

Abstract
Long noncoding RNAs (lncRNAs) have emerged as key coordinators of biological and cellular processes. Characterizing lncRNA expression across cells and tissues is key to understanding their role in determining phenotypes, including human diseases. We present here FC-R2, a comprehensive expression atlas across a broadly defined human transcriptome, inclusive of over 109,000 coding and noncoding genes, as described in the FANTOM CAGE-Associated Transcriptome (FANTOM-CAT) study. This atlas greatly extends the gene annotation used in the original recount2 resource. We demonstrate the utility of the FC-R2 atlas by reproducing key findings from published large studies and by generating new results across normal and diseased human samples. In particular, we (a) identify tissue-specific transcription profiles for distinct classes of coding and noncoding genes, (b) perform differential expression analysis across thirteen cancer types, identifying novel noncoding genes potentially involved in tumor pathogenesis and progression, and (c) confirm the prognostic value for several enhancer lncRNAs expression in cancer. Our resource is instrumental for the systematic molecular characterization of lncRNA by the FANTOM6 Consortium. In conclusion, comprised of over 70,000 samples, the FC-R2 atlas will empower other researchers to investigate functions and biological roles of both known coding genes and novel lncRNAs.
Long noncoding RNAs (lncRNAs) are commonly defined as transcripts longer than 200 nucleotides that are not translated into proteins. This definition is not based on their function, since lncRNAs are involved in distinct molecular processes and biological contexts not yet fully characterized (Batista and Chang 2013). Over the past few years, the importance of lncRNAs has clearly emerged, leading to an increasing focus on decoding the consequences of their modulation and studying their involvement in the regulation of key biological mechanisms during development, normal tissue and cellular homeostasis, and in disease (Esteller 2011; Batista and Chang 2013; Ling et al. 2015).
Given the emerging and previously underestimated importance of noncoding RNAs (ncRNAs), the FANTOM Consortium has initiated the systematic characterization of their biological function. Through the use of Cap Analysis of Gene Expression sequencing (CAGE-seq), combined with RNA-seq data from the public domain, the FANTOM Consortium released a comprehensive atlas of the human transcriptome, encompassing more accurate transcriptional start sites (TSSs) for coding and noncoding genes, including numerous novel long noncoding genes: the FANTOM CAGE-Associated Transcriptome (FANTOM-CAT) (Hon et al. 2017). We hypothesized that these lncRNAs can be measured in many RNA-seq data sets from the public domain and that they have been so far missed by the lack of a comprehensive gene annotation.
Although the systematic analysis of lncRNAs function is being addressed by the FANTOM Consortium in loss-of-function studies, increasing the detection rate of these transcripts combining different studies is difficult because of the heterogeneity of analytic methods employed. Current resources that apply uniform analytic methods to create expression summaries from public data do exist but can miss several lncRNAs because of their dependency on a preexisting gene annotation for creating the gene expression summaries (Tatlow and Piccolo 2016; Lachmann et al. 2018). We recently created recount2 (Collado-Torres et al. 2017b), a collection of uniformly processed human RNA-seq data, wherein we summarized 4.4 trillion reads from over 70,000 human samples from the NCBI Sequence Read Archive (SRA), The Cancer Genome Atlas (TCGA) (The Cancer Genome Atlas Research Network et al. 2013), and the Genotype-Tissue Expression (GTEx) (The GTEx Consortium 2013) projects (Collado-Torres et al. 2017b). Importantly, recount2 provides annotation-agnostic coverage files that allow requantification using a new annotation without having to reprocess the RNA-seq data.
Given the unique opportunity to access the latest results to the most comprehensive human transcriptome (the FANTOM-CAT project) and the recount2 gene agnostic summaries, we addressed the previously described challenges, building a comprehensive atlas of coding and noncoding gene expression across the human genome: the FANTOM-CAT/recount2 expression atlas (FC-R2 hereafter). Our resource contains expression profiles for 109,873 putative genes across over 70,000 samples, enabling an unparalleled resource for the analysis of the human coding and noncoding transcriptome.
Results
Building the FANTOM-CAT/recount2 resource
The recount2 resource includes a coverage track, in the form of a bigWig file, for each processed sample. We built the FC-R2 expression atlas by extracting expression levels from recount2 coverage tracks in regions that overlapped unambiguous exon coordinates for the permissive set of FANTOM-CAT transcripts, according to the pipeline shown in Figure 1. Since recount2’s coverage tracks do not distinguish between genomic strands, we removed ambiguous segments that presented overlapping exon annotations from both strands (see Methods section and Supplemental Methods). After this disambiguation procedure, the remaining 1,066,515 exonic segments mapped back to 109,869 genes in FANTOM-CAT (out of the 124,047 starting ones included in the permissive set [Hon et al. 2017]). Overall, the FC-R2 expression atlas encompasses 2041 studies with 71,045 RNA-seq samples, providing expression information for 22,116 coding genes and 87,763 noncoding genes, such as enhancers, promoters, and other lncRNAs.
Figure 1.
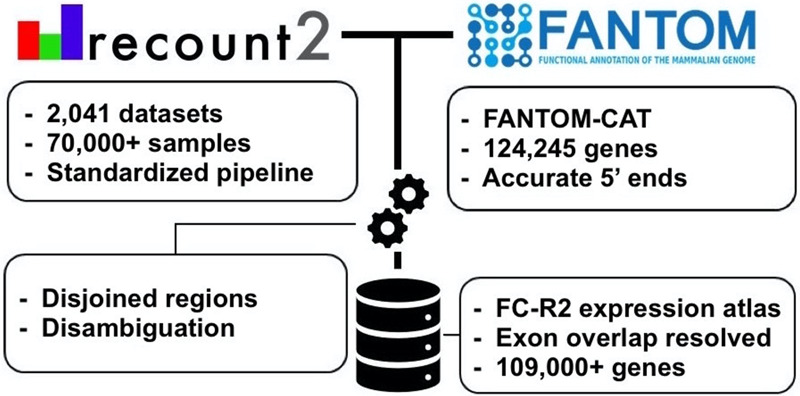
Overview of the FANTOM-CAT/recount2 resource development. FC-R2 leverages two public resources, the FANTOM-CAT gene models and recount2. FC-R2 provides expression information for 109,873 genes, both coding (22,110) and noncoding (87,693). This latter group encompasses enhancers, promoters, and other lncRNAs.
Validating the FANTOM‐CAT/recount2 resource
We first assessed how gene expression estimates in FC-R2 compared to previous gene expression estimates from other projects. Specifically, we considered data from the GTEx Consortium (v6), spanning 9662 samples from 551 individuals and 54 tissues types (The GTEx Consortium 2013). First, we computed the correlation for the GTEx data between gene expression based on the FC-R2 atlas and on the GENCODE (v25) gene model in recount2, which has been already shown to be consistent with gene expression estimates from the GTEx project (Collado-Torres et al. 2017b), observing a median correlation ≥0.986 for the 32,922 genes in common. This result supports the notion that our preprocessing steps to disambiguate overlapping exon regions between strands did not significantly alter gene expression quantification.
Next, we assessed whether gene expression specificity, as measured in FC-R2, was maintained across tissue types. To this end, we selected and compared gene expression for known tissue-specific expression patterns, such as keratin 1 (KRT1), estrogen receptor 1 (ESR1), and neuronal differentiation 1 (NEUROD1) (Fig. 2). Overall, all analyzed tissue-specific markers presented nearly identical expression profiles across GTEx tissue types between the alternative gene models considered (see Fig. 2 and Supplemental Fig. S1), confirming the consistency between gene expression quantification in FC-R2 and those based on GENCODE.
Figure 2.

Tissue-specific expression in GTEx. Log2 expression for three tissue-specific genes (KRT1, NEUROD1, and ESR1) in GTEx data stratified by tissue type using FC-R2- and GENCODE-based quantification. Expression profiles are highly correlated and expressed consistently in the expected tissue types (e.g., KRT1 is most expressed in skin, NEUROD1 in brain, and ESR1 in estrogen-sensitive tissue types like uterus, Fallopian tubes, and breast). Correlations are shown on top for each tissue marker. Center lines, upper/lower quartiles, and whiskers represent the median, 25/75 percentiles, and 1.5 interquartile range, respectively. Additional tissue-specific markers are shown in Supplemental Figure S1.
We also assessed whether there are genes that are not expressed in any of the normal tissues included in GTEx. Out of 109,869 genes, 681 (0.6%) (see Supplemental Figs. S3, S4) were not expressed in any tissue included in GTEx, and they were overrepresented in the FANTOM-CAT permissive set (χ2 test, P-value < 2.2 × 10 16).
Tissue-specific expression of lncRNAs
It has been shown that, although expressed at a lower level, enhancers and promoters are not ubiquitously expressed and are more specific for different cell types than coding genes (Hon et al. 2017). In order to verify this finding, we used GTEx data to assess expression levels and specificity profiles across samples from each of the 54 analyzed tissue types, stratified into four distinct gene categories: coding mRNA, intergenic promoter lncRNA (ip-lncRNA), divergent promoter lncRNA (dp-lncRNA), and enhancer lncRNA (e-lncRNA). Overall, we were able to confirm that these RNA classes are expressed at different levels and that they display distinct specificity patterns across tissues, as shown for primary cell types by Hon et al. (2017), albeit with more variability, likely due to the increased cellular complexity present in tissues. Specifically, coding mRNAs were expressed at higher levels than lncRNAs (log2 median expression of 6.6 for coding mRNAs, and of 4.1, 3.8, and 3.1 for ip-lncRNA, dp-lncRNA, and e-lncRNA, respectively). In contrast, the expression of enhancers and intergenic promoters was more tissue-specific (median = 0.41 and 0.30, respectively) than that observed for divergent promoters and coding mRNAs (median = 0.13 and 0.09, respectively) (Fig. 3A). Finally, when analyzing the percentage of genes expressed across tissues by category, we observed that coding genes are, in general, more ubiquitous, whereas lncRNAs are more specific, with enhancers showing the lowest percentages of expressed genes (mean ranging from 88.42% to 41.98%) (see Fig. 3B), in agreement with the notion that enhancer transcription is tissue-specific (Ong and Corces 2011).
Figure 3.

Expression profiles across GTEx tissues. (A) Expression level and tissue specificity across four distinct RNA categories. The y-axis shows log2 expression levels representing each gene using its maximum expression in GTEx tissues expressed as transcripts per million (TPM). The x-axis shows expression specificity based on entropy computed from median expression of each gene across the GTEx tissue types. Individual genes are highlighted in the figure panels. (B) Percentage of genes expressed for each RNA category stratified by GTEx tissue facets. The dots represent the mean among samples within a facet and the error bars represent 99.99% confidence intervals. Dashed lines represent the means among all samples.
Differential expression analysis of coding and noncoding genes in cancer
We analyzed coding and noncoding gene expression in cancer using TCGA data. To this end, we compared cancer to normal samples separately for 13 tumor types, using FC-R2 requantified data. We further identified the differentially expressed genes (DEGs) in common across the distinct cancer types (see Fig. 4). Overall, the number of DEGs varied across cancer types and by gene class, with a higher number of significant coding than noncoding genes (FDR ≤ 0.01) (see Table 1). A substantial fraction of these genes was exclusively annotated in the FANTOM-CAT meta-assembly, suggesting that relying on other gene models would result in missing many potential important genes (see Table 1). We then analyzed differential gene expression consensus across the considered cancer types. A total of 41 coding mRNAs were differentially expressed across all of the 13 tumor types after global correction for multiple testing (FDR ≤ 10−6) (see Supplemental Table S1). For lncRNAs, a total of 28 divergent promoters, four intergenic promoters, and three enhancers were consistently up- or down-regulated across all the 13 tumor types after global correction for multiple testing (FDR ≤ 0.1) (see Supplemental Tables S2–S4, respectively).
Figure 4.

Differential expression for selected transcripts from distinct RNA classes across tumor types. Box plots for selected differentially expressed genes between tumor and normal samples across all 13 tumor types analyzed. For each tissue of origin, the most up-regulated (on the left) and down-regulated (on the right) gene for each RNA class is shown. Center lines, upper/lower hinges, and the whiskers, respectively, represent the median, the upper and lower quartiles, and 1.5 extensions of the interquartile range. Color coding on the top of the figure indicates the RNA classes (red for mRNA, purple for dp-lncRNA, cyan ip-lncRNA, and green for e-lncRNA). These genes were selected after global multiple testing correction across all 13 tumor types (see Supplemental Tables S1–S4).
Table 1.
Differentially expressed genes in cancer

A usual task performed after differential gene expression analysis is to identify biological processes and pathways associated with the DEGs. To this end, gene set enrichment methods are usually employed; however, this requires detailed gene-to-function annotations, which are mostly lacking for lncRNAs. One possible way to assist prioritizing noncoding transcripts for follow-up functional studies is to identify association with other features along the genome. As an example of this type of analysis, we have assessed the overlap between single-nucleotide polymorphisms (SNPs) associated with cancer in GWAS studies and the list of DEGs we identified. On average, the percentage of DEGs overlapping cancer SNPs ranged from 6.6% in dp-lncRNA to 10.21% in ip-lncRNA across the 13 cancer types (see Supplemental Table S5).
Next, we reviewed the literature to identify functional correlates for these consensus genes. Most of the up-regulated coding genes (Supplemental Table S1) participate in cell cycle regulation, cell division, DNA replication and repair, chromosome segregation, and mitotic spindle checkpoints. Most of the consensus down-regulated mRNAs (Supplemental Table S1) are associated with metabolism and oxidative stress, transcriptional regulation, cell migration and adhesion, and with modulation of DNA damage repair and apoptosis.
Three down-regulated dp-lncRNA genes, GAS1RR, RPL34-DT, and RAP2C-AS1, were reported to be implicated in cancer (Supplemental Table S2). The first one controls epithelial-mesenchymal transition, the second is associated with tumor size increase, whereas the third is associated with urothelial cancer after kidney cancer transplantation (Zhao et al. 2015b; Shang et al. 2016; Zhou et al. 2016). Among the up-regulated dp-lncRNAs (Supplemental Table S2), SNHG1 has been implicated in cellular proliferation and migration and invasion of different cancer types, and to be strongly up-regulated in osteosarcoma, nonsmall lung cancer, and gastric cancer (Cao et al. 2013; Sun et al. 2017).
Among the ip-lncRNAs ubiquitously down-regulated (see Supplemental Table S3), MIR99AHG has been identified in many different tumor types, including leukemia, breast, vulvar, prostate, and bladder cancer (Emmrich et al. 2014; Sun et al. 2014; Gökmen-Polar et al. 2016; Ni et al. 2016; Li et al. 2017). For instance, in vulvar squamous cell carcinoma, MIR99AHG and MIR31HG expressions are correlated and associated with tumor differentiation (Ni et al. 2016). Similarly, MIR99AHG down-regulation in ER-positive breast cancer is associated with progression, recurrence, and metastasis (Gökmen-Polar et al. 2016). In contrast, increased expression of SNHG17 (an ip-lncRNA) (see Supplemental Table S3) was associated with short term survival in breast cancer and with tumor size, stage, and lymph node metastasis in colorectal cancer (Zhao et al. 2015a; Ma et al. 2017). In addition, LINC01311, another ip-lncRNA (Supplemental Table S3), was found to be up-regulated in liver cancer and metastatic prostate cancer (Zhu et al. 2016). Even though we did not identify any cancer association for common e-lncRNAs, one among those we identified, LINC02884, has been previously reported to be up-regulated in late-onset Alzheimer's disease (Humphries et al. 2015). Furthermore, the enhancer lncRNA class also yielded the lowest number of genes in common among all cancer types, reinforcing the concept that enhancers are expressed in a tissue-specific manner (see Fig. 3A and Supplemental Table S4).
Finally, we focused more in depth on prostate cancer (PCa) as a prototypical example, and we were able to confirm previous findings for both coding and noncoding genes (see Supplemental Fig. S2). For coding genes, we confirmed differential expression for known markers of PCa progression and mortality, like ERG, FOXA1, RNASEL, ARVCF, and SLC43A1 (Yu et al. 2010; Lin et al. 2011). Similarly, we also confirmed differential expression for noncoding genes, like PCA3, the first clinically approved lncRNA marker for PCa (Bussemakers et al. 1999; de Kok et al. 2002), PCAT1, a prostate-specific lncRNA involved in disease progression (Prensner et al. 2011), MALAT1, which is associated with PCa poor prognosis (Ren et al. 2013), CDKN2B-AS1, an antisense lncRNA up-regulated in PCa that inhibits tumor suppressor genes activity (Kotake et al. 2011; Gutschner and Diederichs 2012), and the MIR135 host gene, which is associated with castration-resistant PCa (Huang et al. 2015).
Confirming prognostic enhancers
Chen and collaborators have recently surveyed enhancer expression in nearly 9000 patients from TCGA (Chen et al. 2018), using genomic coordinates from the FANTOM5 project (Andersson et al. 2014), identifying 4803 expressed genomic regions with prognostic potential in one or more TCGA tumor types. We therefore leveraged the FC-R2 atlas to identify prognostic coding and noncoding genes using both univariate and multivariate Cox proportional hazard models, comparing our results for e-lncRNAs with those reported by Chen and colleagues. To this end, we started by comparing gene annotations and genomic overlap between the studies. This was necessary because Chen and collaborators relied on the enhancer regions reported by Andersson et al. (2014), which is based on the observation of bidirectional transcription. Our resource, on the contrary, relies on the latest updated FANTOM-CAT annotation, which takes into account other features, such as the epigenetic context, when defining RNA categories. Out of the 4803 genomic regions found prognostic by Chen and collaborators (Chen et al. 2018), we could unambiguously map 1218 regions to exons annotated in the FANTOM-CAT gene models for the four RNA categories we considered in our study (corresponding to a total of 1046 unique genes). Overall, despite the mentioned differences in annotation and quantification (see Supplemental Table S6), we were still able to confirm the prognostic value for 466 genes out of the 1046 reported by Chen et al. (2018), including KLHDC7B-DT (also known as enhancer 22), which was highlighted as a promising prognostic marker for kidney cancer (Supplemental Fig. S5).
We then considered the FANTOM-CAT RNA classes across the different tumor types. We were able to identify a variable number of genes significantly associated with overall survival (FDR ≤ 0.05) in univariate Cox proportional hazards models (see Supplemental Tables S7–S10). Among the consensus DEGs identified across all tumor types, 40 out of 41 coding mRNAs, 25 out of 28 dp-lncRNAs, four out of four ip-lncRNAs, and two out of three e-lncRNAs were found to be associated with survival (see Supplemental Tables S11–S14). Kaplan–Meier curves for selected differentially expressed genes for each RNA category are shown in Supplemental Figure S6. Finally, we performed multivariable analysis controlling for relevant clinical and pathological characteristics in each tumor type. Overall, despite a number of genes being associated with such variables, we obtained similar results (see Supplemental Tables S15–S22).
Discussion
The importance of lncRNAs in cell biology and disease has clearly emerged in the past few years, and different classes of lncRNAs have been shown to play crucial roles in cell regulation and homeostasis (Quinn and Chang 2016). For instance, enhancers—a major category of gene regulatory elements, which has been shown to be expressed (Andersson et al. 2014; Arner et al. 2015)—play a prominent role in oncogenic processes (Herz et al. 2014; Sur and Taipale 2016) and other human diseases (Hnisz et al. 2013). Despite their importance, however, there is a scarcity of large-scale data sets investigating enhancers and other lncRNA categories, in part due to the technical difficulty in applying high-throughput techniques such as ChIP-seq and Hi-C over large cohorts, and to the use of gene models that do not account for them in transcriptomics analyses. Furthermore, the large majority of the lncRNAs that are already known—and that have been shown to be associated with some phenotype—are still lacking functional annotation.
To address these needs, the FANTOM Consortium has first constructed the FANTOM-CAT metatranscriptome, a comprehensive atlas of coding and noncoding genes with robust support from CAGE-seq data (Hon et al. 2017); then, it has undertaken a large scale project to systematically target lncRNAs and characterize their function using a multipronged approach (Ramilowski et al. 2020). In a complementary effort, we have leveraged public domain gene expression data from recount2 (Collado-Torres et al. 2017a,b) to create a comprehensive gene expression compendium across human cells and tissues based on the FANTOM-CAT gene model, with the ultimate goal of facilitating lncRNAs annotation through association studies. To this end, the FC-R2 atlas is already in use in the FANTOM6 project (https://fantom.gsc.riken.jp/6/) to successfully characterize lncRNA expression in human samples (Ramilowski et al. 2020).
In order to validate our resource, we have compared the gene expression summaries based on FANTOM-CAT gene models with previous, well-established gene expression quantifications, demonstrating virtually identical profiles across tissue types overall and for specific tissue markers. We have then confirmed that distinct classes of coding and noncoding genes differ in terms of overall expression level and specificity pattern across cell types and tissues. We also have observed a small subset of genes that were not expressed in the large majority of the samples analyzed in the GTEx project. These genes were mostly classified as small RNAs and enhancers, which was expected given that the RNA-seq libraries included in recount2 did not target small RNAs, and enhancers are usually expressed at a lower level. We further reveal that this subset of genes not expressed in any normal tissue is also associated with a lower level of support of the corresponding FANTOM-CAT gene models (Hon et al. 2017).
Furthermore, using the FC-R2 atlas, we were also able to identify mRNAs, promoters, enhancers, and other lncRNAs that are differentially expressed in cancer, both confirming previously reported findings and identifying novel cancer genes exclusively annotated in the FANTOM-CAT gene models, which have been therefore missed in prior analyses with TCGA data. Finally, we confirmed the prognostic value for some of the enhancer regions recently reported by Chen and colleagues in the TCGA (Chen et al. 2018) by performing a systematic screening for survival association of both coding and noncoding genes that are quantifiable in the FC-R2 resource. Overall, we identified several genes with potential prognostic value across the analyzed cancer types in TCGA; however, further corroboratory studies in independent patient cohorts are necessary to validate these associations.
Collectively, by confirming findings reported in previous studies, our results demonstrate that the FC-R2 gene expression atlas is a reliable and powerful resource for exploring both the coding and noncoding transcriptome, providing compelling evidence and robust support to the notion that lncRNA gene classes, including enhancers and promoters, despite not being yet fully understood, portend significant biological functions. Our resource, therefore, constitutes a suitable and promising platform for future large scale studies in cancer and other human diseases, which in turn hold the potential to reveal important cues to the understanding of their biological, physiological, and pathological roles, potentially leading to improved diagnostic and therapeutic interventions.
Finally, all results, data, and code from the FC-R2 atlas are available as a public tool. With uniformly processed expression data for over 70,000 samples and 109,873 genes ready to analyze, we want to encourage researchers to dive deeper into the study of ncRNAs, their interaction with coding and noncoding genes, and their influence on normal and disease tissues. We hope this new resource will help pave the way to develop new hypotheses that can be followed to unwind the biological role of the transcriptome as a whole.
Methods
Data and preprocessing
The complete FANTOM-CAT gene catalog (inclusive of robust, intermediate, and permissive sets) was obtained from the FANTOM Consortium within the frame of the FANTOM6 project (Ramilowski et al. 2020). The genes were annotated using official HUGO Gene Nomenclature Committee (HGNC) symbols (https://www.genenames.org) when available. For genes without HGNC symbols, we named them according to HGNC instructions (see Supplemental Table S23). The remaining genes were referred to using the official ID from the Consortium that annotated the gene (Ensembl/FANTOM). This catalog accounts for 124,245 genes supported by CAGE peaks, and it includes those described by Hon et al. (2017). In order to remove ambiguity due to overlapping among exons from distinct genes, the BED files containing the coordinates for all genes and exons were processed with the GenomicRanges R/Bioconductor package (Lawrence et al. 2013) to obtain disjoint (nonoverlapping) exon coordinates. To avoid losing strand information from annotation, we processed data using a two-step approach by first disjoining overlapping segments on the same strand and then across strands (Fig. 5). The genomic ranges (disjoint exon segments) that mapped back to more than one gene were discarded. The expression values for these ranges were then quantified using recount.bwtool (Ellis et al. 2018) (code at https://github.com/LieberInstitute/marchionni_projects). The resulting expression quantifications were processed to generate RangedSummarizedExperiment objects compatible with the recount2 framework (Collado-Torres et al. 2017a,b) (code available from https://github.com/eddieimada/fcr2). Thus, the FC-R2 atlas provides expression information for coding and noncoding genes (including enhancers, divergent promoters, and intergenic lncRNAs) for 9662 samples from the GTEx project, 11,350 samples from TCGA, and over 50,000 samples from the SRA.
Figure 5.
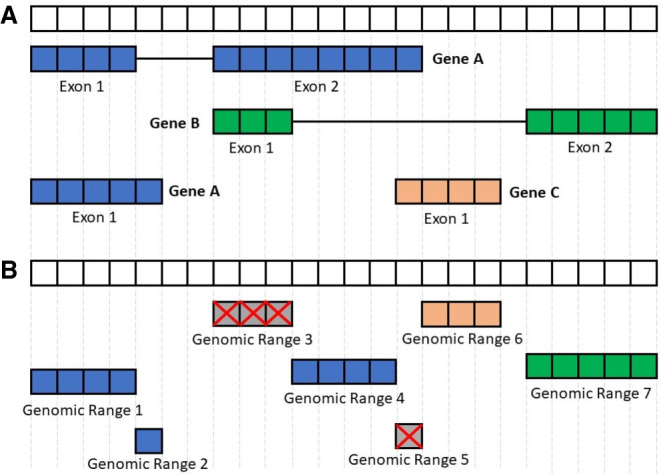
Processing the FANTOM-CAT genomic ranges. This figure summarizes the disjoining and exon disambiguation processes performed before extracting expression information from recount2 using the FANTOM-CAT gene models. (A) Representation of a genomic segment containing three distinct, hypothetical genes: gene A having two isoforms, and genes B and C with one isoform each. Each box can be interpreted as one nucleotide along the genome. Colors indicate the three different genes. (B) Representation of disjoint exon ranges from example in panel A. Each feature is reduced to a set of nonoverlapping genomic ranges. The disjoint genomic ranges mapping back to two or more distinct genes are removed (crossed gray boxes). After removal of ambiguous ranges, the expression information for the remaining ones is extracted from recount2 and summarized at the gene level.
Correlation with other studies
To test if the preprocessing steps used for FC-R2 had a major impact on gene expression quantification, we compared our data to the published GTEx expression values obtained from recount2 (version 2, https://jhubiostatistics.shinyapps.io/recount/). Specifically, we first compared the expression distribution of tissue-specific genes across different tissue types and then computed the Pearson's correlation for each gene in common across the original recount2 gene expression estimates based on GENCODE and our version based on the FANTOM-CAT transcriptome.
Expression specificity of tissue facets
We analyzed the expression level and specificity of each gene stratified by RNA category (i.e., mRNA, e-lncRNA, dp-lncRNA, ip-lncRNA) using the same approach described by Hon et al. (2017) (see Supplemental Methods). Briefly, overall expression levels for each gene were represented by the maximum transcript per million (TPM) values observed across all samples within each tissue type in GTEx. Gene specificity was based on the empirical entropy computed using the mean expression value across tissue types. The 99.99% confidence intervals for the expression of each category by tissue type were calculated based on TPM values. Genes with a TPM greater than 0.01 were considered to be expressed.
Identification of differentially expressed genes
We analyzed differential gene expression in 13 cancer types, comparing primary tumor with normal samples using TCGA data from the FC-R2 atlas. Gene expression summaries for each cancer type were split by RNA category (coding mRNA, intergenic promoter lncRNA, divergent promoter lncRNA, and enhancer lncRNA) and then analyzed independently. A generalized linear model approach, coupled with empirical Bayes moderation of standard errors (Smyth 2004), was used to identify differentially expressed genes between groups. The model was adjusted for the three most relevant coefficients for data heterogeneity as estimated by surrogate variable analysis (SVA) (Leek and Storey 2007). Correction for multiple testing was performed across RNA category by merging the resulting P-values for each cancer type and applying the Benjamini–Hochberg method (Benjamini and Hochberg 1995). Overlapping between DEG and GWAS SNPs was performed using the FANTOM-CAT gene regions coordinates and the SNPs positions obtained from the GWAS catalog (Buniello et al. 2019).
Prognostic analysis
To evaluate the prognostic potential of the genes in FC-R2, we performed both multivariate and univariate Cox proportional hazards regression analysis separately for each RNA class (22,106 mRNAs, 17,404 e-lncRNAs, 6204 dp-lncRNAs, and 1948 ip-lncRNAs) across each of the 13 TCGA cancer types with available survival follow-up information (see Supplemental Methods; Supplemental Table S24). Genes with FDR ≤ 0.05, using the Benjamini–Hochberg correction (Benjamini and Hochberg 1995) within each cancer type and RNA class, were deemed significant prognostic factors. We further analyzed the prognostic value of the consensus differentially expressed genes we identified comparing tumors to normal samples by intersecting the corresponding gene lists with those obtained by Cox proportional regression. Finally, in order to compare our results to previous prognostic analyses, we obtained data on enhancers position and prognostic potential from Chen et al. (2018), performed a liftOver to the hg38 genome assembly to match FC-R2 coordinates, and assessed the overlap between prognostic genes identified in the two studies.
Data access
All data are available from http://marchionnilab.org/fcr2.html. Expression data can be directly accessed through https://jhubiostatistics.shinyapps.io/recount/ and the recount Bioconductor package (v1.9.5 or newer) at https://bioconductor.org/packages/recount as RangedSummarizedExperiment objects organized by the Sequence Read Archive (SRA) study ID. The data can be loaded using R-programming language and are ready to be analyzed using Bioconductor packages, or the data can be exported to other formats for use in another environment. All code used in this manuscript is available for reproducibility and transparency at GitHub (https://github.com/eddieimada/fcr2 and https://github.com/LieberInstitute/marchionni_projects). A compressed archive with all scripts used is also available as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This publication was made possible through support from the National Institutes of Health–National Cancer Institute (NIH-NCI) grants P30CA006973 (L.M. and A.F.), 1U01CA231776 (W.D. and L.M.), U01CA196390 (L.M. and A.S.), and R01CA200859 (W.D. and L.M.); and the National Institutes of Health–National Institute of General Medical Sciences (NIH-NIGMS) grants R01GM118568 (C.W. and B.L.) and R21MH109956-01 (L.C.-T. and A.E.J.); and the U.S. Department of Defense (DoD) office of the Congressionally Directed Medical Research Programs (CDMRP) award W81XWH-16-1-0739 (E.L.I. and L.M.); Russian Academic project 0112-2019-0001 and Russian Foundation for Basic Research project 17-00-00208 (A.F.); and Fundação de Amparo a Pesquisa do Estado de Minas Gerais award BDS-00493-16 (E.L.I. and G.R.F.). recount2 and FC-R2 are hosted on SciServer, a collaborative research environment for large-scale data-driven science. It is being developed at, and administered by, the Institute for Data Intensive Engineering and Science (IDIES) at Johns Hopkins University. SciServer is funded by the National Science Foundation Award ACI-1261715. For more information about SciServer, visit http://www.sciserver.org/.
Author contributions: L.M. conceived the idea; L.M., E.L.I., A.F., and B.L. designed the study; E.L.I., D.F.S., T.M., W.D., A.S., L.C.-T., and L.M. performed the analysis; E.L.I., D.F.S., F.L.-P., G.R.F., and L.M. interpreted the results; L.C.-T., C.W., C-W.Y., K.Y, N.K., M.I., H.S., T.K., C.-C.H., M.J.L.deH., J.W.S., P.C., A.E.J., J.T.L., and B.L. provided the data and tools; E.L.I., D.F.S., L.C.-T., B.L., and L.M. wrote the manuscript; all authors reviewed and approved the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.254656.119.
Freely available online through the Genome Research Open Access option.
References
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. 10.1038/nature12787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. 2015. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347: 1010–1014. 10.1126/science.1259418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Chang HY. 2013. Long noncoding RNAs: cellular address codes in development and disease. Cell 152: 1298–1307. 10.1016/j.cell.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. 2019. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47: D1005–D1012. 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemakers MJ, van Bokhoven A, Verhaegh GW, Smit FP, Karthaus HF, Schalken JA, Debruyne FM, Ru N, Isaacs WB. 1999. DD3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res 59: 5975–5979. [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45: 1113–1120. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao WJ, Wu HL, He BS, Zhang YS, Zhang ZY. 2013. Analysis of long non-coding RNA expression profiles in gastric cancer. World J Gastroentero 19: 3658–3664. 10.3748/wjg.v19.i23.3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Cancer Genome Atlas Research Network, Liang H. 2018. A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell 173: 386–399.e12. 10.1016/j.cell.2018.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Torres L, Nellore A, Jaffe AE. 2017a. recount workflow: accessing over 70,000 human RNA-seq samples with Bioconductor. F1000Res 6: 1558 10.12688/f1000research.12223.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, Jaffe AE, Langmead B, Leek JT. 2017b. Reproducible RNA-seq analysis using recount2. Nat Biotechnol 35: 319–321. 10.1038/nbt.3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW, Swinkels DW, Schalken JA. 2002. DD3PCA3, a very sensitive and specific marker to detect prostate tumors. Cancer Res 62: 2695–2698. [PubMed] [Google Scholar]
- Ellis SE, Collado-Torres L, Jaffe A, Leek JT. 2018. Improving the value of public RNA-seq expression data by phenotype prediction. Nucleic Acids Res 46: e54 10.1093/nar/gky102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmrich S, Streltsov A, Schmidt F, Thangapandi VR, Reinhardt D, Klusmann JH. 2014. LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol Cancer 13: 171 10.1186/1476-4598-13-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12: 861–874. 10.1038/nrg3074 [DOI] [PubMed] [Google Scholar]
- Gokmen-Polar Y, Zavodszky M, Chen X, Gu X, Kodira C, Badve S. 2016. Abstract P2-06-05: LINC00478: a novel tumor suppressor in breast cancer. Cancer Res 76: P2–06–05. [Google Scholar]
- The GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutschner T, Diederichs S. 2012. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol 9: 703–719. 10.4161/rna.20481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Hu D, Shilatifard A. 2014. Enhancer malfunction in cancer. Mol cell 53: 859–866. 10.1016/j.molcel.2014.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. 2013. Super-enhancers in the control of cell identity and disease. Cell 155: 934–947. 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJL, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, et al. 2017. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543: 199–204. 10.1038/nature21374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Yuan T, Liang M, Du M, Xia S, Dittmar R, Wang D, See W, Costello BA, Quevedo F, et al. 2015. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur Urol 67: 33–41. 10.1016/j.eururo.2014.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries CE, Kohli MA, Nathanson L, Whitehead P, Beecham G, Martin E, Mash DC, Pericak-Vance MA, Gilbert J. 2015. Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset Alzheimer's disease. J Alzheimers Dis 44: 977–987. 10.3233/JAD-141989 [DOI] [PubMed] [Google Scholar]
- Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. 2011. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene 30: 1956–1962. 10.1038/onc.2010.568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, Ma'ayan A. 2018. Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun 9: 1366 10.1038/s41467-018-03751-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Storey JD. 2007. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 3: 1724–1735. 10.1371/journal.pgen.0030161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li B, Zheng Y, Li M, Shi L, Pu X. 2017. Exploring functions of long noncoding RNAs across multiple cancers through co-expression network. Sci Rep 7: 754 10.1038/s41598-017-00856-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DW, FitzGerald LM, Fu R, Kwon EM, Zheng SL, Kolb S, Wiklund F, Stattin P, Isaacs WB, Xu J, et al. 2011. Genetic variants in the LEPR, CRY1, RNASEL, IL4, and ARVCF genes are prognostic markers of prostate cancer-specific mortality. Cancer Epidem Biomar 20: 1928–1936. 10.1158/1055-9965.EPI-11-0236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling H, Vincent K, Pichler M, Fodde R, Berindan-Neagoe I, Slack FJ, Calin GA. 2015. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 34: 5003–5011. 10.1038/onc.2014.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Gu S, Song M, Yan C, Hui B, Ji H, Wang J, Zhang J, Wang K, Zhao Q. 2017. Long non-coding RNA SNHG17 is an unfavourable prognostic factor and promotes cell proliferation by epigenetically silencing P57 in colorectal cancer. Mol BioSystems 13: 2350–2361. 10.1039/C7MB00280G [DOI] [PubMed] [Google Scholar]
- Ni S, Zhao X, Ouyang L. 2016. Long non-coding RNA expression profile in vulvar squamous cell carcinoma and its clinical significance. Oncol Rep 36: 2571–2578. 10.3892/or.2016.5075 [DOI] [PubMed] [Google Scholar]
- Ong CT, Corces VG. 2011. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 12: 283–293. 10.1038/nrg2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, Laxman B, Asangani IA, Grasso CS, Kominsky HD, et al. 2011. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 29: 742–749. 10.1038/nbt.1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Chang HY. 2016. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17: 47–62. 10.1038/nrg.2015.10 [DOI] [PubMed] [Google Scholar]
- Ramilowski JA, Yip CW, Agrawal S, Chang JC, Ciani Y, Kulakovskiy IV, Mendez M, Ooi JLC, Ouyang JF, Parkinson N, et al. 2020. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res (this issue) 10.1101/gr.254219.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S, Liu Y, Xu W, Sun Y, Lu J, Wang F, Wei M, Shen J, Hou J, Gao X, et al. 2013. Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. J Urology 190: 2278–2287. 10.1016/j.juro.2013.07.001 [DOI] [PubMed] [Google Scholar]
- Shang D, Zheng T, Zhang J, Tian Y, Liu Y. 2016. Profiling of mRNA and long non-coding RNA of urothelial cancer in recipients after renal transplantation. Tumor Biol 37: 12673–12684. 10.1007/s13277-016-5148-1 [DOI] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3 10.2202/1544-6115.1027 [DOI] [PubMed] [Google Scholar]
- Sun D, Layer R, Mueller AC, Cichewicz MA, Negishi M, Paschal BM, Dutta A. 2014. Regulation of several androgen-induced genes through the repression of the miR-99a/let-7c/miR-125b-2 miRNA cluster in prostate cancer cells. Oncogene 33: 1448–1457. 10.1038/onc.2013.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wei G, Luo H, Wu W, Skogerbø G, Luo J, Chen R. 2017. The long noncoding RNA SNHG1 promotes tumor growth through regulating transcription of both local and distal genes. Oncogene 36: 6774–6783. 10.1038/onc.2017.286 [DOI] [PubMed] [Google Scholar]
- Sur I, Taipale J. 2016. The role of enhancers in cancer. Nat Rev Cancer 16: 483–493. 10.1038/nrc.2016.62 [DOI] [PubMed] [Google Scholar]
- Tatlow PJ, Piccolo SR. 2016. A cloud-based workflow to quantify transcript-expression levels in public cancer compendia. Sci Rep 6: 39259 10.1038/srep39259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, et al. 2010. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17: 443–454. 10.1016/j.ccr.2010.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Luo J, Jiao S. 2015a. Comprehensive characterization of cancer subtype associated long non-coding RNAs and their clinical implications. Sci Rep 4: 6591 10.1038/srep06591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu Y, Zhang W, Zhou Z, Wu J, Cui P, Zhang Y, Huang G. 2015b. Long non-coding RNA Linc00152 is involved in cell cycle arrest, apoptosis, epithelial to mesenchymal transition, cell migration and invasion in gastric cancer. Cell Cycle 14: 3112–3123. 10.1080/15384101.2015.1078034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Hou Y, Yang G, Zhang H, Tu G, Du Y-e, Wen S, Xu L, Tang X, Tang S, et al. 2016. LncRNA-Hh strengthen cancer stem cells generation in twist-positive breast cancer via activation of hedgehog signaling pathway. Stem Cells 34: 55–66. 10.1002/stem.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Li W, Liu J, Chen CH, Liao Q, Xu P, Xu H, Xiao T, Cao Z, Peng J, et al. 2016. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat Biotechnol 34: 1279–1286. 10.1038/nbt.3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.