

Abstract
Background
Porcine epidemic diarrhea (PED) is a highly contagious swine disease caused by the PED virus (PEDV), which is a member of the family Coronaviridae. Since the first outbreaks in Belgium and the United Kingdom were reported in 1971, PED has spread throughout many countries around the world and causing significant economic loss. This study was conducted to investigate the recent distribution of PEDV strains in Vietnam during the 2015–2016 seasons.
Methods
A total of 30 PED‐specific PCR‐positive intestinal and faecal samples were collected from unvaccinated piglets in Vietnam during the 2015–2016 seasons. The full length of the spike (S) gene of these PEDV strains were analysed to determine their phylogeny and genetic relationship with other available PEDV strains globally.
Results
Phylogenetic analysis of the complete S gene sequences revealed that the 28 Vietnamese PEDV strains collected in the northern and central regions clustered in the G2 group (both G2a and G2b sub‐groups), while the other 2 PEDV strains (HUA‐PED176 and HUA‐PED254) collected in the southern region were clustered in the G1/G1b group/sub‐group. The nucleotide (nt) and deduced amino acid (aa) analyses based on the complete S gene sequences showed that the Vietnamese PEDV strains were closely related to each other, sharing nt and aa homology of 93.2%–99.9% and 92.6%–99.9%, respectively. The N‐glycosylation patterns and mutations in the antigenic region were observed in Vietnamese PEDV strains.
Conclusions
This study provides, for the first time, up‐to‐date information on viral circulation and genetic distribution, as well as evidence to assist in the development of effective PEDV vaccines in Vietnam.
Keywords: molecular characterization, phylogenetic analysis, porcine epidemic diarrhea virus (PEDV), spike glycoprotein gene
Porcine epidemic diarrhea viruses cause highly contagious swine disease in Vietnam. The circulating Vietnamese PEDV strains belonged to the G2/G2a and G2b group/sub‐groups and G1/G1b group/sub‐group.

1. INTRODUCTION
Porcine epidemic diarrhea (PED) is a highly contagious swine disease caused by PED virus (PEDV), a member of the family Coronaviridae. Pathologically, the disease is characterized by causing vomiting, enteritis, and watery diarrhoea with high mortality in pigs of all ages, although the most severe signs are reported in piglets less than 2 weeks old (Bridgen, Duarte, Tobler, Laude, & Ackermann, 1993). PEDV is an enveloped coronavirus with a single‐stranded RNA genome about 28 kb in length. The PEDV genome is composed of seven open reading frames (ORFs) encoding for four structural proteins (spike, S; envelope, E; membrane, M; nucleocapsid, N) and three nonstructural proteins (ORF1a, −1b and ORF3) (Song & Park, 2012). Among viral proteins, the S protein is a glycoprotein peplomer on the viral surface which plays an important role in induction of neutralizing antibodies and interaction with cellular receptors in the host. It is cleaved by host‐derived proteases into two subunits, namely S1 (binds to the receptor) and S2 (responsible for fusion activity) (Spaan, Cavanagh, & Horzinek, 1988).
Since the first outbreaks in Belgium and the United Kingdom were reported in 1971, PED has spread throughout many countries in Europe (Pensaert & de Bouck, 1978; Wood, 1977) and Asia (Kusanagi et al., 1992; Lin et al., 2014; Puranaveja et al., 2009). Recently, a new PEDV strain with high morbidity and mortality rates in infected suckling piglets emerged in China in 2010 (Chen et al., 2013) before rapidly spreading to the United States in 2014 (Huang et al., 2013; Jarvis et al., 2016), followed by other countries including Canada (Ojkic et al., 2015), Mexico (Vlasova et al., 2014), Japan (Murakami et al., 2015), Korea (Lee, & Lee., 2014; Sun et al., 2015) and Taiwan (Lin et al., 2014). In addition, new variant of PEDVs, the mild virulence PEDV strains, has insertions and/or deletions (INDEL) in the N‐terminal region of the S genes have been identified in the United States (Wang, Byrum, & Zhang, 2014), South Korea (Park, Kim, Song, & Park, 2014) and Japan (Masuda et al., 2015).
In Vietnam, since the first outbreak reported in 2009, PEDV has spread widely throughout country, causing significant economic loss (Duy, Toan, Puranaveja, & Thanawongnuwech, 2011). Phylogenetic study indicated that PEDV circulating in northern and central Vietnam during 2013–2014 could be divided into two separate groups, G2 and G1 (Kim et al., 2015). On the other hand, these emerging PEDV strains are genetically distant from PEDV vaccine strains at the neutralizing epitope regions, suggesting a novel vaccine is needed to control the current PED outbreaks in Vietnam (Diep et al., 2018).
In this study, for the first time, we investigated the molecular characterization of nearly the full spike glycoprotein gene (S gene) of 30 PEDV strains collected from endemic outbreaks during 2015–2016 in 11 different provinces, representing the three parts of Vietnam (the northern, central and southern regions). Furthermore, phylogenetic trees were constructed to determine the relationship between the Vietnamese PEDV strains and other PEDV strains circulating worldwide. The results of this study provide up‐to‐date information on recent circulating PEDVs in Vietnam, as well as genetic information for downstream vaccine development.
2. MATERIALS AND METHODS
2.1. Sample collection
In order to achieve representative sampling for the entire country, thirty PED‐specific PCR‐positive intestinal and faecal samples were opportunely collected from unvaccinated piglets from 11 different provinces in Vietnam during the 2015–2016 seasons (Table 1, Figure S1). The number of samples from each province was as follows: the northern: Hanoi (n = 2), Hai Phong (n = 4), Hung Yen (n = 4), Lao Cai (n = 2), Thai Nguyen (n = 4), Thai Binh (n = 1), Tuyen Quang (n = 1) and Vinh Phuc (n = 2); the central: Nghe An (n = 1); and the southern: Dong Nai (n = 5), Ho Chi Minh city (n = 4).
Table 1.
Vietnamese PEDV strains used in this study
| No | Sample name | Date of collection | Geographic origin | Type of sample | Accession number | No | Sample name | Date of collection | Geographic origin | Type of sample | Accession number |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HUA‐PED113 | Sep, 2015 | Hanoi | Intestine | KX708896 | 16 | HUA‐PED153 | Jan, 2016 | Thai Nguyen | Intestine | MK435389 |
| 2 | HUA‐PED146 | Dec, 2015 | Hanoi | Feces | MK435391 | 17 | HUA‐PED155 | Jan, 2016 | Thai Binh | Intestine | MK435388 |
| 3 | HUA‐PED86 | Jan, 2015 | Hai Phong | Intestine | KX708902 | 18 | HUA‐PED152 | Jan, 2016 | Tuyen Quang | Intestine | MK435390 |
| 4 | HUA‐PED88 | Jan, 2015 | Hai Phong | Intestine | KX708901 | 19 | HUA‐PED117 | Sep, 2015 | Vinh Phuc | Intestine | KX708900 |
| 5 | HUA‐PED111 | Aug, 2015 | Hai Phong | Feces | KX708903 | 20 | HUA‐PED118 | Sep, 2015 | Vinh Phuc | Intestine | KX708899 |
| 6 | HUA‐PED160 | Feb, 2016 | Hai Phong | Intestine | MK435387 | 21 | HUA‐PED183 | Apr, 2016 | Nghe An | Feces | MK435382 |
| 7 | HUA‐PED94 | Oct, 2015 | Hung Yen | Feces | KX708906 | 22 | HUA‐PED178 | Jan, 2016 | Dong Nai | Intestine | MK435384 |
| 8 | HUA‐PED96 | Oct, 2015 | Hung Yen | Feces | KX708907 | 23 | HUA‐PED192 | Mar, 2016 | Dong Nai | Intestine | MK435381 |
| 9 | HUA‐M17 | Jan, 2016 | Hung Yen | Feces | KX708894 | 24 | HUA‐PED202 | Apr, 2016 | Dong Nai | Intestine | MK435380 |
| 10 | HUA‐PED120 | Dec, 2015 | Hung Yen | Feces | MK435392 | 25 | HUA‐PED203 | Apr, 2016 | Dong Nai | Intestine | MK435379 |
| 11 | HUA‐PED103 | Oct, 2015 | Lao Cai | Intestine | KX708904 | 26 | HUA‐PED230 | Apr, 2016 | Dong Nai | Feces | MK435378 |
| 12 | HUA‐PED106 | Oct, 2015 | Lao Cai | Intestine | KX708905 | 27 | HUA‐PED175 | Oct, 2015 | Ho Chi Minh | Intestine | MK435386 |
| 13 | HUA‐PED114 | Dec, 2015 | Thai Nguyen | Intestine | KX708897 | 28 | HUA‐PED176 | Nov, 2015 | Ho Chi Minh | Intestine | MK435385 |
| 14 | HUA‐PED115 | Dec, 2015 | Thai Nguyen | Intestine | KX708898 | 29 | HUA‐PED180 | Feb, 2016 | Ho Chi Minh | Intestine | MK435383 |
| 15 | HUA‐V2 | Oct, 2015 | Thai Nguyen | Intestine | KX708895 | 30 | HUA‐PED254 | Jul, 2016 | Ho Chi Minh | Intestine | MK435377 |
Abbreviation: PED, porcine epidemic diarrhea; PEDV, porcine epidemic diarrhea virus.
2.2. RNA extraction and reverse transcription polymerase chain reaction
The viral RNA was extracted from infected cell culture supernatants using the QIAamp viral RNA mini kit (Qiagen), according to the manufacturer's instructions. cDNA was synthesized by SuperScript™ III First‐strand Synthesis SuperMix (Invitrogen), according to the manufacturer's instructions. In order to assay the complete S‐gene, a set of previous published primers were used to perform the PCR (Tian et al., 2013). Briefly, the reaction was carried out at 42°C for 60 min (for reverse transcription), 95°C for 5 min (for denaturation), 35 cycles of 95°C for 30 s (for denaturation), 52°C for 30 s (for annealing) and 72°C for 90 s (for extension), followed by 72°C for 10 min (for final extension). The PCR products were separated on 1.2% SeaKem LE agarose gel and viewed on a BioRad Gel Doc XR image analysis system.
2.3. Nucleotide sequencing and sequence analysis
The PCR products were cloned into the pGEM‐T Vector System II (Cat. No. A3610; Promega) according to the manufacturer's instructions. The cloned genes were sequenced with T7 and SP6 sequencing primers on an ABI PRISM® 3730xl DNA Sequencer at Macrogen Institute (Macrogen Co., Ltd.). The raw sequences were assembled by the SeqMan program (DNAstar package). The complete sequences were aligned using BioEdit v7.2.5 (Hall, 1999). The obtained nucleotide sequences were deposited in GenBank database under the accession numbers as shown in Table 1. Glycosylation sites were predicted using the web‐based program N‐Glycosite (http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html) using aa alignment data (Zhang et al., 2004).
2.4. Recombination analysis and phylogenetic analysis
The complete nucleotide sequences of the S‐genes from all 30 PEDV samples examined in this study were compared against a representative S‐gene from the available PEDV sequences in the GenBank database. Recombination Detection Program version 4 (RDP v4) containing seven recombination detection methods (RDP, Bootscan, Geneconv, MaxChi, Chimaera, SISCAN, and 3Seq) was used to predict recombination events (Martin, Murrell, Golden, Khoosal, & Muhire, 2015). Only the recombination events detected by more than five methods implemented in RDP v4 were considered to be reliable. SimPlot 3.5.1 software was further used to confirm putative recombination strains and points (Lole et al., 1999). The analyses were performed with default settings (the window width and the step size to 200 bp and 20 bp, respectively). The phylogenetic tree was reconstructed by the MEGA 6.06 software package using the NEIGHBOR‐JOINING method with the Kimura 2 parameter substitution model (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013). The BOOTSTRAP method, with 1,000 replicates, was used to evaluate the reliability of the phylogenetic tree.
3. RESULTS
3.1. Sequence homology analysis
In this study, 30 completed S‐genes from PEDV collected in 11 different provinces throughout Vietnam were successfully sequenced. The length of these sequences varied between 4,149 and 4,161 nucleotides (nt). The difference in lengths of the S gene among Vietnamese PEDV strains was mostly due to the number of INDEL mutations accumulated in the N‐terminus of the S‐gene (data not shown), indicating the genetic variation of the circulating PEDVs in Vietnam. In particularly, compared to the S gene of the PED prototype strain CV777, the aa deletions and insertions were found at aa 167–168 and 59–62, respectively. In addition, in comparison with the S genes of the other Vietnamese PEDV strains indicated that the HUA‐PED176 and HUA‐PED254 strains have three deletions (a 3‐nt deletion at position 163–164; a 9‐nt deletion at position 173–174; and a 3‐nt deletion at position 405–406) and a 6‐nt insertion at position 406–461. These deletions and insertion were also found in US‐INDEL strains (Iowa106/2013, OH851/2014, Ohio126/2014), suggesting that common origin may have occurred within these strains.
The nucleotide (nt) and amino acid (aa) identity of the Vietnamese PEDV strains active during 2015–2016 were 93.2%–99.9% and 92.6%–99.9%, respectively (Table S1). To be specific, the identity of nucleotides and amino acids among S gene sequences active in the same year were about 93.2%–99.9% and 92.7%–99.9%, respectively. The amino acid similarity among the G1 and G2 groups of the study strains was 98.5%–99.3% and 94.3%–99.9%, respectively.
3.2. Recombination and phylogenetic analysis
Results from RDP v4 and SimPlot 3.5.1 analysis indicated that the PED strains in this study revealed no recombination event among the reference strains (data not shown). Phylogenetic analysis indicated that all PEDV isolates collected in Vietnam during the 2015–2016 outbreaks belonged to two distinct groups, G2 and G1 (Figure 1). In brief, two strains (HUA‐PED176 and HUA‐PED254) collected in Ho Chi Minh City in 2016 were clustered into the G1b sub‐group, while the remaining isolates clustered into the G2 group (both G2a and G2b sub‐groups) which included PEDV strains that caused recent outbreaks in China, United States and Thailand. Interestingly, while HUA‐PED94, HUA‐PED96, HUA‐PED146 and HUA‐PED120 were closed related to PEDV strains recently circulating in United States, the other strains were more genetically related to PEDV strains recently circulating in China (Figure 1).
FIGURE 1.
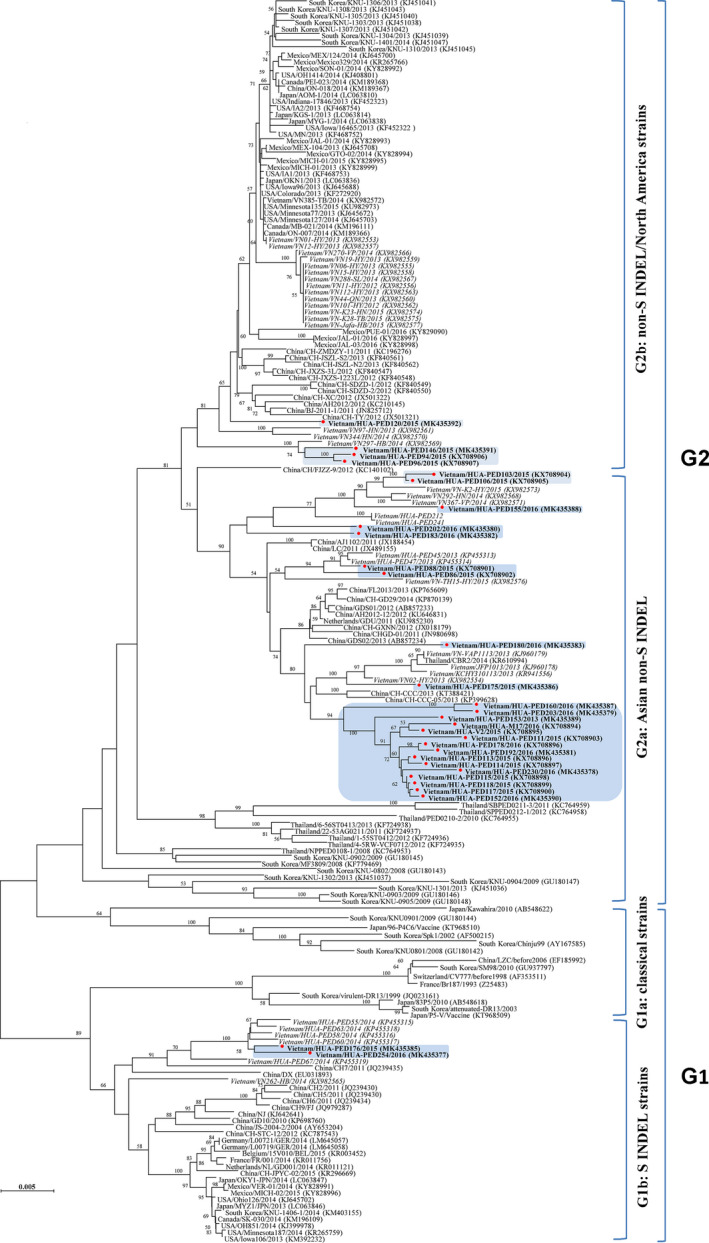
Phylogenetic tree of the Vietnamese PEDV strains (the marked triangle) and other PEDV reference strains used in this study. PEDV, porcine epidemic diarrhea virus
3.3. Analysis of the deduced amino acid sequence of the S genes of Vietnamese PEDV strains
There are four major epitopes capable of inducing neutralizing antibodies located in the S‐protein: the COE region, SS2, SS6 and 2C10. In our study, SS2 and 2C10 motifs were well conserved, with only one change at aa 753C‐R of HUA‐PED103 (collected in Lao Cai in 2015) in the SS2 region, and at aa 1372P‐S of HUA‐PED114 (collected in Thai Nguyen in 2015) in the 2C10 region (Figure 2). In the SS6 region, the Vietnamese PEDV strains as well as topotypes of emergent S‐indel and non‐S‐indel strains shared mutations, as compared to the classical strain CV777, at aa 765D‐S for all strains, and at aa 771A‐V/T for HUA‐PED176 and HUA‐PED254 (Figure 2). In addition, numerous mutations were observed in the COE region (516A‐S/V; 519G‐D, 520H‐S/R/P/Y, 521S‐G, 524N‐D, 526I‐V/L, 537S‐F, 548T‐R/S, 555N‐S, 562K‐N, 565D‐Y, 575V‐I, 580S‐L, 582S‐N, 586V‐A, 590L‐F, 593G‐S, 602Y‐C/H, 604E‐H/A/D, 608G‐S, 609V‐G, 611F‐V/L, 612T‐A, 615Y‐L, 616F‐L, 617Q‐H/S, 618F‐S, 620K‐E, 629K‐T, 632Q‐E/V, 635T‐K) (Figure 2). These results indicate that the recent vaccine strain, DR13, may only partially induce neutralizing antibodies against emergent PEDV strains circulating in Vietnam.
FIGURE 2.
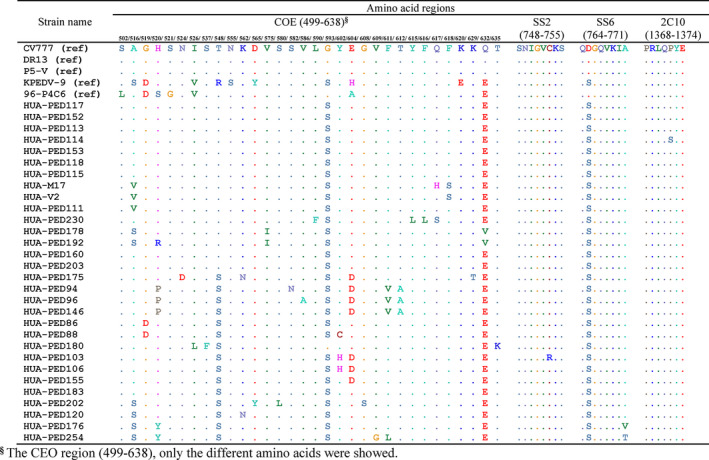
Amino acid alignment of the four major epitopes which are capable of inducing neutralizing antibodies located in S‐protein (COE, SS2, SS6 and 2C10) of the Vietnamese PEDV strains, with topotype of classical PEDV strain CV777, topotype of emergent mild pathogenic strain and topotype of emergent severe pathogenic strains. PEDV, porcine epidemic diarrhea virus
It should be noted that N‐glycosylation affected the cell tropism of viral pathogen in general. The number of N‐glycosylation sites in the study strains varied from 25 to 30 (Table S2). There were 14 significant differences in the N‐glycosylation patterns (5 and 9 N‐glycosylation patterns in the PEDV strains collected in the 2015 and 2016 outbreaks, respectively) which were mainly located in the N‐terminal region of S1 among PEDV strains isolated in Vietnam (Table S2). These results suggest that the effects of changes in N‐glycosylation sites on biological characteristics, pathogenicity and antigenicity of Vietnamese PEDVs need to be evaluated for further information regarding PEDV circulation and control strategies in Vietnam.
4. DISCUSSION
Similar to other coronavirus S proteins, the PEDV S protein, a surface spike glycoprotein, is critical for regulating the interactions of the virus with specific host cell receptor glycoproteins, which mediate viral entry (Bosch, Zee, Haan, & Rottier, 2003). Thus, the S protein is a primary target for vaccines against PEDV. The S protein is also the major envelope glycoprotein of the virion and it is important for understanding the genetic relationships among strains as well as their epidemiological status (Chen et al., 2012; Li, Li, et al., 2012; Li, Zhu, et al., 2012; Song & Park, 2012). In previous studies, phylogenetic analyses showed that PEDV strains could be divided into two main groups, G1 (G1a and G1b sub‐groups) and G2 (G2a and G2b sub‐groups) (Kim et al., 2015; Song, Moon, & Kang, 2015). In this study, the two PEDV strains (HUA‐PED176, HUA‐PED254) collected from Ho Chi Minh city located in the south of Vietnam belonged to sub‐group G1b, which includes mild classical US‐PEDV strains, while the other PEDV strains collected from the northern and central regions of Vietnam belonged to group G2 (G2a and G2b sub‐groups) which includes strains that recently emerged in the United States, China and other countries (Lin, Saif, Marthaler, & Wang, 2016). Interestingly, three PEDV strains isolated from Hung Yen province (HUA‐PED94, HUA‐PED96, HUA‐PED120) and one strain isolated from Hanoi (HUA‐PED146) located in the north of Vietnam were distantly related to strains collected from other northern provinces. Similar results were reported in a pandemic that recently occurred in South Korea (Lee, 2015). These results indicate that emergent, highly virulent PEDV strains could be divided into two different groups, one including US‐like strains appearing currently worldwide and the other tending to restrictively circulate in China, Vietnam and Thailand (Carvajal et al., 2015; Chiou et al., 2017; Lin et al., 2016). As a result, we conclude that PEDV strains belonging to the G2 group were the major agent of PED outbreaks in Vietnam as a whole, but G1b‐PEDV strains still exist in the centre and south of Vietnam. Therefore, further investigation into the molecular characterization of Vietnamese PEDV strains is needed to monitor outbreaks.
In the present study, aa insertions and deletions were found in the S protein of Vietnamese PEDV strains. The PEDV strains in the G2 group contained five aa insertions at residues 59–62 and 143 and two aa deletions at residues 167–168 when compared to the G1 group (G1b sub‐group). Most of the sequence variations were in the N‐terminal region of S1. This finding was similar to previous reports showing that the N‐terminal region of S1 was the most variable region of the S protein (Kim et al., 2015; Lee, Park, Kim, & Lee, 2010; Park et al., 2007). Several regions of the S protein were shown to be related to neutralizing antigenic sites. Antigenic analysis showed that the aa sequences of the SS2 and 2C10 epitopes were conserved among Vietnamese PEDV strains, and they were identical to that of the prototype PEDV CV777 strain. However, the substitution mutations in the SS6 epitope and COE domain changed their antigenic characteristics. The present results suggest that these mutations might be related to virus evolution and immune evasion. This finding is also in agreement with the results of previous studies and suggests that the PED vaccine strains, DR13, e.g., may only induce partial neutralizing antibodies against emergent PED field virus strains (Chung et al., 2016; Hao, Xue, He, Wang, & Cao, 2014; Horie, Kabemura, Masatani, Matsuu, & Ozawa, 2016).
N‐glycosylation of the virus plays an important role in host cell attachment and release, glycan shielding and infectivity (Vigerust & Shepherd, 2007). Site directed mutagenesis of the S gene of SARS‐Coronavirus demonstrated that N‐glycosylation sites were crucial for viral infection (Han, Lohani, & Cho, 2007). However, the function of each N‐glycosylation residue is still unknown. In the present study, the G1 and G2 PEDV strains had different N‐glycosylation patterns. While the G1 strains had similar N‐glycosylation patterns and were closely related genetically to the prototype PEDV CV777 strain, the group G2 strains were genetically distant from the CV777 strain (data not shown). The PEDV G2 strains were closely related to the new emerging PEDV strain USA/Colorado/2013 (isolated in 2013). These Vietnamese PEDV G2 strains collected from the north of Vietnam in 2015–2016 PED outbreaks shared the same N‐glycosylation patterns as the USA/Colorado/2013 strain. This study is the first report of the appearance and prevalence of US‐like PEDV strains in Vietnam. Further study is needed to investigate the prevalence of these PEDV isolates throughout Vietnam.
5. CONCLUSION
Genetic analysis showed that the S gene sequences of Vietnamese PEDV strains collected from three distinct regions (northern, central and southern provinces) of Vietnam in 2015–2016 were heterogeneous. Phylogenetic analysis of the complete S gene sequences showed that there were two types of PEDV strains circulating in Vietnam belonging to the G1 group (G1b sub‐group) and G2 group (both G2a and G2b sub‐groups). Further analysis indicated that there were variations in the antigenic epitopes and N‐glycosylation patterns among the circulating PEDV strains in Vietnam. This study, for the first time, provides up‐to‐date circulation and genetic information of Vietnamese PEDV strains and may contribute to the development of more effective vaccines in the future.
CONFLICT OF INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
AUTHORS’ CONTRIBUTIONS
VPL and VTT conceived and designed the proposal. VPL and TTHV performed the experiments. VTT, SEC, TTHV, TDD, TLN, TTNB, TNM, RMC, DS, DJA and VPL participated in analysing the data. VTT and VPL wrote the paper. All authors have read and approved the final manuscript.
ETHICAL STATEMENT
The authors confirm that the ethical policies of the journal, as noted on the journal's author guidelines page, have been adhered to and the appropriate internal ethics review committee approvals has been received.
Supporting information
FigS1
TableS1
TableS2
ACKNOWLEDGEMENTS
This work was supported by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106‐NN.04‐2014.16.
Than VT, Choe S‐E, Vu TTH, et al. Genetic characterization of the spike gene of porcine epidemic diarrhea viruses (PEDVs) circulating in Vietnam from 2015 to 2016. Vet Med Sci. 2020;6:535–542. 10.1002/vms3.256
REFERENCES
- Bosch, B. J. , van der Zee, R. , de Haan, C. A. , & Rottier, P. J. (2003). The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. Journal of Virology, 77, 8801–8811. 10.1128/JVI.77.16.8801-8811.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgen, A. , Duarte, M. , Tobler, K. , Laude, H. , & Ackermann, M. (1993). Sequence determination of the nucleocapsid protein gene of the porcine epidemic diarrhoea virus confirms that this virus is a coronavirus related to human coronavirus 229E and porcine transmissible gastroenteritis virus. Journal of General Virology, 74(Pt 9), 1795–1804. 10.1099/0022-1317-74-9-1795 [DOI] [PubMed] [Google Scholar]
- Carvajal, A. , Argüello, H. , Martínez‐Lobo, F. J. , Costillas, S. , Miranda, R. , de Nova, P. J. , & Rubio, P. (2015). Porcine epidemic diarrhoea: New insights into an old disease. Porcine Health Management, 1, 12 10.1186/s40813-015-0007-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Liu, X. , Shi, D. , Shi, H. , Zhang, X. , Li, C. , … Feng, L. (2013). Detection and molecular diversity of spike gene of porcine epidemic diarrhea virus in China. Viruses, 5, 2601–2613. 10.3390/v5102601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X. , Yang, J. , Yu, F. , Ge, J. , Lin, T. , & Song, T. (2012). Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) samples from field cases in Fujian, China. Virus Genes, 45, 499–507. 10.1007/s11262-012-0794-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou, H. Y. , Huang, Y. L. , Deng, M. C. , Chang, C. Y. , Jeng, C. R. , Tsai, P. S. , … Chang, H. W. (2017). Phylogenetic analysis of the spike (S) gene of the new variants of porcine epidemic diarrhoea virus in Taiwan. Transboundary and Emerging Diseases, 64, 157–166. 10.1111/tbed.12357 [DOI] [PubMed] [Google Scholar]
- Chung, H. C. , Lee, J. H. , Nguyen, V. G. , Huynh, T. M. L. , Lee, G. E. , Moon, H. J. , … Park, B. K. (2016). New emergence pattern with variant porcine epidemic diarrhea viruses, South Korea, 2012–2015. Virus Research, 226, 14–19. 10.1016/j.virusres.2016.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep, N. V. , Sueyoshi, M. , Izzati, U. , Fuke, N. , Teh, A. P. P. , Lan, N. T. , & Yamaguchi, R. (2018). Appearance of US‐like porcine epidemic diarrhoea virus (PEDV) strains before US outbreaks and genetic heterogeneity of PEDVs collected in Northern Vietnam during 2012–2015. Transboundary and Emerging Diseases, 65, e83–e93. 10.1111/tbed.12681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duy, D. T. , Toan, N. T. , Puranaveja, S. , & Thanawongnuwech, R. (2011). Genetic characterization of porcine epidemic diarrhea virus (PEDV) isolates from Southern Vietnam during 2009–2010 outbreaks. The Thai Journal of Veterinary Medicine, 41, 55–64. [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 95–98.
- Han, D. P. , Lohani, M. , & Cho, M. W. (2007). Specific asparagine‐linked glycosylation sites are critical for DC‐SIGN‐ and L‐SIGN‐mediated severe acute respiratory syndrome coronavirus entry. Journal of Virology, 81, 12029–12039. 10.1128/JVI.00315-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, J. , Xue, C. , He, L. , Wang, Y. , & Cao, Y. (2014). Bioinformatics insight into the spike glycoprotein gene of field porcine epidemic diarrhea strains during 2011–2013 in Guangdong, China. Virus Genes, 49, 58–67. 10.1007/s11262-014-1055-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie, M. , Kabemura, M. , Masatani, T. , Matsuu, A. , & Ozawa, M. (2016). Isolation and molecular characterization of porcine epidemic diarrhea viruses collected in Japan in 2014. Archives of Virology, 161, 2189–2195. 10.1007/s00705-016-2900-1 [DOI] [PubMed] [Google Scholar]
- Huang, Y. W. , Dickerman, A. W. , Pineyro, P. , Li, L. , Fang, L. , Kiehne, R. , … Meng, X. J. (2013). Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. MBio, 4, e00737–e00813. 10.1128/mBio.00737-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis, M. C. , Lam, H. C. , Zhang, Y. , Wang, L. , Hesse, R. A. , Hause, B. M. , … Marthaler, D. (2016). Genomic and evolutionary inferences between American and global strains of porcine epidemic diarrhea virus. Preventive Veterinary Medicine, 123, 175–184. 10.1016/j.prevetmed.2015.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. K. , Lim, S.‐I. , Lim, J.‐A. , Cho, I.‐S. , Park, E.‐H. , Le, V. P. , … An, D.‐J. (2015). A novel strain of porcine epidemic diarrhea virus in Vietnamese pigs. Archives of Virology, 160, 1573–1577. 10.1007/s00705-015-2411-5 [DOI] [PubMed] [Google Scholar]
- Kusanagi, K. , Kuwahara, H. , Katoh, T. , Nunoya, T. , Ishikawa, Y. , Samejima, T. , & Tajima, M. (1992). Isolation and serial propagation of porcine epidemic diarrhea virus in cell cultures and partial characterization of the isolate. Journal of Veterinary Medical Science, 54, 313–318. 10.1292/jvms.54.313 [DOI] [PubMed] [Google Scholar]
- Lee, C. (2015). Porcine epidemic diarrhea virus: An emerging and re‐emerging epizootic swine virus. Virology Journal, 12, 193 10.1186/s12985-015-0421-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. K. , Park, C. K. , Kim, S. H. , & Lee, C. (2010). Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Research, 149, 175–182. 10.1016/j.virusres.2010.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , & Lee, C. (2014). Outbreak‐related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerging Infectious Diseases, 20, 1223–1226. 10.3201/eid2007.140294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Li, H. , Liu, Y. , Pan, Y. , Deng, F. , Song, Y. , … He, Q. (2012). New variants of porcine epidemic diarrhea virus, China, 2011. Emerging Infectious Diseases, 18, 1350–1353. 10.3201/eid1808.120002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. L. , Zhu, L. , Ma, J. Y. , Zhou, Q. F. , Song, Y. H. , Sun, B. L. , … Bee, Y. Z. (2012). Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) field strains in south China. Virus Genes, 45, 181–185. 10.1007/s11262-012-0735-8 [DOI] [PubMed] [Google Scholar]
- Lin, C. N. , Chung, W. B. , Chang, S. W. , Wen, C. C. , Liu, H. , Chien, C. H. , & Chiou, M. T. (2014). US‐like strain of porcine epidemic diarrhea virus outbreaks in Taiwan, 2013–2014. Journal of Veterinary Medical Science, 76, 1297–1299. 10.1292/jvms.14-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. M. , Saif, L. J. , Marthaler, D. , & Wang, Q. (2016). Evolution, antigenicity and pathogenicity of global porcine epidemic diarrhea virus strains. Virus Research, 226, 20–39. 10.1016/j.virusres.2016.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole, K. S. , Bollinger, R. C. , Paranjape, R. S. , Gadkari, D. , Kulkarni, S. S. , Novak, N. G. , … Ray, S. C. (1999). Fulllength human immunodeficiency virus type 1 genomes from subtype C‐infected seroconverters in India, with evidence of intersubtype recombination. Journal of Virology, 73, 152–160. 10.1128/jvi.73.1.152-160.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Murrell, B. , Golden, M. , Khoosal, A. , & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1(1), 1–5. 10.1093/ve/vev003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda, T. , Murakami, S. , Takahashi, O. , Miyazaki, A. , Ohashi, S. , Yamasato, H. , & Suzuki, T. (2015). New porcine epidemic diarrhoea virus variant with a large deletion in the spike gene identified in domestic pigs. Archives of Virology, 160, 2565–2568. 10.1007/s00705-015-2522-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, S. , Miyazaki, A. , Takahashi, O. , Hashizume, W. , Hase, Y. , Ohashi, S. , & Suzuki, T. (2015). Complete genome sequence of the porcine epidemic diarrhea virus variant Tottori2/JPN/2014. Genome Announc, 3(4), e00877‐15 10.1128/genomeA.00877-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojkic, D. , Hazlett, M. , Fairles, J. , Marom, A. , Slavic, D. , Maxie, G. , … Burlatschenko, S. (2015). The first case of porcine epidemic diarrhea in Canada. Canadian Veterinary Journal, 56, 149–152. [PMC free article] [PubMed] [Google Scholar]
- Park, S. , Kim, S. , Song, D. , & Park, B. (2014). Novel porcine epidemic diarrhea virus variant with large genomic deletion, South korea. Emerging Infectious Diseases, 20, 2089–2092. 10.3201/eid2012.131642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S. J. , Moon, H. J. , Yang, J. S. , Lee, C. S. , Song, D. S. , Kang, B. K. , & Park, B. K. (2007). Sequence analysis of the partial spike glycoprotein gene of porcine epidemic diarrhea viruses isolated in Korea. Virus Genes, 35, 321–332. 10.1007/s11262-007-0096-x [DOI] [PubMed] [Google Scholar]
- Pensaert, M. B. , & de Bouck, P. (1978). A new coronavirus‐like particle associated with diarrhea in swine. Archives of Virology, 58, 243–247. 10.1007/BF01317606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puranaveja, S. , Poolperm, P. , Lertwatcharasarakul, P. , Kesdaengsakonwut, S. , Boonsoongnern, A. , Urairong, K. , … Thanawongnuwech, R. (2009). Chinese‐like strain of porcine epidemic diarrhea virus, Thailand. Emerging Infectious Diseases, 15, 1112–1115. 10.3201/eid1507.081256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , Moon, H. , & Kang, B. (2015). Porcine epidemic diarrhea: A review of current epidemiology and available vaccines. Clinical and Experimental Vaccine Research, 4, 166–176. 10.7774/cevr.2015.4.2.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, D. , & Park, B. (2012). Porcine epidemic diarrhoea virus: A comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes, 44, 167–175. 10.1007/s11262-012-0713-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaan, W. , Cavanagh, D. , & Horzinek, M. C. (1988). Coronaviruses: Structure and genome expression. Journal of General Virology, 69(Pt 12), 2939–2952. 10.1099/0022-1317-69-12-2939 [DOI] [PubMed] [Google Scholar]
- Sun, M. , Ma, J. , Wang, Y. , Wang, M. , Song, W. , Zhang, W. , … Yao, H. (2015). Genomic and epidemiological characteristics provide new insights into the phylogeographical and spatiotemporal spread of porcine epidemic diarrhea virus in Asia. Journal of Clinical Microbiology, 53, 1484–1492. 10.1128/JCM.02898-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Y. , Yu, Z. , Cheng, K. , Liu, Y. , Huang, J. , Xin, Y. , … Xia, X. (2013). Molecular characterization and phylogenetic analysis of new variants of the porcine epidemic diarrhea virus in Gansu, China in 2012. Viruses, 5, 1991–2004. 10.3390/v5081991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigerust, D. J. , & Shepherd, V. L. (2007). Virus glycosylation: Role in virulence and immune interactions. Trends in Microbiology, 15, 211–218. 10.1016/j.tim.2007.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova, A. N. , Marthaler, D. , Wang, Q. , Culhane, M. R. , Rossow, K. D. , Rovira, A. , … Saif, L. J. (2014). Distinct characteristics and complex evolution of PEDV strains, North America, May 2013‐February 2014. Emerging Infectious Diseases, 20, 1620–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Byrum, B. , & Zhang, Y. (2014). New variant of porcine epidemic diarrhea virus, United States, 2014. Emerging Infectious Diseases, 20, 917–919. 10.3201/eid2005.140195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, E. N. (1977). An apparently new syndrome of porcine epidemic diarrhoea. The Veterinary Record, 100, 243–244. 10.1136/vr.100.12.243 [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Gaschen, B. , Blay, W. , Foley, B. , Haigwood, N. , Kuiken, C. , & Korber, B. (2004). Tracking global patterns of N‐linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology, 14, 1229–1246. 10.1093/glycob/cwh106 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigS1
TableS1
TableS2