

Abstract
While ketones are among the most versatile functional groups, their synthesis remains reliant upon reactive and low abundance starting materials. In contrast, amide formation is the most-used bond construction in medicinal chemistry because the chemistry is reliable and draws upon large, diverse substrate pools. A new method for the synthesis of ketones is presented here that draws from the same substrates used for amide bond synthesis: amines and carboxylic acids. A nickel terpyridine catalyst couples N-alkyl pyridinium salts with in-situ formed carboxylic acid fluorides or 2-pyridyl esters under reducing conditions (Mn metal). The reaction has broad scope, as demonstrated by the synthesis of 35 different ketones bearing a wide variety of functional groups with an average yield of 60 ± 16%. This approach is capable of coupling diverse substrates, including pharmaceutical intermediates, to rapidly form complex ketones.
Keywords: Ketone Synthesis, Cross-Electrophile Coupling, Pyridinium Salts, Acyl Fluorides, Nickel Catalysis
Graphical Abstract
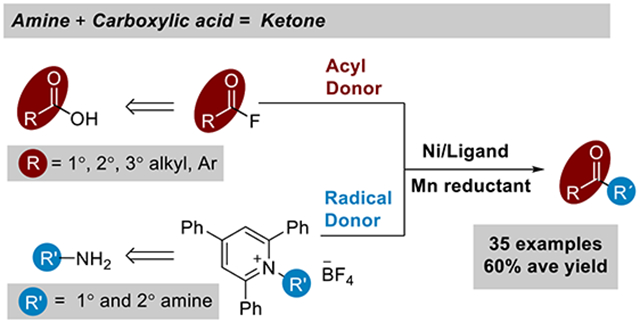
The ketone functional group plays a central role in organic synthesis: it is present in many target molecules (natural products, drugs, materials), and is also a key intermediate in the synthesis of C-C bonds and alcohols.[1] However, the synthesis of ketones continues to rely upon methods that are limited in functional group compatibility and use starting materials of low abundance (Scheme 1). Ketones are most often synthesized by the addition of an organometallic reagent to an aldehyde followed by oxidation[1a] or, more recently, the coupling of an organometallic reagent to an activated ester.[2,3] This latter strategy is useful because there are many more alkanoic acids than aliphatic aldehydes, yet the organometallic coupling partner has limited availability and functional group compatibility.
Scheme 1.

How to make ketones from amine and carboxylic acid derivatives.
In contrast, amide bond formation is among the most reliable coupling reactions.[4] A recent analysis ranked it as the most used reaction in medicinal chemistry, accounting for 25% of all reactions used in drug discovery. This is driven, in part, by the large numbers of amines and carboxylic acids that are commercially available.[5] A ketone synthesis that could utilize these substrate pools would dramatically increase easily available chemical space. In this vein, Matsuo recently reported an exciting advance: the cross-coupling of N-aroylsuccinimides with alkylpyridinium salts [6] This approach worked for benzoic acid derivatives, but was not useful for the larger alkanoic acid substrate pool.
We show here how recent developments in our two research groups can be combined to achieve a general synthesis of dialkyl ketones from alkyl amines and alkyl carboxylic acids (Scheme 1). On one hand, cross-electrophile[7] approaches to ketone synthesis have been developed to eliminate the need for organometallic reagents by coupling of activated esters (-Cl, -SPy, -OCO2R) with alkyl halides[8] or, recently, a second carboxylic acid activated as an N-hydroxyphthalimide ester.[9] On the other hand, deaminative cross-couplings[10] of alkyl pyridinium salts have rapidly progressed, including cross-electrophile couplings with aryl halides.[11] We show here how these advances can be combined to form ketones from amines and carboxylic acid derivatives.[12,13]
The main challenge to realizing this transformation was achieving high cross-selectivity without resorting to a large excess of one coupling partner. Previous cross-electrophile couplings of alkyl pyridinium salts with aryl halides had shown that this balance of reactivity and selectivity could be challenging to achieve.[11] Based upon the hypothesis that the ketone product forms from the coupling of an acylnickel(II) intermediate with an pyridinium-salt-derived alkyl radical (Scheme 1),[8i,12e,14] we sought an acyl donor that would react quickly with the nickel catalyst, would not be easily reduced to form a radical,[15] and could be isolated or formed in situ.
Examination of activating strategies for carboxylic acids led us to acyl fluorides.[16] Acyl fluorides have several advantages: they can be made in situ (an advantage over N-acylimides or imides[17]), the liberated fluoride does not interfere with productive catalysis (a problem for thioesters[8d,8g]), and they can be purified if needed (unlike anhydrides or acid chlorides).
After systematic screening of different ligands, solvents, additives and reaction temperatures, we found optimal conditions for coupling an acyl fluoride with a primary alkyl pyridinium salt (Table 1): NiCl2(dme), L5, and Mn in NMP at 60 °C. These conditions provided ketone product 1 in 79% isolated yield (entry 1, Table 1). While 62-65% yields were obtained with electron-rich bipyridine ligands (L1 and L2, entries 3-4), terpyridine and electron-poor bipyridine ligands were not effective for this transformation (See Tables S2.1-S2.4 in Supporting Information). In contrast to our previous reports [8d, 9b], the analogous S-2-pyridyl thioester (entry 7) or 2-pyridyl ester (entry 8) provided a low yield of product 1 and primarily formed the deaminative alkyl dimer 1,4-diphenylbutane. A lower yield was observed when switching the reductant from Mn to Zn (entry 9). This could be attributed to slower reduction of the catalyst[18] or direct reduction of pyridinium salts by Mn being important for product formation (entry 10).[19] Finally, purified acid fluoride could be replaced by in-situ-generated acid fluoride (entry 2). Among several methods tested, fluoro-N,N,N′,N′-bis(tetramethylene)formamidinium hexafluorophosphate (TFFH) with 1,8-bis(dimethylamino)naphthylene (proton-sponge) was the most useful.[16e] While in-situ activation of the carboxylic acid was successful, we obtained low yields with an unpurified pyridinium salt (22% NMR yield of 1 as in Table 1, entry 2).
Table 1.
Optimal conditions for primary pyridinium salts.
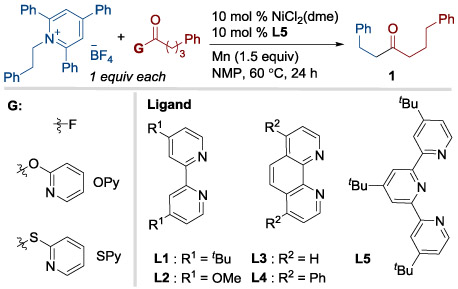 | |||
|---|---|---|---|
| Entry[a] | Change in condition from scheme | G | 1 (%)[b] |
| 1 | None | F | 82 (79) |
| 2[c] | In situ formation of acyl fluoride | F | 75 |
| 3 | L1 instead of L5 | F | 62 |
| 4 | L2 instead of L5 | F | 65 |
| 5 | L3 instead of L5 | F | 53 |
| 6 | L4 instead of L5 | F | 41 |
| 7 | Different G | SPy | 4 |
| 8 | Different G | OPy | 11 |
| 9[e] | Zn instead of Mn | F | 5 |
| 10[d] | No nickel | F | 0 |
| 11[d] | No ligand | F | 0 |
| 12[e] | No Mn reductant | F | 0 |
Pyridinium salt (0.125 mmol, 1 equiv), acyl fluoride (0.125 mmol, 1 equiv), NiCl2(dme) (0.0125 mmol, 10 mol %), ligand (0.0125 mmol, 10 mol %), Mn (0.1875 mmol, 1.5 equiv) was stirred in NMP (0.8 mL) at 60 °C for 24 h.
GC yield vs 1,3.5-trimethoxybenzene standard. Isolated yield in parentheses.
Carboxylic acid (0.125 mmol, 1 equiv), TFFH (0.125 mmol, 1 equiv), 1,8-bis(dimethylamino)naphthylene (0.125 mmol, 1 equiv), NMP (0.8 mL).
Significant amount of acyl fluoride recovered.
Both starting materials recovered.
G = Activating Group.
We found that secondary alkyl pyridinium salts required modified conditions (Table 2 and Tables S2.5-S2.7 in Supporting Information). These secondary alkyl reagents typically undergo more facile C─N bond cleavage, making formation of dihydropyridine byproduct even more competitive.[11a] To avoid this byproduct, these changes were made: 1) using 2-pyridyl esters instead of acyl fluorides; 2) changing the solvent to THF and lowering the temperature; 3) switching the ligand to L4 (entry 2). The use of NiBr2(dme) and a slight increase in the amount of pyridinium salt, from 1.0 to 1.5 equiv, further increased the yield to 72% (entry 4).
Table 2.
Optimal conditions for secondary pyridinium salts.
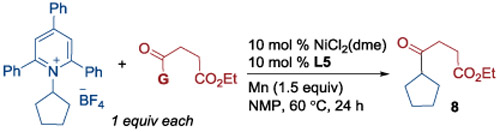 | |||
|---|---|---|---|
| Entry[a] | Change in condition from scheme | G | 8 (%)[b] |
| 1 | None | F | 14 |
| 2[c] | L4 instead of L5 in THF | OPy | 61 |
| 3[c] | As entry 2, NiBr2(dme) instead of NiCl2(dme) | OPy | 66 |
| 4[c] | As entry 3, 1.5 equiv of pyridinum | OPy | 72 |
Pyridinium salt (0.125 mmol, 1 equiv), acyl fluoride or 2-pyridyl ester (0.125 mmol, 1 equiv), NiCl2(dme) (0.0125 mmol, 10 mol%), ligand (0.0125 mmol, 10 mol%), Mn (0.1875 mmol, 1.5 equiv) was stirred in corresponding solvent (0.8 mL) at 60 °C for 24 h.
GC yield vs 1,3.5-trimethoxybenzene standard.
Reaction conducted at r.t.
G = Activating group.
The scope of these conditions is broad, as demonstrated by the 35 examples in Table 3. The majority of these examples used in-situ activation of the carboxylic acid, but in some cases purification of the product from proton-sponge-derived side products was difficult and pre-formed acyl fluorides were used. Pyridinium salts of unbranched and α-branched amines coupled well, but salts of tertiary carbinamines are not suitable substrates.[20] Primary, secondary, and tertiary carboxylic acids coupled in high yield, but a particularly hindered tertiary carboxylic acid, abietic acid, and α-amino acids coupled in low yield (see Supporting Information Table S5 for additional low-yielding substrates). Although not a focus of this study, in a preliminary result, a prototypical aryl carboxylic acid, naphthoic acid, could be coupled via its acid fluoride to give 35 in 41% yield, but a general solution to aroyl acids will require further optimization.[21] Finally, α-alkoxy carboxylic acids, a common motif found in cetirizine and other bioactive compounds,[22] could be coupled with slightly modified conditions (29, 30).
Table 3.
Substrate scope for the deaminative coupling of pyridinium salts with carboxylic acid derivatives.
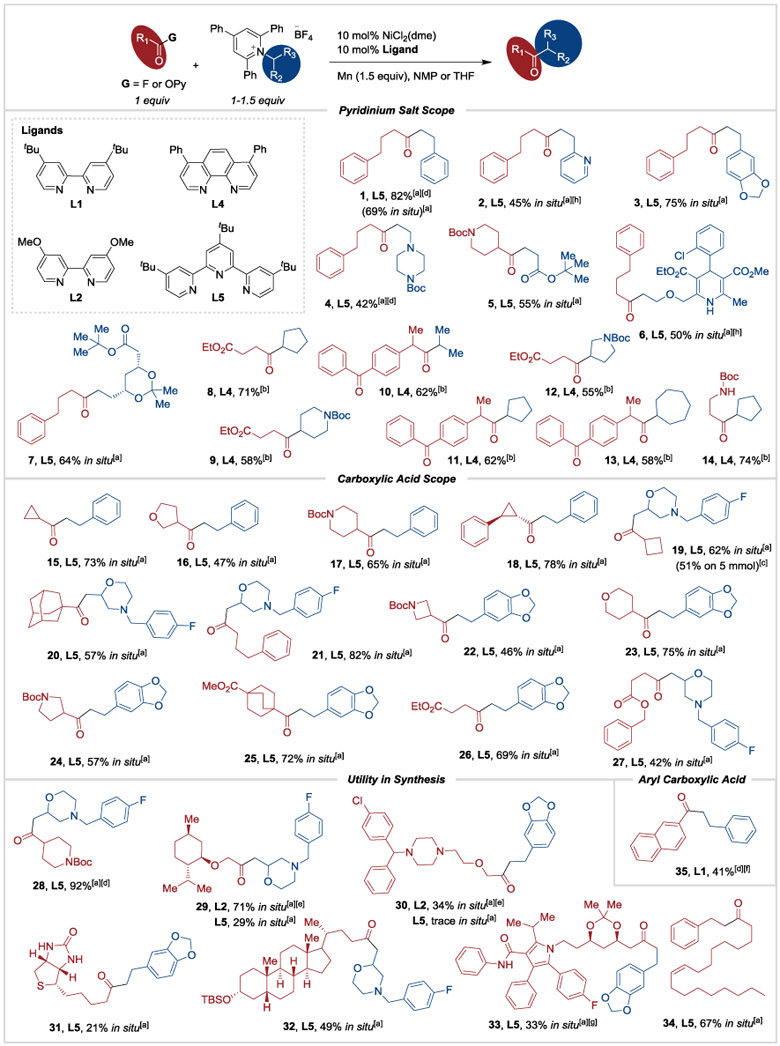 |
After initial acid fluoride formation using carboxylic acid (0.5 mmol, 1 equiv), TFFH (0.5 mmol, 1 equiv) and proton sponge (0.5 mmol, 1 equiv), the coupling was run on 0.5 mmol scale with a 1:1 ratio of starting materials, ligand L5 (0.05 mmol, 10 mol%), Mn (0.75 mmol, 1.5 equiv) in NMP (3 mL) at 60 °C for 24 h.
Reaction run at 0.5 mmol scale using pyridinium salt (0.75 mmol, 1.5 equiv) and pre-formed 2-pyridyl ester (0.5 mmol, 1 equiv) with NiBr2(dme) (0.05 mmol, 10 mol%), ligand L4 (0.05 mmol, 10 mol%), Mn (0.75 mmol, 1.5 equiv) in THF at r.t. for 24 h.
Reaction run on 5 mmol scale on benchtop, see Supporting Information.
Pre-formed acyl fluoride was used.
L2 used instead of L5.
L1 was used instead of L5.
Reaction run on 0.178 mmol scale at standard concentration.
NMR yield shown. Product was not separated from impurities, see Supporting Information for details.
Boc = tert-butoxycarbonyl, TBS = tert-butyldimethylsilyl.
Highlights of the functional group tolerance include basic amines (4, 19, 20, 21, 27, 28, 29, 30, 32), pyridine (2), cis-alkene (34), and even a dihydropyridine (6) that could deactivate a metal catalyst by coordination or be prone to oxidation under photoredox catalysis[23] or electrochemical conditions.[24] Acidic N-H bonds of Boc-β-alanine (14), a urea (31) or an N-aryl amide (33) as well as esters (5-9, 12, 25-27) and ketones (10, 11, 13) were tolerated, but would pose a problem for methods based upon organomagnesium or organolithium reagents.[2] Conveniently, the chemistry can be run preparatively on the benchtop (740 mg of 19) and generally uses a 1:1 or 1:1.5 ratio of starting materials.
Although we have not yet studied the reaction mechanism, the similarities to other cross-electrophile coupling reactions[8i,12e,14] suggest an analogous mechanism: initial oxidative addition of the acyl fluoride to nickel(0)[16e] followed by radical addition to the resulting acylnickel(II) intermediate (Scheme 1). The resulting acyl-alkyl nickel(III) species could reductively eliminate ketone product. Formation of alkyl radicals from N-alkylpyridinium salts can be mediated by nickel or arise from direct reduction with manganese.[6, 10a, 10i, 11a, 11b, 11e] Another possibility is the reduction of an acylnickel(II) intermediate to form an acylnickel(I) species that can then react with pyridinium salts.[16g,25]
The ability to use carboxylic acids and amines as a substrate pool is valuable because these are two of the largest pools of commercially available alkyl fragments.[5] This flows from the reliance of medicinal chemistry on amide bond formation.[4] Indeed, alkyl amines are unique in that there are more listed for sale than reported in the literature![26] In some cases this translates to lower prices (see analysis in the Supporting Information), but it also means that complex amines and acids can be repurposed as starting materials, such as a mosapride intermediate (19-21, 27-29, 32), amlodipine (6), cetirizine (30), an atorvastatin intermediate (33 and a side-chain fragment in 7), biotin (31), and lithocholic acid (32). The ability to leverage these pools to make ketones instead of amides opens up new areas of chemical space with minimal effort, adding another dimension to the medicinal chemist’s toolbox.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R01GM097243 (DJW) and R35GM131816 (MPW) and the University of Wisconsin (DJW). The authors thank Kai Kang, Daniel Enny and Benjamin Chi (Univ. of Wisconsin) for assistance with characterization of compounds. Data were acquired at UD on instruments obtained with assistance of NSF and NIH funding (NSF CHE0421224, CHE1229234, CHE0840401, and CHE1048367; NIH P20 GM104316, P20 GM103541, and S10 OD016267). The following instrumentation in the PBCIC was supported by: Thermo Q Exactive™ Plus by NIH 1S10 OD020022-1; Bruker Avance III 400 by NSF CHE-1048642; Bruker Avance III 500 by a generous gift from Paul J. and Margaret M. Bender. We thank Joe Barendt and Chiral Technologies for the kind donation of achiral SFC columns used in this work.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest: The authors declare no conflict of interest.
References
- [1] a).Lawrence NJ, J. Chem. Soc, Perkin Trans 1 1998, 1739–1750. [Google Scholar]; b) Dieter RK, Tetrahedron 1999, 55, 4177–4236. [Google Scholar]; c) Blangetti M, Rosso H, Prandi C, Deagostino A, Venturello P, Molecules 2013, 18, 1188–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2] a).Nahm S, Weinreb SM, Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar]; b) Balasubramaniam S, Aidhen IS, Synthesis 2008, 3707–3738. [Google Scholar]; c) Senatore R, Ielo L, Monticelli S, Castoldi L, Pace V, Synthesis 2019, 51, 2792–2808. [Google Scholar]; d) Martín R, Romea P, Tey C, Urpí F, Vilarrasa J, Synlett 1997, 1414–1416. [Google Scholar]; e) Kurosu M, Kishi Y, Tetrahedron Lett. 1998, 39, 4793–4796. [Google Scholar]
- [3] a).Tokuyama H, Yokoshima S, Yamashita T, Fukuyama T, Tetrahedron Lett. 1998, 39, 3189–3192. [Google Scholar]; b) Hirschbeck V, Gehrtz PH, Fleischer I, Chem. Eur. J 2018, 24, 7092–7107. [DOI] [PubMed] [Google Scholar]; c) Liebeskind LS, Srogl J, J. Am. Chem. Soc 2000, 122, 11260–11261. [Google Scholar]; d) Prokopcová H, Kappe CO, Angew. Chem. Int. Ed 2009, 48, 2276–2286. [DOI] [PubMed] [Google Scholar]
- [4] a).Cooper TWJ, Campbell IB, Macdonald SJF, Angew. Chem., Int. Ed 2010, 49, 8082–8091. [DOI] [PubMed] [Google Scholar]; b) Roughley SD, Jordan AM, J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]; c) Brown DG, Boström J, J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]; d) Schneider N, Lowe DM, Sayle RA, Tarselli MA, Landrum GA, J. Med. Chem 2016, 59, 4385–4402. [DOI] [PubMed] [Google Scholar]; e) b). Boström J, Brown DG, Young RJ, Keserü GM, Nat. Rev 2018, 17, 709–727. [DOI] [PubMed] [Google Scholar]
- [5].For alkyl-X: X = [ZnX], 443 compounds; X = [B], 4225; X = I, 8621; X = Br, 193284; X = CO2H (1°, 2° and 3°), 1385384; X = NH2 (1° and 2°), 1237920; Data from eMolecules Database accessed via REAXYS (April 8, 2019).
- [6].Yu C-G, Matsuo Y, Org. Lett, 2020, 22, 950–955. [DOI] [PubMed] [Google Scholar]
- [7] a).Goldfogel MJ, Huang L, Weix DJ, in Nickel Catalysis in Organic Synthesis (Ed.: Ogoshi S), Wiley-VCH, Weinheim, 2020, pp. 183–222. [Google Scholar]; b) Weix DJ, Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Knappke CEI, Grupe S, Gärtner D, Corpet M, Gosmini C, Jacobi von Wangelin A, Chem. Eur. J 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]; d) Gu J, Wang X, Xue W, Gong H, Org. Chem. Front 2015, 2, 1411–1421. [Google Scholar]; e) Richmond E, Moran J, Synthesis 2018, 50, 499–513. [Google Scholar]; f) Tasker SZ, Standley EA, Jamison TF, Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8] a).Sato T, Naruse K, Enokiya M, Fujisawa T, Chem. Lett 1981, 10, 1135–1138. [Google Scholar]; b) He J, Song P, Xu X, Zhu S, Wang Y, ACS Catal. 2019, 9, 3253–3259. [Google Scholar]; c) Onaka M, Matsuoka Y, Mukaiyama T, Chem. Lett 1981, 10, 531 – 534. [Google Scholar]; d) Wotal AC, Weix DJ, Org. Lett 2012, 14, 1476 – 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yin H, Zhao C, You H, Lin K, Gong H, Chem. Comm 2012, 48, 7034 – 7036. [DOI] [PubMed] [Google Scholar]; f) Cherney AH, Kadunce NT, Reisman SE, J. Am. Chem. Soc 2013, 135, 7442 – 7445. [DOI] [PubMed] [Google Scholar]; g) Ai Y, Ye N, Wang Q, Yahata K, Kishi Y, Angew. Chem. Int. Ed 2017, 56, 10791 – 10795. [DOI] [PubMed] [Google Scholar]; h) Kumar VP, Babu VS, Yahata K, Kishi Y, Org. Lett 2017, 19, 2766 – 2769. [DOI] [PubMed] [Google Scholar]; i) Zhao C, Jia X, Wang X, Gong H, J. Am. Chem. Soc, 2014, 136, 17645–17651. [DOI] [PubMed] [Google Scholar]; j) Jia X, Zhang X, Qian Q, Gong H, Chem. Comm 2015, 51, 10302–10305. [DOI] [PubMed] [Google Scholar]
- [9] a).Ni S, Muñoz Padial N, Kingston C, Vantourout JC, Schmitt DC, Edwards JT, Kruszyk M, Merchant RR, Mykhailiuk PK, Sanchez B, Yang S, Perry M, Gallego GM, Mousseau JJ, Collins MR, Cherney RJ, Lebed PS, Chen JS, Qin T, Baran PS, J. Am. Chem. Soc 2019, 141, 6726–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang J, Cary BP, Beyer PD, Gellman SH, Weix DJ, Angew. Chem., Int. Ed 2019, 58, 12081–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For representative deaminative cross-couplings under nickel catalysis and photoinduced conditions, see: a) Basch CH, Liao J, Xu J, Piane JJ, Watson MP, J. Am. Chem. Soc 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Klauck FJR, James MJ, Glorius F, Angew. Chem., Int. Ed 2017, 56, 12336–12339. [DOI] [PubMed] [Google Scholar]; c) Hu J, Wang G, Li S, Shi Z, Angew. Chem., Int. Ed 2018, 57, 15227–15231. [DOI] [PubMed] [Google Scholar]; d) Ociepa M, Turkowska J, Gryko D, ACS Catal. 2018, 8, 11362–11367. [Google Scholar]; e) Wu J, He L, Noble A, Aggarwal VK, J. Am. Chem. Soc 2018, 140, 10700–10704. [DOI] [PubMed] [Google Scholar]; f) James MJ, Strieth-Kalthoff F, Sandfort F, Klauck FJR, Wagener F, Glorius F, Chem. Eur. J 2019, 25, 8240–8244. [DOI] [PubMed] [Google Scholar]; g) Jiang X, Zhang MM, Xiong W, Lu LQ, Xiao WJ, Angew. Chem., Int. Ed 2019, 58, 2402–2406. [DOI] [PubMed] [Google Scholar]; h) Klauck FJR, Yoon H, James MJ, Lautens M, Glorius F, ACS Catal. 2019, 9, 236–241. [Google Scholar]; i) Plunkett S, Basch CH, Santana SO, Watson MP, J. Am. Chem. Soc 2019, 141, 2257–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Sun SZ, Romano C, Martin R, J. Am. Chem. Soc 2019, 141, 16197–16201. [DOI] [PubMed] [Google Scholar]; k) Wu J, Grant PS, Li X, Noble A, Aggarwal VK, Angew. Chem., Int. Ed 2019, 58, 5697–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For reductive cross-electrophile couplings of alkyl pyridinium salts with aryl electrophiles, see: a) Liao J, Basch CH, Hoerrner ME, Talley MR, Boscoe BP, Tucker JW, Garnsey MR, Watson MP, Org. Lett 2019, 21, 2941–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Martin-Montero R, Yatham VR, Yin H, Davies J, Martin R, Org. Lett 2019, 21, 2947–2951. [DOI] [PubMed] [Google Scholar]; c) Yi J, Badir SO, Kammer LM, Ribagorda M, Molander GA, Org. Lett 2019, 21, 3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yue H, Zhu C, Shen L, Geng Q, Hock KJ, Yuan T, Cavallo L, Rueping M, Chem. Sci 2019, 10, 4430–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ni S, Li CX, Mao Y, Han J, Wang Y, Yan H, Pan Y, Sci. Adv 2019, 5, DOI 10.1126/sciadv.aaw9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Carbonylative methods are also known, although most require a pre-functionalized organometallic reagent. Carbonylative cross-electrophile ketone syntheses are still relatively underdeveloped; see: a) Oçafrain M, Devaud M, Troupel M, Périchon J, J. Chem. Soc., Chem. Commun 1995, 2331–2332; [Google Scholar]; b) Dolhem E, Oçafrain M, Nédélec JY, Troupel M, Tetrahedron 1997, 53, 17089–17096; [Google Scholar]; c) Oçafrain M, Dolhem E, Nédélec J, Troupel M, J. Organomet. Chem 1998, 571, 37–42; [Google Scholar]; d) Oçafrain M, Devaud M, Nédélec JY, Troupel M, J. Organomet. Chem 1998, 560, 103–107; [Google Scholar]; d) Dolhem E, Barhdadi R, Folest J, Nédélec J, Troupel M, Tetrahedron 2001, 57, 525–529; [Google Scholar]; e) Wotal AC, Ribson RD, Weix DJ, Organometallics 2014, 33, 5874–5881; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Shi R, Hu X, Angew. Chem., Int. Ed 2019, 58, 7454–7458; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131,7532 –7536. [Google Scholar]
- [13].Ketones have been synthesized using aldehydes as starting materials: a) Ishii T, Kakeno Y, Nagao K, Ohmiya H, J. Am. Chem. Soc, 2019, 141, 45. [DOI] [PubMed] [Google Scholar]; b) Huang YC, Majumdar KK, Cheng CH, J. Org. Chem 2002, 67, 1682–1684. [DOI] [PubMed] [Google Scholar]; c) Vandavasi JK, Hua X, Ben Halima H, Newman SG, Angew. Chem. Int. Ed 2017, 56, 15441–15445. [DOI] [PubMed] [Google Scholar]; d) Ruan J, Saidi O, Iggo JA, Xiao J, J. Am. Chem. Soc 2008, 130, 10510–10511. [DOI] [PubMed] [Google Scholar]; e) Ko S, Kang B, Chang S, Angew. Chem. Int. Ed 2005, 44, 455–457. [DOI] [PubMed] [Google Scholar]; f) Pucheault M, Darses S, Genet JP, J. Am. Chem. Soc 2004, 126, 15356–15357. [DOI] [PubMed] [Google Scholar]
- [14].Biswas S, Weix DJ, J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].The reduction potentials in Scheme 1 are from our own measurements and the literature. See Supporting Information, reference [11b], and Grimshaw J, Moore S, Trocha-Grimshaw J. Acta Chem. Scan. B 1983, 37, 485–489 [Google Scholar]
- [16].For selected references, see: a) Zhang Y, Rovis T, J. Am. Chem. Soc 2004, 126, 15964–15965. [DOI] [PubMed] [Google Scholar]; b) Ogiwara Y, Maegawa Y, Sakino D, Sakai N, Chem. Lett 2016, 45, 790–792. [Google Scholar]; c) Ogiwara Y, Sakino D, Sakurai Y, Sakai N, Eur. J. Org. Chem 2017, 2017, 4324–4327. [Google Scholar]; d) Keaveney ST, Schoenebeck F, Angew. Chem. Int. Ed 2018, 57, 4073–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Malapit CA, Bour JR, Brigham CE, Sanford MS, Nature 2018, 563, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Malapit CA, Bour JR, Laursen SR, Sanford MS, J. Am. Chem. Soc 2019, 141, 17322–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Pan F-F, Guo P, Li C-L, Su P, Shu X-Z, Org. Lett 2019, 21, 3701–3705. [DOI] [PubMed] [Google Scholar]; h) Ogiwara Y, Sakai N, Angew. Chem. Int. Ed 2020, 59, 574–594. [DOI] [PubMed] [Google Scholar]
- [17] a).Weires NA, Baker EL, Garg NK, Nat. Chem 2016, 8, 75–79. [DOI] [PubMed] [Google Scholar]; b) Boit TB, Weires NA, Kim J, Garg NK, ACS Catal. 2018, 8, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Meng G, Szostak M, Org. Lett 2015, 17, 4364–4367. [DOI] [PubMed] [Google Scholar]; d) Dardir AH, Melvin PR, Davis RM, Hazari N, Mohadjer Beromi M, J. Org. Chem 2018, 83, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chatupheeraphat A, Liao H-H, Srimontree W, Guo L, Minenkov Y, Poater A, Cavallo L, Rueping M, J. Am. Chem. Soc 2018, 140, 3724–3735. [DOI] [PubMed] [Google Scholar]; f) Shi W, Zou G, Molecules 2018, 23, 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Meng G, Szostak M, Org. Lett 2018, 20, 6789–6793. [DOI] [PubMed] [Google Scholar]; h) Masson-Makdissi J, Vandavasi JK, Newman SG, Org. Lett 2018, 20, 4094–4098. [DOI] [PubMed] [Google Scholar]; i) Liu X, Hsiao C-C, Guo L, Rueping M, Org. Lett 2018, 20, 2976–2979. [DOI] [PubMed] [Google Scholar]
- [18].Huang L, Ackerman LKG, Kang K, Parsons AM, Weix DJ, J. Am. Chem. Soc 2019, 141, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
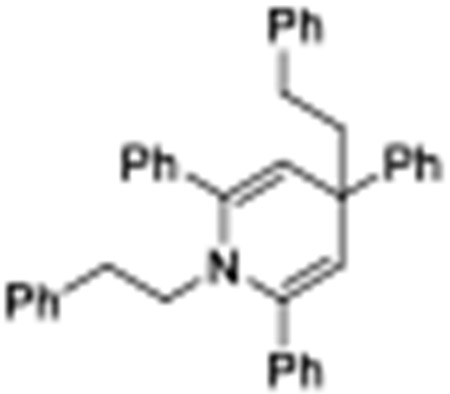
[19].We have observed significant amount of dihydropyridine byproduct from crude NMR. The structure is consistent with our previous observed byproduct (see ref. 11a), and the structure was assigned to be:
- [20].Tertiary carbinamines cannot be converted into 2,4,6-triphenylpyridinium salts, except for cyclopropyl examples, presumably due to steric hindrance between the tertiary alkyl group and 2,6-phenyl groups.
- [21].At this time, reference [6] contains a more general, higher yielding route to aryl alkyl ketones from aryl carboxylic acids and amines.
- [22].A search of the REAXYS database for HO2CCH2OR found 22844 substances with pharmacological data. This includes pesticides such as 2,4-dichlorophenoxyacetic acid (2,4-D) and fluroxypyr as well as drugs such as treprostinil, aceclofenac, cefixime, cetirizine, and rifamycin B.
- [23] a).Nakajima K, Nojima S, Nishibayashi Y, Angew. Chem., Int. Ed 2016, 55, 14106–14110. [DOI] [PubMed] [Google Scholar]; b) Badir SO, Dumoulin A, Matsui JK, Molander GA, Angew. Chem. Int. Ed 2018, 57, 6610–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gandolfo E, Tang X, Raha Roy S, Melchiorre P, Angew. Chem. Int. Ed 2019, 58, 16854–16858. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang X, Macmillan DWC, J. Am. Chem. Soc 2017, 139, 11353–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Joe CL, Doyle AG, Angew. Chem. Int. Ed 2016, 55, 4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24] a).Li H, Breen CP, Seo H, Jamison TF, Fang Y-Q, Bio MM, Org. Lett 2018, 20, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Perkins RJ, Hughes AJ, Weix DJ, Hansen EC, Org. Process Res. Dev 2019, 23, 1746–1751. [Google Scholar]; c) Jiao K-J, Liu D, Ma H-X, Qiu H, Fang P, Mei T-S, Angew. Chem. Int. Ed 2019, DOI 10.1002/anie.201912753 [DOI] [Google Scholar]
- [25].Liu J, Lei C, Gong H, Sci. China. Chem, 2019, 62, 1492–1496. [Google Scholar]
- [26].Alkyl-NH2: 1.26 million in Beilstein Database, 1.40 million listed in eMolecules commercial availability database. Alkyl-Br: 308 thousand in Beilstein, 201 thousand in eMolecules. See also an analysis of substrate availability in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.