

Abstract
Frontotemporal dementias (FTDs) encompass several disorders commonly characterized by progressive frontotemporal lobar degeneration and dementia. Pathologically, TDP-43, FUS, dipeptide repeats, and tau constitute the protein aggregates in FTD, which in turn coincide with heterogeneity in clinical variants. The underlying molecular etiology explaining the formation of each type of protein aggregate remains unclear; however, dysregulated RNA metabolism rises as a common pathogenic factor. Alongside with TDP-43 and FUS, which bind to and regulate RNA dynamics, emerging data suggest that tau may also regulate RNA metabolism and translation. The complex mechanisms that drive translational selectivity in turn regulate the broad clinical presentation of FTDs. Here, we focus on the enigmatic relationship between tau and RNA and review the mechanisms of tau-mediated dysregulation of RNA in tauopathies such as FTD.
Keywords: tau, frontotemporal dementia, RNA, ribosome, translation
Introduction
Frontotemporal dementia (FTD) is an umbrella term for several neurological syndromes that present progressive executive, behavioral, or language dysfunction. Degeneration of the frontotemporal lobe underlies the disease presentation. This cluster of degenerative diseases, collectively and pathologically termed frontal temporal lobar degeneration (FTLD), is predominantly characterized by the deposition of pathological protein inclusions of TDP-43, FUS, dipeptide repeats (which will not be reviewed in this article), and tau (Cairns et al., 2007; Irwin et al., 2015; Mackenzie and Neumann, 2016). Since TDP-43 and FUS pathology are present in a majority of patients with FTD (approximately 50% and 5%, respectively) and in nearly every patient with amyotrophic lateral sclerosis (ALS), a large body of work has focused on understanding how these canonical RNA-binding proteins contribute to RNA metabolism and converge as pathogenic mechanisms in disease [reviewed in(Mackenzie et al., 2010)). However, the role of tau in the FTD etiology is largely suspected to center on tau splicing since many FTD-associated mutations on MAPT, the tau-coding gene, surround exon 10, which is spliced out in 50% of all tau species expressed in the normal adult human brain. The emerging concept of tau serving as a regulator of RNA metabolism now highlight a potentially common pathogenic mechanism in which tau may promote ribonucleopathy.
Historically, tau is classified as an axonal microtubule-associated protein (MAP) that primarily functions to stabilize microtubule dynamics (Brandt and Lee, 1993; Kadavath et al., 2015). In disease conditions, tau undergoes abundant and unbalanced post-translational modifications such as hyper-phosphorylation, which prevent tau binding to microtubules thereby enabling toxic interactions (Sotiropoulos et al., 2017). Tau was discovered to associate with microtubules the same year it was found to associate with RNA, in 1975, and yet the research trajectory of these two interactions have been considerably different (Bryan et al., 1975; Weingarten et al., 1975). Here, we review the growing evidence over the past four decades that tau regulates RNA metabolism, and when dysregulated, this function contributes to disease pathogenesis in FTD and other tauopathies. We suggest that RNA dysfunction links the major proteinopathy variants of FTD, acting as a common mechanism of disease and an important therapeutic target.
Tau associates with RNA: from in vitro solution to stress granules
Tau is a multi-domain protein with considerable chemical complexity, allowing it to associate with many biological species. While overall hydrophilic and basic, tau is asymmetrically charged. While the N terminus is acidic, the central microtubule binding and proline rich regions are basic, and the extreme C-terminus is neutral. Initial work by Bryan, Nagle, and Doegnes in 1975 revealed polyanions such as RNA compete for tau binding with tubulin, inhibiting microtubule assembly (Bryan et al., 1975). This competition with tubulin was not sequence specific and occurred with purified RNA from cell extract and synthetic RNA polynucleotides. This suggests that tau indiscriminately interacts with RNA, potentially due to electrostatic interaction between the negatively charged RNA phosphate backbone and the basic region of tau. Nearly a decade later in 1984, Schröder et. al. discovered that tau isolated from aged bovine brain bound to RNA at nearly half the rate of tau isolated from young brain; moreover, tau’s RNA-binding capacity was altered by its phosphorylation by cAMP-dependent kinase (also known as PKA) (Schroder et al., 1984). PKA phosphorylates tau at multiple sites, many of which are in regions that appear hyper-phosphorylated in disease (Oliveira et al., 2017). This early evidence that tau phosphorylation modifies the tau-RNA interaction dates even before the discovery that tau regulates microtubule assembly in a phosphorylation-dependent manner in the early 1990s (Drechsel et al., 1992).
Aggregated, hyperphosphorylated, and insoluble tau was discovered as the principal constituent of neurofibrillary tangles in Alzheimer’s disease (AD) in the mid-1980s (Brion and Flament-Durand, 1995; Grundke-Iqbal et al., 1986). In the mid-1990s, multiple groups reported the aggregation of tau into paired helical filaments in vitro after incubation with physiological or sub-physiological concentrations of total and tRNA (Hasegawa et al., 1997; Kampers et al., 1996). Importantly, a truncated tau protein containing just three microtubule-binding regions (MTBR) was sufficient to aggregate when co-incubated with tRNA. Decreasing the number of repeats slowed aggregation, and full-length tau with all MTBRs removed did not form filaments with tRNA co-incubation (Kampers et al., 1996). This represents the first attempt to delineate the putative tau-RNA binding region. Soon thereafter, the Trojanowski group identified sequestered RNA in stained neurofibrillary tangles of AD brains (Ginsberg et al., 1997) and other tauopathies (Ginsberg et al., 1997; Ginsberg et al., 1998), detailing in vivo evidence that aggregated tau may associate with RNA. Co-incubation of tau with RNA in vitro was later found to increase tau phosphorylation by GSK3-β and other known tau kinases (Hasegawa et al., 1997), adding to the emerging theory that tau hyper-phosphorylation and other disease conditions are either induced by or causative for aberrant tau-RNA interactions.
Despite studies using truncated and full-length tau to investigate RNA association, the specific site(s) of interaction were not known. In 2006, Wang et al. systematically assessed how various truncated forms of tau interacted with RNA based on gel shift assays (Wang et al., 2006). They found truncated tau constructs containing the proline-rich region or the MTBR associate with RNA and that removal of these domains diminished this interaction, thus supporting the original claim by Schröder et al. that tau has two sites capable of RNA binding (Schroder et al., 1984). Additionally, they reported tau association with rRNA and purified mRNA, which could explain potential nuclear and ribosomal interactions of tau (discussed later) (Wang et al., 2006). Over the next decade, the interaction of tau with dozens of canonical RNA-binding proteins (RBPs) and ribosomal proteins had been reported (Wolozin, 2012), suggesting that tau participates in RNA granule metabolism (discussed later), though the selectivity of which sequences of RNA associate with tau remained undiscerned.
In 2017, Zhang et al., reported the first high-throughput screening of tau-RNA binding utilizing PAR-CLIP, a method of UV cross-linking tau to RNA with subsequent immunoprecipitation for downstream next-generation sequencing (Wolozin, 2012). Zhang and others found that in both human embryonic kidney cells (HEKs) and human-induced pluripotent stem cell (hIPSC)-derived neurons, wild-type and FTD-associated P301L mutant tau bound primarily to tRNA. Interestingly, tau did not equally bind to all tRNAs, enriching the most with tRNAArg. The finding that tau predominantly, though not exclusively, binds tRNA in cells contrasts with the various studies reporting in vitro binding of tau and other forms of RNA. Further studies utilizing CLIP sequencing techniques from in vivo or human tauopathy brain tissue are needed to confirm the selectivity of tau-RNA interactions and their alteration in disease. However, if true, this selectivity for a subset of tRNAs with maximal binding in the anti-codon region over all other RNA species suggests biological specificity beyond simple electrostatic interactions outlined above. Despite continued investigations into the types of RNA that induce tau aggregation (Banerjee et al., 2020), these results by Zhang et al. suggest in vitro experiments conducted using tau and RNA in solution may reveal associations not reflect functional or biological relevance.
Tau also associates with many RNA-binding proteins which regulate RNA metabolism (reviewed in (Cruz et al., 2019)). Spliceosomal RNA-binding proteins such as U1 small nuclear ribonucleoproteins (snRNPs) are dysregulated and undergo aggregation in tauopathies (Bai et al., 2013; Hales et al., 2016; Hales et al., 2014a; Hales et al., 2014b; Johnson et al., 2018). Several groups have shown that these proteins associate with tau or neurofibrillary tangles (Hales et al., 2014b). Recent studies show that tau mediates aberrant splicing defects and neurodegeneration in vivo using models of FTD tauopathy, explained at least in part by tau associating to U1 snRNP or other splicing regulating proteins (Apicco et al., 2019; Hsieh et al., 2019). Whether tau basally alters splicing in humans remains unknown, but these studies suggest a toxic gain of function of tau whereby bona fide splicing regulators become affected due to aberrant tau interactions.
Other studies investigating the link between tau and RNA binding proteins have focused on the liquid-liquid phase separation (LLPS) capacity of tau, whether assisted by polyanionic species such as RNA (Ambadipudi et al., 2017; Lin et al., 2019; Ukmar-Godec et al., 2019; Zhang et al., 2017) or independently (Boyko et al., 2019; Wegmann et al., 2018). The amino acid content of proteins largely drives LLPS, wherein low-complexity domains (such as the proline-rich region of tau, (Ambadipudi et al., 2017) or prion-like domains (such as the MTBR domain of tau, (Ambadipudi et al., 2017; Zhang et al., 2017) facilitate electrostatic assembly that coalesce into phase separated granules [reviewed in (Ukmar-Godec et al., 2020)]. Importantly, both the proline-rich region and the MTBR domain of tau have been reported to bind to RNA (Wang et al., 2006), implying that tau facilitates RNA involvement in granule assembly. Phase separation and protein condensation have been found to be involved in many cellular processes, such as synaptic vesicular release and plasticity (Milovanovic and De Camilli, 2017; Milovanovic et al., 2018; Zeng et al., 2016), nucleolar function (Berry et al., 2015), and even cytoskeletal assembly (Jiang et al., 2015). These findings suggest that tau could (dys)regulate these processes via LLPS interactions, though this has yet to be fully evaluated.
Similar to TDP-43 (Chen et al., 2019; Huang et al., 2013; Zacco et al., 2019) and FUS (Bentmann et al., 2012; Patel et al., 2015), tau interacts with many RNA-binding proteins involved in the formation of ribonucleoprotein granules, such as stress granules (SGs) (Hsieh et al., 2019; Maziuk et al., 2018). These are cytoplasmic complexes of RNA and proteins that assemble as part of the translational stress response, which occurs as a consequence of endoplasmic reticulum (ER) stress and activation of the unfolded protein response (discussed later). During stress granule formation, the actively translating ribosome is stalled and sequestered by RNA-binding proteins until the stress subsides; however, in neurodegenerative disorders, SGs become stable and largely irreversible (reviewed in (Wolozin and Ivanov, 2019)).
Consistent with the findings that translation is dysregulated in tauopathy (discussed later), SGs appear in many tauopathies (reviewed in (Cruz et al., 2019)). Work by the Wolozin group established that interactions between T-cell restricted intracellular antigen-1 (TIA1), a crucial SG protein, and tau are an early and pathogenic contributor in tauopathies (Apicco et al., 2018; Jiang et al., 2019; Maziuk et al., 2018; Vanderweyde et al., 2016; Vanderweyde et al., 2012). The levels and propagation of toxic tau oligomers are mitigated when TIA1 expression is reduced, supporting the findings that phase separated condensates facilitate toxic tau oligomerization (Wegmann et al.). Other studies have similarly found that positive modulators of TIA1/tau stress granule assembly facilitate progression of tau pathology (Piatnitskaia et al., 2019). However, the role of other RNA-binding proteins in promoting tauopathy remains unclear. Recent work by the Kraemer group discovered that a poly(A)-RNA binding protein, mammalian suppressor of tauopathy 2 (MSUT2), enhances hyper-phosphorylated tau accumulation and aggregation through an unknown mechanism (Wheeler et al., 2019). Interestingly, MSUT2 binds and functions in opposition to another nuclear poly(A)-RNA binding protein, PABPN1, which reduces tau pathology. However, the authors found the interplay between these two proteins and tau pathology was unrelated to physico-chemical characteristics of the poly(A)-RNA bound in complex. MSUT2/PABPN1 may instead regulate tau pathology by promoting/negating SG formation or microtubule assembly. Together, these studies highlight the role of RNA-binding proteins and SGs as key pathogenic mediators of tauopathy and as potential targets for therapeutic intervention.
Evidence for translational impairment in tauopathy
The earliest studies investigating defects in protein synthesis in tauopathy, namely AD, largely considered the phenomenon of diminished RNA translation as a product of nucleolar and RNA alterations. In the 1970s and 1980s, several groups reported decreased nucleolar volume and RNA content in AD (Dayan and Ball; Doebler et al., 1987; Guillemette et al., 1986; Mann et al., 1981a; Mann et al., 1981b; Mann and Sinclair, 1978; Mann et al., 1977; Neary et al., 1986; Sajdel-Sulkowska and Marotta, 1984) reviewed in (Marotta et al., 1986). As protein synthesis depends on proper nucleolar activity as the site of ribosome biogenesis and rRNA processing, this implied altered ribosomal function. Some of the earliest and critical evidence of RNA translation defects in AD was reported by the Sajdel-Sulkowska & Marotta group in the early 1980s using the incorporation of radiolabeled methionine in rabbit reticulocyte lysate in vitro as a proxy for translation (Marotta et al., 1981). They reported that polyadenylated mRNA isolated from AD brain was translated less and qualitatively produced fewer proteins relative to non-demented control brain (Sajdel-Sulkowska and Marotta, 1984), reviewed in (Marotta, 1989). Bustany et al. in 1983 reported the first in vivo evidence of altered translation in tauopathy by measuring the uptake of radiolabeled methionine by patients using positron emission tomography, revealing a >50% decrease in uptake in patients with AD. These early results strongly suggested translational impairments in tauopathy, though no direct evidence of ribosomal dysfunction had yet been evaluated.
In 1989, Langstrom et al. isolated polysomes from non-demented control and AD brains and assessed polysomal translational capacity using the in vitro translation assay developed by the Marotta group (Langstrom et al., 1989). Strikingly, not only were less polysomes found in AD brain compared to control, but polysomes taken from the frontal cortex of AD brains had 50% less translational capacity relative to control brain polysomes. This translational difference was not found in isolated polysomes from the cerebellum, suggesting that this apparent ribosomal dysfunction occurs during disease progression and specifically in areas affected by pathology. Several consequent studies expanded on Langstrom et al.’s findings and identified considerable dysregulation of pathways responsible for governing translation in tauopathies (An et al., 2003; Ferrer, 2002; Hoozemans et al., 2005; Langstrom et al., 1989; Li et al., 2004), reviewed in (Ohno, 2014). Though the underlying etiology behind these changes is unclear, the results suggest tauopathies involve complex molecular cascades that converge on the ribosome rather than just a direct effect of a single-factor regulatory system.
Consistent with this theory, increased levels of oxidized rRNA was linked to tauopathy (Nunomura et al., 1999). by directly impairing ribosomal activity, which in turn impaired protein synthesis (Willi et al., 2018). Several reports show that RNA oxidation increases while translational output decreases in the early, mild-cognitive stages of AD (Ding et al.; Ding et al.; Ding et al.; Honda et al., 2005). Interestingly, decreased levels of rRNA and tRNA in regions vulnerable to AD pathology, but not in the cerebellum have been reported, matching the results by Langstrom, et al. and other studies (Pietrzak et al., 2011). Further investigation into alterations of rRNA and ribosomal machinery in AD revealed disease-stage and region-specific decreases of transcripts coding for nucleolar function, rRNA processing, and ribosomal machinery (Hernandez-Ortega et al., 2016). The levels of some of these transcripts increase in AD, possibly suggesting potential compensatory mechanisms for decreased translation (discussed in the next section) or the massive complexity of ribosomal alterations in disease. Importantly, tau regulates nucleolar function (Loomis et al.), reviewed in (Bukar Maina et al., 2016), by remodeling heterochromatin, decreasing rRNA transcription (Maina et al., 2018b), and dysregulating nucleocytosolic transport (Eftekharzadeh et al., 2018), which can also significantly affect the translational landscape.
Studies on alternative mechanisms of translational repression in tauopathy reported an early increase in the phosphorylation of eukaryotic initiation factor 2α (eIF2α) and particularly in regions and neurons with accumulating hyper-phosphorylated tau, indicating the selective attenuation of global translation in these neurons (Hernandez-Ortega et al., 2016; Hoozemans et al.; Lanzillotta et al.; Nijholt et al.; Stutzbach et al.), reviewed in (Bell et al.; Hoozemans and Scheper, 2012). Tau induces eIF2α phosphorylation via stimulating ER stress, activating the unfolded protein response (UPR) and thereby inducing PKR-like endoplasmic reticulum kinase (PERK) to phosphorylate eIF2α (Abisambra et al.; Hoozemans and Scheper, 2012). Translational repression of eIF2α induces SG formation, potentially stimulating tau pathology progression as discussed earlier. Small molecule inhibition of PERK rescued protein synthesis and brain atrophy in various neurodegeneration models (Halliday et al.; Radford et al.), suggesting this pathway is a viable therapeutic target (Halliday et al., 2017). However, the activation of the UPR and the resulting eIF2α phosphorylation in mouse models of tauopathy is contested (Hashimoto and Saido, 2018; Pitera et al., 2019), though consistent reports of UPR activity and eIF2α phosphorylation in human studies suggest these findings may be model artifacts and not reflective of true disease.
In this section, we reviewed many correlational studies of translational dysregulation using AD tissue. It is important to recognize confounding effects of mixed protein pathology found in disease. Though considerably less is known about how amyloid may affect protein synthesis, it is important to note that studies on amyloid-mediated translational changes have reported similar effects as with tau (Maina et al., 2018a). This could be due to strong evidence supporting the amyloid cascade hypothesis, in which formation of amyloid pathology elicits toxic tau events culminating in translational dysfunction. While many of the studies reviewed here evaluate ribosomal function in relation to tauopathies at the disease level (such as in primary tauopathy tissue or tau transgenic mice) or at the cellular level (by correlating findings to same-cell tau pathology), considerable studies have investigated the specific role of tau in regulating translation.
Tau-ribosome association: direct interactions
Shortly after the discovery that tau complexes with RNA, tau was found to associate with ribosomes. Early reports of an in vitro ribosome-tau complex identified that isolated ribosomal fractions from Xenopus oocytes could sequester tau, inhibiting microtubule assembly like RNA (Jessus et al., 1984). Ultrastructural studies by the Papasozomenos and Binder labs in the 1980s and 1990s reported the first evidence of tau-polysome complexes throughout the progression of tangles and AD stage in human brains (Nelson and Saper). In 1988, paired helical filaments were found associated with perinuclear polysomes (Metuzals et al., 1988), suggesting that tau pathologically impacts nuclear function (although it remains to be established whether this relationship is a consequence of the disease process) (reviewed in (Bukar Maina et al.). The first report of tau’s association with polysomes in FTLD was reported in 2002, suggesting tau-ribosome complexing might be a fundamental pathogenic mechanism linking tauopathies (Piao et al., 2002). These ultrastructural studies provided critical evidence that tau can regulate translation by association with ribosomes in vivo and not an artefact found in solution.
Importantly, Nelson, Saper and others published a series of ultrastructural studies in the 1990s that revealed the association between tau and polysomes in dendritic branching points (Nelson et al.; Nelson and Saper). During this time, tau was identified as an axonal protein that mislocalizes to the somatodendritic domain during the progression of tau hyper-phosphorylation and aggregation in disease (Bancher et al., 1989; Binder et al., 1985; Ihara, 1988; Papasozomenos and Binder; Peng et al., 1986). The discovery of tau-polysome complexes in dendrites was originally thought to be a byproduct of local tau translation (Nelson et al.), which seemed reasonable given that tau mRNA was also present in the complexes (Kosik et al., 1989). However, Nelson and Saper later revised this hypothesis to include the potential for tau in “subserving a role in dendritic reorganization” in disease conditions (Nelson and Saper). Only recently, studies confirmed both hypotheses: not only is tau locally translated in somatodendritic compartments (Kobayashi et al., 2017; Li and Gotz, 2017), but tau also regulates the translation of proteins necessary for dendritic maintenance and synaptic plasticity (Evans et al., 2019a; Koren et al.).
The earliest studies of the tau interactome revealed consistent interactions between ribosomes and tau. In 2012, the Gestwicki group published one of the first studies utilizing tau immunoprecipitation-coupled mass spectrometry (IP-MS) in HeLa cells stably expressing a tagged variant of tau (Thompson et al., 2012). They showed that tau associates with over 50 ribosomal proteins and dozens of RNA-binding proteins, and small molecule induction of tau degradation significantly increases association with proteins of these two classes. Three years later, Gunawardana et al., expanded on the tau-associated proteome via IP-MS of SH-SY5Y human neuroblastoma cells stably expressing tagged wild-type tau (Gunawardana et al., 2015). Tau was found to associate with over 50 ribosomal proteins, co-enriching for ribosomal proteins and translational machinery more than any other protein category relative to GFP-only IP-MS controls. Treatment of cell lysate with RNAse to destabilize ribosomal structure decreased the tau-ribosome association. Interestingly, some ribosomal proteins such as rpL10a associated more strongly with tau following RNAse treatment. Considering the developing theory of on-site ribosomal remodeling using locally synthesized rpL10a and other proteins (Shi et al., 2017; Shigeoka et al., 2019), it will be crucial to establish whether tau alters ribosome specialization and remodeling. Lastly, ribosomal protein association was found to be dependent on the domains of tau present in the cell: tau containing the N-terminal projection domain and the proline-rich domain associated more strongly than tau containing the MTBR domain and the C-terminal. This contrasts with earlier studies conducted in solution which found tau bound to RNA at the proline-rich region and the MBTR domain, suggesting separate locations of RNA and ribosome association with tau (Gunawardana et al., 2015).
Work by our lab in the mid-2010s assessed the tau-ribosome interaction network in human tauopathy brains (Meier et al.; Meier et al.). Since tau was consistently found associated with ribosomes on the endoplasmic reticulum (ER) membrane in ultrastructural studies (Abisambra et al.), we sub-cellularly fractionated human AD and age-matched, non-demented control brains to isolate ER microsomes containing ribosomes for tau IP-MS. Semi-quantitative proteomic results showed that tau associates with more ribosomal proteins in AD; importantly, some ribosomal proteins that associate with tau in control brains were distinct when compared to AD samples (Meier et al.). A more recent quantitative study using tau IP-MS from human brain supported these findings, revealing tau has significantly increased association with nearly 50 translational machinery proteins in AD relative to control (Hsieh et al., 2019). Interestingly, these changes in co-immunoprecipitation did not always correspond to differences in protein level, suggesting tau undergoes selective association with ribosomes in tauopathy (Meier et al.) In vitro translation of a GFP reporter plasmid co-incubated with tau oligomers and HEK cells stably expressing tau variants showed tau directly impairs RNA translation, suggesting pathogenic tau can impair ribosomal function (Meier et al.). We reported the unusual finding that PSD95 transcript levels are increased, but protein levels decreased in primary neurons of an FTD tauopathy mouse model, suggesting that tau might regulate the translation of synaptic proteins (Meier et al.). However, it was yet unclear whether tau could directly affect the selectivity of translational output of ribosomes or whether this was a more indirect effect.
In 2019, the Abisambra lab and the Götz group independently published similar findings of tau-mediated translational dysregulation in vivo. Both groups used different methods of assessing translation and mouse models of FTD tauopathy (Evans et al.). The Abisambra group used IP-MS of puromycin, an antibiotic and structural analog of tRNA that tags nascent proteins, Evans et al. labeled nascent proteins with a non-canonical amino acid that readily binds to biotin for affinity purification (reviewed in (Koren et al., 2019a)). Both studies reported that tau accumulation inversely correlates with translational output in vivo. These surprising and independently corroborated findings underscore the importance and consistent finding that tau interferes with translation under pathological conditions. While the translation of some proteins was upregulated (Koren et al.), it is unclear whether this reflects a direct effect of tau on translation, changes in the transcriptome, or artifacts of transgenic tau over-expression. Our earlier work described the association of pathological, oligomeric tau in human AD brain with ribosomal protein S6 (rpS6) (Meier et al.), a protein involved in regulating the translation of a class of RNA transcripts with a 5’ terminal oligopyrimidine (5’TOP) motif (Meyuhas, 2015; Nygard and Nilsson, 1990; Roux et al., 2007). Since most translation machinery proteins have a 5’TOP motif (Yamashita et al., 2008), aberrant tau-rpS6 interactions could underpin our discovery that the translation of ribosomal proteins is down-regulated even though their corresponding transcript levels are unchanged (Koren et al.). Work in human AD brain revealed that tau accumulation coincides with less rpS6 phosphorylation, which the active form; however, this may independently or in combination be the result of altered rpS6 kinase activity (Mueed et al., 2018). We found ribosomal protein levels were significantly decreased, while the corresponding mRNA transcripts for those proteins were increased over five-fold. These data confirmed that down-regulated translation is consistent with decreased rpS6 activity (Koren et al.). Furthermore, rpS6 associated with tau to a greater extent in AD brain compared to age-matched, non-demented controls despite having less total level. This finding was further corroborated by another study using tau IP-MS from human AD brain (Hsieh et al., 2019). This suggests a working model of tau-mediated translational dysregulation wherein tau selectively sequesters rpS6, impairs the translation of ribosomal proteins, and thereby decreases overall translation. However, other mechanisms beyond just rpS6-tau interaction could underlie the translational dysregulation in tauopathy, and more studies are needed to investigate the exact role of tau in these cellular processes.
Consistent with our working model, tau diminishes translation in drosophila (Papanikolopoulou et al., 2019). Tau knock-out flies exhibited elevated translation globally including that of ribosomal proteins including rpS6, and re-introducing tau expression impaired protein-synthesis-dependent long-term memory (Papanikolopoulou et al., 2019). A recent report by Ferrari et al. also showed how an aggregation-prone tau truncation that contains only the MTBR increasingly bound to ribosomal proteins including rpS6 as it aggregated (Ferrari et al., 2020). Interestingly, other ribosomal proteins underwent larger shifts in association with tau during aggregation compared to rpS6, suggesting other potential avenues of tau-mediated translational regulation. As translation is crucial for proper cellular metabolism and synaptic plasticity, this provides a clear link between tau-mediated RNA dysfunction and cognitive decline found across tauopathies.
Concluding remarks
As described in this review, there is considerable evidence for the role of tau in regulating RNA metabolism and mediating RNA dysfunction in disease. This is unique to tau given that it lacks a canonical nucleotide recognition motif, which is distinct from other well-established RNA-binding proteins that adopt aberrant structures in disease. Studies over the last 45 years have elucidated tau as having a functional role in the nucleus, regulating ribosomal biogenesis, RNA transcript splicing, and nucleocytoplasmic transport. In the cytoplasm, tau forms condensates with ribonucleoprotein granules and participate in RNA transport and the translational stress response, potentially in active synaptic dendritic spines and within axons. Tau associates with ribosomes and tRNAs, selectively defining the transcripts to be translated. Together, these studies provide insight into the early pathogenesis of tauopathies beyond the canonical role of tau in regulating microtubule assembly, often with exciting potential for therapeutic interventions for further study (Figure 1). As many of these pathways are similarly dysregulated by TDP-43 and FUS in FTD, tau should be considered a as a mediator of RNA dysfunction in FTD and other tauopathies.
Fig. 1.
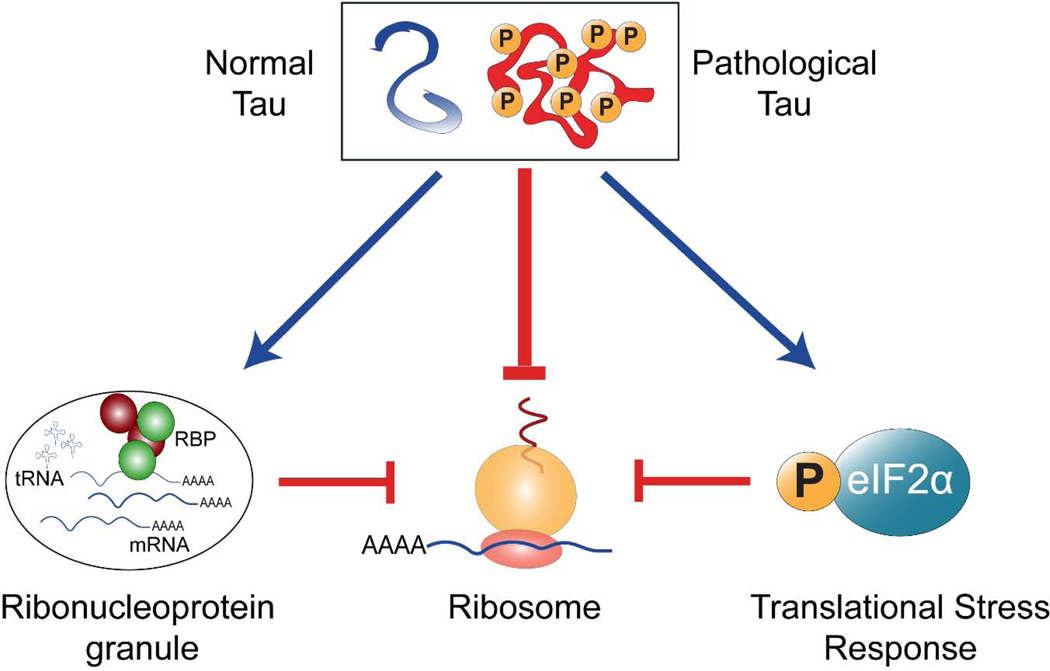
Schematic representation of potential pathological contacts between tau and translation. Evidence suggest that tau directly interacts with various forms of RNA, it can directly impair ribosome function and localization, and it indirectly represses translation by activating the integrated stress response. In doing so, RNA stability and protein synthesis are altered throughout cellular organelles. Finally, sustained changes in translation directly impair cognition. As such, these tau-translation contact points might represent new perspectives on the mechanisms of tau toxicity in FTD-tau and other tauopathies
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abisambra JF, et al. , 2013. Tau Accumulation Activates the Unfolded Protein Response by Impairing Endoplasmic Reticulum-Associated Degradation. J Neurosci. 33, 9498–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S, et al. , 2017. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun. 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WL, et al. , 2003. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am J Pathol. 163, 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicco DJ, et al. , 2018. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci. 21, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicco DJ, et al. , 2019. Dysregulation of RNA Splicing in Tauopathies. Cell Rep. 29, 4377–4388 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, et al. , 2013. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci U S A. 110, 16562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancher C, et al. , 1989. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 477, 90–9. [DOI] [PubMed] [Google Scholar]
- Banerjee S, et al. , 2020. Tau protein-induced sequestration of the eukaryotic ribosome: Implications in neurodegenerative disease. Sci Rep. 10, 5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MC, et al. , 2016. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr Alzheimer Res. 13, 150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentmann E, et al. , 2012. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 287, 23079–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry J, et al. , 2015. RNA transcription modulates phase transition-driven nuclear body assembly. Proc Natl Acad Sci U S A. 112, E5237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, et al. , 1985. The distribution of tau in the mammalian central nervous system. J Cell Biol. 101, 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyko S, et al. , 2019. Liquid-liquid phase separation of tau protein: The crucial role of electrostatic interactions. J Biol Chem. 294, 11054–11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R, Lee G, 1993. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J Biol Chem. 268, 3414–9. [PubMed] [Google Scholar]
- Brion JP, Flament-Durand J, 1995. Distribution and expression of the alpha-tubulin mRNA in the hippocampus and the temporal cortex in Alzheimer’s disease. Pathol Res Pract. 191, 490–8. [DOI] [PubMed] [Google Scholar]
- Bryan JB, et al. , 1975. Inhibition of tubulin assembly by RNA and other polyanions: evidence for a required protein. Proc Natl Acad Sci U S A. 72, 3570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukar Maina M, et al. , 2016. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules. 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, et al. , 2007. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 114, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, et al. , 2019. RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain. 142, 3753–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz A, et al. , 2019. The Pathophysiology of Tau and Stress Granules in Disease. Adv Exp Med Biol. 1184, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayan AD, Ball MJ, 1973. Histometric observations on the metabolism of tangle-bearing neurons. J Neurol Sci. 19, 433–6. [DOI] [PubMed] [Google Scholar]
- Ding Q, et al. , 2006. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem Res. 31, 705–10. [DOI] [PubMed] [Google Scholar]
- Ding Q, et al. , 2005. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 25, 9171–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, et al. , 2012. Increased 5S rRNA oxidation in Alzheimer’s disease. J Alzheimers Dis. 29, 201–9. [DOI] [PubMed] [Google Scholar]
- Doebler JA, et al. , 1987. Neuronal RNA in relation to neuronal loss and neurofibrillary pathology in the hippocampus in Alzheimer’s disease. J Neuropathol Exp Neurol. 46, 28–39. [DOI] [PubMed] [Google Scholar]
- Drechsel DN, et al. , 1992. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 3, 1141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharzadeh B, et al. , 2018. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron. 99, 925–940 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HT, et al. , 2019a. Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J. 38, e101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HT, et al. , 2019b. Decreased synthesis of ribosomal proteins in tauopathy revealed by non-canonical amino acid labelling. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari L, et al. , 2020. Arginine pi-stacking drives binding to fibrils of the Alzheimer protein Tau. Nat Commun. 11, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, 2002. Differential expression of phosphorylated translation initiation factor 2 alpha in Alzheimer’s disease and Creutzfeldt-Jakob’s disease. Neuropathol Appl Neurobiol. 28, 441–51. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, et al. , 1997. Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann Neurol. 41, 200–9. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, et al. , 1998. RNA sequestration to pathological lesions of neurodegenerative diseases. Acta Neuropathol. 96, 487–94. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, et al. , 1986. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 83, 4913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette JG, et al. , 1986. Characterization of messenger RNA from the cerebral cortex of control and Alzheimer-afflicted brain. J Neurochem. 47, 987–97. [DOI] [PubMed] [Google Scholar]
- Gunawardana CG, et al. , 2015. The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol Cell Proteomics. 14, 3000–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, et al. , 2016. Changes in the detergent-insoluble brain proteome linked to amyloid and tau in Alzheimer’s Disease progression. Proteomics. 16, 3042–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, et al. , 2014a. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 24, 344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, et al. , 2014b. U1 small nuclear ribonucleoproteins (snRNPs) aggregate in Alzheimer’s disease due to autosomal dominant genetic mutations and trisomy 21. Mol Neurodegener. 9, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, et al. , 2017. Fine-tuning PERK signaling for neuroprotection. J Neurochem. 142, 812–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday M, et al. , 2015. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 6, e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, et al. , 1997. Alzheimer-like changes in microtubule-associated protein Tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding, stimulation of phosphorylation, and filament assembly depend on the degree of sulfation. J Biol Chem. 272, 33118–24. [DOI] [PubMed] [Google Scholar]
- Hashimoto S, Saido TC, 2018. Critical review: involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Ortega K, et al. , 2016. Altered Machinery of Protein Synthesis in Alzheimer’s: From the Nucleolus to the Ribosome. Brain Pathol. 26, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, et al. , 2005. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem. 280, 20978–86. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Scheper W, 2012. Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol. 44, 1295–8. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, et al. , 2009. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 174, 1241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, et al. , 2005. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 110, 165–72. [DOI] [PubMed] [Google Scholar]
- Hsieh YC, et al. , 2019. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep. 29, 301–316 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YC, et al. , 2013. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS One. 8, e64002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara Y, 1988. Massive somatodendritic sprouting of cortical neurons in Alzheimer’s disease. Brain Res. 459, 138–44. [DOI] [PubMed] [Google Scholar]
- Irwin DJ, et al. , 2015. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 129, 469–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessus C, et al. , 1984. In vitro inhibition of tubulin assembly by a ribonucleoprotein complex associated with the free ribosome fraction isolated from Xenopus laevis oocytes: effect at the level of microtubule-associated proteins. Cell Differ. 14, 179–87. [DOI] [PubMed] [Google Scholar]
- Jiang H, et al. , 2015. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell. 163, 108–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, et al. , 2019. TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 137, 259–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, et al. , 2018. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol Neurodegener. 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadavath H, et al. , 2015. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A. 112, 7501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampers T, et al. , 1996. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 399, 344–9. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, et al. , 2017. Local Somatodendritic Translation and Hyperphosphorylation of Tau Protein Triggered by AMPA and NMDA Receptor Stimulation. EBioMedicine. 20, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren SA, et al. , 2019a. Proteomic Techniques to Examine Neuronal Translational Dynamics. Int J Mol Sci. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren SA, et al. , 2019b. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 137, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, et al. , 1989. Tau in situ hybridization in normal and Alzheimer brain: localization in the somatodendritic compartment. Ann Neurol. 26, 352–61. [DOI] [PubMed] [Google Scholar]
- Langstrom NS, et al. , 1989. Alzheimer’s disease-associated reduction of polysomal mRNA translation. Brain Res Mol Brain Res. 5, 259–69. [DOI] [PubMed] [Google Scholar]
- Lanzillotta C, et al. , 2018. Early and Selective Activation and Subsequent Alterations to the Unfolded Protein Response in Down Syndrome Mouse Models. J Alzheimers Dis. 62, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Gotz J, 2017. Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J. 36, 3120–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. , 2004. Phosphorylated eukaryotic translation factor 4E is elevated in Alzheimer brain. Neuroreport. 15, 2237–40. [DOI] [PubMed] [Google Scholar]
- Lin Y, et al. , 2019. Narrow equilibrium window for complex coacervation of tau and RNA under cellular conditions. Elife. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis PA, et al. , 1990. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci U S A. 87, 8422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, 2016. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem. 138 Suppl 1, 54–70. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, et al. , 2010. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- Maina MB, et al. , 2018a. The Involvement of Abeta42 and Tau in Nucleolar and Protein Synthesis Machinery Dysfunction. Front Cell Neurosci. 12, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maina MB, et al. , 2018b. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun. 6, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, et al. , 1981a. Alterations in protein synthetic capability of nerve cells in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 44, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, et al. , 1981b. Neurofibrillary pathology and protein synthetic capability in nerve cells in Alzheimer’s disease. Neuropathol Appl Neurobiol. 7, 37–47. [DOI] [PubMed] [Google Scholar]
- Mann DM, Sinclair KG, 1978. The quantitative assessment of lipofuscin pigment, cytoplasmic RNA and nucleolar volume in senile dementia. Neuropathol Appl Neurobiol. 4, 129–35. [DOI] [PubMed] [Google Scholar]
- Mann DM, et al. , 1977. Cytophotometric mapping of neuronal changes in senile dementia. J Neurol Neurosurg Psychiatry. 40, 299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta CA, 1989. Molecular biology in psychiatric research: Alzheimer’s disease as a paradigm. Am J Orthopsychiatry. 59, 294–302. [DOI] [PubMed] [Google Scholar]
- Marotta CA, et al. , 1981. In vitro synthesis of human brain proteins including tubulin and actin by purified postmortem polysomes. J Neurochem. 36, 966–75. [DOI] [PubMed] [Google Scholar]
- Marotta CA, et al. , 1986. Transcriptional and translational regulatory mechanisms during normal aging of the mammalian brain and in Alzheimer’s disease. Prog Brain Res. 70, 303–20. [DOI] [PubMed] [Google Scholar]
- Maziuk BF, et al. , 2018. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol Commun. 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, et al. , 2015. Identification of Novel Tau Interactions with Endoplasmic Reticulum Proteins in Alzheimer’s Disease Brain. J Alzheimers Dis. 48, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, et al. , 2016. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J Neurosci. 36, 1001–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metuzals J, et al. , 1988. Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s disease. J Neurocytol. 17, 827–33. [DOI] [PubMed] [Google Scholar]
- Meyuhas O, 2015. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int Rev Cell Mol Biol. 320, 41–73. [DOI] [PubMed] [Google Scholar]
- Milovanovic D, De Camilli P, 2017. Synaptic Vesicle Clusters at Synapses: A Distinct Liquid Phase? Neuron. 93, 995–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milovanovic D, et al. , 2018. A liquid phase of synapsin and lipid vesicles. Science. 361, 604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueed Z, et al. , 2018. Tau and mTOR: The Hotspots for Multifarious Diseases in Alzheimer’s Development. Front Neurosci. 12, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, et al. , 1986. Alzheimer’s disease: a correlative study. J Neurol Neurosurg Psychiatry. 49, 229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. , 1993. Alz-50 immunohistochemistry in the normal sheep striatum: a light and electron microscope study. Brain Res. 600, 285–97. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Saper CB, 1995. Ultrastructure of neurofibrillary tangles in the cerebral cortex of sheep. Neurobiol Aging. 16, 315–23. [DOI] [PubMed] [Google Scholar]
- Nijholt DA, et al. , 2012. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol. 226, 693–702. [DOI] [PubMed] [Google Scholar]
- Nunomura A, et al. , 1999. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 19, 1959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygard O, Nilsson L, 1990. Kinetic determination of the effects of ADP-ribosylation on the interaction of eukaryotic elongation factor 2 with ribosomes. J Biol Chem. 265, 6030–4. [PubMed] [Google Scholar]
- Ohno M, 2014. Roles of eIF2alpha kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci. 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira J, et al. , 2017. Protein Phosphorylation is a Key Mechanism in Alzheimer’s Disease. J Alzheimers Dis. 58, 953–978. [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, et al. , 2019. Drosophila Tau Negatively Regulates Translation and Olfactory Long-Term Memory, But Facilitates Footshock Habituation and Cytoskeletal Homeostasis. J Neurosci. 39, 8315–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasozomenos SC, Binder LI, 1987. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil Cytoskeleton. 8, 210–26. [DOI] [PubMed] [Google Scholar]
- Patel A, et al. , 2015. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 162, 1066–77. [DOI] [PubMed] [Google Scholar]
- Peng I, et al. , 1986. Biochemical and immunological analyses of cytoskeletal domains of neurons. J Cell Biol. 102, 252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao YS, et al. , 2002. Cerebellar cortical tau pathology in progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 103, 469–74. [DOI] [PubMed] [Google Scholar]
- Piatnitskaia S, et al. , 2019. USP10 is a critical factor for Tau-positive stress granule formation in neuronal cells. Sci Rep. 9, 10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzak M, et al. , 2011. Epigenetic silencing of nucleolar rRNA genes in Alzheimer’s disease. PLoS One. 6, e22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitera AP, et al. , 2019. Pathogenic tau does not drive activation of the unfolded protein response. J Biol Chem. 294, 9679–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford H, et al. , 2015. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 130, 633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, et al. , 2007. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 282, 14056–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdel-Sulkowska EM, Marotta CA, 1984. Alzheimer’s disease brain: alterations in RNA levels and in a ribonuclease-inhibitor complex. Science. 225, 947–9. [DOI] [PubMed] [Google Scholar]
- Schroder HC, et al. , 1984. Binding of polyribonucleotides and polydeoxyribonucleotides to bovine brain microtubule protein: age-dependent modulation via phosphorylation of high-molecular-weight microtubule-associated proteins and tau proteins. Mech Ageing Dev. 24, 101–17. [DOI] [PubMed] [Google Scholar]
- Shi Z, et al. , 2017. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol Cell. 67, 71–83 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeoka T, et al. , 2019. On-Site Ribosome Remodeling by Locally Synthesized Ribosomal Proteins in Axons. Cell Rep. 29, 3605–3619 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos I, et al. , 2017. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun. 5, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzbach LD, et al. , 2013. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol Commun. 1, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AD, et al. , 2012. Analysis of the tau-associated proteome reveals that exchange of Hsp70 for Hsp90 is involved in tau degradation. ACS Chem Biol. 7, 1677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukmar-Godec T, et al. , 2019. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat Commun. 10, 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukmar-Godec T, et al. , 2020. Biomolecular condensation of the microtubule-associated protein tau. Semin Cell Dev Biol. 99, 202–214. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, et al. , 2016. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, et al. , 2012. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 32, 8270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. , 2006. The proline-rich domain and the microtubule binding domain of protein tau acting as RNA binding domains. Protein Pept Lett. 13, 679–85. [DOI] [PubMed] [Google Scholar]
- Wegmann S, et al. , 2018. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, et al. , 1975. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 72, 1858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JM, et al. , 2019. Activity of the poly(A) binding protein MSUT2 determines susceptibility to pathological tau in the mammalian brain. Sci Transl Med. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willi J, et al. , 2018. Oxidative stress damages rRNA inside the ribosome and differentially affects the catalytic center. Nucleic Acids Res. 46, 1945–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, 2012. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 7, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Ivanov P, 2019. Stress granules and neurodegeneration. Nat Rev Neurosci. 20, 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R, et al. , 2008. Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res. 36, 3707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacco E, et al. , 2019. RNA as a key factor in driving or preventing self-assembly of the TAR DNA-binding protein 43. J Mol Biol. 431, 1671–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, et al. , 2016. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell. 166, 1163–1175 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, et al. , 2017. RNA stores tau reversibly in complex coacervates. PLoS Biol. 15, e2002183. [DOI] [PMC free article] [PubMed] [Google Scholar]