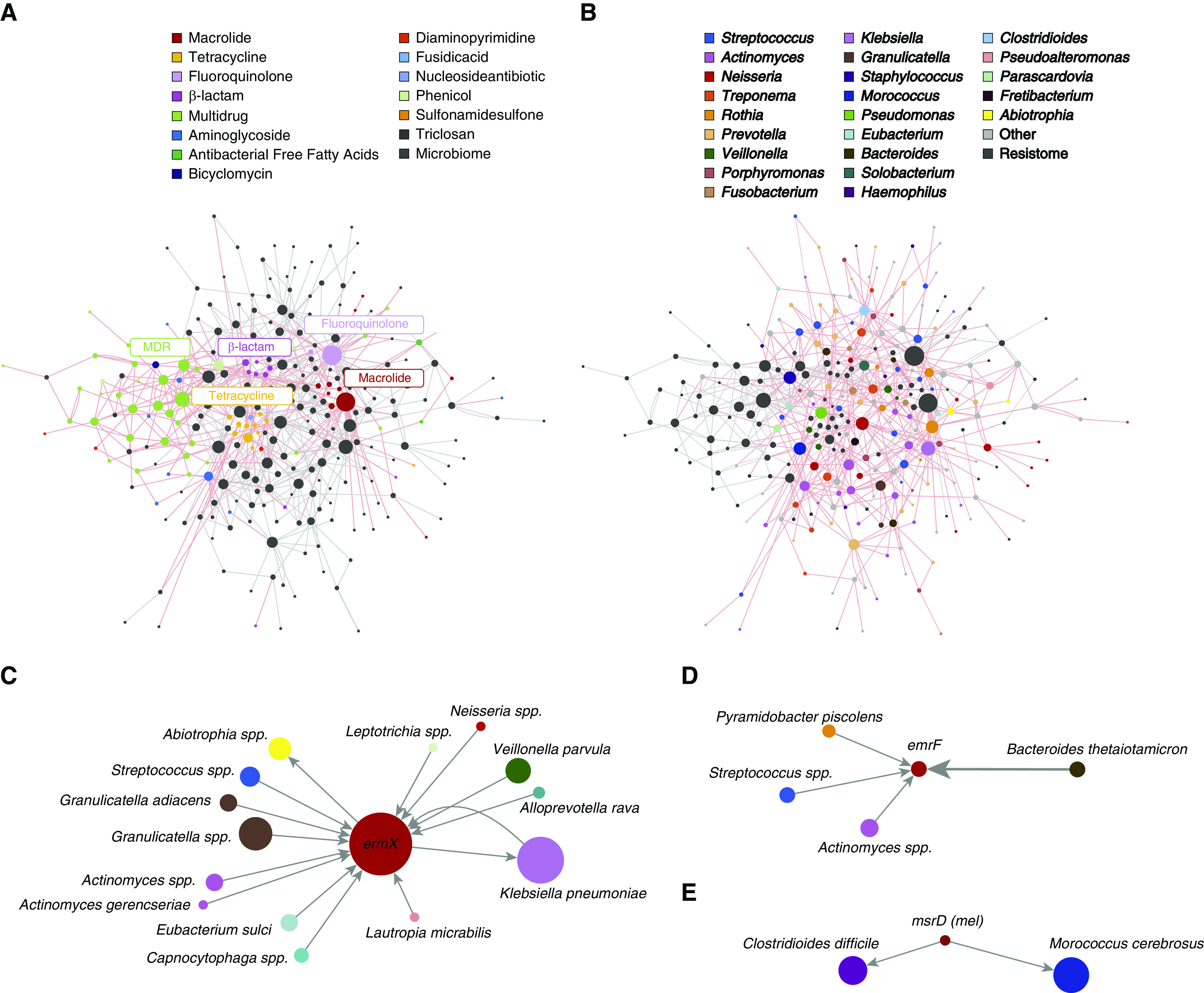
Network inference through co-occurrence analysis reveals gene–microbe associations of the core macrolide resistome. (A) Antibiotic resistance genes within the co-occurrence network are color-coded with respect to antibiotic class, whereas microbes are colored black. Gray lines denote interactions between nodes (representing both microbes and resistance genes), with line thickness reflecting their observed interaction strength. Interactions between resistance genes are highlighted by red lines. (B) Microbes within the co-occurrence network are color-coded with respect to their species, whereas antibiotic resistance genes are colored black. Gray lines denote interactions between nodes (microbes or resistance genes), with thickness reflecting interaction strength. Interactions between species are highlighted by red lines. (C–E) Identified nodes of the macrolide resistome are highlighted, indicating the specific microbes (by species) that associate with ermX (C), ermF (D), and msrD (E). Line thickness reflects the observed interaction strength between microbial nodes and the central resistance gene, whereas arrows depict directionality of the co-occurrence prediction.
Metagenomics Identifies Inhaler Devices as Potential Sinks for Antibiotic Resistance
To assess for potential sites of resistance host–environment transfer, patient-inhaler devices with respectively paired airway specimens were obtained in an independent prospectively recruited cohort of patients with chronic respiratory disease and subjected to metagenomics sequencing and analysis (Table E1). We identify significant overlap between the airway metagenome and that detectable on patient-paired inhaler devices with a significant number of microbial taxa found on both specimens (Figure 5A). This suggests that an inhaler swab, when subjected to metagenomics, could represent a surrogate measure of a patient’s airway microbiome. The number of microbes observed on inhaler devices (n = 207) exceeds that detected from the airways (n = 116), suggestive of other potential environmental influences; however, 80 overlapping microbial taxa were observable in paired airway–inhaler specimens with 36 and 127 microbial taxa found in airway and inhaler devices, respectively, suggesting increased microbial diversity on inhaler devices including microbes of potential environmental origin (Figure 5B). Among the 80 co-occurring microbial taxa, most (63 species and 78.8% of all co-occurring taxa) were detected at the individual level in specimens obtained from the same patient, illustrated for the most abundant taxa in Figure 5C. Similar analyses focused on antibiotic resistance genes reveals comparable metagenomic profiles from paired airway–inhaler device specimens but with lower resistance gene abundance on inhaler devices (Figure 5D). Here, multidrug, macrolide, and tetracycline antibiotic resistance determinants were most frequently observed, a finding consistent with our previously described core resistome (Figure 5D). A slightly higher number of resistance determinants (n = 98) were found associated to inhaler devices as compared with airway specimen (n = 89), a finding consistent with the greater diversity of microbes seen on inhalers, whereas 53 resistance genes were overlapping between inhalers and sputum (Figure 5E). Among overlapping resistance genes between the airway–inhaler device, 86.8% (46 resistance genes) co-occurred in paired airway–inhaler device specimens illustrated for the most abundant resistance genes in Figure 5F. A subset of identified resistance genes was associated with inhaler devices without detectable levels in the sputum of any patient or nondiseased control. These genes may reflect potential environmental sources of resistance gene diversity, 12 of which were independently observed on at least two inhaler devices (Table E2).
Figure 5.
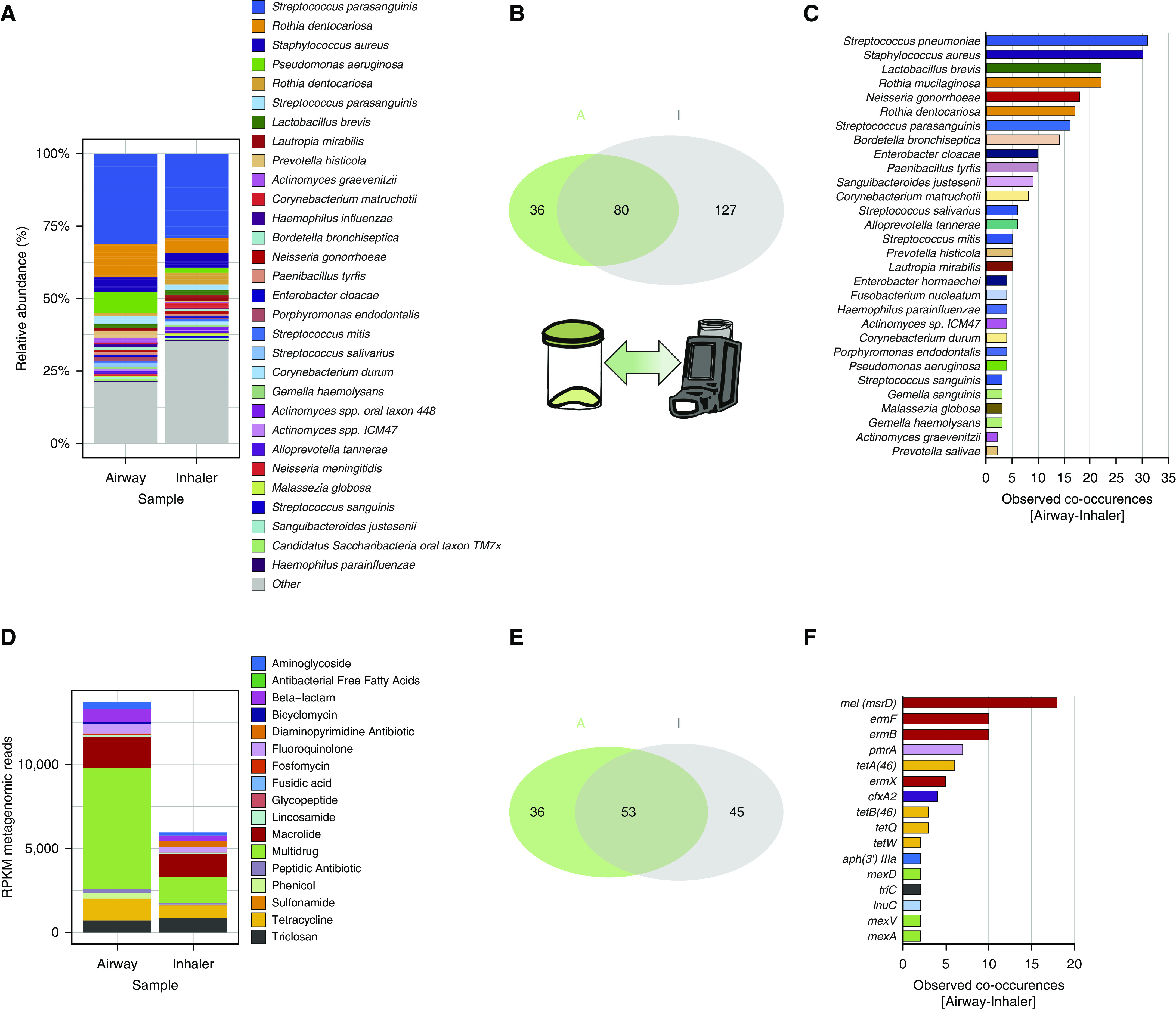
Metagenomics assessment of inhaler devices as potential antibiotic resistance reservoirs. Metagenomics shotgun analyses were performed on paired (airway–inhaler) specimens obtained through sputum collection and swabbing of mouthpieces of patient-inhaler devices. n = total of 31 pairs consisting of n = 16 (severe asthma), n = 11 (chronic obstructive pulmonary disease), and n = 4 (bronchiectasis). (A) Microbiome profiles of paired airway–inhaler devices exhibit a comparable overall pattern illustrated by stacked bar plots of a species-level relative abundance. (B) Venn diagram illustrating the observed metagenomics-derived microbial taxa present in the airway (green set, “A,” n = 116) and inhaler device (gray set, “I,” n = 207) and the co-occurrence of microbial species that are detectable in both groups (intersect, n = 80). Thirty-six and 127 species were therefore unique to the airway sputum and inhaler metagenomics profiles, respectively. (C) Horizontal bar plot indicating microbial species confirmed to co-occur in paired specimens (i.e., species found in both the airway and inhaler device of the same patient) (n = 63 species). (D) Resistance gene profiles for paired airway–inhaler devices demonstrate comparability with a higher abundance of resistance genes (measured in RPKM) detected in airway specimens. (E) Venn diagram illustrating the observed diversity of resistance genes detected in airway specimens (green set, “A,” n = 89) and inhaler devices (gray set, “I,” n = 98) by metagenomics. Co-occurrence of a significant number of microbial species were detected (intersect, n = 53 species). Thirty-six and 45 resistance genes were unique to the airway sputum and inhaler metagenomics profiles, respectively. (F) Horizontal bar plot indicating resistance genes confirmed to co-occur in paired specimens (i.e., genes found in both the airway and inhaler device of the same patient) (n = 46 genes). Gene co-occurrences observed in n ≥ 2 subjects are plotted. RPKM = reads per kilobase million.
Co-occurrence Analysis of Gene–Microbe Associations in Sputum and Inhaler Devices
We next sought to further assess microbial correlates of resistance among the subset of microbes (n = 63) and resistance genes (n = 46) with confirmed co-occurrence detected across the paired airway–inhaler devices (Figures 5C and 5F). Consistent with resistance gene abundance, the most highly correlated gene–microbe pairs included genes conferring resistance to macrolides (n = 4), tetracyclines (n = 4), β-lactams (n = 1), and fluoroquinolones (n = 1). Several species exhibit significant correlation with co-occurring resistance genes and predominantly comprise Firmicutes (Carnobacterium, Granulicatella, Prevotella, and Streptococcus) and Actinobacteria (Actinomyces, Corynebacterium, and Rothia) (Figure 6A). Correlation analysis reveals significant association for all four macrolide genes (msrD, ermF, ermB, and ermX) as well as fluoroquinolone (pmrA), tetracycline (tetA_46 and tetB_46), and β-lactam (cfxA2) resistance determinants. The most highly correlated species associated with msrD was Carnobacterium maltaromanticum; a lactic acid bacterium frequently found in food products including fish, meat, and some dairy. Prevotella intermedia was most closely correlated with ermF, whereas Actinomyces ICM47 was associated with ermF and ermX gene abundance. Streptococci was associated with the presence of fluoroquinolone and tetracycline resistance determinants, whereas Prevotella pallens was identifiable as the most likely microbial source of cfxA2 β-lactamase on the basis of maximal correlation coefficients and significance compared with other taxa. The abundance of these putative microbial resistance vehicles and their associated resistance genes are illustrated in Figure 6B.
Figure 6.
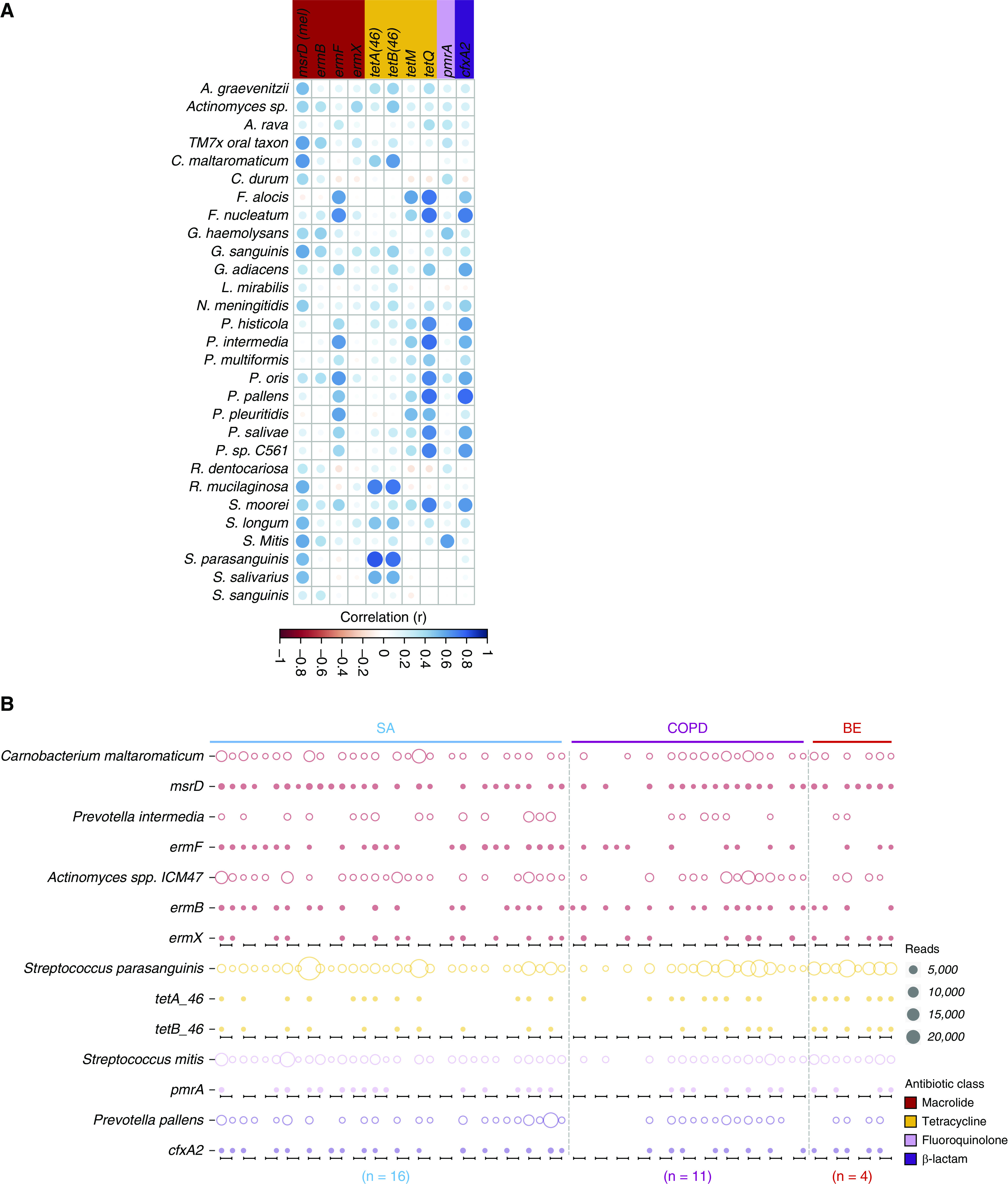
Metagenomic derivation of microbe–gene associations highlighting a potential source of resistance implicated in airway–inhaler device crossover. (A) Correlation plot of microbes and resistance genes coidentified in metagenomic profiles from paired patient airway and inhaler device specimens. The presence of a circle indicates significant association (P < 0.05), whereas circle size and color intensity reflect observed Pearson’s correlation for all pairwise comparisons indicating the strong positive correlations detected between microbes and resistance genes. The antibiotic resistance genes are color-coded according to their respective antibiotic class. (B) Bubble chart illustrating the co-occurrence of the most highly correlated microbe (open circle) and resistance gene (solid circle) combinations illustrated by disease (i.e., SA, COPD, and BE). Bubble size represents the number of classified reads, whereas color indicates the antibiotic class. Black bars along x axis indicate each individual paired airway and inhaler specimen, respectively (from left to right). A. graevenitzii = Actinomyces graevenitzii; A. rava = Alloprevotella rava; BE = bronchiectasis; COPD = chronic obstructive pulmonary disease; C. maltaromaticum = Carnobacterium maltaromaticum; C. durum = Corynebacterium durum; F. alocis = Filifactor alocis; F. nucleatum = Fusobacterium nucleatum; G. adiacens = Granulicatella adiacens; G. haemolysans = Gemella haemolysans; G. sanguinis = Globicatella sanguinis; L. mirabilis = Lautropia mirabilis; N. meningitidis = Neisseria meningitidis; P. histicola = Prevotella histicola; P. intermedia = Prevotella intermedia; P. multiformis = Prevotella multiformis; P. oris = Prevotella oris; P. pallens = Prevotella pallens; P. pleuritidis = Prevotella pleuritidis; P. salivae = Prevotella salivae; P. sp. C561 = Prevotella sp. C561; R. dentocariosa = Rothia dentocariosa; R. mucilaginosa = Rothia mucilaginosa; SA = severe asthma; S. longum = Stomatobaculum longum; S. mitis = Streptococcus mitis; S. moorei = Solobacterium moorei; S. parasanguinis = Streptococcus parasanguinis; S. salivarius = Streptococcus salivarius; S. sanguinis = Streptococcus sanguinis.
Discussion
We describe the largest airway clinical metagenomics analyses performed to date linking the antimicrobial resistome to host microbiomes in chronic respiratory disease. Our work highlights the versatility and usefulness of metagenomics in assessing functional aspects of the human microbiome, including antibiotic resistance. The airway metagenome exhibits functional metabolic dysbiosis including increased antibiotic resistance, predominant in COPD and bronchiectasis. Variability exists in antibiotic-associated functional pathways in healthy and diseased airways, mirrored by a presence and high abundance of resistance genes. Critically, we uncover, even in a healthy state, the presence of a core resistome, dominated by genes from common antibiotic classes including macrolides, β-lactams, and fluroquinolones unrelated to antibiotic exposure. By assessing host microbiomes with increased robustness provided by metagenomics, we link the presence of specific bacterial taxa to the core resistome and specifically genes conferring macrolide resistance. Analysis of paired patient-inhaler metagenomes illustrates significant overlap suggesting the latter as a surrogate for the host microbiome. Patient-inhaler overlap confirms our identified resistance gene–microbe associations as well as identifies resistance determinants unique to inhaler devices aligning with the concept of the wider environment as source of resistance determinants linked to microbial and, therefore, resistance transfer between environment and host.
A key observation from this work is the high abundance of resistance genes within the Macrolide–Lincosamide–Streptogramin (MLS) axis, which dominates the airway resistome. This is important given the widespread and increasing use of macrolides across a variety of clinical respiratory diseases (4, 29–31). Prior work illustrates that macrolide resistance is among the most prevalent in the wider environment, representing a rich source of resistance determinants with strong potential for horizontal transfer (32). Our detection of a core airway resistome dominated by macrolide resistance is therefore of particular concern considering the selective pressure exerted by macrolides toward resistance in both the airway and wider environment (30, 33, 34). Recent work focused on environmental resistomes illustrates their distinct core and discriminatory elements (35). We, in similar fashion, detected core and discriminatory elements within the airway metagenome, where discriminant resistance genes had higher occurrence in diseased states (e.g., multidrug resistance in COPD and bronchiectasis), which associated with their microbiomes. A core airway resistome is also evident comprising genes from the macrolide, fluoroquinolone, β-lactam, and tetracycline antibiotic classes. This core resistome demonstrates stability across all respiratory diseases including healthy individuals and therefore explains the detected relative microbiome stability during exacerbations despite antibiotic pressure (30, 36, 37). Our description of the core airway resistome also provides a novel perspective on reasons why pathogens, expected to exhibit in vivo susceptibility to a particular antibiotic, are not necessarily eradicated by appropriate antimicrobial therapy (38).
Predominance of macrolide resistance within the core resistome is of relevance in respiratory disease. Macrolide use is advocated in severe asthma, COPD, and, more recently, bronchiectasis, particularly for patients demonstrating recurrent and persistent exacerbations (27, 28, 39). The core macrolide resistome includes msrD (mel), ermB, ermX, and ermF genes, which incorporates efflux (msrD) and rRNA methylases (erm), mechanisms previously established in the human gut and sewage effluent (32, 34, 40). Of all macrolide resistance genes identified, ermX exhibits greatest abundance in respiratory disease and presents as a highly connected node in our co-occurrence analysis, its presence relating to several resident airway bacteria. Interestingly, ermF and mstD exhibit association with low-abundance gut microbiota including C. difficile and B. thetaiotaomicron, which are reported to harbor these genes in association with mobile genetic elements (41, 42). This suggests seeding of the airway resistome may occur, through aspiration of gut microbes, in the case of ermF and msrD, contrasting ermX, which strongly associates more directly with the respiratory microbiota. Silent aspiration and/or gastroesophageal reflux is proposed to occur in chronic respiratory disease including asthma, COPD, and bronchiectasis and therefore should be considered as a contributor to the airway resistome (43–46).
To better understand direct relationships and potential transfer between host and environmental resistomes, we next assessed metagenomes in paired patient-inhaler specimens in individual patients. We demonstrate that an inhaler swab, when subjected to metagenomics, may act as a surrogate of the airway microbiome and/or resistome: a feature of relevance in patients with dry nonproductive cough commonly seen in severe asthma but also COPD and bronchiectasis (47). Co-occurrence of microbe-resistance gene combinations was evident, strongly indicative of environmental contributors and microbial sources of the airway resistome. By generating comparable microbiome and resistome profiles between airway and inhalers, we confirm the potential for such therapeutic devices to act as resistance reservoirs, allowing trafficking of pathogenic microbes and resistance determinants between the environment and airway. Recent work illustrates the COPD airway to be an important reservoir for antibiotic resistance genes, linking their abundance to bacterial colonization (11). Interestingly, existing data further propose that macrolide resistance genes, including ermX, may be aerosolized and associate with COPD patient filter masks in the hospital setting (48). Our work further builds on such concepts, demonstrating the potential of clinical metagenomics in inhaler devices to uncover microbe-resistance gene associations through gene–species co-occurrence in environmental and airway specimens. We identify a subset of microbes and genes co-occurring between the airway and inhaler surface when integrated into a co-occurrence network leading to the identification of bacteria highly correlated to genes conferring macrolide, tetracycline, β-lactam, and fluoroquinolone resistance. Interestingly, this reveals taxa previously associated with antimicrobial resistance including Actinomyces, Streptococcus, and Prevotella species, implicating them as potential key vehicles for resistance (36, 49). Prevotella is known to exhibit reduced susceptibility to β-lactams in the CF lung, findings consistent with our observed association between P. pallens and the β-lactamase–encoding cfxA2 gene, whereas the presence of tet genes in Streptococcus is also previously described (50). Association between Carnobacterium maltaromaticum and msrD is not previously described, although the clinical relevance of Carnobacterium species remains to be fully established. Our detected relationship between Actinomyces species and resistance genes appears in contradiction to its described macrolide sensitivity; however, it should be noted that molecular (sequence) data related to Actinomyces spp. ICM47 have yet to be taxonomically confirmed and therefore potentially represent an exception to the general trend in this genera favoring macrolide susceptibility (30).
Our work is novel and represents the largest clinical metagenomics study performed on airway specimens using robust state-of-the-art methodologies effectively applied to low-biomass samples such as outdoor air (23). Using this approach, we uncover a core airway resistome dominated by macrolide resistance linked to the host microbiome. We further illustrate that inhaler devices may act as a surrogate of the host airway microbiome and remain a key resistance reservoir. Despite our study’s strengths and novelty, we acknowledge its limitations. First, despite being the largest clinical metagenomics study to date, we include only 85 individuals, which are further nested into healthy and diseased groups, within a cross-sectional study design requiring validation in larger longitudinal studies given the myriad of confounders that could possibly influence microbiome profiles. For instance, although patients with disease were relatively well matched for age, the healthy cohort were significantly younger. Even with a limited sample size, we could identify microbiome perturbation characteristic of respiratory disease (e.g., COPD and bronchiectasis). Heterogeneity and relationships between the resistome, microbiome, and specific disease phenotypes (e.g., very frequent exacerbators) within the diseased cohorts, most prominent for the bronchiectasis group, could not be fully resolved by this work because of the limited sample size. Recently, Taylor and colleagues have demonstrated the effect of macrolide exposure on the resistome in a longitudinal study of patients with severe asthma. Following azithromycin exposure, the abundance of several macrolide genes (including those observed in our analysis) ermB, ermF, mef, and mel (msrD) were detected, supporting the functional importance of these genes to the resistome in response to antibiotic exposure (30). Interestingly, shifts in the resistome in response to therapy were accompanied by clinical resistance in Haemophilus influenzae isolates, suggesting metagenomic resistome profiling may reflect clinically observed resistance that warrants further analysis in terms of diagnostic implications in the context of the findings from our study. Next, although metagenomic shotgun sequencing is a powerful tool, it remains relatively expensive with slower turnaround times compared with other sequencing approaches. This poses challenges for real-time diagnostic or therapeutic use and translation into everyday clinical practice (51). Although challenging owing to the nature of sample variability in particular conditions such as asthma or (dry) bronchiectasis compared with nondiseased controls, as a matrix, sputum is advantageous in terms of accessibility and scope for broad application in large studies across a range of clinical settings. This clearly contrasts with more invasive BAL or tissue biopsy sampling where sample acquisition and control subject recruitment are major limiting factors. Our study recruited several nondiseased participants, from which airway specimens were readily obtained by applying the huff cough technique in a protocol applied previously to nondiseased control samples in assessment of their airway microbiomes (21). Although clearly important, the degree to which sputum reflects the true microbial ecosystem of the lower airway has been the subject of debate, and its accessibility likely comes at a cost of reduced resolution of lower airway taxa, which may be particularly relevant in diseased states (52). Consistency of sputum sampling in terms of the relative proportions of upper and lower airway biomass in a given sample, variability in sampling during acquisition, and also changes with respect to disease severity are all likely to influence clinical association and remain important areas for future exploration. In our assessment of patient inhalers, we included rigorous swab-sampling contamination controls but lacked the additional experimental control of a swab taken from an unused inhaler device—an important consideration given the sensitivity of metagenomics to detect minute levels of background contamination. The high abundance of human DNA in sputum is an additional hurdle with implications for adequate and unbiased detection of resistance genes, as sequencing depth will influence detection. However, recent work in the area of host DNA removal may address this issue, allowing for greater microbial read depth and scalability of sputum metagenomics in this field (53). Furthermore, metagenomic data processing requires specialized personnel with bioinformatic skills and a facility with the capability to perform high-performance computing, both significant barriers to clinical implementation. The use of short-read sequencing also largely precludes a definitive assessment of mobile genetic elements associated with resistance genes, and long-read metagenomic workflows currently being developed may offer better insights going forward. Finally, our study detected change to genes controlling lipid metabolism across a range of chronic respiratory disease states. Alterations in lipid metabolism are identified in COPD and bronchiectasis and potentially contribute to their pathogenesis through initiation and resolution of inflammation. Emerging data suggest that microbial dysbiosis associates with such metabolic change and remains an important avenue for future study particularly in regard to effect on host immune function (26, 54, 55).
Despite economic, analytical, and resource-related challenges to the application of clinical metagenomics into respiratory practice, its ability to concurrently capture individual microbial taxonomy, function, and resistance in an unbiased robust manner using a single specimen makes it an attractive tool for the delivery of precision respiratory medicine. This is important in the current era of patient endophenotyping including disease overlap. Our identification of a core airway resistome, dominated by macrolide resistance, is an important cautionary warning worthy of clinical consideration. Despite advances in the use of antimicrobials to improve clinical outcomes across a range of chronic respiratory diseases, we must be cognizant of their potential long-term resistance implications and weigh this against the perceived short-term clinical benefit in individual respiratory patients.
Footnotes
Supported by the Singapore Ministry of Health’s National Medical Research Council under its Clinician-Scientist Individual Research Grant (MOH-000141) (S.H.C.) and Research Training Fellowship (NMRC/Fellowship/0049/2017) (P.Y.T.); the Singapore Ministry of Education under its Singapore Ministry of Education Academic Research Fund Tier 1 (2016-T1-001-050) (S.H.C) and Tier 3 (2013-T3-013) (S.C.S. and S.H.C.) and the Nanyang Technological University Integrated Medical, Biological and Environmental Life Sciences (NIMBELS), Nanyang Technological University, Singapore (NIM/03/2018) (S.C.S. and S.H.C.).
Data availability: All sequence data from this study have been uploaded to the National Center for Biotechnology Information sequence read archives under project accession number PRJNA595703.
Author Contributions: M.M.A. and K.J.X.L.: performance and design of experiments, data analysis and interpretation, statistical analysis, and writing the manuscript. Z.C. and J.K.N.: performance of antimicrobial resistance gene analysis and co-occurrence network inference. P.Y.T., T.H.O., and A.L.Y.H.: patient recruitment and procurement of clinical data and specimens. R.W.P. and D.I.D.-M.: metagenomic whole-genome shotgun sequencing and analytics. N.E.G. and T.K.J.: curation of clinical and environmental samples and data. M.S.K. and J.A.A.: intellectual contributions, patient recruitment, and procurement of clinical data and specimens. K.T.-A.: oversight of mathematical methods and statistics. S.C.S. and S.H.C.: conception and design of overall study and experiments, data analysis and interpretation, statistical analysis, writing the manuscript, and procurement of funding.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.201911-2202OC on April 22, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504. doi: 10.1146/annurev-physiol-021115-105238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pitlik SD, Koren O. How holobionts get sick—toward a unifying scheme of disease. Microbiome. 2017;5:64. doi: 10.1186/s40168-017-0281-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sibila O, Laserna E, Shoemark A, Keir HR, Finch S, Rodrigo-Troyano A, et al. Airway bacterial load and inhaled antibiotic response in bronchiectasis. Am J Respir Crit Care Med. 2019;200:33–41. doi: 10.1164/rccm.201809-1651OC. [DOI] [PubMed] [Google Scholar]
- 4.Herath SC, Normansell R, Maisey S, Poole P. Prophylactic antibiotic therapy for chronic obstructive pulmonary disease (COPD) Cochrane Database Syst Rev. 2018;10:CD009764. doi: 10.1002/14651858.CD009764.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miravitlles M, Anzueto A. Antibiotics for acute and chronic respiratory infection in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:1052–1057. doi: 10.1164/rccm.201302-0289PP. [DOI] [PubMed] [Google Scholar]
- 6.Vandeplassche E, Tavernier S, Coenye T, Crabbé A. Influence of the lung microbiome on antibiotic susceptibility of cystic fibrosis pathogens. Eur Respir Rev. 2019;28:190041. doi: 10.1183/16000617.0041-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Surette MD, Wright GD. Lessons from the environmental antibiotic resistome. Annu Rev Microbiol. 2017;71:309–329. doi: 10.1146/annurev-micro-090816-093420. [DOI] [PubMed] [Google Scholar]
- 8.Forbes JD, Knox NC, Peterson CL, Reimer AR. Highlighting clinical metagenomics for enhanced diagnostic decision-making: a step towards wider implementation. Comput Struct Biotechnol J. 2018;16:108–120. doi: 10.1016/j.csbj.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20:341–355. doi: 10.1038/s41576-019-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feigelman R, Kahlert CR, Baty F, Rassouli F, Kleiner RL, Kohler P, et al. Sputum DNA sequencing in cystic fibrosis: non-invasive access to the lung microbiome and to pathogen details. Microbiome. 2017;5:20. doi: 10.1186/s40168-017-0234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramsheh MY, Haldar K, Bafadhel M, George L, Free RC, John C, et al. Resistome analyses of sputum from COPD and healthy subjects reveals bacterial load-related prevalence of target genes. Thorax. 2019;75:8–16. doi: 10.1136/thoraxjnl-2019-213485. [DOI] [PubMed] [Google Scholar]
- 12.Pinto AJ, Raskin L. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One. 2012;7:e43093. doi: 10.1371/journal.pone.0043093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong S, Bunge J, Leslin C, Jeon S, Epstein SS. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009;3:1365–1373. doi: 10.1038/ismej.2009.89. [DOI] [PubMed] [Google Scholar]
- 14.Mac Aogáin M, Zhao C, Purbojati RW, Drautz-Moses DI, Lim AYH, Low TB, et al. The airway ‘resistome’ in chronic respiratory disease: a metagenomics approach. Eur Respir J. 2019;54:OA5141. [Google Scholar]
- 15.GOLD. Global strategy for the diagnosis, management and prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2018. 2018 [accessed 2018 Apr 28]. Available from: http://goldcopd.org/
- 16.Pasteur MC, Bilton D, Hill AT British Thoracic Society Bronchiectasis Non-CF Guideline Group. British Thoracic Society guideline for non-CF bronchiectasis. Thorax. 2010;65:i1–i58. doi: 10.1136/thx.2010.142778. [DOI] [PubMed] [Google Scholar]
- 17.Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J. 2014;43:343–373. doi: 10.1183/09031936.00202013. [DOI] [PubMed] [Google Scholar]
- 18.Global Initiative for Asthma. Global management and prevention 2019. [accessed 2019 Oct 17]. Available from: www.ginasthma.org.
- 19.Chalmers JD, Goeminne P, Aliberti S, McDonnell MJ, Lonni S, Davidson J, et al. The bronchiectasis severity index: an international derivation and validation study. Am J Respir Crit Care Med. 2014;189:576–585. doi: 10.1164/rccm.201309-1575OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathan RA, Sorkness CA, Kosinski M, Schatz M, Li JT, Marcus P, et al. Development of the asthma control test: a survey for assessing asthma control. J Allergy Clin Immunol. 2004;113:59–65. doi: 10.1016/j.jaci.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Mac Aogáin M, Chandrasekaran R, Lim AYH, Low TB, Tan GL, Hassan T, et al. Immunological corollary of the pulmonary mycobiome in bronchiectasis: the CAMEB study. Eur Respir J. 2018;52:1800766. doi: 10.1183/13993003.00766-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luhung I, Wu Y, Ng CK, Miller D, Cao B, Chang VW-C. Protocol improvements for low concentration DNA-based bioaerosol sampling and analysis. PLoS One. 2015;10:e0141158. doi: 10.1371/journal.pone.0141158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gusareva ES, Acerbi E, Lau KJX, Luhung I, Premkrishnan BNV, Kolundžija S, et al. Microbial communities in the tropical air ecosystem follow a precise diel cycle. Proc Natl Acad Sci USA. 2019;116:23299–23308. doi: 10.1073/pnas.1908493116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019;7:907–920. doi: 10.1016/S2213-2600(18)30510-1. [DOI] [PubMed] [Google Scholar]
- 27.Wedzicha JA, Ritchie AI, Martinez FJ. Can macrolide antibiotics prevent hospital readmissions? Am J Respir Crit Care Med. 2019;200:796–798. doi: 10.1164/rccm.201905-0957ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chalmers JD, Boersma W, Lonergan M, Jayaram L, Crichton ML, Karalus N, et al. Long-term macrolide antibiotics for the treatment of bronchiectasis in adults: an individual participant data meta-analysis. Lancet Respir Med. 2019;7:845–854. doi: 10.1016/S2213-2600(19)30191-2. [DOI] [PubMed] [Google Scholar]
- 29.Laska IF, Crichton ML, Shoemark A, Chalmers JD. The efficacy and safety of inhaled antibiotics for the treatment of bronchiectasis in adults: a systematic review and meta-analysis. Lancet Respir Med. 2019;7:855–869. doi: 10.1016/S2213-2600(19)30185-7. [DOI] [PubMed] [Google Scholar]
- 30.Taylor SL, Leong LEX, Mobegi FM, Choo JM, Wesselingh S, Yang IA, et al. Long-term azithromycin reduces Haemophilus influenzae and increases antibiotic resistance in severe asthma. Am J Respir Crit Care Med. 2019;200:309–317. doi: 10.1164/rccm.201809-1739OC. [DOI] [PubMed] [Google Scholar]
- 31.Welte T. Azithromycin: the holy grail to prevent exacerbations in chronic respiratory disease? Am J Respir Crit Care Med. 2019;200:269–270. doi: 10.1164/rccm.201903-0706ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendriksen RS, Munk P, Njage P, van Bunnik B, McNally L, Lukjancenko O, et al. Global Sewage Surveillance Project Consortium. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat Commun. 2019;10:1124. doi: 10.1038/s41467-019-08853-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milaković M, Vestergaard G, González-Plaza JJ, Petrić I, Šimatović A, Senta I, et al. Pollution from azithromycin-manufacturing promotes macrolide-resistance gene propagation and induces spatial and seasonal bacterial community shifts in receiving river sediments. Environ Int. 2019;123:501–511. doi: 10.1016/j.envint.2018.12.050. [DOI] [PubMed] [Google Scholar]
- 34.Roberts MC. Environmental macrolide-lincosamide-streptogramin and tetracycline resistant bacteria. Front Microbiol. 2011;2:40. doi: 10.3389/fmicb.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta S, Arango-Argoty G, Zhang L, Pruden A, Vikesland P. Identification of discriminatory antibiotic resistance genes among environmental resistomes using extremely randomized tree algorithm. Microbiome. 2019;7:123. doi: 10.1186/s40168-019-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choo JM, Abell GCJ, Thomson R, Morgan L, Waterer G, Gordon DL, et al. Impact of long-term erythromycin therapy on the oropharyngeal microbiome and resistance gene reservoir in non-cystic fibrosis bronchiectasis. mSphere. 2018;3:e00103-18. doi: 10.1128/mSphere.00103-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox MJ, Turek EM, Hennessy C, Mirza GK, James PL, Coleman M, et al. Longitudinal assessment of sputum microbiome by sequencing of the 16S rRNA gene in non-cystic fibrosis bronchiectasis patients. PLoS One. 2017;12:e0170622. doi: 10.1371/journal.pone.0170622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chalmers JD. Macrolide resistance in Pseudomonas aeruginosa: implications for practice. Eur Respir J. 2017;49:1700689. doi: 10.1183/13993003.00689-2017. [DOI] [PubMed] [Google Scholar]
- 39.Holguin F, Cardet JC, Chung KF, Diver S, Ferreira DS, Fitzpatrick A, et al. Management of severe asthma: a European Respiratory Society/American Thoracic Society guideline. Eur Respir J. 2020;55:1900508. doi: 10.1183/13993003.00588-2019. [DOI] [PubMed] [Google Scholar]
- 40.Ohashi Y, Fujisawa T. Detection of antibiotic resistance genes in the feces of young adult Japanese. Biosci Microbiota Food Health. 2017;36:151–154. doi: 10.12938/bmfh.17-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Löfmark S, Jernberg C, Jansson JK, Edlund C. Clindamycin-induced enrichment and long-term persistence of resistant Bacteroides spp. and resistance genes. J Antimicrob Chemother. 2006;58:1160–1167. doi: 10.1093/jac/dkl420. [DOI] [PubMed] [Google Scholar]
- 42.Isidro J, Menezes J, Serrano M, Borges V, Paixão P, Mimoso M, et al. Genomic study of a Clostridium difficile multidrug resistant outbreak-related clone reveals novel determinants of resistance. Front Microbiol. 2018;9:2994. doi: 10.3389/fmicb.2018.02994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raghu G, Meyer KC. Silent gastro-oesophageal reflux and microaspiration in IPF: mounting evidence for anti-reflux therapy? Eur Respir J. 2012;39:242–245. doi: 10.1183/09031936.00211311. [DOI] [PubMed] [Google Scholar]
- 44.Lee AL, Goldstein RS. Gastroesophageal reflux disease in COPD: links and risks. Int J Chron Obstruct Pulmon Dis. 2015;10:1935–1949. doi: 10.2147/COPD.S77562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mandal P, Morice AH, Chalmers JD, Hill AT. Symptoms of airway reflux predict exacerbations and quality of life in bronchiectasis. Respir Med. 2013;107:1008–1013. doi: 10.1016/j.rmed.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 46.Leggett JJ, Johnston BT, Mills M, Gamble J, Heaney LG. Prevalence of gastroesophageal reflux in difficult asthma: relationship to asthma outcome. Chest. 2005;127:1227–1231. doi: 10.1378/chest.127.4.1227. [DOI] [PubMed] [Google Scholar]
- 47.Aliberti S, Lonni S, Dore S, McDonnell MJ, Goeminne PC, Dimakou K, et al. Clinical phenotypes in adult patients with bronchiectasis. Eur Respir J. 2016;47:1113–1122. doi: 10.1183/13993003.01899-2015. [DOI] [PubMed] [Google Scholar]
- 48.Kennedy M, Ramsheh MY, Williams CML, Auty J, Haldar K, Abdulwhhab M, et al. Face mask sampling reveals antimicrobial resistance genes in exhaled aerosols from patients with chronic obstructive pulmonary disease and healthy volunteers. BMJ Open Respir Res. 2018;5:e000321. doi: 10.1136/bmjresp-2018-000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baron S, Diene S, Rolain J-M. Human microbiomes and antibiotic resistance. Hum Microbiome. 2018;10:43–52. [Google Scholar]
- 50.Montanari MP, Cochetti I, Mingoia M, Varaldo PE. Phenotypic and molecular characterization of tetracycline- and erythromycin-resistant strains of Streptococcus pneumoniae. Antimicrob Agents Chemother. 2003;47:2236–2241. doi: 10.1128/AAC.47.7.2236-2241.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greninger AL. The challenge of diagnostic metagenomics. Expert Rev Mol Diagn. 2018;18:605–615. doi: 10.1080/14737159.2018.1487292. [DOI] [PubMed] [Google Scholar]
- 52.Carney SM, Clemente JC, Cox MJ, Dickson RP, Huang YJ, Kitsios GD, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62:283–299. doi: 10.1165/rcmb.2019-0273TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nelson MT, Pope CE, Marsh RL, Wolter DJ, Weiss EJ, Hager KR, et al. Human and extracellular DNA depletion for metagenomic analysis of complex clinical infection samples yields optimized viable microbiome profiles. Cell Rep. 2019;26:2227–2240, e5. doi: 10.1016/j.celrep.2019.01.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, Li Z, Dong L, Wu Y, Shen H, Chen Z. Lipid metabolism in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2019;14:1009–1018. doi: 10.2147/COPD.S196210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olveira G, Olveira C, Dorado A, García-Fuentes E, Rubio E, Tinahones F, et al. Cellular and plasma oxidative stress biomarkers are raised in adults with bronchiectasis. Clin Nutr. 2013;32:112–117. doi: 10.1016/j.clnu.2012.06.002. [DOI] [PubMed] [Google Scholar]