

Abstract
Background: This guideline addresses the diagnosis of hypersensitivity pneumonitis (HP). It represents a collaborative effort among the American Thoracic Society, Japanese Respiratory Society, and Asociación Latinoamericana del Tórax.
Methods: Systematic reviews were performed for six questions. The evidence was discussed, and then recommendations were formulated by a multidisciplinary committee of experts in the field of interstitial lung disease and HP using the GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) approach.
Results: The guideline committee defined HP, and clinical, radiographic, and pathological features were described. HP was classified into nonfibrotic and fibrotic phenotypes. There was limited evidence that was directly applicable to all questions. The need for a thorough history and a validated questionnaire to identify potential exposures was agreed on. Serum IgG testing against potential antigens associated with HP was suggested to identify potential exposures. For patients with nonfibrotic HP, a recommendation was made in favor of obtaining bronchoalveolar lavage (BAL) fluid for lymphocyte cellular analysis, and suggestions for transbronchial lung biopsy and surgical lung biopsy were also made. For patients with fibrotic HP, suggestions were made in favor of obtaining BAL for lymphocyte cellular analysis, transbronchial lung cryobiopsy, and surgical lung biopsy. Diagnostic criteria were established, and a diagnostic algorithm was created by expert consensus. Knowledge gaps were identified as future research directions.
Conclusions: The guideline committee developed a systematic approach to the diagnosis of HP. The approach should be reevaluated as new evidence accumulates.
Keywords: hypersensitivity pneumonitis, fibrotic hypersensitivity pneumonitis, nonfibrotic hypersensitivity pneumonitis, interstitial lung disease, pulmonary fibrosis
Contents
Summary of Recommendations
Introduction
How to Use These Guidelines
Methods
Definition
Clinical Manifestations
Subtypes of HP
Symptoms and Signs
Natural History and Prognosis
Epidemiology
Pathogenesis
Inciting Agents
Immunological Dysregulation
Genetic/Host Susceptibility
Radiological Features
Chest HRCT Scanning Protocol
Radiological Features of HP
Histopathological Features
Histopathological Features of Nonfibrotic, or Cellular, HP
Histopathological Features of Fibrotic HP
Diagnostic Criteria
Diagnostic Interventions
Question 1: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without an overt history of exposures capable of causing ILD in the patient’s environment at home, work, or elsewhere, be subjected to formal questioning using a questionnaire to raise the possibility that a) potential inciting agents of HP are the etiology of the ILD and b) the diagnosis of the ILD is HP?
Question 2: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without an overt history of exposures capable of causing ILD in the patient’s environment at home, work, or elsewhere, undergo serum testing for IgG antibodies against specific antigens to raise the possibility that a) potential inciting agents of HP are the etiology of the ILD and b) the diagnosis of the ILD is HP?
Question 3: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo BAL fluid lymphocyte cellular analysis to diagnose HP?
Question 4: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo transbronchial forceps lung biopsy to diagnose HP?
Question 5: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo transbronchial lung cryobiopsy to diagnose HP?
Question 6: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo SLB to diagnose HP?
Future Directions
Conclusions
Summary of Recommendations
-
1.
Hypersensitivity pneumonitis (HP) must be considered in the differential diagnosis for patients with newly identified interstitial lung disease (ILD).
-
2.The guideline committee categorized HP into two clinical phenotypes—nonfibrotic and fibrotic HP—and made separate recommendations for each:
-
a.For patients with clinical and radiographic manifestations suggestive of nonfibrotic HP (i.e., patients without radiological and/or histopathological evidence of fibrosis), the guideline committee:
-
a.
i. makes no recommendation or suggestion for or against the use of a questionnaire to identify potential HP inciting agents and sources; instead, the guideline committee recommends development and validation of a questionnaire. Remark: Pending the availability of a validated questionnaire, the guideline committee advocates that clinicians take a thorough history to identify potential exposures and sources in the patient’s environment that are known to be associated with HP.
-
ii.
suggests performing serum IgG testing that targets potential antigens associated with HP (suggestion, very low confidence in the estimated effects).
-
iii.
recommends obtaining bronchoalveolar lavage (BAL) fluid for lymphocyte cellular analysis (recommendation, very low confidence in the estimated effects).
-
iv.
suggests transbronchial forceps lung biopsy (suggestion, very low confidence in the estimated effects).
-
v.
makes no recommendation or suggestion for or against transbronchial lung cryobiopsy.
-
vi.suggests surgical lung biopsy only when all other diagnostic testing has not yielded a diagnosis (suggestion, very low confidence in the estimated effects).
- b. For patients with clinical and radiographic manifestations suggestive of fibrotic HP (i.e., patients with radiological and/or histopathological evidence of fibrosis), the guideline committee:
- i. makes no recommendation or suggestion for or against the use of a questionnaire to identify potential HP inciting agents and sources; instead, the guideline committee recommends development and validation of a questionnaire. Remark: Pending the availability of a validated questionnaire, the guideline committee advocates that clinicians take a thorough history to identify potential exposures and sources in the patient’s environment that are known to be associated with HP.
- ii. suggests performing serum IgG testing that targets potential antigens associated with HP (suggestion, very low confidence in the estimated effects).
- iii. suggests obtaining BAL fluid for lymphocyte cellular analysis (suggestion, very low confidence in the estimated effects).
- iv. makes no recommendation or suggestion for or against transbronchial forceps lung biopsy.
- v. suggests transbronchial lung cryobiopsy (suggestion, very low confidence in the estimated effects).
- vi. suggests surgical lung biopsy; this recommendation is intended to apply when all other diagnostic testing has not yielded a diagnosis (suggestion, very low confidence in the estimated effects).
Introduction
HP is typically an immune-mediated disease that manifests as ILD in susceptible individuals after exposure to an identified or unidentified factor (1). Various alternative definitions of HP have been proposed, but agreement among experts regarding disease definition, diagnostic criteria, and diagnostic approach is lacking, despite efforts by international groups (2–8). Without a consensus definition, it is challenging to diagnose and research HP (7–11). Recent articles have highlighted substantial gaps in our knowledge about the epidemiology, pathogenesis, optimal diagnostic approach, classification, treatment, and follow-up of HP (9–11).
HP shares features of other acute and chronic pulmonary diseases; as a result, fibrotic/chronic HP can be misdiagnosed as idiopathic pulmonary fibrosis (IPF) or another idiopathic interstitial pneumonia (IIP) (12). Many inciting agents have been associated with HP since its recognition in 1700 (13), but the antigen and exposure are not identified in up to 60% of patients with HP, despite a thorough history (14–18). This highlights the difficulty in identifying a culprit exposure and raises the possibility that HP can occur in the absence of an inhalational exposure. It also emphasizes the difficulty in making a definitive diagnosis of HP (particularly fibrotic/chronic HP), which is the reason that a diagnosis of HP requires a multidisciplinary approach that includes radiologists and pathologists. There are many questions about the identification, duration, quantity, frequency, intensity of exposure to the inciting agent and its source that is required to induce HP, and factors that may predispose people to develop HP.
Clinical practice guidelines (CPGs) for the diagnosis and management of HP are lacking. As a result, clinical practice varies substantially from region to region and among countries, agreement on HP diagnosis is poor (19), and some clinicians continue to use a consensus statement from nearly 30 years ago for guidance (6). This CPG was developed by an ad hoc committee of experts appointed by the American Thoracic Society (ATS), the Japanese Respiratory Society (JRS), and the Asociación Latinoamericana del Tórax (ALAT), as well as European and Australian experts in HP. The target audience of this CPG is clinicians (i.e., pulmonologists, radiologists, and pathologists) who care for adults with ILD. The main objective is to help clinicians who are evaluating patients with newly identified ILD to accurately recognize nonfibrotic HP and fibrotic HP in a timely manner that will lead to avoidance of culprit environmental factors and potentially change the disease course. It is also hoped that the CPG will stimulate research into environmental factors and measures to avoid exposure to factors known to induce HP in genetically susceptible persons, decreasing the incidence of HP and more severe forms of the disease.
How to Use These Guidelines
There are many similarities in the initial presentation of patients with fibrotic ILD. This similarity lends itself to the question, “When should clinicians use these guidelines and when should they use the 2018 ATS/European Respiratory Society (ERS)/JRS/ALAT guidelines on the diagnosis of IPF (20)?” because both guidelines address patients with newly identified fibrotic ILD.
Most patients with fibrotic ILD present with an insidious onset of cough, exertional dyspnea, and bibasilar crackles with radiological evidence of fibrosis in lower lobes. Both CPGs are applicable to such patients. Additional history is the first step in evaluating such patients and is essential to deciding which guideline to follow. If the patient has a potential culprit exposure, this CPG should be followed, which means that the initial steps include a high-resolution computed tomography (HRCT) scan and BAL fluid lymphocyte cellular analysis, followed by a multidisciplinary discussion (MDD). If the patient has no culprit exposures and is a male former smoker >60 years old, the 2018 ATS/ERS/JRS/ALAT guidelines on the diagnosis of IPF (20) should be followed, which means that the initial steps include an HRCT scan followed by an MDD. For all other patients with newly identified fibrotic ILD, the decision of which CPG to initially follow should be made on a case-by-case basis. Regardless of which CPG is followed, the initial steps are similar, and ongoing diagnostic evaluation may be redirected on the basis of the MDD.
It should be emphasized that clinicians should apply the recommendations within this CPG in the clinical context of each individual patient, considering the patient’s values and preferences, and should not consider any recommendations as mandates. No CPG or recommendation can consider all potential clinical circumstances.
Methods
A multidisciplinary (pulmonologists, radiologists, methodologists, pathologists, and patient) panel of experts from the ATS, JRS, and ALAT was composed to identify clinically important questions about diagnostic testing for HP among patients with newly identified ILD. The CPG was created in two parts. The first portion describes clinical, radiological, and pathological features of HP while proposing a definition, diagnostic criteria, and a diagnostic algorithm. It was approached in a consensus fashion and informed by a nonsystematic review of the literature. The second portion makes graded recommendations that answer questions about whether to perform a diagnostic intervention. It was informed by National Academy of Medicine–adherent guideline methodology, including a full systematic review for each question and the formulation, writing, and grading of recommendations using the GRADE (Grading of Recommendations, Assessment, Development, and Evaluation) approach. For a detailed description of the methods, see the online supplement.
Implications of the different degrees of recommendation are described in Table 1. Using the GRADE approach, each recommendation was rated as either a “recommendation” or a “suggestion.” The meaning of a recommendation is the same as a strong recommendation in typical GRADE nomenclature, and the meaning of a suggestion is the same as a weak or conditional recommendation in typical GRADE nomenclature. Typical GRADE nomenclature was altered for this guideline to address prior criticism that the term “conditional” created uncertainty in the context of translation into non-English languages.
Table 1.
Strengths of Recommendations
| From the GRADE working group | Recommendation (“We recommend . . .”) | Suggestion (“We suggest . . .”) |
|---|---|---|
| For patients | The overwhelming majority of individuals in this situation would want the recommended course of action and only a small minority would not. | The majority of individuals in this situation would want the suggested course of action, but a sizable minority would not. |
| For clinicians | The overwhelming majority of individuals should receive the recommended course of action. Adherence to this recommendation according to the guideline could be used as a quality criterion or performance indicator. Formal decision aids are not likely to be needed to help individuals make decisions consistent with their values and preferences. | Different choices will be appropriate for different patients, and you must help each patient arrive at a management decision consistent with her or his values and preferences. Decision aids may be useful to help individuals make decisions consistent with their values and preferences. Clinicians should expect to spend more time with patients when working toward a decision. |
| For policy-makers | The recommendation can be adapted as policy in most situations, including for use as performance indicators. | Policy-making will require substantial debates and involvement of many stakeholders. Policies are also more likely to vary between regions. Performance indicators would have to focus on the fact that adequate deliberation about the management options has taken place. |
| From the ATS/JRS/ALAT Diagnosis of Hypersensitivity Pneumonitis Guidelines panel discussion | ||
| It is the right course of action for >95% of patients. | It is the right course of action for >50% of patients. | |
| “Just do it.” | “Slow down, think about it, discuss it with the patient.” | |
| You would be willing to tell a colleague who did not follow the recommendation that he/she did the wrong thing. | You would not be willing to tell a colleague who did not follow the recommendation that he/she did the wrong thing; it is “style” or “equipoise.” | |
| The recommended course of action may be an appropriate performance measure. | The recommended course of action is not appropriate for a performance measure. |
Definition of abbreviations: ALAT = Asociación Latinoamericana del Tórax; ATS = American Thoracic Society; GRADE = Grading of Recommendations, Assessment, Development, and Evaluation; JRS = Japanese Respiratory Society.
The meaning of a suggestion is the same as a weak or conditional recommendation in typical GRADE nomenclature.
Definition
HP is an inflammatory and/or fibrotic disease affecting the lung parenchyma and small airways. It typically results from an immune-mediated reaction provoked by an overt or occult inhaled antigen in susceptible individuals.
HP was historically termed “extrinsic allergic alveolitis” and categorized as acute, subacute, or chronic. However, these categories are not easily demarcated, and their delineation has been variable and arbitrary in many studies. Because the presence of radiographic or histopathological fibrosis is the primary determinant of prognosis (3, 21–29), the guideline committee decided unanimously to categorize HP as either fibrotic (i.e., mixed inflammatory plus fibrotic or purely fibrotic) or nonfibrotic (i.e., purely inflammatory), given the greater clinical utility of this stratification. Some patients may have mixed features; in such circumstances, the categorization is determined by the predominance of features.
Although HP is characteristically associated with an inhaled antigen, exposures may not be identified, despite a thorough evaluation in patients with otherwise typical features of HP (some experts have used the term “cryptogenic HP” or “HP of undetermined cause”) (9, 14, 15, 21, 30, 31). It is unknown whether these situations represent unidentified exposure or whether these patients instead have features of HP that are primarily due to an independent, intrinsic/primary process. Although virtually all diseases occur in “susceptible individuals,” this phrase was included in the definition of HP to emphasize the critical importance of sensitization in the pathogenesis of HP.
Clinical Manifestations
Subtypes of HP
HP is a disease with heterogeneous clinical presentations and outcomes, with subtypes historically categorized by disease duration at the time of presentation (i.e., acute, subacute, or chronic) (4). These categories were vaguely defined in the existing literature and were not consistently associated with outcomes; some patients have a benign course with complete recovery once the relevant exposure has been eliminated, whereas others do not recover and progress to respiratory failure, irrespective of their classification as having acute, subacute, or chronic HP (1, 14). On the basis of evolving knowledge and clinical experience, the guideline committee concluded that patients should be classified as having fibrotic HP or nonfibrotic HP, as determined by the predominant presence or absence of radiological and/or histopathological fibrosis. This new approach reflects the consensus that classification as fibrotic or nonfibrotic HP is more objective, may reflect disease presentation, and is likely to be more consistently associated with the clinical course and other outcomes (9, 10).
Symptoms and Signs
Common symptoms and signs of both nonfibrotic and fibrotic HP include dyspnea, cough, and midinspiratory squeaks (or chirping rales or squawks) (32). Less frequently, there may be constitutional symptoms such as weight loss, flu-like symptoms (chills, low-grade fever, and malaise), chest tightness, and wheezing, as well as physical examination findings of rales and cyanosis (1). Onset may be acute (developing over days to weeks, occasionally with pleural effusion) or may also be insidious (developing and worsening over months to years); episodes may be recurrent. Although an acute presentation with or without constitutional symptoms seems more consistent with nonfibrotic HP and the insidious presentation seems more consistent with fibrotic HP, duration of symptoms has not been rigorously characterized with respect to fibrosis status (1, 33).
Prevalence of HP is highest among older individuals (i.e., 65 yr and older, with the average patient receiving a diagnosis in their fifth or sixth decade) (34). It can also be diagnosed among younger adults and children (14, 35). Patients with fibrotic HP are more likely to be older, have an unidentified inciting agent, and have a lower vital capacity (VC), diffusion capacity, and percentage of lymphocytes in their BAL fluid than patients with nonfibrotic HP (36).
Natural History and Prognosis
The natural history of HP ranges from improvement to progressive decline and death due to respiratory failure (15). Patients with nonfibrotic HP who avoid ongoing exposure to the inciting agent may have a favorable prognosis with the possibility of stabilization or full recovery (15, 37, 38). Patients with fibrotic HP, particularly those with a usual interstitial pneumonia (UIP)-like pattern, have reduced survival (15, 22, 23, 25, 29, 30, 38–42). Other features associated with poor prognosis include cigarette smoking, lower baseline VC, lack of BAL lymphocytosis (29, 42, 43), persistent exposure to the inciting agent, and/or inability to identify an inciting agent (15). Notably, it has been reported that an inciting agent is not identified in 30–50% of cases evaluated at ILD referral centers (15, 36, 44).
Epidemiology
The prevalence of HP varies with regional disparities in climate, occupational exposures, and environmental exposures (see Table E1 in the online supplement) (34, 45–57). Available studies estimate an incidence between 0.3 and 0.9 per 100,000 individuals (34, 45–57), although the incidence may be even higher according to one study that reported bird breeder’s disease in 4.9 per 100,000 individuals over a 10-year period or 54.6 per 100,000 bird breeders (58). Insurance claims–based analyses conducted between 2004 and 2013 estimated 1-year prevalence to be 1.67–2.71 per 100,000 in the U.S. population (34). The proportion of HP among all ILD cases varies tremendously, ranging from 2% to 47% in studies and registries (35, 59–67). Childhood HP is uncommon but may represent 50% of all childhood ILDs (68–70). Sporadic outbreaks of HP have been reported in a variety of exposed groups, including lifeguards at swimming pools (71), automobile workers exposed to polyurethane (72), and office workers exposed to a contaminated humidifier (73) or forced-air climate control (74).
Pathogenesis
Inciting Agents
HP develops in susceptible individuals after repeated exposure to one or more inciting agents. Several potential inciting agents and hundreds of sources of such agents have been reported (11) (Table 2). These inciting agents are diverse, vary by geographic region, and are usually protein antigens derived from microorganisms, fungi, or animals (e.g., avian antigens). They may also be polysaccharides or low-molecular-weight nonprotein chemicals (e.g., isocyanates) (9, 11, 12, 15, 75). The location of exposure can be occupational, household related, or recreational. In many cases, an exposure is not identified (12, 15). Relationships between exposure-specific factors (e.g., concentration, duration, frequency of exposure, particle size, and particle solubility) and clinical course are frequently observed but are not well delineated (4, 11, 76–78). It has been hypothesized that the inciting agent can be part of a mixture of microbes, proteins, or other matter (e.g., dust). Common antigenic motifs (epitopes) have also been hypothesized; under this theory, sensitization to one antigen may result in hypersensitivity to multiple inciting agents (79–85). It is unknown why some exposed individuals also develop other types of lung pathology (e.g., the higher-than-expected prevalence of emphysema among patients with HP, independent of smoking status) (86, 87).
Table 2.
Sources of Antigens Known to Cause HP
| Matter | Typical Sources | HP “Disease” |
|---|---|---|
| Organic particulate matter |
|
|
| I. Microbes |
|
|
| Fungi/molds |
|
|
| Aspergillus spp. | Contaminated plant material | Farmer’s lung |
| Alternaria alternata, Aureobasidium spp. | Contaminated water | Humidifier lung |
| Botrytis cinerea | Contaminated houses (flooded) | Malt worker’s lung |
| Cephalosporium spp. | Upholstered furniture | Woodworker’s lung |
| Cladosporium spp. | Contaminated stucco | Indoor-air alveolitis (domestic HP) |
| Cryptococcus spp. | Contaminated raw materials in food-processing industry | Compost lung |
| Fusarium spp. | Organic wastes | Mushroom grower’s lung |
| Graphium spp. | Contaminated sawdust | Malt worker’s lung |
| Mucor spp. | Moldy wood | Stucco worker’s lung |
| Penicillium spp. | Aspergillus enzyme in baking agents | Suberosis |
| Rhizopus spp. | Contaminated domestic ventilation and cooling systems | Baker’s lung |
| Trichoderma spp. | Potted flowers, greenhouses | Waste sorter’s lung |
| Phytase (enzyme from Aspergillus or Trichoderma) | Mold on grapes | Sauna taker’s lung |
| Contaminated wind instruments | Wine grower’s lung | |
| Contaminated soil | Wind-instrument alveolitis | |
| Peat | Sequoiosis | |
| Peat worker’s lung | ||
| Cheese washer’s lung | ||
| Salami producer’s lung | ||
| Phytase alveolitis | ||
| Yeasts | ||
| Candida spp. | Contaminated misting fountains and humidifiers | Humidifier lung |
| Geotrichum candidum | Moldy hay, compost, mushrooms | Farmer’s lung |
| Saccharomyces cerevisiae | Contaminated swimming pools | Footcare alveolitis |
| Saccharomonospora viridis | Contaminated wind instruments | Candida alveolitis |
| Saccharopolysporarectivirgula | Human intestine, fingernails, and skin | Indoor-air alveolitis |
| Torulopsis glabrata | Milk mold | Yeast-powder alveolitis |
| Trichosporon cutaneum | Baker’s yeast, brewer’s yeast, wine yeasts | Thatched-roof lung |
| Contaminated houses | Mushroom worker’s lung | |
| Dried grasses, leaves | Summer-type HP | |
| Compost | Wind-instrument lung | |
| Mushrooms | ||
| Edible mushrooms | ||
| Mushrooms (shiitake, bunashimeji, Pleurotus, Pholiota, Lyophyllum, Agaricus) | Mushrooms growing in indoor environments | Mushroom grower’s lung |
| Bacteria | ||
| Acinetobacter spp. | Contaminated water, whirlpools | Machine operator’s lung |
| Bacillus spp. | Contaminated machine fluid | Humidifier lung |
| Klebsiella spp. | Sewage treatment plants | Woodworker’s lung |
| Nontuberculous mycobacteria | Sawdust | Detergent worker’s alveolitis |
| Phoma spp. | Moist wood | Summer-type HP |
| Pseudomonas spp. | Detergents | Farmer’s lung |
| Stenotrophomonas spp. | Biological cleaning agents | Hot-tub lung |
| Staphylococcus spp. | Washing powders | Whirlpool alveolitis |
| Streptomyces spp. | Contaminated houses | Wind-instrument alveolitis |
| Thermoactinomyces spp. | Moldy plants | Indoor-air alveolitis |
| Endotoxin from pool-water sprays and fountains | Contaminated wind instruments | Steam-iron alveolitis |
| Bacillus subtilis enzymes (subtilisin) | Moldy shower curtains | Mushroom grower’s lung |
| Compost | Thatched-roof disease | |
| Edible mushroom manure | Bagassosis | |
| Contaminated soil | Compost lung | |
| Moldy thatched roofs | ||
| Protozoa | ||
| Amoebae | Contaminated humidifiers and air-conditioning systems | Humidifier lung |
| Nematodes | ||
| Nematodes | Contaminated humidifiers and air-conditioning systems | Humidifier lung |
| Mite | ||
| Acarus siro | Contaminated cheese | — |
| II. Proteins/enzymes | ||
| Animal proteins | ||
| Animal fur dust | Animal pelts | Furrier’s lung |
| Avian droppings, serum, and feathers | Parakeets, canaries, budgerigars, pigeons, parrots, chicken, turkeys, geese, ducks, wild birds, pheasants | Bird fancier’s disease, bird breeder’s disease, pigeon breeder’s lung, chicken breeder’s lung |
| Avian feathers | Feather beds, pillows, duvets | Feather-duvet lung |
| Bats | Contact with bats | — |
| Carmine (from Coccus cacti) | Food and cosmetics | Carmine alveolitis, dyer’s lung |
| Cow milk | Cow milk | Heiner syndrome |
| Fish feed | Daphnia, meat, mosquito larvae | Fish-feed alveolitis |
| Fish meal | Animal feed | Fish-meal alveolitis |
| Shell protein (oyster, sea snail, mussels) | Oyster-shell powder | Shellfish alveolitis, oyster-shell HP, mollusk-shell HP |
| Pig pancreas | Animal extracts | — |
| Pituitary proteins | Pituitary powder | Pituitary snuff-taker’s lung |
| Rat and desert mouse (gerbil) urine, serum, pelts | Rats, gerbils | Alveolitis due to rat and mouse proteins |
| Silkworm proteins | Dust from silkworm larvae and cocoon | Silkworm rearer’s lung |
| Weevils (corn, wheat) (Sitophilus spp.) | Contaminated grain or flour | Corn (wheat)–weevil lung |
| Plant proteins | ||
| Alginate | Seaweed | — |
| Argan cake | Cosmetics, unsaturated fatty acids, phytosterol | — |
| Catechin | Green-tea powder | — |
| Esparto dust | Esparto grass | Esparto lung, plasterer’s lung |
| Grain flour (wheat, rye, oats, maize) | Flour dust | Flour-dust alveolitis |
| Malt | Food-processing industry | — |
| Legumes (soy) | Legumes (soya) flour dust | Soya-dust alveolitis |
| Paprika | Paprika dust | Paprika splitter’s lung |
| Pyrethrum | Plant-based insecticide | — |
| Spinach | Spinach powder | — |
| Tiger nut | Horchata (drink) | Tiger-nut alveolitis |
| Wood (cabreuva, cedar, mahogany, pine, ramin, umbrella pine) | Wood particles | Wood fiber alveolitis |
| Inorganic particulate matter | ||
| I. Chemicals | ||
| Acid anhydrides (pyromellitic and trimellitic anhydrides) | Polyurethane foams, spray paints, elastomers, glues, adhesives, mattresses, car parts, shoes, imitation leather, rubber products, chipboards, elastic synthetic fibers, electrical insulations | Acid anhydride alveolitis |
| Acrylate compounds (methyl methacrylate) | Dental materials, lacquer, resin, glues | Methacrylate alveolitis |
| Copper sulfate | Copper-sulfate Bordeaux mixture | Vineyard sprayer’s lung |
| Chloroethylene (trichlorethylene) | Degreasing agents, cleaning agents, extraction agents | Chemical alveolitis |
| Dimethyl phthalate and styrene | Industrial solvents, plasticizers | — |
| HFC-134a | Coolant fluid in laser hair-removal devices | Hair-remover lung |
| Isocyanates (toluene diisocyanate, methylene diphenyl diisocyanate, hexamethylene diisocyanate, MIC, NDI, polyisocyanate) | As in acid anhydrides | Isocyanate alveolitis |
| Tetrachlorophthalic and hexahydrophthalic acid | Hardener for epoxy resin | Acid anhydride alveolitis |
| Sodium diazobenzene sulfate | Laboratory reagent, chromatography | Chemical alveolitis |
| Triglycidyl isocyanurate | Polyester powder (powder paints) | Painter’s lung |
| | ||
| II. Pharmaceutical agents | ||
| Penicillins, cephalosporins | Antibiotics | Drug-induced HP |
| Methotrexate | Immunosuppressive agents | |
| α-IFN | Immunomodulatory agents | |
| Lenalidomide | Hypolipidemics | |
| Pravastatin | Antidepressants | |
| Venlafaxine | Alkylating agents | |
| Temozolomide | ||
| III. Metals | ||
| Cobalt | Hard metals, alloys | Giant cell pneumonitis |
| Zinc (tungsten and alloys) | Zinc fumes | Zinc-fumes alveolitis |
| Zirconium | Zircon | Zirconium alveolitis |
| Beryllium | Batteries, computers, neons | Beryllium HP |
| TMI | Organometallic compound for semiconductors used in industry | — |
Definition of abbreviations: HFC-134a = hydrofluorocarbon 134a; HP = hypersensitivity pneumonitis; MIC = methylisocyanate; NDI = naphtylene-1,5-diisocyanate; TMI = trimethylindium.
Adapted from Reference 11.
Immunological Dysregulation
In sensitized individuals, the immune reaction after exposure to an antigen appears to consist of both humoral (i.e., antigen-specific IgG antibodies) and T-helper cell type 1 (Th1) cellular immune responses (83, 88). These responses lead to a predominantly lymphocytic inflammatory pattern and granulomatous inflammation (11, 75, 89). Neutrophilic inflammation may play a role early in the disease course and during subsequent fibrosis (90, 91), whereas impaired function of T regulatory cells may play a role in the exaggerated immune response (92). Some evidence suggests that a relative switch from Th1 to Th2 activity (93–95) as well as augmented epithelial apoptosis and abnormal fibroblast activity (96, 97) contribute to pulmonary fibrosis that may mimic patterns of fibrotic IIPs, including, most importantly, UIP. A subgroup of patients with HP has been shown to have concurrent autoimmune features, although the underlying mechanisms are still not known (98).
Genetic/Host Susceptibility
The best-studied host factor that creates a predisposition for HP development is genetic variation. Variants in genes involved with innate and adaptive immunity may enable sensitization to inciting agents (Table E2). Polymorphisms in major histocompatibility complex class II, proteasomes, transporter proteins, and tissue inhibitors of matrix metalloproteinases have been associated with HP (99–106). Among patients with fibrotic HP, the MUC5B (mucin 5B) promoter polymorphism is more prevalent than in the general population and is associated with shortened survival (107). HP has also been described in probands of familial pulmonary fibrosis, including those with telomere-related gene mutations (108, 109). A study performed in two cohorts of patients with chronic HP revealed that around 10% of the patients had rare, protein-altering variants in telomere-related genes, which were associated with short telomere length and significantly reduced transplant-free survival (110). Microchimerism has been identified in a larger fraction of patients with HP compared with patients with IPF and healthy women; among women with HP, microchimerism is associated with a lower diffusion capacity (111). Preceding respiratory viral infection is another proposed host-sensitizing factor (112–114), and exposure to pesticides seems to increase the risk of HP in farmers (115).
Radiological Features
Chest HRCT Scanning Protocol
The scanning protocol for the evaluation of suspected HP is identical to the protocol described in the ATS/ERS/JRS/ALAT diagnosis-of-IPF guidelines (Table 3) (20). It is based on high-resolution volumetric scanning of the chest, with special attention to the selection of parameters ensuring creation of motion-free images and adequate image quality at a reduced radiation dose. In both fibrotic and nonfibrotic HP, two series of images acquired in the supine position are obtained: one at deep inspiration and a second after prolonged expiration. All features of lung infiltration can be depicted on the inspiratory images, except for air trapping, which is an expiratory HRCT finding. Analysis of lung changes at expiration may increase diagnostic confidence in nonfibrotic HP and is necessary for better characterization of heterogeneous lung attenuation in both forms of the disease. Owing to the widespread distribution of lung changes in HP, a third acquisition in the prone position is usually not necessary. The optimal chest HRCT scan for characterizing HP should be a noncontrast examination, except in the context of acute respiratory decline, in which case CT angiography may be justified to detect acute pulmonary embolisms. CT angiography should be preceded by a noncontrast chest HRCT scan to detect new ground-glass changes that raise the probability of acute exacerbation in the absence of pulmonary embolisms.
Table 3.
Recommended Chest HRCT Scanning Parameters in the Diagnostic Approach of HP
| 1. Noncontrast examination |
| 2. Volumetric acquisition with selection of: |
| • Submillimetric collimation |
| • Shortest rotation time |
| • Highest pitch |
| • Tube potential and tube current appropriate to patient size: |
| ✓ Typically: 120 kVp and ≤240 mAs |
| ✓ Lower tube potentials (e.g., 100 kVp) with adjustment of tube current encouraged for thin patients |
| ✓ Use of techniques available to avoid unnecessary radiation exposure (e.g., tube current modulation) |
| 3. Reconstruction of thin-section CT images (≤1.5 mm): |
| • Contiguous or overlapping |
| • Using a high-spatial-frequency algorithm |
| • Iterative reconstruction algorithm if validated on the CT unit (if not, filtered back projection) |
| 4. Number of acquisitions |
| • Supine position: inspiratory (volumetric) and expiratory (sequential or volumetric) acquisitions |
| • Prone (optional): only inspiratory scans (can be sequential or volumetric) |
| • Inspiratory scans obtained at full inspiration |
| 5. Recommended radiation dose for the inspiratory volumetric acquisition: |
| • 1–3 mSv (i.e., “reduced” dose) |
| • Strong recommendation to avoid “ultra–low-dose CT” (<1 mSv) |
Definition of abbreviations: CT = computed tomography; HP = hypersensitivity pneumonitis; HRCT = high-resolution CT; kVp = kilovolt peak.
Adapted from Reference 20.
Radiological Features of HP
The imaging features of HP are influenced by the histopathological stage of disease at the time of diagnosis. Our proposed approach reconciles the committee’s preferred two-pattern description (i.e., nonfibrotic and fibrotic HP) with the three subtypes of HP previously considered (i.e., acute, subacute, and chronic forms). For nonfibrotic HP, we use consensus descriptions of the inflammatory and often reversible changes established in the literature (116–119). For fibrotic HP, we provide a novel approach that integrates 1) HRCT scan findings previously described as chronic HP (116–121) and 2) recent data on the diagnostic impact of several radiological patterns (7, 122, 123). Fibrotic HP is widely recognized to have a variable radiological appearance, and the approach proposed here does not consider potential geographical specificities that may influence the most prevalent HRCT pattern.
The following descriptions are intended to provide a summary of HRCT findings that are 1) highly suggestive of HP, which we categorize as “typical HP”; 2) less frequently reported but compatible with HP, which we refer to as “compatible with HP”; or 3) “indeterminate for HP” when the HRCT findings are neither suggestive nor compatible with features of HP. Radiological terms related to the heterogenous lung attenuation are defined in Table 4.
Table 4.
Radiological Terms for Heterogenous Lung Attenuation
| Terminology | Significance | Description |
|---|---|---|
| Mosaic attenuation* | • Generic term referring to a patchwork of regions of differing attenuation on inspiratory CT images | • Term only used for description of inspiratory CT images |
| • Can reflect the presence of vascular disease, airway abnormalities, or ground-glass interstitial or airspace infiltration | • Combination of areas of low and high attenuation that can correspond to two main situations: | |
| a. Areas of GGO (“high”) and normal lung (“low”) or | ||
| b. Areas of normal lung (“high”) and areas of decreased attenuation (“low”) | ||
| • Areas of GGO reflect an infiltrative lung disease | ||
| Air trapping* | • Abnormal retention of air distal to airway obstruction | • Term exclusively used for description of expiratory CT images |
| • Recognized as parenchymal areas that lack the normal increase in attenuation and the volume reduction of normally ventilated lung | • Air trapping appears as focal zones of hypoattenuation in the background of hyperattenuating normal lung on expiratory CT images | |
| • Mosaic attenuation and air trapping are not synonymous and cannot be used interchangeably | ||
| Mosaic perfusion† | • Regional differences in lung attenuation secondary to regional differences in lung perfusion | • Term used for description of inspiratory CT images |
| • May be seen in vascular (exclusive perfusion abnormalities) or airway (perfusion abnormalities resulting from abnormal regional lung ventilation) diseases | • Presence of decreased vascular sections within areas of low attenuation in comparison with areas of normal lung | |
| • Differential diagnosis facilitated by expiratory scans: | ||
| a. In case of vascular disease: same gradient of attenuation between areas of low and high attenuation | ||
| b. In case of airways disease: the attenuation differences are accentuated due to the additional depiction of air trapping | ||
| “Three-density pattern”‡ | • Term coined to replace the “headcheese” sign, as most individuals worldwide do not relate to the headcheese sign | • Combination of three attenuations on inspiratory CT images: |
| a. Normal-appearing lung | ||
| • Indicative of a mixed obstructive and infiltrative process: | b. High attenuation (GGO) | |
| a. The obstructive abnormality (seen in small airway disease) is manifested by areas of decreased attenuation and decreased vascularity | c. Lucent lung (i.e., regions of decreased attenuation and decreased vascular sections) | |
| b. The infiltrative disorder results in GGO surrounding preserved normal lobules | ||
| • Sharply demarcated from each other | ||
| • Highly specific for fibrotic HP; has not been shown to be specific for nonfibrotic HP |
Definition of abbreviations: CT = computed tomography; GGO = ground-glass opacity; HP = hypersensitivity pneumonitis.
See Reference 326.
See Reference 327.
The term “three-density pattern” was coined by this committee. This descriptive pattern was unanimously determined by the committee to be the preferred term. This pattern has been shown to differentiate fibrotic HP from idiopathic pulmonary fibrosis (123) and, thus, raises the index of suspicion for the diagnosis of fibrotic HP whenever present; however, it is unknown whether the pattern is also present in nonfibrotic HP. Some radiologists relate this pattern to the appearance of headcheese and, therefore, it has been referred to as the “headcheese sign” in the literature(328, 329). The guideline committee strongly discourages the use of the term “headcheese” to describe this pattern.
Nonfibrotic HP
The typical HP pattern (Table 5) relies on the identification of diffusely distributed HRCT findings that include features of lung infiltration (i.e., ground-glass opacity [GGO], mosaic attenuation) plus at least one HRCT abnormality suggestive of small airway disease. HRCT features of small airway disease include ill-defined, small (<5 mm) centrilobular nodules on inspiratory images and air trapping on expiratory images. Mosaic attenuation refers to coexisting areas of varying attenuation within the lung parenchyma on inspiratory HRCT images (Figures 1, E1, and E2). In nonfibrotic HP, mosaic attenuation typically reflects coexistent lobules affected by pneumonitis (increased attenuation) interspersed with lobules of normal or slightly decreased attenuation (due to bronchiolar obstruction). These parenchymal patterns are usually bilateral and symmetric with a diffuse distribution, both axially and craniocaudally. Although a combination of parenchymal abnormalities and features of small airway disease is highly suggestive of nonfibrotic HP, isolated air trapping is another pattern that may be seen with HP. Three additional HRCT features have also been described in nonfibrotic HP: uniform and subtle GGO, airspace consolidation, and lung cysts (124–126). Each of these features is nonspecific but can be compatible with nonfibrotic HP in the appropriate clinical context.
Table 5.
Chest HRCT Scan Features of the Nonfibrotic HP Pattern
| HRCT Pattern | Typical HP | Compatible with HP | Indeterminate for HP |
|---|---|---|---|
| Description | The “typical HP” pattern is suggestive of a diagnosis of HP. It requires a) at least one HRCT abnormality indicative of parenchymal infiltration and b) at least one HRCT abnormality indicative of small airway disease, both in a diffuse distribution | “Compatible-with-HP” patterns are nonspecific patterns that have been described in HP | N/A |
| Relevant radiological findings | HRCT abnormalities indicative of parenchymal infiltration: | Parenchymal abnormalities: | N/A |
| • GGOs | • Uniform and subtle GGOs | ||
| • Mosaic attenuation* | • Airspace consolidation | ||
| • Lung cysts | |||
| HRCT abnormalities indicative of small airway disease: | |||
| • Ill-defined, centrilobular nodules | Distribution of parenchymal abnormalities: | ||
| • Air trapping | • Craniocaudal: diffuse (variant: lower lobe predominance) | ||
| • Axial: diffuse (variant: peribronchovascular) | |||
| Distribution of parenchymal abnormalities: | |||
| • Craniocaudal: diffuse (with or without some basal sparing) | |||
| • Axial: diffuse |
Definition of abbreviations: GGO = ground-glass opacity; HP = hypersensitivity pneumonitis; HRCT = high-resolution computed tomography; N/A = not applicable.
Mosaic attenuation corresponding to parenchymal infiltration is created by GGOs adjacent to normal-appearing lung.
Figure 1.

“Typical hypersensitivity pneumonitis (HP)” and “compatible-with-HP” high-resolution computed tomography patterns. The nonfibrotic typical HP pattern is characterized by (A) centrilobular nodules, (B) mosaic attenuation on an inspiratory scan, and (C) air trapping on an expiratory scan. (D) The nonfibrotic compatible-with-HP pattern is exemplified by uniform and subtle ground-glass opacity and cysts. The fibrotic typical HP pattern consists of (E) coarse reticulation and minimal honeycombing in a random axial distribution with no zonal predominance in association with (F) small airway disease. The fibrotic compatible-with-HP pattern varies in the patterns and/or distribution of lung fibrosis (e.g., basal and subpleural predominance, [G] upper-lung-zone predominance, [H] central [or peribronchovascular] predominance [arrows], or [I] fibrotic ground-glass attenuation seen alone or in association with small airway disease). The fibrotic indeterminate-for-HP pattern includes the usual interstitial pneumonia pattern, nonspecific interstitial pneumonia pattern, organizing pneumonia–like pattern, or truly indeterminate findings.
Fibrotic HP
Coexisting lung fibrosis and signs of bronchiolar obstruction are highly suggestive of fibrotic HP (Table 6) (7, 40, 121, 127, 128).
Table 6.
Chest HRCT Scan Features of the Fibrotic HP Pattern
| HRCT Pattern | Typical HP | Compatible with HP | Indeterminate for HP |
|---|---|---|---|
| Description | The “typical HP” pattern is suggestive of a diagnosis of HP. It requires a) an HRCT pattern of lung fibrosis (as listed below) in one of the distributions and b) at least one abnormality that is indicative of small airway disease | “Compatible-with-HP” patterns exist when the HRCT pattern and/or distribution of lung fibrosis varies from that of the typical HP pattern; the variant fibrosis should be accompanied by signs of small airway disease | The “indeterminate-for-HP” pattern exists when the HRCT is neither suggestive nor compatible with a typical and probable HP pattern |
| Relevant radiological findings | HRCT abnormalities indicative of lung fibrosis are most commonly composed of irregular linear opacities/coarse reticulation with lung distortion; traction bronchiectasis and honeycombing may be present but do not predominate | Variant patterns of lung fibrosis: | Lone patterns (i.e., not accompanied by other findings suggestive of HP) of: |
| • UIP pattern: basal and subpleural distribution of honeycombing with/without traction bronchiectasis (per 2018 diagnosis of IPF guidelines [20]) | • UIP pattern (as per 2018 IPF diagnosis guidelines [20]) | ||
| The distribution of fibrosis may be: | |||
| • Random both axially and craniocaudally or | • Extensive GGOs with superimposed subtle features of lung fibrosis | • Probable UIP pattern (as per 2018 IPF diagnosis guidelines [20]) | |
| • Mid lung zone–predominant or | • Indeterminate pattern for UIP (as per 2018 IPF diagnosis guidelines [20]) | ||
| • Relatively spared in the lower lung zones | Variant (predominant) distributions of lung fibrosis: | • Fibrotic NSIP pattern | |
| • Axial: peribronchovascular, subpleural areas | • Organizing pneumonia–like pattern | ||
| HRCT abnormalities indicative of small airway disease: | • Craniocaudal: upper lung zones | • Truly indeterminate HRCT pattern | |
| • Ill-defined, centrilobular nodules and/or GGOs | |||
| • Mosaic attenuation, three-density pattern,* and/or air trapping (often in a lobular distribution) | HRCT abnormalities indicative of small airway disease: | ||
| • Ill-defined centrilobular nodules, or | |||
| • Three-density pattern* and/or air trapping |
Definition of abbreviations: GGO = ground-glass opacity; HP = hypersensitivity pneumonitis; HRCT = high-resolution computed tomography; IPF = idiopathic pulmonary fibrosis; NSIP = nonspecific interstitial pneumonia; UIP = usual interstitial pneumonia.
Rarely, fibrotic HP may be seen 1) as a component of combined pulmonary fibrosis and emphysema or pleuroparenchymal fibroelastosis with emphysema, 2) as a pure emphysematous form of HP, or 3) in acute exacerbation.
The three-density pattern was formerly called the “headcheese sign.” It is described in detail in Table 4.
Lung fibrosis in HP most frequently manifests as irregular fine or coarse reticulation with architectural lung distortion, sometimes with septal thickening, that can be seen alone or in association with traction bronchiectasis in areas of GGO. Honeycombing can be present and is often described as minimal, but extensive honeycombing in severe forms of fibrotic HP may also occur. Lung fibrosis is most severe in the mid or mid and lower lung zones or equally distributed in the three lung zones with relative basal sparing. On axial images, there is often no central or peripheral predominance of lung fibrosis (Figures 1 and E3).
Bronchiolar obstruction manifests with several HRCT features in fibrotic HP. Like that observed in nonfibrotic HP, ill-defined centrilobular nodules and mosaic attenuation can be seen (7, 122). Bronchiolar obstruction is also present in an HRCT pattern combining three different lung densities (GGO, lobules of decreased attenuation and vascularity, and normal-appearing lung) that is highly specific to fibrotic HP (123). We coined the term “three-density pattern” to describe the presence of these three different lung densities, which some radiologists have referred to as the “headcheese sign” (Figure 2 and Table 4). This pattern emphasizes the diagnostic value of lobules with decreased attenuation and vascularity on inspiratory HRCT images, especially when concomitant with air trapping at expiration, both suggesting the presence of severe bronchiolar obstruction. These two individual HRCT features were the highest-ranked radiological features for fibrotic HP in an International Modified Delphi Survey (7).
Figure 2.

Three-density pattern. High-resolution computed tomography (A) inspiratory and (B) expiratory images from a patient with hypersensitivity pneumonitis demonstrating the three different densities: high attenuation (ground-glass opacity) (red stars), lucent lung (regions of decreased attenuation and decreased vascular sections) (red arrows), and normal lung (black arrows), which are sharply demarcated from each other.
In the context of fibrotic HP, mosaic attenuation is often described as “extensive” (128) and “marked” (20), but these descriptors do not state specific numerical values. In a recent study, the threshold of five or more lobules of mosaic attenuation in each of three or more lobes bilaterally was found to have the highest specificity for fibrotic HP and helped differentiate this disorder from IPF (123). Because air trapping is a nonspecific finding reflecting small airway alterations of variable cause and/or severity, it is not surprising that it is also found in non-HP ILDs (8), especially connective tissue disease (CTD)-associated ILD (CTD-ILD) (129) and sarcoidosis (130).
Some variants in the distribution of fibrosis are compatible with fibrotic HP, although they are less frequent (Figures 1 and E4). These include fibrosis with an axially peripheral (subpleural) or central (peribronchovascular) distribution, as well as basal-predominant disease. Although upper zone–predominant fibrosis has been described as a feature that may separate fibrotic HP from IPF (127), only a small proportion of patients with fibrotic HP (<10%) have upper lung–preponderant disease (35, 119, 121).
Fibrotic HP may also present with HRCT patterns that are neither suggestive nor compatible with features of HP; these HRCT patterns should be classified as indeterminate for fibrotic HP (Figures 1 and E5). They include the patterns of UIP alone (i.e., no other features of HP accompanying the UIP pattern), fibrotic nonspecific interstitial pneumonia (NSIP), and organizing pneumonia (40, 127). The UIP pattern is recognized by honeycombing with or without peripheral bronchiolectasis, with a subpleural and basal predominance. Fibrotic NSIP is suggested by the presence of bilateral, predominantly lower-lung-zone GGO with fine reticulation and traction bronchiectasis, with peribronchovascular predominance in the axial distribution. The pattern of organizing pneumonia relies on the presence of consolidation in a peribronchovascular and/or peripheral distribution, often seen with GGO and sometimes associated with a reverse halo pattern. The presence of a reticular pattern superimposed on parenchymal consolidation suggests an “organizing pneumonia–like” pattern of fibrotic HP. As in other ILDs, HP may also present with a truly indeterminate HRCT pattern.
Combined pulmonary fibrosis and emphysema (82) and pleuroparenchymal fibroelastosis with emphysema (87) can also occur in HP (Figure E6), although they are infrequent. Purely emphysematous forms of HP can be seen independently of smoking history (Figure E7) (85, 116, 131, 132), and fibrotic HP may also be diagnosed at the time of an acute exacerbation (Figure E8) (133).
Histopathological Features
Lung biopsy often plays a pivotal role in ascertaining a diagnosis of HP. The criteria proposed for diagnosis are valid to any biopsy type but are based on historical descriptions of the histopathological findings in surgical lung biopsy (SLB) specimens (1, 3, 12, 23, 28, 39, 40, 42, 134–142). The findings that make a diagnosis of HP likely apply to both nonfibrotic (i.e., cellular) and fibrotic variants, the difference being the presence or absence of a fibrotic pattern that may show histological overlap with fibrotic IIP (Table 7).
Table 7.
Histopathological Criteria for the Diagnosis of HP (Other than “Hot-Tub Lung”*)
| HP | Probable HP | Indeterminate for HP |
|---|---|---|
|
Nonfibrotic HP (cellular HP) | ||
| Typical histopathological features of nonfibrotic HP; at least one biopsy site showing all three of the following features: | Both of the following features (1 and 2 from first column) in at least one biopsy site: | At least one biopsy site showing one of the following: |
| 1. Cellular interstitial pneumonia | 1. Cellular interstitial pneumonia | • 1 or 2 from the first column |
| • Bronchiolocentric (airway-centered) | • Bronchiolocentric (airway-centered) | • Selected IIP patterns |
| • Cellular NSIP-like pattern | • Cellular NSIP-like pattern | ○ Cellular NSIP pattern |
| • Lymphocyte-predominant | • Lymphocyte-predominant | ○ Organizing pneumonia pattern |
| ○ Peribronchiolar metaplasia without other features to suggest fibrotic HP and | ||
| 2. Cellular bronchiolitis | 2. Cellular bronchiolitis | Absence of features in any biopsy site to suggest an alternative diagnosis |
| • Lymphocyte-predominant (lymphs > plasma cells) with no more than focal peribronchiolar lymphoid aggregates with germinal centers | • Lymphocyte-predominant (lymphs > plasma cells) with no more than focal peribronchiolar lymphoid aggregates with germinal centers | • Plasma cells > lymphs |
| • ±Organizing pneumonia pattern with Masson bodies | • ±Organizing pneumonia pattern with Masson bodies | • Extensive lymphoid hyperplasia |
| • ±Foamy macrophages in terminal air spaces | • ±Foamy macrophages in terminal air spaces and | • Extensive well-formed sarcoidal granulomas and/or necrotizing granulomas |
| Absence of features in any biopsy site to suggest an alternative diagnosis | • Aspirated particulates | |
| 3. Poorly formed nonnecrotizing granulomas† | • Plasma cells > lymphs | |
| • Loose clusters of epithelioid cells and/or multinucleated giant cells ± intracytoplasmic inclusions | • Extensive lymphoid hyperplasia | |
| • Situated in peribronchiolar interstitium, terminal air spaces, and/or organizing pneumonia (Masson bodies) and | • Extensive well-formed sarcoidal granulomas and/or necrotizing granulomas | |
| Absence of features in any biopsy site to suggest an alternative diagnosis | • Aspirated particulates | |
| • Plasma cells > lymphs | ||
| • Extensive lymphoid hyperplasia | ||
| • Extensive well-formed sarcoidal granulomas and/or necrotizing granulomas | ||
| • Aspirated particulates | ||
|
Fibrotic HP‡ | ||
| Typical histopathological features of fibrotic HP; 1 or 2 and 3 in at least one biopsy site: | Both of the following features (1 or 2 from first column) in at least one biopsy site: | Either one of the following features in at least one biopsy site: |
| 1. Chronic fibrosing interstitial pneumonia | 1. Chronic fibrosing interstitial pneumonia | |
| 1. Chronic fibrosing interstitial pneumonia | • Architectural distortion, fibroblast foci ± subpleural honeycombing | • Architectural distortion, fibroblast foci ± honeycombing |
| • Fibrotic NSIP-like pattern | ||
| • Architectural distortion, fibroblast foci ± subpleural honeycombing | • Fibrotic NSIP-like pattern | ±Cellular interstitial pneumonia |
| • Fibrotic NSIP-like§ pattern | 2. Airway-centered fibrosis • ±Peribronchiolar metaplasia |
±Cellular bronchiolitis |
| • ±Bridging fibrosis‖ | ±Organizing pneumonia pattern and | |
| ±Cellular interstitial pneumonia | Absence of features in any biopsy site to suggest an alternative diagnosis | |
| 2. Airway-centered fibrosis • ±Peribronchiolar metaplasia | ±Cellular bronchiolitis | • Plasma cells > lymphs |
| • ±Bridging fibrosis‖ | ±Organizing pneumonia pattern and | • Extensive lymphoid hyperplasia |
| Absence of features in any biopsy site to suggest an alternative diagnosis | • Extensive well-formed sarcoidal granulomas and/or necrotizing granulomas | |
| 3. Poorly formed nonnecrotizing granulomas† | • Plasma cells > lymphs | • Aspirated particulates |
| ±Cellular interstitial pneumonia | • Extensive lymphoid hyperplasia | |
| ±Cellular bronchiolitis | • Extensive well-formed sarcoidal granulomas and/or necrotizing granulomas | |
| ±Organizing pneumonia pattern and | • Aspirated particulates | |
| Absence of features in any biopsy site to suggest an alternative diagnosis | ||
| • Plasma cells > lymphs | ||
| • Extensive lymphoid hyperplasia | ||
| • Extensive well-formed sarcoidal granulomas and/or necrotizing granulomas | ||
| • Aspirated particulates | ||
Definition of abbreviations: HP = hypersensitivity pneumonitis; IIP = idiopathic interstitial pneumonias; lymphs = lymphocytes; NSIP = nonspecific interstitial pneumonia; UIP = usual interstitial pneumonia.
Histological findings in hot-tub lung are distinctly different from nonfibrotic and fibrotic forms of classic HP.
Granulomas in HP are smaller, less tightly clustered, and lack the perigranulomatous hyaline fibrosis commonly seen in sarcoidosis.
Fibrotic HP may show classic features of nonfibrotic HP (cellular HP) in less fibrotic or nonfibrotic areas; if present, this combination of findings is a histological clue to the diagnosis of HP.
Updates to the classification of IIPs by Travis and colleagues (330) and diagnostic guidelines for idiopathic pulmonary fibrosis (20, 128) tightly link a UIP pattern with idiopathic pulmonary fibrosis and an NSIP pattern with idiopathic NSIP.
Bridging fibrosis spans subpleural and centriacinar or neighboring centriacinar fibrotic foci.
Histopathological Features of Nonfibrotic, or Cellular, HP
A confident histopathological diagnosis of nonfibrotic HP requires the presence of typical histopathological features. These include 1) a cellular interstitial pneumonia accentuated around small airways (“bronchiolocentric”) accompanied by 2) a cellular chronic bronchiolitis, 3) a distinctive pattern of granulomatous inflammation, and 4) no histopathological features to suggest a more likely alternative (Figure 3 and Table 7) (3, 39, 44, 137, 141–144). This combination of findings is often present in a single biopsy specimen. In other patients, each of several biopsy sites may demonstrate only a subset of findings, requiring review of all specimens to appreciate the complete set of features required for a confident histological diagnosis of HP.
Figure 3.

Surgical lung biopsy specimen from a patient with nonfibrotic hypersensitivity pneumonitis (HP). (A) Low-magnification photomicrograph showing preservation of lung architecture and a cellular chronic interstitial pneumonia that is accentuated around bronchioles (asterisks). Magnification, 20×. (B) Higher-magnification photomicrograph showing expansion of distal acinar and peribronchiolar interstitium by a cellular infiltrate of mononuclear inflammatory cells. Magnification, 88×. (C) Photomicrograph showing a cellular bronchiolitis in which the peribronchiolar interstitium is expanded by cellular infiltrate, predominantly comprising lymphocytes without lymphoid aggregates or follicles. Magnification, 108×. (D) Higher-magnification view of airway illustrated in C, demonstrating a poorly formed nonnecrotizing granuloma (arrow) characteristic of HP comprising loose clusters of epithelioid cells (macrophages). Magnification, 400×. (E) High-magnification photomicrograph illustrating another poorly formed nonnecrotizing granuloma (arrows) in the same biopsy specimen from a patient with nonfibrotic HP. Magnification, 264×. Hematoxylin and eosin staining was used.
The interstitial pneumonia is bronchiolocentric in distribution and comprises predominantly small lymphocytes. The inflammatory infiltrate is typically polymorphic in that it includes smaller numbers of plasma cells and occasionally eosinophils in some patients. Lymphoid aggregates, especially those with secondary germinal centers, are either absent or very focal and relatively inconspicuous. Follicular lymphoid hyperplasia and a plasma cell–predominant infiltrate suggest other possibilities, including underlying CTD or various forms of immunodeficiency. Prominent peribronchiolar lymphoid hyperplasia accompanied by granulomatous inflammation should raise concern for the possibility of granulomatous-lymphocytic ILD, which is characteristic of common variable immunodeficiency and is a lesion type usually separable from HP by the extent of the lymphoid hyperplasia, which tends to more closely resemble lymphoid interstitial pneumonia and/or low-grade lymphoma (145).
The chronic bronchiolitis characteristic of nonfibrotic HP is a continuum with bronchiolocentric interstitial pneumonia and comprise expansion of the peribronchiolar interstitium by the same lymphocyte-predominant inflammatory infiltrate, without or with only focal lymphoid aggregates that generally lack secondary germinal centers. Affected small airways may show associated organizing pneumonia that is exquisitely bronchiolocentric. Foamy alveolar macrophages may be conspicuous in peribronchiolar air spaces and are a form of microscopic obstructive pneumonia that reflects small airway dysfunction.
Granulomatous inflammation completes the triad that allows a confident diagnosis of HP on the basis of histology alone, but the diagnostic value is heavily dependent on the qualitative features of the granulomas (Figure 4) (44, 142, 144). The granulomas of HP are typically small and poorly formed, comprising loose, poorly circumscribed clusters of epithelioid and multinucleated cells (macrophages) that tend to be most prevalent in the peribronchiolar interstitium. Isolated multinucleated giant cells are common and often show nonspecific cytoplasmic inclusions such as Schaumann bodies, asteroid bodies, or cholesterol-like clefts. The poorly formed granulomas and multinucleated giant cells spill into peribronchiolar air spaces, where they may be intimately associated with organizing pneumonia but should also involve the peribronchiolar interstitium (144). Well-formed granulomas resembling those seen in sarcoidosis and granulomatous infections are uncommon and should raise the likelihood of other conditions if they predominate (134, 135). Aspiration is another important consideration that is characterized by well-formed intraluminal granulomas, often with small foci of central necrosis and associated neutrophils. The granulomas are often affiliated with aspirated foreign material, including a combination of organic and/or nonorganic particulates such as excipients used in oral medications (146). This is true of “hot-tub lung,” a diffuse lung disease (DLD) associated with Mycobacterium avium complex with clinical and radiological findings that overlap with classical types of HP, in which well-formed granulomas with or without central necrosis tend to be limited to the lumens of distal bronchioles (Figure E9) (147).
Figure 4.

(A–C) Poorly formed granulomas characteristic of hypersensitivity pneumonitis (HP) contrasted with (D and E) well-formed granulomas more typical of sarcoidosis. (A) High-magnification photomicrograph illustrating isolated multinucleated giant cells in a surgical lung biopsy specimen from a patient with nonfibrotic HP. Magnification, 400×. (B) Another photomicrograph illustrating giant cells in a patient with HP. These giant cells are distinguished by cytoplasmic cholesterol-like clefts, a nonspecific but common finding. Magnification, 400×. (C) In this high-magnification photomicrograph of a surgical lung biopsy specimen, the giant cells are largely obscured by cytoplasmic Schaumann bodies (arrow), another nonspecific but characteristic feature of the granulomatous response in HP. Magnification, 400×. (D) Low-magnification photomicrograph of surgical lung biopsy specimen from a patient with sarcoidosis showing characteristic “lymphangitic” distribution, in which the granulomas are limited to the interstitium and involve visceral pleura (asterisk), interlobular septa (arrow), and bronchovascular bundles. Magnification, 20×. (E) High-magnification photomicrograph showing a well-formed nonnecrotizing granuloma in a surgical lung biopsy specimen from a patient with sarcoidosis. The well-circumscribed, tight cluster of epithelioid cells (macrophages) is affiliated with a characteristic pattern of circumferential lamellar fibrosis. Magnification, 400×. Hematoxylin and eosin staining was used. B = bronchovascular bundle.
Probable HP refers to cases in which only some of the features described above are present. It requires the presence of both a lymphocyte-rich, bronchiolocentric interstitial pneumonia and an associated bronchiolitis, but without the granulomatous inflammation characteristic of classical HP. Indeterminate HP refers to cases in which either a cellular bronchiolocentric interstitial pneumonia or an otherwise unexplained cellular chronic bronchiolitis is present, but without the characteristic granulomatous inflammation. The chronic bronchiolitis may include peribronchiolar metaplasia (PBM), characterized by expansion of the peribronchiolar interstitium by mild, nondistorting fibrosis that extends into contiguous alveolar septa in which lining pneumocytes have been replaced by a columnar bronchiolar epithelium, without any of the other features to suggest fibrotic HP, as discussed below. Foci of organizing pneumonia may also be present in these categories.
Histopathological Features of Fibrotic HP
Fibrotic HP differs from nonfibrotic HP in that the underlying chronic interstitial pneumonia and/or bronchiolitis is complicated by fibrosis. Typical histopathological features of fibrotic HP include subpleural and centriacinar fibrosis, with or without bridging fibrosis that spans both subpleural and centriacinar regions, or with neighboring centriacinar fibrotic lesions (Figure 5) (23, 137, 140). The pattern of fibrotic interstitial pneumonia may include features that overlap with a UIP pattern, including patchy collagen fibrosis, fibroblast foci, and associated subpleural-dominant honeycombing (3, 12, 39, 40, 42, 142, 143, 148, 149). Some have applied the term “UIP-like” to draw attention to the histological overlap with a UIP pattern, which frequently poses problems in the differential diagnosis (3, 23, 28, 39). Given the potentially confusing nature of the term “UIP-like” we have chosen not to apply it in this manuscript, although we acknowledge the histological overlaps and highlight those histological features helpful in distinguishing fibrotic HP from other diffuse fibrotic lung diseases. In others, the interstitial pneumonia may have a more uniform and diffuse distribution without honeycomb change and may more closely resemble a fibrotic NSIP pattern (“NSIP-like”). Bronchiolar fibrosis typically takes the form of PBM with fibrosis, a finding that shows significant histological overlap with descriptions of interstitial airway-centered fibrosis (150, 151). Neither PBM nor airway-centered fibrosis is unique to HP, and they therefore do not by themselves establish the diagnosis (140, 152), but they are characteristic and tend to be more profuse in patients with fibrotic HP compared with patients with fibrotic IIPs (140).
Figure 5.

Photomicrographs of surgical lung biopsy specimens from two different sites in a patient with fibrotic hypersensitivity pneumonitis. (A) Scanning magnification view showing multiple sections of a right-lower-lobe biopsy specimen. There is patchy fibrosis with architectural distortion, a combination of findings that resembles usual interstitial pneumonia. Magnification, 6×. (B) Low-magnification photomicrograph showing one of the sections illustrated in A, characterized by a pattern of patchy fibrosis with subpleural honeycomb change that resembles usual interstitial pneumonia. Magnification, 17×. (C) Higher-magnification view showing expansion of the peribronchiolar interstitium by a cellular infiltrate of mononuclear inflammatory cells (upper left) and isolated Schaumann bodies (arrows) at the edge of the biopsy specimen. Magnification, 46×. (D) High-magnification photomicrograph showing one of the isolated Schaumann bodies illustrated in C. Magnification, 400×. (E) Photomicrograph from another section illustrated in A showing an isolated Schaumann body (arrow) in the fibrotic peribronchiolar interstitium. Magnification, 63×. (F) Low-magnification photomicrograph of a right-middle-lobe biopsy specimen from the same patient showing features more closely resembling nonfibrotic hypersensitivity pneumonitis. There is a more cellular chronic interstitial pneumonia accentuated around bronchioles with scattered calcified Schaumann bodies (arrows) marking isolated multinucleated giant cells. Magnification, 43×. Hematoxylin and eosin staining was used. B = bronchiole.
Distinguishing fibrotic HP from fibrotic IIPs requires identification of centriacinar fibrotic lesions and the features described in nonfibrotic HP. The latter features are usually observed in less fibrotic lung tissue. This often requires sampling of more than one site. One site may show findings indistinguishable from a fibrotic interstitial pneumonia, whereas another may show features typical of nonfibrotic HP, including those that might be more appropriately characterized as “probable” or “indeterminate” (143). This sort of diagnostic discordance between sites is analogous to the histopathological variability documented in patients with IPF, in whom NSIP-like changes are common and may be the sole finding in some samples (153). In other patients, much of a single-site biopsy specimen may mimic a fibrotic IIP, whereas the evidence in support of HP is patchy and often limited to less fibrotic lung tissue. Centriacinar fibrotic lesions, in addition to the subpleural-dominant fibrotic lesions with or without honeycombing, prominent PBM, and/or isolated peribronchiolar giant cells, often with conspicuous Schaumann bodies, may be the clues to search more diligently for the features that would make a diagnosis of HP more likely. It is important to document the fibrotic component when diagnosing HP, as this is an adverse prognostic factor.
Diagnostic Criteria
The diagnosis of HP requires integration of multiple domains that are ideally considered in the context of an MDD. Given the multitude of presenting features, fibrotic HP should be considered in the differential diagnosis for all patients with a fibrotic ILD. This is particularly challenging, given the absence of an identifiable exposure in up to 50% of patients with fibrotic HP (87, 122, 131–133). Nonfibrotic HP is usually associated with a clear exposure and less frequently poses a diagnostic dilemma, but it similarly lacks a single diagnostic pathway. For these reasons, a comprehensive multidisciplinary approach is important in diagnosing HP, particularly fibrotic HP; however, there remains substantial diagnostic disagreement across experienced MDD teams that likely reflects the absence of standardized diagnostic criteria (17).
Previous studies have identified features that increase the likelihood of HP, with diagnostic algorithms or criteria proposed by multiple groups (1, 5–10). The studies on which these proposals are based all have methodological limitations, most notably incorporation bias (e.g., serum IgG and BAL studies), incomplete consideration of all potentially informative features, absence of appropriate control groups, and inadequate validation (e.g., questionnaires). Despite these limitations, some key features are consistently identified as increasing the likelihood of an HP diagnosis, including exposure to a known offending agent (1, 7, 8), typical imaging findings (7, 8, 122, 154), and typical biopsy findings (7). BAL lymphocytosis is an important feature (1, 7); serum-specific immunoglobulins might also be helpful (1, 155, 156). Female sex, midinspiratory squeaks (or chirping rales or squawks) (157, 158), absence of a smoking history, and obstructive or mixed restrictive/obstructive physiology have also been identified as potential predictors of an HP diagnosis, but with more limited diagnostic utility. Other features are less frequently identified (e.g., episodes of symptoms and symptoms 4–8 h after exposure) (12), likely reflecting variable proportions of fibrotic and nonfibrotic HP in previous studies.
Although the diagnosis of HP is predominantly based on exposure identification, chest HRCT scan pattern, and bronchoscopic/histopathological findings, a major challenge is that no individual feature is sufficient in isolation, nor are any mandatory. This results in the potential for multiple combinations of abnormalities that can result in a diagnosis of HP. Although a single diagnostic algorithm may be applied to both fibrotic and nonfibrotic HP, these populations have frequent differences in their underlying features. For example, patients with nonfibrotic HP more often have an acute and identifiable exposure, rapid onset of both pulmonary and systemic symptoms, presence of centrilobular nodularity on chest CT scans, and lymphocytosis on BAL cellular analysis (3, 25, 105–107). Conversely, patients with fibrotic HP are less likely to have an identified exposure and more frequently have an insidious and chronic onset of isolated pulmonary symptoms, fibrotic changes with or without more specific features of HP on chest imaging, and a nonspecific differential cell profile on BAL analysis (4, 105–107). Additional features may be useful in the context of an MDD to increase or decrease the diagnostic confidence of HP on a case-by-case basis, but these are not sufficiently sensitive or specific to justify inclusion in formal diagnostic criteria.
There is often substantial uncertainty in the diagnosis of HP. This occurs most frequently in the distinction between fibrotic HP and IPF (12), reflecting the overlapping features and lack of a single, definitive gold-standard test for both diagnoses. The diagnostic criteria for HP provided in this guideline emphasize the importance of three primary domains: 1) exposure identification (e.g., clinical history with or without a questionnaire, serum IgG testing against potential antigens associated with HP, and/or specific inhalational challenge), 2) imaging pattern, and 3) BAL lymphocytosis/histopathological findings, with each described in detail in the corresponding sections of this document. Although the specific features that satisfy each domain are different for fibrotic and nonfibrotic HP, a single approach is used for all patients who have a clinical presentation consistent with HP.
The diagnostic criteria are presented in a way that explicitly conveys the diagnostic confidence associated with common combinations of specific features. We used an approach similar to the approach proposed by an international working group, which categorized ILD diagnoses on the basis of confidence (159). We categorized diagnoses as definite (≥90% confidence), high-confidence (80–89%), moderate-confidence (70–79%), and low-confidence (51–69%) diagnoses. This approach is supported by recent studies suggesting the potential therapeutic and prognostic utility of assigning diagnostic confidence in this manner (160, 161).
Criteria and an algorithm for establishing a diagnosis of HP are provided in Figures 6 and 7, which may be applied to patients with a clinical presentation consistent with either fibrotic or nonfibrotic HP. Both were developed through iterative discussion and consensus by the full guideline committee on the basis of the evidence syntheses and recommendations presented below, supplemented by the guideline committee’s collective clinical experience.
Figure 6.
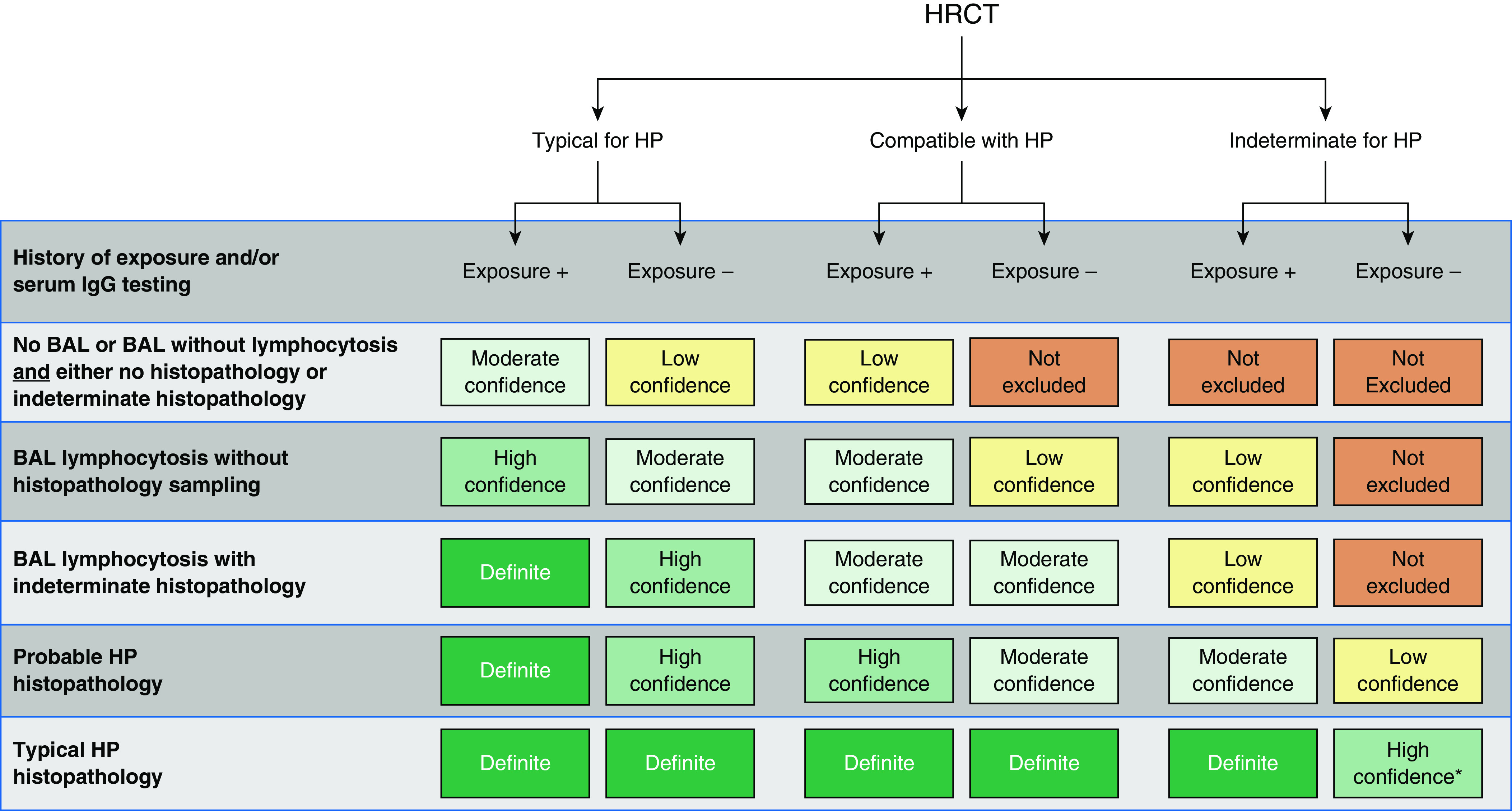
Hypersensitivity pneumonitis diagnosis based on incorporation of imaging, exposure assessment, BAL lymphocytosis, and histopathological findings. All confidence levels are subject to multidisciplinary discussion. *Confidence may increase to “definite” if the pathologist’s conclusion persists after reevaluation in the context of additional clinical information or an expert second opinion on histopathology. HP = hypersensitivity pneumonitis; HRCT = high-resolution computed tomography.
Figure 7.
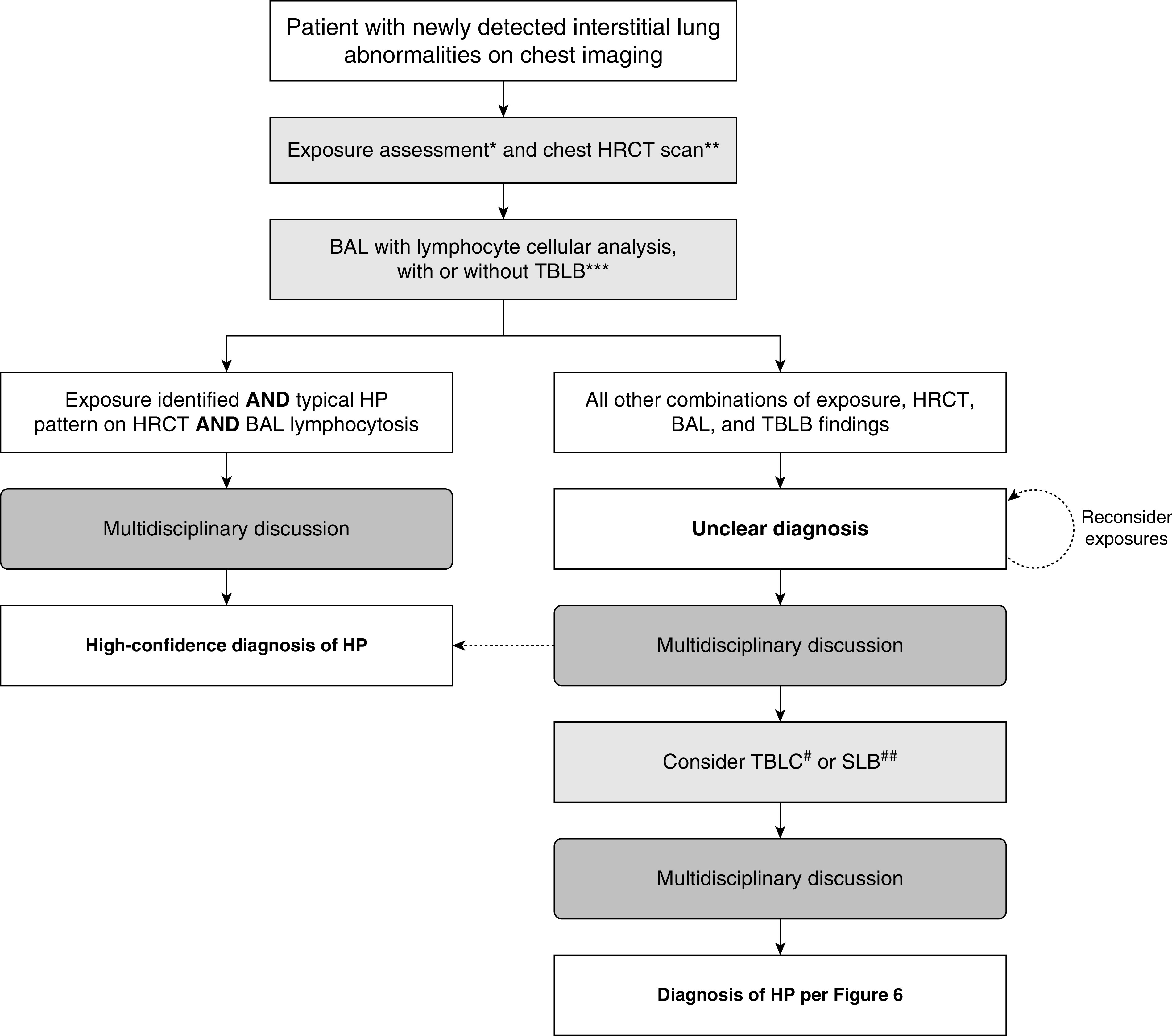
Algorithm for the diagnostic evaluation of possible hypersensitivity pneumonitis (HP). Specific features are described for all steps of the algorithm in the corresponding sections of the manuscript. A provisional diagnosis may be adequate in patients for whom the differential diagnosis has been sufficiently narrowed such that further investigations are unlikely to alter management, when invasive testing has unacceptable risks, or when such tests are declined by the patient. *Exposure assessment includes a thorough clinical history and/or serum IgG testing against potential antigens associated with HP and/or, in centers with the appropriate expertise and experience, specific inhalational challenge testing as described in References 9, 323, 324, and 325. **High-resolution computed tomography should be performed using the technique described in Table 3 and then reviewed with a thoracic radiologist. ***Transbronchial lung biopsy is suggested for patients with potential nonfibrotic HP (see question 4, recommendation 1). #TBLC is suggested for patients with potential nonfibrotic HP, depending on local expertise (see question 5, recommendation 2). ##SLB is infrequently considered in patients with nonfibrotic HP. HRCT = high-resolution computed tomography; SLB = surgical lung biopsy; TBLB = transbronchial lung biopsy; TBLC = transbronchial lung cryobiopsy.
The primary goal in the diagnosis of ILD is to make a confident diagnosis using the least invasive approach. HP can be diagnosed with high confidence in patients in whom an exposure has been identified and who have a typical HP pattern at HRCT and have BAL lymphocytosis; such patients do not require additional testing. Patients with any other combination of exposure history, HRCT pattern, and BAL results should undergo an MDD that includes an experienced expert in ILD (pulmonologist), a chest radiologist, and, if transbronchial lung biopsies were performed at the time of BAL, a pathologist familiar with histopathological features of interstitial pneumonias and HP. Additional histopathological sampling should be considered after the MDD in some patients with a high-confidence diagnosis, moderate-confidence diagnosis, or low-confidence diagnosis or in patients for whom an alternative diagnosis has not been established (161). A low-confidence diagnosis may be adequate in patients for whom the differential diagnosis has been sufficiently narrowed such that further investigations are unlikely to alter management, when invasive testing has unacceptable risks, or when such tests are declined by the patient. The diagnosis should be reconsidered at subsequent visits, particularly for patients without a definite diagnosis.
Diagnostic Interventions
Question 1: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without an overt history of exposures capable of causing ILD in the patient’s environment at home, work, or elsewhere, be subjected to formal questioning using a questionnaire to raise the possibility that a) potential inciting agents of HP are the etiology of the ILD and b) the diagnosis of the ILD is HP?
Summary of evidence
A systematic search of the literature identified 1,141 potentially relevant articles. The full text of 32 articles was reviewed, and 2 observational studies were selected to inform the guideline committee (12, 162). One study enrolled 19 patients with definite or probable HP and used clinical history, a 22-item questionnaire, and serum IgG testing against HP-associated antigens to identify potential inciting agents of HP. The environments of patients with positive findings were sampled, and potential inciting agents were confirmed or excluded (162). Another study enrolled 46 patients with IPF and used a nine-item questionnaire, serum IgG testing against HP-associated antigens, and bronchial-challenge testing to identify potential inciting agents of HP. BAL was performed, and histopathological specimens were revisited to confirm IPF or reclassify the condition as chronic HP (12). Neither questionnaire had been evaluated in previous studies.
In the former study, the questionnaire identified a potential inciting agent in 19 out of 19 (100%) patients; only 7 patients underwent subsequent environmental testing, with the potential inciting agent confirmed in 5 out of the 7 (71%) (162). In the latter study, the questionnaire identified a potential inciting agent in 27 out of 46 (59%) patients; the final diagnosis was reclassifed from IPF to chronic HP in 18 out of 27 (67%) (12). A questionnaire was more likely to identify a potential inciting agent when compared with clinical history (relative risk [RR], 3.80; 95% confidence interval [CI], 1.79–8.06) or serum IgG testing (RR, 1.58; 95% CI, 1.12–2.23), but there was no difference when a questionnaire was compared with the combination of serum IgG testing against potential antigens associated with HP plus bronchial-challenge testing (RR, 0.90; 95% CI, 0.65–1.24).
Committee discussion
The guideline committee appreciated the potential of a questionnaire to facilitate the systematic identification of inciting agents of HP in a patient’s environment, compared with clinical history-taking alone, which may be less systematic. However, there is no questionnaire that has been validated for this purpose, and it seems likely that different questionnaires are appropriate for different locations and populations (163). Thus, the guideline committee concluded that it is premature to recommend using a specific questionnaire but that use of a questionnaire may be an important adjunct to the care of patients with newly detected ILD in the future. In the interim, the committee encourages clinicians to take a thorough history to identify potential exposures and to develop and use their own questionnaires to ensure routine inquiry about potential exposures whenever they evaluate a patient with newly detected ILD. Such questionnaires should include ambient and occupational causes of HP relevant to the patient’s geographical location and cultural habits.
Recommendation
For patients with newly identified ILD whose differential diagnosis includes nonfibrotic HP or fibrotic HP, the guideline committee makes no recommendation or suggestion for or against the use of a specific questionnaire to identify potential inciting agents of HP; instead, the guideline committee recommends the development and validation of a questionnaire. Voting results: unanimous, no recommendation or suggestion. Remark: Pending the availability of a validated questionnaire, the guideline committee advocates that clinicians take a thorough history to identify potential exposures and sources in the patient’s environment that are known to be associated with HP.
Question 2: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without an overt history of exposures capable of causing ILD in the patient’s environment at home, work, or elsewhere, undergo serum testing for IgG antibodies against specific antigens to raise the possibility that a) potential inciting agents of HP are the etiology of the ILD and b) the diagnosis of the ILD is HP?
Summary of evidence
A systematic search of the literature identified 926 potentially relevant articles. The full text of 49 articles was reviewed, and 15 observational studies were selected to inform the guideline committee (1, 12, 155, 164–175). Most studies enrolled patients with known HP, usually farmer’s lung disease or bird fancier’s disease, and determined the sensitivity and specificity of serum IgG testing against potential antigens associated with HP using ELISA or precipitins, the latter including double diffusion, immunoelectrophoresis, or electrosyneresis. The antigens to which serum IgG were measured varied from study to study. Many studies included exposed and unexposed control groups, the latter most often comprising blood donors.
Serum IgG testing against potential antigens associated with HP distinguished HP from other ILDs with a sensitivity and specificity of 83% and 68%, respectively, derived from bivariate analysis of the summary receiver operator curve created by pooling four studies. Serum IgG testing against potential antigens associated with HP performed best when ELISA was the method used.
Serum IgG testing against potential antigens associated with HP distinguished patients with HP from exposed control subjects with a sensitivity and specificity of 90% and 91%, respectively, derived by pooling eight studies. Serum IgG testing against potential antigens associated with HP performed similarly for ELISA and precipitin testing but performed best for patients with metal worker’s lung, followed by farmer’s lung, bird fancier’s lung, and bagassosis.
Serum IgG testing against potential antigens associated with HP distinguished patients with HP from unexposed control subjects with a sensitivity and specificity of 93% and 100%, respectively, derived by pooling seven studies. Serum IgG testing against potential antigens associated with HP performed similarly for ELISA and precipitin testing, and among all types of HP.
A sensitivity analysis was performed to assess potential incorporation bias (overestimation of sensitivity and specificity due to the inclusion of studies that used a positive serum IgG test result as a diagnostic criterion for HP). Five such studies were removed from the analyses, and the results remained similar. Therefore, the studies were kept in the final analysis.
Committee discussion
The evidence synthesis estimated that serum IgG testing against HP-associated antigens distinguishes HP from other ILDs with a sensitivity and specificity of 83% and 68%, respectively. The committee was unanimous in the opinion that both test characteristics are suboptimal. Most committee members considered testing convenient and adequate for generating supportive data; however, they acknowledged that testing is insufficient for confirming or excluding a diagnosis of HP because the test characteristics are inferior to most screening tests currently in use.
It was emphasized that a positive serum IgG result does not mean that the exposure is the cause of the lung condition; it only indicates that the patient has likely been exposed to a potential cause of HP at some point in his or her life, and it may be worthy of further consideration to explore the source of the potential causative agent in the patient’s domestic, social, and/or work environment before assignment of a diagnosis of HP, particularly when other diagnostic findings of HP are less certain (e.g., no BAL lymphocytosis, probable or indeterminate CT or biopsy patterns).
The committee acknowledged the lack of standardization of serum IgG testing against potential antigens associated with HP, with no standardized, internationally accepted “HP panel” and different commercial kits being used by different laboratories. In addition, there was discussion of how the performance of serum IgG testing may vary with some serum IgG testing being more helpful with some antigens than others and in some parts of the world. Development and validation of a standard or personalized hypersensitivity panel was considered a priority for future clinical investigation.
Recommendations
1. For patients with newly identified ILD whose differential diagnosis includes nonfibrotic HP, the guideline committee suggests performing serum IgG testing that targets potential antigens associated with HP (suggestion, very low confidence in the estimated effects). Voting results: recommendation for, 8; suggestion for, 12; no recommendation or suggestion, 3; suggestion against, 5; recommendation against, 0.
2. For patients with newly identified ILD whose differential diagnosis includes fibrotic HP, the guideline committee suggests performing serum IgG testing that targets potential antigens associated with HP (suggestion, very low confidence in the estimated effects). Voting results for fibrotic HP: recommendation for, 8; suggestion for, 14; no recommendation or suggestion, 3; suggestion against, 3; recommendation against, 0.
Question 3: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo BAL fluid lymphocyte cellular analysis to diagnose HP?
Summary of evidence
A systematic search of the literature identified 1,500 potentially relevant articles. The full text of 340 articles was reviewed, and 84 observational studies were selected to inform the guideline committee (12, 105, 176–255). Most studies enrolled patients with known HP or other types of ILD, performed BAL with lymphocyte cellular analysis, and compared the percentage of lymphocytes among patients with different conditions. Farmer’s lung and bird fancier’s lung disease were the most common types of HP, whereas IPF and sarcoidosis were the most common non-HP ILDs enrolled.
A meta-analysis of 36 studies (1,643 patients) demonstrated that patients with HP had a higher proportion of BAL fluid lymphocytes than patients with IPF (mean difference [MD], 30%; 95% CI, 27–34%). This effect was seen regardless of whether the study enrolled patients with nonfibrotic HP (9 studies; MD, 34%; 95% CI, 29–40%), fibrotic HP (12 studies; MD, 21%; 95% CI, 14–27%), or mixed populations with both nonfibrotic and fibrotic HP (15 studies; MD, 36%; 95% CI, 32–40%).
Similarly, a meta-analysis of 53 studies (3,112 patients) demonstrated that patients with HP had a higher proportion of BAL fluid lymphocytes than patients with sarcoidosis (MD, 19%; 95% CI, 17–21%). This effect was seen regardless of whether the study enrolled patients with nonfibrotic HP (17 studies; MD, 25%; 95% CI, 22–27%), fibrotic HP (16 studies; MD, 16%; 95% CI, 11–20%), or mixed populations with both nonfibrotic and fibrotic HP (21 studies; MD, 18%; 95% CI, 15–20%).
In studies with few patients, HP was compared with other ILDs, including CTD-ILD, idiopathic NSIP, and cryptogenic organizing pneumonia, but the number of patients was too small to make meaningful comparisons.
For distinguishing fibrotic HP from IPF, BAL fluid lymphocyte thresholds of 20%, 30%, and 40% yielded sensitivities of 69%, 55%, and 41%, respectively, and specificities of 61%, 80%, and 93% respectively, with an area under the curve of 0.54 (95% CI, 0.51–0.58). For distinguishing fibrotic HP from sarcoidosis, BAL fluid lymphocyte thresholds of 20%, 30%, and 40% yielded sensitivities of 69%, 55%, and 41%, respectively, and specificities of 26%, 43%, and 61% respectively, with an area under the curve of 0.44 (95% CI, 0.41–0.47). Finally, for distinguishing nonfibrotic HP from sarcoidosis, BAL fluid lymphocyte thresholds of 20%, 30%, and 40% yielded sensitivities of 95%, 88%, and 76%, respectively, and specificities of 26%, 43%, and 61% respectively, with an area under the curve of 0.71 (95% CI, 0.67–0.74). A sensitivity analysis was performed to assess potential inclusion bias (overestimation of sensitivity and specificity due to the inclusion of studies that used BAL fluid lymphocytosis as a diagnostic criterion for HP); the results remained the same, and the studies were therefore kept in the final analysis.
Committee discussion
The large MD identified when the proportion of BAL fluid lymphocytes among patients with HP was compared with the proportion of BAL fluid lymphocytes among patients with IPF or sarcoidosis led most of the guideline committee to conclude that BAL fluid cellular lymphocyte analysis can play a key role in distinguishing fibrotic HP from IPF and sarcoidosis and in distinguishing nonfibrotic HP from sarcoidosis. The committee was unwilling to extrapolate the results to other ILDs, given the paucity of data, and identified such comparisons as a key research priority.
The committee did not identify a threshold proportion of BAL fluid lymphocytes that distinguishes HP from non-HP ILD, given the poor area under the curve for each comparison. The seeming discordance of the MD and the area under the curve was attributed to the large standard deviation of many studies. In the absence of empirical evidence, the committee’s collective clinical experience indicated that healthy nonsmokers have a proportion of BAL fluid lymphocytes of 10–15% and, therefore, the committee considered a 30% threshold to be reasonable (256).
Although the guideline committee concluded that BAL fluid cellular lymphocyte analysis is indicated to increase the diagnostic likelihood of HP, the committee made a stronger recommendation for BAL in patients with suspected nonfibrotic HP than for patients with suspected fibrotic HP because there is an additional reason for BAL in patients with suspected nonfibrotic HP—to identify or exclude pulmonary infection, especially M. tuberculosis in patients from endemic areas with a high prevalence of M. tuberculosis, which can progress to death if untreated.
Recommendations
1. For patients with newly identified ILD whose differential diagnosis includes nonfibrotic HP, the guideline committee recommends BAL with lymphocyte cellular analysis (recommendation, very low confidence in the estimated effects). Voting results: recommendation for, 19; suggestion for, 11; no recommendation or suggestion, 0; suggestion against, 1; recommendation against, 0.
2. For patients with newly identified ILD whose differential diagnosis includes fibrotic HP, the guideline committee suggests BAL with lymphocyte cellular analysis (suggestion, very low confidence in the estimated effects). Voting results: recommendation for, 10; suggestion for, 18; no recommendation or suggestion, 0; suggestion against, 3; recommendation against, 0.
Question 4: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo transbronchial forceps lung biopsy to diagnose HP?
Summary of evidence
A systematic search of the literature identified 2,465 potentially relevant articles. The full text of 24 articles was reviewed, and 13 observational studies were selected to inform the guideline committee (14, 35, 257–267). Four studies enrolled patients with known HP, six enrolled patients with ILD, and three enrolled patients with DLD. We initially considered ILD and DLD to be synonymous, but, on noting different diagnostic results in the two groups, we opted to analyze each separately. All studies performed transbronchial forceps lung biopsy (TBBx), and most reported the histopathological diagnostic yield of the procedure; some also reported the frequency of adverse effects.
Adequate specimens were obtained in 87% (95% CI, 79–96%) of sampling procedures. The diagnostic yield (defined as the number of procedures that yielded a histopathological diagnosis among the total number of procedures performed) among patients with ILD was 37% (95% CI, 32–42%). Inclusion of TBBx in the multimodality diagnostic approach of patients with ILD increased the likelihood of arriving at a diagnosis compared with an approach without TBBx (RR, 1.67; 95% CI, 1.21–2.30). The diagnostic yield among patients with DLD was 68% (95% CI, 50–86%). Patients with DLD were more likely than patients with ILD to receive a non-ILD diagnosis like malignancy, infection, etc. The diagnostic yield could not be calculated for patients with known HP, but 41% (95% CI, 25–56%) of such patients who underwent TBBx were confirmed to have HP.
Among the studies that enrolled patients with ILD or DLD, two studies reported the pneumothorax rate (7%; 95% CI, 1–13%), two studies reported periprocedural mortality (no cases; one study monitored for 24 h and the other monitored for 6 mo), two studies reported respiratory exacerbation or failure (no cases), four studies reported moderate-to-severe bleeding (4%; 95% CI, 0–8%), and six studies reported severe bleeding (no cases). One study reported the number of pneumothoraces that required a chest tube (6%; 95% CI, 0–13%), with none lasting longer than 72 hours. The studies that enrolled patients with known HP did not report adverse outcomes.
Committee discussion
There was general agreement that the diagnostic yield of TBBx was suboptimal, with only half of procedures resulting in a diagnosis and with the exact frequency depending on the population. However, the guideline committee was divided regarding the interpretation of the evidence. Some members argued that roughly half of patients who undergo TBBx will be spared an SLB, and other members contended that roughly half of patients will require two sampling procedures instead of one, with the former favoring TBBx and the latter arguing against TBBx. An additional concern is the potential for diagnostic misclassification based on potentially suboptimal TBBx specimens.
Notably, the guideline committee observed that the diagnostic yield was substantially higher among patients with DLD than among patients with ILD (68% vs. 37%). This was extrapolated to the notion that diagnostic yield may be higher among patients with suspected nonfibrotic HP than among patients with suspected fibrotic HP. This potential difference was supported by nonsystematic clinical observations that granulomas are more likely to be detected and to be diagnostic among patients with nonfibrotic HP than among patients with fibrotic HP. Thus, TBBx was judged to be worthwhile in patients with suspected nonfibrotic HP, but not in patients with suspected fibrotic HP.
The committee acknowledged that TBBx is safe, available in most institutions, minimally burdensome, and inexpensive and that most pulmonary clinicians have experience performing the procedure. The patient representative indicated that he would choose a TBBx in the hope of avoiding SLB, even with the knowledge that a second procedure would be necessary if the TBBx were nondiagnostic.
Recommendations
1. For patients with newly identified ILD whose differential diagnosis includes nonfibrotic HP, the guideline committee suggests TBBx (suggestion, very low confidence in the estimated effects). Voting results: recommendation for, 1; suggestion for, 20; no recommendation or suggestion, 4; suggestion against, 3; recommendation against, 0.
2. For patients with newly identified ILD whose differential diagnosis includes fibrotic HP, the guideline committee makes no recommendation or suggestion for or against TBBx. Voting results: recommendation for, 1; suggestion for, 13; no recommendation or suggestion, 6; suggestion against, 7; recommendation against, 1.
Question 5: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo transbronchial lung cryobiopsy to diagnose HP?
Summary of evidence
A systematic search of the literature identified 695 potentially relevant articles. The full text of 34 articles was reviewed, and 24 observational studies were selected to inform the guideline committee (257, 261, 262, 264, 268–287). One study enrolled patients with known or suspected HP, 19 enrolled patients with ILD, and 4 enrolled patients with DLD. All studies performed transbronchial lung cryobiopsy (TBLC) and reported the diagnostic yield of the procedure; some also reported the frequency of adverse effects.
The diagnostic yields were 91% (95% CI, 83–99%), 82% (95% CI, 78–86%), and 82% (95% CI, 73–90%) among patients with known or suspected HP, ILD, and DLD, respectively. Among patients with known or suspected HP in whom a diagnosis was made by TBLC, 100% (95% CI, 91.4–100%) were confirmed to have HP and none had an alternative ILD or a non-ILD diagnosis. Among patients with ILD in whom a diagnosis was made by TBLC, 7.2% (95% CI, 5.6–9.2%) were determined to have HP, 77.5% (95% CI, 74.5–80.3%) were found to have an alternative type of ILD, and 15.3% (95% CI, 13–18%) were determined to have a non-ILD diagnosis, such as an infection. Among patients with DLD in whom a diagnosis was made by TBLC, 13.4% (95% CI, 10.9–16.2%) were determined to have HP, 71.5% (95% CI, 67.9–74.9%) were found to have an alternative type of ILD, and 15.1% (95% CI, 12.5–18.1%) were determined to have a non-ILD diagnosis.
Several studies reported adverse events of TBLC. In the only study that enrolled patients with known or suspected HP, bleeding of any severity occurred in 4% (95% CI, 0–10%) and pneumothoraces occurred in 27% (95% CI, 14–40%). Among studies that enrolled patients with ILD or DLD, 16 studies reported procedural mortality (rare cases; 95% CI, 0–1%); 11 studies did not specify the follow-up duration, three studies employed 30 days of follow-up, one study used 90 days of follow-up, and one study had both 30 days and 90 days of follow-up. Nine studies reported post-procedural exacerbation or respiratory failure (rare cases; 95% CI, 0–1%), 17 studies reported bleeding of any severity (11%; 95% CI, 7–15%), 18 studies reported severe bleeding (0%; 95% CI, 0–1%), and 23 studies reported pneumothoraces (10%; 95% CI, 8–13%).
Notably, several studies directly evaluated TBLC and TBBx within the same populations (257, 261, 262). The studies reported higher diagnostic yield with TBLC, although the incidence of bleeding was also higher with TBLC.
Committee discussion
There was general agreement among members of the guideline committee that the diagnostic yield of TBLC is favorable, with 82–91% of patients potentially avoiding SLB. Histopathological patterns of UIP identified by TBLC have been shown to have substantial agreement with findings from SLB, including both histopathological agreement (kappa, 0.70; 95% CI, 0.55–0.86) and diagnostic agreement at MDD (kappa, 0.62; 95% CI, 0.47–0.78) (288). The safety profile of TBLC is also favorable compared with SLB. However, the committee acknowledged that most medical centers, particularly community medical centers, currently cannot provide TBLC due to lack of equipment and expertise, nor do they have a large enough volume to justify establishing a TBLC program.
The guideline committee related the evidence from studies that enrolled patients with ILD to its recommendations for patients with suspected fibrotic HP and related the evidence from studies that enrolled patients with known or suspected HP to its recommendations for patients with suspected nonfibrotic HP. They concluded that, in TBLC-capable medical centers, TBLC should be offered to those with suspected fibrotic HP because it may lead to avoidance of a more burdensome, expensive, uncomfortable, and potentially harmful SLB. However, there was lack of agreement about whether TBLC should be similarly offered to patients with suspected nonfibrotic HP because most centers that offer TBLC can also perform TBBx; TBBx probably has fewer complications and was recommended for this patient group as described above.
Recommendations
1. For patients with newly identified ILD whose differential diagnosis includes nonfibrotic HP, the guideline committee makes no recommendation or suggestion for or against TBLC. Voting results: recommendation for, 4; suggestion for, 10; no recommendation or suggestion, 7; suggestion against, 6; recommendation against, 0.
2. For patients with newly identified ILD whose differential diagnosis includes fibrotic HP, the guideline committee suggests TBLC (suggestion, very low confidence in estimated effects). Voting results: recommendation for, 8; suggestion for, 11; no recommendation or suggestion, 5; suggestion against, 3; recommendation against, 0.
Question 6: Should patients with newly detected ILD on chest radiographs or a CT scan of the chest, with or without a history of exposure capable of causing HP, undergo SLB to diagnose HP?
Summary of evidence
A systematic search of the literature identified 501 potentially relevant articles. The full text of 56 articles was reviewed, and 34 nonrandomized studies were selected to inform the guideline committee (14, 289–322). One study enrolled patients with known or suspected HP, 27 enrolled patients with ILD, and 6 enrolled patients with DLD. All studies performed SLB and reported the diagnostic yield of the procedure; some also reported the frequency of adverse effects.
The diagnostic yields were 96% (95% CI, 90–100%), 98% (95% CI, 98–99%), and 96% (95% CI, 93–99%) among patients with known or suspected HP, ILD, and DLD, respectively. Among patients with known or suspected HP in whom a diagnosis was made by SLB, 91% (95% CI, 79.3–96.5%) were confirmed to have HP and 9% (95% CI, 3.5–20.7%) had an alternative ILD. Among patients with ILD (not specifically suspected or known to have HP) in whom a diagnosis was made by SLB, 9% (95% CI, 5.9–13.1%) were determined to have HP, 61% (95% CI, 54.3–66.4%) were found to have an alternative type of ILD, and 31% (95% CI, 25.1–36.6%) were determined to have a non-ILD diagnosis, such as an infection. Among patients with DLD in whom a diagnosis was made by SLB, 9% (95% CI, 8.1–10.3%) were determined to have HP, 61% (95% CI, 59.2–62.9%) were found to have an alternative type of ILD, and 30% (95% CI, 28.1–31.6%) were determined to have a non-ILD diagnosis.
Many studies reported adverse events of SLB. Procedural mortality was 2% (95% CI, 1–3%; after 30 days of follow-up). Post-procedural exacerbations or respiratory failure occurred in 2% (95% CI, 1–3%). Bleeding was detected in 1% (95% CI, 0–1%). Pneumothorax persistence after chest tube removal was observed in 4% (95% CI, 2–5%), with a prolonged air leak occurring in 3% (95% CI, 2–4%). Respiratory infection and delayed wound healing were complications in 3% (95% CI, 2–3%) and 3% (95% CI, 1–5%), respectively.
Committee discussion
The differential diagnosis for ILD is wide, and management varies substantially among the causes of ILD. Thus, making a definitive or highly confident diagnosis is usually beneficial to patients. The guideline committee concluded that the diagnostic yield was sufficiently large and the adverse events sufficiently few to warrant SLB, despite its burdens and costs, once other diagnostic tests had failed to result in a definitive diagnosis (including other forms of biopsy). The committee emphasized that the decision to perform a SLB should be made after a comprehensive assessment of all available data, ideally in the context of an MDD, as well as after a thorough discussion with the patient about the potential benefits and risks of this procedure.
Recommendations
1. For patients with newly identified ILD whose differential diagnosis includes nonfibrotic HP, the guideline committee suggests SLB; this recommendation is intended for after alternative diagnostic options have been exhausted (suggestion, very low confidence in estimated effects). Voting results: recommendation for, 1; suggestion for, 20; no recommendation or suggestion, 1; suggestion against, 7; recommendation against, 1.
2. For patients with newly identified ILD whose differential diagnosis includes fibrotic HP, the guideline committee suggests SLB; this recommendation is intended for after alternative diagnostic options have been exhausted (suggestion, very low confidence in estimated effects). Voting results: recommendation for, 6; suggestion for, 23; no recommendation or suggestion, 1; suggestion against, 0; recommendation against, 0.
Future Directions
The guideline committee recognized an urgent need to improve knowledge on several topics, including 1) understanding the nature and pathophysiology of HP, 2) diagnostic approaches, 3) disease behavior and natural history, and 4) therapeutic approaches. Key questions on pathophysiology include genetic susceptibility and both host and environmental factors. For diagnostic approaches, important needs include the validation and standardization of questionnaires, BAL lymphocytosis threshold, specific antibodies, and biomarkers. In addition, new techniques like genomic classifiers and artificial intelligence to improve diagnosis and prognosis need to be assessed. Questions about disease behavior range from prevention to the phenotyping of HP. Regarding phenotyping, the differences between predominantly inflammatory and fibrotic subtypes are important questions. Finally, now that this guideline has established a standardized diagnostic approach to HP, future work needs to address the management of the different subtypes of HP in clinical trials and other research. For a full list of questions deemed important by the guideline committee, see the online supplement.
Registries can play an important role in acquiring knowledge. To maximize their potential, HP-specific registries are needed. Such registries would be particularly helpful in determining the worldwide and national incidence and prevalence of all HP, subtypes of HP (i.e., fibrotic and nonfibrotic HP), and phenotypes of HP (e.g., HP with autoimmune features, HP with progressive fibrotic behavior, etc.), including those without an identifiable inciting agent by serum IgG testing against potential antigens associated with HP and a thorough evaluation. Registries can also help us understand the frequencies of various detectable and nondetectable exposures and can serve as a biobank for blood and tissue specimens, chest HRCT images, and antigen quantification from the home and workplace.
Conclusions
When a patient presents with newly detected ILD identified by HRCT, the clinician should be prompted to elicit a careful and thorough history and possibly utilize formal questionnaires to reveal the possibility of a temporal relationship between environmental factors/exposures and the onset of symptoms. Serum IgG antibody testing against potential antigens associated with HP may also be performed to detect exposure to potential inciting agents of HP. Many patients should proceed to BAL with lymphocyte cellular analysis with or without transbronchial lung biopsy, and if this information, accompanied by the HRCT patterns, is insufficient to make a definitive diagnosis, they should undergo an MDD with consideration of TBBx, TBLC, or SLB. This approach is recommended by experts in HP on the basis of the best available evidence. The approach should be reevaluated as new evidence becomes available and should be modified as needed.
Acknowledgments
This official clinical practice guideline was prepared by an ad hoc subcommittee of the ATS, JRS, and ALAT.
Members of the subcommittee are as follows:
Ganesh Raghu, M.D. (Co-Chair)1*
Kevin C. Wilson, M.D. (Co-Chair)2,3‡
Elena Bargagli, M.D., Ph.D.4
Elisabeth Bendstrup, M.D., Ph.D.5
Hassan A. Chami, M.D., M.Sc.6§
Abigail T. Chua, M.D.7§
Jonathan H. Chung, M.D.8
Bridget F. Collins, M.D.1
Tamera J. Corte, M.B. B.S., Ph.D.9
Jean-Charles Dalphin, M.D., Ph.D.10†
Sonye K. Danoff, M.D., Ph.D.11
Javier Diaz-Mendoza, M.D.12§
Abhijit Duggal, M.D., M.S., M.P.H.13§
Ryoko Egashira, M.D., Ph.D.14
Thomas Ewing, B.S.N.||
Mridu Gulati, M.D., M.P.H.15
Yoshikazu Inoue, M.D., Ph.D.16
Alex R. Jenkins, Ph.D.17§
Kerri A. Johannson, M.D., M.P.H.18
Takeshi Johkoh, M.D., Ph.D.19
Masanori Kitaichi, M.D., Ph.D.20
Shandra L. Knight, M.S.21§
Dirk Koschel, M.D.22
Michael Kreuter, M.D.23¶
David J. Lederer, M.D., M.S.24,25
Yolanda Mageto, M.D., M.P.H.26
Lisa A. Maier, M.D., M.S.P.H.27,28
Carlos Matiz, M.D.29
Ferran Morell, M.D., Ph.D.30
Jeffrey L. Myers, M.D.31**
Andrew G. Nicholson, D.M.32,33
Setu Patolia, M.D., M.P.H.34§
Carlos A. Pereira, M.D.35
Martine Remy-Jardin, M.D., Ph.D.36‡‡
Elisabetta A. Renzoni, M.D., Ph.D.37
Christopher J. Ryerson, M.D., M.A.S.38§§
Margaret L. Salisbury, M.D., M.S.39
Moises Selman, M.D.40
Maximiliano Tamae-Kakazu, M.D.41§
Martina Vasakova, M.D., Ph.D.42||||
Simon L. F. Walsh, M.D.43
Wim A. Wuyts, M.D., Ph.D.44
1Center for Interstitial Lung Diseases, Department of Medicine and Department of Laboratory Medicine and Pathology (Adjunct), University of Washington, Seattle, Washington; 2Department of Medicine, School of Medicine, Boston University, Boston, Massachusetts; 3American Thoracic Society, New York, New York; 4Department of Clinical Sciences, Surgery and Neurosciences, University of Siena, Siena, Italy; 5Center for Rare Lung Diseases, Department of Respiratory Diseases and Allergy, Aarhus University Hospital, Aarhus University, Aarhus, Denmark; 6Department of Medicine, American University of Beirut, Beirut, Lebanon; 7Division of Pulmonary, Critical Care, and Sleep Medicine, Department of Medicine, Stony Brook University Hospital, Stony Brook University, Stony Brook, New York; 8Department of Radiology, University of Chicago, Chicago, Illinois; 9Centre of Research Excellence in Pulmonary Fibrosis, Royal Prince Alfred Hospital, University of Sydney, Sydney, Australia; 10Service de Pneumologie, Centre Hospitalier Universitaire Besançon, Universitaire Besançon, Besançon, France; 11Division of Pulmonary and Critical Care Medicine, Department of Medicine, School of Medicine, John Hopkins University, Baltimore, Maryland; 12Division of Pulmonary and Critical Care Medicine, Henry Ford Hospital–Wayne State University, Detroit, Michigan; 13Department of Critical Care, Respiratory Institute, Cleveland Clinic, Cleveland, Ohio; 14Department of Radiology, Faculty of Medicine, Saga University, Saga, Japan; 15Section of Pulmonary, Critical Care, and Sleep Medicine–Yale Occupational and Environmental Medicine Program, School of Medicine, Yale University, New Haven, Connecticut; 16Clinical Research Center, National Hospital Organization Kinki-Chuo Chest Medical Centre, Osaka, Japan; 17Division of Respiratory Medicine, University of Nottingham, Nottingham, United Kingdom; 18Department of Medicine and Department of Community Health Sciences, University of Calgary, Calgary, Alberta, Canada; 19Department of Radiology, Kansai Rosai Hospital, Amagaski, Japan; 20Department of Pathology, National Hospital Organization Minami Wakayama Medical Center, Tanabe, Wakayama, Japan; 21Library and Knowledge Sciences and 27Division of Environmental and Occupational Health Sciences, Department of Medicine, National Jewish Health, Denver, Colorado; 22Zentrum für Pneumologie und Bereich Pneumologie, Fachkrankenhaus Coswig–Uniklinikum Dresden, Coswig, Germany; 23Center for Interstitial and Rare Lung Diseases, Pneumology and Respiratory Critical Care Medicine, Thoraxklinik, University of Heidelberg–German Center for Lung Research, Heidelberg, Germany; 24Regeneron Pharmaceuticals, Inc., Tarrytown, New York; 25Irving Medical Center, Columbia University, New York, New York; 26Center for Advanced Heart and Lung Disease, Baylor University Medical Center, Baylor University, Dallas, Texas; 28Department of Medicine, School of Medicine and Department of Environmental and Occupational Health, School of Public Health, University of Colorado, Aurora, Colorado; 29Departamento de Medicina Pulmonar y Interna, Fundación Santa Fe de Bogotá–Universidad de los Andes, Bogotá, Colombia; 30Institut de Recerca Vall d’Hebron, CIBER de Malalties Respiratòries (CIBERES), Institut de Salut Carles III, Servei de Pneumologia–Universitat Autònoma de Barcelona, Catalonia, Spain; 31Department of Pathology, University of Michigan, Ann Arbor, Michigan; 32Department of Histopathology, Royal Brompton and Harefield National Health Service Foundation Trust, London, United Kingdom; 33National Heart and Lung Institute, 37Interstitial Lung Disease Unit, Royal Brompton Hospital, and 43Department of Radiology, National Heart and Lung Institute, Imperial College London, London, England, United Kingdom; 34Division of Pulmonary, Critical Care, and Sleep Medicine, Saint Louis University, St. Louis, Missouri; 35Interstitial Lung Disease Program, Federal University of São Paulo, São Paulo, Brazil; 36Department of Thoracic Imaging, University of Lille, Lille, France; 38Department of Medicine, University of British Columbia, Vancouver, British Columbia, Canada; 39Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee; 40Instituto Nacional de Enfermedades Respiratorias, Ismael Cosio Villegas, Mexico; 41Spectrum Health–Michigan State University, Grand Rapids, Michigan; 42Department of Respiratory Medicine, First Faculty of Medicine, Charles University–Thomayer Hospital, Prague, Czech Republic; and 44Unit for Interstitial Lung Diseases, Department of Respiratory Medicine, University Hospitals Leuven, Leuven, Belgium
*Content Co-Chair.
†Deceased.
‡Methodology Co-Chair.
§Methodologist.
||Patient representative.
¶Future directions section leader.
**Pathology section leader.
‡‡Radiology section leader.
§§Definition and Diagnostic Criteria sections leader.
||||Clinical Manifestations, Epidemiology, and Pathogenesis sections leader.
Footnotes
Supported by the American Thoracic Society, Japanese Respiratory Society, and Asociación Latinoamericana de Tórax.
This Official clinical practice guideline was approved by the American Thoracic Society, Japanese Respiratory Society, and Asociación Latinoamericana de Tórax May 2020
An Executive Summary of this document is available at http://www.atsjournals.org/doi/suppl/10.1164/rccm.202005-2032ST.
This document has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202005-2032ST on July 24, 2020
Author Disclosures: G.R. served as a consultant for Bellerophon, Biogen, BMS, Boehringer Ingelheim, Fibrogen, Genentech, Gilead, Nitto, Promedior, Pure Tech Health, Respivant, Roche, Veracyte, and Zambon; and served on a data and safety monitoring board for Avalyn. K.C.W. is employed by the American Thoracic Society as Chief of Documents and Documents Editor with interest in the success of ATS guidelines. H.A.C. served on an advisory committee for AstraZeneca, Boehringer Ingelheim, Merck Sharp & Dohme, Novartis, and Pfizer; served as a speaker for Mundipharma, Novartis, and Pfizer; and received research support from Pfizer. J.H.C. served as a consultant for Veracyte; served as a speaker for Boehringer Ingelheim and Genentech; and received author royalties from Elsevier. T.J.C. served on an advisory committee for Ad Alta, Boehringer Ingelheim, Bristol Myers Squibb, Promedior, and Roche; served as a consultant for Boehringer Ingelheim and Roche; and received research support from Actelion, Avalyn Pharma, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Galapagos, and Roche. J.-C.D. served on an advisory committee for Chiesi, Novartis, and Teva; served as a speaker for Boehringer Ingelheim and Menarini; received research support from Asten France and SOS Oxygene; and received other transfers of value from GlaxoSmithKline, Novartis, and Roche. S.K.D. received research support from Boehringer Ingelheim and Genentech/Roche; served on a data and safety monitoring board for Galapagos and Galecto; and is an employee of the Pulmonary Fibrosis Foundation. R.E. served as a speaker for AstraZeneca, Bayer, Boehringer Ingelheim, and Shionogi. M.G. served on an advisory committee and received other transfers of value from Boehringer Ingelheim and the France Foundation; received research support from Boehringer Ingelheim; and received travel expenses from the Pulmonary Fibrosis Foundation. Y.I. served as a consultant for Boehringer Ingelheim and Promedior; served on an advisory committee for Asahi Kasei, Galapagos, and Shionogi; received research support from Sekisui Medical; and received honoraria from Boehringer Ingelheim and Shionogi. K.A.J. served on an advisory committee for Blade Therapeutics, Boehringer Ingelheim, Roche, and Theravance; served as a consultant for Blade Therapeutics, Boehringer Ingelheim, Theravance, and Three Lakes Foundation; served as a speaker for Boehringer Ingelheim and Roche; and received research support from UCB. D.K. served on an advisory committee and received research support from Boehringer Ingelheim and Roche. M. Kreuter served on an advisory committee for Boehringer Ingelheim, Galapagos, and Roche; and received research support from Boehringer Ingelheim and Roche. D.J.L. served on an advisory committee for Boehringer Ingelheim; served as a consultant for Galapagos, Galecto, Genentech/Roche, Patara, Pulmonary Fibrosis Foundation, and Veracyte; and is an employee of Regeneron. Y.M. received honoraria from Boehringer Ingelheim and Genentech. C.M. received research support from AstraZeneca, Boehringer Ingelheim, Celgene, Merck, and Sanofi; and his spouse is an employee of Merck. A.G.N. served as a consultant for eResearch Technology and MedQIA; and served as a speaker for Boehringer Ingelheim. E.A.R. served on an advisory committee for Roche; served as a speaker for Boehringer Ingelheim, Mundipharma, and Roche; and received travel support from Boehringer Ingelheim. C.J.R. received research support and served as a speaker for Boehringer Ingelheim and Roche. M.L.S. served on an advisory committee, served as a speaker, and received research support from Boehringer Ingelheim; and served as a consultant for Boehringer Ingelheim and Orinove. M.S. served as a consultant for Boehringer Ingelheim and Celgene. M.V. served on an advisory committee and as a consultant for Boehringer Ingelheim; and received research support from Roche. S.L.F.W. served on an advisory committee for Boehringer Ingelheim and Roche; served as a consultant for Galapagos, OSIC, and Sanofi; served as a speaker for Bracco; and received research support from Boehringer Ingelheim. W.A.W. received research support from Boehringer Ingelheim and Roche. E. Bargagli, E. Bendstrup, A.T.C., B.F.C., J.D.-M., A.D., T.E., A.R.J., T.J., M. Kitaichi, S.L.K., L.A.M., F.M., J.L.M., S.P., C.A.P., M.R.-J., and M.T.-K. reported no relationships with relevant commercial interests.
Contributor Information
Collaborators: on behalf of the American Thoracic Society, Japanese Respiratory Society, and Asociación Latinoamericana de Tórax
References
- 1.Lacasse Y, Selman M, Costabel U, Dalphin JC, Ando M, Morell F, et al. HP Study Group. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168:952–958. doi: 10.1164/rccm.200301-137OC. [DOI] [PubMed] [Google Scholar]
- 2.Elicker BM, Jones KD, Henry TS, Collard HR. Multidisciplinary approach to hypersensitivity pneumonitis. J Thorac Imaging. 2016;31:92–103. doi: 10.1097/RTI.0000000000000186. [DOI] [PubMed] [Google Scholar]
- 3.Churg A, Muller NL, Flint J, Wright JL. Chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2006;30:201–208. doi: 10.1097/01.pas.0000184806.38037.3c. [DOI] [PubMed] [Google Scholar]
- 4.Fink JN, Ortega HG, Reynolds HY, Cormier YF, Fan LL, Franks TJ, et al. Needs and opportunities for research in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2005;171:792–798. doi: 10.1164/rccm.200409-1205WS. [DOI] [PubMed] [Google Scholar]
- 5.Schuyler M, Cormier Y. The diagnosis of hypersensitivity pneumonitis. Chest. 1997;111:534–536. doi: 10.1378/chest.111.3.534. [DOI] [PubMed] [Google Scholar]
- 6.Richerson HB, Bernstein IL, Fink JN, Hunninghake GW, Novey HS, Reed CE, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis: report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84:839–844. doi: 10.1016/0091-6749(89)90349-7. [DOI] [PubMed] [Google Scholar]
- 7.Morisset J, Johannson KA, Jones KD, Wolters PJ, Collard HR, Walsh SLF, et al. HP Delphi Collaborators. Identification of diagnostic criteria for chronic hypersensitivity pneumonitis: an international modified Delphi survey. Am J Respir Crit Care Med. 2018;197:1036–1044. doi: 10.1164/rccm.201710-1986OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johannson KA, Elicker BM, Vittinghoff E, Assayag D, de Boer K, Golden JA, et al. A diagnostic model for chronic hypersensitivity pneumonitis. Thorax. 2016;71:951–954. doi: 10.1136/thoraxjnl-2016-208286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity pneumonitis: perspectives in diagnosis and management. Am J Respir Crit Care Med. 2017;196:680–689. doi: 10.1164/rccm.201611-2201PP. [DOI] [PubMed] [Google Scholar]
- 10.Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ, Flaherty KR. Diagnosis and treatment of fibrotic hypersensitivity pneumonia: where we stand and where we need to go. Am J Respir Crit Care Med. 2017;196:690–699. doi: 10.1164/rccm.201608-1675PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasakova M, Selman M, Morell F, Sterclova M, Molina-Molina M, Raghu G. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. Am J Respir Crit Care Med. 2019;200:301–308. doi: 10.1164/rccm.201903-0541PP. [DOI] [PubMed] [Google Scholar]
- 12.Morell F, Villar A, Montero MÁ, Muñoz X, Colby TV, Pipvath S, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1:685–694. doi: 10.1016/S2213-2600(13)70191-7. [DOI] [PubMed] [Google Scholar]
- 13.Ramazzini B. De morbis artificium diatriba. Modena, Italy: Antonio Capponi; 1700. [Google Scholar]
- 14.Hanak V, Golbin JM, Ryu JH. Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. Mayo Clin Proc. 2007;82:812–816. doi: 10.4065/82.7.812. [DOI] [PubMed] [Google Scholar]
- 15.Fernández Pérez ER, Swigris JJ, Forssén AV, Tourin O, Solomon JJ, Huie TJ, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144:1644–1651. doi: 10.1378/chest.12-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inase N, Ohtani Y, Sumi Y, Umino T, Usui Y, Miyake S, et al. A clinical study of hypersensitivity pneumonitis presumably caused by feather duvets. Ann Allergy Asthma Immunol. 2006;96:98–104. doi: 10.1016/S1081-1206(10)61047-2. [DOI] [PubMed] [Google Scholar]
- 17.Jordan LE, Guy E. Paediatric feather duvet hypersensitivity pneumonitis. BMJ Case Rep. 2015;2015:bcr2014207956. doi: 10.1136/bcr-2014-207956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145:723–728. doi: 10.1378/chest.13-1474. [DOI] [PubMed] [Google Scholar]
- 19.Walsh SLF, Wells AU, Desai SR, Poletti V, Piciucchi S, Dubini A, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4:557–565. doi: 10.1016/S2213-2600(16)30033-9. [DOI] [PubMed] [Google Scholar]
- 20.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 21.Mooney JJ, Elicker BM, Urbania TH, Agarwal MR, Ryerson CJ, Nguyen MLT, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144:586–592. doi: 10.1378/chest.12-2623. [DOI] [PubMed] [Google Scholar]
- 22.Salisbury ML, Gu T, Murray S, Gross BH, Chughtai A, Sayyouh M, et al. Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest. 2019;155:699–711. doi: 10.1016/j.chest.2018.08.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiba S, Tsuchiya K, Akashi T, Ishizuka M, Okamoto T, Furusawa H, et al. Chronic hypersensitivity pneumonitis with a usual interstitial pneumonia-like pattern: correlation between histopathologic and clinical findings. Chest. 2016;149:1473–1481. doi: 10.1016/j.chest.2015.12.030. [DOI] [PubMed] [Google Scholar]
- 24.Ojanguren I, Morell F, Ramón MA, Villar A, Romero C, Cruz MJ, et al. Long-term outcomes in chronic hypersensitivity pneumonitis. Allergy. 2019;74:944–952. doi: 10.1111/all.13692. [DOI] [PubMed] [Google Scholar]
- 25.Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM. Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol. 2012;22:1672–1679. doi: 10.1007/s00330-012-2427-0. [DOI] [PubMed] [Google Scholar]
- 26.Hanak V, Golbin JM, Hartman TE, Ryu JH. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest. 2008;134:133–138. doi: 10.1378/chest.07-3005. [DOI] [PubMed] [Google Scholar]
- 27.Lima MS, Coletta EN, Ferreira RG, Jasinowodolinski D, Arakaki JS, Rodrigues SC, et al. Subacute and chronic hypersensitivity pneumonitis: histopathological patterns and survival. Respir Med. 2009;103:508–515. doi: 10.1016/j.rmed.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 28.Ohtani Y, Saiki S, Kitaichi M, Usui Y, Inase N, Costabel U, et al. Chronic bird fancier’s lung: histopathological and clinical correlation. An application of the 2002 ATS/ERS consensus classification of the idiopathic interstitial pneumonias. Thorax. 2005;60:665–671. doi: 10.1136/thx.2004.027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vourlekis JS, Schwarz MI, Cherniack RM, Curran-Everett D, Cool CD, Tuder RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med. 2004;116:662–668. doi: 10.1016/j.amjmed.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 30.Kern RM, Singer JP, Koth L, Mooney J, Golden J, Hays S, et al. Lung transplantation for hypersensitivity pneumonitis. Chest. 2015;147:1558–1565. doi: 10.1378/chest.14-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters GI, Mokhlis JM, Moore VC, Robertson AS, Burge GA, Bhomra PS, et al. Characteristics of hypersensitivity pneumonitis diagnosed by interstitial and occupational lung disease multi-disciplinary team consensus. Respir Med. 2019;155:19–25. doi: 10.1016/j.rmed.2019.06.026. [DOI] [PubMed] [Google Scholar]
- 32.Reich JM. Chirping rales in bird-fancier’s lung. Chest. 1993;104:326–327. doi: 10.1378/chest.104.1.326b. [DOI] [PubMed] [Google Scholar]
- 33.Ohtani Y, Saiki S, Sumi Y, Inase N, Miyake S, Costabel U, et al. Clinical features of recurrent and insidious chronic bird fancier’s lung. Ann Allergy Asthma Immunol. 2003;90:604–610. doi: 10.1016/S1081-1206(10)61863-7. [DOI] [PubMed] [Google Scholar]
- 34.Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc. 2018;15:460–469. doi: 10.1513/AnnalsATS.201704-288OC. [DOI] [PubMed] [Google Scholar]
- 35.Morell F, Roger A, Reyes L, Cruz MJ, Murio C, Muñoz X. Bird fancier’s lung: a series of 86 patients. Medicine (Baltimore) 2008;87:110–130. doi: 10.1097/MD.0b013e31816d1dda. [DOI] [PubMed] [Google Scholar]
- 36.De Sadeleer LJ, Hermans F, De Dycker E, Yserbyt J, Verschakelen JA, Verbeken EK, et al. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. J Clin Med. 2018;8:14. doi: 10.3390/jcm8010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung. Am Rev Respir Dis. 1992;145:3–5. doi: 10.1164/ajrccm/145.1.3. [DOI] [PubMed] [Google Scholar]
- 38.Gimenez A, Storrer K, Kuranishi L, Soares MR, Ferreira RG, Pereira CAC. Change in FVC and survival in chronic fibrotic hypersensitivity pneumonitis. Thorax. 2018;73:391–392. doi: 10.1136/thoraxjnl-2017-210035. [DOI] [PubMed] [Google Scholar]
- 39.Churg A, Sin DD, Everett D, Brown K, Cool C. Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2009;33:1765–1770. doi: 10.1097/PAS.0b013e3181bb2538. [DOI] [PubMed] [Google Scholar]
- 40.Pérez-Padilla R, Salas J, Chapela R, Sánchez M, Carrillo G, Pérez R, et al. Mortality in Mexican patients with chronic pigeon breeder’s lung compared with those with usual interstitial pneumonia. Am Rev Respir Dis. 1993;148:49–53. doi: 10.1164/ajrccm/148.1.49. [DOI] [PubMed] [Google Scholar]
- 41.Sahin H, Brown KK, Curran-Everett D, Hale V, Cool CD, Vourlekis JS, et al. Chronic hypersensitivity pneumonitis: CT features comparison with pathologic evidence of fibrosis and survival. Radiology. 2007;244:591–598. doi: 10.1148/radiol.2442060640. [DOI] [PubMed] [Google Scholar]
- 42.Wang P, Jones KD, Urisman A, Elicker BM, Urbania T, Johannson KA, et al. Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest. 2017;152:502–509. doi: 10.1016/j.chest.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Ohtsuka Y, Munakata M, Tanimura K, Ukita H, Kusaka H, Masaki Y, et al. Smoking promotes insidious and chronic farmer’s lung disease, and deteriorates the clinical outcome. Intern Med. 1995;34:966–971. doi: 10.2169/internalmedicine.34.966. [DOI] [PubMed] [Google Scholar]
- 44.Coleman A, Colby TV. Histologic diagnosis of extrinsic allergic alveolitis. Am J Surg Pathol. 1988;12:514–518. doi: 10.1097/00000478-198807000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med. 1994;150:967–972. doi: 10.1164/ajrccm.150.4.7921471. [DOI] [PubMed] [Google Scholar]
- 46.Solaymani-Dodaran M, West J, Smith C, Hubbard R. Extrinsic allergic alveolitis: incidence and mortality in the general population. QJM. 2007;100:233–237. doi: 10.1093/qjmed/hcm008. [DOI] [PubMed] [Google Scholar]
- 47.Kornum JB, Christensen S, Grijota M, Pedersen L, Wogelius P, Beiderbeck A, et al. The incidence of interstitial lung disease 1995-2005: a Danish nationwide population-based study. BMC Pulm Med. 2008;8:24. doi: 10.1186/1471-2466-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hendrick DJ, Faux JA, Marshall R. Budgerigar-fancier’s lung: the commonest variety of allergic alveolitis in Britain. BMJ. 1978;2:81–84. doi: 10.1136/bmj.2.6130.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grant IW, Blyth W, Wardrop VE, Gordon RM, Pearson JC, Mair A. Prevalence of farmer’s lung in Scotland: a pilot survey. BMJ. 1972;1:530–534. doi: 10.1136/bmj.1.5799.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christensen LT, Schmidt CD, Robbins L. Pigeon breeders’ disease: a prevalence study and review. Clin Allergy. 1975;5:417–430. doi: 10.1111/j.1365-2222.1975.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 51.Fenclová Z, Pelclová D, Urban P, Navrátil T, Klusácková P, Lebedová J. Occupational hypersensitivity pneumonitis reported to the Czech National Registry of Occupational Diseases in the period 1992–2005. Ind Health. 2009;47:443–448. doi: 10.2486/indhealth.47.443. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida K, Suga M, Nishiura Y, Arima K, Yoneda R, Tamura M, et al. Occupational hypersensitivity pneumonitis in Japan: data on a nationwide epidemiological study. Occup Environ Med. 1995;52:570–574. doi: 10.1136/oem.52.9.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dalphin JC. Extrinsic allergic alveolitis in agricultural environment [in French] Rev Prat. 1992;42:1790–1796. [PubMed] [Google Scholar]
- 54.Ando M, Suga M, Nishiura Y, Miyajima M. Summer-type hypersensitivity pneumonitis. Intern Med. 1995;34:707–712. doi: 10.2169/internalmedicine.34.707. [DOI] [PubMed] [Google Scholar]
- 55.Griese M, Haug M, Hartl D, Teusch V, Glöckner-Pagel J, Brasch F National EAA Study Group. Hypersensitivity pneumonitis: lessons for diagnosis and treatment of a rare entity in children. Orphanet J Rare Dis. 2013;8:121. doi: 10.1186/1750-1172-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Griese M, Irnstetter A, Hengst M, Burmester H, Nagel F, Ripper J, et al. Categorizing diffuse parenchymal lung disease in children. Orphanet J Rare Dis. 2015;10:122. doi: 10.1186/s13023-015-0339-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buchvald F, Petersen BL, Damgaard K, Deterding R, Langston C, Fan LL, et al. Frequency, treatment, and functional outcome in children with hypersensitivity pneumonitis. Pediatr Pulmonol. 2011;46:1098–1107. doi: 10.1002/ppul.21479. [DOI] [PubMed] [Google Scholar]
- 58.Morell F, Villar A, Ojanguren I, Munoz X, Cruz MJ, Sansano I, et al. Hypersensitivity pneumonitis and (idiopathic) pulmonary fibrosis due to feather duvets and pillows [in English, Spanish] Arch Bronconeumol. [online ahead of print] 11 Feb 2020. [DOI] [PubMed]
- 59.Thomeer MJ, Costabe U, Rizzato G, Poletti V, Demedts M. Comparison of registries of interstitial lung diseases in three European countries. Eur Respir J Suppl. 2001;32:114s–118s. [PubMed] [Google Scholar]
- 60.Schweisfurth H. Report by the scientific working group for therapy of lung diseases: German fibrosis register with initial results [in German] Pneumologie. 1996;50:899–901. [PubMed] [Google Scholar]
- 61.Thomeer M, Demedts M, Vandeurzen K VRGT Working Group on Interstitial Lung Diseases. Registration of interstitial lung diseases by 20 centres of respiratory medicine in Flanders. Acta Clin Belg. 2001;56:163–172. doi: 10.1179/acb.2001.026. [DOI] [PubMed] [Google Scholar]
- 62.Fisher J, Shapera S, Algamdi M, Morisset J, Johannson KA, Fell CD, et al. Baseline characteristics and comorbidities in the Canadian registry for pulmonary fibrosis [abstract] Am J Respir Crit Care Med. 2020;201:A7478. doi: 10.1186/s12890-019-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kreuter M, Herth FJ, Wacker M, Leidl R, Hellmann A, Pfeifer M, et al. Exploring clinical and epidemiological characteristics of interstitial lung diseases: rationale, aims, and design of a nationwide prospective registry: the EXCITING-ILD registry. BioMed Res Int. 2015;2015:123876. doi: 10.1155/2015/123876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karakatsani A, Papakosta D, Rapti A, Antoniou KM, Dimadi M, Markopoulou A, et al. Hellenic Interstitial Lung Diseases Group. Epidemiology of interstitial lung diseases in Greece. Respir Med. 2009;103:1122–1129. doi: 10.1016/j.rmed.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 65.Ansarie M. A national guideline and ILD PAK Registry Report: recent landmarks in the understanding of interstitial lung diseases in Pakistan. J Pak Med Assoc. 2016;66:1050–1053. [PubMed] [Google Scholar]
- 66.Strambu I, Salmen T, Traila D, Croitoru A. Romanian Registry for Interstitial Lung Diseases (REGIS): inclusion of patients in 3 years. Eur Respir J Suppl. 2017;50:PA868. [Google Scholar]
- 67.Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, Katiyar S, et al. Interstitial lung disease (ILD) in India: results of a prospective registry. Am J Respir Crit Care Med. 2017;195:801–813. doi: 10.1164/rccm.201607-1484OC. [DOI] [PubMed] [Google Scholar]
- 68.Xaubet A, Ancochea J, Morell F, Rodriguez-Arias JM, Villena V, Blanquer R, et al. Spanish Group on Interstitial Lung Diseases, SEPAR. Report on the incidence of interstitial lung diseases in Spain. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21:64–70. [PubMed] [Google Scholar]
- 69.Alhamad EH. Interstitial lung diseases in Saudi Arabia: a single-center study. Ann Thorac Med. 2013;8:33–37. doi: 10.4103/1817-1737.105717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.PFF patient registry. Chicago, IL: Pulmonary Fibrosis Foundation; [accessed 2020 May 4]. Available from: https://www.pulmonaryfibrosis.org/medical-community/pff-patient-registry. [Google Scholar]
- 71.Rose CS, Martyny JW, Newman LS, Milton DK, King TE, Jr, Beebe JL, et al. “Lifeguard lung”: endemic granulomatous pneumonitis in an indoor swimming pool. Am J Public Health. 1998;88:1795–1800. doi: 10.2105/ajph.88.12.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simpson C, Garabrant D, Torrey S, Robins T, Franzblau A. Hypersensitivity pneumonitis-like reaction and occupational asthma associated with 1,3-bis(isocyanatomethyl) cyclohexane pre-polymer. Am J Ind Med. 1996;30:48–55. doi: 10.1002/(SICI)1097-0274(199607)30:1<48::AID-AJIM8>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 73.Ganier M, Lieberman P, Fink J, Lockwood DG. Humidifier lung: an outbreak in office workers. Chest. 1980;77:183–187. doi: 10.1378/chest.77.2.183. [DOI] [PubMed] [Google Scholar]
- 74.Banaszak EF, Thiede WH, Fink JN. Hypersensitivity pneumonitis due to contamination of an air conditioner. N Engl J Med. 1970;283:271–276. doi: 10.1056/NEJM197008062830601. [DOI] [PubMed] [Google Scholar]
- 75.Selman M, Pardo A, King TE., Jr Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186:314–324. doi: 10.1164/rccm.201203-0513CI. [DOI] [PubMed] [Google Scholar]
- 76.Gbaguidi-Haore H, Roussel S, Reboux G, Dalphin JC, Piarroux R. Multilevel analysis of the impact of environmental factors and agricultural practices on the concentration in hay of microorganisms responsible for farmer’s lung disease. Ann Agric Environ Med. 2009;16:219–225. [PubMed] [Google Scholar]
- 77.Saltoun CA, Harris KE, Mathisen TL, Patterson R. Hypersensitivity pneumonitis resulting from community exposure to Canada goose droppings: when an external environmental antigen becomes an indoor environmental antigen. Ann Allergy Asthma Immunol. 2000;84:84–86. doi: 10.1016/S1081-1206(10)62745-7. [DOI] [PubMed] [Google Scholar]
- 78.Koschel D, Wittstruck H, Renck T, Müller-Wening D, Höffken G. Presenting features of feather duvet lung. Int Arch Allergy Immunol. 2010;152:264–270. doi: 10.1159/000283036. [DOI] [PubMed] [Google Scholar]
- 79.Mizobe T, Ando M, Yamasaki H, Onoue K, Misaki A. Purification and characterization of the serotype-specific polysaccharide antigen of Trichosporon cutaneum serotype II: a disease-related antigen of Japanese summer-type hypersensitivity pneumonitis. Clin Exp Allergy. 1995;25:265–272. doi: 10.1111/j.1365-2222.1995.tb01039.x. [DOI] [PubMed] [Google Scholar]
- 80.Mundt C, Becker WM, Schlaak M. Farmer’s lung: patients’ IgG2 antibodies specifically recognize Saccharopolyspora rectivirgula proteins and carbohydrate structures. J Allergy Clin Immunol. 1996;98:441–450. doi: 10.1016/s0091-6749(96)70169-0. [DOI] [PubMed] [Google Scholar]
- 81.Todd A, Coan RM, Allen A. Pigeon breeders’ lung: pigeon intestinal mucin, an antigen distinct from pigeon IgA. Clin Exp Immunol. 1991;85:453–458. doi: 10.1111/j.1365-2249.1991.tb05748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baldwin CI, Todd A, Bourke SJ, Allen A, Calvert JE. Pigeon fanciers’ lung: identification of disease-associated carbohydrate epitopes on pigeon intestinal mucin. Clin Exp Immunol. 1999;117:230–236. doi: 10.1046/j.1365-2249.1999.00981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hisauchi-Kojima K, Sumi Y, Miyashita Y, Miyake S, Toyoda H, Kurup VP, et al. Purification of the antigenic components of pigeon dropping extract, the responsible agent for cellular immunity in pigeon breeder’s disease. J Allergy Clin Immunol. 1999;103:1158–1165. doi: 10.1016/s0091-6749(99)70193-4. [DOI] [PubMed] [Google Scholar]
- 84.Seed MJ, Enoch SJ, Agius RM. Chemical determinants of occupational hypersensitivity pneumonitis. Occup Med (Lond) 2015;65:673–681. doi: 10.1093/occmed/kqv143. [DOI] [PubMed] [Google Scholar]
- 85.Seed MJ, Agius RM. Progress with structure-activity relationship modelling of occupational chemical respiratory sensitizers. Curr Opin Allergy Clin Immunol. 2017;17:64–71. doi: 10.1097/ACI.0000000000000355. [DOI] [PubMed] [Google Scholar]
- 86.Soumagne T, Chardon ML, Dournes G, Laurent L, Degano B, Laurent F, et al. Emphysema in active farmer’s lung disease. PLoS One. 2017;12:e0178263. doi: 10.1371/journal.pone.0178263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jacob J, Odink A, Brun AL, Macaluso C, de Lauretis A, Kokosi M, et al. Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir Med. 2018;138:95–101. doi: 10.1016/j.rmed.2018.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morell F, Jeanneret A, Aïache JM, Molina C. Leukocyte migration inhibition in farmer’s lung. J Allergy Clin Immunol. 1982;69:405–409. doi: 10.1016/0091-6749(82)90114-2. [DOI] [PubMed] [Google Scholar]
- 89.Burrell R, Rylander R. A critical review of the role of precipitins in hypersensitivity pneumonitis. Eur J Respir Dis. 1981;62:332–343. [PubMed] [Google Scholar]
- 90.Vogelmeier C, Krombach F, Münzing S, König G, Mazur G, Beinert T, et al. Activation of blood neutrophils in acute episodes of farmer’s lung. Am Rev Respir Dis. 1993;148:396–400. doi: 10.1164/ajrccm/148.2.396. [DOI] [PubMed] [Google Scholar]
- 91.Pardo A, Barrios R, Gaxiola M, Segura-Valdez L, Carrillo G, Estrada A, et al. Increase of lung neutrophils in hypersensitivity pneumonitis is associated with lung fibrosis. Am J Respir Crit Care Med. 2000;161:1698–1704. doi: 10.1164/ajrccm.161.5.9907065. [DOI] [PubMed] [Google Scholar]
- 92.Girard M, Israël-Assayag E, Cormier Y. Impaired function of regulatory T-cells in hypersensitivity pneumonitis. Eur Respir J. 2011;37:632–639. doi: 10.1183/09031936.00055210. [DOI] [PubMed] [Google Scholar]
- 93.Barrera L, Mendoza F, Zuñiga J, Estrada A, Zamora AC, Melendro EI, et al. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2008;177:44–55. doi: 10.1164/rccm.200701-093OC. [DOI] [PubMed] [Google Scholar]
- 94.Kishi M, Miyazaki Y, Jinta T, Furusawa H, Ohtani Y, Inase N, et al. Pathogenesis of cBFL in common with IPF? Correlation of IP-10/TARC ratio with histological patterns. Thorax. 2008;63:810–816. doi: 10.1136/thx.2007.086074. [DOI] [PubMed] [Google Scholar]
- 95.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, et al. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182:657–665. [PMC free article] [PubMed] [Google Scholar]
- 96.Jinta T, Miyazaki Y, Kishi M, Akashi T, Takemura T, Inase N, et al. The pathogenesis of chronic HP in common with IPF. Am J Clin Pathol. 2010;134:613–620. doi: 10.1309/AJCPK8RPQX7TQRQC. [DOI] [PubMed] [Google Scholar]
- 97.García de Alba C, Buendia-Roldán I, Salgado A, Becerril C, Ramírez R, González Y, et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am J Respir Crit Care Med. 2015;191:427–436. doi: 10.1164/rccm.201407-1334OC. [DOI] [PubMed] [Google Scholar]
- 98.Adegunsoye A, Oldham JM, Demchuk C, Montner S, Vij R, Strek ME. Predictors of survival in coexistent hypersensitivity pneumonitis with autoimmune features. Respir Med. 2016;114:53–60. doi: 10.1016/j.rmed.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rittner C, Sennekamp J, Mollenhauer E, Rösinger N, Niese D, Lüttkenhorst M, et al. Pigeon breeder’s lung: association with HLA-DR 3. Tissue Antigens. 1983;21:374–379. doi: 10.1111/j.1399-0039.1983.tb00186.x. [DOI] [PubMed] [Google Scholar]
- 100.Selman M, Terán L, Mendoza A, Camarena A, Martínez-Cordero E, Lezama M, et al. Increase of HLA-DR7 in pigeon breeder’s lung in a Mexican population. Clin Immunol Immunopathol. 1987;44:63–70. doi: 10.1016/0090-1229(87)90052-3. [DOI] [PubMed] [Google Scholar]
- 101.Ando M, Hirayama K, Soda K, Okubo R, Araki S, Sasazuki T. HLA-DQw3 in Japanese summer-type hypersensitivity pneumonitis induced by Trichosporon cutaneum. Am Rev Respir Dis. 1989;140:948–950. doi: 10.1164/ajrccm/140.4.948. [DOI] [PubMed] [Google Scholar]
- 102.Camarena A, Juárez A, Mejía M, Estrada A, Carrillo G, Falfán R, et al. Major histocompatibility complex and tumor necrosis factor-alpha polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163:1528–1533. doi: 10.1164/ajrccm.163.7.2004023. [DOI] [PubMed] [Google Scholar]
- 103.Falfán-Valencia R, Camarena A, Pineda CL, Montaño M, Juárez A, Buendía-Roldán I, et al. Genetic susceptibility to multicase hypersensitivity pneumonitis is associated with the TNF-238 GG genotype of the promoter region and HLA-DRB1*04 bearing HLA haplotypes. Respir Med. 2014;108:211–217. doi: 10.1016/j.rmed.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 104.Camarena A, Aquino-Galvez A, Falfán-Valencia R, Sánchez G, Montaño M, Ramos C, et al. PSMB8 (LMP7) but not PSMB9 (LMP2) gene polymorphisms are associated to pigeon breeder’s hypersensitivity pneumonitis. Respir Med. 2010;104:889–894. doi: 10.1016/j.rmed.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 105.Aquino-Galvez A, Camarena A, Montaño M, Juarez A, Zamora AC, González-Avila G, et al. Transporter associated with antigen processing (TAP) 1 gene polymorphisms in patients with hypersensitivity pneumonitis. Exp Mol Pathol. 2008;84:173–177. doi: 10.1016/j.yexmp.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 106.Hill MR, Briggs L, Montaño MM, Estrada A, Laurent GJ, Selman M, et al. Promoter variants in tissue inhibitor of metalloproteinase-3 (TIMP-3) protect against susceptibility in pigeon breeders’ disease. Thorax. 2004;59:586–590. doi: 10.1136/thx.2003.012690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5:639–647. doi: 10.1016/S2213-2600(17)30216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48:1710–1720. doi: 10.1183/13993003.00308-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Okamoto T, Miyazaki Y, Tomita M, Tamaoka M, Inase N. A familial history of pulmonary fibrosis in patients with chronic hypersensitivity pneumonitis. Respiration. 2013;85:384–390. doi: 10.1159/000338123. [DOI] [PubMed] [Google Scholar]
- 110.Ley B, Torgerson DG, Oldham JM, Adegunsoye A, Liu S, Li J, et al. Rare protein-altering telomere-related gene variants in patients with chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2019;200:1154–1163. doi: 10.1164/rccm.201902-0360OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bustos ML, Frías S, Ramos S, Estrada A, Arreola JL, Mendoza F, et al. Local and circulating microchimerism is associated with hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2007;176:90–95. doi: 10.1164/rccm.200608-1129OC. [DOI] [PubMed] [Google Scholar]
- 112.Cormier Y, Isräel-Assayag E, Fournier M, Tremblay GM. Modulation of experimental hypersensitivity pneumonitis by Sendai virus. J Lab Clin Med. 1993;121:683–688. [PubMed] [Google Scholar]
- 113.Dakhama A, Hegele RG, Laflamme G, Isräel-Assayag, Cormier Y. Common respiratory viruses in lower airways of patients with acute hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1999;159:1316–1322. doi: 10.1164/ajrccm.159.4.9807085. [DOI] [PubMed] [Google Scholar]
- 114.Gudmundsson G, Monick MM, Hunninghake GW. Viral infection modulates expression of hypersensitivity pneumonitis. J Immunol. 1999;162:7397–7401. [PubMed] [Google Scholar]
- 115.Hoppin JA, Umbach DM, Kullman GJ, Henneberger PK, London SJ, Alavanja MC, et al. Pesticides and other agricultural factors associated with self-reported farmer’s lung among farm residents in the Agricultural Health Study. Occup Environ Med. 2007;64:334–341. doi: 10.1136/oem.2006.028480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Remy-Jardin M, Remy J, Wallaert B, Müller NL. Subacute and chronic bird breeder hypersensitivity pneumonitis: sequential evaluation with CT and correlation with lung function tests and bronchoalveolar lavage. Radiology. 1993;189:111–118. doi: 10.1148/radiology.189.1.8372179. [DOI] [PubMed] [Google Scholar]
- 117.Lynch DA, Newell JD, Logan PM, King TE, Jr, Müller NL. Can CT distinguish hypersensitivity pneumonitis from idiopathic pulmonary fibrosis? AJR Am J Roentgenol. 1995;165:807–811. doi: 10.2214/ajr.165.4.7676971. [DOI] [PubMed] [Google Scholar]
- 118.Hansell DM, Wells AU, Padley SPG, Müller NL. Hypersensitivity pneumonitis: correlation of individual CT patterns with functional abnormalities. Radiology. 1996;199:123–128. doi: 10.1148/radiology.199.1.8633133. [DOI] [PubMed] [Google Scholar]
- 119.Tateishi T, Ohtani Y, Takemura T, Akashi T, Miyazaki Y, Inase N, et al. Serial high-resolution computed tomography findings of acute and chronic hypersensitivity pneumonitis induced by avian antigen. J Comput Assist Tomogr. 2011;35:272–279. doi: 10.1097/RCT.0b013e318209c5a6. [DOI] [PubMed] [Google Scholar]
- 120.Adler BD, Padley SPG, Müller NL, Remy-Jardin M, Remy J. Chronic hypersensitivity pneumonitis: high-resolution CT and radiographic features in 16 patients. Radiology. 1992;185:91–95. doi: 10.1148/radiology.185.1.1523340. [DOI] [PubMed] [Google Scholar]
- 121.Chung JH, Montner SM, Adegunsoye A, Oldham JM, Husain AN, Vij R, et al. CT findings associated with survival in chronic hypersensitivity pneumonitis. Eur Radiol. 2017;27:5127–5135. doi: 10.1007/s00330-017-4936-3. [DOI] [PubMed] [Google Scholar]
- 122.Salisbury ML, Gross BH, Chughtai A, Sayyouh M, Kazerooni EA, Bartholmai BJ, et al. Development and validation of a radiological diagnosis model for hypersensitivity pneumonitis. Eur Respir J. 2018;52:1800443. doi: 10.1183/13993003.00443-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Barnett J, Molyneaux PL, Rawal B, Abdullah R, Hare SS, Vancheeswaran R, et al. Variable utility of mosaic attenuation to distinguish fibrotic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis. Eur Respir J. 2019;54:1900531. doi: 10.1183/13993003.00531-2019. [DOI] [PubMed] [Google Scholar]
- 124.Hansell DM, Moskovic E. High-resolution computed tomography in extrinsic allergic alveolitis. Clin Radiol. 1991;43:8–12. doi: 10.1016/s0009-9260(05)80345-9. [DOI] [PubMed] [Google Scholar]
- 125.Franquet T, Müller NL. Disorders of the small airways: high-resolution computed tomographic features. Semin Respir Crit Care Med. 2003;24:437–444. doi: 10.1055/s-2003-42378. [DOI] [PubMed] [Google Scholar]
- 126.Franquet T, Hansell DM, Senbanjo T, Remy-Jardin M, Müller NL. Lung cysts in subacute hypersensitivity pneumonitis. J Comput Assist Tomogr. 2003;27:475–478. doi: 10.1097/00004728-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 127.Silva CIS, Müller NL, Lynch DA, Curran-Everett D, Brown KK, Lee KS, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology. 2008;246:288–297. doi: 10.1148/radiol.2453061881. [DOI] [PubMed] [Google Scholar]
- 128.Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med. 2018;6:138–153. doi: 10.1016/S2213-2600(17)30433-2. [DOI] [PubMed] [Google Scholar]
- 129.Tokura S, Okuma T, Akira M, Arai T, Inoue Y, Kitaichi M. Utility of expiratory thin-section CT for fibrotic interstitial pneumonia. Acta Radiol. 2014;55:1050–1055. doi: 10.1177/0284185113512300. [DOI] [PubMed] [Google Scholar]
- 130.Miller WT, Jr, Chatzkel J, Hewitt MG. Expiratory air trapping on thoracic computed tomography: a diagnostic subclassification. Ann Am Thorac Soc. 2014;11:874–881. doi: 10.1513/AnnalsATS.201311-390OC. [DOI] [PubMed] [Google Scholar]
- 131.Lalancette M, Carrier G, Laviolette M, Ferland S, Rodrique J, Bégin R, et al. Farmer’s lung: long-term outcome and lack of predictive value of bronchoalveolar lavage fibrosing factors. Am Rev Respir Dis. 1993;148:216–221. doi: 10.1164/ajrccm/148.1.216. [DOI] [PubMed] [Google Scholar]
- 132.Baqir M, White D, Ryu JH. Emphysematous changes in hypersensitivity pneumonitis: a retrospective analysis of 12 patients. Respir Med Case Rep. 2018;24:25–29. doi: 10.1016/j.rmcr.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest. 2008;134:1265–1270. doi: 10.1378/chest.08-0866. [DOI] [PubMed] [Google Scholar]
- 134.Kitaichi M. Comparative pulmonary histopathology of sarcoidosis, chronic beryllium disease and hypersensitivity pneumonitis [in Japanese] Nihon Kyobu Shikkan Gakkai Zasshi. 1984;22:769–782. [PubMed] [Google Scholar]
- 135.Kitaichi M.Pathology of pulmonary sarcoidosis Izumi T.Sarcoidosis, Clinics in Dermatology Philadelphia, PA: JB Lippincott; 1986. p108–115. [DOI] [PubMed] [Google Scholar]
- 136.American Thoracic Society; European Respiratory Society; World Association of Sarcoidosis and Other Granulomatous Disorders. Statement on sarcoidosis: joint statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160:736–755. doi: 10.1164/ajrccm.160.2.ats4-99. [DOI] [PubMed] [Google Scholar]
- 137.Kitaichi M, Shimizu S, Tamaya M, Takaki M, Inoue Y. Pathology of hypersensitivity pneumonitis. In: Sharma OP, editor. Clinical focus series, hypersensitivity pneumonitis. New Delhi, India: Jaypee Brothers Medical Publishers; 2013. p. 22–32. [Google Scholar]
- 138.Minomo S, Tachibana K, Tsuyuguchi K, Akira M, Kitaichi M, Suzuki K. A unique case of hot tub lung worsening during the winter. Intern Med. 2015;54:491–495. doi: 10.2169/internalmedicine.54.3394. [DOI] [PubMed] [Google Scholar]
- 139.Fujimura N, Kino T, Nagai S, Kitaichi M, Izumi T, Oshima S, et al. Hypersensitivity pneumonitis in a polyurethane paint sprayer [in Japanese] Nihon Kyobu Shikkan Gakkai Zasshi. 1984;22:506–513. [PubMed] [Google Scholar]
- 140.Akashi T, Takemura T, Ando N, Eishi Y, Kitagawa M, Takizawa T, et al. Histopathologic analysis of sixteen autopsy cases of chronic hypersensitivity pneumonitis and comparison with idiopathic pulmonary fibrosis/usual interstitial pneumonia. Am J Clin Pathol. 2009;131:405–415. doi: 10.1309/AJCPNWX4SLZRP9SW. [DOI] [PubMed] [Google Scholar]
- 141.Takemura T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Pathology of hypersensitivity pneumonitis. Curr Opin Pulm Med. 2008;14:440–454. doi: 10.1097/MCP.0b013e3283043dfa. [DOI] [PubMed] [Google Scholar]
- 142.Herbst JB, Myers JL. Hypersensitivity pneumonia: role of surgical lung biopsy. Arch Pathol Lab Med. 2012;136:889–895. doi: 10.5858/arpa.2012-0201-CR. [DOI] [PubMed] [Google Scholar]
- 143.Trahan S, Hanak V, Ryu JH, Myers JL. Role of surgical lung biopsy in separating chronic hypersensitivity pneumonia from usual interstitial pneumonia/idiopathic pulmonary fibrosis: analysis of 31 biopsies from 15 patients. Chest. 2008;134:126–132. doi: 10.1378/chest.08-0033. [DOI] [PubMed] [Google Scholar]
- 144.Castonquay M, Ryu J, Yi E, Tazelaar H. Granulomas and giant cells in hypersensitivity pneumonitis. Hum Pathol. 2015;46:607–613. doi: 10.1016/j.humpath.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 145.Hurst JR, Verma N, Lowe D, Baxendale HE, Jolles S, Kelleher P, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network consensus statement on the definition, diagnosis, and management of granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2017;5:938–945. doi: 10.1016/j.jaip.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 146.Mukhopadhyay S, Katzenstein A-L. Pulmonary disease due to aspiration of food and other particulate matter: a clinicopathologic study of 59 cases diagnosed on biopsy or resection specimens. Am J Surg Pathol. 2007;31:752–759. doi: 10.1097/01.pas.0000213418.08009.f9. [DOI] [PubMed] [Google Scholar]
- 147.Khoor A, Leslie KO, Tazelaar HD, Helmers RA, Colby TV. Diffuse pulmonary disease caused by nontuberculous mycobacteria in immunocompetent people (hot tub lung) Am J Clin Pathol. 2001;115:755–762. doi: 10.1309/JRDC-0MJV-ACA3-2U9L. [DOI] [PubMed] [Google Scholar]
- 148.Takemura T, Akashi T, Kamiya H, Ikushima S, Ando T, Oritsu M, et al. Pathological differentiation of chronic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histopathology. 2012;61:1026–1035. doi: 10.1111/j.1365-2559.2012.04322.x. [DOI] [PubMed] [Google Scholar]
- 149.Churg A, Bilawich A, Wright JL. Pathology of chronic hypersensitivity pneumonitis what is it? What are the diagnostic criteria? Why do we care? Arch Pathol Lab Med. 2018;142:109–119. doi: 10.5858/arpa.2017-0173-RA. [DOI] [PubMed] [Google Scholar]
- 150.Fukuoka J, Franks TJ, Colby TV, Flaherty KR, Galvin JR, Hayden D, et al. Peribronchiolar metaplasia: a common histologic lesion in diffuse lung disease and a rare cause of interstitial lung disease: clinicopathologic features of 15 cases. Am J Surg Pathol. 2005;29:948–954. doi: 10.1097/01.pas.0000168177.71405.ac. [DOI] [PubMed] [Google Scholar]
- 151.Churg A, Myers J, Suarez T, Gaxiola M, Estrada A, Mejia M, et al. Airway-centered interstitial fibrosis: a distinct form of aggressive diffuse lung disease. Am J Surg Pathol. 2004;28:62–68. doi: 10.1097/00000478-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 152.Tanizawa K, Ley B, Vittinghoff E, Elicker BM, Henry TS, Wolters PJ, et al. Significance of bronchiolocentric fibrosis in patients with histopathological usual interstitial pneumonia. Histopathology. 2019;74:1088–1097. doi: 10.1111/his.13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164:1722–1727. doi: 10.1164/ajrccm.164.9.2103074. [DOI] [PubMed] [Google Scholar]
- 154.Rival G, Manzoni P, Lacasse Y, Polio JC, Westeel V, Dubiez A, et al. High-resolution CT predictors of hypersensitivity pneumonitis. Sarcoidosis Vasc Diffuse Lung Dis. 2016;33:117–123. [PubMed] [Google Scholar]
- 155.Fenoglio CM, Reboux G, Sudre B, Mercier M, Roussel S, Cordier JF, et al. Diagnostic value of serum precipitins to mould antigens in active hypersensitivity pneumonitis. Eur Respir J. 2007;29:706–712. doi: 10.1183/09031936.00001006. [DOI] [PubMed] [Google Scholar]
- 156.Reboux G, Piarroux R, Mauny F, Madroszyk A, Millon L, Bardonnet K, et al. Role of molds in farmer’s lung disease in eastern France. Am J Respir Crit Care Med. 2001;163:1534–1539. doi: 10.1164/ajrccm.163.7.2006077. [DOI] [PubMed] [Google Scholar]
- 157.Pereira CAC, Soares MR, Boaventura R, Castro MDC, Gomes PS, Gimenez A, et al. Squawks in interstitial lung disease prevalence and causes in a cohort of one thousand patients. Medicine (Baltimore) 2019;98:e16419. doi: 10.1097/MD.0000000000016419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bohadana A, Izbicki G, Kraman SS. Fundamentals of lung auscultation. N Engl J Med. 2014;370:744–751. doi: 10.1056/NEJMra1302901. [DOI] [PubMed] [Google Scholar]
- 159.Ryerson CJ, Corte TJ, Lee JS, Richeldi L, Walsh SLF, Myers JL, et al. A standardized diagnostic ontology for fibrotic interstitial lung disease: an international working group perspective. Am J Respir Crit Care Med. 2017;196:1249–1254. doi: 10.1164/rccm.201702-0400PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Walsh SLF, Maher TM, Kolb M, Poletti V, Nusser R, Richeldi L, et al. IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: an international case-cohort study. Eur Respir J. 2017;50:1700936. doi: 10.1183/13993003.00936-2017. [DOI] [PubMed] [Google Scholar]
- 161.Walsh SLF, Lederer DJ, Ryerson CJ, Kolb M, Maher TM, Nusser R, et al. Diagnostic likelihood thresholds that define a working diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:1146–1153. doi: 10.1164/rccm.201903-0493OC. [DOI] [PubMed] [Google Scholar]
- 162.Millerick-May ML, Mulks MH, Gerlach J, Flaherty KR, Schmidt SL, Martinez FJ, et al. Hypersensitivity pneumonitis and antigen identification: an alternate approach. Respir Med. 2016;112:97–105. doi: 10.1016/j.rmed.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 163.Polke M, Kirsten D, Teucher B, Kahn N, Geissler K, Costabel U, et al. A comparison of existing questionnaires for identifying the causes of interstitial and rare lung diseases. Respiration. 2020;99:119–124. doi: 10.1159/000504677. [DOI] [PubMed] [Google Scholar]
- 164.Andersen P, Christensen KM, Jensen BE, Axél K, Laursen JC, Geday H, et al. Antibodies to pigeon antigens in pigeon breeders: detection of antibodies by an enzyme-linked immunosorbent assay. Eur J Respir Dis. 1982;63:113–121. [PubMed] [Google Scholar]
- 165.Aznar C, Andre PM, Deunff J, Robert R. Investigation of human immune response to Micropolyspora faeni antigens by enzyme-linked immunoelectrodiffusion assay and immunoblotting. J Clin Microbiol. 1988;26:443–447. doi: 10.1128/jcm.26.3.443-447.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Boiron P, Drouhet E, Dupont B. Enzyme-linked immunosorbent-assay (ELISA) for IgG in bagasse workers’ sera: comparison with counter-immunoelectrophoresis. Clin Allergy. 1987;17:355–363. doi: 10.1111/j.1365-2222.1987.tb02025.x. [DOI] [PubMed] [Google Scholar]
- 167.Barrera C, Millon L, Rognon B, Quadroni M, Roussel S, Dalphin JC, et al. Immunoreactive proteins of Saccharopolyspora rectivirgula for farmer’s lung serodiagnosis. Proteomics Clin Appl. 2014;8:971–981. doi: 10.1002/prca.201400024. [DOI] [PubMed] [Google Scholar]
- 168.Hébert J, Beaudoin J, Laviolette M, Beaudoin R, Bélanger J, Cormier Y. Absence of correlation between the degree of alveolitis and antibody levels to Micropolysporum faeni. Clin Exp Immunol. 1985;60:572–578. [PMC free article] [PubMed] [Google Scholar]
- 169.Huizinga M, Berrens L. Detection of class-specific antibodies against Micropolyspora faeni antigens in farmers’ lung. Clin Allergy. 1985;15:139–145. doi: 10.1111/j.1365-2222.1985.tb02265.x. [DOI] [PubMed] [Google Scholar]
- 170.Reboux G, Piarroux R, Roussel S, Millon L, Bardonnet K, Dalphin JC. Assessment of four serological techniques in the immunological diagnosis of farmers’ lung disease. J Med Microbiol. 2007;56:1317–1321. doi: 10.1099/jmm.0.46953-0. [DOI] [PubMed] [Google Scholar]
- 171.Rodrigo MJ, Benavent MI, Cruz MJ, Rosell M, Murio C, Pascual C, et al. Detection of specific antibodies to pigeon serum and bloom antigens by enzyme linked immunosorbent assay in pigeon breeder’s disease. Occup Environ Med. 2000;57:159–164. doi: 10.1136/oem.57.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Roussel S, Reboux G, Rognon B, Monod M, Grenouillet F, Quadroni M, et al. Comparison of three antigenic extracts of Eurotium amstelodami in serological diagnosis of farmer’s lung disease. Clin Vaccine Immunol. 2010;17:160–167. doi: 10.1128/CVI.00129-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sandoval J, Bañales JL, Cortés JJ, Mendoza F, Selman M, Reyes PA. Detection of antibodies against avian antigens in bronchoalveolar lavage from patients with pigeon breeder’s disease: usefulness of enzyme-linked immunosorbent assay and enzyme immunotransfer blotting. J Clin Lab Anal. 1990;4:81–85. doi: 10.1002/jcla.1860040202. [DOI] [PubMed] [Google Scholar]
- 174.Simpson C, Shirodaria PV, Evans JP, Simpson DIH, Stanford CF. Comparison of immunodiffusion and enzyme linked immunosorbent assay in the detection of abnormal antibodies in pigeon breeder’s disease. J Clin Pathol. 1992;45:490–493. doi: 10.1136/jcp.45.6.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tillie-Leblond I, Grenouillet F, Reboux G, Roussel S, Chouraki B, Lorthois C, et al. Hypersensitivity pneumonitis and metalworking fluids contaminated by mycobacteria. Eur Respir J. 2011;37:640–647. doi: 10.1183/09031936.00195009. [DOI] [PubMed] [Google Scholar]
- 176.Bellanger AP, Gbaguidi-Haore H, Gondoin A, Pallandre JR, Vacheyrou M, Valot B, et al. Positive fungal quantitative PCR and Th17 cytokine detection in bronchoalveolar lavage fluids: complementary biomarkers of hypersensitivity pneumonitis? J Immunol Methods. 2016;434:61–65. doi: 10.1016/j.jim.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 177.Furasawa H, Saito H, Sema M, Tateishi T, Miyazaki Y, Inase N. Bronchoalveolar lavage in chronic hypersensitivity pneumonitis and IPF [abstract] Am J Respir Crit Care Med. 2017;195:A5429. [Google Scholar]
- 178.Groot Kormelink T, Pardo A, Knipping K, Buendía-Roldán I, García-de-Alba C, Blokhuis BR, et al. Immunoglobulin free light chains are increased in hypersensitivity pneumonitis and idiopathic pulmonary fibrosis. PLoS One. 2011;6:e25392. doi: 10.1371/journal.pone.0025392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lehtonen ST, Karvonen HM, Harju T, Sormunen R, Lappi-Blanco E, Hilli M, et al. Stromal cells can be cultured and characterized from diagnostic bronchoalveolar fluid samples obtained from patients with various types of interstitial lung diseases. APMIS. 2014;122:301–316. doi: 10.1111/apm.12146. [DOI] [PubMed] [Google Scholar]
- 180.Markart P, Luboeinski T, Korfei M, Schmidt R, Wygrecka M, Mahavadi P, et al. Alveolar oxidative stress is associated with elevated levels of nonenzymatic low-molecular-weight antioxidants in patients with different forms of chronic fibrosing interstitial lung diseases. Antioxid Redox Signal. 2009;11:227–240. doi: 10.1089/ars.2008.2105. [DOI] [PubMed] [Google Scholar]
- 181.Nukui Y, Miyazaki Y, Masuo M, Okamoto T, Furusawa H, Tateishi T, et al. Periostin as a predictor of prognosis in chronic bird-related hypersensitivity pneumonitis. Allergol Int. 2019;68:363–369. doi: 10.1016/j.alit.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 182.Okamoto T, Fujii M, Furusawa H, Tsuchiya K, Miyazaki Y, Inase N. The usefulness of KL-6 and SP-D for the diagnosis and management of chronic hypersensitivity pneumonitis. Respir Med. 2015;109:1576–1581. doi: 10.1016/j.rmed.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 183.Przybylski G, Chorostowska-Wynimko J, Dyczek A, Wędrowska E, Jankowski M, Szpechciński A, et al. Studies of hepatocyte growth factor in bronchoalveolar lavage fluid in chronic interstitial lung diseases. Pol Arch Med Wewn. 2015;125:260–271. doi: 10.20452/pamw.2784. [DOI] [PubMed] [Google Scholar]
- 184.Schmidt R, Meier U, Markart P, Grimminger F, Velcovsky HG, Morr H, et al. Altered fatty acid composition of lung surfactant phospholipids in interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1079–L1085. doi: 10.1152/ajplung.00484.2001. [DOI] [PubMed] [Google Scholar]
- 185.Martina S, Martina V, Monika M, Jan P, Libor K, Ilja S. Angiostatic versus angiogenic chemokines in IPF and EAA. Respir Med. 2009;103:1651–1656. doi: 10.1016/j.rmed.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 186.Vasakova M, Sterclova M, Kolesar L, Slavcev A, Pohunek P, Sulc J, et al. Bronchoalveolar lavage fluid cellular characteristics, functional parameters and cytokine and chemokine levels in interstitial lung diseases. Scand J Immunol. 2009;69:268–274. doi: 10.1111/j.1365-3083.2008.02222.x. [DOI] [PubMed] [Google Scholar]
- 187.Willems S, Verleden SE, Vanaudenaerde BM, Wynants M, Dooms C, Yserbyt J, et al. Multiplex protein profiling of bronchoalveolar lavage in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Ann Thorac Med. 2013;8:38–45. doi: 10.4103/1817-1737.105718. [Published erratum appears in Ann Thorac Med 8:85.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ohshimo S, Bonella F, Cui A, Beume M, Kohno N, Guzman J, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:1043–1047. doi: 10.1164/rccm.200808-1313OC. [DOI] [PubMed] [Google Scholar]
- 189.Pesci A, Bertorelli G, Gabrielli M, Olivieri D. Mast cells in fibrotic lung disorders. Chest. 1993;103:989–996. doi: 10.1378/chest.103.4.989. [DOI] [PubMed] [Google Scholar]
- 190.Bertorelli G, Pesci A, Consigli GF, Minisini R, Mori PA, Dall’Aglio PP, et al. Evaluation of some immunological parameters in interstitial lung disease by discriminant analysis. Respiration. 1988;54:23–29. doi: 10.1159/000195493. [DOI] [PubMed] [Google Scholar]
- 191.Drent M, van Nierop MA, Gerritsen FA, Wouters EF, Mulder PG. A computer program using BALF-analysis results as a diagnostic tool in interstitial lung diseases. Am J Respir Crit Care Med. 1996;153:736–741. doi: 10.1164/ajrccm.153.2.8564126. [DOI] [PubMed] [Google Scholar]
- 192.Haslam PL, Parker DJ, Townsend PJ. Increases in HLA-DQ, DP, DR, and transferrin receptors on alveolar macrophages in sarcoidosis and allergic alveolitis compared with fibrosing alveolitis. Chest. 1990;97:651–661. doi: 10.1378/chest.97.3.651. [DOI] [PubMed] [Google Scholar]
- 193.Hoogsteden HC, van Hal PT, Wijkhuijs JM, Hop W, Hilvering C. Differences in expression of monocyte/macrophage surface antigens in peripheral blood and bronchoalveolar lavage cells in interstitial lung diseases. Lung. 1993;171:149–160. doi: 10.1007/BF00183944. [DOI] [PubMed] [Google Scholar]
- 194.Kucejko W, Chyczewska E, Naumnik W, Ossolińska M. Concentration of surfactant protein D, Clara cell protein CC-16 and IL-10 in bronchoalveolar lavage (BAL) in patients with sarcoidosis, hypersensivity pneumonitis and idiopathic pulmonary fibrosis. Folia Histochem Cytobiol. 2009;47:225–230. doi: 10.2478/v10042-009-0028-9. [DOI] [PubMed] [Google Scholar]
- 195.Papakosta D, Manika K, Gounari E, Kyriazis G, Kontakiotis T, Spyropoulos G, et al. Bronchoalveolar lavage fluid and blood natural killer and natural killer T-like cells in cryptogenic organizing pneumonia. Respirology. 2014;19:748–754. doi: 10.1111/resp.12305. [DOI] [PubMed] [Google Scholar]
- 196.Pérez-Arellano JL, Pedraz MJ, Fuertes A, de la Cruz JL, González de Buitrago JM, Jiménez A. Laminin fragment P1 is increased in the lower respiratory tract of patients with diffuse interstitial lung diseases. Chest. 1993;104:1163–1169. doi: 10.1378/chest.104.4.1163. [DOI] [PubMed] [Google Scholar]
- 197.Robinson BW, Rose AH, Thompson PJ, Hey A. Comparison of bronchoalveolar lavage helper/suppressor T-cell ratios in sarcoidosis versus other interstitial lung diseases. Aust N Z J Med. 1987;17:9–15. doi: 10.1111/j.1445-5994.1987.tb05041.x. [DOI] [PubMed] [Google Scholar]
- 198.Yamashita M, Mouri T, Niisato M, Nitanai H, Kobayashi H, Ogasawara M, et al. Lymphangiogenic factors are associated with the severity of hypersensitivity pneumonitis. BMJ Open Respir Res. 2015;2:e000085. doi: 10.1136/bmjresp-2015-000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Costabel U, Bross KJ, Rühle KH, Löhr GW, Matthys H. Ia-like antigens on T-cells and their subpopulations in pulmonary sarcoidosis and in hypersensitivity pneumonitis: analysis of bronchoalveolar and blood lymphocytes. Am Rev Respir Dis. 1985;131:337–342. doi: 10.1164/arrd.1985.131.3.337. [DOI] [PubMed] [Google Scholar]
- 200.Krombach F, Gerlach JT, Padovan C, Burges A, Behr J, Beinert T, et al. Characterization and quantification of alveolar monocyte-like cells in human chronic inflammatory lung disease. Eur Respir J. 1996;9:984–991. doi: 10.1183/09031936.96.09050984. [DOI] [PubMed] [Google Scholar]
- 201.Radomska-Leœniewska DM, Sobiecka G, Bia B. Relationship between IL-8, IL-10, MMP-9 level and morphological pattern of BAL fluid in interstitial lung diseases patients. Cent Eur J Immunol. 2003;28:155–159. [Google Scholar]
- 202.Pforte A, Gerth C, Voss A, Beer B, Häussinger K, Jütting U, et al. Proliferating alveolar macrophages in BAL and lung function changes in interstitial lung disease. Eur Respir J. 1993;6:951–955. [PubMed] [Google Scholar]
- 203.Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J. 1999;14:376–382. doi: 10.1034/j.1399-3003.1999.14b23.x. [DOI] [PubMed] [Google Scholar]
- 204.Bargagli E, Penza F, Vagaggini C, Magi B, Perari MG, Rottoli P. Analysis of carbonylated proteins in bronchoalveolar lavage of patients with diffuse lung diseases. Lung. 2007;185:139–144. doi: 10.1007/s00408-007-9001-6. [DOI] [PubMed] [Google Scholar]
- 205.Cai M, Bonella F, He X, Sixt SU, Sarria R, Guzman J, et al. CCL18 in serum, BAL fluid and alveolar macrophage culture supernatant in interstitial lung diseases. Respir Med. 2013;107:1444–1452. doi: 10.1016/j.rmed.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 206.Espoladore LM, Gregório BB, Lima MS, de Pereira CA, Soares MR, Coletta EN. Cytological analysis of bronchoalveolar lavage in patients with interstitial lung diseases and the relation of cytological analysis to fibrosis in high-resolution computed tomography. Anal Quant Cytopathol Histpathol. 2014;36:206–212. [PubMed] [Google Scholar]
- 207.Fireman E, Ben Efraim S, Greif J, Peretz H, Kivity S, Topilsky M, et al. Differential proliferative characteristics of alveolar fibroblasts in interstitial lung diseases: regulative role of IL-1 and PGE(2) Mediators Inflamm. 1994;3:445–452. doi: 10.1155/S0962935194000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hoogsteden HC, van Dongen JJ, van Hal PT, Delahaye M, Hop W, Hilvering C. Phenotype of blood monocytes and alveolar macrophages in interstitial lung disease. Chest. 1989;95:574–577. doi: 10.1378/chest.95.3.574. [DOI] [PubMed] [Google Scholar]
- 209.Jara-Palomares L, Martín-Juan J, Gómez-Izquierdo L, Cayuela-Domínguez A, Rodríguez-Becerra E, Rodríguez-Panadero F. Bronchoalveolar lavage findings in patients with diffuse interstitial lung disease: prospective study of a cohort of 562 patients [in Spanish] Arch Bronconeumol. 2009;45:111–117. doi: 10.1016/j.arbres.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 210.Kopiński P, Balicka-Ślusarczyk B, Dyczek A, Szpechciński A, Przybylski G, Jarzemska A, et al. Enhanced expression of Fas ligand (FasL) in the lower airways of patients with fibrotic interstitial lung diseases (ILDs) Folia Histochem Cytobiol. 2011;49:636–645. doi: 10.5603/fhc.2011.0087. [DOI] [PubMed] [Google Scholar]
- 211.Pardo A, Smith KM, Abrams J, Coffman R, Bustos M, McClanahan TK, et al. CCL18/DC-CK-1/PARC up-regulation in hypersensitivity pneumonitis. J Leukoc Biol. 2001;70:610–616. [PubMed] [Google Scholar]
- 212.Phelps DS, Umstead TM, Mejia M, Carrillo G, Pardo A, Selman M. Increased surfactant protein-A levels in patients with newly diagnosed idiopathic pulmonary fibrosis. Chest. 2004;125:617–625. doi: 10.1378/chest.125.2.617. [DOI] [PubMed] [Google Scholar]
- 213.Sherson D, Nielsen H, Frederiksen J, Milman N, Struve-Christensen E, Petersen BN. Superoxide anion release from blood monocytes and alveolar macrophages in patients with diffuse lung fibrosis. APMIS. 1992;100:408–414. doi: 10.1111/j.1699-0463.1992.tb00891.x. [DOI] [PubMed] [Google Scholar]
- 214.Sokhatska O, Padrão E, Sousa-Pinto B, Beltrão M, Mota PC, Melo N, et al. NK and NKT cells in the diagnosis of diffuse lung diseases presenting with a lymphocytic alveolitis. BMC Pulm Med. 2019;19:39. doi: 10.1186/s12890-019-0802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Thomeer MJ, Vansteenkiste J, Verbeken EK, Demedts M. Interstitial lung diseases: characteristics at diagnosis and mortality risk assessment. Respir Med. 2004;98:567–573. doi: 10.1016/j.rmed.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 216.Welker L, Jörres RA, Costabel U, Magnussen H. Predictive value of BAL cell differentials in the diagnosis of interstitial lung diseases. Eur Respir J. 2004;24:1000–1006. doi: 10.1183/09031936.04.00101303. [DOI] [PubMed] [Google Scholar]
- 217.Wojtan P, Mierzejewski M, Osińska I, Domagała-Kulawik J. Macrophage polarization in interstitial lung diseases. Cent Eur J Immunol. 2016;41:159–164. doi: 10.5114/ceji.2016.60990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zissel G, Bäumer I, Schlaak M, Müller-Quernheim J. In vitro release of interleukin-15 by broncho-alveolar lavage cells and peripheral blood mononuclear cells from patients with different lung diseases. Eur Cytokine Netw. 2000;11:105–112. [PubMed] [Google Scholar]
- 219.Günther A, Schmidt R, Nix F, Yabut-Perez M, Guth C, Rosseau S, et al. Surfactant abnormalities in idiopathic pulmonary fibrosis, hypersensitivity pneumonitis and sarcoidosis. Eur Respir J. 1999;14:565–573. doi: 10.1034/j.1399-3003.1999.14c14.x. [DOI] [PubMed] [Google Scholar]
- 220.Oshima M, Maeda A, Ishioka S, Hiyama K, Yamakido M. Expression of C-C chemokines in bronchoalveolar lavage cells from patients with granulomatous lung diseases. Lung. 1999;177:229–240. doi: 10.1007/pl00007643. [DOI] [PubMed] [Google Scholar]
- 221.Raulf M, Liebers V, Steppert C, Baur X. Increased gamma/delta-positive T-cells in blood and bronchoalveolar lavage of patients with sarcoidosis and hypersensitivity pneumonitis. Eur Respir J. 1994;7:140–147. doi: 10.1183/09031936.94.07010140. [DOI] [PubMed] [Google Scholar]
- 222.Agostini C, Trentin L, Zambello R, Luca M, Masciarelli M, Cipriani A, et al. Pulmonary alveolar macrophages in patients with sarcoidosis and hypersensitivity pneumonitis: characterization by monoclonal antibodies. J Clin Immunol. 1987;7:64–70. doi: 10.1007/BF00915427. [DOI] [PubMed] [Google Scholar]
- 223.Agostini C, Zambello R, Sancetta R, Cerutti A, Milani A, Tassinari C, et al. Expression of tumor necrosis factor-receptor superfamily members by lung T lymphocytes in interstitial lung disease. Am J Respir Crit Care Med. 1996;153:1359–1367. doi: 10.1164/ajrccm.153.4.8616567. [DOI] [PubMed] [Google Scholar]
- 224.Bäumer I, Zissel G, Schlaak M, Müller-Quernheim J. Soluble intercellular adhesion molecule 1 (sICAM-1) in bronchoalveolar lavage (BAL) cell cultures and in the circulation of patients with tuberculosis, hypersensitivity pneumonitis and sarcoidosis. Eur J Med Res. 1998;3:288–294. [PubMed] [Google Scholar]
- 225.Cantin A, Bégin R, Boileau R, Drapeau G, Rola-Pleszczynski M. Features of bronchoalveolar lavage differentiating hypersensitivity pneumonitis and pulmonary sarcoidosis at time of initial presentation. Clin Invest Med. 1984;7:89–94. [PubMed] [Google Scholar]
- 226.Couto M, Palmares C, Beltrão M, Neves S, Mota P, Morais A, et al. Integrin α E β 7 (CD103) expression in bronchoalveolar lymphocytes of patients with hypersensitivity pneumonitis. Int Arch Occup Environ Health. 2015;88:167–173. doi: 10.1007/s00420-014-0947-4. [DOI] [PubMed] [Google Scholar]
- 227.Fujishima S, Nakamura M, Nakamura H, Inoue T, Yogo Y, Okubo Y, et al. Flow cytometric detection of cell-associated interleukin-8 in alveolar macrophages in vivo from patients with hypersensitivity pneumonitis and sarcoidosis. Scand J Clin Lab Invest. 2004;64:237–243. doi: 10.1080/00365510410006018. [DOI] [PubMed] [Google Scholar]
- 228.Hamagami S, Miyagawa T, Ochi T, Tsuyuguchi I, Kishimoto S. A raised level of soluble CD8 in bronchoalveolar lavage fluid in summer-type hypersensitivity pneumonitis in Japan. Chest. 1992;101:1044–1049. doi: 10.1378/chest.101.4.1044. [DOI] [PubMed] [Google Scholar]
- 229.Heron M, Claessen AM, Grutters JC, van den Bosch JM. T-cell activation profiles in different granulomatous interstitial lung diseases: a role for CD8+CD28(null) cells? Clin Exp Immunol. 2010;160:256–265. doi: 10.1111/j.1365-2249.2009.04076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ohtsuka M, Yoshizawa Y, Naitou T, Yano H, Sato T, Hasegawa S. The motility of lung lymphocytes in hypersensitivity pneumonitis and sarcoidosis. Am J Respir Crit Care Med. 1994;149:455–459. doi: 10.1164/ajrccm.149.2.8306045. [DOI] [PubMed] [Google Scholar]
- 231.Shinohara T, Tsuji S, Okano Y, Machida H, Hatakeyama N, Ogushi F. Elevated levels of intelectin-1, a pathogen-binding lectin, in the BAL fluid of patients with chronic eosinophilic pneumonia and hypersensitivity pneumonitis. Intern Med. 2018;57:3507–3514. doi: 10.2169/internalmedicine.0841-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Taniuchi N, Ghazizadeh M, Enomoto T, Matsuda K, Sato M, Takizawa Y, et al. Evaluation of fractional analysis of bronchoalveolar lavage combined with cellular morphological features. Int J Med Sci. 2009;6:1–8. doi: 10.7150/ijms.6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Unoura K, Miyazaki Y, Sumi Y, Tamaoka M, Sugita T, Inase N. Identification of fungal DNA in BALF from patients with home-related hypersensitivity pneumonitis. Respir Med. 2011;105:1696–1703. doi: 10.1016/j.rmed.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 234.Uzaslan E, Guzman J, Costabel U. Cockade-like structures in alveolar macrophages in extrinsic allergic alveolitis. Respiration. 2005;72:46–51. doi: 10.1159/000083400. [DOI] [PubMed] [Google Scholar]
- 235.Valenti S, Scordamaglia A, Crimi P, Mereu C. Bronchoalveolar lavage and transbronchial lung biopsy in sarcoidosis and extrinsic allergic alveolitis. Eur J Respir Dis. 1982;63:564–569. [PubMed] [Google Scholar]
- 236.Delacroix DL, Marchandise FX, Francis C, Sibille Y. Alpha-2-macroglobulin, monomeric and polymeric immunoglobulin A, and immunoglobulin M in bronchoalveolar lavage. Am Rev Respir Dis. 1985;132:829–835. doi: 10.1164/arrd.1985.132.4.829. [DOI] [PubMed] [Google Scholar]
- 237.Hamm H, Lührs J, Guzman y Rotaeche J, Costabel U, Fabel H, Bartsch W. Elevated surfactant protein A in bronchoalveolar lavage fluids from sarcoidosis and hypersensitivity pneumonitis patients. Chest. 1994;106:1766–1770. doi: 10.1378/chest.106.6.1766. [DOI] [PubMed] [Google Scholar]
- 238.Korosec P, Osolnik K, Kern I, Silar M, Mohorcic K, Kosnik M. Expansion of pulmonary CD8+CD56+ natural killer T-cells in hypersensitivity pneumonitis. Chest. 2007;132:1291–1297. doi: 10.1378/chest.07-0128. [DOI] [PubMed] [Google Scholar]
- 239.Leatherman JW, Michael AF, Schwartz BA, Hoidal JR. Lung T cells in hypersensitivity pneumonitis. Ann Intern Med. 1984;100:390–392. doi: 10.7326/0003-4819-100-3-390. [DOI] [PubMed] [Google Scholar]
- 240.Sharma SK, Pande JN, Verma K, Guleria JS. Bronchoalveolar lavage fluid (BALF) analysis in interstitial lung diseases: a 7-year experience. Indian J Chest Dis Allied Sci. 1989;31:187–196. [PubMed] [Google Scholar]
- 241.Sterclova M, Matej R, Mandakova P, Skibova J, Vasakova M. Role of interleukin 4 and its receptor in clinical presentation of chronic extrinsic allergic alveolitis: a pilot study. Multidiscip Respir Med. 2013;8:35. doi: 10.1186/2049-6958-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Fujimori Y, Kataoka M, Tada S, Takehara H, Matsuo K, Miyake T, et al. The role of interleukin-8 in interstitial pneumonia. Respirology. 2003;8:33–40. doi: 10.1046/j.1440-1843.2003.00420.x. [DOI] [PubMed] [Google Scholar]
- 243.Godard P, Clot J, Jonquet O, Bousquet J, Michel FB. Lymphocyte subpopulations in bronchoalveolar lavages of patients with sarcoidosis and hypersensitivity pneumonitis. Chest. 1981;80:447–452. doi: 10.1378/chest.80.4.447. [DOI] [PubMed] [Google Scholar]
- 244.Milman N, Graudal N, Jacobsen GK. Bronchoalveolar lavage in radiologically detected diffuse lung disease: diagnostic value of total and differential cell count in a series of 130 patients. APMIS. 1995;103:764–768. [PubMed] [Google Scholar]
- 245.Nagata N, Takayama K, Nikaido Y, Yokosaki Y, Kido M. Comparison of alveolar septal inflammation to bronchoalveolar lavage in interstitial lung diseases. Respiration. 1996;63:94–99. doi: 10.1159/000196525. [DOI] [PubMed] [Google Scholar]
- 246.Satake N, Nagai S, Kawatani A, Kaneshima H, Tanaka S, Takeuchi M, et al. Density of phenotypic markers on BAL T-lymphocytes in hypersensitivity pneumonitis, pulmonary sarcoidosis and bronchiolitis obliterans with organizing pneumonia. Eur Respir J. 1993;6:477–482. [PubMed] [Google Scholar]
- 247.Schildge J, Nagel C, Grun C. Bronchoalveolar lavage in interstitial lung diseases: does the recovery rate affect the results? Respiration. 2007;74:553–557. doi: 10.1159/000102890. [DOI] [PubMed] [Google Scholar]
- 248.Semenzato G, Chilosi M, Ossi E, Trentin L, Pizzolo G, Cipriani A, et al. Bronchoalveolar lavage and lung histology. Comparative analysis of inflammatory and immunocompetent cells in patients with sarcoidosis and hypersensitivity pneumonitis. Am Rev Respir Dis. 1985;132:400–404. doi: 10.1164/arrd.1985.132.2.400. [DOI] [PubMed] [Google Scholar]
- 249.Shiota Y. Clinical studies on human alveolar macrophages: II. Functions of alveolar macrophages collected by bronchoalveolar lavage. Nippon Ketsueki Gakkai Zasshi. 1987;50:79–88. [PubMed] [Google Scholar]
- 250.Soler P, Nioche S, Valeyre D, Basset F, Benveniste J, Burtin C, et al. Role of mast cells in the pathogenesis of hypersensitivity pneumonitis. Thorax. 1987;42:565–572. doi: 10.1136/thx.42.8.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tøndell A, Rø AD, Børset M, Moen T, Sue-Chu M. Activated CD8+ T cells and natural killer T cells in bronchoalveolar lavage fluid in hypersensitivity pneumonitis and sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2015;31:316–324. [PubMed] [Google Scholar]
- 252.Drent M, Grutters JC, Mulder PG, van Velzen-Blad H, Wouters EF, van den Bosch JM. Is the different T helper cell activity in sarcoidosis and extrinsic allergic alveolitis also reflected by the cellular bronchoalveolar lavage fluid profile? Sarcoidosis Vasc Diffuse Lung Dis. 1997;14:31–38. [PubMed] [Google Scholar]
- 253.Sterclova M, Vasakova M, Pavlicek J, Metlicka M, Krasna E, Striz I. Chemokine receptors in a regulation of interstitial lung fibrosis and inflammation. Exp Lung Res. 2009;35:514–523. doi: 10.1080/01902140902759282. [DOI] [PubMed] [Google Scholar]
- 254.Sterclova M, Paluch P, Skibova J, Vasakova M. Influence of age on manifestation, VC and TLCO values, and bronchoalveolar lavage cell counts of sarcoidosis and extrinsic allergic alveolitis. Clin Respir J. 2015;9:39–44. doi: 10.1111/crj.12102. [DOI] [PubMed] [Google Scholar]
- 255.Dai H, Guzman J, Chen B, Costabel U. Production of soluble tumor necrosis factor receptors and tumor necrosis factor-alpha by alveolar macrophages in sarcoidosis and extrinsic allergic alveolitis. Chest. 2005;127:251–256. doi: 10.1378/chest.127.1.251. [DOI] [PubMed] [Google Scholar]
- 256.Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. American Thoracic Society Committee on BAL in Interstitial Lung Disease. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004–1014. doi: 10.1164/rccm.201202-0320ST. [DOI] [PubMed] [Google Scholar]
- 257.Pajares V, Puzo C, Castillo D, Lerma E, Montero MA, Ramos-Barbón D, et al. Diagnostic yield of transbronchial cryobiopsy in interstitial lung disease: a randomized trial. Respirology. 2014;19:900–906. doi: 10.1111/resp.12322. [DOI] [PubMed] [Google Scholar]
- 258.Sindhwani G, Shirazi N, Sodhi R, Raghuvanshi S, Rawat J. Transbronchial lung biopsy in patients with diffuse parenchymal lung disease without ‘idiopathic pulmonary fibrosis pattern’ on HRCT scan: Experience from a tertiary care center of North India. Lung India. 2015;32:453–456. doi: 10.4103/0970-2113.164148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Morell F, Reyes L, Doménech G, De Gracia J, Majó J, Ferrer J. Diagnoses and diagnostic procedures in 500 consecutive patients with clinical suspicion of interstitial lung disease [in Spanish] Arch Bronconeumol. 2008;44:185–191. [PubMed] [Google Scholar]
- 260.Sheth JS, Belperio JA, Fishbein MC, Kazerooni EA, Lagstein A, Murray S, et al. Utility of transbronchial vs surgical lung biopsy in the diagnosis of suspected fibrotic interstitial lung disease. Chest. 2017;151:389–399. doi: 10.1016/j.chest.2016.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pourabdollah M, Shamaei M, Karimi S, Karimi M, Kiani A, Jabbari HR. Transbronchial lung biopsy: the pathologist’s point of view. Clin Respir J. 2016;10:211–216. doi: 10.1111/crj.12207. [DOI] [PubMed] [Google Scholar]
- 262.Ramaswamy A, Homer R, Killam J, Pisani MA, Murphy TE, Araujo K, et al. Comparison of transbronchial and cryobiopsies in evaluation of diffuse parenchymal lung disease. J Bronchology Interv Pulmonol. 2016;23:14–21. doi: 10.1097/LBR.0000000000000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Babiak A, Hetzel J, Krishna G, Fritz P, Moeller P, Balli T, et al. Transbronchial cryobiopsy: a new tool for lung biopsies. Respiration. 2009;78:203–208. doi: 10.1159/000203987. [DOI] [PubMed] [Google Scholar]
- 264.Hetzel J, Eberhardt R, Petermann C, Gesierich W, Darwiche K, Hagmeyer L, et al. Bleeding risk of transbronchial cryobiopsy compared to transbronchial forceps biopsy in interstitial lung disease: a prospective, randomized, multicentre cross-over trial. Respir Res. 2019;20:140. doi: 10.1186/s12931-019-1091-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Casoni GL, Gurioli C, Chhajed PN, Chilosi M, Zampatori M, Olivieri D, et al. The value of transbronchial lung biopsy using jumbo forceps via rigid bronchoscope in DILD. Monaldi Arch Chest Dis. 2008;69:59–64. doi: 10.4081/monaldi.2008.397. [DOI] [PubMed] [Google Scholar]
- 266.Adams TN, Newton CA, Batra K, Abu-Hijleh M, Barbera T, Torrealba J, et al. Utility of bronchoalveolar lavage and transbronchial biopsy in patients with hypersensitivity pneumonitis. Lung. 2018;196:617–622. doi: 10.1007/s00408-018-0139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lacasse Y, Fraser RS, Fournier M, Cormier Y. Diagnostic accuracy of transbronchial biopsy in acute farmer’s lung disease. Chest. 1997;112:1459–1465. doi: 10.1378/chest.112.6.1459. [DOI] [PubMed] [Google Scholar]
- 268.Cascante JA, Cebollero P, Herrero S, Yagüe A, Echegoyen A, Elizalde J, et al. Transbronchial cryobiopsy in interstitial lung disease: are we on the right path? J Bronchology Interv Pulmonol. 2016;23:204–209. doi: 10.1097/LBR.0000000000000292. [DOI] [PubMed] [Google Scholar]
- 269.Fruchter O, Fridel L, El Raouf BA, Abdel-Rahman N, Rosengarten D, Kramer MR. Histological diagnosis of interstitial lung diseases by cryo-transbronchial biopsy. Respirology. 2014;19:683–688. doi: 10.1111/resp.12296. [DOI] [PubMed] [Google Scholar]
- 270.Griff S, Schönfeld N, Ammenwerth W, Blum TG, Grah C, Bauer TT, et al. Diagnostic yield of transbronchial cryobiopsy in non-neoplastic lung disease: a retrospective case series. BMC Pulm Med. 2014;14:171. doi: 10.1186/1471-2466-14-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Hernández-González F, Lucena CM, Ramírez J, Sánchez M, Jimenez MJ, Xaubet A, et al. Cryobiopsy in the diagnosis of diffuse interstitial lung disease: yield and cost-effectiveness analysis. Arch Bronconeumol. 2015;51:261–267. doi: 10.1016/j.arbres.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 272.Kronborg-White S, Folkersen B, Rasmussen TR, Voldby N, Madsen LB, Rasmussen F, et al. Introduction of cryobiopsies in the diagnostics of interstitial lung diseases: experiences in a referral center. Eur Clin Respir J. 2017;4:1274099. doi: 10.1080/20018525.2016.1274099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Kropski JA, Pritchett JM, Mason WR, Sivarajan L, Gleaves LA, Johnson JE, et al. Bronchoscopic cryobiopsy for the diagnosis of diffuse parenchymal lung disease. PLoS One. 2013;8:e78674. doi: 10.1371/journal.pone.0078674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ussavarungsi K, Kern RM, Roden AC, Ryu JH, Edell ES. Transbronchial cryobiopsy in diffuse parenchymal lung disease: etrospective analysis of 74 cases. Chest. 2017;151:400–408. doi: 10.1016/j.chest.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 275.Gershman E, Fruchter O, Benjamin F, Nader AR, Rosengarten D, Rusanov V, et al. Safety of cryo-transbronchial biopsy in diffuse lung diseases: analysis of three hundred cases. Respiration. 2015;90:40–46. doi: 10.1159/000381921. [DOI] [PubMed] [Google Scholar]
- 276.Ravaglia C, Wells AU, Tomassetti S, Gurioli C, Gurioli C, Dubini A, et al. Diagnostic yield and risk/benefit analysis of trans-bronchial lung cryobiopsy in diffuse parenchymal lung diseases: a large cohort of 699 patients. BMC Pulm Med. 2019;19:16. doi: 10.1186/s12890-019-0780-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Hagmeyer L, Theegarten D, Wohlschläger J, Hager T, Treml M, Herkenrath SD, et al. Transbronchial cryobiopsy in fibrosing interstitial lung disease: modifications of the procedure lead to risk reduction. Thorax. 2019;74:711–714. doi: 10.1136/thoraxjnl-2018-212095. [DOI] [PubMed] [Google Scholar]
- 278.Vlacic G, Kern I, Rozman A. Transbronchial cryobiopsy: a single-center experience. Virchows Arch. 2018;473:s18. [Google Scholar]
- 279.Gnass M, Filarecka A, Pankowski J, Soja J, Bugalho A, Szlubowski A. Transbronchial lung cryobiopsy guided by endobronchial ultrasound radial miniprobe in interstitial lung diseases: preliminary results of a prospective study. Pol Arch Intern Med. 2018;128:259–262. doi: 10.20452/pamw.4253. [DOI] [PubMed] [Google Scholar]
- 280.Dhooria S, Mehta RM, Srinivasan A, Madan K, Sehgal IS, Pattabhiraman V, et al. The safety and efficacy of different methods for obtaining transbronchial lung cryobiopsy in diffuse lung diseases. Clin Respir J. 2018;12:1711–1720. doi: 10.1111/crj.12734. [DOI] [PubMed] [Google Scholar]
- 281.Morais AMM, Melo N, Mota P, Magalhães A, Guimarães S, Moura CS. Transbronchial criobiopsy (TCB) in two lung lobes-diagnostic accuracy. Eur Respir J. 2017;50:PA860. [Google Scholar]
- 282.Li YS, Guo SL, Yi XH, Xiao ML, Jin XX, Xiao Y, et al. Efficacy and safety of transbronchial cryobiopsy in the etiologic diagnosis of diffuse lung disease [in Chinese] Zhonghua Yi Xue Za Zhi. 2017;97:3617–3623. doi: 10.3760/cma.j.issn.0376-2491.2017.46.004. [DOI] [PubMed] [Google Scholar]
- 283.de Sousa Antunes Dias Padrão EF, Mota PC, Melo N, Guimarães S, Moura CS, Magalhães A, et al. Transbronchial lung cryobiopsy in the diagnosis of hypersensitivity pneumonitis. Eur Respir J. 2017;50:PA3018. [Google Scholar]
- 284.Colella S, Massaccesi C, Fioretti F, Panella G, Primomo GL, D'Emilio V, et al. Transbronchial lung cryobiopsy in lung diseases: diagnostic yield and safety. Eur Respir J. 2017;50:PA3025. [Google Scholar]
- 285.Bondue B, Pieters T, Alexander P, De Vuyst P, Ruiz Patino M, Hoton D, et al. Role of transbronchial lung cryobiopsies in diffuse parenchymal lung diseases: interest of a sequential approach. Pulm Med. 2017;2017:6794343. doi: 10.1155/2017/6794343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wälscher J, Groß B, Eberhardt R, Heussel CP, Eichinger M, Warth A, et al. Transbronchial cryobiopsies for diagnosing interstitial lung disease: real-life experience from a tertiary referral center for interstitial lung disease. Respiration. 2019;97:348–354. doi: 10.1159/000493428. [DOI] [PubMed] [Google Scholar]
- 287.Romagnoli M, Colby TV, Berthet JP, Gamez AS, Mallet JP, Serre I, et al. Poor concordance between sequential transbronchial lung cryobiopsy and surgical lung biopsy in the diagnosis of diffuse interstitial lung diseases. Am J Respir Crit Care Med. 2019;199:1249–1256. doi: 10.1164/rccm.201810-1947OC. [DOI] [PubMed] [Google Scholar]
- 288.Troy LK, Grainge C, Corte TJ, Williamson JP, Vallely MP, Cooper WA. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective comparative study. Lancet Respir Med. 2019;8:171–181. doi: 10.1016/S2213-2600(19)30342-X. [DOI] [PubMed] [Google Scholar]
- 289.Ayed AK, Raghunathan R. Thoracoscopy versus open lung biopsy in the diagnosis of ILD: a randomised controlled trial. J R Coll Surg Edinb. 2000;45:159–163. [PubMed] [Google Scholar]
- 290.Miller JD, Urschel JD, Cox G, Olak J, Young JE, Kay JM, et al. A randomized, controlled trial comparing thoracoscopy and limited thoracotomy for lung biopsy in interstitial lung disease. Ann Thorac Surg. 2000;70:1647–1650. doi: 10.1016/s0003-4975(00)01913-5. [DOI] [PubMed] [Google Scholar]
- 291.Blewett CJ, Bennett WF, Miller JD, Urschel JD. Open lung biopsy as an outpatient procedure. Ann Thorac Surg. 2001;71:1113–1115. doi: 10.1016/s0003-4975(00)02657-6. [DOI] [PubMed] [Google Scholar]
- 292.Qureshi RA, Ahmed TA, Grayson AD, Soorae AS, Drakeley MJ, Page RD.Does lung biopsy help patients with interstitial lung disease? Eur J Cardiothorac Surg 200221621–626.[Discussion, p. 626.] [DOI] [PubMed] [Google Scholar]
- 293.Ayed AK. Video-assisted thoracoscopic lung biopsy in the diagnosis of diffuse interstitial lung disease: a prospective study. J Cardiovasc Surg (Torino) 2003;44:115–118. [PubMed] [Google Scholar]
- 294.Yamaguchi M, Yoshino I, Suemitsu R, Osoegawa A, Kameyama T, Tagawa T, et al. Elective video-assisted thoracoscopic lung biopsy for interstitial lung disease. Asian Cardiovasc Thorac Ann. 2004;12:65–68. doi: 10.1177/021849230401200116. [DOI] [PubMed] [Google Scholar]
- 295.Ooi A, Iyenger S, Ferguson J, Ritchie AJ. VATS lung biopsy in suspected, diffuse interstitial lung disease provides diagnosis, and alters management strategies. Heart Lung Circ. 2005;14:90–92. doi: 10.1016/j.hlc.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 296.Lettieri CJ, Veerappan GR, Helman DL, Mulligan CR, Shorr AF. Outcomes and safety of surgical lung biopsy for interstitial lung disease. Chest. 2005;127:1600–1605. doi: 10.1378/chest.127.5.1600. [DOI] [PubMed] [Google Scholar]
- 297.Park JH, Kim DK, Kim DS, Koh Y, Lee SD, Kim WS, et al. Mortality and risk factors for surgical lung biopsy in patients with idiopathic interstitial pneumonia. Eur J Cardiothorac Surg. 2007;31:1115–1119. doi: 10.1016/j.ejcts.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 298.Kreider ME, Hansen-Flaschen J, Ahmad NN, Rossman MD, Kaiser LR, Kucharczuk JC, et al. Complications of video-assisted thoracoscopic lung biopsy in patients with interstitial lung disease. Ann Thorac Surg. 2007;83:1140–1144. doi: 10.1016/j.athoracsur.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 299.Coutinho GF, Pancas R, Magalhães E, Bernardo JE, Eugénio L, Antunes MJ. Diagnostic value of surgical lung biopsy: comparison with clinical and radiological diagnosis. Eur J Cardiothorac Surg. 2008;33:781–785. doi: 10.1016/j.ejcts.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 300.Sigurdsson MI, Isaksson HJ, Gudmundsson G, Gudbjartsson T. Diagnostic surgical lung biopsies for suspected interstitial lung diseases: a retrospective study. Ann Thorac Surg. 2009;88:227–232. doi: 10.1016/j.athoracsur.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 301.Bando M, Ohno S, Hosono T, Yanase K, Sato Y, Sohara Y, et al. Risk of acute exacerbation after video-assisted thoracoscopic lung biopsy for interstitial lung disease. J Bronchology Interv Pulmonol. 2009;16:229–235. doi: 10.1097/LBR.0b013e3181b767cc. [DOI] [PubMed] [Google Scholar]
- 302.Guerra M, Miranda JA, Leal F, Vouga L. Interstitial lung disease: diagnostic accuracy and safety of surgical lung biopsy. Rev Port Pneumol. 2009;15:433–442. [PubMed] [Google Scholar]
- 303.Ishie RT, Cardoso JJ, Silveira RJ, Stocco L. Video-assisted thoracoscopy for the diagnosis of diffuse parenchymal lung disease. J Bras Pneumol. 2009;35:234–241. doi: 10.1590/s1806-37132009000300007. [DOI] [PubMed] [Google Scholar]
- 304.Fibla JJ, Molins L, Blanco A, Royo I, Martínez Vallina P, Martínez N, et al. Video-assisted thoracoscopic lung biopsy in the diagnosis of interstitial lung disease: a prospective, multi-center study in 224 patients. Arch Bronconeumol. 2012;48:81–85. doi: 10.1016/j.arbres.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 305.Fibla JJ, Brunelli A, Cassivi SD, Deschamps C. Aggregate risk score for predicting mortality after surgical biopsy for interstitial lung disease. Interact Cardiovasc Thorac Surg. 2012;15:276–279. doi: 10.1093/icvts/ivs174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Blackhall V, Asif M, Renieri A, Civitelli S, Kirk A, Jilaihawi A, et al. The role of surgical lung biopsy in the management of interstitial lung disease: experience from a single institution in the UK. Interact Cardiovasc Thorac Surg. 2013;17:253–257. doi: 10.1093/icvts/ivt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Blanco M, Obeso GA, Durán JC, Rivo JE, García-Fontán E, Peña E, et al. Surgical lung biopsy for diffuse lung disease: our experience in the last 15 years. Rev Port Pneumol. 2013;19:59–64. doi: 10.1016/j.rppneu.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 308.Kayatta MO, Ahmed S, Hammel JA, Fernandez F, Pickens A, Miller D, et al. Surgical biopsy of suspected interstitial lung disease is superior to radiographic diagnosis. Ann Thorac Surg. 2013;96:399–401. doi: 10.1016/j.athoracsur.2013.04.065. [DOI] [PubMed] [Google Scholar]
- 309.Pompeo E, Rogliani P, Cristino B, Schillaci O, Novelli G, Saltini C. Awake thoracoscopic biopsy of interstitial lung disease. Ann Thorac Surg. 2013;95:445–452. doi: 10.1016/j.athoracsur.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 310.Luo Q, Han Q, Chen X, Xie J, Wu L, Chen R. The diagnosis efficacy and safety of video-assisted thoracoscopy surgery (VATS) in undefined interstitial lung diseases: a retrospective study. J Thorac Dis. 2013;5:283–288. doi: 10.3978/j.issn.2072-1439.2013.04.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Findikcioglu A, Karadayi S. Is surgical biopsy necessary for diagnosis of interstitial lung diseases: a retrospective clinical study. J Clin Anal Med. 2014;5:204–208. [Google Scholar]
- 312.Morris D, Zamvar V. The efficacy of video-assisted thoracoscopic surgery lung biopsies in patients with interstitial lung disease: a retrospective study of 66 patients. J Cardiothorac Surg. 2014;9:45. doi: 10.1186/1749-8090-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Sonobe M, Handa T, Tanizawa K, Sato M, Sato T, Chen F, et al. Videothoracoscopy-assisted surgical lung biopsy for interstitial lung diseases. Gen Thorac Cardiovasc Surg. 2014;62:376–382. doi: 10.1007/s11748-014-0383-0. [DOI] [PubMed] [Google Scholar]
- 314.Bagheri R, Haghi SZ, Attaran D, Hashem Asnaashari AM, Basiri R, Rajabnejad A. Efficacy of minimally invasive surgery in diagnosis of interstitial lung disease. Asian Cardiovasc Thorac Ann. 2015;23:851–854. doi: 10.1177/0218492315593694. [DOI] [PubMed] [Google Scholar]
- 315.Rotolo N, Imperatori A, Dominioni L, Facchini A, Conti V, Castiglioni M, et al. Efficacy and safety of surgical lung biopsy for interstitial disease: experience of 161 consecutive patients from a single institution in Italy. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32:251–258. [PubMed] [Google Scholar]
- 316.Samejima J, Tajiri M, Ogura T, Baba T, Omori T, Tsuboi M, et al. Thoracoscopic lung biopsy in 285 patients with diffuse pulmonary disease. Asian Cardiovasc Thorac Ann. 2015;23:191–197. doi: 10.1177/0218492314550724. [DOI] [PubMed] [Google Scholar]
- 317.Knipscheer BJ, van Moorsel CH, Grutters JC. Non-specific and usual interstitial pneumonia, short-term survival after surgical biopsy. Lung. 2015;193:449–450. doi: 10.1007/s00408-015-9721-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Khalil M, Cowen M, Chaudhry M, Loubani M. Single versus multiple lung biopsies for suspected interstitial lung disease. Asian Cardiovasc Thorac Ann. 2016;24:788–791. doi: 10.1177/0218492316665551. [DOI] [PubMed] [Google Scholar]
- 319.Tomassetti S, Wells AU, Costabel U, Cavazza A, Colby TV, Rossi G, et al. Bronchoscopic lung cryobiopsy increases diagnostic confidence in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193:745–752. doi: 10.1164/rccm.201504-0711OC. [DOI] [PubMed] [Google Scholar]
- 320.Ravaglia C, Bonifazi M, Wells AU, Tomassetti S, Gurioli C, Piciucchi S, et al. Safety and diagnostic yield of transbronchial lung cryobiopsy in diffuse parenchymal lung diseases: a comparative study versus video-assisted thoracoscopic lung biopsy and a systematic review of the literature. Respiration. 2016;91:215–227. doi: 10.1159/000444089. [DOI] [PubMed] [Google Scholar]
- 321.Ahmed S, El Hindawi A, Mashhour S. Spectrum of diffuse parenchymal lung diseases using medical thoracoscopic lung biopsy: an experience with 55 patients during 2013–2015. Egypt J Chest Dis Tuberc. 2016;65:717–722. [Google Scholar]
- 322.Lieberman S, Gleason JB, Ilyas MIM, Martinez F, Mehta JP, Savage EB. Assessing the safety and clinical impact of thoracoscopic lung biopsy in patients with interstitial lung disease. J Clin Diagn Res. 2017;11:OC57–OC59. doi: 10.7860/JCDR/2017/20281.9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Morell F, Villar A, Ojanguren I, Muñoz X, Cruz MJ. Hypersensitivity pneumonitis: challenges in diagnosis and management, avoiding surgical lung biopsy. Semin Respir Crit Care Med. 2016;37:395–405. doi: 10.1055/s-0036-1580692. [DOI] [PubMed] [Google Scholar]
- 324.Ishizuka M, Miyazaki Y, Tateishi T, Tsutsui T, Tsuchiya K, Inase N. Validation of inhalation provocation test in chronic bird-related hypersensitivity pneumonitis and new prediction score. Ann Am Thorac Soc. 2015;12:167–173. doi: 10.1513/AnnalsATS.201408-350OC. [DOI] [PubMed] [Google Scholar]
- 325.Munoz X, Sanchez-Ortiz M, Torres F, Villar A, Morell F, Cruz MJ. Diagnostic yield of specific inhalation challenge in hypersensitivity pneumonitis. Eur Respir J. 2014;44:1658–1665. doi: 10.1183/09031936.00060714. [DOI] [PubMed] [Google Scholar]
- 326.Hansell DM, Bankier AA, MacMahon H, McLoud TC, Muller NL, Remy J. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246:697–722. doi: 10.1148/radiol.2462070712. [DOI] [PubMed] [Google Scholar]
- 327.Webb WR, Muller NL, Naidich DP. Standardized terms for high-resolution computed tomography of the lung: a proposed glossary. J Thorac Imaging. 1993;8:167–175. doi: 10.1097/00005382-199322000-00002. [DOI] [PubMed] [Google Scholar]
- 328.Chung MH, Edinburgh KJ, Webb EM, McCowin M, Webb WR. doi: 10.1097/00005382-200104000-00001. Mixed infiltrative and obstructive disease on high-resolution CT: differential diagnosis and functional correlates in a consecutive series. J Thorac Imaging 2001;16:69–75. [DOI] [PubMed] [Google Scholar]
- 329.Chong BJ, Kanne JP, Chung JH.Headcheese sign. J Thorac Imaging 2014;29:W13. [DOI] [PubMed] [Google Scholar]
- 330.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, et al. ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]