

Abstract
We have produced RNA sequencing data for 53 primary cells from different locations in the human body. The clustering of these primary cells reveals that most cells in the human body share a few broad transcriptional programs, which define five major cell types: epithelial, endothelial, mesenchymal, neural, and blood cells. These act as basic components of many tissues and organs. Based on gene expression, these cell types redefine the basic histological types by which tissues have been traditionally classified. We identified genes whose expression is specific to these cell types, and from these genes, we estimated the contribution of the major cell types to the composition of human tissues. We found this cellular composition to be a characteristic signature of tissues and to reflect tissue morphological heterogeneity and histology. We identified changes in cellular composition in different tissues associated with age and sex, and found that departures from the normal cellular composition correlate with histological phenotypes associated with disease.
Transcriptional profiles reflect cell type, condition, and function. In tissues and organs, they are monitored in RNA extracted from millions to billions of cells (116–109) (Haque et al. 2017), likely including multiple cell types. As a consequence, the transcriptional profiles obtained from tissue samples represent the average expression of genes across heterogeneous cellular collections, and gene expression differences measured in bulk tissue transcriptomes may thus reflect changes in cellular composition rather than changes in the expression of genes in individual cells. Single-cell RNA sequencing (scRNA-seq) has indeed revealed large cellular heterogeneity in many tissues and organs (Trapnell 2015), and the Human Cell Atlas (HCA) project (Regev et al. 2017) has been recently initiated to define all human cell types and to infer the cellular taxonomy of the human body. As a step in that direction and to bridge the transcriptomes of tissues with the transcriptomes of the constituent primary cells, and to understand how these impact tissue phenotypes, we have generated bulk expression profiles of 53 primary cell lines isolated from 10 different anatomical sites in the human body. These profiles include long- and short-strand-specific RNA-seq and RAMPAGE data (Fig. 1A; Supplemental Tables S1–S4).
Figure 1.

Basic transcriptional programs of human primary cells. (A) Overview of primary cells analyzed in this study and the body location they are extracted from. (B) Hierarchical clustering of human primary cells based on the correlation of gene expression. The clustering in four major clusters is supported by the silhouette analysis and the elbow method (Supplemental Fig. S2A,B). t-SNE of human primary cells based on gene expression measured here (C), on gene expression measured by CAGE by the FANTOM Consortium (D), and on candidate regulatory elements (cREs) by the ENCODE Encyclopedia scored DNase I hypersensitivity signal (E). (F) Correspondence between transcriptionally derived major cell types and classical histological types.
Results
Major cell types in the human body
Clustering of the primary cells based on gene expression profiles revealed a number of well-defined clusters (Fig. 1B,C; Supplemental Figs. S1, S2A,B; Supplemental Methods 1). One cluster was composed of endothelial cells; a second large cluster included a mixture of cell types: fibroblasts, stem cells, and muscle cells, among others, which we collectively termed as mesenchymal. Two smaller clusters, which clustered together, were composed of epithelial cells; finally, the melanocytes clustered separately. Almost all of the individual primary cells are assigned to the proper major cell type. The exceptions are renal mesangial cells, which have contractile properties but are classified as epithelial, and lung epithelial cells, that are classified as mesenchymal. These two cell types, however, are of embryonic origin—in contrast to the vast majority of primary cells in our study, which are adult (Supplemental Table S1)—and their transcriptomes may not reflect the transcriptomes of fully differentiated cells.
The clustering of primary cells does not appear to be dominated by body location or embryological origin. Body location contributes very little to the expression profile of primary cells, explaining only ∼4% of the variance in gene expression (Supplemental Fig. S2C). Variation of gene expression among organs is similar for the different clusters (Supplemental Fig. S2D). The transcriptional diversity among cells within a given organ can be as high as that across the entire human body (Supplemental Fig. S2E). A similar clustering is obtained using FANTOM CAGE-based transcriptomic data on 105 primary cells (Fig. 1D; Supplemental Fig. S3A,B; Supplemental Table S5; Supplemental Methods 2; The FANTOM Consortium and the RIKEN PMI and CLST (DGT) 2014), which reveals, in addition, two clusters corresponding to blood and neural cells, which were not represented in our set of primary cells. The analysis of a different set of primary cells from the ENCODE Encyclopedia Registry of candidate regulatory elements (cREs) (Supplemental Table S6; The ENCODE Project Consortium 2020), based on DNase I hypersensitive sites (DHSs), also recapitulates the clustering (Fig. 1E; Supplemental Fig. S3C; Supplemental Methods 3). The clustering remains in the set of 146 nonredundant primary cells that results from merging the RNA-seq, the CAGE, and the DHS data. The clustering is thus conserved despite the heterogeneity of the underlying assays and experimental protocols used to generate these different data sets (Supplemental Fig. S4). In the clustering, neural cells (mostly astrocytes from different brain regions and neurons) cluster together with a few neuroepithelial primary cells (we labeled them epithelial, but they are mostly ciliated cells from different sites in the eye). Although the neural cells profiled by CAGE seem to have a distinct transcriptional signature (Supplemental Fig. S3A), neural cells profiled by DNase-seq show a gene expression pattern similar to mesenchymal cells (Supplemental Fig. S3C). However, the neural cells profiled by DNase-seq are, in contrast to most primary cells investigated here, of embryonic origin; thus, they are unlikely to express the transcriptional program characteristic of adult neural cells. The analysis of publicly available transcriptomics data from nervous tissues, including single-cell and bulk RNA-seq, strongly support that the neural cell type is a proper major type differentiated from the other types (Supplemental Figs. S5–S7; Supplemental Methods 1).
Comparable multitissue RNA-seq data have become recently available at the single-cell level for 20 mouse organs and tissues through the Tabula Muris project (The Tabula Muris Consortium 2018). Principal component analysis (PCA) of the individual cells and hierarchical clustering of the primary cell types show that most individual cells, and most cell types, clustered into the aforementioned five major cell types, irrespective of the organ of origin (Supplemental Figs. S8, S9; Supplemental Methods 4). As in the case of melanocytes, we also found a few specialized cell types which do not properly belong to these types. Hepatocytes are a notable example (Supplemental Figs. S8A, S9A). Although closer to the epithelial cells than to cells of other types, they seem to have a quite specialized transcriptional program.
Altogether, these results suggest the existence of a limited number of core transcriptional programs encoded in the human genome, and likely in mammalian genomes, in general. These programs underlie the morphology and function common to a few major cellular types, which are at the root of the hierarchy of the many cell types that exist in the human body (Table 1). They all show similar transcriptional heterogeneity, with blood and epithelial within the solid tissues being the most transcriptionally diverse (Supplemental Fig. S10). These transcriptionally defined major cell types correspond broadly, but not exactly, the basic histological types in which tissues are usually classified (e.g., Eroschenko 2013; Mescher 2013; Young et al. 2013): epithelial, of which endothelial is often considered a subtype; muscular; connective, which includes blood; and neural. However, from the transcriptional standpoint, endothelial constitutes a separate type, closer, if any, to the mesenchymal than to the epithelial type. Blood is also a separate major cell type, and the connective (but not blood) and the muscular histological types cluster together into a single mesenchymal transcriptional type (Fig. 1F).
Table 1.
Cell types in the human body
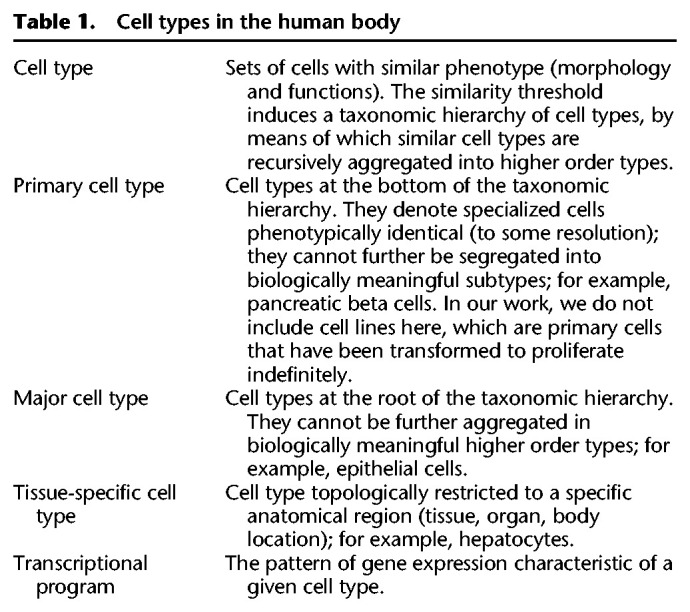
Within each of the major types, further hierarchical organization of cell types may exist. Although we have not profiled enough diversity of primary cells to resolve the taxonomic substructure within each major cell type, hints of this substructure can be seen in the epithelial type. Within the epithelial cluster, two well-defined subclusters can be identified (Fig. 1B–E; Supplemental Fig. S2A). One of the clusters is made mostly by renal cells, indicating that body location may play a role in subtype specialization. The epithelial cluster includes primary cells of all embryonic origins (ectoderm, endoderm, and mesoderm), suggesting that the transcriptional programs of cells may not be fully inherited through development, but partially adopted through function. The more heterogeneous composition of the epithelial type is also apparent in the mouse scRNA-seq (Supplemental Figs. S8, S9).
Our results also suggest that although many cells are likely to adhere to these basic transcriptional programs, many other primary cells are likely highly specialized and very tissue-specific. As with melanocytes and hepatocytes in our analyses, these specialized cells are likely to have their unique transcriptional program.
Cell-type-specific genes
We identified a total of 2871 genes (including 2463 protein-coding genes, 283 long noncoding RNAs, and 125 pseudogenes), the expression of which is specific to epithelial, endothelial, mesenchymal or melanocyte cell types (Fig. 2A; Supplemental Fig. S11; Supplemental Table S7). These cell-type-specific genes include nearly all genes that we identified as the major drivers of the clustering (Supplemental Fig. S12; Supplemental Methods 1). Examples of these genes include collagen (COL1A2, COL3A1, COL6A1/A2/A3), expressed in mesenchymal cells; epithelial transcription factors genes OVOL1/2; VWF gene encoding for the endothelial marker von Willebrand Factor; and TYR gene encoding for the melanocyte-specific enzyme tyrosinase (for a list of manually curated driver genes, see Supplemental Table S8). Figure 2B shows the expression pattern of LINC01235, an endothelial-specific long noncoding RNA (lncRNA) of unknown function. The gene is expressed in nearly all endothelial cells analyzed here, but not in cells from other types, and its expression is correlated to protein-coding genes with endothelial-related functions (Supplemental Fig. S13A). The gene, however, is expressed in multiple tissues; therefore, it is not tissue specific.
Figure 2.

Cell-cluster-specific genes. (A) Expression of 2871 genes specific to major cell types. (B) Expression of the endothelial-specific lncRNA LINC01235. Separate strand-specific signal tracks are shown for endothelial cells, and the other tracks contain overlaid signal for each cell type. The lncRNA has highly correlated (correlation coefficient >0.9) expression with 72 protein-coding genes across our set of primary cells. Nearly all these genes are endothelial specific, and they are functionally enriched for vessel development and angiogenesis (Supplemental Fig. S13A). The gene appears to be under relatively strong regulation, because it has almost 1500 eQTLs across multiple tissues in GTEx (v7), well above the average eQTLs for lncRNAs (about 450). (C) Network of the most strongly coexpressed (Pearson's r > 0.85) cell-type-specific transcription factors (TFs). Nodes are colored according to the cell type specificity of the TF, and they shaped based on the availability of sequence motif: (square) available; (circle) not available. (D) Proportion of cell-type-specific genes with predicted TF binding over cell-type-specific genes that harbor a DHS around their TSS (−10 kb/+5 kb), individually for each cell-type-specific TF (with binding motif available) and cell line for which DNase-seq data was available. In general, we found that genes specific to a given type are enriched for binding motifs for TFs specific to that type. For example, the proportion of endothelial-specific genes with DHS sites that harbor motifs for the endothelial-specific TF ERG in dermal blood endothelial cells (HDBEC) is larger than the proportion of genes with DHS sites specific of other major cell types. Primary cells highlighted in red, although included within the epithelial major cell type, they have been labeled as neural/epithelial in Figure 1D, and they are therefore not proper epithelial; consistently, they do not show the enrichment in binding motifs for epithelial-specific transcription factors. Refer to Supplemental Table S6 for a complete description of the acronyms. Enrichment adjusted P-values: (*) <0.05; (**) <0.01; (***) <0.001.
The functions of annotated tissue-specific genes closely match the expected biology of the primary cells in each type (Supplemental Fig. S13B). Cell-type-specific genes show consistent restricted expression in the FANTOM CAGE data (Supplemental Fig. S14), and they are enriched for encyclopedia cREs (Sheffield et al. 2013) specifically in the primary cells of that type (Supplemental Fig. S15). Using ChIP-seq histone modification data obtained in a number of primary cells (Supplemental Table S9; Supplemental Methods 5; The ENCODE Project Consortium 2012), we found the promoters of genes specific to a given type to be enriched for activating chromatin marks in primary cells of that type compared with primary cells of different type (Supplemental Fig. S16A). However, overall, except for H3K4me1, we found low levels of most activating marks in the promoters of cell-type-specific genes compared with all genes, even after controlling for differences in gene expression. In contrast, the promoters of cell-type-specific genes show similar or higher levels of repressive histone modifications compared to all genes (Supplemental Fig. S16B). This is consistent with previous reports showing that genes under tighter regulation show lower levels of activating histone modifications than broadly expressed genes (e.g., Rach et al. 2011; Pervouchine et al. 2015).
Among cell-type-specific genes, we identified 167 transcription factors (TFs) from a total of 1544 TFs annotated in the human genome (Zhang et al. 2012). We focused on 56 that showed the strongest coexpression patterns (Pearson's r ≥ 0.8) (Fig. 2C; Supplemental Fig. S17). They include previously annotated cell-type-specific transcriptional regulators, such as ERG, which has been shown to regulate endothelial cell differentiation (McLaughlin et al. 2001), and TP63, which is an established regulator of epithelial cell fate and is often altered in tumor cells (Yoh and Prywes 2015). Consistent with the hypothesis that the cell-type-specific TFs might regulate cell type specificity, we found that genes specific to a given type are enriched for binding motifs for TFs specific to that type in most cell lines (Fig. 2D). The enrichment arises specifically when the motifs occur in open chromatin domains in primary cells of that type (e.g., in epithelial primary cells, epithelial-specific genes are enriched, compared to genes specific to other types, in epithelial-specific TF motifs occurring in open chromatin domains) (Fig. 2; Supplemental Fig. S18).
We found that transcriptional regulation appears to play a major role compared to post-transcriptional (splicing) regulation, both in defining the major cell types as well as the individual primary cells within the types. We estimated the fraction of the variation in isoform abundance explained by variation in gene expression (Gonzalez-Porta et al. 2012) to be on average 67% across transcriptional types and 55% across primary cells (Fig. 3A). The lower proportion of variance explained across primary cells suggests that splicing plays a comparatively more important role in defining the transcriptomes of primary cells within a given type than in setting the transcriptional programs of the major cell types. In additional support of this conclusion, we found that although the number of differentially expressed genes in pairwise comparisons of primary cells is much larger between than within cell types, the number of differentially spliced genes is similar (Fig. 3B; Supplemental Fig. S19; Supplemental Methods 6).
Figure 3.

Transcriptional complexity of human primary cells and evolutionary conservation of cell-type-specific genes. (A) Distribution of the relative contribution of gene expression to the variation in isoform abundance between major cell types (blue) and between all primary cells. Large values of the contribution of gene expression indicate that changes in isoform abundance from one condition (primary cell, cell type) to another can be simply explained by changes in gene expression. Small values, in contrast, indicate that changes of isoform abundance are mostly independent of changes in gene expression and can obey changes in the relative abundance of the isoform. (B) Number of differentially expressed genes (DE, y-axis) versus the number of genes with differentially spliced exons (DS, x-axis), between pairs of samples of the same cell type (within, blue), or different cell types (between, red). DS genes have been obtained using IPSA (https://github.com/pervouchine/ipsa-full). See also Supplemental Figure S19. (C) Fraction of 1 to 1 orthologs between each species and human for major cell-type-specific genes and for protein-coding genes overall. Species are sorted by increasing evolutionary distance from human. The black line is given as a reference, and it indicates the proportion of six-way orthologs (chimpanzee, rhesus, mouse, opossum, platypus, and chicken) that are present in each species. The proportion is not 100% in these species because different versions of the GENCODE gene set reference were used. The genes in this set of six-way orthologs are used for the comparison of gene expression in Supplemental Figure S22A. See also Supplemental Figure S22C. (D) Pearson's r between gene expression in each human organ and the corresponding one in every other species. The correlation is computed across all the genes in each major cell type separately. See also Supplemental Figure S23.
Although bulk gene expression is the main contributor to define cell-type specificity, other transcriptional events are also cell-type specific. First, using the RNA-seq data, we identified cell-type-specific splicing events, independent of the tissue of origin (Supplemental Fig. S20; Supplemental Table S10; Supplemental Methods 6). Second, using the RAMPAGE data, we identified cell-type-specific TSSs (Supplemental Fig. S21; Supplemental Table S11; Supplemental Methods 7).
The basic human transcriptional programs seem to have been established early in vertebrate evolution: genes orthologous of cell-type-specific genes are underrepresented compared to orthologs of all genes in invertebrate genomes (Supplemental Fig. S22A,B), but they are overrepresented in vertebrates, as early as in tetrapoda. One exception is epithelial genes, which are overrepresented only in mammals (Fig. 3C; Supplemental Fig. S22C). Within the set of orthologous genes across tetrapoda (Barbosa-Morais et al. 2012), the expression of cell-type-specific genes is less conserved than that of protein-coding genes overall, especially at larger evolutionary distances (Fig. 3D; Supplemental Figs. S22D, S23; Supplemental Methods 1). This suggests an important role in the evolution of gene expression regulation in shaping the basic transcriptional programs in the human genome. Epithelial-specific genes also show the lowest conservation of expression levels. The transcriptional program characteristic of the epithelium appears to be, therefore, the most dynamic evolutionarily—possibly reflecting a greater need for adaptation of the epithelial layer in constant interaction with the environment—and it is also consistent with the greater transcriptional heterogeneity of this major cell type.
Estimation of the cellular composition of complex organs from the expression of cell-type-specific genes
We used the patterns of expression of cell-type-specific genes to estimate the cellular composition of human tissues and organs from GTEx bulk tissue transcriptome data (version 6, 8555 samples, 31 tissues, 544 individuals) (The GTEx Consortium 2017). We used xCell (Aran et al. 2017), using the sets of genes specific to epithelial, endothelial, and mesenchymal major cell types derived from ENCODE, and specific to brain (neural) and blood derived from GTEx (Yang et al. 2018) as signatures, and computed the enrichments of these cell types in each GTEx tissue sample (Supplemental Methods 8).
The xCell enrichments (Fig. 4A; Supplemental Table S12) are largely consistent with the histology of the tissues. For example, esophagus mucosa is enriched for epithelial cells, whereas esophagus muscularis is enriched for mesenchymal cells. Skin (both exposed and unexposed) is enriched in epithelial cells and fibroblasts in mesenchymal cells. Blood and brain are only enriched in blood and neural cells, respectively. Most other tissues are not enriched in these two major cell types, with the expected exceptions of spleen enriched in blood cells and pituitary enriched in neural cells. Testis, which has widespread transcription (Soumillon et al. 2013), is also enriched in neural cells, a reflection of the similarity of the expression programs of these two organs (Guo et al. 2005). Consistent with previous observations (Frontini et al. 2012), we found enrichment of cells of endothelial type in adipose tissue. The analysis of the pathology reports of the subcutaneous adipose tissue shows that it is often contaminated with other tissues, in particular blood vessels, which would explain the enrichment in cells of the endothelial type. We have further processed and analyzed the histopathology images available from the GTEx adipose samples (Supplemental Methods 8) and estimated that, on average, ∼84% of the adipose tissue corresponds to adipocytes (Supplemental Fig. S24), which would explain the endothelial enrichment. In skeletal muscle, we do not observe a particularly large enrichment in cells of the mesenchymal type, in apparent contradiction with our initial classification (Fig. 1B,F). The samples in GTEx, however, are all from differentiated skeletal muscle, whereas the ENCODE primary cells that we used to identify the mesenchymal-specific genes are undifferentiated satellite cells (SkMC) and smooth muscle cells (Supplemental Table S1). We analyzed single-cell RNA-seq data produced during skeletal myoblast differentiation (Trapnell et al. 2014) and found that differentiating skeletal muscle cells retain the mesenchymal signature through most of the differentiation pathway, acquiring only the GTEx muscle specific signature when fully differentiated (Supplemental Fig. S25A–C). Further supporting that muscle is indeed of mesenchymal type, potentially forming a well-defined subtype, gene expression profiles cluster together myoblast differentiating single cells with ENCODE mesenchymal cells, rather than with epithelial or endothelial cells, or forming a separate cluster (Supplemental Fig. S25D).
Figure 4.

Expression of cell-type-cluster-specific genes in GTEx organs. (A) Enrichment of each major cell type in GTEx tissues, estimated from bulk tissue RNA-seq using the xCell method. As a control, we also include, at the bottom of the plot, the enrichments of the endothelial, epithelial, and mesenchymal primary cells monitored here (Fig. 1B). As expected, since the gene signatures have been derived from these very same cells, endothelial primary cells are heavily enriched in the endothelial type, but not in the other types, epithelial cells in the epithelial type and mesenchymal cells in the mesenchymal type. (B) Example of stomach histological slides, which represent the two main tissue layers and the procedure for the manual annotation of the images based on the presence of those layers. Each GTEx histological image displays up to six tissue slices. For the stomach samples, we scored each slice for the presence (1) or absence (0) of the muscularis and mucosa layers, summed up the values for each layer separately and divided by the number of slices. If the proportion of slices with mucosa layer, or muscularis layer, is more than 50% we classify the entire slide as mc1, or ms1, respectively. If the proportion is lower, we classify the slide as mc0 or ms0. A combined class, for example mc0ms1, is assigned to the slides. Thus, samples labeled mc0ms1 are mostly muscularis, and samples labeled mc1ms0 are mostly mucosa. (C) Enrichment of cells of epithelial and mesenchymal types in stomach samples containing mostly the mucosa (green) or mostly the muscularis (purple) layer. (D) Expression of the cell-type-specific genes that drive the separation of stomach samples in mostly muscularis or mostly mucosa samples. Among discriminant cell-type-specific genes, mucosa-only samples express almost exclusively epithelial-specific genes, whereas muscularis-only samples express exclusively mesenchymal-specific genes.
To independently assess the xCell enrichments, we analyzed the histological images of the few tissues in which samples were obtained from different subregions. These are most notable in the case of transverse colon and stomach. The GTEx stomach samples are all from the gastric body, whose walls consist of two broad layers: the mucosa, which is mostly epithelial, and the muscularis, which is smooth muscle (Fig. 4B). We processed the histological images and identified a subset of samples that presented mostly the muscularis or the mucosa layer (Supplemental Methods 8). The enrichment of epithelial cells in the samples from the muscularis layer is much lower than in the samples from the mucosa layer; conversely, the enrichment of mesenchymal cells is much higher in the muscularis than in the mucosa layer. The two sets of samples are almost perfectly separated by our cellular enrichments (Fig. 4C), explaining the bimodality in the distribution of cell type enrichments observed specifically in the stomach samples (Fig. 4A). Consistently, we found that epithelial-specific genes were exclusively expressed in the mucosa layer, and mesenchymal-specific genes were exclusively expressed in the muscularis layer (Fig. 4D). Next, we used the classification of stomach images to train an SVM model (Supplemental Fig. S26A,B) and used this model to predict the presence of the two layers in 196 transverse colon samples, with histology similar to that of stomach (Supplemental Methods 8). The SVM-predicted classification closely matches the differences observed at the transcriptional level and confirms that the bimodality of cellular composition (Fig. 4A) is again related to the unbalanced presence of the two tissue layers across samples (Supplemental Fig. S26C). Considering that stomach and colon were not represented in our primary cell collection, this constitutes a strong validation of our estimates of the cellular enrichments in tissues.
Alterations of cellular composition in pathological states
We projected the solid non-neural GTEx tissue samples on a three-dimensional space according to the enrichments of epithelial, endothelial, and mesenchymal cell types in each sample (Fig. 5A; Supplemental Fig. S27). The spatial arrangement of the samples recapitulates tissue type as strongly as the clustering based on gene expression (Supplemental Fig. S28). This suggests that the basic cell type composition is a characteristic signature of tissues and that departures from this composition may reflect pathological or diseased states. To assess this hypothesis, we analyzed the histological reports associated with the GTEx images (7911 reports). We used fuzzy string search and parse trees to convert the natural language annotations produced by the pathologists to annotations in a controlled vocabulary that can be analyzed automatically (Supplemental Methods 8; Supplemental Table S13). In this way, we identified 19 histological phenotypes affecting one or more tissues for which there were at least 30 affected samples. From these, we identified six conditions with significant (FDR < 0.01) altered contributions of major cell types when comparing the composition of affected and normal tissue (Fig. 5B–E). Atherosclerosis in the tibial artery, which is more prevalent in older donors (Supplemental Fig. S29A), is associated with an increase in endothelial cells (Fig. 5B); this might be attributed to endothelial proliferation stimulated in peripheral artery occlusion (Ziegler et al. 2010). Atrophic skeletal muscle, a phenotype that is also correlated with age (Supplemental Fig. S29B), is associated with an increase in mesenchymal cells, which is consistent with the reported increase of connective tissue (Appell 1990) and intermuscular fat (Manini et al. 2007; Addison et al. 2014) in atrophy (Fig. 5C). Indeed, analysis of the pathology reports of GTEx muscle histological images reveals that the proportion of fat is almost twice as high in atrophic than in non-atrophic muscle (24% vs. 13%) (Supplemental Methods 8). Elevated enrichments of mesenchymal cells are also observed in liver congestion (Supplemental Fig. S30A), a condition that often precedes fibrosis, which is characterized by an activation of matrix-producing cells, including fibroblasts, fibrocytes, and myofibroblasts (Elpek 2014). Despite the low presence of cells of the major cell types in the testis, we found a further reduction of enrichment of endothelial cells in testis undergoing spermatogenesis (Supplemental Fig. S30B). In lung pneumonia, we also observe alteration of all cell types (Supplemental Fig. S30C). The sixth condition is gynecomastia, a pathology that is characterized by ductal epithelial hyperplasia (Cuhaci et al. 2014). We investigated differences in cellular composition between males and females and found them significant only in mammary tissue, where female breasts show much higher enrichment in epithelial cells than male breasts, possibly owing to the presence of epithelial ducts and lobules (Fig. 5D). Males diagnosed with gynecomastia show a cellular composition similar to that of females, mirroring tissue morphology.
Figure 5.

Alterations of the contributions of the major cell types to tissues in histological phenotypes. (A) GTEx samples represented in a 3D space in which the axes are the enrichments of endothelial, epithelial, and mesenchymal cells. (B,C) Differences in xCell enrichments of major cell types (Mann–Whitney U test, adjusted P-values as FDR) between affected and normal states. Histological images of affected and normal tissues are displayed (see text for details): (Athr) atherosclerosis (n = 31); (Atrp) atrophy (n = 34); (Nrml) normal (n = 285 and n = 388, respectively). (D) Major cell type xCell enrichments in female breast samples (Feml, n = 85), and male breast samples with (MlGy, n = 36) or without gynecomastia (Male, n = 85). Only significant FDR (≤0.05) are shown, all of them being between female and male without gynecomastia (left, FDR) and between male without gynecomastia and male with gynecomastia (right, FDR). (E) Changes in major cell type xCell enrichments in lung samples with age. Pearson's r and adjusted P-values as FDR: endothelial r = 0.17 and FDR = 3.2 × 10−3; epithelial r = −0.23 and FDR = 6 × 10−5; mesenchymal r = 0.25 and FDR = 2.4 × 10−5.
We also observed specific age-related changes in cellular composition in lung and ovarian tissues. In lung samples we observe changes of all cell types, in particular, a significant reduction of epithelial cells in older donors (Fig. 5E), which is consistent with the impaired recellularization of lung epithelium that has been observed in decellularized lungs of aged mice (Sokocevic et al. 2013). Consistently, a similar pattern can be observed in the lungs of the individuals that died of respiratory-related causes (Supplemental Fig. S30D,E). In ovarian samples of women older than 48, a lower bound for menopause occurrence, we observe a decrease in endothelial cells (Supplemental Fig. S30F), potentially related to an age-dependent decline in ovarian follicle vascularity (Tatone et al. 2008).
Altered cellular composition is likely to be particularly relevant in cancer. Therefore, we analyzed transcriptome data from The Cancer Genome Atlas Pan-Cancer Analysis of Whole Genomes Project (PCAWG) (The Cancer Genome Atlas Research Network et al. 2013) for 19 cancers affecting tissues also profiled in the GTEx collection and estimated the cellular enrichments of the major cell types (Supplemental Fig. S31; Supplemental Methods 9). In some cases, there is also transcriptome data for normal samples from the same cancer project, which serves as a control for the highly different methodologies used in GTEx and the cancer projects. Thus, in lung cancer, there is an increase in epithelial cells (Fig. 6A,B), likely reflecting the epithelial origin of most lung cancers. In kidney primary tumors, in contrast, there is an overall increase of endothelial cells across most cancer subtypes, consistent with the increased vascularity associated with the cancer (Fig. 6C,D). The exceptions are renal papillary cell carcinomas, which instead present reduced vascularity (Aziz et al. 2013). In both cases, the cellular composition of GTEx samples and normal samples from the cancer projects are similar, supporting the robustness of our cellular characterization. Alterations in cellular composition can also reflect cancer progression. For ovary, even though we lack a comparable set of normal samples from the cancer projects, there are data on different stages of the disease, which serve as an internal control (Fig. 6E,F). Compared to GTEx normal data, there is an increase in epithelial cells in cancer, which is more evident as the severity of the cancer progresses, from primary to recurrent.
Figure 6.

Alterations of the contributions of the major cell types to tissues in cancer. (A) xCell enrichments in epithelial cells in lung cancers and matched normal controls from the PCAWG project separated by cancer project: (LUAD-US) lung adenocarcinoma, TCGA, USA; (LUSC-US) lung squamous cell carcinoma, TCGA, USA. (B) Enrichment in matched normal and cancer lung samples by donor, pooled across the cancer projects. The P-value for the Mann–Whitney U test for the differences in epithelial contribution between normal and cancer samples in the LUAD-US project is: 8.1 × 10−6. (C) xCell enrichment in endothelial cells in kidney cancers and matched normal controls from the PCAWG project separated by cancer project. (RECA-EU) renal cell cancer, France, EU; (KIRP-US) kidney renal papillary cell carcinoma, TCGA, USA; (KIRC-US) kidney renal clear cell carcinoma, TCGA, USA; (KICH-US) kidney chromophobe, TCGA, USA. (D) xCell enrichments in matched normal and cancer kidney samples by donor. The adjusted P-values for the Mann–Whitney U test for the differences in endothelial contribution between normal and cancer samples in the RECA-EU, KIRC-US, KICH-US projects are respectively 3.8 × 10−12, 0.0024, and 0.65. (E,F) xCell enrichments in epithelial cells in ovarian cancers from the PCAWG project separated by cancer project (E) or by donor for matched primary and recurrent samples (F): (OV-AU) ovarian cancer, Austria; (OV-US) ovarian serous cystadenocarcinoma, TCGA, USA. The P-value for the Mann–Whitney U test for the differences in endothelial contribution between primary and recurrent samples in the OV-AU project is 3.6 × 10−27. The donors in B, D, and F are sorted based on the difference between the enrichments. The dashed lines in D and F separate the matched samples in which the enrichment of endothelial (epithelial) cells is larger in the cancer sample from those in which it is larger in the normal sample.
Discussion
The ultimate aim of human genetics is to understand how variations in the sequence of DNA impact organismal traits. However, the path connecting the DNA sequence of the genome to the phenotypic traits of the organism remains mostly unknown, involving a hierarchy of levels of increasing organizational complexity. This path, which unfolds during development, initiates with the transcription of DNA into RNA and its subsequent processing to functional mature RNAs. These, mostly through translation to proteins, determine cell morphology and function. Cells with similar functions aggregate to form tissues, and tissues organize into organs. Systems are made of different types of organs that work cooperatively to perform a particular function. Owing mostly to genome-wide association studies (GWASs), thousands of genetic variants have been connected to human traits and diseases. GWASs, however, uncover only statistical association. Fully understanding the causes and the mechanisms through which DNA variation impacts organismal phenotypes requires understanding how this variation impacts through each of the intermediate levels of organizational complexity. The advent of high throughput technologies to monitor transcription—microarrays first, then RNA sequencing—made possible the identification of genetic variants affecting gene expression. However, how DNA variants and the resulting molecular phenotypes propagate through intermediate levels of biological organization, namely, cells and tissues (or organs), is largely unknown. The reason has been the lack of phenotypic data on cells and tissues with associated genomic, epigenomic, and transcriptomic data.
Very recently, however, mostly through advances both in single-cell sequencing and in digital imaging technologies, data have started to become available, which can be used to connect the molecular to the cellular level, and this, in turn, to the tissue level. In this regard, the data collected here on the transcriptomics of human primary cells, and the links that we have established between these data and the phenotypic traits of organs constitute a unique resource, serving as an intermediate resolution of complexity between single-cell and whole-organ transcriptomics. This resource will contribute to the understanding of how the interplay between transcription and cellular composition shapes tissue histology and ultimately impacts human phenotypes. Our analyses suggest that a large fraction of human cells and cell types in tissues belong to a few major cell types, providing a high-level transcriptionally based hierarchical classification of human cells. Extending the variety of profiled cell types, achieving single-cell resolution, and integrating expression data with epigenetics data, as proposed in the HCA project (Regev et al. 2017), will enrich our understanding of the constitutive cell types in the human body and their functional relationship.
Methods
RNA isolation, library construction, and sequencing
For each cell type to be made into a library, we obtained cell pellets that were stored in RNAlater (Thermo Fisher Scientific) as catalog items from PromoCell (https://www.promocell.com) and ScienCell (https://www.sciencellonline.com/) (for a list of primary cells, see Supplemental Table S1). In short, the RNA was isolated from sorted cells based on cell morphology and cell surface markers. Each cell type was passaged to expand the cell numbers for 24–48 h (1-2 doublings) before total RNA extraction and shipping. Thus, this protocol represents a minimum of exposure to non-native conditions. The cell morphologies are checked at this time. Although it is clear that the molecular context (influence of external cytokins and neighboring cells) of these cells has changed, they cluster in a very similar fashion to profiles shown by single-cell isolates of the corresponding types. Thus, the limited passage has an unlikely effect on the gene expression program. We rely on the providers’ standards for quality assurance. Quality sheets are available through the ENCODE portal (https://www.encodeproject.org/search/?type=Biosample&organism.scientific_name=Homo+sapiens&biosample_ontology.classification=primary+cell&lab.title=Thomas+Gingeras%2C+CSHL&source.title=PromoCell&award.rfa=ENCODE3). We ordered three vials per cell type per donor for a total of 3 million cells. The three vials were combined, and we isolated total RNA from them using the Ambion mirVana miRNA Isolation kit (AM1561). The rRNA was removed using the RiboZero Gold Protocol (RZG1224). The libraries are made using a homebrew “dUTP” protocol (Parkhomchuk et al. 2009), which generates stranded libraries. They were sequenced on the Illumina platform in mate-pair fashion and processed through the data processing pipeline at the ENCODE DCC. Additional information about each of these steps, metadata, and files can be found at https://www.encodeproject.org/.
RAMPAGE sample preparation
Isolation of RNA is described in the preceding section. The RAMPAGE protocol (Batut and Gingeras 2013) was used to make libraries. Each library was sequenced in mate-pair fashion on the Illumina platform. Detailed protocol and quality-control images and metrics on a per library basis can be found in the “Production Documents” appended to each RAMPAGE assay at the ENCODE Data Coordination Center (https://www.encodeproject.org/).
Small RNA isolation, library construction, and sequencing
Isolation of RNA is described in the preceding section. The Illumina TruSeq protocol was used to make libraries. Each library was sequenced in single end fashion on the Illumina platform. Detailed protocol and quality-control images and metrics on a per library basis can be found in the “Production Documents” appended to each Small RNA assay at the ENCODE Data Coordination Center (https://www.encodeproject.org/).
RNA-seq processing pipeline
Raw reads from the 106 RNA-seq libraries (for a list of ENCODE library IDs, see Supplemental Table S1; for submitted FASTQ files, see https://www.encodeproject.org/) were aligned with STAR v2.3.1z (Dobin et al. 2013) to the human genome assembly hg19. Reads mapping to more than 20 multiple positions were discarded. Read counts for all long genes annotated in GENCODE v19 (Harrow et al. 2012) were computed with RSEM 1.2.19 (expected read counts) (Li and Dewey 2011). Statistics on the number of reads and mapping are available on Supplemental Table S14. Furthermore, we verified using liftOver that the cell-type-specific genes are consistent between GRCh37/hg19 and GRCh38/hg38, with a successful conversion of 2855 of the 2871 genes.
For most of the analyses, we average expression values for a given pair of replicates and sometimes the two biological replicates are from donors of the opposite sex; therefore, we remove genes on Chromosome Y. The lack of an enrichment step for polyadenylated transcripts preserves the presence of some short biotype genes, which are still longer than 200 bp. Thus, we remove genes with at least one transcript annotated as short RNA in GENCODE. These genes are often of repetitive nature, which makes the quantification of their expression problematic; this is why we decided to remove them.
Read counts which are not reproducible between two replicates (npIDR > 0.1) (Djebali et al. 2012) are set to 0. The matrix of read counts after npIDR is provided as Supplemental Table S2. After filtering for reproducibility, read counts are normalized to a slightly modified version of RPKM (reads per kilobase of exon model per million mapped reads) (Mortazavi et al. 2008). Specifically, read counts were first normalized to counts per million (cpm), in which the library sizes are the trimmed mean of M values (TMM) (Robinson and Oshlack 2010) scaled sums of exonic reads, and then normalized by gene length. Finally, RPKM values from the two replicates were averaged, and genes with RPKM < 1 in all samples were discarded, resulting in 16,265 genes, including 13,990 protein coding, 1380 long noncoding RNAs, and 895 pseudogenes. Statistical analyses were performed with R version 3.6.1 (R Core Team 2019).
As the samples were prepared and sequenced in three known distinct batches (Supplemental Table S1), we used the removeBatchEffect() function from R limma package (Ritchie et al. 2015) to build a linear model with the batch information and the cell types on log10-transformed RPKM (with a pseudocount of 0.01), and we regressed out the batch variable.
Data access
All experimental protocols for the samples described here, and all data generated for this study, are publicly available on the ENCODE portal (https://www.encodeproject.org/). GTEx gene expression is available in the GTEx portal (https://www.gtexportal.org).
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This project was supported by awards U54HG007004, U41HG007234, and R01MH101814 from the National Human Genome Research Institute of the National Institutes of Health, as well as from the Spanish Ministry of Economy and Competitiveness, Centro de Excelencia Severo Ochoa 2013–2017, SEV-2012-0208, Programa de Ayudas FPI del Ministerio de Economía y Competitividad BES-2012-055848 to A.B., and Ministerio de Educación, Cultura y Deporte, under the FPU programme (Formación de Profesorado Universitario) with predoctoral fellowship FPU15/03635 to M.M.A., as well as the support of the CERCA programme/Generalitat de Catalunya. D.G.M. is supported by a “la Caixa”-Severo Ochoa predoctoral fellowship LCF/BQ/SO15/52260001. We also acknowledge support from the European Research Council (ERC) under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement 294653. We thank Kristin Ardlie and Detlev Arendt for useful discussions. We acknowledge and thank the donors and their families for their generous gifts of organ donation for transplantation and tissue donations for the GTEx research study. The Genotype-Tissue Expression (GTEx) project was supported by the Common Fund of the Office of the Director of the National Institutes of Health (https://commonfund.nih.gov/GTEx). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We acknowledge the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) to the EMBL partnership. Figure 1A was created with https://biorender.com/. R.G. dedicates this work to the Catalan leaders and the people in jail and exile for defending freedom and democracy, without which science cannot flourish.
Author contributions: A.B., C.A.D., M.M.A., V.W., R.G., and T.R.G. conceived and designed the experiments and analyses. J.D., C.A.D., A.S., and C.D. performed the experiments. A.B., M.M.A., V.W., and D.G.M. analyzed the data. J.G., D.D.P., A.V., A.D., C.Z., D.G.M., F.R., and M.P.S. contributed ideas and statistical advice. A.B., M.M.A., V.W., R.G., and T.R.G. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.263186.120.
Freely available online through the Genome Research Open Access option.
References
- Addison O, Marcus RL, LaStayo PC, Ryan AS. 2014. Intermuscular fat: a review of the consequences and causes. Int J Endocrinol 2014: 309570 10.1155/2014/309570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appell HJ. 1990. Muscular atrophy following immobilisation. Sports Med 10: 42–58. 10.2165/00007256-199010010-00005 [DOI] [PubMed] [Google Scholar]
- Aran D, Hu Z, Butte AJ. 2017. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 18: 220. 10.1186/s13059-017-1349-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz SA, Sznol J, Adeniran A, Colberg JW, Camp RL, Kluger HM. 2013. Vascularity of primary and metastatic renal cell carcinoma specimens. J Transl Med 11: 15 10.1186/1479-5876-11-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- Batut P, Gingeras TR. 2013. RAMPAGE: promoter activity profiling by paired-end sequencing of 5′-complete cDNAs. Curr Protoc Mol Biol 104 10.1002/0471142727.mb25b11s104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The cancer genome atlas pan-cancer analysis project. Nat Genet 45: 1113–1120. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuhaci N, Polat S, Evranos B, Ersoy R, Cakir B. 2014. Gynecomastia: clinical evaluation and management. Indian J Endocrinol Metab 18: 150–158. 10.4103/2230-8210.129104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elpek GÖ. 2014. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J Gastroenterol 20: 7260–7276. 10.3748/wjg.v20.i23.7260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, et al. 2020. Expanded encyclopedias of DNA elements in the human and mouse genomes. Nature (in press) doi:10.1038/s41586-020-2493-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroschenko VP. 2013. DiFiore's atlas of histology with functional correlations. Lippincott Williams & Wilkins, Baltimore. [Google Scholar]
- The FANTOM Consortium and the RIKEN PMI and CLST (DGT). 2014. A promoter-level mammalian expression atlas. Nature 507: 462–470. 10.1038/nature13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frontini A, Giordano A, Cinti S. 2012. Endothelial cells of adipose tissues: a niche of adipogenesis. Cell Cycle 11: 2765–2766. 10.4161/cc.21255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Porta M, Calvo M, Sammeth M, Guigo R. 2012. Estimation of alternative splicing variability in human populations. Genome Res 22: 528–538. 10.1101/gr.121947.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2017. Genetic effects on gene expression across human tissues. Nature 550: 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JH, Huang Q, Studholme DJ, Wu CQ, Zhao Z. 2005. Transcriptomic analyses support the similarity of gene expression between brain and testis in human as well as mouse. Cytogenet Genome Res 111: 107–109. 10.1159/000086378 [DOI] [PubMed] [Google Scholar]
- Haque A, Engel J, Teichmann SA, Lönnberg T. 2017. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 9: 75. 10.1186/s13073-017-0467-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774. 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. 10.1186/1471-2105-12-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manini TM, Clark BC, Nalls MA, Goodpaster BH, Ploutz-Snyder LL, Harris TB. 2007. Reduced physical activity increases intermuscular adipose tissue in healthy young adults. Am J Clin Nutr 85: 377–384. 10.1093/ajcn/85.2.377 [DOI] [PubMed] [Google Scholar]
- McLaughlin F, Ludbrook VJ, Cox J, von Carlowitz I, Brown S, Randi AM. 2001. Combined genomic and antisense analysis reveals that the transcription factor Erg is implicated in endothelial cell differentiation. Blood 98: 3332–3339. 10.1182/blood.V98.12.3332 [DOI] [PubMed] [Google Scholar]
- Mescher AL. 2013. Junqueira's basic histology: text and atlas. McGraw-Hill Medical, New York. [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37: e123 10.1093/nar/gkp596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervouchine DD, Djebali S, Breschi A, Davis CA, Barja PP, Dobin A, Tanzer A, Lagarde J, Zaleski C, See LH, et al. 2015. Enhanced transcriptome maps from multiple mouse tissues reveal evolutionary constraint in gene expression. Nat Commun 6: 5903. 10.1038/ncomms6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rach EA, Winter DR, Benjamin AM, Corcoran DL, Ni T, Zhu J, Ohler U. 2011. Transcription initiation patterns indicate divergent strategies for gene regulation at the chromatin level. PLoS Genet 7: e1001274 10.1371/journal.pgen.1001274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. 2017. The human cell atlas. eLife 6: e27041 10.7554/eLife.27041.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, Furey TS. 2013. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res 23: 777–788. 10.1101/gr.152140.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokocevic D, Bonenfant NR, Wagner DE, Borg ZD, Lathrop MJ, Lam YW, Deng B, DeSarno MJ, Ashikaga T, Loi R, et al. 2013. The effect of age and emphysematous and fibrotic injury on the re-cellularization of de-cellularized lungs. Biomaterials 34: 3256–3269. 10.1016/j.biomaterials.2013.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthès P, Kokkinaki M, Nef S, Gnirke A, et al. 2013. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3: 2179–2190. 10.1016/j.celrep.2013.05.031 [DOI] [PubMed] [Google Scholar]
- The Tabula Muris Consortium. 2018. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562: 367–372. 10.1038/s41586-018-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatone C, Amicarelli F, Carbone MC, Monteleone P, Caserta D, Marci R, Artini PG, Piomboni P, Focarelli R. 2008. Cellular and molecular aspects of ovarian follicle ageing. Hum Reprod Update 14: 131–142. 10.1093/humupd/dmm048 [DOI] [PubMed] [Google Scholar]
- Trapnell C. 2015. Defining cell types and states with single-cell genomics. Genome Res 25: 1491–1498. 10.1101/gr.190595.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RY, Quan J, Sodaei R, Aguet F, Segrè AV, Allen JA, Lanz TA, Reinhart V, Crawford M, Hasson S, et al. 2018. A systematic survey of human tissue-specific gene expression and splicing reveals new opportunities for therapeutic target identification and evaluation. bioRxiv 10.1101/311563 [DOI] [Google Scholar]
- Yoh K, Prywes R. 2015. Pathway regulation of p63, a director of epithelial cell fate. Front Endocrinol (Lausanne) 6: 51 10.3389/fendo.2015.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B, O'Dowd G, Woodford P. 2013. Wheater's functional histology. Elsevier Health Sciences, New York. [Google Scholar]
- Zhang HM, Chen H, Liu W, Liu H, Gong J, Wang H, Guo AY. 2012. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res 40: D144–D149. 10.1093/nar/gkr965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler MA, Distasi MR, Bills RG, Miller SJ, Alloosh M, Murphy MP, Akingba AG, Sturek M, Dalsing MC, Unthank JL. 2010. Marvels, mysteries, and misconceptions of vascular compensation to peripheral artery occlusion. Microcirculation 17: 3–20. 10.1111/j.1549-8719.2010.00008.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.