

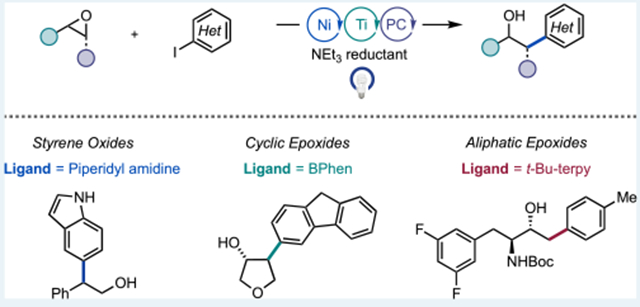
Abstract
A cross-electrophile coupling reaction of epoxides and (hetero)aryl iodides that operates via the merger of three catalytic cycles involving a Ni-, Ti-, and organic photoredox catalyst has been developed. Three distinct classes of epoxides, styrene oxides, cyclic epoxides, and terminal aliphatic epoxides, all undergo coupling in moderate to good yield and high regioselectivity with the use of three different nitrogen-based ligands for Ni under otherwise identical reaction conditions. The mild reaction conditions accommodate a broad scope of abundant and complex coupling partners. Mechanistic studies suggest that when styrene oxides are employed radical intermediates are involved via Ti-radical ring-opening of the epoxide. Conversely, for terminal aliphatic epoxides, involvement of an iodohydrin intermediate enables the formation of the unexpected linear product.
Keywords: epoxides, arylation, cross-electrophile coupling, photoredox catalysis, nickel catalysis, titanium catalysis, radical chemistry, photochemistry
Graphical Abstract
In recent years, Ni-photoredox catalysis has emerged as a powerful approach for forging C(sp2)–C(sp3) bonds that are otherwise challenging to prepare via traditional cross-coupling strategies.1 Recently, researchers have applied Ni-photoredox catalysis to the area of cross-electrophile reactions, enabling the coupling of organohalides in the absence of a stoichiometric metal reductant (Scheme 1A).2,3 Most of the photoredox-assisted reductive cross-coupling (PARC) reactions rely on the coupling of aryl and alkyl halide electrophiles, with the exception of reports from the Amgoune lab using amides as the C(sp2)-component3l and from the Molander lab using Katritzky salts as the C(sp3)-component.3g Notably, the use of distinct and abundant aliphatic electrophile classes could afford new PARC reactions and potentially broaden the scope of cross-electrophile couplings. Epoxides, in particular, are important and versatile building blocks in organic synthesis,4 but have seen limited application as partners in traditional cross-coupling reactions.5 For these reasons, and based on our interest in cross-coupling reactions with 3-membered ring heterocycles,6 we questioned whether a photoredox-assisted coupling approach could be utilized to develop a general, mild, and selective cross-electrophile coupling of epoxides.
Scheme 1.
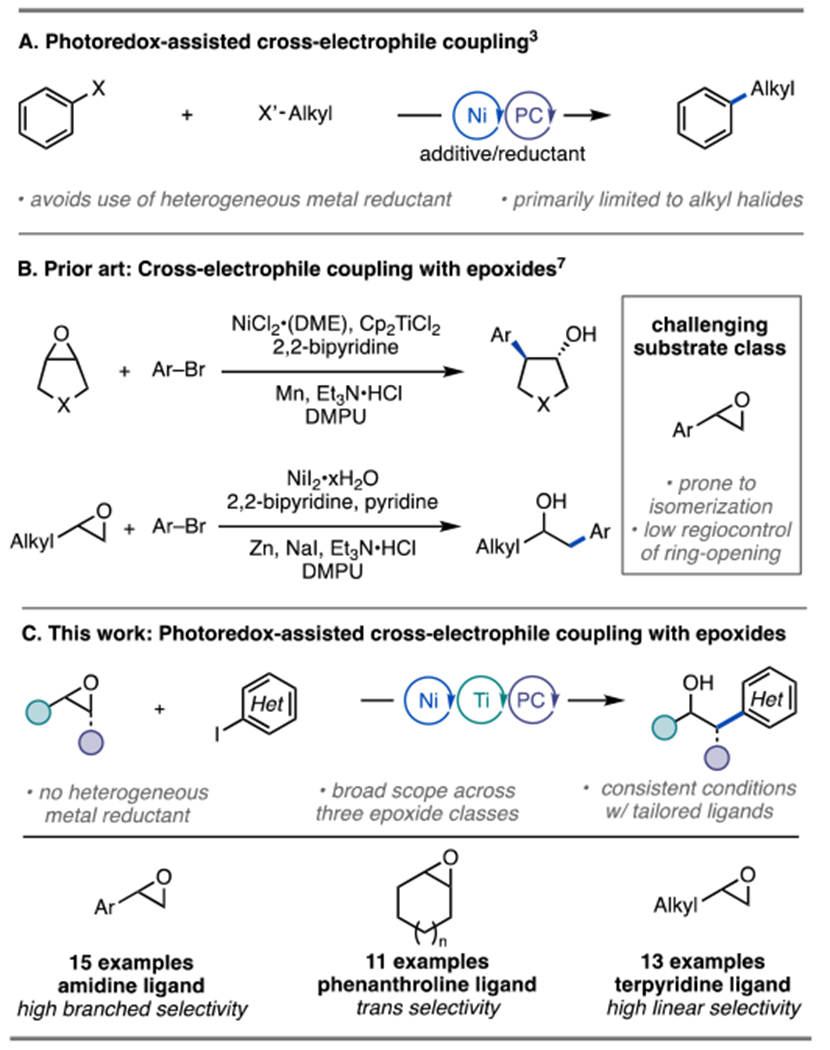
Weix and co-workers have previously described a Ni-catalyzed cross-electrophile coupling reaction of epoxides and aryl halides using Mn as a stoichiometric reductant (Scheme 1B).7 In combination with a Ti co-catalyst to assist with ring-opening, the reaction proceeds efficiently for cyclic epoxides. The use of terminal aliphatic epoxides resulted in mixtures of regioisomers favoring the branched isomer in low to moderate yield. These results prompted their identification of an alternative strategy featuring distinct reaction conditions for the coupling of terminal aliphatic epoxides via the intermediacy of an iodohydrin, which delivers the linear isomer selectively. For both activation strategies, styrene oxides were challenging substrates as they afforded low yield and low regioselectivity. Subsequently, Yamamoto and co-workers developed an asymmetric cross-electrophile coupling of styrene oxide derivatives possessing an ethyl alcohol-directing group; however, reactions of unsubstituted styrene oxides resulted in low yield and low regioselectivity.8 Hence, the development of an approach for the selective cross-coupling of styrene oxides is an unmet need.
Here, we demonstrate that the combination of a Ni-, Ti-, and organic photoredox catalyst promotes highly regioselective and high-yielding cross-coupling of three distinct classes of epoxides: styrenyl-, cyclic-, and terminal aliphatic epoxides (Scheme 1C). Key to this generality is the identification of distinct ligands (holding the reaction conditions otherwise identical) that suppress undesired homocoupling and epoxide rearrangement pathways for each class of epoxides.6b,9 While most Ni-photoredox reactions have been restricted to a single ligand, 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbbpy), thereby limiting opportunities for reaction optimization and translation to enantioselective reactions, this study represents a relatively rare example of a Ni-photoredox reaction that shows broad ligand response.3f,10 Notably, under otherwise similar conditions, opposite regiochemical outcomes were observed for the cases of styrenyl and terminal aliphatic epoxides. Mechanistic studies are presented to explain these observations.
Our investigation began by examining the Ni-photoredox coupling of 4-iodotoluene with three representatives of distinct epoxide classes: styrene oxide 1a, cyclohexene oxide 2a, and 1-dodecene oxide 3a (Table 1). Gansäuer and Shi have recently reported Ti-catalyzed reductive reactions of aliphatic epoxides in the presence of an Ir(dF-CF3-ppy)2(dtbbpy)PF6 and 4CzIPN photocatalyst, respectively.11 Photophysical studies by both groups have shown that the active Ti(III)-complex can be formed via single electron transfer (SET) from a photoredox catalyst, thereby obviating the use of stoichiometric heterogeneous reductants. On the basis of these results and the precedent from Weix, we selected a starting set of reaction conditions: 5 mol % 4CzIPN as a photocatalyst, 10 mol % NiBr2·diglyme as a precatalyst, 20 mol % dtbbpy as a ligand, 5 equiv Et3N as a soluble reductant, and 25 mol % Cp2TiCl212 as a co-catalyst. For styrene oxide 1a, the branched coupling product (4a) was formed as the sole regioisomer in 33% yield (entry 1). Conversely, the linear coupling product (6a) was formed selectively from 3a in 38% yield under the same conditions (entry 3). Use of cyclohexene oxide 2a resulted in the formation of 24% yield of the trans coupling product 5a (entry 2). However, the formation of various side products was observed under these reaction conditions. Biaryl (4,4′-dimethyl-1,1′-biphenyl) was detected in 14%, 24%, and 40% yield for the reaction of epoxides 1a–3a, respectively. The formation of biaryl is a common side reaction in cross-electrophile coupling reactions. For 1a and 3a, the corresponding aldehydes, formed via Ti/Ni-catalyzed isomerization, were also detected in 20% and 12% yield, respectively.
Table 1.
Optimization of the Reaction Parametersa
 |
Reactions were conducted on a 0.05 mmol scale. n/a = not applicable.
Reaction time with 1a = 24 h; with 2a = 36 h; and with 3a = 60 h.
Branched (b):linear (l) as determined by gas chromatography (GC).
Aldehyde was formed via isomerization of the epoxide.
4,4′-Dimethyl-1,1′-biphenyl.
GC yields were calibrated using pentadecane as an internal standard. GC yield of the major isomer is shown.
MeCN was used as solvent.
10 mol % of L2 was used.
We sought to evaluate whether the use of an alternate ligand could be used to suppress these side pathways to improve the efficiency of the epoxide coupling. From our screening studies, three ligands emerged as successful: with phenanthroline ligand BPhen (L1), reactions of 1a and 3a resulted in low reaction yield with significant generation of biaryl and isomerization byproducts (entries 4 and 6). However, reaction of 2a with L1 generated the desired coupling product 5a in 90% yield with minor side product formation (entry 5). With tridentate ligand t-Bu-terpy (L2), 3a afforded the linear coupling product 6a in 85% yield with trace amounts of the reaction byproducts (entry 9). The reactions of 1a and 2a, however, resulted in low yields under the reaction conditions with L2 (entries 7 and 8). Finally, using amidine ligand L3, the branched coupling product 4a was obtained in high yield and selectivity from epoxide 1a (entry 10). Interestingly, 2a and 3a were poor-performing substrates under the reaction conditions with L3 (entries 11 and 12). Control studies for each class of epoxides, with their respective ligand, indicated that all of the reaction parameters were necessary (entries 13–15 and Supporting Information (SI)).
A few general considerations emerge from these optimization studies. First, linear isomeric product 6a was obtained from aliphatic epoxide 3a independent of the ligand identity, whereas the branched isomeric product 4a was formed from styrene oxide 1a, suggestive of a change in mechanism (vide infra). Second, dtbbpy was not optimal for any epoxide class but showed intermediate reactivity in each case, an illustration of how it may have emerged as the most general ligand in Ni-photoredox catalysis (entries 1–3). While our future efforts will be directed at gaining a detailed understanding of the origin of ligand specificity in these reactions, it is clear from the data that use of a tridentate ligand suppresses biaryl formation, consistent with literature reports on Ni cross-electrophile coupling reactions.13 However, t-Bu-terpy is only effective for coupling with epoxide 3a, presumably because the ligand is too hindered to facilitate productive bond formation in the cases of epoxides 1a and 2a, which both afford secondary as compared to primary C(sp3)–C(sp2) bonds. In the case of styrene oxide 1a, amidine ligand L3 is optimal instead. While amidine ligands have been employed for Ni-catalyzed reductive coupling reactions,14 to the best of our knowledge, they have not been used in Ni-photoredox catalysis. We propose that amidine ligand L3 is effective because it affords an electron-rich Ni-complex that suppresses Ni-mediated isomerization of 1a. Among the three epoxide classes, cyclohexene oxide 2a represents the most challenging from the perspective of C-C reductive elimination. As such, we expect that the enhanced π-acidity of BPhen could accelerate reduction steps at Ni such as reductive elimination, which may be responsible for its role in this coupling reaction.15
Next, the scope of the transformation was investigated (Table 2), beginning with an examination of different styrene oxides (1). Under the standard reaction conditions using L3, both electron-rich and electron-deficient aryl iodides were well tolerated, affording branched products 4a–e in good yields. Employment of an aryl iodide possessing a BPin substituent that can be further functionalized by traditional cross-coupling methods produced 4f in 65% yield. Heteroaryl iodides, such as 2-fluoro-5-iodopyridine and 5-iodoindole, afforded the corresponding alcohols 4g and 4h in moderate yield. In varying the styrene oxide framework, we found that sterically hindered 2-(o-tolyl)oxirane 1k underwent coupling regioselectively, albeit in 23% yield (4k). Coupling of electronically diverse styrene oxides (1l–1n) with a range of aryl iodides generated the corresponding branched products in moderate to good yields (4l–n). Finally, heterocyclic styrene oxide 1o produced 4o in 43% yield under the reaction conditions.
Table 2.
Scope of the Cross-Electrophile Coupling of Epoxidesa
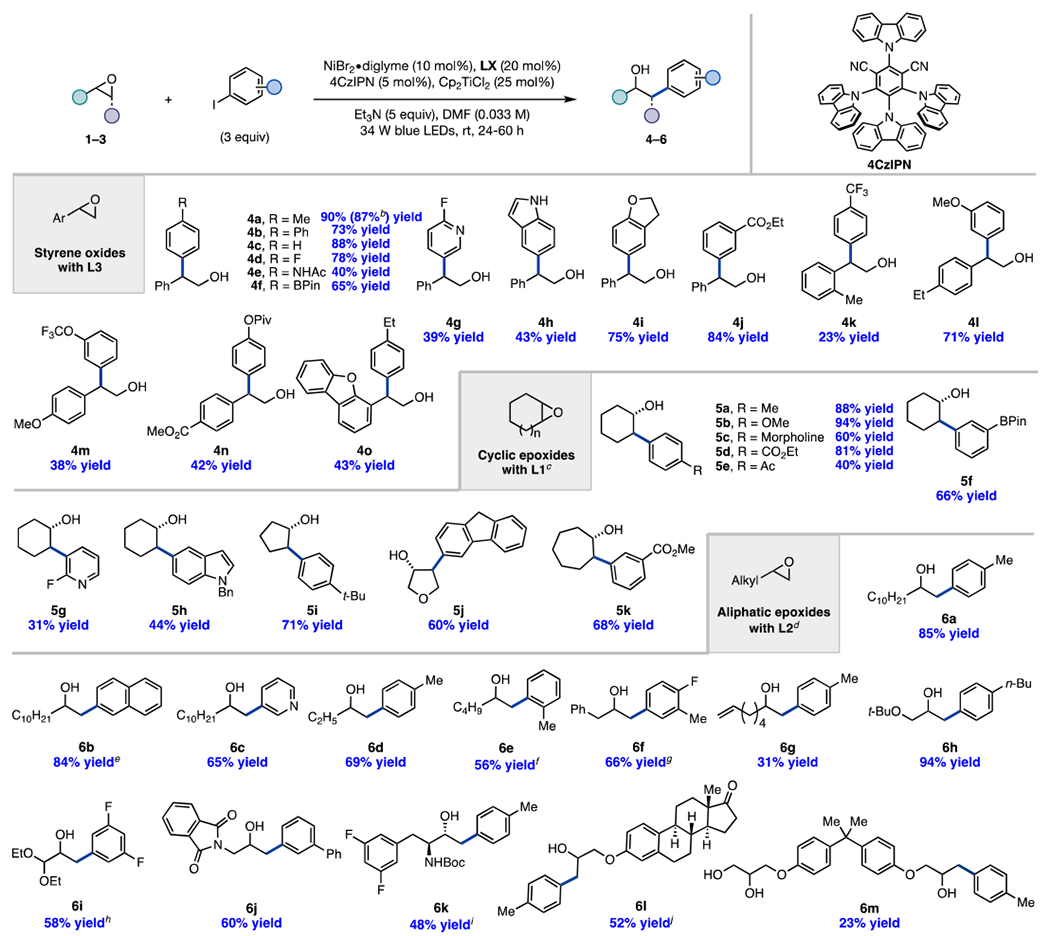 |
Isolated yields are reported based on an average of two runs (0.2 mmol scale). b:l = branched:linear ratio. Unless otherwise noted, the regioselectivity is ≥20:1. Reaction time with 1 = 24 h; with 2 = 36 h; and with 3 = 60 h.
1 mmol scale.
MeCN was used instead of DMF.
10 mol % of L3 was used.
b:l = 1:17.
dtbbpy was used instead of L2.
b:l = 1:11.4.
b:l = 1:15.
Single diastereomer.
b:l = 1:7.3; regioisomers are separable.
In exploring the scope of cyclic epoxides using ligand L1, we found that cyclohexene oxide reacted with electron-rich and electron-deficient aryl iodides, resulting in trans products 5a–e in 40–94% yield. Of note are products 5d and 5e, since their synthesis via traditional ring-opening of cyclohexene oxide with Grignard reagents would lead to low chemoselectivity due to the pendant ester and ketone functionalities. In addition, 3-BPin-aryl iodide and heteroaryl iodides coupled well with cyclohexene oxide (5f–h). Notably, the 5-membered cyclic epoxides, such as cyclopentene oxide and epoxy tetrahydrofuran, were high-performing substrates (5i–j). A 7-membered cyclic epoxide 2k was also amenable to this cross-coupling reaction (5k).
Next, the scope of terminal aliphatic epoxides was examined with L2. The reaction of electronic rich, neutral, and heteroaryl iodides with 1-dodecene oxide provided linear products (6a–c) in high yield and regioselectivity. Coupling of sterically hindered 2-iodotoluene with 1-hexene oxide resulted in no reaction; however, using less sterically hindered dtbbpy in place of L2 afforded product 6e in 56% yield. Benzyl epoxide reacted well under the reaction conditions, producing 6f in 66% yield, with slightly diminished regioselectivity (b:l = 1:11.4). Employment of aliphatic epoxide 3g, possessing sensitive olefin functionality, generated coupling product 6g in 31% yield. Epoxides possessing glycidyl ether, acetal, and phthalimide functional groups were well tolerated (6h–j), highlighting the broad functional group tolerance of the method.
Notably, this protocol was amenable to late-stage coupling as showcased by the following reactions. Protease inhibitor precursor16 3k produced amino alcohol 6k in 48% yield as a single diastereomer under the reaction conditions. Glycidyl estrone ether17 3l reacted smoothly, producing 6l in 52% yield and 1:7.3 b:l regioselectivity. Bisphenol A glycidyl ether, an industrial resin,18 was subjected to the reaction conditions, affording 6m in 23% yield.
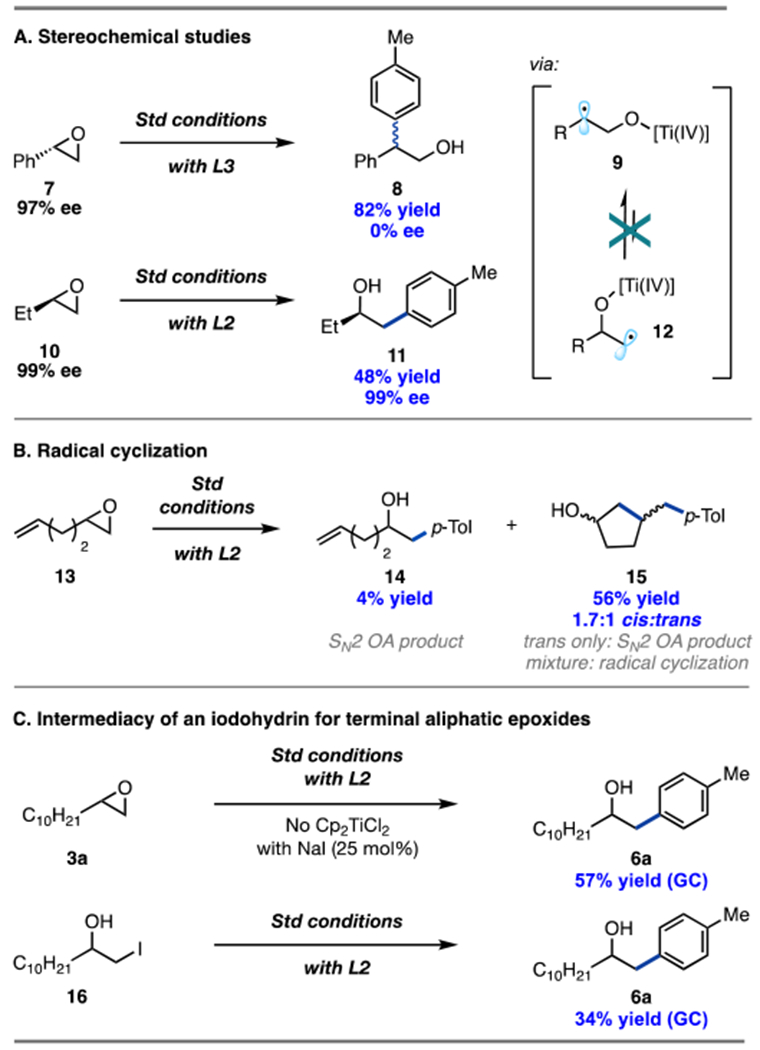
The mechanism of the reaction was interrogated next. Given the difference in regioselectivity for styrenyl and aliphatic epoxides, tandem mechanistic experiments were performed with these two substrate classes. Control experiments for all classes of epoxides revealed that the Ti-catalyst was an important parameter (Table 1, entries 13–15). Thus, we considered radical ring-opening of the epoxide via SET with the Ti-catalyst as a possible mechanistic pathway.12 To probe this possibility further, stereochemical studies were conducted involving enantioenriched styrene oxide 7 and aliphatic epoxide 10 (Scheme 2A). Subjecting 7 to the reaction conditions resulted in coupling product 8 with erosion of the stereocenter, supporting the formation of radical intermediate 9. Conversely, 10 generated product 11 with conservation of ee under the reaction conditions, suggesting that reversible Ti-mediated ring-opening, leading to the primary alkyl radical 12, is highly unlikely (9 → 12).12
Scheme 2.
Mechanistic Studies
Since arylation occurs at the terminal end of the aliphatic epoxide, we considered that an SN2 oxidative addition (OA) mechanism of Ni(0) with the epoxide could instead be operative.5i To probe if epoxide ring-opening occurs via Ni-activation or a radical pathway, radical clock substrate 13 was tested (Scheme 2B).19 If an SN2-type OA by Ni(0) were occurring, then 14 and/or the trans diastereomer of 15 would be expected as the major product(s).5i However, upon exposure of 13 to the reaction conditions, the cyclization product 15 was formed as a mixture of cis/trans isomers, providing evidence for the formation of a radical species at the terminal position of the epoxide. Next, a control study was conducted by replacing Cp2TiCl2 with a catalytic amount of NaI (Scheme 2C).7a The reaction of 3a under these modified conditions resulted in significant yield of 6a, suggesting the intermediacy of an iodohydrin (16).7 Furthermore, submission of independently synthesized iodohydrin 16 to the standard reaction conditions generated linear product 6a in 34% yield. Hence, for aliphatic epoxides, formation of an iodohydrin intermediate is highly probable.
Based on the above mechanistic studies, two distinct mechanisms are proposed for styrenyl and aliphatic epoxides (Scheme 3). In both cases, irradiation of 4CzIPN (PC) under blue light generates photoexcited 4CzIPN*,11 which undergoes oxidative quenching with Ti(IV) to generate Ti(III) (E1/2 PC•+/PC* = −1.18 V vs SCE,20 E1/2 Ti(IV)/Ti(III) = −0.57 V vs SCE11). This quenching pathway was supported by Stern–Volmer quenching studies (see SI) and recent literature reports. For styrene oxides, the formed Ti(III) species engages in a radical ring-opening event with 1 to form intermediate 9. This intermediate reacts with the Ni(II) complex 1821 to form branched Ni(III) intermediate 19. Facile reductive elimination of 19 results in the branched coupling product 4 and Ni(I) intermediate 20. Complex 20 undergoes reduction with 4CzIPN•− (E1/2 PC/PC•− = −1.24 V vs SCE,20 E1/2 Ni(I)/Ni(0) = −1.17 V vs SCE22), which was generated via reductive quenching with triethylamine (Et3N) (E1/2 PC*/PC•− = +1.43 V vs SCE,20 E1/2 Et3N•+/Et3N = +0.93 V vs SCE23) to turn over the Ni-cycle and the photocatalytic cycle.23 The mechanism for cyclic epoxides is speculated to be analogous to that of styrene oxides based on current (see SI) and previous mechanistic studies.7b
Scheme 3.
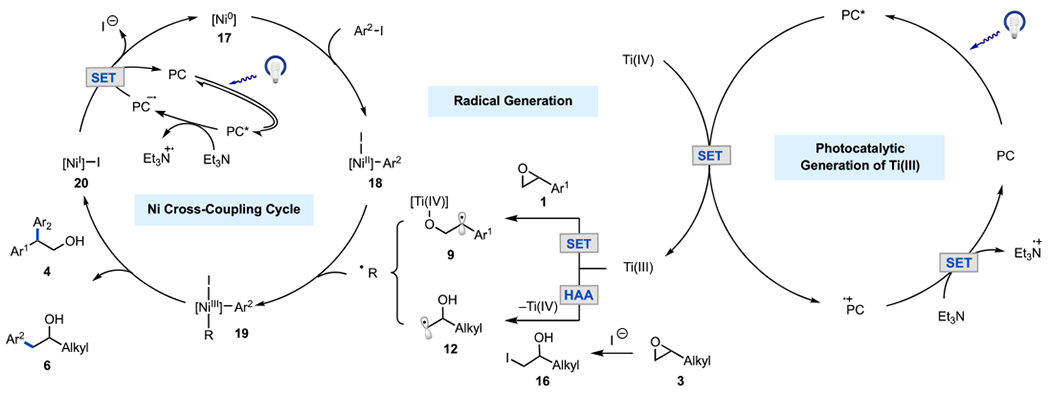
Proposed Mechanisms
For terminal aliphatic epoxides, formation of the iodohydrin intermediate 16 is proposed to occur via the nucleophilic addition of in situ generated iodide with the aliphatic epoxide (3). Catalytic amounts of iodide are likely formed via reduction of Ni(I)–I intermediate 20 with 4CzIPN•−. Next, the iodohydrin intermediate 16 participates in a halogen atom abstraction24 (HAA) event with Ti(III) to generate primary alkyl radical 12,25 which undergoes radical addition to Ni(II) complex 18 to form linear Ni(III) species 19. Subsequent reductive elimination of the latter generates the linear coupling product 6. An alternative pathway, where the iodohydrin intermediate is formed via Lewis acid-mediated iodide ring-opening of the epoxide with Ti, cannot be ruled out (see SI).
In conclusion, we have developed a photocatalytic cross-electrophile coupling of three different classes of epoxides with (hetero)aryl iodides. The transformation exhibits interesting ligand effects, where a different amine-based ligand was required for the efficient coupling of a particular class of epoxide. The scope of the transformation was found to be general, as a range of electronically and sterically diverse epoxides and aryl iodides coupled efficiently. Notably, reaction of styrene epoxides led to the branched coupling products selectively, whereas terminal aliphatic epoxides generated the unexpected linear isomer under otherwise similar reaction conditions. Mechanistic studies revealed that linear selectivity from aliphatic epoxides arises due to the involvement of an iodohydrin intermediate. In contrast, for styrene epoxides, branched products are formed via Ti-mediated radical epoxide ring-opening. Based on the broad ligand response of this transformation, future efforts will be focused on understanding ligand effects for related Ni-photoredox reactions.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from Pfizer, NIGMS (5R35GM126986-02), and NIH F32 Ruth L. Kirschstein NRSA Fellowship (1F32GM129910-01A1) (M.P.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c01199.
Full optimization of the reaction parameters; experimental details; general procedures; isolated yields and characterization data; mechanistic studies; and NMR spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.0c01199
The authors declare no competing financial interest.
Contributor Information
Marvin Parasram, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States.
Benjamin J. Shields, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States
Omar Ahmad, Blueprint Medicines, Cambridge, Massachusetts 02139, United States.
Thomas Knauber, Worldwide Research and Development, Pfizer, Inc., Groton, Connecticut 06340, United States.
Abigail G. Doyle, Department of Chemistry, Princeton University, Princeton, New Jersey 08544, United States.
REFERENCES
- (1).For reviews on TM/Photoredox catalysis, see:; (a) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lang X; Zhao J; Chen X Cooperative Photoredox Catalysis. Chem. Soc. Rev 2016, 45, 3026–3038. [DOI] [PubMed] [Google Scholar]; (c) Tellis JC; Kelly CB; Primer DN; Jouffroy M; Patel NR; Molander GA Single-Electron Transmetalation via Photoredox/Nickel Dual Catalysis: Unlocking a New Paradigm for sp3–sp2 Cross-Coupling. Acc. Chem. Res 2016, 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1, No. 0052. [Google Scholar]; (e) Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).For reviews and selected papers on Ni-catalyzed cross-electrophile reactions, see:; (a) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shu W; García-Domínguez A; Quirós MT; Mondal R; Cárdenas DJ; Nevado C Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights. J. Am. Chem. Soc 2019, 141, 13812–13821. [DOI] [PubMed] [Google Scholar]
- (3).For recent reports on cross-electrophile reactions under Ni-photoredox catalysis, see:; (a) Duan Z; Li W; Lei A Nickel-Catalyzed Reductive Cross-Coupling of Aryl Bromides with Alkyl Bromides: Et3N as the Terminal Reductant. Org. Lett 2016, 18, 4012–4015. [DOI] [PubMed] [Google Scholar]; (b) Paul A; Smith MD; Vannucci AK Photoredox-Assisted Reductive Cross-Coupling: Mechanistic Insight into Catalytic Aryl–Alkyl Cross-Couplings. J. Org. Chem 2017, 82, 1996–2003. [DOI] [PubMed] [Google Scholar]; (c) Zhang P; Le CC; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith RT; Zhang X; Rincón JA; Agejas J; Mateos C; Barberis M; Garcia-Cerrada S; de Frutos O; MacMillan DWC Metallaphotoredox-Catalyzed Cross-Electrophile Csp3–Csp3 Coupling of Aliphatic Bromides. J. Am. Chem. Soc 2018, 140, 17433–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang R; Li G; Wismer M; Vachal P; Colletti SL; Shi Z-C Profiling and Application of Photoredox C(sp3)–C(sp2) Cross-Coupling in Medicinal Chemistry. ACS Med. Chem. Lett 2018, 9, 773–777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yu W; Chen L; Tao J; Wang T; Fu J Dual Nickel- and Photoredox-Catalyzed Reductive Cross-Coupling of Aryl Vinyl Halides and Unactivated Tertiary Alkyl Bromides. Chem. Commun 2019, 55, 5918–5921. [DOI] [PubMed] [Google Scholar]; (g) Yi J; Badir SO; Kammer LM; Ribagorda M; Molander GA Deaminative Reductive Arylation Enabled by Nickel/Photoredox Dual Catalysis. Org. Lett 2019, 21, 3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Pomberger A; Mo Y; Nandiwale KY; Schultz VL; Duvadie R; Robinson RI; Altinoglu EI; Jensen KF A Continuous Stirred-Tank Reactor (CSTR) Cascade for Handling Solid-Containing Photochemical Reactions. Org. Process Res. Dev 2019, 23, 2699–2706. [Google Scholar]; (i) Brill ZG; Ritts CB; Mansoor UF; Sciammetta N Continuous Flow Enables Metallaphotoredox Catalysis in a Medicinal Chemistry Setting: Accelerated Optimization and Library Execution of a Reductive Coupling between Benzylic Chlorides and Aryl Bromides. Org. Lett 2020, 22, 410–416. [DOI] [PubMed] [Google Scholar]; (j) Dewanji A; Bulow RF; Rueping M Photoredox/Nickel Dual-Catalyzed Reductive Cross Coupling of Aryl Halides Using an Organic Reducing Agent. Org. Lett 2020, 22, 1611–1617. [DOI] [PubMed] [Google Scholar]; (k) Guan H; Zhang Q; Walsh PJ; Mao J Nickel/Photoredox-Catalyzed Asymmetric Reductive Cross-Coupling of Racemic α-Chloro Esters with Aryl Iodides. Angew. Chem., Int. Ed 2020, 59, 5172–5177. [DOI] [PubMed] [Google Scholar]; (l) Kerackian T; Reina A; Bouyssi D; Monteiro N; Amgoune A Silyl Radical Mediated Cross-Electrophile Coupling of N-Acyl-imides with Alkyl Bromides under Photoredox/Nickel Dual Catalysis. Org. Lett 2020, 22, 2240–2245. [DOI] [PubMed] [Google Scholar]
- (4).(a) Hanson RM The Synthetic Methodology of Nonracemic Glycidol and Related 2,3-Epoxy Alcohols. Chem. Rev 1991, 91, 437–475. [Google Scholar]; (b) Herzberger J; Niederer K; Pohlit H; Seiwert J; Worm M; Wurm FR; Frey H Polymerization of Ethylene Oxide, Propylene Oxide, and Other Alkylene Oxides: Synthesis, Novel Polymer Architectures, and Bioconjugation. Chem. Rev 2016, 116, 2170–2243. [DOI] [PubMed] [Google Scholar]
- (5).(a) Lu X-Y; Yan L-Y; Li J-S; Li J-M; Zhou H.-p.; Jiang R-C; Liu C-C; Lu R; Hu R Base-free Ni-catalyzed Suzuki-type cross-coupling reactions of epoxides with boronic acids. Chem. Commun 2020, 56, 109–112. [DOI] [PubMed] [Google Scholar]; (b) Ye K-Y; McCallum T; Lin S Bimetallic Radical Redox-Relay Catalysis for the Isomerization of Epoxides to Allylic Alcohols. J. Am. Chem. Soc 2019, 141, 9548–9554. [DOI] [PubMed] [Google Scholar]; (c) Ahmad A; Burtoloso ACB Total Synthesis of (±)-Brussonol and (±)-Komaroviquinone via a Regioselective Cross-Electrophile Coupling of Aryl Bromides and Epoxides. Org. Lett 2019, 21, 6079–6083. [DOI] [PubMed] [Google Scholar]; (d) Lu X-Y; Li J-S; Wang S-Q; Zhu Y-J; Li Y-M; Yan L-Y; Li J-M; Wang J-Y; Zhou H-P; Ge X-T Pd-Catalyzed Decarboxylative Cross-Coupling Reactions of Epoxides with α,β-Unsaturated Carboxylic Acids. Chem. Commun 2019, 55, 11123–11126. [DOI] [PubMed] [Google Scholar]; (e) Min L; Wu G; Liu M; Gao W; Ding J; Chen J; Huang X; Wu H Copper-Catalyzed Oxirane-Opening Reaction with Aryl Iodides and Se Powder. J. Org. Chem 2016, 81, 7584–7590. [DOI] [PubMed] [Google Scholar]; (f) Ebrahim-Alkhalil A; Zhang Z-Q; Gong T-J; Su W; Lu X-Y; Xiao B; Fu Y Copper-Catalyzed Cross-Coupling Reactions of Epoxides with Gem-diborylmethane: Access to γ-Hydroxyl Boronic Esters. Chem. Commun 2016, 52, 4891–4893. [DOI] [PubMed] [Google Scholar]; (g) Lu X-Y; Yang CT; Liu J-H; Zhang Z-Q; Lu X; Lou X; Xiao B; Fu Y Cu-Catalyzed Cross-Coupling Reactions of Epoxides with Organoboron Compounds. Chem. Commun 2015, 51, 2388–2391. [DOI] [PubMed] [Google Scholar]; (h) Beaver MG; Jamison TF Ni(II) Salts and 2-Propanol Effect Catalytic Reductive Coupling of Epoxides and Alkynes. Org. Lett 2011, 13, 4140–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Molinaro C; Jamison TF Nickel-Catalyzed Reductive Coupling of Alkynes and Epoxides. J. Am. Chem. Soc 2003, 125, 8076–8077. [DOI] [PubMed] [Google Scholar]
- (6).(a) Huang C-Y; Doyle AG The Chemistry of Transition Metals with Three-Membered Ring Heterocycles. Chem. Rev 2014, 114, 8153–8198. [DOI] [PubMed] [Google Scholar]; (b) Nielsen DK; Doyle AG Nickel-Catalyzed Cross-Coupling of Styrenyl Epoxides with Boronic Acids. Angew. Chem., Int. Ed 2011, 50, 6056–6059. [DOI] [PubMed] [Google Scholar]; (c) Woods BP; Orlandi M; Huang C-Y; Sigman MS; Doyle AG Nickel-Catalyzed Enantioselective Reductive Cross-Coupling of Styrenyl Aziridines. J. Am. Chem. Soc 2017, 139, 5688–5691. [DOI] [PubMed] [Google Scholar]; (d) Huang C-Y; Doyle AG Electron-Deficient Olefin Ligands Enable Generation of Quaternary Carbons by Ni-Catalyzed Cross-Coupling. J. Am. Chem. Soc 2015, 137, 5638–5641. [DOI] [PubMed] [Google Scholar]; (e) Nielsen DK; Huang C-Y; Doyle AG Directed Nickel-Catalyzed Negishi Cross Coupling of Alkyl Aziridines. J. Am. Chem. Soc 2013, 135, 13605–13609. [DOI] [PubMed] [Google Scholar]; (f) Huang C-Y; Doyle AG Nickel-Catalyzed Negishi Alkylations of Styrenyl Aziridines. J. Am. Chem. Soc 2012, 134, 9541–9544. [DOI] [PubMed] [Google Scholar]
- (7).(a) Zhao Y; Weix DJ Nickel-Catalyzed Regiodivergent Opening of Epoxides with Aryl Halides: Co-Catalysis Controls Regioselectivity. J. Am. Chem. Soc 2014, 136, 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao Y; Weix DJ Enantioselective Cross-Coupling of meso-Epoxides with Aryl Halides. J. Am. Chem. Soc 2015, 137, 3237–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Banerjee A; Yamamoto H Nickel Catalyzed Regio-, Diastereo-, and Enantioselective Cross-Coupling of 3,4-Epoxyalcohol with Aryl Iodides. Org. Lett 2017, 19, 4363–4366. [DOI] [PubMed] [Google Scholar]
- (9).(a) Ogoshi S; Tonomori K-I; Oka M.-a.; Kurosawa H Reversible Carbon–Carbon Bond Formation between 1,3-Dienes and Aldehyde or Ketone on Nickel(0). J. Am. Chem. Soc 2006, 128, 7077–7086. [DOI] [PubMed] [Google Scholar]; (b) Desnoyer AN; Geng J; Drover MW; Patrick BO; Love JA Catalytic Functionalization of Styrenyl Epoxides via 2-Nickela(II)oxetanes. Chem. - Eur. J 2017, 23, 11509–11512. [DOI] [PubMed] [Google Scholar]
- (10).Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Zhang Z; Richrath RB; Gansäuer A Merging Catalysis in Single Electron Steps with Photoredox Catalysis—Efficient and Sustainable Radical Chemistry. ACS Catal 2019, 9, 3208–3212. [Google Scholar]; (b) Lin S; Chen Y; Li F; Shi C; Shi L Visible-light-driven Spirocyclization of Epoxides via Dual Titanocene and Photoredox Catalysis. Chem. Sci 2020, 11, 839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) RajanBabu TV; Nugent WA Selective Generation of Free Radicals from Epoxides Using a Transition-Metal Radical. A Powerful New Tool for Organic Synthesis. J. Am. Chem. Soc 1994, 116, 986–997. [Google Scholar]; (b) Gansäuer A; Narayan S Regiodivergent Epoxide Opening: A Concept in Stereoselective Catalysis beyond Classical Kinetic Resolutions and Desymmetrizations. Adv. Synth. Catal 2002, 344, 465–475. [DOI] [PubMed] [Google Scholar]; (c) Gansäuer A; Fan C-A; Keller F; Karbaum P Regiodivergent Epoxide Opening: A Concept in Stereoselective Catalysis beyond Classical Kinetic Resolutions and Desymmetrizations. Chem. - Eur. J 2007, 13, 8084–8090. [DOI] [PubMed] [Google Scholar]; (d) Gansäuer A; Shi L; Otte M Catalytic Enantioselective Radical Cyclization via Regiodivergent Epoxide Opening. J. Am. Chem. Soc 2010, 132, 11858–11859. [DOI] [PubMed] [Google Scholar]; (e) González-Delgado JA; Arteaga JF Control of Homocoupling Versus Reduction in Titanium(III)-Mediated Radical Opening of Styrene Oxides. Eur. J. Org. Chem 2019, 7864–7869. [Google Scholar]; (f) Gansäuer A; Hildebrandt S; Vogelsang E; Flowers RA II. Tuning the Redox Properties of the Titanocene(III)/(IV)-Couple for Atom-Economical Catalysis in Single Electron Steps. Dalton Trans 2016, 45, 448–452. [DOI] [PubMed] [Google Scholar]; (g) Gansäuer A; von Laufenberg D; Kube C; Dahmen T; Michelmann A; Behlendorf M; Sure R; Seddiqzai M; Grimme S; Sadasivam DV; Fianu GD; Flowers RA II. Mechanistic Study of the Titanocene(III)-Catalyzed Radical Arylation of Epoxides. Chem. - Eur. J 2015, 21, 280–289. [DOI] [PubMed] [Google Scholar]
- (13).Budnikova YH; Vicic DA; Klein A Exploring Mechanisms in Ni Terpyridine Catalyzed C–C Cross-Coupling Reactions—A Review. Inorganics 2018, 6, 18. [Google Scholar]
- (14).For thermal reactions involving amidine ligands, see:; (a) Hansen EC; Pedro DJ; Wotal AC; Gower NJ; Nelson JD; Caron S; Weix DJ New Ligands for Nickel Catalysis from Diverse Pharmaceutical Heterocycle Libraries. Nat. Chem 2016, 8, 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hansen EC; Li C; Yang S; Pedro D; Weix DJ Coupling of Challenging Heteroaryl Halides with Alkyl Halides via Nickel-Catalyzed Cross-Electrophile Coupling. J. Org. Chem 2017, 82, 7085–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereo-convergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ghosh AK; Cárdenas EL; Brindisi M Highly Stereoselective Asymmetric Aldol Routes to Tert-butyl-2-(3,5-difluorophenyl)-1-oxiran-2-yl)ethyl)carbamates: Building Blocks for Novel Protease Inhibitors. Tetrahedron Lett 2017, 58, 4062–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Morzycki JW; Maj J; Nikitiuk A; Kwolek G; Malinowska B Three New Derivatives of 3-amino-1,2-propanediol; Their Spectral Properties and Biological Evaluation. Acta Pol. Pharm 2001, 58, 249–256. [PubMed] [Google Scholar]
- (18).Pham H-Q; Marks MJ Epoxy Resins In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2005; pp 171–175. [Google Scholar]
- (19).Newcomb M; Toy PH Hypersensitive Radical Probes and the Mechanisms of Cytochrome P450-Catalyzed Hydroxylation Reactions. Acc. Chem. Res 2000, 33, 449–455. [DOI] [PubMed] [Google Scholar]
- (20).Speckmeier E; Fischer TG; Zeitler K A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- (21).Formation of complex 18 via oxidative addition of Ni(0) (17) with aryl halides was supported by additional mechanistic studies, involving stoichiometric and crossover studies, as well as Hammett studies. Please see the Supporting Information for details.
- (22).Shields BJ; Doyle AG Direct C(sp3)–H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shang T-Y; Lu L-H; Cao Z; Liu Y; He W-M; Yu B Recent Advances of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) in Photocatalytic Transformations. Chem. Commun 2019, 55, 5408–5419. [DOI] [PubMed] [Google Scholar]
- (24).Wu X; Hao W; Ye K-Y; Jiang B; Pombar G; Song Z; Lin S Ti-Catalyzed Radical Alkylation of Secondary and Tertiary Alkyl Chlorides Using Michael Acceptors. J. Am. Chem. Soc 2018, 140, 14836–14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Alternatively, formation of primary radical intermediate 12 could occur via a SET or HAA event with Ni(I) complex 20 and iodohydrin intermediate 16 as part of a radical chain process (see ref 6 for further details).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.