

Abstract
Understanding the relationship between the metallocofactor and its protein environment is the key to uncovering the mechanism of metalloenzymes. PqqE, a radical S-adenosylmethionine (RS) enzyme in pyrroloquinoline quinone biosynthesis, contains three iron-sulfur cluster binding sites. Two auxiliary iron-sulfur cluster binding sites, designated as AuxI and AuxII, use distinctive ligands compared to other proteins in the family while their functions remain unclear. Here, we investigate the electronic properties of these iron-sulfur clusters and compare the catalytic efficiency of wild-type (WT) Methylorubrum extorquens AM1 PqqE to a range of mutated constructs. Using native mass spectrometry, protein film electrochemistry, and electron paramagnetic resonance spectroscopy, we confirm the previously proposed incorporation of a mixture of [2Fe-2S] and [4Fe-4S] clusters at the AuxI site and are able to assign redox potentials to each of the three iron-sulfur clusters. Significantly, a conservative mutation at AuxI, C268H, shown to selectively incorporate a [4Fe-4S] cluster, catalyzes an enhancement of uncoupled S-adenosylmethionine cleavage relative to WT, together with the elimination of detectable peptide cross-linked product. While a [4Fe-4S] cluster can be tolerated at the AuxI site, the aggregate findings suggest a functional [2Fe-2S] configuration within the AuxI site. PqqE variants with non-destructive ligand replacements at AuxII also show that the reduction potential at this site can be manipulated by changing the electronegativity of the unique aspartate ligand. A number of novel mechanistic features are proposed based on the kinetic and spectroscopic data. Additionally, bioinformatic analyses suggest that the unique ligand environment of PqqE may be relevant to its role in PQQ biosynthesis within an oxygen-dependent biosynthetic pathway.
Keywords: Pyrroloquinoline Quinone PQQ Biosynthesis, SPASM-Domain Enzyme, Radical SAM Enzyme, PqqE, Ribosomally-Synthesized and Post-Translationally-Modified Peptide RiPP, PqqA, Iron-Sulfur Cluster, Carbon-Carbon Crosslink Reaction, Native Mass Spectrometry, Protein Film Electrochemistry, Electron Paramagnetic Resonance, Catalytic Mechanism
Graphical Abstract

Introduction
Iron-sulfur cluster-containing proteins, such as nitrogenase,1 hydrogenase,2 ferredoxin,3 and mitochondrial respiration complexes4 sustain fundamental biological processes within cells. The radical S-adenosylmethionine (RS) superfamily represents one of the major iron-sulfur cluster-containing enzymes that are widely distributed across all life kingdoms and catalyze stunning transformations in highly diverse biosynthetic pathways.5 A common feature of canonical RS enzymes is the CxxxCxxC motif in a (α/β)6 TIM barrel, for which the cysteines coordinate to three irons in a [4Fe-4S] cluster, leaving one iron site open for coordination to the co-substrate S-adenosylmethionine (SAM).5,6 Upon anaerobic reduction of the iron-sulfur site, from its resting [4Fe-4S]2+ state to [4Fe-4S]1+, an intramolecular electron transfer to SAM generates a 5′-deoxyadenosyl (5′-dA) radical, initiating a cascade of free radical chemistry.7,8 Currently, over 100,000 homologs catalyzing more than 80 different types of reactions have been found within this superfamily, the majority of which encode unassigned functions.9–11
Among the RS superfamily, a significant number of enzymes contain SPASM/twitch domains that were initially identified in the maturases of Subtilosin, Pyrroloquinoline quinone (PQQ), Anaerobic Sulfatases, and Mycofactocin.9 Structurally, the SPASM/twitch domain is a C-terminal hairpin loop extension that coordinates either one (twitch) or two (SPASM) additional iron-sulfur clusters.12 Many enzymes of this subfamily (cf. SkfB,13 CteB,14 AlbA,15 MftC,16 WkgB,17 and SuiB18) catalyze new thioether or challenging carbon-carbon bond formation within ribosomally-synthesized and post-translationally modified peptides (RiPPs). Possible roles for these auxiliary clusters include substrate binding,14,19,20 structural stability,21,22 and electron transfer.23,24
PQQ, a prokaryotic redox cofactor, arises from a heavily processed RiPP.25 It is found in plants and animals, including humans, as a result of symbiotic interspecies relationships and/or dietary influx.26–28 Besides its role in bacteria, PQQ serves as a plant growth promoter, enhancing mitochondria-related functions, as well as preventing hepatic lipotoxicity and inflammation in humans.29,30 The pathway for the production of PQQ in bacteria has been studied over several decades31–44 yet details of catalysis in each enzyme in the pathway still require further investigation (Figure 1). In this paper, we focus on the first step of PQQ biosynthesis catalyzed by the RS-SPASM enzyme, PqqE, in complex with a chaperone protein, PqqD, which binds and positions the substrate PqqA, leading to the formation of a carbon-carbon cross-link between the side chains of Glu-16 and Tyr-20 (residue number in Methylorubrum extorquens AM1) in the peptide substrate, PqqA.35
Figure 1. Biosynthesis of PQQ in Methylobacterium extorquens AM1.

PQQ biosynthesis requires 7 genes pqqABCDEFG, pqqF/G present outside of the operon. The first step of PQQ biosynthesis is the C-C crosslink of glutamate and tyrosine of PqqA. The reaction is catalyzed by PqqE in the presence of chaperone PqqD, reducing reagent and SAM. The crosslinked PqqA is shown undergoing digestion by proteinase PqqF/G41 prior to the action of PqqB. By analyzing the activity of substrate analogues, an excised cross-linked Glu-Tyr is proposed to generate oxidation by PqqB41, followed by spontaneous cyclization, and an eight-electron oxidation by PqqC40 finally forming PQQ.
The X-ray crystal structure of recombinant Methylorubrum extorquens PqqE shows two unique features of the SPASM domain.32 First, as predicted by sequence alignment,40,44 PqqE harbors an absolutely conserved aspartate ligand, Asp-319, at its AuxII site in contrast to the remainder of the RS-SPASM proteins (e.g., anSME and lysine-tryptophan cyclase) that employ only cysteines.44–48 In an earlier work, an alanine variant of Asp-319 in PqqE was shown to impair the integrity of the AuxII iron-sulfur cluster and abolish peptide modification activity, suggesting the catalytic importance of the AuxII site.32 The evolutionary benefit of using aspartate as a ligand in AuxII of PqqE has been unclear. Second, the structure of PqqE shows a well-defined [2Fe-2S] cluster that is ligated by four cysteines at the AuxI site. (Figure 2A) The available EPR and Mössbauer spectra of PqqE have further supported the existence of a [2Fe-2S] cluster at AuxI.22,32 Other RS-SPASM proteins that catalyze a C-C bond crosslink on RiPPs, such as MftC,16 WgkB17 and StrB/SuiB,18,47–49 universally contain a [4Fe-4S] cluster at AuxI (as well as AuxII) (Figure 2B). Recently, a [2Fe-2S] cluster was found in the twitch domain of SkfB, a sporulation killing factor maturation protein50 (Figure 2C), while an earlier study suggested that an [4Fe-4S] cluster could be incorporated at this site.13,50 A common feature of PqqE and SkfB is that the [2Fe-2S] cluster is liganded by a CXC motif, where each Cys coordinates to a different iron atom in the center. Analogous to SkfB,50 computer modeling22 and spectroscopy data51 have also provided evidence that AuxI in PqqE can exist as a [4Fe-4S]. Using Ti(III) citrate and Eu(II)-DTPA as reducing reagents, a new EPR active species with g = 2.104 was recently assigned as a four-cysteine liganded [4Fe-4S] cluster in the AuxI site of PqqE.51 (Figure S1).
Figure 2. Crystal structure of PqqE and other SPASM/twitch auxiliary iron-sulfur clusters.

A) Crystal structure of PqqE (PDB 6C8V). The iron-sulfur cluster binding sites are shown in a dashed rectangle to the right. The zoom-in structure shows the ligand environment of each iron-sulfur cluster in PqqE. The structural model of the RS [4Fe-4S] cluster was built by SWISS-MODEL (https://swissmodel.expasy.org/) using CteB (PDB 5WGG) as a structural template. B) The crystal structure of SuiB (PDB 5V1Q) and its [4Fe-4S] AuxI site. C) The crystal structure of SkfB (PDB 6EFN) and its auxiliary [2Fe-2S] cluster.
To better understand how the unusual auxiliary iron-sulfur clusters and their ligand environment contribute to the function of PqqE, we have pursued a detailed examination of the iron-sulfur clusters at AuxI and AuxII, using native mass spectrometry (native MS), protein film electrochemistry (PFE) and electron paramagnetic resonance (EPR) spectroscopy. These studies are focused not only on iron-sulfur cluster knockouts, but on several variants with non-destructive ligand replacements that preserve iron-sulfur cluster integrity. The findings support earlier reports that either a [4Fe-4S] or [2Fe-2S] cluster can be incorporated into the AuxI site of WT PqqE.51 The use of a single-site Cys to His replacement within AuxI restricts the iron-sulfur center to [4Fe-4S] while abrogating peptide substrate cross-linking activity. This unexpected observation implicates [2Fe-2S] at AuxI may support the catalytic activity of PqqE. Pursuing the replacement of side chains that bind iron at AuxII, we further identify mutations that fine-tune the redox properties at this cluster as well as catalytic activity. The present combination of site-specific mutagenesis, electrochemistry and spectroscopy measurements expands our understanding of the uniquely evolved and conserved SPASM domain of PqqE, leading to new mechanistic perspectives regarding function.
Results and Discussion
Protein preparation does not affect the presence of the [2Fe-2S] cluster.
Intrigued by the unique [2Fe-2S] cluster in the AuxI site in the crystal structure of PqqE among functionally related enzymes, we first asked whether the incorporation of the [2Fe-2S] could be an artifact of the iron-sulfur cluster assembly machinery that is co-expressed in the production of PqqE. In previous studies, an N-terminal His-tagged PqqE was used as the standard construct for protein expression and purification.33 The plasmid pPH149 or pPH151 containing E. coli IscSUA-HscBA-Fd or E. coli sufABCDE genes was expressed along with PqqE, to support iron-sulfur cluster biogenesis during PqqE over-expression; no difference was found using either plasmid.40 Since these two iron-sulfur cluster assembly systems are both from E. coli, to further test for possible artifacts we co-expressed the His-tagged PqqE with pDB1282, a plasmid containing the isc operon for iron-sulfur cluster assembly machinery from Azotobacter vinelandii.52 In the presence of pDB1282, the expression of PqqE in E. coli BL21(DE3)Gold yielded 10 ± 1 Fe per monomer in the as-purified protein, which is similar to the previously reported data using protein co-expressed with pPH151.32 When the as-purified protein sample was reduced by dithionite (DTH), EPR spectra were also similar to those of the His-tagged PqqE co-expressed with pPH151 (Figure S2). Therefore, it is unlikely that the [2Fe-2S] cluster observed in PqqE is a result of unsuitable iron-sulfur assembly machinery for cluster insertion.
We next modified the purification procedure by introducing a Strep-tag at the N-terminus of PqqE. As demonstrated in our previous EPR study,51 the N-terminal Strep-tagged PqqE benefits from a straightforward two-step purification protocol (QIAGEN, MD) with minimal disruption to the iron-sulfur clusters.53 For this reason, subsequent studies herein are restricted to the use of the Strep-tagged PqqE construct. We note that the [4Fe-4S] clusters in the Strep-tagged PqqE are comparable with the chemically reconstituted His-tagged protein and that a [2Fe-2S] cluster remains in the PqqE sample as detected by X-band EPR.51 This indicates that the [2Fe-2S] cluster is unlikely to be an artifact of the preparation and purification of His-tagged protein.
In M-cluster maturation studies of nitrogenase, synthetic [4Fe-4S] clusters have been successfully reconstituted to apo-protein as a replacement of endogenous iron-sulfur clusters.54 We then tested whether we could remove the [2Fe-2S] cluster inserted during expression and reconstitute the apo-PqqE with a chemically synthesized [4Fe-4S] cluster. When PqqE was treated with the iron chelator, bathophenanthroline disulfonate, for 1 hour on ice, PqqE formed aggregates and did not reconstitute with any synthetic [4Fe-4S] clusters. Shortening the chelating time to 15 min resulted in PqqE containing only [2Fe-2S] cluster, which was confirmed by the DTH-reduced sample in EPR spectra (Figure S4). We rationalized that apo-protein unfolds and cannot be recovered by the addition of a synthetic [4Fe-4S] cluster when the iron-sulfur clusters are completely removed by the iron chelator. When chelating is incomplete, only the [2Fe-2S] cluster-containing protein survived the harsh process of cluster removal and reconstitution. This suggests that a [2Fe-2S] cluster at AuxI may be structurally stabilized by PqqE. The above observations point toward a [2Fe-2S] center as being an intrinsic property to PqqE.
Native mass spectrometry reveals the existence of both [2Fe-2S] and [4Fe-4S] cluster-containing PqqE.
The evidence that a [2Fe-2S] center is unlikely to be an artifact of protein preparation together with the recent attribution of a novel, low potential EPR signal to [4Fe-4S] at AuxI in PqqE, supports the view51 that either [2Fe-2S] or [4Fe-4S] can be inserted into this site. We sought to assess the molecular components of isolated PqqE using native mass spectrometry (MS) measurements. This technique takes advantage of the production of intact gaseous ions of protein together with its non-covalently bound binding partner by nanoelectrospray ionization,55,56 and has been effectively applied to proteins containing iron,57 copper,58 and iron-sulfur clusters (e.g., BioB,59 NsrR,60 and WhiB,60 O2-sensing fumarate and nitrate reduction protein,61 IscU62 and ferredoxin63,64). The goal of these experiments was to confirm that both the three [4Fe-4S] configuration and the two [4Fe-4S] and one [2Fe-2S] cluster configuration co-exist in preparations of holo-PqqE.
The as-purified PqqE was exchanged into ammonium carbonate buffer anaerobically prior to native MS measurement using positive ion mode.65 Multiple charge states of WT PqqE were observed in the resulting mass spectrum, the most abundant of which was the 13+, which was our main focus for molecular mass assignments. Multiple peaks with different mass-to-charge ratio (m/z) values were resolved in the 13+ charge state group, indicating the presence of complexes with different molecular mass values (Figure 3). The measured molecular mass values were all higher than the mass of apo-PqqE, 42987 Da, and a component having the molecular mass of apo-PqqE was not observed in the mass spectrum, suggesting that native MS retained iron-sulfur clusters in PqqE during the measurement. The iron-sulfur content of each species was calculated from the molecular mass difference between each detected species and apo-PqqE (See Supporting Information). As shown in Table 1, the as-purified PqqE solution is heterogeneous, harboring multiple iron and sulfur atoms. Among these species, we identified species F (Figure 3 and Table 1) with a molecular weight of 43857 ± 4 Da. This molecular weight corresponds to 10 added iron-sulfur pairs to the apo-PqqE with 10 deprotonated cysteine ligands (3 in the RS site, 4 in the AuxI site and 3 in the AuxII site), consistent with the presence of one [2Fe-2S] and two [4Fe-4S] clusters. This agrees with the presence of one [4Fe-4S] cluster each at the RS site and the AuxII site, and a [2Fe-2S] cluster at the AuxI site, as seen in the reported X-ray structure of PqqE32 and assigned by EPR.51 In the same spectrum, another species (H in Figure 3) was detected at much lower abundance (molecular weight of 44056 ± 4 Da, 1078 Da heavier than apo-PqqE). This mass difference corresponds to 12 iron and 12 sulfur atoms, and we ascribe H to a species with three [4Fe-4S] clusters and an additional sodium ion. This approach to iron and sulfur counting supports the presence of some [4Fe-4S] cluster in the AuxI site as well as [4Fe-4S] clusters in the RS and AuxII sites, as proposed previously.22,51 Species A, with 6 iron and 6 sulfur atoms, is assigned as the degradation product of species F containing one [4Fe-4S] and one [2Fe-2S] cluster, which represents the configuration with the missing RS site as observed in the X-ray crystal structure.32 Species B-E with lower iron and sulfur contents may be derived from species F-I via degradation of the iron-sulfur centers during protein purification and measurements (See Supporting Information).
Figure 3. Native MS confirms that the as-purified PqqE contains a mixture of iron-sulfur clusters.

A) Mass spectrum showing charge states of PqqE formed in native MS. The 13+ charge state is formed at the highest abundance. B) View of the mass spectrum showing detail for the 13+ charge state. Species are labeled alphabetically and the molecular weights of each species are listed in Table 1.
Table 1. Native MS confirms that the as-purified PqqE contains the previously proposed two [4Fe-4S] and one [2Fe-2S] species as well as a form with three [4Fe-4S] species.
The ligand mass is calculated from the differences of molecular weights between deconvoluted holo-PqqE from MS and the apo-PqqE (42987 Da) when 10 cysteines are deprotonated as ligands (42977 Da). Species that are shaded (A, F and H) have been observed previously by X-ray crystallography, and by EPR and Mössbauer spectroscopy. A more detailed analysis of the proposed iron and sulfur content and the proposed Fe-S cluster configurations is given in the Supporting Information.
| Deconvoluted holo-protein mass (Da) | Ligand mass (Da) | Proposed Fe/S content | Possible Fe-S cluster configuration | |
|---|---|---|---|---|
| I | 44094 ± 4 | 1117 | 12Fe-13S | 3[4Fe-4S]/Na/S |
| H | 44056 ± 4 | 1078 | 12Fe-12S | 3[4Fe-4S]/Na |
| G | 44021 ± 4 | 1044 | 12Fe-11S | 2[4Fe-4S]/[4Fe-3S]/Na |
| F | 43857 ± 4 | 880 | 10Fe-10S | 2[4Fe-4S]/[2Fe-2S] |
| E | 43820 ± 4 | 842 | 10Fe-9S | [4Fe-4S]/[4Fe-3S]/[2Fe-2S] |
| D | 43783 ± 4 | 806 | 10Fe-8S | 2[4Fe-3S]/[2Fe-2S] |
| C | 43625 ± 4 | 648 | 7Fe-8S | 2[2Fe-2S]/[3Fe-4S] |
| B | 43589 ± 4 | 611 | 7Fe-7S | 2[2Fe-2S]/[3Fe-3S] |
| A | 43551 ± 4 | 574 | 6Fe-6S | [4Fe-4S]/[2Fe-2S]/2Na |
The native MS results presented herein are fully consistent with PqqE accommodating either a [4Fe-4S] or [2Fe-2S] cluster at AuxI while maintaining [4Fe-4S] clusters at the RS and AuxII site. Significantly, the major components observed by MS, species D, E and F, can be ascribed to the enzyme form containing one [2Fe-2S] and two [4Fe-4S] clusters (F) or degradation products thereof (E and D). This is in agreement with the finding that the [2Fe-2S] cluster-containing PqqE was crystallized.32 While the greater abundance of species E and F than species H may argue that [2Fe-2S] is the dominant form of PqqE, it is also possible that species H is less stable having undergone degradation to species F in the course of purification and analysis. To our knowledge, this is the first time that native MS has been applied to a protein containing multiple iron-sulfur clusters with the objective of identifying the iron-sulfur cluster(s) in RS-SPASM/twitch proteins.
Bioinformatic analyses are more consistent with a functional [2Fe-2S] than [4Fe-4S] cluster in AuxI.
After experimentally confirming that either [2Fe-2S] or [4Fe-4S] can be incorporated at the AuxI site of PqqE, we sought structural support from the iron-sulfur cluster-containing proteins in the Protein Data Bank (PDB). Sequence alignment of PqqE across various species shows that the CRC motif in the AuxI site of the SPASM domain is fully conserved (Figure S5). It is generally believed that a CXC motif prefers to accommodate a [2Fe-2S] cluster.44 Here we examine the ligand environment of [2Fe-2S] and [4Fe-4S] clusters among existing structures in the PDB of unique protein sequences containing a CXC motif at an iron-sulfur cluster (Table S1).
CXC motif and [2Fe-2S] cluster.
Among 637 unique proteins associated with one or more [2Fe-2S] clusters, 47 proteins use CXC motifs as [2Fe-2S] cluster ligands. Among these 47 proteins, 27 of them employ both cysteines in the CXC motif as [2Fe-2S] cluster ligands, and 20 proteins use only one of the cysteines in the CXC motif (Figure 4A). For the 20 proteins that use only one cysteine as the [2Fe-2S] cluster ligand, there is either a cysteine or histidine located close to the iron (Table S1). For the 47 proteins that use cysteines as the [2Fe-2S] cluster ligands, 18 use each cysteine as a ligand to different irons within the cluster, which is similar to the ligand geometries of PqqE and SkfB (Figure 4A). Of potential significance, almost all the organisms that utilize a CXC motif (42/47) are found to grow aerobically or as facultative anaerobes (Figure 4B). The middle X residue is arginine in the majority of proteins (Figure 4C).
Figure 4. Bioinformatic analysis of the iron-sulfur cluster-containing proteins in the PDB that use a CXC motif in iron-sulfur cluster binding.

A) Among 637 [2Fe-2S] cluster-containing entries and 1016 [4Fe-4S] cluster-containing entries in the PDB, CXC motifs were found as ligands for 47 of [2Fe-2S] and 9 of [4Fe-4S] clusters. B) The majority of proteins containing a CXC binding [2Fe-2S] cluster are from aerobes or facultative anaerobes. C) The variation of the middle residue in CXC shows that CRC is found preferentially in [2Fe-2S]. D) The crystal structure of Ralstonia solanacearum CDGSH iron-sulfur protein (PDB 3TBM) shows a CRC motif binding to a [2Fe-2S] cluster using two cysteines to coordinate one iron. E) Crystal structure of E. coli aldehyde oxidase (PDB 5G5G) shows a CRC motif binding to a [2Fe-2S] cluster using two cysteines to coordinate two irons. F) Crystal structure of Methanothermococcus thermolithotrophicus heterodisulfide reductase (PDB 5ODC) shows a CHC motif binding to a [4Fe-4S] cluster using two cysteines to coordinate two irons and the presence of a fifth cysteine near the cluster.
CXC motif and [4Fe-4S] cluster.
A small set (9) of characterized proteins have been found to utilize CXC motifs within a [4Fe-4S] cluster (out of a total of 1016 [4Fe-4S] cluster-containing protein structures in the PDB) (Figure 4A). Only 2 of these 9 proteins use both cysteines in their CXC motifs as ligands to different irons, as observed for PqqE and SkfB, (Figure 4A) and the sole CRC containing protein does not use both cysteines as ligands (Figure 4C and Table S1). Further, among the identified proteins with CXC motifs where either one or both cysteines are ligated to a [4Fe-4S] cluster, the majority (6 out of 9) are present in strict anaerobes (Figure 4B).
[2Fe-2S] vs. [4Fe-4S] in PqqE.
The above comparative analysis shows that the CXC motif presents in both [2Fe-2S] and [4Fe-4S] clusters, with a strong preference for [2Fe-2S] cluster in proteins characterized from aerobic organisms. [4Fe-4S] clusters can convert to [2Fe-2S], and the latter form is more stable to oxygen.61,66 We note that the later steps of PQQ biosynthesis require dioxygen in the reactions catalyzed by PqqB43 and PqqC,41,42 while the PqqE-catalyzed reaction is a classic anaerobic process. Although we purify recombinant PqqE anaerobically, it is likely that PqqE will need to function under aerobic conditions in the native organism. This is consistent with our bioinformatic analysis, which shows a much greater presence of CXC/[2Fe-2S] cluster-containing enzymes among bacteria that grow aerobically. An earlier experimental study had suggested that PqqE is more oxygen tolerant in relation to other RS enzymes.67 The geometric dictates for the formation of a [2Fe-2S] (Figure 4D & E) vs. [4Fe-4S] (Figure 4F) cluster are distinct, raising the question of how a protein such as PqqE can accomplish binding of [4Fe-4S] as well. Two cysteines derived from outside of the CXC motif at the AuxI site of PqqE are both located in loop regions, and it is possible that small loop movements could create the geometry required for a [4Fe-4S] cluster.
Protein film electrochemistry allows the assignment of a reduction potential to each cluster within PqqE.
Protein film electrochemistry (PFE) has become a central tool in the investigation of the electronic properties of iron-sulfur clusters in proteins and in establishing structure-function relationships for complicated biological systems.68,69 Due to the complexity of the multiple iron-sulfur clusters in PqqE, we systematically generated a series of single and double cluster-knockout variants and used these as references to deconvolute the reduction potential of each cluster and to illustrate possible electronic interactions between each cluster (Table 2). The variants for which ligands have been altered at the auxiliary clusters are also listed in Table 2.
Table 2. Nomenclature of PqqE and its variants.
The name of each protein indicates the cluster(s) present in the protein. Detailed information on variants is given in the Supporting Information.
| Nomenclature | Description | Replaced residues |
|---|---|---|
| WT | As-purified wild-type PqqE | - |
| WT(R) | Chemically reconstituted wild-type PqqE | - |
| AuxI/AuxII | RS knockout | C28A/C32A/C35A |
| RS/AuxII | AuxI knockout | C248A/C268A |
| RS/AuxI | AuxII knockout | C310A/C313A |
| RS only | AuxI/AuxII double knockout | C310A/C313A/C323A/C325A |
| AuxI only | RS/AuxII double knockout | C28A/C32A/C35A /C310A/C313A |
| RS/AuxI/D319H | Single site mutation at Asp-319 | D319H |
| RS/AuxI/D319C | Single site mutation at Asp-319 | D319C |
| RS/D319C | D319C and AuxI knockout | C323A/C325A/D319C |
| AuxI/D319C | D319C and RS knockout | C28A/C32A/C35A/D319C |
| RS/C268H/AuxII | Single site mutation at Cys-268 | C268H |
Reduction potential assignment of each iron-sulfur cluster in PqqE.
Our native MS results show a major species in PqqE samples that contains [4Fe-4S] clusters at the RS and AuxII sites and a [2Fe-2S] cluster at the AuxI site. Previous EPR analyses51 have confirmed all three of these clusters can be reduced by DTH, consistent with reduction potentials that are more positive than DTH (−660 mV at pH 770). Cyclic and square-wave voltammetry measurements were performed on the WT PqqE and cluster knockouts using an approach recently employed for other SPASM/twitch domain proteins of the RS superfamily24,71,72 (Table 3). The cyclic voltammogram of the WT PqqE can be fit to three independent one-electron transfers with reduction potentials of −535 mV, −485 mV, and −435 mV vs. SHE (Figure 5A). Removal of single iron-sulfur clusters provides a further simplification: the cyclic voltammetry of the RS cluster knockout, which contains both AuxI and AuxII clusters (AuxI/AuxII), was fit to two independent one-electron transfers, with reduction potentials of −510 mV and −440 mV vs. SHE (Figure 5B). The cyclic voltammetry of the AuxI cluster knockout, which contains RS and AuxII clusters (RS/AuxII), was fit to two independent one-electron transfers, with reduction potentials of −525 mV and −460 mV vs. SHE (Figure 5C). The AuxII cluster knockout, which contains AuxI and RS clusters (RS/AuxI), was fit to two independent one-electron transfers, with reduction potentials of −495 mV and −435 mV vs. SHE (Figure 5D). The RS only variant, which contains only the [4Fe-4S] cluster located in the RS site, showed a potential of −455 mV vs. SHE, which is similar to the redox potential of the RS cluster assigned in other proteins (Figure 5E). The AuxI only variant (Figure 5F) showed the mid-point potential at −370 mV vs. SHE, which corresponds to the [2Fe-2S] cluster form in the AuxI site, as the EPR spectra of AuxI only suggest that there is only a [2Fe-2S] cluster present in this variant (Figure 6). This leads to the assignment of the −535 mV feature in the WT PqqE voltammogram to the AuxII cluster, the −485mV feature to the RS cluster, and the −435 mV feature to AuxI.
Table 3. Assignment of the reduction potential of each cluster in PqqE and its variants.
The reported reduction potentials of other SPASM/twitch domain proteins are listed for comparison. Uncertainties of the experimental redox potentials are ± 3 mV for the single cluster variants, and ± 5 mV for the other variants.
| Reduction potential (mV vs. S. H. E.) | |||
|---|---|---|---|
| PqqE variants | (RS [4Fe-4S]) | (AuxI [2Fe-2S]) | (AuxII [4Fe-4S]) |
| WT | −485 | −435 | −535 |
| AuxI/AuxII | - | −440 | −510 |
| RS/AuxII | −460 | - | −525 |
| RS/AuxI | −495 | −435 | - |
| RS only | −455 | - | - |
| AuxI only | - | −370 | - |
| RS/AuxI/D319H | −465 | −405 | −520 |
| RS/AuxI/D319C | −500 | −445 | −555 |
| RS/D319C | −440 | - | −495 |
| AuxI/D319C | - | −500 | −560 |
| RS/C268H/AuxII | −475 | - | −520 |
| Other SPASM/twitch | E1 | E2 | E3 |
Figure 5. Cyclic and square-wave voltammetry of PqqE and its cluster knockout variants.

For each protein, the cyclic voltammogram is shown on the left panel and the square-wave voltammogram is shown on the right panel. Measurements and the signal after baseline subtraction are shown in colored solid lines. Baseline measurements are shown in gray. A) WT signal (black) was fitted to three one-electron transfers: AuxII (green dashed line), AuxI (dark red dashed line) and RS (blue dashed line). B) AuxI/AuxII signal (purple) was fitted to two one-electron transfers (dashed line). C) RS/AuxII signal (pink) was fitted to two one-electron transfers (dashed line). D) RS/AuxI signal (magenta) was fitted to two one-electron transfers (dashed line). Reduction potentials obtained from cyclic and square-wave voltammetry of E) AuxI only (brown) and F) RS only (blue) were used for fitting the reduction potentials in single cluster knockouts. Cyclic voltammogram measured with a scan rate of 50 mV/s and square-wave voltammogram measured with a frequency of 15 Hz and amplitude of 50 mV using variants at 4 °C and pH 7.5 on pencil graphite electrode (PGE) modified with multiwall carbon nanotube (MWCNT).
Figure 6. X-band CW EPR spectra of AuxI only variant.
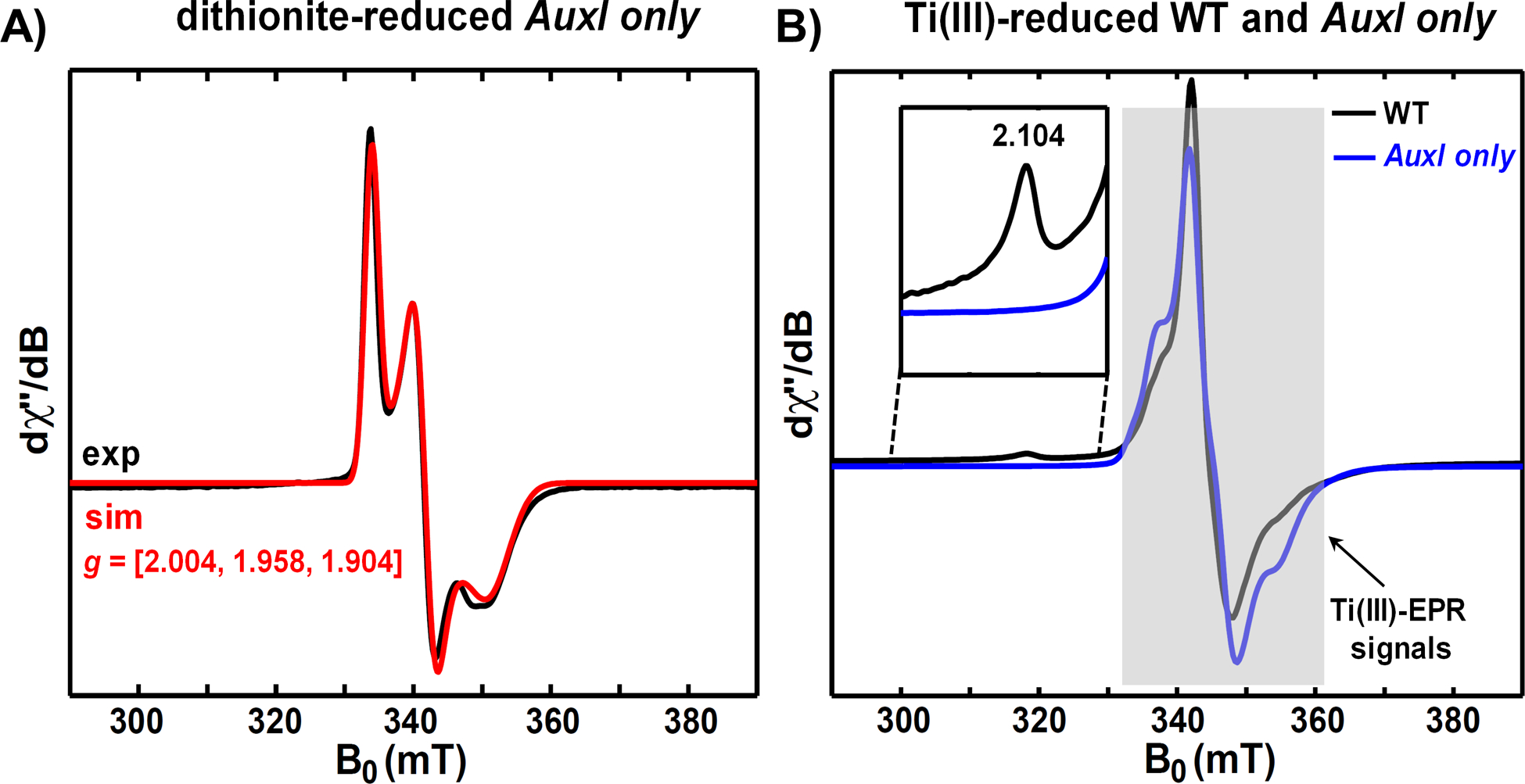
A) Low-temperature (15 K) X-band EPR spectrum of dithionite-reduced AuxI only variant, which contains only the reduced [2Fe-2S]+ cluster with g-values = [2.004, 1.958, 1.904]. Experimental data are shown in black and the simulation is shown in red. B) Low-temperature (10 K) X-band EPR spectrum of Ti(III) citrate-reduced AuxI only variant (blue), in which no g1 = 2.104 signal corresponding to the reduced [4Fe-4S]+ cluster in the AuxI site is observed. The WT PqqE signal (black) obtained under the same condition shows the g1 = 2.104 signal that corresponds to the reduced [4Fe-4S]+ cluster in the AuxI site.51
The reduction potential of the cluster in the presence of SAM binding was also analyzed using the RS only variant. The resulting square-wave voltammogram shows a 20–25 mV potential alteration in a more positive direction compared to RS only in the absence of SAM (Figure S6). This result is consistent with other RS enzymes, such as LAM73 and BtrN,72 where the binding of SAM moves the reduction potential toward a more positive value.
When we compared the reduction potential of the iron-sulfur clusters in PqqE with those of MftC,24 Tte1186,71 and BtrN,72 we noticed that, in all cases but PqqE, the AuxI cluster shows a lower reduction potential than the RS cluster. In the SPASM proteins, where two auxiliary clusters have been reported, the potential differences between the two clusters are around 45 mV to 50 mV, and the AuxII clusters in MftC and Tte1186 are both more negative than the AuxI cluster. However, in PqqE, the AuxI shows a 50 mV more positive potential compared to the RS cluster. This also leads to a 100 mV difference between the two auxiliary clusters in the SPASM domain of PqqE. Furthermore, the dramatic shift of the reduction potential of the AuxI site in the absence of both RS and AuxII clusters is also unique compared to what has been observed for the single cluster-containing variants in the case of Tte1168, in which the potential of the auxiliary [4Fe-4S] cluster only shifted to 15 mV more positive as a result of loss of the two other [4Fe-4S] clusters.71 This further supports the unique properties of the PqqE AuxI site among all the RS-SPASM enzymes.
Reduction potential impact by altering AuxII ligand Asp-319.
To understand the functional role of the unique Asp-319 ligand in PqqE, we replaced Asp-319 by either cysteine or histidine. In our previous EPR analysis, the replacement of Asp-319 by Cys or His changed the electronic properties of the AuxII cluster while retaining the integrity of the iron-sulfur cluster.51 The cyclic voltammogram of the D319C variant, RS/AuxI/D319C, was fit to three independent one-electron transfers with reduction potentials of −555 mV, −500 mV, and −445 mV vs. SHE (Figure 7A). Additionally, the cyclic voltammogram of the D319H variant, RS/AuxI/D319H, was fit to three independent one-electron transfers with reduction potentials of −520 mV, −465 mV, and −405 mV vs. SHE (Figure 7B). We also incorporated the D319C variants on top of either RS or AuxI cluster knockouts, RS/D319C and AuxI/D319C, in which the cysteine ligands at RS or AuxI sites were replaced by alanines, so that we could assess interactions solely between two clusters (Figure 7C & D). This also allowed us to assign the reduction potentials to RS, AuxI and AuxII sites in RS/AuxI/D319C (Table 3). The replacement of Asp-319 by histidine showed an overall shift to a more positive potential (15 mV to 30 mV) for all three clusters compared to the WT PqqE, and RS/AuxI/D319C showed an overall shift to a more negative potential (15 mV to 20 mV) for all three clusters. Alterations in potential as a result of mutating the Asp residue to Cys have been previously observed in other proteins with the same ligation pattern, ranging between 30–60 mV.74–76 The trend of the positive shift in potential upon the switch of Cys to His is also similar to recently reported data for the [FeFe] hydrogenase.77 The different trends in reduction potential for Cys to Asp vs. Cys to His are consistent with the differences in side-chain electronegativity among these residues.
Figure 7. Cyclic voltammetry of PqqE variants with single ligand replacement.
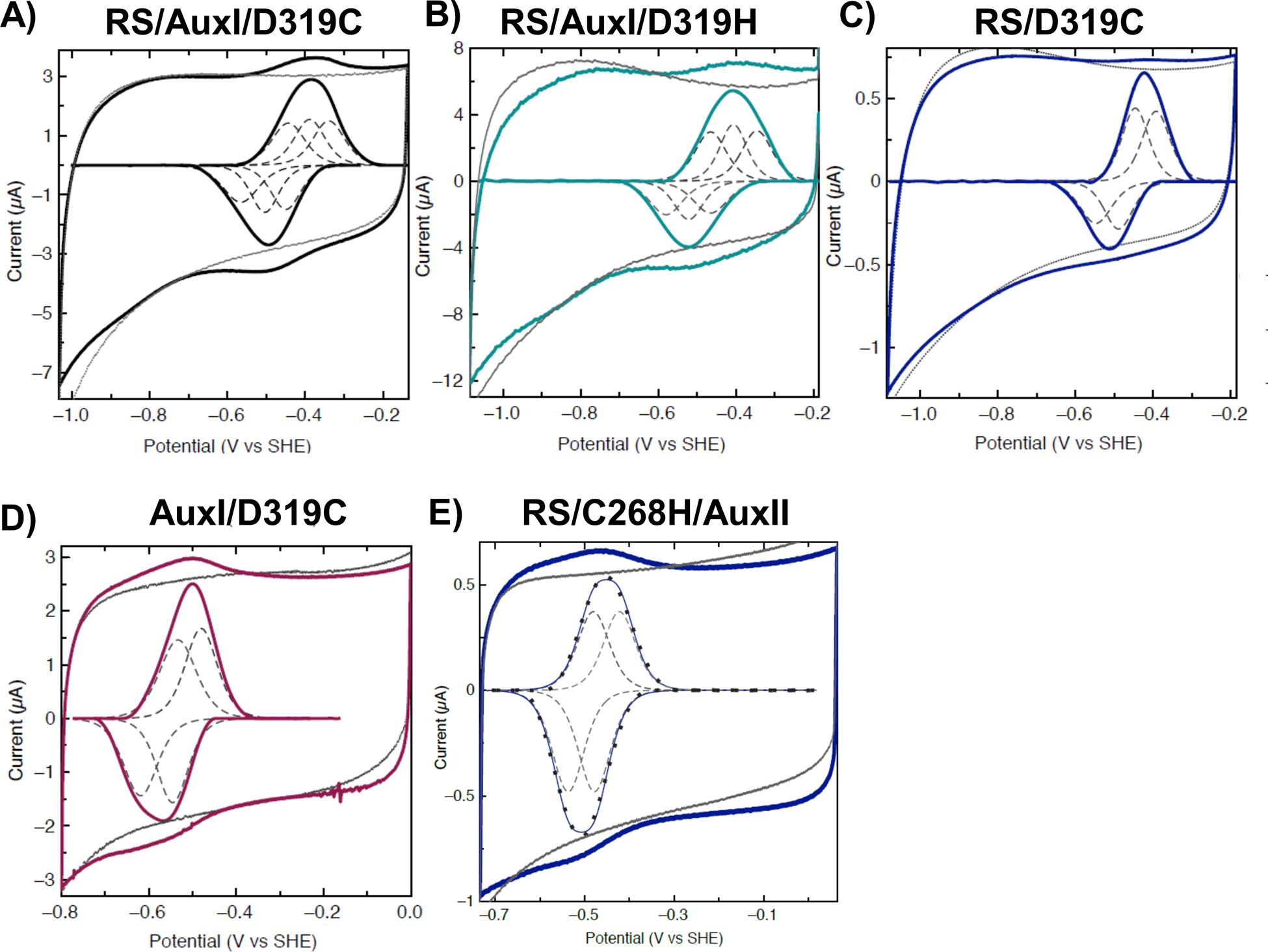
For each protein, measurements and the signals after baseline subtraction are shown in colored solid lines. Baselines are in gray. Species fittings are in dashed lines. A) RS/AuxI/D319C signal (black) was fitted by three one-electron transfers (dashed line). B) RS/AuxI/D319H signal (cyan) was fitted by three one-electron transfers (dashed line). C) RS/D319C signal (purple) was fitted by two one-electron transfers (dashed line). D) AuxI/D319C signal (wine) was fitted by two one-electron transfers (dashed line). E) RS/C268H/AuxII signal (dark blue) was able to be fitted by two one-electron transfers (dashed line). Cyclic voltammograms were measured with a scan rate of 50 mV/s at 4 °C and pH 7.5 on PGE modified with MWCNT.
Reduction potential impact by altering AuxI ligand Cys-268.
We generated a single-point variant C268H, RS/C268H/AuxII, in which one cysteine ligand at the AuxI site is replaced by histidine. This species contains no detectable [2Fe-2S] cluster when reduced by DTH and analyzed by EPR (Figure 8) yet reveals an EPR feature at g = 2.104 when reduced by Ti(III) citrate; the latter has been attributed to a [4Fe-4S] cluster in the WT. Similarly, evidence for a [2Fe-2S] cluster feature was not observed in the UV-vis absorption spectrum of this variant (Figure S7). The cyclic voltammograms of RS/C268H/AuxII were therefore measured to determine possible changes in redox characteristics. Comparing the RS/C268H/AuxII to WT PqqE it was clear that RS/C268H/AuxII displays no obvious changes in the voltammetric data (Figure 7E). Yet, unlike WT the data were fit best by two one-electron features only, with deconvoluted reduction potentials of −475 mV and −520 mV, respectively, assigned to RS and AuxII sites (Table 3). The lack of needing to fit these data to a third feature, and our inability to detect another redox couple suggests that the AuxI cluster, in this case, is of exceedingly low potential. While searching for a lower potential AuxI [4Fe-4S] cluster redox couple that was proposed in the previous EPR analysis,51 we observed a voltammetric feature of variable intensity at ~ −800 mV in PqqE and many of its variants (Figure S8 and Table S4). As this lowest redox potential feature displayed an intensity that is highly variable between experiments and constructs, and does not appear to correlate with activity in a clear manner, its nature awaits further characterization (See Supporting Information for further discussion).
Figure 8. X-band CW EPR spectra of RS/C268H/AuxII variant.

A) Low-temperature (10 K) X-band EPR spectrum of dithionite-reduced RS/C268H/AuxII (black), which contains the corresponding signals from the reduced RS [4Fe-4S]+ cluster (g = [2.040, 1.927, 1.897]) and the reduced AuxII [4Fe-4S]+ cluster (g = [2.059, 1.940, 1.903]). The simulated signal for RS and AuxII clusters is shown in black. B) High-temperature (60 K) X-band EPR spectrum of dithionite-reduced RS/C268H/AuxII (blue), which shows no [2Fe-2S] cluster signal. C) Low-temperature (10 K) X-band EPR spectrum of Ti(III) citrate-reduced RS/C268H/AuxII (black), which shows the g1 = 2.104 signal corresponding to the reduced [4Fe-4S]+ cluster in the AuxI site.51 The shaded area is due to the Ti(III) EPR signal.
Catalytic Activity of PqqE.
The catalytic activity of SPASM proteins can be evaluated by the product formation from either an uncoupled SAM cleavage reaction and/or the peptide modification reaction. Assaying in the presence of the Methylorubrum extorquens ferredoxin and ferredoxin-NADP+ reductase (see Supporting Information), we determined SAM cleavage activity by monitoring the formation of the 5’-dA and peptide modification activity from the detection of crosslinked PqqA using mass spectrometry. Results for WT PqqE and the variants are shown in Figure 9.
Figure 9. Comparison of 5’-dA and crosslinked PqqA production between WT PqqE and its variants.

Activities of WT PqqE (red), reconstituted WT PqqE (orange), RS/AuxI/D319C (green), RS/AuxI/D319H (blue), RS/AuxI (magenta), RS/AuxII (wheat), AuxI/AuxII (cyan), RS only (yellow), and RS/C268H/AuxII (gray), are plotted based on their production of 5’-dA (left panel) and peptide modification (right panel). All the experiments were performed in duplicate. The production of 5’-dA and the crosslinked PqqA is determined after a 16-hour reaction at room temperature in the anaerobic chamber.
We observed a similar level of peptide modification with either the WT or D319 variants, together with a similar trend in the uncoupled SAM cleavage reaction. The reconstituted PqqE did not significantly affect either the SAM cleavage reaction or the peptide modification reaction. This is consistent with the previous EPR results51 that the as-purified Strep-tagged PqqE was loaded with as much iron-sulfur clusters as the reconstituted sample. Results show that the detection of cross-linking activity required the presence of all three clusters. The SAM cleavage activity is also imparied by removal of one or both of the iron-sulfur clusters in the SPASM domain. RS/AuxII abolished the SAM cleavage, while RS/AuxI and RS only maintain a much lower level of 5’-dA production compared to that of WT.
For the single point mutation at AuxII site, RS/AuxI/D319H maintains both activities at a similar or higher level than the WT protein, while RS/AuxI/D319C exhibits slightly lower SAM cleavage and peptide modification activity. This can be well explained by the reduction potential measurements. The RS reduction potential in RS/AuxI/D319H is more positive than WT, suggesting that the capacity of the enzyme to be reduced by exogenous ferredoxin and ferredoxin reductase is greater, while the more negative reduction potential of the RS site in RS/AuxI/D319C would have the opposite effect. In each case, it is noteworthy that the ratio of uncoupling to cross-linking is unaltered relative to WT and WT(R).
Turning to the single point mutation at the AuxI site, RS/C268H/AuxII shows higher SAM cleavage activity compared to WT while the peptide modification activity is below the detection limit. Given measurable reduction potentials at the residual RS and AuxII cluster that are essentially the same as WT PqqE (Table 3), the behavior of RS/C268H/AuxII can be attributed to a perturbation at AuxI upon substitution of Cys-286 with histidine. A salient feature of this substitution is the loss of any evidence for a [2Fe-2S] EPR signal, together with a g = 2.104 EPR signal in the presence of a stronger reductant than dithionite (Figure 8). Analogous EPR behavior has been previously published for as-isolated PqqE and attributed to the fraction of enzyme that contains [4Fe-4S] at AuxI.51 The available spectroscopic and reduction potential data for RS/C268H/AuxII are, thus, indicative of reconstitution by a low potential [4Fe-4S] center at the AuxI site. The fact that RS/C268H/AuxII remains able to catalyze uncoupled reductive cleavage of SAM at a rate comparable to or faster than WT indicates an intact protein that can perform SAM binding and reductive cleavage. In contrast, the presence of [4Fe-4S] in AuxI renders the enzyme completely inactive toward peptide cross-linking, leading us to propose that a [2Fe-2S] cluster within the AuxI site is essential for productive/coupled catalysis.
Mechanistic Implications.
The growing body of structural, kinetic and spectroscopic data for PqqE provides a platform for beginning to understand the mechanistic impact of the features that distinguish it from other RS-SPASM enzymes. These features include the presence of Asp319 as a fourth ligand to the iron-sulfur cluster in AuxII and the persistent evidence for a [2Fe-2S] cluster at AuxI. The functional role of the unique aspartate ligand in AuxII of PqqE had been probed earlier via the generation of RS/AuxI/D319A, indicating damage to the [4Fe-4S] binding capacity at AuxII site, and the loss of peptide modification activity.32 Elimination of the iron-sulfur cluster at AuxII also abolishes StrB peptide modification activity, while this is not the case for SuiB crosslinking activity, even though these two enzymes catalyze essentially the same reaction.48,49 In order to explore further the origin of the impaired activity of the RS/AuxI/D319A variant, the properties of more conservative ligand replacements at D319 site in PqqE, RS/AuxI/D319H and RS/AuxI/D319C, were tested. These variants maintain the integrity of their [4Fe-4S] clusters and display alterations to their EPR spectra that are indicative of (expected) changes in electronic properties.51 Yet, their impacts on redox potential (Table 3) and catalytic activity (Figure 9) are subtle in comparison to cluster-knockouts. Further, from the present study, the small impact of the AuxII knockout on the reduction potentials at the RS and AuxI sites (Table 3) appears to rule out significant electronic coupling between AuxII and the other Fe-S sites. We conclude that AuxII is both critical for enzyme stability and fine-tunes local electronic properties that impact catalysis. Additionally, as indicated from our previous EPR study,51 Asp-319 can be replaced by a strong-field ligand (e.g. KCN). It has been reported that Asp is a labile iron-sulfur cluster ligand in the Bacillus subtilis O2-sensing fumarate and nitrate reduction protein and the Streptomyces coelicolor [4Fe-4S] NO-sensing response regulator.81 Both enzymes use an Asp ligand as a potential replacement binding site for gas molecules, such as O2 and NO, which once bound initiate changes in protein conformation. These precedents raise the intriguing possibility that Asp-319 in PqqE may behave as a response element at elevated O2 concentration.
Turning to the role of AuxI, a number of mechanistic proposals have emerged for RS-SPASM domain enzymes. These include the maintenance of an open coordination site for substrate binding in NxxcB, a β-thioether bond formation RS-SPASM enzyme20 and, more generally, as in the case of MftC,24 an electron hopping mechanism that arises from a gradient of reduction potentials from the most surface-exposed AuxII site to the RS site. In anSME and AlbA, the two auxiliary [4Fe-4S] clusters have further been proposed as electron conduits to transfer the electron during the catalysis.78,79 The functional importance of the AuxI site in PqqE is supported by the inactivity of RS/AuxII toward both SAM cleavage and peptide crosslinking (Figure 9). The incorporated changes in C248A/C268A that lead to loss of the [2Fe-2S] cluster at the AuxI site may be expected to disrupt the electron transfer during the catalysis as well as impair the protein stability. On the other hand, even though RS only, like RS/AuxII, does not contain a [2Fe-2S] cluster at AuxI, the 5’-dA production remains detectable. We note that the alanine-replaced AuxI ligands in RS/AuxII are C248A/C268A, which differ from the ones replaced in RS only, C323A/C325A (Table 1 and Supporting Information). In the PqqE crystal structure, C248 and C268 face toward the SAM cleavage site near the RS [4Fe-4S] cluster, while C323 and C325 are closer in space to the AuxII site. The observed difference in the 5’-dA production between RS only and RS/AuxII may, thus, be expected to reflect, in part, the specified sites of mutation. The pathway for reduction of the RS site in each construct by ferredoxin is likely to deviate from that of WT, with a possibility of reduction occuring at the RS, AuxI or AuxII site depending on the nature of the mutant undergoing characterization.
From the reduction potentials observed herein for the RS cluster of PqqE (−485 mV, Table 3) and the ~25 mV positive-shift of SAM-bound RS cluster (Figure S6), the PqqE RS site will be considerably more positive than its AuxII site. While this feature can support a long-range electron hopping mechanism in PqqE, an unusual feature of the AuxI cluster of PqqE is its more positive potential relative to the other two iron-sulfur sites (Table 3). As already discussed,51 [4Fe-4S] clusters are most commonly seen in the auxiliary sites of radical SAM enzymes, with a few exceptions, such as SkfB and BioB.80 However, based on the EPR-detected, very low redox potential assigned to a WT AuxI [4Fe-4S] site,51 the redox potentials measured for AuxI (Table 3) are attributed to a [2Fe-2S] cluster.
These features set the stage for distinguishing the catalytic relevance of a [4Fe-4S] vs. a [2Fe-2S] cluster at the AuxI site as a step toward a working mechanism for PqqE. Important observations include the finding that the g value of the AuxI [4Fe-4S] cluster (g = 2.104) remains unchanged in Ti(III)-reduced EPR samples for RS/C268H/AuxII relative to WT (Figure 6B and 8C), implying similar [4Fe-4S] clusters at AuxI in both forms of the enzyme. Additionally, the measured reduction potentials at the AuxII and RS sites (Table 3 & Figure 7E) are unaltered in RS/C268H/AuxII, arguing against the impact of this mutation on the reducing environments at RS and AuxII in this mutant. There is also the significant kinetic observation for RS/C268H/AuxII of a higher yield of uncoupled products from SAM than for WT, together with a failure to produce any detectable cross-linked peptide product. We propose that the presence of a [4Fe-4S] cluster at the AuxI site is incapable of supporting a peptide modification reaction, despite no evidence for alterations in the electronic properties of the site. These kinetic findings (Figure 9), in the context of the spectroscopic data, point toward other explanations for the highly uncoupled activity seen with RS/C268H/AuxII. This is reminiscent of early findings of PqqE behavior in the absence of PqqD39 and the fact that it was only possible to demonstrate peptide cross-linking in the presence of the (tight) complex between PqqA and PqqD.35,36 Thus, in addition to the absence of the more positive potential cluster at AuxI detected in WT (Table 3), the inability of RS/C268H/AuxII to catalyze detectable substrate functionalization may be a consequence of its impaired ability to bind the PqqD/A complex. The latter possibility will form the basis of future studies.
Based on the aggregate properties now available for PqqE, a plausible reaction mechanism for the cross-linking reaction of PqqE is presented in Figure 10. As illustrated, the reduction of SAM at the RS site by a biological reducing system (e.g., ferredoxin and NADP+-ferredoxin reductase) generates the 5’-dA radical to initiate hydrogen atom abstraction from Glu and de novo C-C bond formation between Glu and Tyr in PqqA. The latter generates a radical anion within the Tyr ring, with a strong driving force toward oxidation and concomitant recovery of its aromaticity. It is possible that the oxidation of this Tyr radical occurs rapidly and directly within the adjacent oxidized RS site or, based on the new data presented, that the RS and AuxI sites act in equilibrium, to reinforce each other as the electron sink. In the optimal condition, no extra reducing power may be needed for turnover, as the first electron that has entered the active site can be recycled from the reduced RS/AuxI pair to facilitate multiple catalytic cycles. Since the reduction potential difference between AuxII [4Fe-4S] and AuxI [2Fe-2S] is 2-fold larger than the potential difference between the AuxI [2Fe-2S] and RS [4Fe-4S] cluster, it is unlikely that AuxII will play a role in this portion of the catalytic reaction. In the case of protein containing a [4Fe-4S] cluster, RS/C268H/AuxII, the very low potential at AuxI intercepts the ability of the enzyme to drive the reaction to completion, resulting in a breakdown of productive communication between the RS and AuxI sites (Figure 10). Such ineffective engagement will lead to an uncoupling of peptide cross-linking from SAM cleavage as well as the need for the extra input of an electron from the external reducing system. We note that the origin of the high ratio of uncoupled SAM cleavage to peptide crosslink activity in WT enzyme itself remains to be resolved, and suggest that this may be due to the fact that the as-isolated enzyme is always a mixture of [4Fe-4S] and [2Fe-2S] clusters at the AuxI site. We further note that a quantitative correlation between the relative amount of [2Fe-2S] vs. [4Fe-4S] within AuxI and the extent of full peptide cross-linking vs. uncoupled SAM cleavage may not be possible at the current level of understanding; for example, if the rate limiting step of cross-linked peptide formation from enzyme with [2Fe-2S] at AuxI occurs subsequent to the initial SAM cleavage reaction, uncoupling of these two processes would give rise to much faster SAM cleavage. Additionally, any interference from [4Fe-4S] at AuxI with PqqA/D binding would lead to a different enzyme form catalyzing SAM cleavage, with an inherent rate constant that may be distinct from the SAM cleavage portion of the net reaction catalyzed by a productive PqqE/D/A complex. What remains clear is that it will be thermodynamically preferable to involve a [2Fe-2S] cluster with a reduction potential of −435 mV than the putative [4Fe-4S] cluster of a potential of −800 mV in the oxidation of an unstable aromatic free radical intermediate.
Figure 10. Proposed mechanism of PqqE.

The biological reduction system used in this study includes [2Fe-2S]-ferredoxin (darker gray), ferredoxin-NADP+ reductase (lighter gray), and NADPH (blue spheres). The electron generated from the oxidation of NADPH (blue spheres) by flavin (yellow spheres) is transferred via a ferredoxin [2Fe-2S] center (yellow and orange spheres) to PqqE, which is used for a reductive SAM cleavage. Generation of the 5′-dA radical at the RS site is followed by hydrogen atom abstraction at Glu-16 on PqqA. The glutamyl radical attacks the Tyr-20 and forms a de novo C−C bond in PqqA. This step, coupled to proton transfer, generates a highly reactive tyrosine radical within the Tyr ring, leading to a return of an electron back to the iron−sulfur cluster(s) in PqqE. This electron can be recycled in the next round of the catalysis when new SAM and PqqA/D bind to PqqE. An uncoupling reaction (gray dashed arrows) will occur when this electron is lost, which leads to the need for the exogenous reduction system in the next turnover. The boxed gray areas indicate the location of the proposed electron hopping within the system.
Conclusions
RS-SPASM enzymes that work on RiPPs catalyze fascinating and biologically important chemical conversions, using multiple auxiliary [4Fe-4S] clusters in addition to their RS site. Among the known RS-dependent pathways, the PQQ biosynthetic pathway is unique in that its initial RS process, catalyzed by PqqE, is accompanied by a series of subsequent O2-dependent enzymatic transformations. In this work, we demonstrate how the protein ligands of PqqE manipulate the properties of the iron-sulfur clusters, presenting evidence in favor of a functional [2Fe-2S] cluster within AuxI of the enzyme. A mild ligand replacement, C268H, at the AuxI is shown to produce only the [4Fe-4S] cluster while abolishing both the [2Fe-2S] cluster binding and peptide modification. We introduce the use of native MS as an informative complement to other well-established methods, such as Mossbauer82 and EPR83 spectroscopy, in the characterization of multi-center iron-sulfur proteins. While comparative bioinformatics and native MS studies can rationalize either [2Fe-2S] or [4Fe-4S] at the AuxI site, these analyses also predict a preference for [2Fe-2S] at AuxI in PqqE. By systematic generation of a series of variants at each iron-sulfur cluster binding site, we have been able to tease apart reduction potentials for the three iron-sulfur clusters. Using non-destructive ligand replacements, we show that the reduction potential at AuxII is sensitive to replacement of the highly conserved Asp-319 by side chains of varying electronegativity. These studies emphasize the important relationship between protein structure/function and the ligands at the iron-sulfur centers. The unusual role for the proposed [2Fe-2S] at AuxI is discussed in the context of PqqE stability in O2, the protein’s ability to interact with the PqqD-PqqA complex, and the detailed mechanistic requirements of the catalyzed peptide cross-linking reaction.
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. Squire Booker (Penn State University) for generously distributing the pDB1282 plasmid that was originally constructed by Prof. Dennis Dean (Virginia Tech). We acknowledge Prof. Troy Stich (Wake Forest College) for preliminary EPR measurements in the project. We thank Prof. Yilin Hu (UC Irvine) for generously providing the synthetic [4Fe-4S] cluster. Supported by the National Institutes of Health (GM118117 to J.P.K; GM120283 to SJE; GM104543, 1R35GM126961-01 to R.D.B.). The EPR spectrometers at the CalEPR facility (UC Davis) used in this study were funded by the NIH (S10-RR021075) and the NSF (CHE-1048671) faculty grants. The mass spectrometer at UC Berkeley was purchased with NIH support (grant 1S10OD020062-01).
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
REFERENCES
- (1).Peters J; Fisher K; Dean D Nitrogenase Structure and Function: A Biochemical-Genetic Perspective. Annu Rev Microbiol 1995, 49 (1), 335–366. [DOI] [PubMed] [Google Scholar]
- (2).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chem Rev 2014, 114 (8), 4081–4148. [DOI] [PubMed] [Google Scholar]
- (3).Orme-Johnson W Iron-Sulfur Proteins: Structure and Function. Annu Rev Biochem 1973, 42 (1), 159–204. [DOI] [PubMed] [Google Scholar]
- (4).Hirst J Mitochondrial Complex I. Annu Rev Biochem 2013, 82 (1), 551–575. [DOI] [PubMed] [Google Scholar]
- (5).Broderick JB; Duffus BR; Duschene KS; Shepard EM Radical S-Adenosylmethionine Enzymes. Chem Rev 2014, 114 (8), 4229–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Walsby CJ; Ortillo D; Broderick WE; Broderick JB; Hoffman BM An Anchoring Role for FeS Clusters: Chelation of the Amino Acid Moiety of S-Adenosylmethionine to the Unique Iron Site of the [4Fe-4S] Cluster of Pyruvate Formate-Lyase Activating Enzyme. J Am Chem Soc 2002, 124 (38), 11270–11271. [DOI] [PubMed] [Google Scholar]
- (7).Sayler RI; Stich TA; Joshi S; Cooper N; Shaw JT; Begley TP; Tantillo DJ; Britt DR Trapping and Electron Paramagnetic Resonance Characterization of the 5′dAdo• Radical in a Radical S-Adenosyl Methionine Enzyme Reaction with a Non-Native Substrate. ACS Central Sci 2019, 5 (11), 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yang H; McDaniel EC; Impano S; Byer AS; Jodts RJ; Yokoyama K; Broderick WE; Broderick JB; Hoffman BM The Elusive 5′-Deoxyadenosyl Radical: Captured and Characterized by Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Spectroscopies. J Am Chem Soc 2019, 141 (30), 12139–12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Holliday GL; Akiva E; Meng EC; Brown SD; Calhoun S; Pieper U; Sali A; Booker SJ; Babbitt PC Chapter One Atlas of the Radical SAM Superfamily: Divergent Evolution of Function Using a “Plug and Play” Domain. Methods Enzymol 2018, 606, 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Byer AS; Yang H; McDaniel EC; Kathiresan V; Impano S; Pagnier A; Watts H; Denler C; Vagstad AL; Piel J & Duschene KS Paradigm Shift for Radical S-adenosyl-L-Methionine Reactions: The Organometallic Intermediate Ω is Central to Catalysis. J Am Chem Soc 2018, 140 (28), 8634–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yokoyama K, & Lilla EA C–C Bond Forming Radical SAM Enzymes Involved in the Construction of Carbon Skeletons of Cofactors and Natural Products. Nat prod Rep 2018, 35 (7), 660–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Grell TA; Goldman PJ; Drennan CL SPASM and Twitch Domains in S-Adenosylmethionine (SAM) Radical Enzymes. J Biol Chem 2015, 290 (7), 3964–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Flühe L; Burghaus O; Wieckowski BM; Giessen TW; Linne U; Marahiel MA Two [4Fe-4S] Clusters Containing Radical SAM Enzyme SkfB Catalyze Thioether Bond Formation during the Maturation of the Sporulation Killing Factor. J Am Chem Soc 2013, 135 (3), 959–962. [DOI] [PubMed] [Google Scholar]
- (14).Grove TL; Himes P; Hwang S; Yumerefendi H; Bonanno JB; Kuhlman B; Almo SC; Bowers AA Structural Insights into Thioether Bond Formation in the Biosynthesis of Sactipeptides. J Am Chem Soc 2017, 139 (34), 11734–11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Benjdia A; Guillot A; Lefranc B; Vaudry H; Leprince J; Berteau O Thioether Bond Formation by SPASM Domain Radical SAM Enzymes: C α H-Atom Abstraction in Subtilosin A Biosynthesis. Chem Comm 2016, 52 (37), 6249–6252. [DOI] [PubMed] [Google Scholar]
- (16).Bruender NA; Bandarian V The Radical S-Adenosyl-l-Methionine Enzyme MftC Catalyzes an Oxidative Decarboxylation of the C-Terminus of the MftA Peptide. Biochem 2016, 55 (20), 2813–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bushin LB; Clark KA; Pelczer I; Seyedsayamdost MR Charting an Unexplored Streptococcal Biosynthetic Landscape Reveals a Unique Peptide Cyclization Motif. J Am Chem Soc 2018, 140 (50), 17674–17684. [DOI] [PubMed] [Google Scholar]
- (18).Davis K; Schramma KR; Hansen WA; Bacik JP; Khare SD; Seyedsayamdost MR; Ando N Structures of the Peptide-Modifying Radical SAM Enzyme SuiB Elucidate the Basis of Substrate Recognition. Proc Natl Acad Sci 2017, 114 (39), 10420–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hänzelmann P; Schindelin H Binding of 5′-GTP to the C-Terminal FeS Cluster of the Radical S-Adenosylmethionine Enzyme MoaA Provides Insights into Its Mechanism. Proc Natl Acad Sci 2006, 103 (18), 6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Caruso A; Bushin LB; Clark KA; Martinie RJ; Seyedsayamdost MR A Radical Approach to Enzymatic β-Thioether Bond Formation. J Am Chem Soc 2018, 141 (2), 990–997. [DOI] [PubMed] [Google Scholar]
- (21).Schramma KR, & Seyedsayamdost MR Lysine-tryptophan-crosslinked Peptides Produced by Radical SAM Enzymes in Pathogenic Streptococci . ACS Chem Biol 2017, 12 (4), 922–927. [DOI] [PubMed] [Google Scholar]
- (22).Saichana N; Tanizawa K; Ueno H; Pechoušek J; Novák P; Frébortová J Characterization of Auxiliary Iron–Sulfur Clusters in a Radical S‐adenosylmethionine Enzyme PqqE from Methylobacterium Extorquens AM1. Febs Open Bio 2017, 7 (12), 1864–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kühner M; Schweyen P; Hoffmann M; Ramos J; Reijerse EJ; Lubitz W; Bröring M; Layer G The Auxiliary [4Fe–4S] Cluster of the Radical SAM Heme Synthase from Methanosarcina Barkeri is Involved in Electron Transfer. Chem Sci 2016, 7 (7), 4633–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ayikpoe R; Ngendahimana T; Langton M; Bonitatibus S; Walker LM; Eaton SS; Eaton GR; Pandelia M-E; Elliott SJ; Latham JA Spectroscopic and Electrochemical Characterization of the Mycofactocin Biosynthetic Protein, MftC, Provides Insight into Its Redox Flipping Mechanism. Biochem 2019, 58 (7), 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Latham JA; Barr I; Klinman JP At the Confluence of Ribosomally Synthesized Peptide Modification and Radical S-Adenosylmethionine (SAM) Enzymology. J Biol Chem 2017, 292 (40), 16397–16405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kumazawa T; Sato K; Seno H; Ishii A; Suzuki O Levels of Pyrroloquinoline Quinone in Various Foods. Biochem J 1995, 307 (2), 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kumazawa T; Seno H; Urakami T; Matsumoto T; Suzuki O Trace Levels of Pyrroloquinoline Quinone in Human and Rat Samples Detected by Gas Chromatography/Mass Spectrometry. BBA - Gen Subj 1992, 1156 (1), 62–66. [DOI] [PubMed] [Google Scholar]
- (28).Akagawa M; Minematsu K; Shibata T; Kondo T; Ishii T; Uchida K Identification of Lactate Dehydrogenase as a Mammalian Pyrroloquinoline Quinone (PQQ)-Binding Protein. Sci Rep 2016, 6 (1), srep26723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jonscher KR; Stewart MS; Alfonso-Garcia A; DeFelice BC; Wang XX; Luo Y; Levi M; Heerwagen MJ; Janssen RC; de la Houssaye BA; et al. Early PQQ Supplementation Has Persistent Long-Term Protective Effects on Developmental Programming of Hepatic Lipotoxicity and Inflammation in Obese Mice. Faseb J 2016, 31 (4), 1434–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Harris CB; Chowanadisai W; Mishchuk DO; Satre MA; Slupsky CM; Rucker RB Dietary Pyrroloquinoline Quinone (PQQ) Alters Indicators of Inflammation and Mitochondrial-Related Metabolism in Human Subjects. J Nutr Biochem 2013, 24 (12), 2076–2084. [DOI] [PubMed] [Google Scholar]
- (31).Duine JA The PQQ Story. Journal of Biosci Bioeng 1999, 88 (3), 231–236. [DOI] [PubMed] [Google Scholar]
- (32).Barr I; Stich TA; Gizzi AS; Grove TL; Bonanno JB; Latham JA; Chung T; Wilmot CM; Britt R; Almo SC; et al. X-Ray and EPR Characterization of the Auxiliary Fe-S Clusters in the Radical SAM Enzyme PqqE. Biochem 2018, 57 (8), 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Evans RL; Latham JA; Xia Y; Klinman JP; Wilmot CM Nuclear Magnetic Resonance Structure and Binding Studies of PqqD, a Chaperone Required in the Biosynthesis of the Bacterial Dehydrogenase Cofactor Pyrroloquinoline Quinone. Biochem 2017, 56 (21), 2735–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Evans RL; Latham JA; Klinman JP; Wilmot CM; Xia Y 1H, 13C, and 15N Resonance Assignments and Secondary Structure Information for Methylobacterium Extorquens PqqD and the Complex of PqqD with PqqA. Biomol NMR Assign 2016, 10 (2), 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Barr I; Latham JA; Iavarone AT; Chantarojsiri T; Hwang JD; Klinman JP Demonstration That the Radical S-Adenosylmethionine (SAM) Enzyme PqqE Catalyzes de Novo Carbon-Carbon Cross-Linking within a Peptide Substrate PqqA in the Presence of the Peptide Chaperone PqqD. J Biol Chem 2016, 291 (17), 8877–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Latham JA; Iavarone AT; Barr I; Juthani PV; Klinman JP PqqD is a Novel Peptide Chaperone that forms a Ternary Complex with the Radical S-Adenosylmethionine Protein PqqE in the Pyrroloquinoline Quinone Biosynthetic Pathway. J Biol Chem 2015, 290 (20), 12908–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Shen Y-Q; Bonnot F; Imsand EM; RoseFigura JM; Sjölander K; Klinman JP Distribution and Properties of the Genes Encoding the Biosynthesis of the Bacterial Cofactor, Pyrroloquinoline Quinone. Biochem 2012, 51 (11), 2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).RoseFigura JM; Puehringer S; Schwarzenbacher R; Toyama H; Klinman JP Characterization of a Protein-Generated O₂ Binding Pocket in PqqC, a Cofactorless Oxidase Catalyzing the Final Step in PQQ Production. Biochem 2011, 50 (9), 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wecksler SR; Stoll S; Iavarone AT; Imsand EM; Tran H; Britt DR; Klinman JP Interaction of PqqE and PqqD in the Pyrroloquinoline Quinone (PQQ) Biosynthetic Pathway Links PqqD to the Radical SAM Superfamily. Chem Comm 2010, 46 (37), 7031–7033. [DOI] [PubMed] [Google Scholar]
- (40).Wecksler SR; Stoll S; Tran H; Magnusson OT; Wu S; King D; Britt DR; Klinman JP Pyrroloquinoline Quinone Biogenesis: Demonstration That PqqE from Klebsiella Pneumoniae is a RadicalS-Adenosyl-l-Methionine Enzyme. Biochem 2009, 48 (42), 10151–10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Magnusson O; Toyama H; Saeki M; Rojas A; Reed JC; Liddington RC; Klinman JP; Schwarzenbacher R Quinone Biogenesis: Structure and Mechanism of PqqC, the Final Catalyst in the Production of Pyrroloquinoline Quinone. Proc Natl Acad Sci 2004, 101 (21), 7913–7918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Magnusson OT; Toyama H; Saeki M; Schwarzenbacher R; Klinman JP The Structure of a Biosynthetic Intermediate of Pyrroloquinoline Quinone (PQQ) and Elucidation of the Final Step of PQQ Biosynthesis. J Am Chem Soc 2004, 126 (17), 5342–5343. [DOI] [PubMed] [Google Scholar]
- (43).Koehn EM; Latham JA; Armand T; Evans RL; Tu X; Wilmot CM; Iavarone AT; Klinman JP Discovery of Hydroxylase Activity for PqqB Provides a Missing Link in the Pyrroloquinoline Quinone Biosynthetic Pathway. J Am Chem Soc 2019, 141 (10), 4398–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Martins AM; Latham JA; Martel PJ; Barr I; Iavarone AT; Klinman JP A Two-Component Protease in Methylorubrum extorquens with High Activity Toward the Peptide Precursor of the Redox Cofactor Pyrroloquinoline Quinone. J Biol Chem 2019, 294 (41), 15025–15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Benjdia A, Subramanian S, Leprince J, Vaudry H, Johnson MK, Berteau O Anaerobic Sulfatase‐maturating Enzyme–A Mechanistic Link with Glycyl Radical‐Activating Enzymes? FEBS J 2010, 277 (8), 1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Benjdia A, Leprince J, Sandstrom C, Vaudry H, Berteau O Mechanistic Investigations of Anaerobic Sulfatase-maturating Enzyme: Direct Cβ H-atom Abstraction Catalyzed by a Radical AdoMet Enzyme. J Am Chem Soc 2009, 131 (24), 8348–8349. [DOI] [PubMed] [Google Scholar]
- (47).Benjdia A, Decamps L, Guillot A, Kubiak X, Ruffié P, Sandström C, Berteau O Insights into the Catalysis of a Lysine-tryptophan Bond in Bacterial Peptides by a SPASM Domain Radical S-adenosylmethionine (SAM) Peptide Cyclase. J Biol Chem 2017, 292 (26), 10835–10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Schramma KR; Forneris CC; Caruso A; Seyedsayamdost MR Mechanistic Investigations of Lysine-Tryptophan Cross-Link Formation Catalyzed by Streptococcal Radical S-Adenosylmethionine Enzymes. Biochem 2018, 57 (4), 461–468. [DOI] [PubMed] [Google Scholar]
- (49).Schramma KR; Bushin LB; Seyedsayamdost MR Structure and Biosynthesis of a Macrocyclic Peptide Containing an Unprecedented Lysine-to-Tryptophan Crosslink. Nat Chem 2015, 7 (5), 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Grell TA; Kincannon WM; Bruender NA; Blaesi EJ; Krebs C; Bandarian V; Drennan CL Structural and Spectroscopic Analyses of the Sporulation Killing Factor Biosynthetic Enzyme SkfB, a Bacterial AdoMet Radical Sactisynthase. J Biol Chem 2018, 293 (45), 17349–17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tao L; Zhu W; Klinman JP; Britt DR Electron Paramagnetic Resonance Spectroscopic Identification of the Fe-S Clusters in the SPASM Domain-Containing Radical SAM Enzyme PqqE. Biochem 2019, 58 (51), 5173–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Frazzon J; Fick J; Dean D Biosynthesis of Iron-Sulphur Clusters Is a Complex and Highly Conserved Process. Biochem Soc Trans 2002, 30 (4), 680–685. [DOI] [PubMed] [Google Scholar]
- (53).Kuchenreuther JM; Shiigi SA; Swartz JR Cell-free Synthesis of the H-cluster: a Model for the in vitro Assembly of Metalloprotein Metal Centers In Metalloproteins (pp. 49–72) 2014, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- (54).Tanifuji K; Lee C; Sickerman NS; Tatsumi K; Ohki Y; Hu Y; Ribbe MW Tracing the ‘Ninth Sulfur’ of the Nitrogenase Cofactor via a Semi-Synthetic Approach. Nat Chem 2018, 10 (5), 568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Hernandez H; Hewitson KS; Roach P; Shaw NM; Baldwin JE; Robinson CV Observation of the Iron-Sulfur Cluster in Escherichia c oli Biotin Synthase by Nanoflow Electrospray Mass Spectrometry. Anal Chem 2001, 73 (17), 4154–4161. [DOI] [PubMed] [Google Scholar]
- (56).Sterling HJ; Cassou CA; Susa AC; Williams ER Electrothermal Supercharging of Proteins in Native Electrospray Ionization. Anal Chem 2012, 84 (8), 3795–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kenney GE; Dassama LM; Pandelia M-E; Gizzi AS; Martinie RJ; Gao P; DeHart CJ; Schachner LF; Skinner OS; Ro SY; et al. The Biosynthesis of Methanobactin. Science 2018, 359 (6382), 1411–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ro SY; Schachner LF; Koo CW; Purohit R; Remis JP; Kenney GE; Liauw BW; Thomas PM; Patrie SM; Kelleher NL; et al. Native Top-down Mass Spectrometry Provides Insights into the Copper Centers of Membrane-Bound Methane Monooxygenase. Nat Commun 2019, 10 (1), 2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hernández H; Hewitson K; Roach P; Naw; Baldwin J; Robinson C Observation of the Iron-Sulfur Cluster in Escherichia Coli Biotin Synthase by Nanoflow Electrospray Mass Spectrometry. Anal Chem 2001, 73 (17), 4154–4161. [DOI] [PubMed] [Google Scholar]
- (60).Crack JC; Brun NE Mass Spectrometric Identification of [4Fe–4S](NO)x Intermediates of Nitric Oxide Sensing by Regulatory Iron–Sulfur Cluster Proteins. Chem European J 2019, 25 (14), 3675–3684. [DOI] [PubMed] [Google Scholar]
- (61).Crack JC; Thomson AJ; Brun NE Mass Spectrometric Identification of Intermediates in the O2-Driven [4Fe-4S] to [2Fe-2S] Cluster Conversion in FNR. Proc Natl Acad Sci 2017, 114 (16), E3215–E3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Lin CW; McCabe JW; Russell DH; Barondeau DP Molecular Mechanism of ISC Iron-Sulfur Cluster Biogenesis Revealed by High-resolution Native Mass Spectrometry. J Am Chem Soc 2020, 142 (13), 6018–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Jia M; Sen S; Wachnowsky C; Fidai I; Cowan JA; Wysocki VH Characterization of [2Fe-2S]-cluster-bridged Protein Complexes and Reaction Intermediates by Use of Native Mass Spectrometric Methods. Angew Chem Int Ed 2020, 59 (17), 6724–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Johnson KA; Verhagen MF; Brereton PS; Adams MW; Amster IJ Probing the Stoichiometry and Oxidation States of Metal Centers in Iron−Sulfur Proteins Using Electrospray FTICR Mass Spectrometry. Analy Chem 2000, 72 (7), 1410–1418. [DOI] [PubMed] [Google Scholar]
- (65).Liu TY; Iavarone AT; Doudna JA RNA and DNA Targeting by a Reconstituted Thermus Thermophiles Type III-A CRISPR-Cas System. Plos One 2017, 12 (1), e0170552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Holm R; Lo W Structural Conversions of Synthetic and Protein-Bound Iron–Sulfur Clusters. Chem Rev 2016, 116 (22), 13685–13713. [DOI] [PubMed] [Google Scholar]
- (67).Saichana N; Tanizawa K; Pechoušek J; Novák P, Yakushi T; Toyama H; Frébortová J PqqE from Methylobacterium extorquens AM1: a Radical S-adenosyl-l-Methionine Enzyme with an Unusual Tolerance to Oxygen. J Biochem 2016, 159 (1), 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Maiocco SJ; Arcinas AJ; Booker SJ; Elliott SJ Parsing Redox Potentials of Five Ferredoxins Found within Thermotoga Maritima. Prot Sci 2019, 28 (1), 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Armstrong FA; Evans RM; Megarity CF Protein Film Electrochemistry of Iron–Sulfur Enzymes. Methods Enzymol 2017, 599, 387–407. [DOI] [PubMed] [Google Scholar]
- (70).Mayhew SG The Redox Potential of Dithionite and SO2- from Equilibrium Reactions with Flavodoxins, Methyl Viologen and Hydrogen plus Hydrogenase. Eur J Biochem 1978, 85 (2), 535–547. [DOI] [PubMed] [Google Scholar]
- (71).Walker LM; Kincannon WM; Bandarian V; Elliott SJ Deconvoluting the Reduction Potentials for the Three [4Fe-4S] Clusters in an AdoMet Radical SCIFF Maturase. Biochem 2018, 57 (42), 6050–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Maiocco SJ; Grove TL; Booker SJ; Elliott SJ Electrochemical Resolution of the [4Fe-4S] Centers of the AdoMet Radical Enzyme BtrN: Evidence of Proton Coupling and an Unusual, Low-Potential Auxiliary Cluster. J Am Chem Soc 2015, 137 (27), 8664–8667. [DOI] [PubMed] [Google Scholar]
- (73).Hinckley GT; Frey PA Cofactor Dependence of Reduction Potentials for [4Fe-4S]2+/1+in Lysine 2,3-Aminomutase. Biochem 2006, 45 (10), 3219–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Busch JL; Breton JL; Bartlett BM; Armstrong FA; James R; Thomson AJ [3Fe-4S]↔[4Fe-4S] Cluster Interconversion in Desulfovibrio Africanus Ferredoxin III: Properties of an Asp14→Cys Mutant. Biochem J 1997, 323 (1), 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Brereton PS; Verhagen MF; Zhou ZH; Adams MW Effect of Iron-Sulfur Cluster Environment in Modulating the Thermodynamic Properties and Biological Function of Ferredoxin FromPyrococcus Furiosus†. Biochem 1998, 37 (20), 7351–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chen K; Tilley GJ; Sridhar V; Prasad GS; Stout CD; Armstrong FA; Burgess BK Alteration of the Reduction Potential of the [4Fe-4S]2+/+ Cluster of Azotobacter Vinelandii Ferredoxin I. J Biol Chem 1999, 274 (51), 36479–36487. [DOI] [PubMed] [Google Scholar]
- (77).Rodríguez-Maciá P; Kertess L; Burnik J; Birrell JA; Hofmann E; Lubitz W; Happe T; Rüdiger O His-Ligation to the [4Fe-4S] Sub-Cluster Tunes the Catalytic Bias of [FeFe] Hydrogenase. J Am Chem Soc 2018, 141 (1), 472–481. [DOI] [PubMed] [Google Scholar]
- (78).Benjdia A, Balty C, & Berteau O Radical SAM Enzymes in the Biosynthesis of Ribosomally Synthesized and Post-translationally Modified Peptides (RiPPs). Front Chem 2017, 5, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Lanz ND, & Booker SJ Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes. Biochim Biophys Acta 2015, 1853 (6), 1316–1334. [DOI] [PubMed] [Google Scholar]
- (80).Tao L, Stich TA, Fugate CJ, Jarrett JT, Britt RD (2018). EPR-derived structure of a paramagnetic intermediate generated by biotin synthase BioB. J Am Chem Soc 2018, 140 (40), 12947–12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Crack JC; Green J; Thomson AJ;Le Brun NE Iron–sulfur Cluster Sensor-Regulators. Curr Opin Chem Biol 2012, 16 (1–2), 35–44. [DOI] [PubMed] [Google Scholar]
- (82).Pandelia M-E; Lanz ND; Booker SJ; Krebs C Mössbauer Spectroscopy of Fe/S Proteins. Biochimica Et Biophysica Acta Bba - Mol Cell Res 2015, 1853 (6), 1395–1405. [DOI] [PubMed] [Google Scholar]
- (83).Hagen WR EPR Spectroscopy of Complex Biological Iron–Sulfur Systems. J Biol Inorg Chem 2018, 23 (4), 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.