

Abstract
Reactive nitrogen emissions into the atmosphere are increasing due to human activities, affecting nitrogen deposition to the surface and impacting the productivity of terrestrial and marine ecosystems. An atmospheric chemistry-transport model (TM4-ECPL) is here used to calculate the global distribution of total nitrogen deposition, accounting for the first time for both its inorganic and organic fractions in gaseous and particulate phases, and past and projected changes due to anthropogenic activities. The anthropogenic and biomass burning ACCMIP historical and RCP6.0 and RCP8.5 emissions scenarios are used. Accounting for organic nitrogen (ON) primary emissions, the present-day global nitrogen atmospheric source is about 60% anthropogenic, while total N deposition increases by about 20% relative to simulations without ON primary emissions. About 20–25% of total deposited N is ON. About 10% of the emitted nitrogen oxides are deposited as ON instead of inorganic nitrogen (IN) as is considered in most global models. Almost a 3-fold increase over land (2-fold over the ocean) has been calculated for soluble N deposition due to human activities from 1850 to present. The investigated projections indicate significant changes in the regional distribution of N deposition and chemical composition, with reduced compounds gaining importance relative to oxidized ones, but very small changes in the global total flux. Sensitivity simulations quantify uncertainties due to the investigated model parameterizations of IN partitioning onto aerosols and of N chemically fixed on organics to be within 10% for the total soluble N deposition and between 25–35% for the dissolved ON deposition. Larger uncertainties are associated with N emissions.
1. Introduction
Nitrogen (N) is an important constituent of any form of life, including plant tissues and proteins. Reactive N availability in the environment together with other nutrients like iron, phosphorus and silica, control ecosystem productivity (Mills et al. 2004). Nitrogen availability is thus linked to the carbon cycle and in particular to carbon dioxide (CO2) removal from the atmosphere by the terrestrial and marine ecosystems and to food production [Duce et al. (2008); Galloway et al. (2008)]. Atmospheric deposition of reactive nitrogen (N) compounds fertilizes ecosystems or has negative impacts due to acidification and accumulation of excess nutrients (Driscoll et al. 2003), while reactive nitrogen chemically trapped on pollen particles (Franze et al. 2005) or forming secondary gaseous and particulate pollutants in the atmosphere (Lelieveld et al. 2015) can be harmful for human health. Reactive nitrogen compounds are emitted to the atmosphere both in oxidized or reduced inorganic (IN) or organic (ON) forms by natural (e.g. soils, lightning, plants, bacteria, viruses) and anthropogenic sources (e.g. industries, transportation, domestic wood burning) [Neffe et al. (2002); Dentener et al. (2006); Galloway et al. (2008) and references therein]. Recent observations and modeling studies have shown that ON is a significant fraction of total nitrogen (TN) deposition, however the chemical characterization of this fraction remains a challenge [Cornell (2010); Cape et al. (2011); Altieri et al. (2012); Kanakidou et al. (2012) and references therein].
Since the beginning of industrialization, human activities have dramatically increased the amounts of N emitted to the atmosphere and deposited to the surface, modifying the biogeochemical cycle of N (Galloway et al. 2008). Duce et al. (2008) estimated that the human-induced increase in atmospheric N deposition to the oceans may account globally for up to ~3% of the annual new oceanic primary productivity. Higher contributions have been estimated for semi-enclosed marine ecosystems such as the Mediterranean Sea, for which atmospheric deposition of N may account for up to 35–60% of new production (Christodoulaki et al. 2013). Reactive nitrogen is also an important driver of atmospheric chemistry, since ozone production is controlled by nitrogen oxides availability. Nitric acid is a major contributor to the atmospheric acidity, ranking second after sulfuric acid, and NH3 is the main neutralizing gas for these acidic compounds (Seinfeld and Pandis, 2006). Thus, removal of atmospheric N species by deposition also impacts atmospheric chemistry. Consequently, evaluation of the impact of N emissions on the environment and climate requires the estimation of past and future changes in N atmospheric deposition. Projections for N emissions in the atmosphere are based on different scenarios that assume control of nitrogen oxide emissions, but not those of ammonia [Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Lamarque et al. (2013a,b)], pointing to expected changes in the chemical composition and thus possibly in the bioavailability of deposited N.
The present study uses the global atmospheric chemistry/transport model TM4-ECPL [Myriokefalitakis et al. (2015) and references therein] to evaluate past and future changes in atmospheric deposition of N that are driven by human activities, accounting for ON in the aerosol phase that has been neglected in most of the earlier studies [e.g. Lamarque et al. (2013)]. Thus, the present study accounts for ON primary emissions as well as chemical formation of ON in secondary organic aerosol (SOA). Unlike recent ON global modeling studies [Kanakidou et al. (2012); Ito et al. (2014, 2015)] the N-containing SOA formation, when it is not originating from amines, is here considered to occur only when sufficient nitrogen oxides (NOx) are available. It is also the first time that expected future changes in ON atmospheric deposition based on human-driven emission scenarios are evaluated. A global picture of the changes in the atmospheric deposition of nitrogen and its composition, in particular the ON component, due to changes in anthropogenic emissions, and subsequently atmospheric chemistry, is here provided together with an uncertainty estimate.
2. Methodology- Model description
The TM4-ECPL model accounts for gas and multiphase oxidants, volatile organic compounds (VOC) chemistry in the troposphere and all major primary and secondary aerosol components, including SOA formation [Tsigaridis and Kanakidou (2003 and 2007); Myriokefalitakis et al. (2011); Kanakidou et al. (2012)]. The present model configuration explicitly considers the atmospheric iron cycle (Myriokefalitakis et al. 2015). It uses the ISORROPIA II thermodynamic equilibrium module (Fountoukis and Nenes 2007) to calculate the formation of ammonium (NH4+) and nitrate (NO3−), assuming thermodynamically stable conditions (Karydis et al. 2015) and accounting for the impact of mineral dust and sea-salt elements on nitrate and ammonium partitioning to the aerosol phase [see Myriokefalitakis et al. (2015)]. In-cloud pH is controlled by strong acids (sulphuric acid, (H2SO4), methanesulphonic acid (MSA) and nitric acid (HNO3)), bases (ammonia (NH3)) and by the dissociation of hydrated CO2, SO2 and oxalic acid (Myriokefalitakis et al. 2011).
The photochemical degradation of VOC in the atmosphere forms secondary products, carbon monoxide, and, ultimately, CO2. Thus, only part of the emitted organics are ultimately deposited to the surface by dry or wet deposition processes in either the gas or particulate form. The deposition parameterization in TM4-ECPL uses solubility estimates for the individual compounds [Tsigaridis et al. (2006); Myriokefalitakis et al. (2011); Kanakidou et al. (2012)].
The model uses primary emissions of N oxides (NOx), ammonia, marine amines (that are 2 orders of magnitude weaker than ammonia marine emissions [Duce et al. (1983); Duce et al. (1991); Facchini et al. (2008)]) and emissions of particulate ON from various natural and anthropogenic sources [Table S1; see also Kanakidou et al. (2012)]. These particulate ON emissions contain both oxygenated and reduced nitrogen species although no specific distinction is made in the model between the two categories of ON due to insufficient knowledge. Moreover, marine amines (ON) in the gas phase are here considered to form amine salts (ON in the particulate phase) consistent with the ON characterization by Altieri et al. (2012) as predominantly containing reduced nitrogen. Amines of continental origin are not considered here due to the uncertainty and the weakness of their sources both of human and natural origin [Schade and Crutzen (1995); Ge et al. (2011); Karl et al. (2014); Sindermann and Neftel (2015)]. The organic nitrates that are of secondary origin and oxygenated inorganic nitrogen compounds both in the gas and particulate phases are explicitly calculated (Myriokefalitakis et al. 2011). An increasing number of global atmospheric chemistry transport models account for the formation of organic nitrate during VOC oxidation using chemical schemes of various complexity. However, they do not explicitly account for the production of other forms of ON, particularly those associated with the primary and secondary organic aerosol (OA). Thus, these models underestimate the total atmospheric burden of ON and its deposition to the surface. In this study, to account for particulate ON primary emissions (Table S1), particulate ON concentrations are linked to source-specific OA tracers using varying N:C molar ratios at emission time as measured in the organic matter from different source types and detailed in Kanakidou et al. (2012). Two different N:C molar ratios have been used for fossil fuel and biomass burning sources (0.16 and 0.30) within the range of uncertainty (Kanakidou et al. 2012). Furthermore, it is here assumed that SOA formed under high NOx-to-VOC conditions [as these are defined in Tsigaridis et al. (2006)] by VOC other than amines, contains N, and thus during this SOA formation IN is converted into ON. Sensitivity simulations have also been performed neglecting this NOx-to-VOC dependence and the mixing with dust and sea-salt aerosol in ISORROPIA II, for comparison to the earlier study by Kanakidou et al. (2012). The present model set up differs also from that earlier study in the meteorological fields used (ECMWF ERA interim here instead of ECMWF operational data) and the emission datasets as described below. The results of all simulations are provided in Table 1.
Table 1.
Atmospheric total deposition of reactive N calculated by the TM4-ECPL model in Tg-N y−1 calculated based on different anthropogenic emission inventories. In parentheses the respective deposition fluxes over the oceans are provided. The estimate from the ACCMIP multi-model simulations (Lamarque et al. 2013a,b), from the PhotoComp simulations (Dentener et al. 2006) and from the Vet et al. (2014) for the year 2000 are provided for comparison. For the Vet et al. (2014) estimates number in parenthesis provides deposition on coastal and ocean systems. Non-marked (base case) simulations are done with N:C of 0.16 for combustion sources and sea-salt and dust components taken into account in ISORROPIA II; crosses (+) mark simulations as base case but with N:C ratio of 0.3 and stars (*) mark simulations as base case but without NOx-to-VOC dependence of N-SOA formation and neglecting sea-salt and dust aerosol components in the ISORROPIA II thermodynamic equilibrium model. &NOy=sum of NOx,HNO3, NO3p; NHx = sum of NH3,NH4+
| Deposition | ACCMIP | RCP6.0 | RCP6.0 | RCP8.5 | ACCMIP | PhotoComp | Vet et al (2014) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1850 | 1850+ | 1850* | 2005 | 2005+ | 2005* | 2050 | 2050+ | 2050* | 2050 | 2000 | 2000 | 2000 | |
| NHx & | 17 (10) | 17 (10) | 17 (10) | 53 (19) | 53 (19) | 53 (20) | 68 (21) | 68 (21) | 64 (22) | 70 (21) | 49±1 | 63 | |
| NOy & | 12 (6) | 12 (5) | 6 (3) | 46 (22) | 46 (22) | 40 (20) | 37 (18) | 37 (18) | 30 (15) | 39 (20) | 51±4 | 51 | |
| DON | 14 (9) | 13 (8) | 21 (10) | 22 (12) | 28 (14) | 29 (13) | 22 (12) | 28 (14) | 29 (13) | 21 (12) | - | - | |
| ON insoluble | 5 (2) | 7 (2) | 7 (1) | 5 (2) | 5 (2) | 7 (1) | 5 (2) | 5 (2) | 7 (1) | 5 (2) | |||
| Total N | 48 (27) | 49 (23) | 51 (24) | 126 (55) | 132 (58) | 129 (54) | 132 (53) | 138 (55) | 129 (50) | 135 (55) | 101±2 | 114 | 122.2 (39) |
Natural emissions of reactive gases and aerosols from both terrestrial and oceanic sources as well as lightning used in the model are as reported in Myriokefalitakis et al. (2015) but for the year 2005. Primary biogenic particle emissions are distributed according to the Leaf Area Index spatial and temporal distribution with 25% in the fine and 75% in the coarse mode [as in Kanakidou et al. (2012)]. Anthropogenic and biomass burning emissions of NOx, carbonaceous aerosols, SO2 and organics come from the ACCMIP database: for 1850 (past) the historical ACCMIP inventories; for 2005 (present day) first scenario year of the Representative Concentration Pathway 6.0 (RCP6.0) inventory; for 2050 (future) both RCP6.0 and RCP8.5 inventories have been used (Lamarque et al. 2013a) (Fig. 1, columns marked with ‘xx_emi’).
FIG. 1.
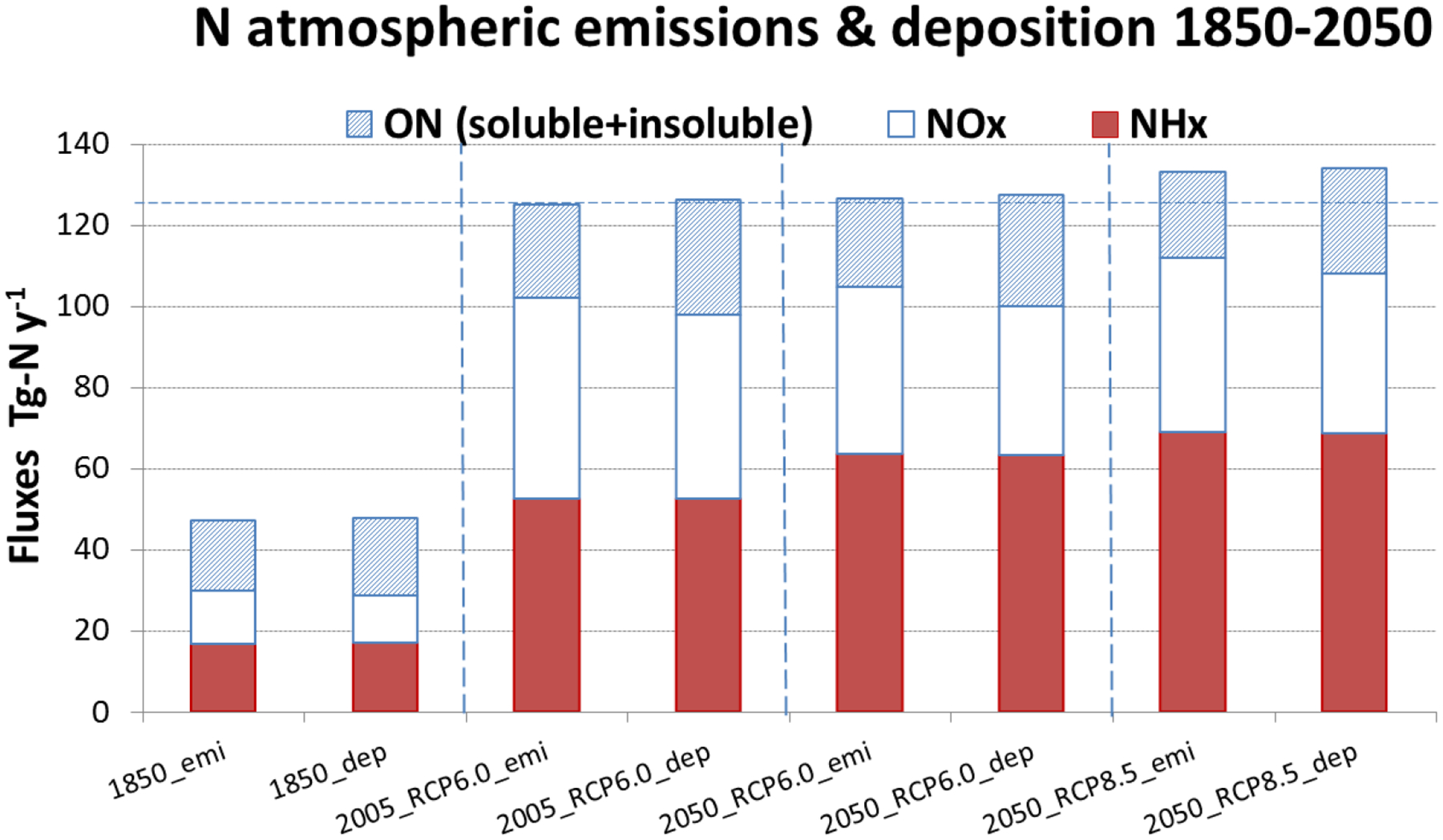
Anthropogenic emissions of NH3 and NOx and those of particulate ON used for this study for 1850, 2005 and 2050 from historical ACCMIP database and RCP6.0, RCP8.5 emissions (marked by ‘year’_emi). Particulate ON emissions are derived in the present study from the particulate OC corresponding emissions based on the methodology developed by Kanakidou et al. (2012). Atmospheric deposition of reactive nitrogen as computed by TM4-ECPL based on these emissions (marked by ‘year’_dep). N flux as inorganic reduced N (NHx) (solid), as inorganic oxygenated N (NOy) (open) and as ON (hashed). Noticeable are differences in the projections between RCPs. NOy is calculated as the sum of NOx, HNO3, NO3−, HONO, HNO4 and N2O5.
While past ON emissions are estimated to be of the same magnitude as alkaline (NHx) and acidic (NOx) compounds emissions (Fig. 1), projections (van Vuuren et al. 2011) show that NHx emissions will gain importance compared to NOx emissions, overall reducing atmospheric acidity. There is no clear trend in future estimates of ON emissions. All simulations presented here have been performed using present-day ECMWF ERA-interim meteorology (Dee et al. 2011) and natural emissions of the year 2005. Thus, the calculated changes are only due to anthropogenic emission changes. Henry Law coefficients for organic nitrates are taken from Sander (1999) and the soluble fractions of primary particulate ON by Kanakidou et al. (2012) are used. Simulations are performed with the model horizontal resolution of 3° longitude × 2° latitude and 34 vertical layers. Atmospheric deposition fluxes are computed in this horizontal resolution and have been further interpolated to 1° x1° grid for further analysis.
3. Results
The global distributions of organic and inorganic (IN) fractions of nitrogen atmospheric deposition are computed as the sum of the corresponding terms of the individual ON and IN model tracers calculated by TM4-ECPL. The results are summarized in Table 1 and Fig. 1 (columns marked with ‘XX_dep’) that show the global changes in N atmospheric deposition due to human activities computed using different parameterizations and emission inventories (Fig. 1). Figure 2 compares the computed TN and dissolved ON (DON) deposition fluxes for the base simulation [see Table 1] with observationally-derived fluxes [compilation in Kanakidou et al. (2012)]. Modeled TN deposition fluxes are in good agreement with observations [see also supplementary figures S2 and S3 using observations from Vet et al. (2014)]. The highest observed DON fluxes seem to be underestimated by the model by less than an order of magnitude, indicating a potential underestimate of the contribution of DON, relative to IN, in TN deposition fluxes. According to our model estimates, primary sources dominate DON emissions, increasing the total present-day N sources commonly taken into account in global models by about 20%. Thus no direct link is expected between ON and NOx deposition. Secondary sources of ON (both gases and aerosols) are estimated to be low, ranging from 4 to 13 Tg-N y−1, i.e. about 15% to 35% of the global present-day sources of ON and converting about 10% of primary NOx emissions to ON. From this secondary source of ON, most global models account only for the gaseous organic nitrate and PAN formation (Lamarque et al. 2013) that in our model amounts to between 0.9 and 2.9 Tg-N y−1, i.e. 3–10% of the total ON source.
FIG. 2.
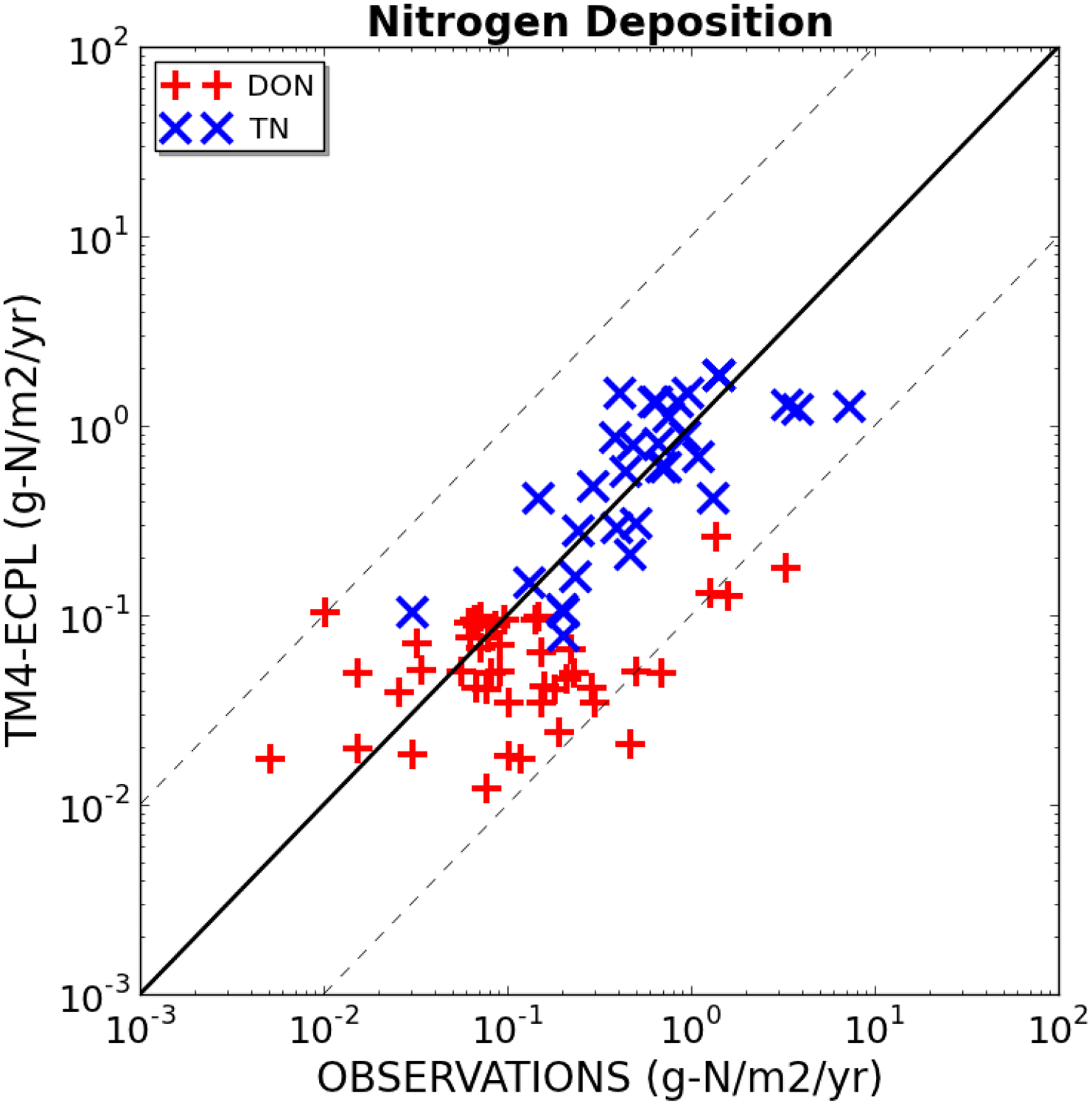
Comparison between modelled (TM4-ECPL) and observationally derived (OBSERVATIONS) atmospheric deposition annual fluxes of total nitrogen (x) and DON (+) in g-N m−2y−1 (compilation of data by Kanakidou et al. 2012 and references therein). Solid line indicates the 1:1 and dashed lines the 10:1 and 1:10 model-to-observation ratios.
Additional comparisons between model results and observations are provided in the supplement (Figures S1–S4). Comparisons of model results with atmospheric concentrations of N-containing species from the EBAS database indicate an overestimate by the base case simulation of NO3− (Normalized Mean Bias, NMB=115%, see supplement) and NH4+ (NMB=54%) concentrations in the PM10, while HNO3 and NO2 concentrations compare well (Figures S4). Simulated atmospheric deposition fluxes are compared with data compiled by Baker et al (2016) from oceanographic cruises (Figure S1; dry deposition fluxes) and by Vet et al (2014) from continental networks (Figure S2; wet deposition fluxes and Figure S3; sum of wet and dry deposition fluxes). Fluxes of NOy are biased high for the base case simulation (NMB=17%) and low for the simulations without NOx-to-VOC dependence (marked with * in Table 1) (NMB=−28%) compared to deposition fluxes derived from observations over land (Figures S2 and S3) and over the ocean (Figure S1). NHx deposition fluxes seem to be overestimated by the model (NMB=17%, Figure S3a) while for NH4+ wet deposition no systematic bias has been found. This difference in the model performance between NHx and NH4+ deposition indicates a potential overestimate in NH3 deposition. (Figures S1 and S3). The here computed deposition fluxes have been also compared with those of the ACCMIP multi model simulations in Figures S5 and S6. These comparisons show that the base case simulation is very close to the 1:1 line with the mean of ACCMIP models, although an underestimate of the low NOy and NHx deposition fluxes can be seen.
As seen in Table 1 and Fig. 1b, NH3 and NOx emissions increased from 1850 to the present leading to similar (2- to 3-fold) increases in their respective N atmospheric deposition. In the future (2050) however, NOx emissions are projected to decline while NH3 emissions will continue to grow. These changes are reflected in the simulated deposition fluxes that are projected to become less acidic (due to 22% increase in NHx and 20% decrease in NOy deposition fluxes). Overall, projected changes in global TN deposition fluxes are small, indicating a 5% to 10% increase for RCP6.0 and RCP8.5, respectively.
3.1. Past-present-future changes of atmospheric nitrogen deposition
Figures 3 (a–d) show the computed spatial distribution of NOy (oxidized IN) and NHx (reduced IN), DON and TN atmospheric deposition for the year 2005. TN atmospheric deposition (Fig. 3d) is the sum of IN, DON and insoluble ON (Kanakidou et al. 2012) as seen in Table 1. NOy deposition (Fig. 3a) shows the highest fluxes over industrial areas of the northern hemisphere and tropical biomass burning regions, while NHx deposition (Fig. 3b) maximizes over Europe, China and Indonesia reflecting the important NH3 emissions in these regions. Smaller deposition fluxes are computed over oceanic regions, most of them related to recycling of NH3 oceanic emissions that in our model amount to 8.15 Tg-N y−1 (Bouwman et al. 1997). The here computed global annual present-day NOy and NHx deposition fluxes are well within the range of multimodel estimates that neglect the presence of ON originating from primary ON emissions and SOA formation [Table 1 and references therein]. As seen in Fig. 3d, TN deposition exceeds the 1 g-N.m−2y−1 critical load for vegetation at several locations over the eastern USA, Europe, India, China and Indonesia. High DON deposition flux (Fig. 3c) in the tropics is due to the large contribution by primary biogenic particles, biomass burning and SOA formation. The DON maxima computed for China and southeast Asia indicate, in addition to these sources, the importance of anthropogenic emissions in Asia. According to our calculations the contribution of DON to the global soluble N deposition amounts to 15–20% globally and to 20–35% over the ocean. The overall estimated range, including previous studies, in present-day DON atmospheric deposition is 20–32 Tg-N.y−1 globally and 10–20 Tg-N.y−1 over the ocean (Table 2), with about 30–50% of this ammount associated with human activities. This range reflects the uncertainties in the sources and chemical characterisation of ON and indicate that further studies are required to improve the understanding of ON in the atmosphere.
FIG. 3.
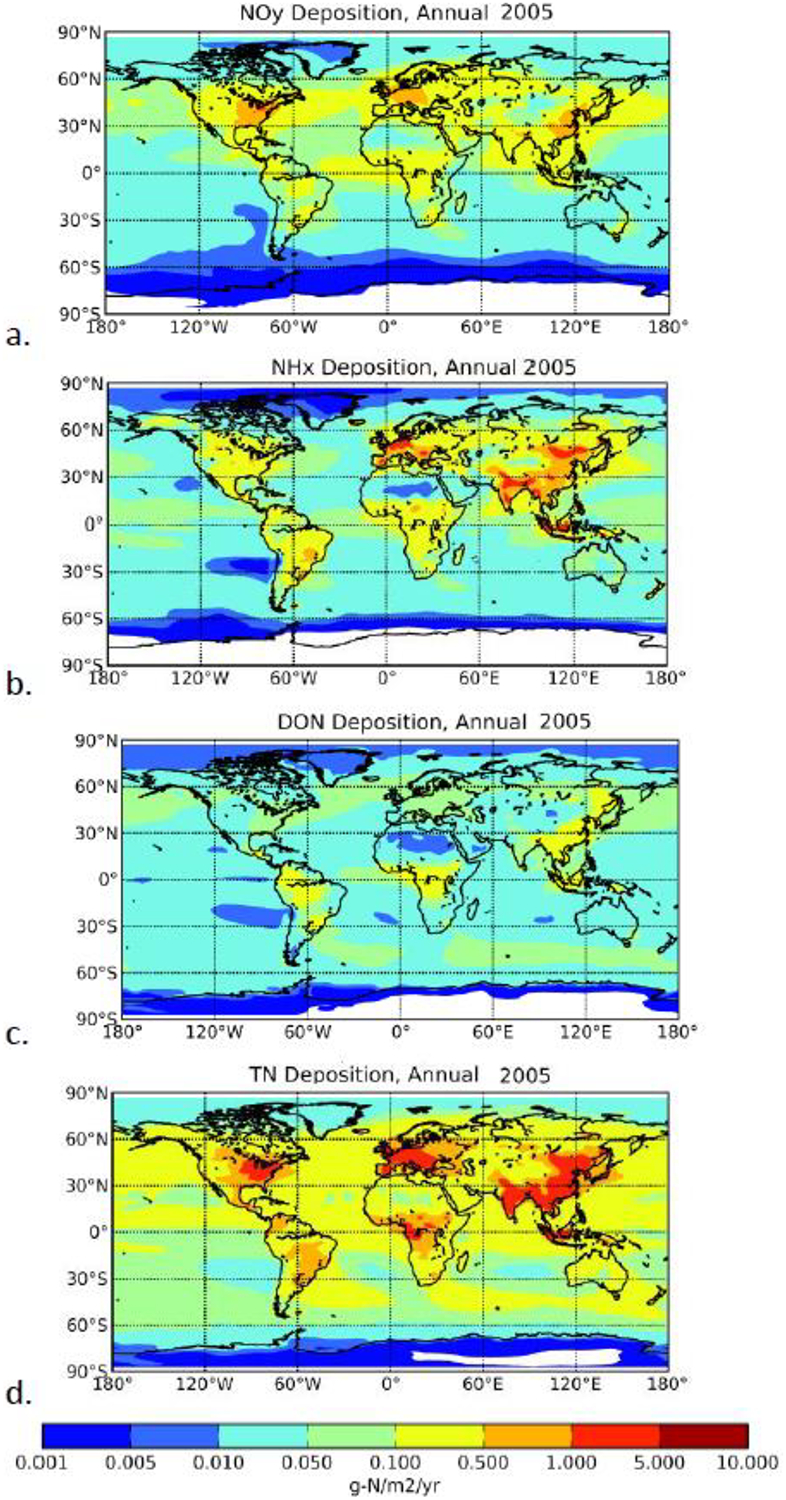
Atmospheric deposition of N in g-N.m−2y−1 computed by TM4-ECPL for NOy (a) NHx, (b) DON (c) and TN (d), for 2005.
Table 2.
DON annual deposition fluxes estimates (in Tg-N.y−1) for present day.
| DON deposition global | DON deposition ocean | Reference |
|---|---|---|
| 9 (up to 50) | Neff et al 2002 | |
| 20 | Duce et al 2008 | |
| 32 | 16 | Kanakidou et al 2012 |
| 26 | 1.1–10 | Ito et al 2014 |
| 19–30 | 9–14 | Ito et al 2015 |
| 20–29 | 10–13 | This work |
According to our calculations, the anthropogenic contribution to TN emissions increased from 15% in 1850 (mainly in the form of NHx) to 60% at present (2005, in the form of NHx, 50%, NOx, 40%, ON, 10%) and in the future (2050, in the form of NHx, 60%, NOx, 30%, ON, 10%), with similar changes in deposition fluxes. The atmospheric soluble N deposition increased since the preindustrial period (Table 1) by a factor of 3 mainly due to a larger (3.5-fold) increase in IN deposition (Fig. 4 a,c) and is expected to further change for 2050 (Fig. 4 b,d), although this estimated change is smaller than 15%. These changes are 15% to about 150% larger over land where most anthropogenic N sources are located than over the ocean. NHx deposition (Fig. 4b) is calculated to have increased globally since 1850 emissions (except for a few locations over the eastern USA, south India, the Bengal Bay, the Arabian Sea and southeast Austalia, where significant land use changes have occurred), and is expected to further increase over densily populated regions (Fig. 4d). Large increases have occurred over the heavily industrialized areas of the northern hemisphere for all soluble reactive N fractions as depicted in Figures 4 a,b,c and are projected to occur in the future mainly in Asia (Fig. 4 b,d,f), while NOy deposition will be reduced everywhere except over Asia (Fig. 4b). Projected DON deposition changes show similar patterns with those of NOy, with significant reductions over Europe and increases over Asia.
FIG. 4.
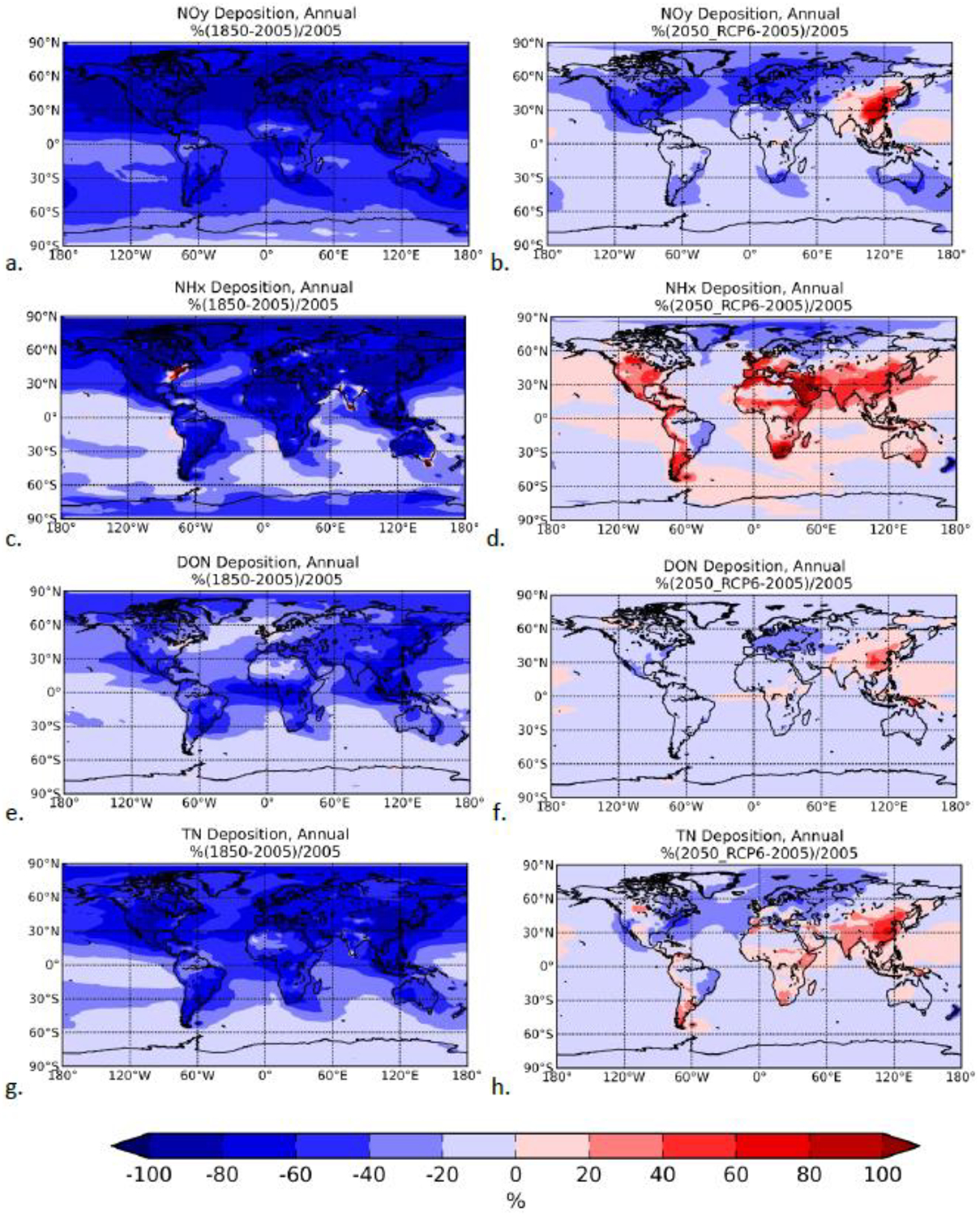
Percent changes relative to 2005 annual deposition flux computed by TM4-ECPL for NOy, NHx, DON and TN due to pre-industrial emissions (left panels) and due to anthropogenic RCP6.0 emissions (right panels).
The comprehensive inclusion of ON in the model increases the total soluble N deposition (since additional primary (ON) sources are taken into account), but also converts NOx to ON, thus changing the chemical composition of TN deposition. However the amount of ON originating from NOx chemistry remains uncertain. According to the present-day estimates it can vary between 4 and 13 Tg-N y−1.
3.2. Uncertainties
In the present study the fraction of ON secondary formation from non-amine VOC that is driven by NOx chemistry leads to about 3 Tg-N y−1 of organic nitrates and 2 Tg-N y−1 of N-SOA formation in the present-day. This present-day N-SOA flux estimate (2 Tg-N.y−1) is 5- to 8-fold lower than earlier estimates, which neglect the NOx dependence [10 Tg-N.y−1 (Kanakidou et al. 2012) and 17.6 Tg-N.y−1 (Ito et al. 2014)]. This indicates the large uncertainty associated with the chemistry of ON aerosols, and this introduces an uncertainty of at least 25–35% in the global deposition fluxes of soluble ON and about 10% in the global TN deposition. Significantly larger uncertainties exist also in the primary emission of ON, as is the case for other pollutant primary emissions (Granier et al. 2011). For instance, when using a N:C ratio for anthropogenic aerosol varying from 0.16 (base case) to 0.3 (simulations marked with star in Table 1), the primary ON emissions are evaluated between 23 and 30 Tg-N.y−1, respectively, i.e. about 30% different. Thus, the largest uncertainties in the deposition fluxes are found to be associated with the primary and secondary sources of ON (that can vary by at least a factor of two) and to the projected anthropogenic emissions. The latter vary in magnitude by about 50% and can even show different trends [Stohl et al. (2015) and references therein]. Kanakidou et al. (2014) investigated the impact of the use of different anthropogenic emission inventories [GAINSv4a (Klimont et al. 2013) versus RCPs (Lamarque et al. 2013)] on the calculated global annual deposition fluxes of N. They found higher N-deposition fluxes by 17% for present-day and 30% for 2050 when using GAINSv4a emissions compared with RCP6.0 emissions. In addition, the higher increase in N-deposition computed for the year 2050 using GAINSv4a emission inventories is mainly associated with the stronger projected increase in NH3 emissions in GAINSv4a than in RCPs. Thus while using RCP6.0 or RCP 8.5 projections the calculated N-deposition future change is small (<3%), but when using GAINS projections, N-deposition is calculated to further increase in the future (+12%). In addition, natural emissions have significant natural variability that has been neglected here. The deposition fluxes to the ocean have also been found to be sensitive to the model horizontal resolution, most probably because increasing resolution leads to better separation between ocean and land grid boxes. Thus, a 4-times higher horizontal resolution (smaller grid) leads to about 20% increase in atmospheric deposition of reactive nitrogen over the oceans, while, as expected, global deposition fluxes remain practically unchanged. This uncertainty associated with the limitations of numerical models in separating coastal from ocean regions has to be taken into account when studing the impact of atmospheric deposition to marine ecosystems.
4. Conclusions
For the present study, anthropogenic and biomass burning emissions were taken from the RCP6.0 database for the year 2005, from ACCMIP historical emissions for the year 1850 and from RCP6.0 and RCP8.5 projections for the year 2050. Present-day global TN deposition is estimated between 125 and 132 Tg-N.y−1. ON deposition (27–36 Tg-N y−1) is about 20–30% of the TN deposition (Figure S6), about 30–50% of which is associated with human activities. From this ON deposition only 25–35% that is associated with the secondary ON source has been accounted for in earlier studies as gaseous organic nitrates or IN deposition. However, targeted observational experiments are needed to improve the parameterization of the chemical bonding of N on OA under clean and polluted atmospheric conditions as well as the estimate of the primary emissions of ON. Large areas in the northern hemisphere are subject to TN deposition higher than the critical load for vegetation. Atmospheric TN deposition has increased by a factor of about 3 since 1850 (calculated using ACCMIP historical emissions), mainly due to the large increase of IN. This also led to a decrease in the ratio of dissolved ON to dissolved TN from 0.77 in 1850 to 0.30 nowadays [in agreement with Duce et al. (2008) estimate] on an annual global mean basis while regionally significant deviations from these mean ratios are computed (Figure S6). For the future, using RCP6.0 and RCP8.5 scenarios, the reduction in NOx emissions is projected to be compensated by the continuing increase in NH3 emissions, and as a result the global TN deposition is not expected to change much while its acidity will be significantly reduced. Regionally significant changes are projected, in particular over Asia. These results neglect future changes in biogenic emissions driven by climate. However changes in natural emissions are expected to have much smaller impact on N deposition than changes in anthropogenic N emissions. The evaluation of the impact of the estimated changes in TN fluxes on the ecosystems requires dedicated biogeochemical ecosystem studies.
Supplementary Material
Acknowledgments.
This research has been co-financed by the European Union (European Social Fund - ESF) and Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) - Research Funding Program ARISTEIA I - PANOPLY. AB and MK have been partially supported by ADAMANT project. The authors gratefully acknowledge the sources of the precipitation chemistry and deposition data listed on page 92 of Vet et al. (2014). This is a contribution to GESAMP WG 38. We thank the anonymous reviewers for their pertinent and useful comments.
REFERENCES
- Altieri KE, Hastings MG, Peters AJ, and Sigman DM, 2012: Molecular characterization of water soluble organic nitrogen in marine rainwater by ultra-high resolution electrospray ionization mass spectrometry, Atmos. Chem. Phys, 12, 3557–3571, doi: 10.5194/acp-12-3557-2012. [DOI] [Google Scholar]
- Bouwman AF, Lee DS, Asman WAH, Dentener FJ, Van Der Hoek KW, Olivier JGJ, 1997: A global high-resolution emission inventory for ammonia. Global Biogeochem. Cycles 11, 561–587. doi: 10.1029/97GB02266. [DOI] [Google Scholar]
- Cape JN, Cornell SE, Jickells TD, and Nemitz E: Organic nitrogen in the atmosphere – where does it come from? A review of sources and methods, Atmos. Res, 102, 30–48, doi: 10.1016/j.atmosres.2011.07.009, 2011 [DOI] [Google Scholar]
- Christodoulaki S, Petihakis G, Kanakidou M, Mihalopoulos N, Tsiaras K and Triantafyllou G, 2013: Atmospheric deposition in the Eastern Mediterranean, A driving force for ecosystem dynamics. Journal of Marine Systems, 109–110, 78–93. [Google Scholar]
- Cornell SC 2010: Atmospheric nitrogen deposition: Revisiting the question of the importance of the organic component, Environmental Pollution, 159(10), 2214–2222, doi: 10.1016/j.envpol.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Dee DP, Uppala SM, and Coauthors, 2011: The ERA-Interim reanalysis: configuration and performance of the data assimilation system. Q. J. Roy. Meteor. Soc 137, 553–597. doi: 10.1002/qj.828 [DOI] [Google Scholar]
- Dentener FJ, Drevet J, and Coauthors, 2006: Nitrogen and sulfur deposition on regional and global scales: a multimodel evaluation, Global Biogeochem. Cycles, 20, GB4003, doi: 10.1029/2005GB002672. [DOI] [Google Scholar]
- Driscoll CT, Whitall D, and Coauthors 2003: Nitrogen pollution in the northeastern United States: sources, effects, and management options. BioScience, 53, 357–374. [Google Scholar]
- Duce RA, Mohnen VA, and Coauthors, 1983: Organic material in the global troposphere, Review of Geophysics and Space Physics, 21, 921–952. [Google Scholar]
- Duce RA, Liss PS, and Coauthors, 1991: The Atmospheric Input of trace species to the world ocean, Global Biogeochemical Cycles 5 (3), 193–25. [Google Scholar]
- Duce RA, LaRoche J, and Coauthors 2008: Impacts of Atmospheric Anthropogenic Nitrogen on the Open Ocean. Science, 320, 893–897. [DOI] [PubMed] [Google Scholar]
- Facchini MC, Decesari S, and Coauthors, 2008: Important source of marine secondary organic aerosol from biogenic amines, Environmental Science and Technology, 42(24), 9116–9121, doi: 10.1021/es8018385. [DOI] [PubMed] [Google Scholar]
- Franze T, Weller MG, Niessner R, Pöschl U, 2005: Protein Nitration by Polluted Air, Environmental Science and Technology, 39, 1673–1678, DOI: 10.1021/es0488737. [DOI] [PubMed] [Google Scholar]
- Fountoukis C, and Nenes A, 2007: ISORROPIA II, a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+–NH4+–Na+–SO42−–NO3−–Cl−–H2O aerosols, Atmos. Chem. Phys, 7, 4639–4659, doi: 10.5194/acp-7-4639-2007. [DOI] [Google Scholar]
- Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA, 2008: Transformation of the Nitrogen Cycle: Recent Trends, Questions, and Potential Solutions. Science, 320, 889–892. [DOI] [PubMed] [Google Scholar]
- Granier C, Bessagnet B, and Coauthors, 2011: Evolution of anthropogenic and biomass burning emissions of air pollutants at global and regional scales during the 1980–2010 period, Climatic Change, 109, 163–190. [Google Scholar]
- Ito A, Lin G, Penner JE, 2014: Reconciling modeled and observed atmospheric deposition of soluble organic nitrogen at coastal locations. Glob. Biogeochem. Cycles, 28, 10.1002/2013GB004721. [DOI] [Google Scholar]
- Ito A, Lin G, Penner JE, 2015: Global modeling study of soluble organic nitrogen from open biomass burning, Atmospheric Environment, 121, 103–112, 10.1016/j.atmosenv.2015.01.031. [DOI] [Google Scholar]
- Karydis VA, Tsimpidi AP, Pozzer A, Astitha M, and Lelieveld J, 2015: Effects of mineral dust on global atmospheric nitrate concentrations, Atmos. Chem. Phys. Discuss, 15, 11525–11572, doi: 10.5194/acpd-15-11525-2015. [DOI] [Google Scholar]
- Kanakidou M, Duce RA, and Coauthors 2012: Atmospheric fluxes of organic N and P to the global ocean. Global Biogeochemical Cycles, 26, GB3026, doi: 10.1029/2011GB004277. [DOI] [Google Scholar]
- Lamarque J-F, Shindell DT, and Coauthors, 2013a: The Atmospheric Chemistry and Climate Model Intercomparison Project, ACCMIP: overview and description of models, simulations and climate diagnostics, Geosci. Model Dev, 6, 179–206. [Google Scholar]
- Lamarque J-F, Dentener FJ, and Coauthors, 2013b: Multi-model mean nitrogen and sulfur deposition from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP): evaluation of historical and projected future changes, Atmos. Chem. Phys, 13, 7997–8018, doi: 10.5194/acp-13-7997-2013. [DOI] [Google Scholar]
- Lelieveld J, Evans JS, Giannadaki D, Fnais M, Pozzer A, 2015: The contribution of outdoor air pollution sources to premature mortality on a global scale Nature, 525, 367–371, doi: 10.1038/nature15371 [DOI] [PubMed] [Google Scholar]
- Mills MM, Ridame C, Davey M, La Roche J, Geider RJ, 2004: Iron and phosphorus co-limit nitrogen fixation in the eastern tropical North Atlantic, Nature, 429 (6989), 292–294. [DOI] [PubMed] [Google Scholar]
- Myriokefalitakis S, Vignati E, and Coauthors, 2010: Global modelling of the oceanic source of organic aerosols, Advances in Meteorology, 2010, 16p., doi: 10.1155/2010/939171. [DOI] [Google Scholar]
- Myriokefalitakis S, Tsigaridis K, Mihalopoulos N, Sciare J, Nenes A, Kawamura K, Segers A, and Kanakidou M, 2011: In-cloud oxalate formation in the global troposphere: a 3-D modeling study, Atmos. Chem. Phys, 11, 5761–5782. [Google Scholar]
- Myriokefalitakis S, Daskalakis N, Mihalopoulos N, Baker AR, Nenes A, and Kanakidou M, 2015: Changes in dissolved iron deposition to the oceans driven by human activity: a 3-D global modelling study, Biogeosciences, 12, 3973–3992, doi: 10.5194/bg-12-3973-2015. [DOI] [Google Scholar]
- Neff JC, Holland EA, Dentener FJ, McDowell WH, and Russell KM, 2002: The origin, composition and rates of organic nitrogen deposition: a missing piece of the nitrogen cycle?, Biogeochemistry 57, 99–136. [Google Scholar]
- Sander R, 1999: Compilation of Henry’s Law Constants for Inorganic and Organic Species of Potential Importance in Environmental Chemistry [WWW Document]. Air Chem. Dep. Max-Planck Inst. Chem URL http://www.mpch-mainz.mpg.de/~sander/res/henry.html [Google Scholar]
- Seinfeld JH, and Pandis SN, 2006: Atmospheric chemistry and physics: From air pollution to climate change, Third edition, John Wiley, New York. [Google Scholar]
- Sindelarova K, Granier G, and Coauthors, 2014: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys, 14, 9317–9341, doi: 10.5194/acp-14-9317-2014. [DOI] [Google Scholar]
- Sintermann J and Neftel A, 2015: Ideas and perspectives: on the emission of amines from terrestrial vegetation in the context of new atmospheric particle formation, Biogeosciences, 12, 3225–3240, doi: 10.5194/bg-12-3225-2015. [DOI] [Google Scholar]
- Stohl A, and Coauthors, 2015: Evaluating the climate and air quality impacts of short-lived pollutants, Atmos. Chem. Phys, 15, 10529–10566, doi: 10.5194/acp-15-10529-2015. [DOI] [Google Scholar]
- Tsigaridis K and Kanakidou M, 2003: Global modelling of secondary organic aerosol in the troposphere: A sensitivity analysis, Atmos. Chem. Phys, 3, 1849–1869. [Google Scholar]
- Tsigaridis K and Kanakidou M, 2007: Secondary organic aerosol importance in the future atmosphere, Atmos. Environ, 41, 4682–4692, doi: 10.1016/j.atmosenv.2007.03.045. [DOI] [Google Scholar]
- Tsigaridis K, Krol M, Dentener FJ, Balkanski Y, Lathière J, Metzger S, Hauglustaine DA, and Kanakidou M, 2006: Change in global aerosol composition since preindustrial times, Atmos. Chem. Phys, 6, 5143–5162, doi: 10.5194/acp-6-5143-2006. [DOI] [Google Scholar]
- Tsigaridis K, Daskalakis N, Kanakidou M, and Coauthors, 2014: The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem.Phys 14, 10845–10895. [Google Scholar]
- van Vuuren DP, Edmonds J, and Coauthors 2011: The representative concentration pathways: an overview, Clim. Change, 109, 5–31, doi: 10.1007/s10584-011-0148-z. [DOI] [Google Scholar]
- Vet R, Artz RS, and Coauthors, 2014: A global assessment of precipitation chemistry and deposition of sulfur, nitrogen, sea salt, base cations, organic acids, acidity and pH, and phosphorus, Atmos. Environ, 93, 3–100. [Google Scholar]
- Vignati E, Facchini MC, and Coauthors, 2010: Global scale emission and distribution of sea spray aerosol: Sea-salt and organic enrichment, Atmos. Environ, 44, 670–677 doi: 10.1016/j.atmosenv.2009.11.013. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.