

1. Introduction
Within the lung vasculature, reactive oxygen species (ROS) are critical signaling intermediates in both health and disease. ROS are thought to play a particularly important role in the pathobiology of pulmonary hypertension (PH) [1, 2]. Alterations in ROS homeostasis in the pulmonary vasculature, specifically in pulmonary smooth muscle (SMC) and endothelial (EC) cells, lead to a variety of signaling events that promote enhanced vasoreactivity, increased cellular migration and proliferation, and vascular remodeling of the pulmonary arteries. For instance increased ROS activate multiple kinase pathways and, of particular interest to this review, has been shown to increase intracellular Ca2+ concentration ([Ca2+]i) in various vascular beds [3]. It is now appreciated that pulmonary vascular ECs exhibit considerable phenotypic heterogeneity based on whether they reside in the large, conduit vessels, i.e., the pulmonary artery, or the smaller vessels in the microvasculature [4]. Although a significant amount is known regarding the role of ROS in abnormal EC function in the systemic circulation, it is not clear which of these pathways may also play a role in promoting changes in EC phenotype in different types of lung ECs. While more recent studies have begun utilizing ECs isolated from specific sites within the lung (i.e., conduit lung vessels versus microvasculature) in order to better understand the specific pathways underlying lung EC dysfunction in PH [5–7], significant gaps in our understanding of the specific molecular mechanisms governing lung EC function remain. In this chapter, we will review the role played by elevations in ROS, and in particular ROS-induced changes [Ca2+]i, in promoting abnormal vasoreactivity and EC function in PH.
2. Pulmonary Hypertension
PH refers to a heterogeneous condition defined as increased (≥25 mmHg at rest) mean pulmonary arterial pressure (mPAP), leading to progressive right ventricular overload and, in some cases, failure [8]. PH is diagnosed based on clinical examination and right heart catheterization, and is classified into five main groups based on hemodynamic and pathologic differences (Table 1). Group 1 PH, also called pulmonary arterial hypertension (PAH), is characterized by very high right-sided pressures and development of occlusive lesions in the pulmonary vasculature. The etiology of PH in Group 1 includes inherited and idiopathic causes as well as PAH due to drugs and toxins, connective tissue disease, HIV, and chronic liver disease (portal hypertension). Group 2 refers to PH that occurs as a consequence of left heart dysfunction, while Group 3 is defined as PH occurring in the context of chronic hypoxia, often in the setting of parenchymal lung disease. Group 4 and Group 5 refer to elevated PPA due to chronic thromboembolic and systemic diseases, respectively.
Table 1.
Classification of PH (adapted from Galie et al., Eur Heart J 2015)
| Classification of PH |
|---|
| Group 1: Pulmonary Arterial Hypertension (PAH) |
| Idiopathic PAH |
| Heritable PAH due to known (e.g., BMPR2) or other mutations |
| Drug and toxin induced |
| PAH associated with systemic disease |
| Connective tissue disease |
| HIV |
| Portal hypertension (liver disease) |
| Congenital heart disease |
| Schistosomiasis |
| Group 1′: Pulmonary veno-occlusive disease Pulmonary capillary hemangiomatosis |
| Idiopathic |
| Heritable (EIF2AK4 or other mutations) |
| Drugs, toxins and radiation-induced |
| Associated with connective tissue disease or HIV infection |
| Group 1′′: Persistent PH of the newborn |
| Group 2: PH due to left heart disease |
| Left ventricular systolic or diastolic dysfunction |
| Valvular disease |
| Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies |
| Congenital/acquired pulmonary vein stenosis |
| Group 3: PH due to lung diseases and/or hypoxia |
| PH due to chronic hypoxia (COPD, interstitial lung disease) |
| PH due to sleep-disordered breathing |
| Alveolar hypoventilation disorders |
| Chronic high altitude exposure |
| Developmental lung disease |
| Group 4: PH due to chronic thromboembolic disease and other pulmonary artery obstructions |
| Chronic thromboembolic pulmonary hypertension |
| Other pulmonary artery obstructions (e.g., Angiosarcoma, arteritis, parasites) |
| Group 5: PH due to unclear mechanism |
| Hematologic disorders (e.g., chronic hemolytic anemia) |
| Systemic disorders (e.g., sarcoidosis, LAM) |
| Metabolic disorders (e.g., glycogen storage disease) |
| Other (e.g., tumoral thrombotic microangiopathy, fibrosing mediastinitis. Chronic renal failure, segmental PH) |
While each group in the PH classification arises from varied etiologies, in each case the underlying pulmonary vascular abnormalities fall into three main categories: (1) increased vascular tone due to sustained contraction of the arterial tree, (2) vascular wall thickening due to uncontrolled EC, SMC, and fibroblast migration and proliferation, and in the case of PAH, (3) development of occlusive lesions in the distal arterioles that obstruct blood flow. These occlusive lesions of PAH comprise multiple cell types, including ECs that are monoclonal, apoptosis-resistant and hyperproliferative. The fundamental cell signaling abnormalities that lead to this aberrant EC phenotype are still not clear, with many potential pathways remaining under investigation. Over the past decade, there has been continued interest in the role of ROS in regulating both abnormal pulmonary vascular tone and promoting migration and proliferation.
3. ROS in Pulmonary Hypertension
Accumulating evidence supports a role for increased ROS in the development and progression of PH [1, 9]. Urinary and plasma levels of oxidative stress markers have been detected in PAH patients [10, 11] while histological examination of lung sections from patients displayed increased nitrotyrosine and 8-hydroxyguanosine residues, which are by-products of oxidative stress [12]. Experimentally decreasing ROS levels with antioxidants augmented pulmonary arterial responsiveness to the vasodilator, nitric oxide (NO), in rat pulmonary arterial rings [13, 14]. Similarly, inhibition of xanthine oxidase or treatment with the ROS scavenger TEMPOL, attenuated development of chronic hypoxic PH. These data, collectively suggest that elevated ROS are involved in the pathobiology of PH, particularly by influencing Ca2+-dependent pulmonary vasoreactivity and increased migration and proliferation of ECs.
4. Sources of ROS in PH
ROS can arise from multiple extracellular (i.e., infiltrating inflammatory cells) and intracellular sources [1]. Generation of intracellular ROS in the lung vasculature occurs via mechanisms similar to those described in other cell types. For example, membrane bound NOX produce ROS in response to external agonists, while non membrane-bound enzymes, such as xanthine oxidase, aid in generation of additional cytosolic ROS [2]. Perhaps the most common source of ROS generation is mitochondria [15].
NAPDH Oxidase.
NOX is a complex enzyme comprising several protein subunits [16]. It catalyzes the transfer of two electrons from NADPH to two successive oxygen molecules, generating superoxide anions. In a variety of cell types, including ECs, NOX generates superoxide that is quickly converted to hydrogen peroxide (H2O2), allowing the enzyme to exert significant signaling effects on ROS-sensitive targets. In phagocytic cells, NOX-derived ROS forms part of the defense mechanism against pathogens whereas in non-phagocytic cells NOX is thought to serve primarily in a signaling capacity. The catalytic subunit has several isoforms (NOX1–5, DUOX1–2) and associates with multiple binding partners that enhance enzymatic activity (reviewed in [2, 16, 17]). Some of these binding partners are isoform specific; for instance, NOX2 associates with several “phox” proteins, including p47 and p67, and Rac1 while NOX4 associates with Poldip2 and p22 [18]. NOX1–4 are expressed in ECs, whereas both NOX2 and NOX4 have both been shown to contribute to EC proliferation [19]. NOX4 may be particularly important to EC function, as specific deletion of NOX4 inhibits microvascular and umbilical vein EC migration and proliferation. Conversely, NOX4 overexpression is sufficient to increase migration and proliferation [20].
Evidence suggests that NOX expression and activity is in fact increased in PH, which leads to pulmonary vascular hyperreactivity. For example, studies performed on homogenates of endothelium-intact pulmonary arteries isolated from a model of fetal/newborn PH demonstrated that increased NOX expression and uncoupling of endothelial nitric oxide synthase (eNOS; see Fig. 1) leads to elevated ROS production [21, 22]. Moreover, lung tissue expression of NOX subunits was increased in a murine model of chronic intermittent hypoxia (modeling PH due to obstructive sleep apnea) [23]. PH induced by either 3 or 10 days of hypoxia also increased NOX1 expression in pulmonary resistance arteries of newborn piglets [24]. On the other hand, mice with deficiency of the gp91phox (i.e., NOX2) subunit failed to develop PH with chronic sustained [25] or intermittent hypoxia [23]. Loss of gp91phox also attenuated hypoxia-induced, NO-dependent relaxation in isolated pulmonary arteries [26]. While all of these studies suggest a role for increased NOX expression/activity in PH, a caveat with the use of whole tissue homogenates or global knockouts is that the exact contribution of ECs (vs. SMCs) cannot be fully ascertained.
Fig. 1.
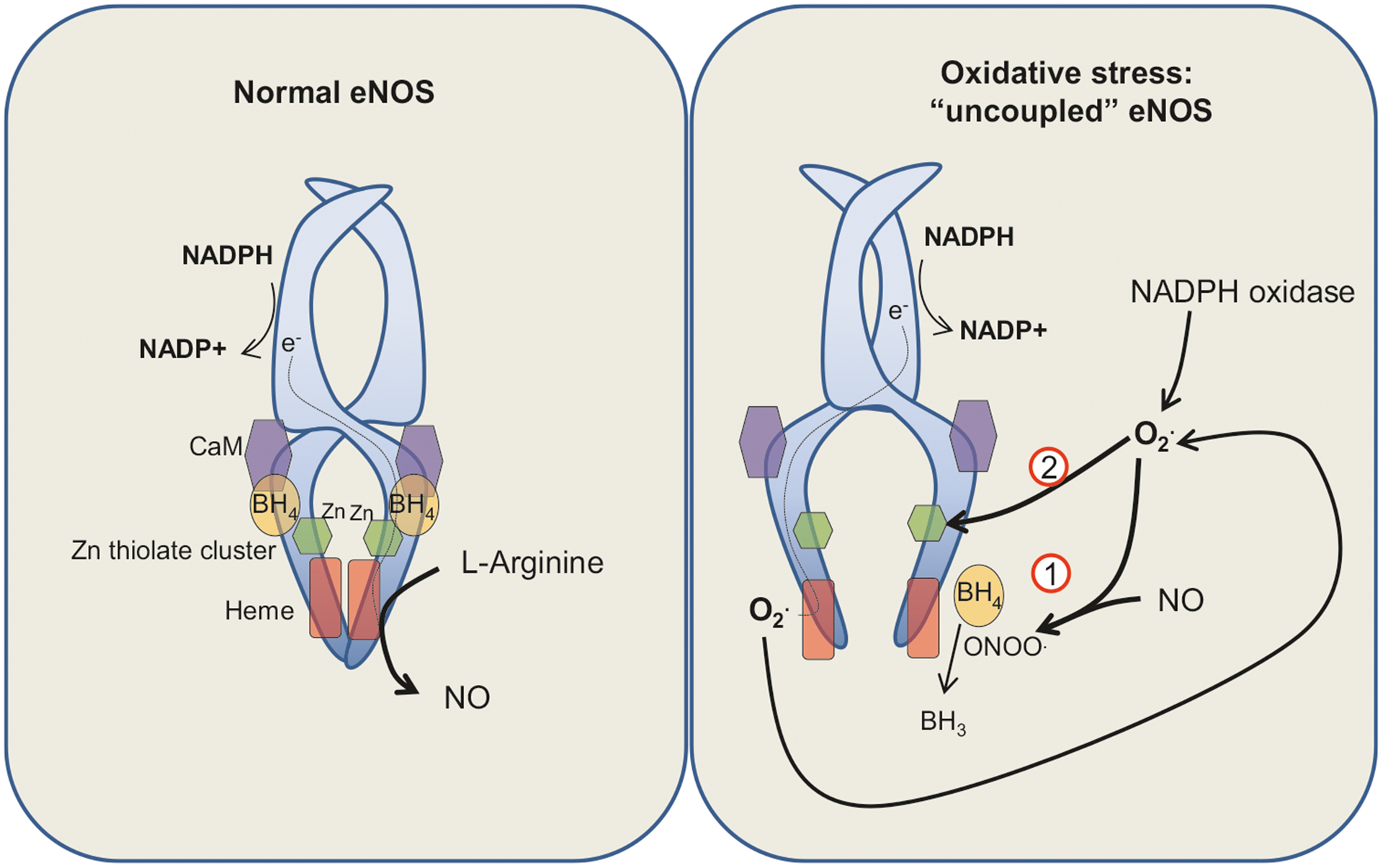
Reactive oxygen species (ROS) and endothelial nitric oxide synthase (eNOS) uncoupling. Under normal circumstances (left panel), eNOS utilizes various cofactors including l-arginine, molecular O2 and (6R-)5,6,7,8-tetrahydrobiopterin (BH4) to catalyze nitric oxide (NO) formation (at the heme site). The eNOS enzyme includes various regulatory subunits including a calmodulin (CaM) binding site. In situations of oxidative stress (right panel), local increases in superoxide (O2•) lead to: (1) formation of peroxinitrites (ONOO−) due to reactions with NO, causing oxidation of BH4 (to BH3) and inactivation of this critical cofactor and (2) oxidative modification of the Zn-thiolate cluster. Together, these events lead to decreased catalytic efficiency of eNOS and generation of O2• instead of NO. Increased O2• in turn can augment further uncoupling of eNOS via a feed-forward mechanism
NOX is activated by ligation of VEGF receptor-2 (VEGFR2) via Rac1 [27]. In addition, NOX can be activated by a myriad other injurious stimuli including angiotensin II [27], tumor necrosis factor-alpha (TNF-α) [28] and shear stress [29]. The functional consequences of increased NOX-derived ROS in ECs are incompletely understood. Based on work performed in systemic ECs, basal ROS generation by NOX likely contributes to EC proliferation and angiogenesis [19, 20], although these effects are mild. The growth response is enhanced when NOX activity is stimulated by agonists known to be involved in PH, including hypoxia [30] and vascular endothelial growth factor (VEGF) [31]. Regarding the latter, NOX-generated ROS may serve as a signal for VEGF-induced changes in EC growth [31, 32].
Once generated, NOX-derived ROS activate a variety of targets, including signaling molecules (e.g., Src, Akt, and MAP kinases) and transcription factors (e.g., NF-kB and AP-1) [28]. ROS serve as an activation signal by increasing leukocyte recruitment to the endothelium [33, 34] and initiating and propagating EC migration [35, 36]. In response to VEGF, localized production of ROS by NOX mediates interactions with TRAF4 and PAK1, two cytoskeletal proteins involved in cell motility [37, 38]. NOX-derived ROS are also critical for EC proliferation [39] via activation of Ras/ERK and JNK pathways in response to the viral protein, Tat, in a HIV-induced model of EC migration and proliferation. These data suggest that ROS play a critical role in EC movement and division, especially in response to injurious stimuli.
Mitochondrial ROS (mtROS).
Mitochondria are major sources of ROS in all cell types, and have recently come under increased scrutiny as drivers of ROS-induced EC dysfunction in PH. Complexes I and III of the electron transport chain are known mitochondrial sites of ROS generation [15, 40, 41]. Superoxide produced by complex I is released into the mitochondrial matrix, while superoxide produced by complex III can be released into either the intermembrane space or the mitochondrial matrix. Upon release, superoxide released into the intermembrane space or matrix is converted to H2O2 by superoxide dismutase (SOD)-1 or SOD-2, respectively. The site-specific superoxide release in mitochondria can modulate different signaling pathways [40]. Since mitochondria are thought to function as cellular oxygen sensors, mtROS may link changes in oxygen tension and hypoxia-induced alterations in cell signaling and function. In part due to the limitations surrounding ROS-measurement methods, difficulty of measuring mitochondrial O2 in vivo and the differential effects of superoxide due to compartmentalization, the exact relationship between hypoxia and mtROS remains an area of active investigation, with lines of evidence pointing towards both decreased [42–44] and increased ROS [45, 46] in the setting of acute hypoxia (also reviewed in [1, 45, 47]).
With respect to the role of mtROS in prolonged hypoxia, which is more relevant to PH, most studies have been performed in pulmonary arterial SMCs, where one of the main consequences of altered mtROS following hypoxia is stabilization of hypoxia-inducible factor-1α (HIF-1α), the oxygen-sensitive subunit of the HIF-1 transcription factor (Fig. 2). HIF-1, a master regulator of adaptive cell responses to hypoxia [48], is constitutively ubiquitinated and degraded due to hydroxylation by proline hydroxylase domain (PHD) proteins. Since this reaction requires O2, hypoxia leads to loss of hydroxylation secondary to decreased substrate availability and stabilization of HIF-1α [49]. HIF-1α/HIF-1β heterodimers transcriptionally regulate a variety of cellular responses to hypoxia [50]. Although the mechanisms are not yet fully clear, regulation of HIF-1α by mtROS likely involves inhibition of enzymes (i.e., PHDs) that normally aid in HIF-1α degradation [51]. For instance, quenching to H2O2 in the intermembrane space of the mitochondrion before it is able to reach the cytosol attenuates hypoxia induced HIF-1α stabilization [52]. Less is known regarding the relationship between hypoxia and mtROS in pulmonary ECs. However, Al-Mehdi et al. [53] reported that, in PA ECs, acute hypoxia increased mtROS and mobilized mitochondria to the perinuclear space, leading to diffusion of mtROS into the nucleus. Taken together with reports describing induction of HIF-1α [54] expression in response to hypoxia in PA ECs, it is likely that signaling pathways similar to those described in SMCs, involving mtROS generation and HIF-1α stabilization, exist in PA ECs as well.
Fig. 2.
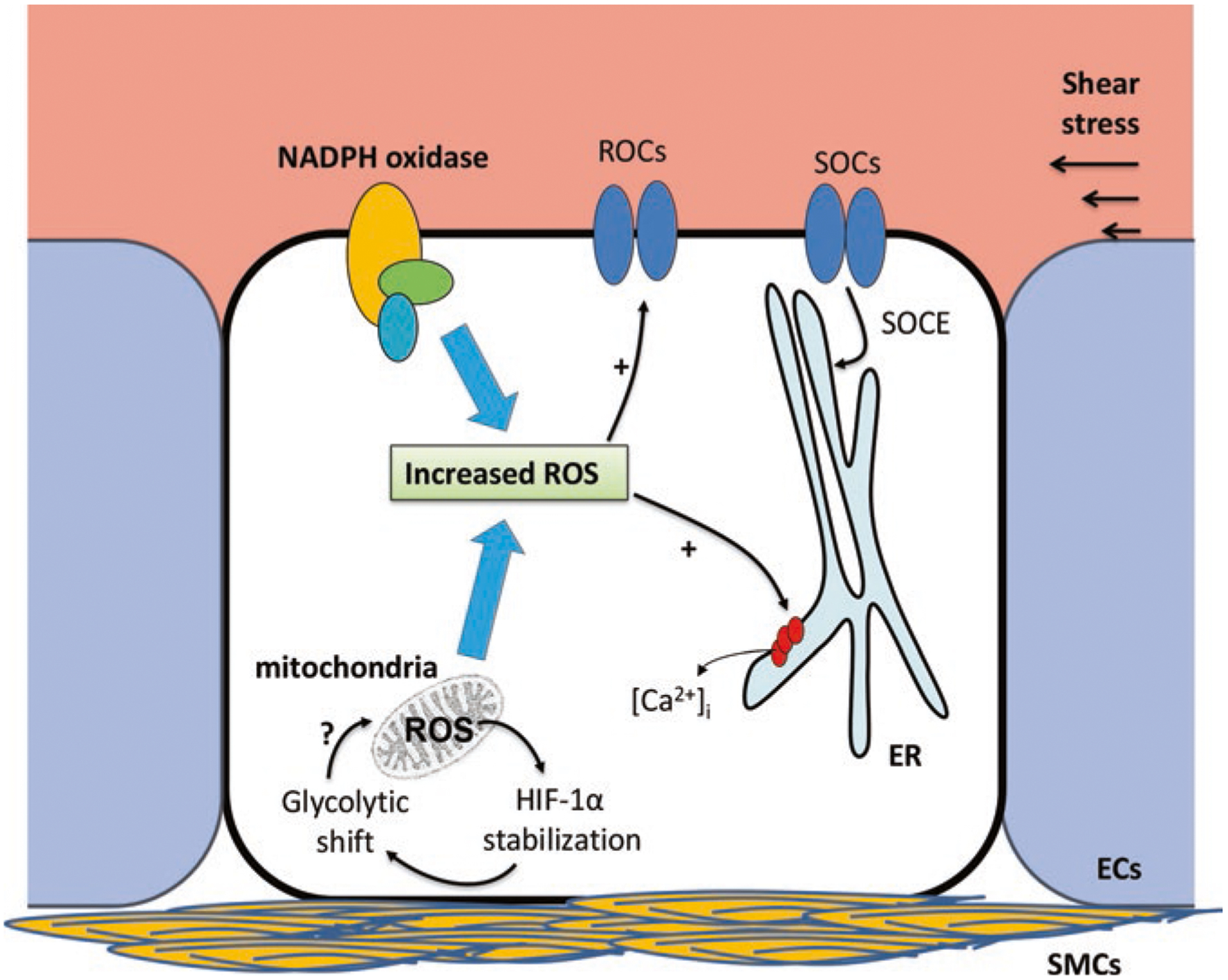
Mechanisms of reactive oxygen species (ROS)-induced Ca2+ entry in endothelial cells (ECs) in pulmonary arterial hypertension (PAH). Increases in ROS from cytosolic (primarily NADPH oxidase) and mitochondrial sources can lead to both release of Ca2+ from internal endoplasmic reticulum (ER) stores as well as influx through receptor-operated (ROC) and store-operated (SOC) channels on the cell membrane. Release of Ca2+ from the ER can activate SOCs in order to replete ER Ca2+ stores via store-operated Ca2+ entry (SOCE). The sources of increased ROS in ECs include: NADPH oxidase, mitochondria and physical stimuli like shear stress. Based on data from both ECs and smooth muscle cells (SMCs) in PAH, mitochondrial ROS generation may be linked to abnormal HIF-1α stabilization and glycolytic shift
Main transcriptional targets of HIF-1 include metabolic enzymes that control cellular shifts from oxidative phosphorylation towards glycolysis [44]. This shift, called the Warburg effect, was initially described in cancer cells and, while less efficient in terms of ATP production, allows for increased production of other macromolecules required for cellular growth and proliferation. Under conditions of tissue hypoxia, glycolysis is a normal response to cellular stress (i.e., the Pasteur effect). However, as increasingly seen in the context of PAH, abnormal HIF stabilization during normoxic conditions may induce a metabolic shift and consequent changes in cell function. Histologic sections of lungs from patients with PAH that stain positive for markers of oxidative stress also exhibit increased HIF-1α expression [55], while pulmonary ECs from patients with PAH display glycolytic shift [56]. These data indicate that HIF is abnormally stabilized in PAH and ROS, most likely mitochondrial in origin, are probably involved. It is likely that mtROS, maladaptive HIF stabilization and glycolytic shift are related processes that participate in a feed-forward mechanism such that changes in mtROS may enhance HIF stabilization, which in turn promotes glycolytic shift, altering mitochondrial redox state. However, much remains unknown with regards to the specifics of mtROS in PAH, particularly in ECs.
While little is known about mtROS levels in lung endothelium in the context of changes in mitochondrial respiration, data collected in systemic ECs may provide important clues [41]. In general, vascular ECs do not use oxidative phosphorylation as primary source of ATP [57]. Agonists, such as glucose, Ang II, and VEGF, increase EC mtROS production [41] and also activate NOX, arguing for the possibility that EC ROS production in PH may occur at multiple sites and may actually be linked, as significant cross talk likely exists between these pathways. As an example, mitochondrial biogenesis [58] and ROS production induced by VEGF, may in turn increase NOX expression/activity, further promoting oxidative stress [59].
Cyclic stretch provides one model where the interplay between mtROS and HIF-1α stabilization has been recently investigated. Unlike shear stress, which results from flow of fluid along (or parallel to) the endothelium in a single direction, stretch is primarily determined by the intraluminal vascular pressure [60], and exerts force perpendicular to the endothelial surface. Most often used in the context of ventilator-associated lung injury, in vitro models of stretch may mimic pressure-related changes in the PA that occur during PH. While the effect of increased strain has not been evaluated in pulmonary ECs, PASMCs undergoing cyclic stretch exhibit increased mtROS and HIF-1α activity; interestingly, these findings mirror those found in SMCs isolated from newborn lambs in a pediatric PH model induced by in utero ligation of the ductus arteriosus [61, 62]. Finally, HUVECs undergoing cyclic strain generated ROS, a response that was absent in cells depleted of mitochondria [63].
Although much remains unknown regarding the role of mtROS in promoting functional changes in the pulmonary vasculature in PH, a recent report noted that induction of mtROS promoted pulmonary arterial EC migration and proliferation in a process that involved p38 MAP kinases [64]. In the systemic circulation, the role of mtROS in EC-mediated vascular dysfunction has been studied more in more detail, with mtROS promoting EC migration and proliferation directly [40, 65]. Moreover, H2O2 generated by mitochondria was required for EC-mediated flow-induced coronary resistance artery dilation [66]. Taken together, these data suggest that EC mtROS are likely to play a role in ROS-induced EC dysfunction during PH.
5. EC [Ca2+]i in Pulmonary Hypertension
In non-excitable cells, Ca2+ homeostasis is maintained via several mechanisms. Ca2+ can enter cells from the extracellular space through plasma membrane channels or be pumped from the cell by Ca2+-ATPases. Ca2+ is also sequestered within, and released from, organelles. In the endoplasmic reticulum (ER) , Ca2+ is tightly regulated by release channels and Ca2+ transporters on the ER membrane, while similar channels control mitochondrial Ca2+ influx and efflux (Fig. 2). [Ca2+]i homeostasis between the cytosol and ER can be pharmacologically disrupted using agents such as thapsigargin (Tg), an agent that inhibits Ca2+ uptake into the ER and depletes ER stores. A full listing of endothelial Ca2+ channels/transporters is beyond the scope of this chapter; however, in general, plasmalemmal Ca2+ channels can be receptor-operated (e.g., transient receptor potential [TRP]), voltage-gated (e.g., T-type) or activated by depletion of ER Ca2+ stores (store-operated) [67–69], while ER release channels belong to inositol triphosphate or ryanodine receptor families.
At baseline, lung EC cytosolic Ca2+ levels are tightly controlled, typically averaging 100 nM [70]. Stimuli can increase [Ca2+]i by directly binding receptor-operated channels on the cell membrane, activating metabolites that subsequently agonize channels, or stimulating Ca2+ efflux from the ER. Limited information is available regarding changes in, and the specific role of, basal EC [Ca2+]i in PH. Globally, whole genome profiling approaches showed that genes encoding Ca2+ signaling pathways were significantly dysregulated in ECs in a model of PH induced by the diet drug, fenfluramine [71]. Exposure of pulmonary ECs to either acute [72] or chronic hypoxia [73] increased [Ca2+]i, with the latter mediated via upregulation of store-operated TRP channels. Together, these data suggest a role for increased [Ca2+]i in PAH EC pathobiology, but as detailed below, the specific source of [Ca2+]i and the channel involved remain under investigation.
One Ca2+ channel that has recently come into focus in PH studies is a member of the transient receptor potential (canonical) family, TRPC4. A store-operated Ca2+ channel, TRPC4 plays a key role in increasing endothelial permeability in the large vessels of the lung [74], leading to the formation of perivascular cuffing [75–77]. Interestingly, rats subjected to the SU5416 plus hypoxia (SuHx) protocol, which creates a severe experimental model of PAH, displayed increased EC Ca2+ transients through TRPC4 [78] and perivascular edema in response to Tg compared to normoxic controls, suggesting a higher sensitivity to induction of EC hyperpermeability through activation of SOCE [79]. These responses were attenuated in TRPC4−/− rats even though PA pressures were similar to wild-type animals [79]. Loss of TRPC4 also conferred a survival benefit in SuHx rats [80], although the exact cell type (EC or SMC) responsible for this benefit remains to be determined. These data suggest that changes in SOCE mediated changes in EC permeability may contribute to mortality in PAH, though it is unclear whether the same calcium channels also contribute to other aspects of PAH pathogenesis such as formation of vaso-occlusive lesions.
6. Links Between ROS and [Ca2+]i in the Lung Endothelium
ROS-induced Ca2+ entry.
Elevations in ROS have been shown to increase [Ca2+]i and, conversely, increased [Ca2+]i participates in ROS generation [3]. With respect to the former, ROS can activate Ca2+ channels by two mechanisms [81–83]: via direct activation through redox sensitive cysteine residues on the channels (i.e., TRPA1, TRPV4, TRPM4) or via activation by metabolites generated by elevations in ROS (i.e., TRPM2) [81, 84–86, 87]. In addition to modulating the activity of plasma membrane channels, ROS can also induce release from internal stores, presumably through an effect on ER Ca2+ transporters [88–91]. The proteins responsible for replenishing ER Ca2+ stores following depletion (i.e., STIM and Orai) are also redox sensitive [92]. It should be noted, however, that the specific radical generated and/or compartmentalization may result in different Ca2+ responses [93]. Further complicating ROS-mediated Ca2+ responses is the fact that the increase in [Ca2+]i likely represents a summation of multiple, temporally distinct Ca2+ release phenomena, with a mix of release and influx events.
mtROS and [Ca2+]:
Mitochondria also possess distinct Ca2+ channels that regulate Ca2+ uptake and release [94]. The mitochondrial Ca2+ uniporter (MCU) is responsible for Ca2+ transport into the mitochondria while various other pumps are responsible for Ca2+ efflux. In addition to buffering cytosolic Ca2+ levels, proximity of mitochondria to the ER allows for Ca2+ ion transfer between these organelles [95]. Several enzymatic reactions that occur in the mitochondria, including the portions of the Krebs cycle, require Ca2+ and large increases in mitochondrial Ca2+ can also act as a signal for impending cell failure and activate cell death [94]. Increased mitochondrial Ca2+ levels and mtROS are often observed under similar conditions, and mice deficient for UCP2, a mitochondrial Ca2+ transport protein, developed PH, suggesting a common pathway between mtROS and [Ca2+]i. Furthermore, pulmonary SMCs isolated from UCP2 mice exhibit stabilization of HIF under normoxic conditions. While these changes mimic those observed with hypoxia [96] or in ECs from PAH patients [97] and animal models [98], it is not clear if similar HIF stabilization was also present in ECs of UCP2-deficient mice. Thus, the role for EC UCP2 in modulating maladaptive mitochondrial function in PH remains unknown.
ROS and [Ca2+]i in EC migration and proliferation.
Migration and proliferation are dependent on both ROS and [Ca2+]i in a variety of tissue types [3]. In PH, increased migration and proliferation occur in both SMCs and ECs, and the anatomic site for these functional changes is at the level of the resistance arteries [68]. Unlike SMCs, where significantly more is known regarding migration and proliferation following ROS-generating stimuli, considerably less is known about the interplay of ROS and Ca2+ in EC, particularly lung EC. Data from systemic ECs demonstrated that exposure to VEGF increased NADPH-derived ROS, leading to posttranslational modification of SERCA2b (S-glutathiolation), increased [Ca2+]i, and migration. Notably, removal of a reactive cysteine on SERCA2b attenuated increases in migration by either VEGF or exogenous H2O2 [91, 99]. Interplay between ECs and SMCs exists as well; for example, endothelial store-operated [Ca2+]i entry is important for transcriptional activation of various inflammatory factors that in turn drive pulmonary smooth muscle proliferation and vascular remodeling [73].
ROS and [Ca2+]i in EC shear stress responses.
A phenomenon of particular interest in PH is shear stress. Recent studies using magnetic resonance imaging elegantly demonstrated the presence of complex flow patterns in the pulmonary arteries of patients with PAH [100, 101]. ECs are uniquely positioned to serve as sensors of changes in adjacent blood flow, with EC dysfunction occurring as a consequence of both changes in laminar flow and formation of non-laminar flow patterns. Changes in shear stress cause a wide variety of responses in ECs, from alterations in gene expression [102] to cytoskeletal reorganization [103]. With respect to ROS, both increased and abrupt cessation of blood flow triggered radical production [104–106] and increased [Ca2+]i [107–109], suggesting the possibility of cross talk between these signaling pathways. Supporting this notion, flow-adapted microvascular ECs subjected to sudden disruption in flow exhibited augmented baseline expression of T-type Ca2+ channels and NOX-dependent ROS generation [110, 111] that was followed sequentially by an increase [Ca2+]i [112, 113] that was inhibited by depletion of ER stores. However, the effect of changes in shear stress due to the complex flow patterns observed in PH has not been fully elucidated. Interestingly, high shear stress increased [Ca2+]i to a greater extent in SMC from PAH patients compared to normal controls [114] through a mechanism involving increased expression of TRPM7 and TRPV4 channels. Further studies will be required to determine whether a similar response occurs through the same mechanisms in ECs.
ROS and Ca2+ in pulmonary vasoreactivity.
Interactions between ROS and Ca2+ have been implicated in several stimulus-specific mechanisms of increased vasoreactivity including: (1) hypoxic pulmonary vasoconstriction (HPV), a well-described pulmonary vascular response to acute hypoxia, (2) nitric oxide production, a key step in endothelium-mediated pulmonary vascular vasodilation, and (3) pulmonary arterial constriction in chronic hypoxia, a known mediator of PH, particularly in Class III disease.
(a). Hypoxic Vasoconstriction:
It is well documented that decreased oxygen tension causes HPV in an effort to shunt blood away from poorly oxygenated alveoli. HPV may also play some role in the development of PH, although the exact cellular mechanisms underlying contraction in response to acute and chronic hypoxia may differ depending on the length of hypoxic exposure. Nonetheless, both ROS and increased [Ca2+]i are involved in HPV [115–117]. ROS are sufficient to induce pulmonary vasoconstriction; in studies utilizing endothelium-intact pulmonary arterial rings, contraction induced by provisioning exogenous ROS was not attenuated by removal of extracellular Ca2+ or blocking of membrane channels, arguing for store release as the mechanism of ROS-induced Ca2+ influx mediating PA contraction [118–120] (Fig. 3). However, an important caveat to these studies is that it is not possible to delineate which effects were mediated by the endothelium and which might have occurred in SMCs.
Fig. 3.
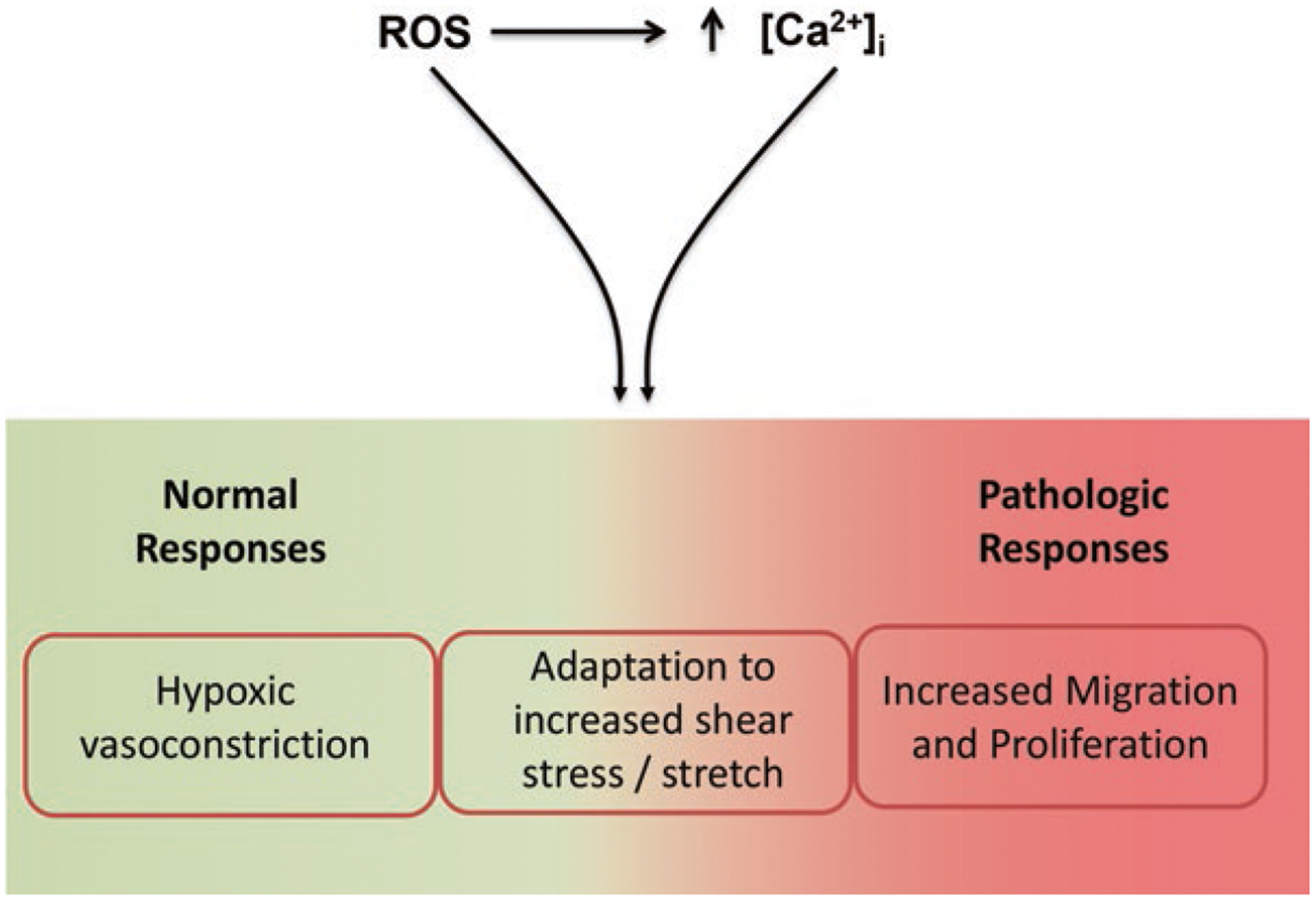
Functional consequences of elevated reactive oxygen species (ROS) and intracellular Ca2+ concentration ([Ca2+]i) in pulmonary endothelial cells (ECs). Elevations of both ROS and [Ca2+]i promote normal physiologic responses, such as hypoxic vasoconstriction, maladaptive responses such as EC adaptation to shear stress and stretch and lastly, pathologic responses such as increased migration and proliferation
(b). Nitric Oxide Production:
Nitric oxide (NO) production by ECs is a key defense mechanism against vascular dysfunction across multiple vascular beds, including the pulmonary circulation. In ECs, NO production occurs primarily via endothelial nitric oxide synthase (eNOS). Dimeric units of eNOS catalyze electron transfer from NADPH to various electron carrying intermediates to eventually produce NO from l-arginine in a process that requires molecular O2, l-arginine and (6R-)5,6,7,8-tetrahydrobiopterin (BH4). This process occurs in two steps. First, electrons are transferred from NAPDH to a heme group within eNOS where they are stabilized with the aid of O2. Next, the electrons are used to catalyze NO production from l-arginine in a process that requires BH4. eNOS activity is regulated by a variety of posttranslational modifications, including phosphorylation and oxidative modification of Cys residues. NO production is also determined by the availability of the critical cofactors such that when BH4 is unavailable, eNOS cannot successfully complete electron transfer from NADPH to l-arginine, resulting in formation of superoxide radicals. Under these conditions, BH4 deficiency uncouples the first half of the reaction (electron transfer from NADPH to the heme group) from the second half (electron transfer from the heme group to l-arginine).
Increases in cytosolic ROS decrease the ability of eNOS to efficiently produce NO primarily by decreasing BH4 availability [121]. NOX-generated superoxide can react with NO to produce peroxinitrites (ONOO•) which can oxidize BH4 to a product (BH3) that cannot serve as a eNOS cofactor [122]. NOX-generated ROS can also oxidize cysteine residues on eNOS to decrease BH4 binding [121] and/or oxidize a moiety that is critical for NO generation (i.e., the Zn-thiolate cluster) [123]. Through these mechanisms, increased cytosolic ROS facilitate uncoupling of eNOS, leading to a feed-forward mechanism of ROS-induced ROS generation in ECs.
While increases in ROS disrupt eNOS function via oxidative modification of critical motifs and cofactors, increases in [Ca2+]i modulate eNOS function via calmodulin (CaM). As reviewed in detail elsewhere [124, 125], the sensitivity of NOS to changes in [Ca2+]i is isoform-dependent. For the purposes of this review, we will limit our discussion to the calcium-sensitive, constitutively expressed eNOS; however, inducible NOS (iNOS), which is not Ca2+ dependent, has also been implicated in oxidative stress. Although controversy remains regarding the specifics of eNOS subcellular localization and the role of various posttranslational modifications on eNOS activity, it is generally accepted that increased [Ca2+]i leads to CaM binding which in turn facilitates electron transfer towards the heme moiety where NO synthesis occurs. Thus, while increased ROS leads to uncoupling, increases in [Ca2+]i activate NO production. The net result of increases in both ROS and [Ca2+]i on NO production in pulmonary ECs may vary with source of ROS, and compartmentalization; studies conducted in ECs from PAH patients show both oxidant stress [12] and increased [Ca2+]i [73], with decreased NO production [126], suggesting that in PH the net effect shifts towards uncoupling/decreased NO production. Consistent with these findings, Ghosh et al. [127] recently showed that abnormal phosphorylation of eNOS contributed to decreased NO production in PAH ECs and may contribute to eNOS uncoupling [128], but further work will be needed to elucidate whether this mechanism is related to ROS and/or Ca2+ levels.
Once produced, NO acts within the vessel lumen, with anti-inflammatory effects on leukocytes and platelets to decrease adherence of inflammatory cells to the endothelium and intracellularly, to induce production of cyclic guanosine monophosphate (cGMP). In SMCs, cGMP promotes vasodilation. Indeed, targeting cGMP availability, by either inhibiting degradation or stimulating production, are two treatment avenues currently utilized in the treatment of PAH.
(c). Chronic Hypoxia:
Increased [Ca2+]i is responsible for enhanced pulmonary vasoreactivity and development of abnormal EC function that occur with chronic hypoxia [129, 130]. For instance, chronic hypoxia increased agonist-induced vasoconstriction via both ROS generation and enhancement of Ca2+-sensitivity of the contractile apparatus due to ROS-induced activation of RhoA/ROCK [131]. However, most of these studies were performed either in SMCs or preparations that included ECs and SMC; thus, the EC-specific roles of ROS and Ca2+ in regulating vasoreactivity during chronic hypoxia remain unclear.
In summary, it is evident that inhibition of either ROS or Ca2+ entry invokes largely similar responses with regards to vasoreactivity in the pulmonary vasculature. However, significant gaps still exist in our understanding as to whether ROS directly activates Ca2+ entry in pulmonary ECs, and if so, the exact channels that are involved in this response.
7. Conclusions
Based on available evidence, elevations in ROS and [Ca2+]i play key roles in the pathobiology of PH, mainly because increased ROS generation elevates EC [Ca2+]i. While it seems clear that elevations in ROS from NOX and mitochondria are important for migration, proliferation, and vaso-reactive responses, the role played by ROS-induced Ca2+ influx on these pathways, particularly in ECs, remains to be determined. Continued investigation is necessary to better understand the role of both ROS and [Ca2+]i in promoting vascular reactivity, migration, and proliferation in pulmonary ECs. The hope is that this will lead to targeted therapies aimed at reversing or slowing progression of this devastating disease.
Contributor Information
Karthik Suresh, Division of Pulmonary and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21224, USA; Johns Hopkins Asthma and Allergy Center, 5501 Hopkins Bayview Circle, Baltimore, MD 21224, USA.
Larissa A. Shimoda, Division of Pulmonary and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21224, USA
References
- 1.Aggarwal S, Gross CM, Sharma S, Fineman JR, & Black SM (2013). Reactive oxygen species in pulmonary vascular remodeling. Comprehensive Physiology, 3, 1011–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damico R, Zulueta JJ, & Hassoun PM (2012). Pulmonary endothelial cell NOX. American Journal of Respiratory Cell and Molecular Biology, 47, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Touyz RM (2005). Reactive oxygen species as mediators of calcium signaling by angiotensin II: Implications in vascular physiology and patho-physiology. Antioxidants & Redox Signaling, 7, 1302–1314. [DOI] [PubMed] [Google Scholar]
- 4.Gebb S, & Stevens T (2004). On lung endothelial cell heterogeneity. Microvascular Research, 68, 1–12. [DOI] [PubMed] [Google Scholar]
- 5.Drake KM, Comhair SA, Erzurum SC, Tuder RM, & Aldred MA (2015). Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dys-regulates SMAD9 signaling. American Journal of Respiratory and Critical Care Medicine, 191, 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duong HT, Comhair SA, Aldred MA, et al. (2011). Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulmonary Circulation, 1, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevens T (2011). Functional and molecular heterogeneity of pulmonary endothelial cells. Proceedings of the American Thoracic Society, 8, 453–457. [DOI] [PubMed] [Google Scholar]
- 8.Galie N, Humbert M, Vachiery JL, et al. (2016). 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). European Heart Journal, 37, 67–119. [DOI] [PubMed] [Google Scholar]
- 9.Wong CM, Bansal G, Pavlickova L, Marcocci L, & Suzuki YJ (2013). Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxidants & Redox Signaling, 18, 1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cracowski JL, Degano B, Chabot F, et al. (2012). Independent association of urinary F2-isoprostanes with survival in pulmonary arterial hypertension. Chest, 142, 869–876. [DOI] [PubMed] [Google Scholar]
- 11.Irodova NL, Lankin VZ, Konovalova GK, Kochetov AG, & Chazova IE (2002). Oxidative stress in patients with primary pulmonary hypertension. Bulletin of Experimental Biology and Medicine, 133, 580–582. [DOI] [PubMed] [Google Scholar]
- 12.Bowers R, Cool C, Murphy RC, et al. (2004). Oxidative stress in severe pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine, 169, 764–769. [DOI] [PubMed] [Google Scholar]
- 13.Wanstall JC, Kaye JA, & Gambino A (1997). The in vitro pulmonary vascular effects of FK409 (nitric oxide donor): A study in normotensive and pulmonary hypertensive rats. British Journal of Pharmacology, 121, 280–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jernigan NL, Walker BR, & Resta TC (2004). Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. American Journal of Physiology. Lung Cellular and Molecular Physiology, 287, L801–L808. [DOI] [PubMed] [Google Scholar]
- 15.Murphy MP (2009). How mitochondria produce reactive oxygen species. The Biochemical Journal, 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lassegue B, & Clempus RE (2003). Vascular NAD(P)H oxidases: Specific features, expression, and regulation. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 285, R277–R297. [DOI] [PubMed] [Google Scholar]
- 17.Nauseef WM (2008). Biological roles for the NOX family NADPH oxidases. The Journal of Biological Chemistry, 283, 16961–16965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drummond GR, & Sobey CG (2014). Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends in Endocrinology and Metabolism: TEM, 25, 452–463. [DOI] [PubMed] [Google Scholar]
- 19.Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, & Gorlach A (2006). NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxidants & Redox Signaling, 8, 1473–1484. [DOI] [PubMed] [Google Scholar]
- 20.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, & Jiang F (2007). Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology, 27, 2319–2324. [DOI] [PubMed] [Google Scholar]
- 21.Brennan LA, Steinhorn RH, Wedgwood S, et al. (2003). Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: A role for NADPH oxidase. Circulation Research, 92, 683–691. [DOI] [PubMed] [Google Scholar]
- 22.Grobe AC, Wells SM, Benavidez E, et al. (2006). Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: Role of NADPH oxidase and endothelial NO synthase. American Journal of Physiology. Lung Cellular and Molecular Physiology, 290, L1069–L1077. [DOI] [PubMed] [Google Scholar]
- 23.Nisbet RE, Graves AS, Kleinhenz DJ, et al. (2009). The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. American Journal of Respiratory Cell and Molecular Biology, 40, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dennis KE, Aschner JL, Milatovic D, et al. (2009). NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. American Journal of Physiology. Lung Cellular and Molecular Physiology, 297, L596–L607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu JQ, Zelko IN, Erbynn EM, Sham JS, & Folz RJ (2006). Hypoxic pulmonary hypertension: Role of superoxide and NADPH oxidase (gp91phox). American Journal of Physiology. Lung Cellular and Molecular Physiology, 290, L2–10. [DOI] [PubMed] [Google Scholar]
- 26.Fresquet F, Pourageaud F, Leblais V, et al. (2006). Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. British Journal of Pharmacology, 148, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimmeler S, & Zeiher AM (2000). Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regulatory Peptides, 90, 19–25. [DOI] [PubMed] [Google Scholar]
- 28.Ushio-Fukai M (2006). Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovascular Research, 71, 226–235. [DOI] [PubMed] [Google Scholar]
- 29.Duerrschmidt N, Stielow C, Muller G, Pagano PJ, & Morawietz H (2006). NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. The Journal of Physiology, 576, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schafer M, Schafer C, Ewald N, Piper HM, & Noll T (2003). Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circulation Research, 92, 1010–1015. [DOI] [PubMed] [Google Scholar]
- 31.Colavitti R, Pani G, Bedogni B, et al. (2002). Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. The Journal of Biological Chemistry, 277, 3101–3108. [DOI] [PubMed] [Google Scholar]
- 32.Ushio-Fukai M (2007). VEGF signaling through NADPH oxidase-derived ROS. Antioxidants & Redox Signaling, 9, 731–739. [DOI] [PubMed] [Google Scholar]
- 33.Marui N, Offermann MK, Swerlick R, et al. (1993). Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. The Journal of Clinical Investigation, 92, 1866–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook-Mills JM, Marchese ME, & Abdala-Valencia H (2011). Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxidants & Redox Signaling, 15, 1607–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moldovan L, Moldovan NI, Sohn RH, Parikh SA, & Goldschmidt-Clermont PJ (2000). Redox changes of cultured endothelial cells and actin dynamics. Circulation Research, 86, 549–557. [DOI] [PubMed] [Google Scholar]
- 36.van Wetering S, van Buul JD, Quik S, et al. (2002). Reactive oxygen species mediate Rac-induced loss of cell-cell adhesion in primary human endothelial cells. Journal of Cell Science, 115, 1837–1846. [DOI] [PubMed] [Google Scholar]
- 37.Wu RF, Xu YC, Ma Z, Nwariaku FE, Sarosi GA Jr., & Terada LS (2005). Subcellular targeting of oxidants during endothelial cell migration. The Journal of Cell Biology, 171, 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu RF, Gu Y, Xu YC, Nwariaku FE, & Terada LS (2003). Vascular endothelial growth factor causes translocation of p47phox to membrane ruffles through WAVE1. The Journal of Biological Chemistry, 278, 36830–36840. [DOI] [PubMed] [Google Scholar]
- 39.Wu RF, Ma Z, Myers DP, & Terada LS (2007). HIV-1 Tat activates dual Nox pathways leading to independent activation of ERK and JNK MAP kinases. The Journal of Biological Chemistry, 282, 37412–37419. [DOI] [PubMed] [Google Scholar]
- 40.Zhang DX, & Gutterman DD (2007). Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology, 292, H2023–H2031. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Fang P, Li Y, et al. (2016). Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arteriosclerosis, Thrombosis, and Vascular Biology, 36, 1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Archer SL, Huang J, Henry T, Peterson D, & Weir EK (1993). A redox-based O2 sensor in rat pulmonary vasculature. Circulation Research, 73, 1100–1112. [DOI] [PubMed] [Google Scholar]
- 43.Dunham-Snary KJ, Hong ZG, Xiong PY, et al. (2016). A mitochondrial redox oxygen sensor in the pulmonary vasculature and ductus arteriosus. Pflugers Archiv: European Journal of Physiology, 468, 43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, & Weir EK (2008). Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. American Journal of Physiology. Heart and Circulatory Physiology, 294, H570–H578. [DOI] [PubMed] [Google Scholar]
- 45.Waypa GB, & Schumacker PT (2005). Hypoxic pulmonary vasoconstriction: Redox events in oxygen sensing. Journal of Applied Physiology (1985), 98, 404–414. [DOI] [PubMed] [Google Scholar]
- 46.Waypa GB, & Schumacker PT (2010). Hypoxia-induced changes in pulmonary and systemic vascular resistance: Where is the O2 sensor? Respiratory Physiology & Neurobiology, 174, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolin MS, Ahmad M, & Gupte SA (2005). Oxidant and redox signaling in vascular oxygen sensing mechanisms: Basic concepts, current controversies, and potential importance of cytosolic NADPH. American Journal of Physiology. Lung Cellular and Molecular Physiology, 289, L159–L173. [DOI] [PubMed] [Google Scholar]
- 48.Semenza GL (2011). Oxygen sensing, homeostasis, and disease. The New England Journal of Medicine, 365, 537–547. [DOI] [PubMed] [Google Scholar]
- 49.Pisarcik S, Maylor J, Lu W, et al. (2013). Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. American Journal of Physiology. Lung Cellular and Molecular Physiology, 304, L549–L561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tuder RM, Davis LA, & Graham BB (2012). Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine, 185, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waypa GB, Smith KA, & Schumacker PT (2016). O2 sensing, mitochondria and ROS signaling: The fog is lifting. Molecular Aspects of Medicine, 47–48, 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabharwal SS, Waypa GB, Marks JD, & Schumacker PT (2013). Peroxiredoxin-5 targeted to the mitochondrial intermembrane space attenuates hypoxia-induced reactive oxygen species signalling. The Biochemical Journal, 456, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryan JM (1979). Effect of different fetal bovine serum concentrations on the replicative life span of cultured chick cells. In Vitro, 15, 895–899. [DOI] [PubMed] [Google Scholar]
- 54.Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, & Semenza GL (1998). Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. The American Journal of Physiology, 275, L818–L826. [DOI] [PubMed] [Google Scholar]
- 55.Tuder RM, Chacon M, Alger L, et al. (2001). Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. The Journal of Pathology, 195, 367–374. [DOI] [PubMed] [Google Scholar]
- 56.Xu W, Koeck T, Lara AR, et al. (2007). Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proceedings of the National Academy of Sciences of the United States of America, 104, 1342–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quintero M, Colombo SL, Godfrey A, & Moncada S (2006). Mitochondria as signaling organelles in the vascular endothelium. Proceedings of the National Academy of Sciences of the United States of America, 103, 5379–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wright GL, Maroulakou IG, Eldridge J, et al. (2008). VEGF stimulation of mitochondrial biogenesis: Requirement of AKT3 kinase. FASEB Journal, 22, 3264–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen K, Thomas SR, Albano A, Murphy MP, & Keaney JF Jr. (2004). Mitochondrial function is required for hydrogen peroxide-induced growth factor receptor transactivation and downstream signaling. The Journal of Biological Chemistry, 279, 35079–35086. [DOI] [PubMed] [Google Scholar]
- 60.Birukov KG (2009). Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxidants & Redox Signaling, 11, 1651–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wedgwood S, Lakshminrusimha S, Schumacker PT, & Steinhorn RH (2015). Hypoxia inducible factor signaling and experimental persistent pulmonary hypertension of the newborn. Frontiers in Pharmacology, 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farrow KN, Wedgwood S, Lee KJ, et al. (2010). Mitochondrial oxidant stress increases PDE5 activity in persistent pulmonary hypertension of the newborn. Respiratory Physiology & Neurobiology, 174, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ali MH, Pearlstein DP, Mathieu CE, & Schumacker PT (2004). Mitochondrial requirement for endothelial responses to cyclic strain: Implications for mechanotransduction. American Journal of Physiology. Lung Cellular and Molecular Physiology, 287, L486–L496. [DOI] [PubMed] [Google Scholar]
- 64.Li Q, Mao M, Qiu Y, et al. (2016). Key role of ROS in the process of 15-lipoxygenase/15-hydroxyeicosatetraenoiccid-induced pulmonary vascular remodeling in hypoxia pulmonary hypertension. PloS One, 11, e0149164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Zang QS, Liu Z, et al. (2011). Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. American Journal of Physiology. Cell Physiology, 301, C695–C704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, & Gutterman DD (2003). Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circulation Research, 93, 573–580. [DOI] [PubMed] [Google Scholar]
- 67.Cioffi DL, Wu S, & Stevens T (2003). On the endothelial cell I(SOC). Cell Calcium, 33, 323. [DOI] [PubMed] [Google Scholar]
- 68.Shimoda LA, Wang J, & Sylvester JT (2006). Ca2+ channels and chronic hypoxia. Microcirculation, 13, 657–670. [DOI] [PubMed] [Google Scholar]
- 69.Ying X, Minamiya Y, Fu C, & Bhattacharya J (1996). Ca2+ waves in lung capillary endothelium. Circulation Research, 79, 898. [DOI] [PubMed] [Google Scholar]
- 70.Tiruppathi C, Minshall RD, Paria BC, Vogel SM, & Malik AB (2002). Role of Ca2+ signaling in the regulation of endothelial permeability. Vascular Pharmacology, 39, 173–185. [DOI] [PubMed] [Google Scholar]
- 71.Yao W, Mu W, Zeifman A, et al. (2011). Fenfluramine-induced gene dysregulation in human pulmonary artery smooth muscle and endothelial cells. Pulmonary Circulation, 1, 405–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hampl V, Cornfield DN, Cowan NJ, & Archer SL (1995). Hypoxia potentiates nitric oxide synthesis and transiently increases cytosolic calcium levels in pulmonary artery endothelial cells. The European Respiratory Journal, 8, 515–522. [PubMed] [Google Scholar]
- 73.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, & Yuan JX (2003). Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. American Journal of Physiology. Lung Cellular and Molecular Physiology, 285, L1233–L1245. [DOI] [PubMed] [Google Scholar]
- 74.Cioffi DL, Lowe K, Alvarez DF, Barry C, & Stevens T (2009). TRPing on the lung endothelium: Calcium channels that regulate barrier function. Antioxidants & Redox Signaling, 11, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Townsley MI, King JA, & Alvarez DF (2006). Ca2+ channels and pulmonary endothelial permeability: Insights from study of intact lung and chronic pulmonary hypertension. Microcirculation, 13, 725–739. [DOI] [PubMed] [Google Scholar]
- 76.Lowe K, Alvarez D, King J, & Stevens T (2007). Phenotypic heterogeneity in lung capillary and extra-alveolar endothelial cells. Increased extra-alveolar endothelial permeability is sufficient to decrease compliance. The Journal of Surgical Research, 143, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alvarez DF, King JA, Weber D, Addison E, Liedtke W, & Townsley MI (2006). Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: A novel mechanism of acute lung injury. Circulation Research, 99, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Francis M, Xu N, Zhou C, & Stevens T (2016). Transient receptor potential channel 4 encodes a vascular permeability defect and high-frequency Ca(2+) transients in severe pulmonary arterial hypertension. The American Journal of Pathology, 186, 1701–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou C, Townsley MI, Alexeyev M, Voelkel NF, & Stevens T (2016). Endothelial hyperpermeability in severe pulmonary arterial hypertension: Role of store operated calcium entry. American Journal of Physiology. Lung Cellular and Molecular Physiology, 311(3), L560–L569. doi: 10.1152/ajplung.00057.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alzoubi A, Almalouf P, Toba M, et al. (2013). TRPC4 inactivation confers a survival benefit in severe pulmonary arterial hypertension. The American Journal of Pathology, 183, 1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, & Niemeyer BA (2011). Redox regulation of calcium ion channels: Chemical and physiological aspects. Cell Calcium, 50, 407. [DOI] [PubMed] [Google Scholar]
- 82.Brini M, Cali T, Ottolini D, & Carafoli E (2012). Calcium pumps: Why so many? Comprehensive Physiology, 2, 1045–1060. [DOI] [PubMed] [Google Scholar]
- 83.Parekh AB, & Putney JW Jr. (2005). Store-operated calcium channels. Physiological Reviews, 85, 757. [DOI] [PubMed] [Google Scholar]
- 84.Chuang HH, & Lin S (2009). Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proceedings of the National Academy of Sciences of the United States of America, 106, 20097–20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Macpherson LJ, Xiao B, Kwan KY, et al. (2007). An ion channel essential for sensing chemical damage. The Journal of Neuroscience, 27, 11412–11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simon F, Leiva-Salcedo E, Armisen R, et al. (2010). Hydrogen peroxide removes TRPM4 current desensitization conferring increased vulnerability to necrotic cell death. The Journal of Biological Chemistry, 285, 37150–37158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kolisek M, Beck A, Fleig A, & Penner R (2005). Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Molecular Cell, 18, 61–69. [DOI] [PubMed] [Google Scholar]
- 88.Volk T, Hensel M, & Kox WJ (1997). Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: Role of hydrogen peroxide. Molecular and Cellular Biochemistry, 171, 11–21. [DOI] [PubMed] [Google Scholar]
- 89.Dreher D, Jornot L, & Junod AF (1995). Effects of hypoxanthine-xanthine oxidase on Ca2+ stores and protein synthesis in human endothelial cells. Circulation Research, 76, 388–395. [DOI] [PubMed] [Google Scholar]
- 90.Graier WF, Hoebel BG, Paltauf-Doburzynska J, & Kostner GM (1998). Effects of superoxide anions on endothelial Ca2+ signaling pathways. Arteriosclerosis, Thrombosis, and Vascular Biology, 18, 1470–1479. [DOI] [PubMed] [Google Scholar]
- 91.Evangelista AM, Thompson MD, Bolotina VM, Tong X, & Cohen RA (2012). Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radical Biology & Medicine, 53, 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bogeski I, Kilch T, & Niemeyer BA (2012). ROS and SOCE: Recent advances and controversies in the regulation of STIM and Orai. The Journal of Physiology, 590, 4193–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dreher D, & Junod AF (1995). Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. Journal of Cellular Physiology, 162, 147–153. [DOI] [PubMed] [Google Scholar]
- 94.Contreras L, Drago I, Zampese E, & Pozzan T (2010). Mitochondria: The calcium connection. Biochimica et Biophysica Acta, 1797, 607–618. [DOI] [PubMed] [Google Scholar]
- 95.Rizzuto R, Marchi S, Bonora M, et al. (2009). Ca(2+) transfer from the ER to mitochondria: When, how and why. Biochimica et Biophysica Acta, 1789, 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, & Michelakis ED (2013). Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circulation Research, 113, 126–136. [DOI] [PubMed] [Google Scholar]
- 97.Fijalkowska I, Xu W, Comhair SA, et al. (2010). Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. The American Journal of Pathology, 176, 1130–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bonnet S, Michelakis ED, Porter CJ, et al. (2006). An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation, 113,2630–2641. [DOI] [PubMed] [Google Scholar]
- 99.Evangelista AM, Thompson MD, Weisbrod RM, et al. (2012). Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxidants & Redox Signaling, 17, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barker AJ, Roldan-Alzate A, Entezari P, et al. (2015). Four-dimensional flow assessment of pulmonary artery flow and wall shear stress in adult pulmonary arterial hypertension: Results from two institutions. Magnetic Resonance in Medicine, 73, 1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kheyfets VO, Schafer M, Podgorski CA, et al. (2016). 4D magnetic resonance flow imaging for estimating pulmonary vascular resistance in pulmonary hypertension. Journal of Magnetic Resonance Imaging, 44, 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garcia-Cardena G, Comander JI, Blackman BR, Anderson KR, & Gimbrone MA (2001). Mechanosensitive endothelial gene expression profiles: Scripts for the role of hemodynamics in atherogenesis? Annals of the New York Academy of Sciences, 947, 1–6. [PubMed] [Google Scholar]
- 103.Barakat AI (1999). Responsiveness of vascular endothelium to shear stress: Potential role of ion channels and cellular cytoskeleton (review). International Journal of Molecular Medicine, 4, 323–332. [DOI] [PubMed] [Google Scholar]
- 104.Laurindo FR, Pedro Mde A, Barbeiro HV, et al. (1994). Vascular free radical release. Ex vivo and in vivo evidence for a flow-dependent endothelial mechanism. Circulation Research, 74, 700–709. [DOI] [PubMed] [Google Scholar]
- 105.Chiu JJ, Wung BS, Shyy JY, Hsieh HJ, & Wang DL (1997). Reactive oxygen species are involved in shear stress-induced intercellular adhesion molecule-1 expression in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 17, 3570–3577. [DOI] [PubMed] [Google Scholar]
- 106.Wei Z, Costa K, Al-Mehdi AB, Dodia C, Muzykantov V, & Fisher AB (1999). Simulated ischemia in flow-adapted endothelial cells leads to generation of reactive oxygen species and cell signaling. Circulation Research, 85, 682–689. [DOI] [PubMed] [Google Scholar]
- 107.Ando J, Komatsuda T, & Kamiya A (1988). Cytoplasmic calcium response to fluid shear stress in cultured vascular endothelial cells. In Vitro Cellular & Developmental Biology, 24, 871–877. [DOI] [PubMed] [Google Scholar]
- 108.Mo M, Eskin SG, & Schilling WP (1991). Flow-induced changes in Ca2+ signaling of vascular endothelial cells: Effect of shear stress and ATP. The American Journal of Physiology, 260, H1698–H1707. [DOI] [PubMed] [Google Scholar]
- 109.Shen J, Luscinskas FW, Connolly A, Dewey CF Jr., & Gimbrone MA Jr. (1992). Fluid shear stress modulates cytosolic free calcium in vascular endothelial cells. The American Journal of Physiology, 262, C384–C390. [DOI] [PubMed] [Google Scholar]
- 110.Zhang Q, Matsuzaki I, Chatterjee S, & Fisher AB (2005). Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. American Journal of Physiology. Lung Cellular and Molecular Physiology, 289, L954–L961. [DOI] [PubMed] [Google Scholar]
- 111.Al-Mehdi AB, Zhao G, Dodia C, et al. (1998). Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circulation Research, 83, 730–737. [DOI] [PubMed] [Google Scholar]
- 112.Song C, Al-Mehdi AB, & Fisher AB (2001). An immediate endothelial cell signaling response to lung ischemia. American Journal of Physiology. Lung Cellular and Molecular Physiology, 281, L993–1000. [DOI] [PubMed] [Google Scholar]
- 113.Tozawa K, al-Mehdi AB, Muzykantov V, & Fisher AB (1999). In situ imaging of intracellular calcium with ischemia in lung subpleural microvascular endothelial cells. Antioxidants & Redox Signaling, 1, 145–154. [DOI] [PubMed] [Google Scholar]
- 114.Song S, Yamamura A, Yamamura H, et al. (2014). Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. American Journal of Physiology. Cell Physiology, 307, C373–C383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu Q, Sham JS, Shimoda LA, & Sylvester JT (2001). Hypoxic constriction of porcine distal pulmonary arteries: Endothelium and endothelin dependence. American Journal of Physiology. Lung Cellular and Molecular Physiology, 280, L856–L865. [DOI] [PubMed] [Google Scholar]
- 116.Sylvester JT, Shimoda LA, Aaronson PI, & Ward JP (2012). Hypoxic pulmonary vasoconstriction. Physiological Reviews, 92, 367–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goldenberg NM, Wang L, Ranke H, Liedtke W, Tabuchi A, & Kuebler WM (2015). TRPV4 is required for hypoxic pulmonary vasoconstriction. Anesthesiology, 122, 1338–1348. [DOI] [PubMed] [Google Scholar]
- 118.Pelaez NJ, Braun TR, Paul RJ, Meiss RA, & Packer CS (2000). H(2)O(2) mediates Ca(2+)- and MLC(20) phosphorylation-independent contraction in intact and permeabilized vascular muscle. American Journal of Physiology. Heart and Circulatory Physiology, 279, H1185–H1193. [DOI] [PubMed] [Google Scholar]
- 119.Pourmahram GE, Snetkov VA, Shaifta Y, et al. (2008). Constriction of pulmonary artery by peroxide: Role of Ca2+ release and PKC. Free Radical Biology & Medicine, 45, 1468–1476. [DOI] [PubMed] [Google Scholar]
- 120.Sheehan DW, Giese EC, Gugino SF, & Russell JA (1993). Characterization and mechanisms of H2O2-induced contractions of pulmonary arteries. The American Journal of Physiology, 264, H1542–H1547. [DOI] [PubMed] [Google Scholar]
- 121.Forstermann U, & Munzel T (2006). Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation, 113, 1708–1714. [DOI] [PubMed] [Google Scholar]
- 122.Kuzkaya N, Weissmann N, Harrison DG, & Dikalov S (2003). Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. The Journal of Biological Chemistry, 278, 22546–22554. [DOI] [PubMed] [Google Scholar]
- 123.Zou MH, Shi C, & Cohen RA (2002). Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. The Journal of Clinical Investigation, 109, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fleming I, & Busse R (1999). Signal transduction of eNOS activation. Cardiovascular Research, 43, 532–541. [DOI] [PubMed] [Google Scholar]
- 125.Busse R, & Fleming I (1995). Regulation and functional consequences of endothelial nitric oxide formation. Annals of Medicine, 27, 331–340. [DOI] [PubMed] [Google Scholar]
- 126.Xu W, Kaneko FT, Zheng S, et al. (2004). Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB Journal, 18, 1746–1748. [DOI] [PubMed] [Google Scholar]
- 127.Ghosh S, Gupta M, Xu W, et al. (2016). Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology, 310, L1199–L1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen F, Kumar S, Yu Y, et al. (2014). PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+ -toxins. PloS One, 9, e99823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tang H, Yamamura A, Yamamura H, et al. (2016). Pathogenic role of calcium-sensing receptors in the development and progression of pulmonary hypertension. American Journal of Physiology. Lung Cellular and Molecular Physiology, 310, L846–L859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wan J, Yamamura A, Zimnicka AM, et al. (2013). Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. American Journal of Physiology. Lung Cellular and Molecular Physiology, 305, L154–L164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Broughton BR, Jernigan NL, Norton CE, Walker BR, & Resta TC (2010). Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. American Journal of Physiology. Lung Cellular and Molecular Physiology, 298, L232–L242. [DOI] [PMC free article] [PubMed] [Google Scholar]