

Abstract
Atrial fibrillation (AF) is a highly prevalent arrhythmia, with substantial associated morbidity and mortality. There have been significant management advances over the past 2 decades, but the burden of the disease continues to increase and there is certainly plenty of room for improvement in treatment options. A potential key to therapeutic innovation is a better understanding of underlying fundamental mechanisms. This article reviews recent advances in understanding the molecular basis for AF, with a particular emphasis on relating these new insights to clinical translation opportunities. We first review the evidence relating basic electrophysiological mechanisms to the characteristics of clinical AF. We then discuss the molecular control of factors leading to some of the principal determinants, including abnormalities in impulse conduction (such as tissue fibrosis and other extra-cardiomyocyte alterations, connexin dysregulation and Na+-channel dysfunction), electrical refractoriness and impulse generation. We then consider the molecular drivers of AF progression, including a range of Ca2+-dependent intracellular processes, microRNA changes and inflammatory signaling. The concept of key interactome-related nodal points is then evaluated, dealing with systems like those associated with Ca2+/calmodulin-dependent protein kinase-II, NLRP3 (NACHT, LRR and PYD domains-containing protein-3) and transcription-factors like TBX5 and PitX2c.We conclude with a critical discussion of therapeutic implications, knowledge gaps and future directions, dealing with such aspects as drug repurposing, biologics, multispecific drugs, the targeting of cardiomyocyte inflammatory signaling and potential considerations in intervening at the level of interactomes and gene-regulation. The area of molecular intervention for AF management presents exciting new opportunities, along with substantial challenges.
Keywords: atrial fibrillation, mechanisms, personalized therapy, translational research
Subject codes: [106], [130], [132]
Introduction
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia and is associated with considerable morbidity and mortality.1 Presently available therapies have many limitations, including limited efficacy and significant adverse-effect potential for antiarrhythmic drugs1,2 and recurrences and potential complications for AF-ablation.1 The progressive nature of the arrhythmic substrate is a major problem limiting the long-term success of both pharmacological and ablation therapies.3 Preventive approaches targeting risk factors have shown promise,4 but a better understanding of the mechanisms underlying AF and its progression is needed to improve both understanding and our ability to create and exploit novel therapeutic avenues.2 The goal of the present narrative review is to provide a portrait of the molecular mechanisms underlying AF, with a particular focus on clinically-relevant mechanisms that are relevant both to understanding the clinical features of AF and to the development of novel treatments.
The area addressed is vast- a single Medline search with the term “molecular mechanisms atrial fibrillation” revealed 611 papers. We have therefore chosen to focus on clinically-relevant mechanisms with potential translational significance. We begin with an analysis of the available information from patient data (in vivo studies, analyses of atrial-tissue samples) and animal models regarding the mechanisms responsible for initial AF-occurrence. We then consider the molecular basis for some of the key macroscopic mechanisms like spontaneous atrial ectopic firing and the development of a reentry substrate. After examining the sources of initial AF-occurrence, we discuss the molecular mechanisms underlying the atrial remodeling resulting from AF that causes substrate progression once AF occurs. Following that, we detail the evolving concept of central interactome nexuses that are involved in producing patterns of molecular response leading to the arrhythmic substrate. We conclude with a discussion of the potential therapeutic relevance of understanding the molecular mechanisms, an analysis of the challenges to developing new therapeutic approaches targeting molecular processes and a consideration of potential future directions.
Electrophysiological Mechanisms Implicated in Initial AF Occurrence
AF is known to promote its own maintenance and to be progressive in nature;3,5 a great deal has been written about the mechanisms and clinical importance of AF-induced remodeling.6 However, before AF can become self-promoting, there must be mechanistic factors that determine its initial onset. In considering the molecular pathways leading to AF, it is important to have a global sense of the pathophysiological mechanisms leading to the occurrence of AF prior to its progression.
Studies in patients and patient tissue-samples provide insights into the pathophysiology of AF-occurrence, as summarized in Figure 1. Ectopic activity, particularly occurring in the pulmonary veins (PVs), is centrally involved in AF-onset.7 A number of factors predispose the PVs to the generation of ectopic activity, including both ion-channel and structural features.8,9 The canine PVs have smaller inward-rectifier K+-current (IK1) and L-type Ca2+-current (ICa,L), as well as larger delayed-rectifier K+-currents, compared to left-atrial cells.8 These properties reduce action-potential (AP) duration (APD), making reentry more likely, and increase the likelihood of spontaneous ectopy due to delayed afterdepolarizations (DADs). Furthermore, the PVs have a unique anatomical structure, composed of branching fibers with limited lateral coupling and abrupt fiber-orientation change,9 which also increases their ability to generate spontaneous activity and support reentry.9,10 Finally, the PVs are situated very close to major cardiac autonomic ganglia, which strongly modulate their electric properties as well as those of other atrial regions and thereby contribute to determining AF-susceptibility.11,12
Figure 1. Overview of mechanisms linked to AF-occurrence.
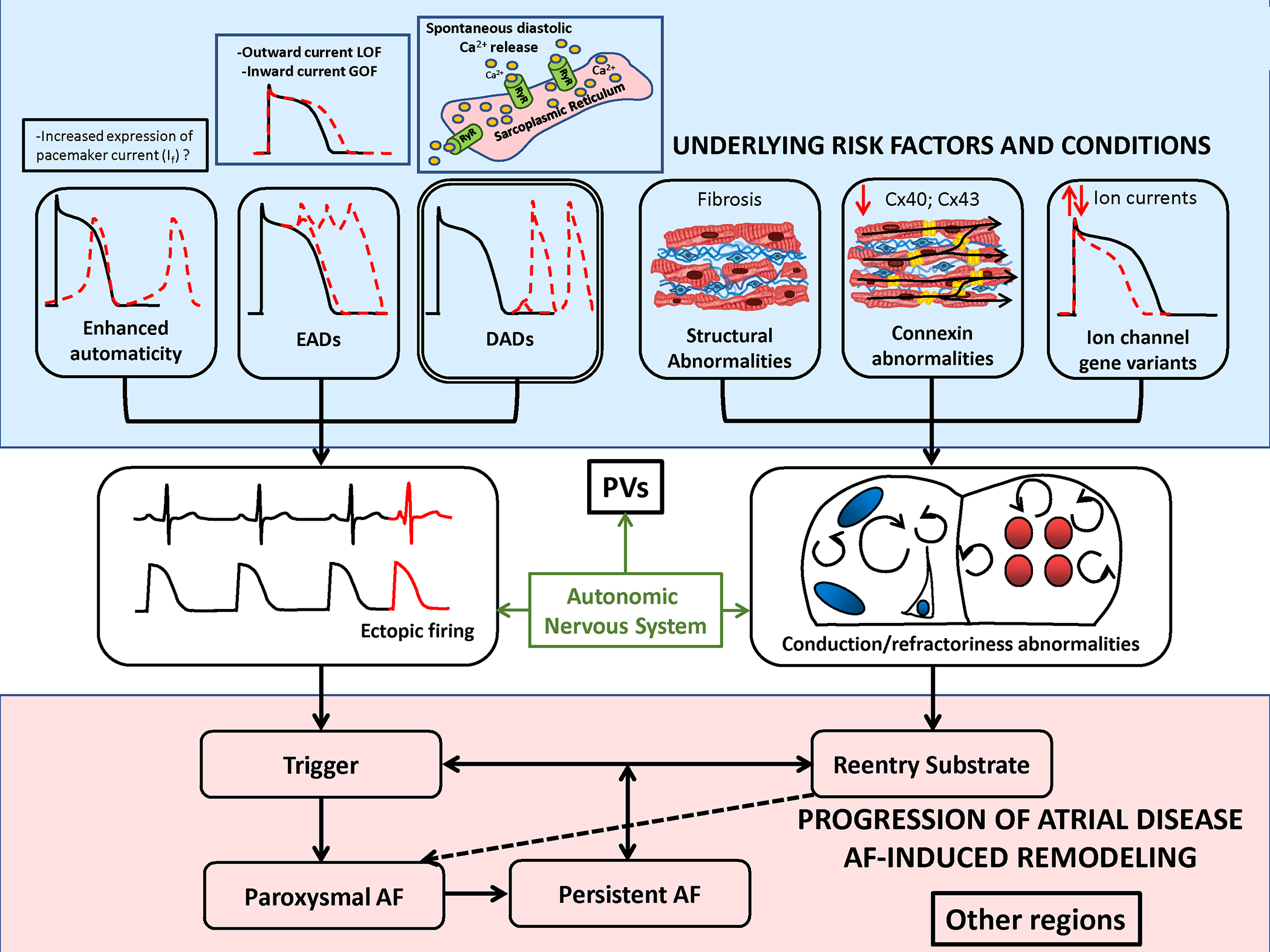
AF AF-triggers result from focal ectopic firing. Ectopic activity is most clearly linked to spontaneous diastolic Ca2+-release from the sarcoplasmic reticulum Ca2+-stores via leaky ryanodine-receptor (RyR2) Ca2+-release channels. Early afterdepolarizations (EADs) due to loss-of-function (LOF) outward-current mutations or gain-of-function (GOF) inward-current mutations have also been linked to spontaneous ectopy. Enhanced automaticity, for example due to pacemaker current expression, is another possible cause of ectopic activitty, but has not been definitively demonstrated. AF-persistence is linked to AF-maintaining reeentry that requires both trigger and a vulnerable reentrant substrate. The latter can be caused by abbreviated refractoriness (e.g. due to a GOF K+-channel mutation or to enhanced vagal tone) or by conduction abnormalities due to tissue fibrosis, connexin (Cx) dysfunction or LOF Na+-channel mutations. Ectopic firing typically originates from the pulmonary veins (PVs), but the PVs are also a priviledged site for reentry susceptibility.
Clinical studies of electrophysiological indices and imaging analyses also provide insights into the mechanisms associated with AF-occurrence. Abnormalities are consistently noted in PVs of patients with paroxysmal AF (pAF), including smaller electrogram voltages, slowed conduction, shorter effective refractory periods and a greater vulnerability to AF-induction during programmed electrical stimulation, indicating a reentry-prone substrate.10,13 Atrial fibrous-tissue content is increased in pAF patients and atrial scarring correlates with clinical outcomes.14 Thus, a reentry-prone substrate clearly favors AF-occurrence. At the same time, spontaneous atrial ectopic activity appears to be an important trigger for reentry. Studies employing ambulatory monitoring reveal atrial premature complexes initiating arrhythmia for >95% of pAF episodes.15
Additional insights are provided by the analysis of gene-variants associated with AF and the cellular electrophysiology of pAF patients.13 Short-QT syndromes due to gain-of-function K+-channel mutations have been implicated in pAF, likely due to reentry facilitated by abbreviated refractoriness.16–19 In addition to reentry facilitated by reduced refractoriness, gene-variants causing reentry-promoting conduction-abnormalities also cause AF. For example, gene-variants in the gap-junction connexin ion-channels impair conduction and are associated with AF.20 Finally, atrial fibrosis, which causes conduction-abnormalities and creates a substrate for AF,21 is present by voltage-mapping or delayed gadolinium-enhancement magnetic resonance imaging in pAF patients.22
There are also data pointing to mechanisms increasing spontaneous atrial activity in AF-onset. A variety of mutations that promote spontaneous Ca2+-release events (SCaEs) during diastole and the formation of DAD-mediated ectopic activity have also been implicated in spontaneous AF-onset.23–25 Long-QT syndrome has also been associated with spontaneous AF,26 apparently by inducing early afterdepolarization (EAD)-mediated ectopic activity.27 Abnormal atrial automaticity has been indirectly implicated in AF-associated ectopy via altered expression of HCN-channels and automaticity-modulating microRNAs in tissue-samples from AF-patients,28 but direct evidence is lacking. Analyses of cellular electrophysiology in right-atrial samples from patients with pAF show no AF-induced remodeling of action potential (AP) or ion-current properties;29 however, left-atrial inward-rectifier K+-current is upregulated,30 potentially contributing to the stabilization of reentry. SCaEs and DADs associated with increased sarcoplasmic-reticulum (SR) Ca2+-content, cardiac ryanodine-receptor channel type-2 (RyR2) expression and open-probability are present in right-atrial cardiomyocytes from pAF patients.29
Ca2+-handling dysregulation can also promote beat-to-beat alternation in APD (alternans) that favors reentry.31 In patients studied in the electrophysiology laboratory, APD alternans precedes pacing-induced AF, and is produced most readily in persistent-AF patients, less readily in pAF and least in sinus-rhythm controls.31 In sheep, aging is associated with increased susceptibility to APD-alternans and persistence of AF.32 In dogs, AF-induced remodeling delays recovery of the cellular Ca2+-transient, which causes increased magnitude and spatial dispersion of susceptibility to Ca2+- and repolarization-alternans.33 Consequently, rapid pacing leads to spatially-discordant alternans in the presence of AF-induced remodeling, causing reentrant rotor formation and enhancing vulnerability to initiation and maintenance of AF.33
Clinical Perspectives
In summary, there is extensive evidence that AF-initiation involves atrial ectopic triggers and a reentry-prone substrate. The PVs play a central role as both ectopic sources and zones of reentry, with autonomic tone being a key regulator. Disturbances in conduction related to tissue fibrosis and/or connexin-abnormalities predispose to reentry, with abbreviated refractoriness being a potential contributor, particularly among individuals with gene-variants that accelerate atrial repolarization. Repolarization alternans is often a path to reentry. Novel therapeutic priorities might be the identification of tractable molecular targets at the level of ectopic-beat generation, mechanisms leading to conduction abnormalities and the early detection and targeted therapy of gene-based pathways.
Molecular Determinants of AF-promoting Atrial Ectopy
Ca2+-handling Abnormalities and DADs
Atrial cardiomyocyte excitation-contraction coupling is unique by virtue of a lack of a fully developed t-tubular system, although axial tubules are present in atrial cardiomyocytes of several species, including humans.34–36 Upon cell-depolarization, Ca2+ influx via L-type Ca2+-channels triggers a much larger Ca2+ release from neighboring SR sites via RyR2 channels. The resulting intracellular Ca2+ waves propagate to the cell center, activating non-junctional SR Ca2+ release sites, thereby causing a small delay in the rise of Ca2+ in the cell center. During diastole, SCaEs can activate the cardiac Na/Ca2+-exchanger type-1 (NCX1), leading to a depolarizing transient-inward current, largely carried by INCX. Ectopic/triggered activity occurs when the resulting DAD is sufficiently large to overcome the electrotonic load and activate Na+-channels, but in silico studies suggest that even sub-threshold DADs can have proarrhythmic effects by promoting dispersion of excitability with subsequent conduction block due to heterogenous voltage-dependent Na+-channel inactivation (Figure 2).37 Besides SCaEs, Ca2+ waves triggered by L-type Ca2+-channels at the cell boundary might activate Ca2+-overloaded SR sites at the cardiomyocyte center, leading to large triggered Ca2+ waves (TCWs) during the AP. TCWs may cause subcellular Ca2+ instability with sub-threshold EADs increasing the dispersion of refractoriness38 and can be observed in atrial cardiomyocytes from failing canine and human hearts.39
Figure 2. Atrial ectopy. Molecular pathways promoting Ca2+-mediated ectopy.
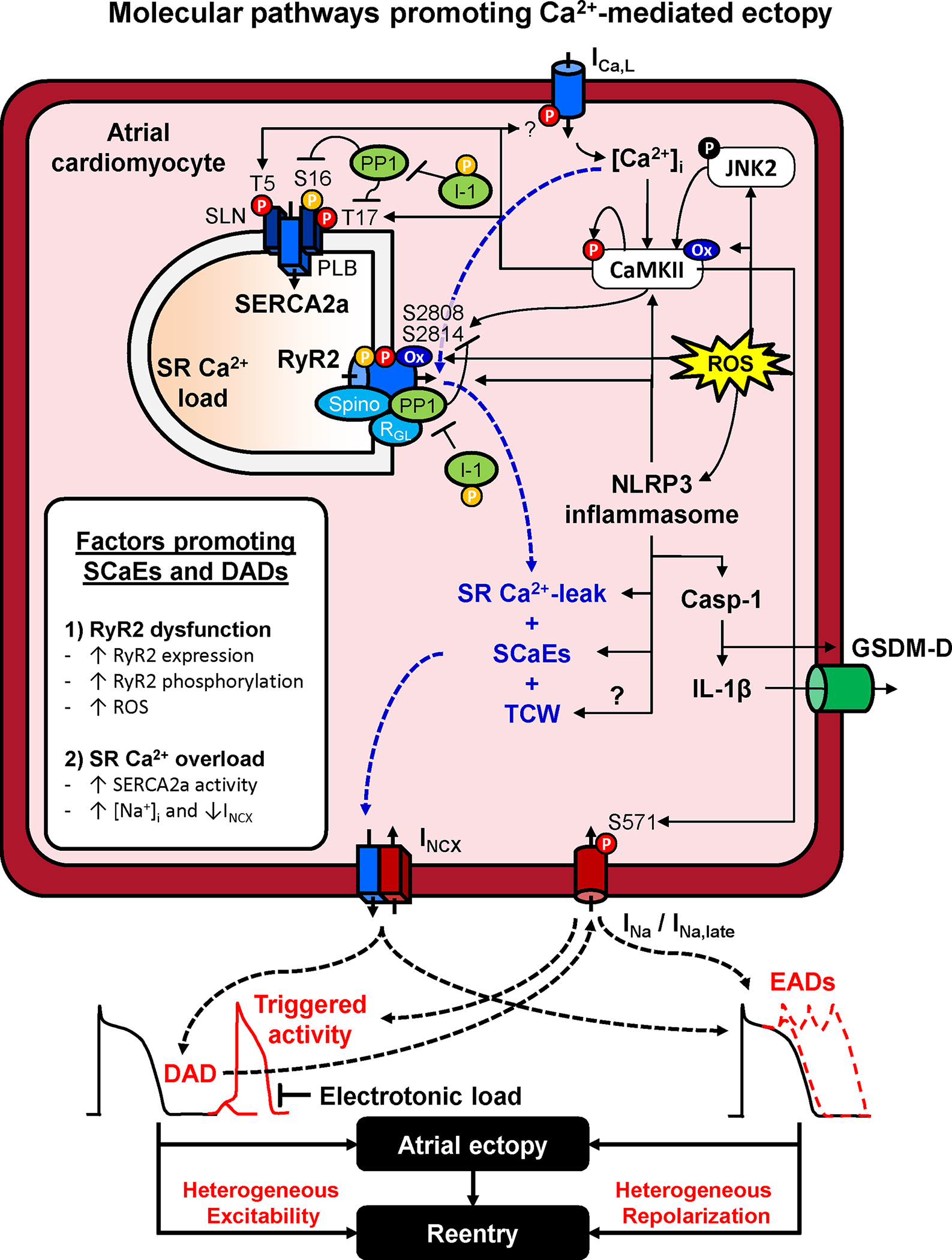
Increased sarcoplasmic reticulum (SR) Ca2+ leak and spontaneous SR Ca2+-release events (SCaEs) primarily result from dysfunction of the cardiac ryanodine receptor type-2 (RyR2) channel or SR Ca2+-overload. RyR2 dysfunction is promoted by increased RyR2 expression, hyperphosphorylation (e.g., due to increased Ca2+/calmodulin-dependent protein kinase-II, CaMKII, activity or improper targeting of protein phosphatase-1, PP1), or RyR2 oxidation due to increased reactive oxygen species (ROS). ROS mediated NLRP3 inflammasome activation amplifies the Ca2+-handling abnormalities and activates caspase-1 (Casp-1) which increases interleukin (IL)-1β generation and the formation of gasdermin-D-derived plasmamembrane-pores, allowing the release of IL-1β out of the cell, spreading inflammatory signaling. SR Ca2+-overload is promoted by increased activity of the SR Ca2+-ATPase-2a (SERCA2a) or elevated intracellular Na+, reducing Ca2+-extrusion via the Na+/Ca2+ exchanger type-1 (NCX1). SR Ca2+ overload also promotes L-type Ca2+-current (ICa,L)-dependent triggered Ca2+ waves (TCW). SCaEs and TCW activate a transient-inward current mediated by NCX (INCX) resulting in DADs or EADs, depending on their timing relative to the atrial action potential. DADs and EADs can promoted atrial ectopy, as well as reentry through increased heterogeneity of excitability and repolarization. Abbreviations: GSDM-D, N-terminal Gasdermin-D fragment; I-1, inhibitor-1 of PP1; JNK2, c-Jun N-terminal kinases-2; NLRP3, NACHT, LRR and PYD domains-containing protein 3; PLB, phospholamban; SLN, sarcolipin.
SCaEs are promoted by RyR2-dysfunction and SR Ca2+-overload. The RyR2 is a large macromolecular complex and its dysfunction is often due to impaired interaction between stabilizing regulatory subunits and the pore-forming α-subunit, leading to increased open probability.40 For example, loss of RyR2-associated calmodulin, the FK506-binding protein 12.6 or junctophilin-2 leads to RyR2-dysfunction and increased AF susceptibility in mouse and canine hearts.24,41–44 RyR2 hyperphosphorylation by Ca2+/calmodulin-dependent protein kinase-II (CaMKII) at Ser2814 and at Ser2808 or Ser2030 by protein kinase-A (PKA), along with RyR2 oxidation, also promote RyR2 dysfunction in human atrial samples and animal models.45–50 Under physiological conditions, CaMKII is activated by binding of Ca2+/calmodulin to its regulatory domain, which and leads to sustained, Ca2+-independent activity due to autophosphorylation at Thr287, which accumulates between beats at faster heart rates.51 Besides local CaMKII and PKA activity, RyR2 phosphorylation is controlled by the amount of RyR2-associated protein phosphatases type-1 (PP1) and type-2a. Accordingly, loss of subunits responsible for targeting PP1 to the RyR2 (e.g., spinophilin or PP1R3A, also known as RGL) results in RyR2 dysfunction and increased AF susceptibility in mice52,53 and PP1-complex expression, including the expression of PP1-subunits and of endogenous PP1 regulators, is altered in atria of AF patients.54–56
SR Ca2+-overload is promoted by increased activity of the SR Ca2+-ATPase-2a (SERCA2a), e.g., due to reduced expression or hyperphosphorylation of the inhibitory interacting proteins phospholamban or atrial-specific sarcolipin.49 Furthermore, SR Ca2+-load is increased by elevated intracellular Na+-concentration. This occurs, for example in the presence of Na+-K+-ATPase inhibition by cardiac glycosides, which impair Ca2+-extrusion via NCX1.57 Increased Na+-influx through non-inactivating Na+-channels (persistent/late Na+ current, INa,late) also promotes Ca2+-handling abnormalities and AF inducibility in small-animal models58,59. CaMKII-dependent Ser571-hyperphosphorylation of the pore-forming subunit NaV1.5 increases INa,late, potentially creating an AF-promoting positive feed-back loop.59 Consistent with this idea, SR Ca2+-overload and CaMKII hyperactivity can also cause TCWs and CaMKII-dependent dysregulation of INa,late, producing proarrhythmic activity in atrial cardiomyocytes from patients with sleep-disordered breathing,60 who are at an increased risk for developing AF.4 On the other hand, no significant INa,late could be detected under physiological conditions in atrial cardiomyocytes from patients in sinus rhythm or with AF in a different study.61 The potential role of INa,late in AF pathophysiology needs further elaboration and validation.
Ca2+ handling abnormalities are a common finding in atrial cardiomyocytes of AF patients,29,46,47,62,63 as well as in patients with risk factors and comorbidities promoting AF. For example, SR Ca2+-load and the incidence of spontaneous INCX is increased in heart failure (HF) patients,63 and patients with sleep-disordered breathing have increased CaMKII activity, DADs and EADs.60 Although the exact molecular mechanisms differ between different forms of AF and different comorbidities, CaMKII activation appears to be a central element controlled by several upstream regulators. The autonomic nervous system plays a major role in AF-pathophysiology, with combined sympatho-vagal activation producing both a substrate and triggers for AF initiation.64 Sympathetic stimulation results in PKA-dependent phosphorylation of numerous Ca2+-handling proteins through activation of β-adrenoceptors and cyclic adenosine monophosphate (cAMP) production.64 The resultant increase in SR Ca2+-load contributes to the positive inotropic effects of sympathetic stimulation and together with RyR2 hyperphosphorylation can promote proarrhythmic Ca2+-handling abnormalities.49,64 The elevated Ca2+-levels and increased heart rate during sympathetic stimulation also sustains CaMKII activation.51 Furthermore, cAMP production during β-adrenoceptor stimulation in mice can activate CaMKII via exchange-protein activated by cAMP65 and cAMP/PKA-dependent phosphorylation of the inhibitor-1 protein that regulates PP1, preventing dephosphorylation of CaMKII-Thr287.66
Oxidative stress is common in a variety of cardiovascular diseases and has been associated with Ca2+-handling abnormalities and increased risk of AF. In turn, rapid atrial activation during AF can further promote oxidative stress via CD44/NOX4 signaling.67 Oxidative stress activates CaMKII through oxidation at Met281/282, which is increased in AF patients.68 Oxidized CaMKII creates a substrate for TCWs and reentry in the atria of HF dogs50 and has been implicated in AF promotion by the tyrosine kinase inhibitor ibrutinib used in cancer treatment.69 Oxidative stress also activates the stress-response kinase c-Jun N-terminal kinase-2 (JNK2), which phosphorylates CaMKII, leading to increased CaMKII activity (Figure 2).70 In addition, JNK2 activation upregulates CaMKII expression, further contributing to CaMKII-mediated arrhythmogenesis.71 JNK2-dependent CaMKII activation has been implicated in several AF-promoting conditions, including advanced age70,71 and binge-alcohol exposure.72
Atrial inflammatory signaling is increased in several conditions predisposing patients to AF and inflammatory markers such as interleukin (IL)-6, IL-1β and tumor necrosis factor-α correlate with AF progression.73 Recent work has indicated that cardiomyocyte NACHT, LRR and PYD domains-containing protein-3 (NLRP3)-inflammasome signaling plays a causative role in AF development in genetically-targeted mouse-models.74 Expression of NLRP3-inflammasome components (‘priming’) is increased in atrial samples of AF patients,74 as well as in atrial samples from patients that subsequently develop post-operative AF,75 or that have type-2 diabetes and an increased risk of AF.76 Diabetic rabbits that show increased activity of the atrial NLRP3-infllammasome system, along with slowed heterogeneous conduction and increased atrial fibrosis and fibrotic markers, exhibit increased AF inducibility, with changes being reversed by the NLRP3-inflammasome inhibitor (and hypoglycemic agent) glyburide.77 Assembly (‘triggering’) of the NLRP3-inflammasome complex consisting of NLRP3, apoptosis-associated speck-like protein containing a C-terminal caspase activation and recruitment domain (ASC) and pro-caspase-1 promotes auto-cleavage of pro-caspase-1. The resulting increase in active caspase-1 results in cleavage of inactive precursors of IL-1β and IL-18 to their active forms and of gasdermin-D, releasing the pore-forming N-terminal fragment that allows IL-1β and IL-18 to leave the cardiomyocyte to exert autocrine and paracrine effects.73 Mice with cardiomyocyte-restricted constitutive activation of the NLRP3-inflammasome have increased atrial ectopy associated with Ca2+-handling abnormalities due to increased RyR2 expression, as well as reentry-promoting APD abbreviation (due to increased atrial-selective ultra-rapid delayed-rectifier K+-current, IKur and acetylcholine-activated inward-rectifier K+-current, IK,ACh) and structural remodeling (hypertrophy and fibrosis).74 Similarly, short-term (hours) exposure to IL-1β and tumor necrosis factor-α produces proarrhythmic Ca2+-handling abnormalities in rodent cardiomyocytes,78,79 highlighting the potential to trigger AF. CaMKII activation in response to pressure overload or chronic angiotensin-II (Ang-II) exposure activates NLRP3 in mice cardiomyocytes,80,81 NLRP3-inflammasome-dependent activation of IL-18 upon acute β-adrenoceptor stimulation triggers pathological cardiac remodeling,82 and oxidative stress drives NLRP3-inflammasome activation during high-glucose and ischemia/reperfusion injury in H9C2 rat cardiomyocytes.83 While the recent evidence in favor of NLRP3-inflammasome signaling as a central proarrhythmic mediator of multiple pathophysiological signals in AF is intriguing, the relative contribution of these pathways to atrial ectopy and AF in humans remains to be conclusively established.
Early Afterdepolarizations
EADs are promoted by excessive APD prolongation, providing time for recovery of L-type Ca2+-currents (ICa,L) from voltage- and Ca2+-dependent inactivation during the plateau phase of the AP. The resulting increase in ICa,L during the AP-plateau or phase-3 produces membrane depolarization and EAD-formation. EADs are a major arrhythmogenic mechanism in the ventricles, e.g., in the setting of (drug-induced) long-QT syndrome, where they promote ectopy, increased dispersion of repolarization and beat-to-beat repolarization alternans, all of which are proarrhythmic.84,85 Long-QT syndrome can also promote AF.86 Mice with long-QT syndrome type-3 due to a gain-of-function mutation in the Na+-channel show excessive atrial APD-prolongation and EADs.27,85,87 Similarly, acute CaMKII activation due to oxidative stress initiates AF through EAD-mediated triggered activity in old rats, which can be suppressed by inhibition of INa,late.88 Nonetheless, there is little direct evidence for a major role of EADs in atrial arrhythmogenesis in patients with AF in the absence of repolarization-delaying mutations.
Abnormal Automaticity
The sinoatrial node (SAN) acts as the primary pacemaker via spontaneous AP generation (“automaticity”). In SAN cells, automaticity is controlled by a coupled-clock system involving hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and SR Ca2+-leak activating depolarizing INCX.89 On a background of low-level IK1 expression in SAN cells, these systems produce spontaneous diastolic depolarization leading to AP firing. The relevance of abnormal automaticity for atrial ectopy is unclear. The expression of HCN in the healthy atrium is limited, although an age-dependent increase has been observed in dogs.90 Atrial HCN expression is also increased in end-stage failing human hearts compared to non-failing hearts,91 as well as in AF-patients compared to sinus rhythm controls, possibly due to a reduction in inhibitory microRNA-1 and microRNA-133.28 Normally, however, atrial IK1 appears to be sufficiently large to maintain a stable resting membrane potential that prevents substantial automaticity. Consistent with this notion, abnormal automaticity in canine PV sleeves can only be observed in the presence of IK1 inhibition.92
Clinical Perspectives
The mechanistic basis for spontaneous atrial ectopy is not fully established, but the weight of evidence points to DAD-related activity due to aberrant Ca2+-release as the most likely causative paradigm. Increased RyR2 open-probability as a result of CaMKII-mediated hyperphosphorylation is widely seen in studies of atrial samples from AF-patients and in animal models of AF. CaMKII-activation results from a variety of molecular pathways, including enhanced oxidative stress and inflammatory signaling, which could constitute novel therapeutic targets. EADs are also capable of mediating atrial ectopic firing, but to date the most convincing evidence is limited to contexts with clearly delayed atrial repolarization like Long-QT Syndromes.
Molecular Control of Factors Leading to Conduction Abnormalities
Conduction abnormalities are a ubiquitous feature in patients with AF-substrates,93,94 presumably by increasing susceptibility to reentry initiation and maintenance. The most important determinants of conduction are structural integrity of atrial tissue (importantly disturbed by fibrosis), effective cell-to-cell coupling (principally determined by connexin hemichannels in intercalated disks) and the integrity of the rapid phase-0 Na+-current (INa) which provides the electrical energy for conduction.
Molecular Determinants of Structural Remodeling
Atrial structural remodeling, prominently including tissue fibrosis, is associated with conduction abnormalities that create a substrate for AF-maintenance.21 Atrial fibrosis is a common feature in AF-patients and the extent of fibrosis is a predictor of AF-recurrence after catheter ablation.95 A vast body of research has established a wide range of molecular mechanisms controlling atrial fibrosis.96 The principal determinants are illustrated in Figure 3. A variety of cell-membrane receptor systems cause fibroblasts to proliferate and differentiate into profibrotic collagen-secreting myofibroblasts. These include receptors for Ang-II, platelet-derived growth factor, connective-tissue growth factor, and transforming growth-factor β. The receptor systems are coupled to second-messenger systems, which activate transcription-factors (TFs) that modify the mRNA-transcription of proteins like procollagen, fibronectin, matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases that govern extracellular-matrix remodeling and lead to fibrosis. Reactive oxygen species (ROS) are generated by reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase coupled to type-1 Ang-II receptors and transforming growth-factor β-receptors and play a prominent role in AF.97 ROS are important stimulants to tissue-fibrosis, acting via downstream systems like mitogen-activated protein kinases98 and NLRP3-inflammasome formation and profibrotic inflammatory signaling.74,99,100 All of these molecular systems eventually lead to increased synthesis of procollagen α- and β-chains in the endoplasmic reticulum. These trimerize to form procollagen molecules, which are secreted into the extracellular space where the N-terminal and C-terminal ends are removed by PNPase and PCPase enzymes, respectively, to produce mature collagen molecules. Self-assemble generates collagen myofibrils that are crosslinked by lysine-oxidase enzymes to form mature collagen. Fibroblasts also contain membrane ion-channels that mediate (receptor- and store-operated transient-receptor potential [TRP] channels) or regulate (IK1 and Ca2+-dependent K+-channels) transmembrane Ca2+-entry, which acts as a key signal for fibroblast-activation.101–103
Figure 3. Molecular determinants of tissue fibrosis.

The main pathways governing profibrotic signaling. Extracellular profibrotic signaling molecules like angiotensin-II (Ang-II), transforming growth factor-β1 (TGFβ), platelet-derived growth-factor (PDGF) and connective- tissue growth-factor (CTGF) activate membrane receptors coupled to downstream signaling which leads to enhanced gene-transcription to increase extracellular matrix (ECM) production. Fibroblast ion-channels control Ca2+-entry to regulate fibroblast activation. For additional discussion, see text. Abbreviations: bb, integrin receptor oblast activation; Ang-II, angiotensin- II; AP, activator-protein; AT1R, angiotensin-II type-1 receptor; CTGF, connective-tissue growth- factor; ER, endoplasmic reticulum; ERK1/2, extracellular signal-related kinase-1/2; ERK-P, phosphorylated extracellular signal-related kinase; Grb2, growth-factor receptor binding-protein 2; IK1, inward-rectifier K+-channel; IP3, inositol 1,4,5-trisphosphate; JAK, Janus kinase; JNK, c-jun N-terminal kinase; LOX, lysyl oxidase; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; NADPH, nicotine adenine dinucleotide-phosphate; NF-κB, nuclear factor-kappa B; NLRP3, NACHT-, LRR- and PYD domains-containing protein 3; PKC, protein-kinase C; PDGF, platelet-derived growth factor; PDGFR, PDGF-receptor; PIP2, phosphatidylinositol bisphosphate; PLC, phospholipase-C; ROC, receptor-operated channel; ROS, reactive-oxygen species; Shc, src homologous and collagen protein; SMAD, sma- and mad-related proteins; SOC, store-operated channel; SOS, son of sevenless protein; Src, sarcoma proto-oncogene tyrosine kinase; STAT, signal transducers and activators of transcription; TAK1, TGFTGF TAK1, ed kinase-1; TF, transcription factor; TGFβR, transforming growth factor β receptor; TIMP, tissue inhibitor of matrix metalloproteinase; TRP, transient receptor potential; TSS, transcriptional start site.
Besides atrial fibrosis, there are additional changes in extracellular-tissue properties that have been implicated in AF-promotion. For example, in one study the presence of atrial amyloidosis correlated better with AF-occurrence in patients than that of fibrosis.104 Human atrial adipose tissue secretome induced rat atrial-tissue fibrosis in organo-culture,105 and AF is associated with fibrosis in subepicardial fatty infiltrates in humans and sheep.106 The importance of the extracellular-tissue changes associated with AF are reflected in the fact that they are incorporated in 3 of the 4 classes of atrial cardiomyopathy of the EHRAS classification system.107
While the molecular determinants of atrial fibrosis have been extensively investigated, the links between clinical conditions leading to AF and extracellular-matrix remodeling are poorly understood. Atrial stretch, for example due to valvular heart disease, HF or diastolic dysfunction, is commonly assumed to lead to atrial fibrosis. However, the underlying molecular mechanisms are poorly studied, particularly at the atrial level, and therapeutic manipulation has proven quite challenging.108 Cardiomyocyte NLRP3-inflammasome activation appears to be a common feature in patients prone to AF and to lead to atrial fibrosis,74,75,99,100 but the factors causing NLRP3-inflammasome activation in the atria are poorly understood. Clearly, much more work needs to be done in this area.
Molecular Determinants of Connexin Dysfunction
Connexins are ion-channel proteins that form hemichannels specialized to ensure cell-to-cell communication at the intercalated disks.109 Connexins have a large single-channel conductance and allow cardiac tissue to function as an electrically-continuous syncytium. The principal atrial connexins are connexin-43 (Cx43) and connexin-40 (Cx40), encoded by GJA1 and GJA5 respectively. While Cx43 is expressed ubiquitously in cardiac tissues, Cx40 is expressed in the atria, but not ventricles. Connexins are located primarily in “connexons”, 6-hemichannel complexes generally localized to gap junctions in intercalated disks connecting cells at their cell-to-cell junctions. Each connexon connects to a complementary connexon from the next cell, with the hemichannels coupling to form cell-to-cell channels. Abnormalities in connexin number, function or localization impair electrical propagation and can lead to initiation and/or stabilization of reentry circuits that support AF (Figure 1). Figure 4 illustrates the molecular determinants of connexin function that, when disturbed, may lead to AF. GJA5 polymorphisms are well-recognized to be associated with AF in man, affecting conduction via reduced expression,110 reduced single-channel open probability,20 and impaired channel formation or trafficking.111 The expression of AF-associated mutated channels in mice slows atrial conduction and increases AF-vulnerability.20,112 Atrial tissues from pAF patients show reduced Cx40 expression and increased heterogeneity of Cx40-distribution, while persistent (chronic)-AF (cAF) patients show severe reductions of Cx40-immunostaining.113 Goats with AF-induced atrial remodeling show markedly-increased Cx40-distribution heterogeneity.114 Connexin downregulation and/or relocalization from cell-ends to lateral margins might play a role in AF-promoting conduction abnormalities due to obstructive sleep apnea,115 acute inflammation116, aging117 and chronic kidney disease.99 Assessing the relative importance of connexin changes in AF may be difficult when connexin-changes occur along with other aspects of atrial remodeling. In at least one context, that of HF, connexin alterations occur (dephosphorylation, increased Cx40/Cx43 protein-ratios, lateralization), but appear to be of much less functional significance than concomitant fibrosis-development.118 An additional layer of complexity is added by the observation that reduced Cx43-expression can enhance the fibrotic response by increasing fibroblast activity.119
Figure 4. Connexin dysregulation in AF.
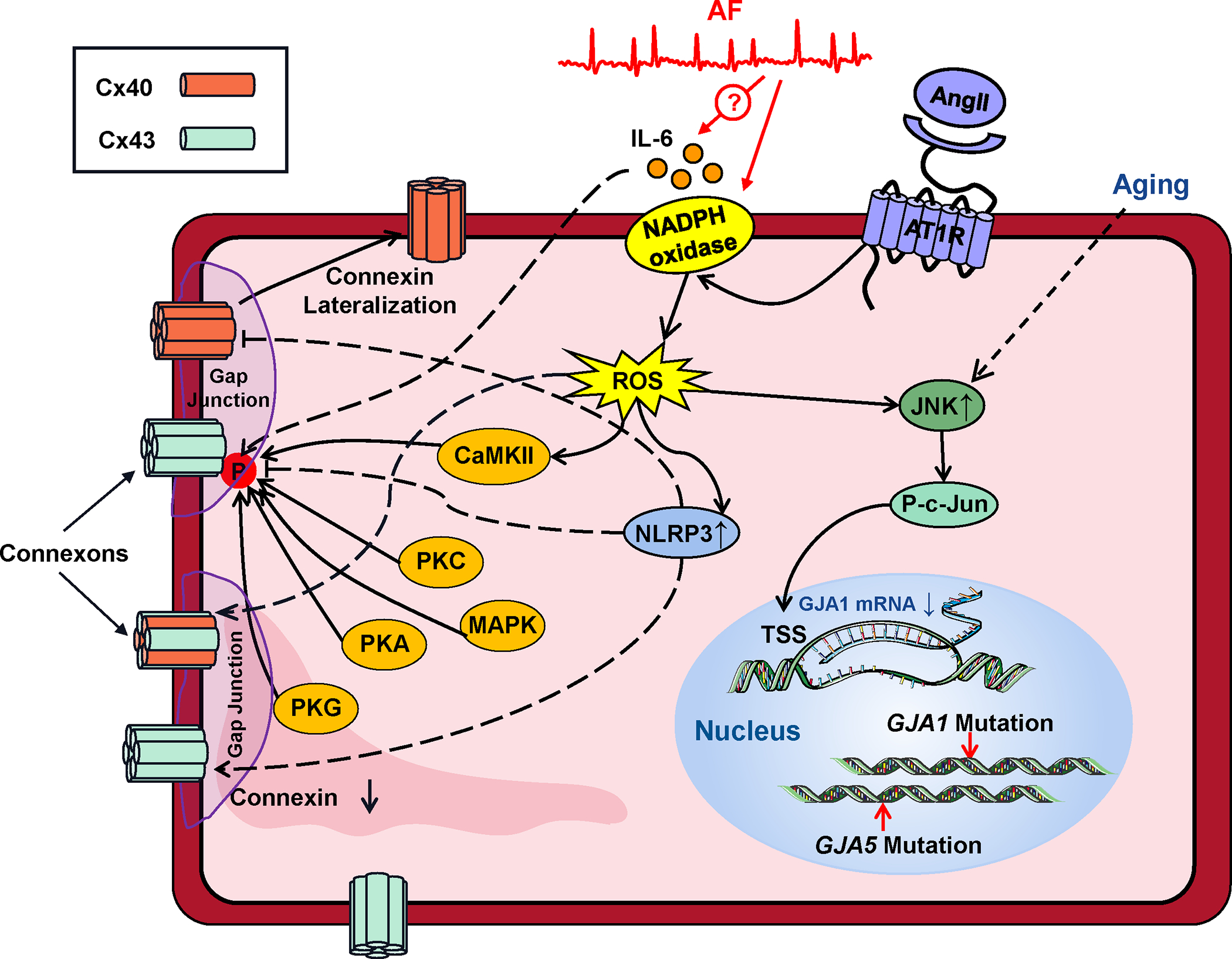
Dysfunction of the connexins that ensure cell-to-cell coupling in gap junctions results from connexin (Cx) downregulation or lateralization to transverse cell-borders. The moecular mechanisms governing these changes are illustrated. Abbreviations: Ang-II, angiotensin-II, AT1R, angiotensin-receptor type-1; CaMKII, Ca2+-calmodulin dependent kinase type-II; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein-kinase; P, phosphate; PKA, PKC, PKG, protein-kinases A, C, G respectively; ROS, reactive-oxygen species.
Phosphorylation regulates connexin channel function, membrane expression and cellular localization in complex ways. Cx43 has at least 21 discrete phosphorylation sites, which are phosphorylated specifically by a variety of kinases including PKA, protein kinase C (PKC), CaMKII and mitogen-activated protein kinases.120 Phosphorylation can increase or decrease gap-junctional conductance, alter connexin transport to the cell membrane, affect connexin degradation in the proteasome and affect gap-junction assembly, resulting in a potentially wide range of functional effects.120 In addition, phosphorylation of transcription factors like c-Jun (AP-1) affects connexin production by entering the nucleus and regulating transcription to decrease GJA1 mRNA-production.117 Intracellular ROS production alters connexin function, either directly or via the activation of kinases like CaMKII or JNK.120
Connexin-dysfunction in AF has a number of potential therapeutic implications. Cx43 gene-therapy has been used effectively to suppress AF-progression in a porcine model,121,122 but the barriers to applying cardiac gene-therapy in man are substantial. An antiarrhythmic peptide has been developed that enhances gap-junction conductivity. Detailed study in experimental AF-models of atrial-tachycardia, HF and ischemic atrial remodeling shows improved conduction in all, but antiarrhythmic efficacy limited to the acute ischemic paradigm.123
Changes in Na+-channel Function
INa is another important determinant of cardiac conduction-properties. Experimental atrial-tachycardia remodeling reduces atrial INa and slows conduction over a time-frame of weeks.124 Studies of AF-associated INa-changes in man have provided varying results, with early studies showing only a depolarizing voltage-shift in voltage-dependence125 and more recent work pointing to a decrease in peak-INa and increased INa,late.126 The latter effect was associated with increased susceptibility to Ca2+-related ectopic firing that could be suppressed by the INa,late-blocker ranolazine.126 Finally, CaMKII-regulation of INa appears to be an upstream-event to AF-related Ca2+-handling abnormalities and associated ectopic activity59 and CaMKII-dependent dysregulation of INa,late results in proarrhythmic activity in atrial cardiomyocytes from patients with sleep-disordered breathing,60, who are prone to AF. Overall, INa-changes appear to have functional significance in AF, but are much less studied than other molecular components of the system.
Clinical Perspectives
Conduction abnormalities are common in AF and play an important role in AF-maintaining reentry. Structural remodeling is a central motif, with tissue-fibrosis being a common finding but also potential contributions from other extra-cardiomyocyte components like adipose tissue and amyloid-proteins. Many AF-associated molecular signaling systems like those associated with the renin-angiotensin-aldosterone axis, platelet-derived growth factors, connective-tissue growth factor, and transforming growth-factor β, as well as heart-disease risk factors, promote the development of atrial fibrosis. There is evidence that oxidative stress and proinflammatory signaling may be significant contributors. Cell-to-cell conduction depends on connexin-proteins, which are susceptible to genetic abnormalities and remodeling by heart disease leading to AF-promoting conduction abnormalities. The prevention of structural remodeling is potentially a very interesting therapeutic approach, but the ubiquitousness of many of the mechanisms makes safe and specific targeting a challenge.
Molecular Drivers of AF Progression
Progression of the AF-supporting substrate is recognized to be an important factor leading to therapeutic resistance and failure.3,127 Progression of the AF-substrate can be caused by remodeling associated with the arrhythmia or through the effects of predisposing heart disease and/or risk factors.3,127,128 Risk-factor control helps to prevent AF-progression;4,128 while it is likely that effective management of heart-disease can also prevent progression of the AF-substrate, this is difficult to address and to our knowledge there is no confirmatory evidence to date from well-controlled randomized trials.
Here we will focus on the AF-associated molecular factors that lead to progression of the AF-maintaining reentrant substrate. Figure 5 illustrates some of the key pathways shown to be involved in this process, which involve electrophysiological, Ca2+-handling and structural remodeling.
Figure 5. Molecular pathways involved in AF-progression.
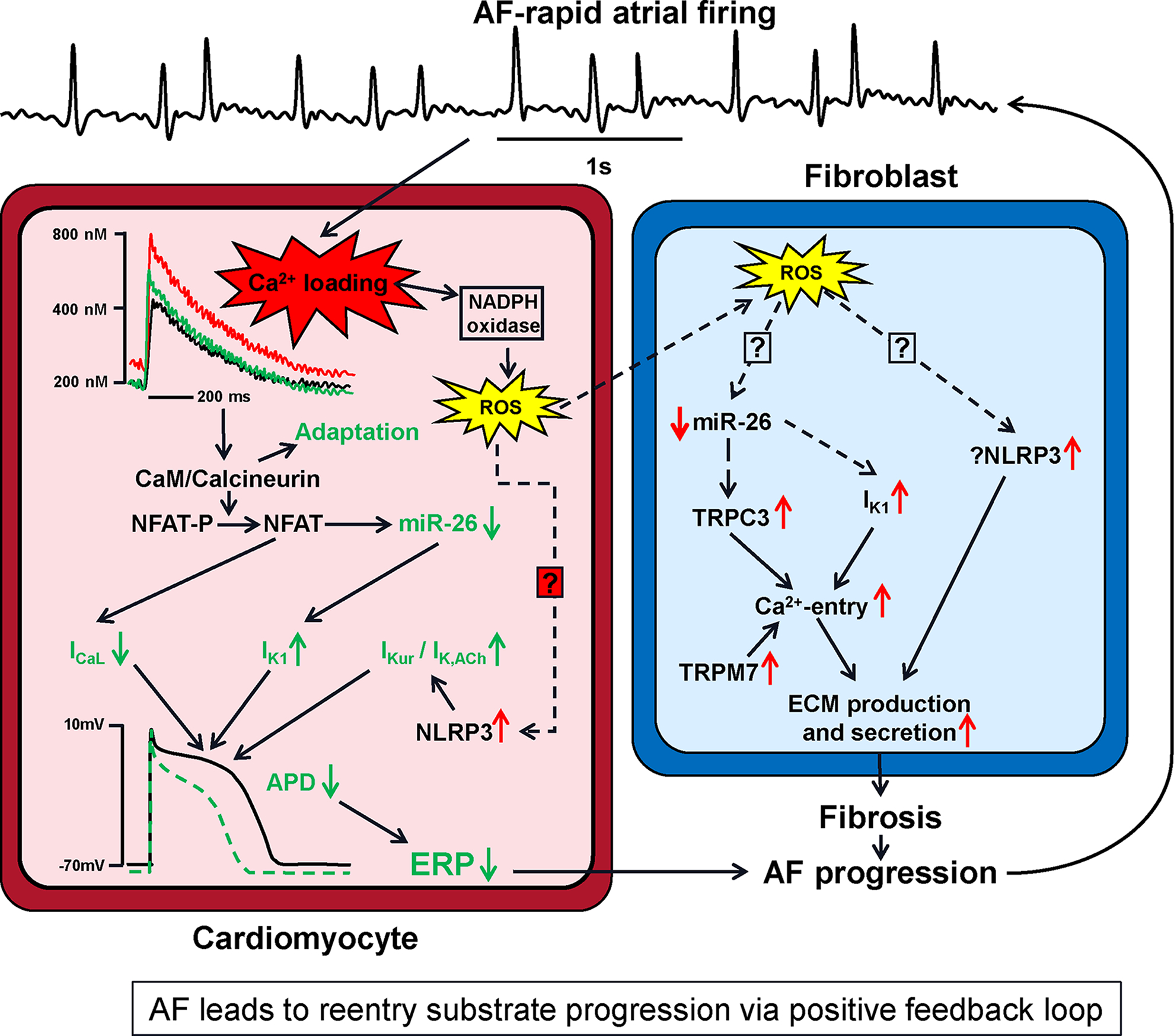
The rapid atrial firing in AF leads to cellular Ca2+-loading, which engages compensatory mechanisms (shown in green) that attenuate Ca2+-loading at the price of action-potential duration (APD) abbreviation that favors reentry. Ca2+-loading is a proximal signal to this and other processes resulting ultimately in APD and refractoriness-abbreviation in cardiomyocytes, along with enhanced collagen-production in fibroblasts, to cause progression of the reentry substrate and greater resistance of AF to therapy. A positive feedback loop results, wherein AF causes changes that increase AF-vulnerability and perpetuate the events that cause progression. Abbreviations: ECM, extracellular matrix; ERP, effective refractory period; ICa,L, L-type Ca2+-current; IK1, inward-rectifier K+-current; miR, microRNA; NLRP3, NACHT-, LRR- and PYD domains-containing protein 3; TRPC3, transient-receptor channel potential current canonical type-3; TRPM7, transient-receptor channel potential current melastatin type-7; ROS, reactive-oxygen species.
Ca2+-handling Changes and AF-progression
The rapid rate of atrial-cardiomyocyte firing in AF leads to Ca2+-loading in canine atrial cardiomyocytes.129 The atrial arrhythmia associated with AF, even in the absence of any alterations in cardiac function, contributes to progression of the AF-substrate due to both electrophysiological and structural remodeling in dogs.130 Differences in cardiomyocyte Ca2+-handling properties between pAF and cAF patients,29,47 as well as animal models with atrial tachycardia-related remodeling131,132 also point towards progressive changes in atrial Ca2+-handling due to AF itself. Ca2+-transient amplitude is increased in pAF, but decreased in cAF and animal models of atrial tachycardia-related remodeling, at least when studied at slow pacing rates ex vivo, potentially reflecting a protective mechanism against Ca2+-overload induced cell death at fast rates.127 This reduction in Ca2+-transient amplitude contributes to prothrombotic atrial hypocontractility.131 Consistent with its role as frequency sensor, CaMKII autophosphorylation and CaMKII-dependent RyR2 phosphorylation are elevated in cAF but not pAF. In addition to the rapid activation rate itself, variability in beating rates contributes to AF-related Ca2+-handling remodeling, with irregular 3-Hz pacing producing more Ca2+-handling abnormalities and CaMKII activation than regular 3-Hz pacing in neonatal rat cardiomyocytes.133 Despite the reduction in Ca2+-transient amplitude in cAF, elevated diastolic Ca2+-levels persist due to increased CaMKII-dependent SR Ca2+-leak, particularly during tachycardia, and may trigger proarrhythmic TCWs, SCaEs and drive Ca2+-dependent structural remodeling, thus contributing to AF maintenance.
Electrophysiological Changes that Promote Reentry
Canine atrial cardiomyocytes adapt to Ca2+-loading by engaging intracellular pathways that reduce cell-[Ca2+]i directly via reduced Ca2+-influx through decreased ICa,L and indirectly via abbreviated APD (since a large portion of Ca2+-entry occurs during the AP-plateau).134 Rate-induced Ca2+-loading causes enhanced binding of Ca2+ to calmodulin, which activates the phosphatase calcineurin. Calcineurin then dephosphorylates the nuclear factor of activated T-cells (NFAT), which translocates into the nucleus and inhibits production of CACNA1C mRNA, decreasing the message for the ICa,L α-subunit, decreasing its protein and ion-transport function.134 In parallel, NFAT suppresses the production of microRNA (miR)-26 by binding to and negatively regulating sites upstream to the transcriptional start site in human and mouse atrium.135 One of the binding targets of the miR-26 seed site is KCNJ2, the gene encoding the IK1 channel. Reduced miR-26 expression caused by AF removes miR-26-induced destabilization of the KCNJ2 message and inhibition of its translation.135 Inward-rectifier current functional expression is enhanced by this mechanism, as well as by increased constitutive acetylcholine-dependent current (IK,ACh) in human and canine models,136,137 leading to acceleration of repolarization and resting-membrane-potential hyperpolarization, both of which stabilize AF-maintaining rotors and promote AF-maintenance.138 Recent work also shows that NLRP3 inflammasome activity is increased via increases in both triggering and priming in patients with persistent AF, and that NLRP3 activation increases the gene expression of the channels subunits underlying the atrial-selective currents IKur and IK,ACh.74 The molecular basis for AF-associated NLRP3-activation, on one hand, and the mechanisms by which NLRP3 regulates IKur and IK,ACh, on the other, remain to be established. In addition to the pathways illustrated in Figure 5, some other molecular candidate mechanisms have been reported to contribute to APD-shortening in AF. These include miR-328 dysregulation of ICa,L,139 protein-kinase isoform switches that upregulate the constitutive activity of IK,ACh,140 and upregulation of 2-pore and Ca+-dependent K+-channels.141,142
AF-induced Structural Changes that Promote Reentry
There is now clear evidence that AF induces atrial structural remodeling, particularly fibrosis, some of which can be attributed to poor control of the ventricular rate, but a component of which is clearly due to the atrial tachyarrhythmia per se.130 Rapid cardiomyocyte firing leads to fibroblast activation via a diffusible substance in HL-1 atrial-derived cardiomyocytes,143 which appears to be ROS-derived from cardiomyocyte NADPH oxidase stimulated by Ca2+-loading.144 The mechanisms through which ROS diffusing from cardiomyocytes to fibroblasts causes their activation are unknown. ROS are known to activate NLRP3-inflammasomes in other systems145 and NLRP3-inflammasome activation is known to cause atrial fibrosis.74 Whether this system is in fact operative in atrial fibroblasts in AF remains to be defined. As in cardiomyocytes, fibroblast miR-26 is downregulated by AF in a canine model,101 possibly by ROS. There is evidence that TRP channels, both TRPC3 in canine, goat and human models101 and TRPM7 in human AF,146 are upregulated in AF-fibroblasts and enhance fibroblast Ca2+-entry; TRPC3 is under the control of miR-26 and its upregulation is related to miR-26 downregulation.101 Once again similar to the system in cardiomyocytes, fibroblast IK1 is upregulated in response to miR-26 decreases in canine AF.102 Fibroblast IK1-upregulation increases the driving force for Ca2+-entry, activating fibroblasts by enhancing fibroblast-stimulating Ca2+-influxes through store-operated channels.102 Ca2+-loading appears to be the proximal signal for many of the AF-induced remodeling processes that cause progression of the arrhythmic substrate.147 Interestingly, an increased intracellular Ca2+-load resulting from leaky RyR2 also appears to promote progression of the AF-substrate via CaMKII-activation.148
Clinical Perspectives
Progression of the AF-substrate is a major clinical problem. Aggressive early reversal of AF and sinus-rhythm maintenance might be a successful strategy, but this is as yet unproven. There is much evidence favoring vigorous control of risk factors in preventing progression and possibly even reversing the AF-substrate. Key components identified in substrate progression include Ca2+-loading of both cardiomyocytes and fibroblasts, oxidative stress and low-grade local inflammation. Further research into underlying molecular mechanisms and specific therapeutic targeting approaches has the potential to provide new and helpful treatments.
Interactomes relevant for AF
Nodal Points
Targeting nodal intersection points in the signaling pathways underlying atrial remodeling might constitute a promising strategy to prevent or halt AF-progression. AF-related Ca2+-overload and Ca2+-handling abnormalities contribute to the triggers and substrates that maintain AF.147 CaMKII is a central nodal point integrating electrical, Ca2+-handling and structural atrial remodeling (Figure 6).49–51,59,70,88,131,149–151 CaMKII senses and integrates signals via receptor systems leading to oxidative stress and ROS generation.50,67,68
Figure 6. Atrial fibrillation-promoting Ca2+/calmodulin-dependent protein kinase-II (CaMKII)- and NACHT, LRR and PYD domains-containing protein 3 (NLRP3)-inflammasome feed-forward signaling network.
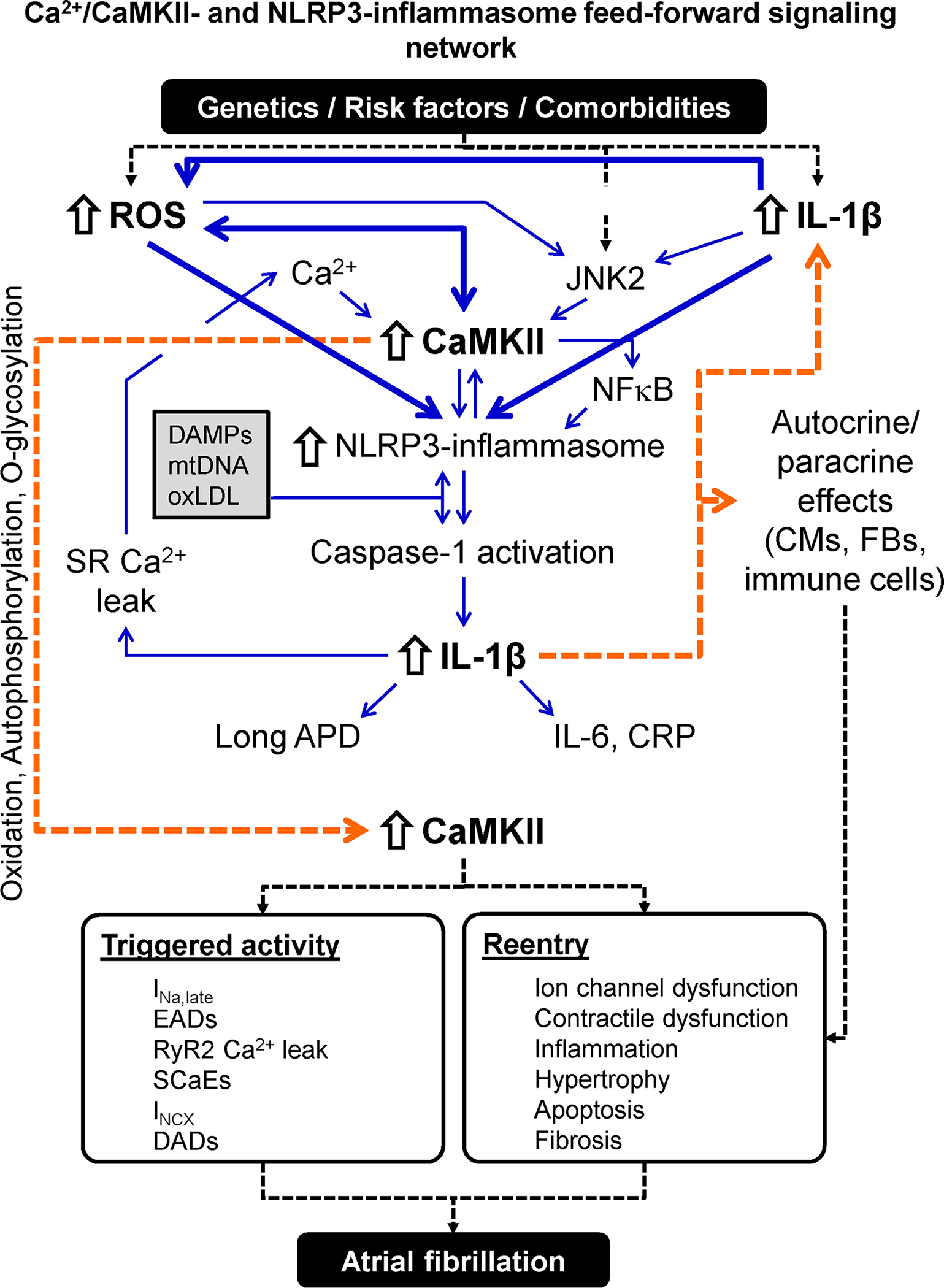
Risk factors and comorbidities create an environment in which danger-associated molecular patterns (DAMPs), mitochondrial DNA (mtDNA) and oxLDL activates the atrial NLRP3 inflammasome. Cardiac-restricted increases in reactive oxygen species (ROS) production and c-Jun N-terminal kinase-2 (JNK2) activity further stimulate the NLRP3 inflammasome via CaMKII-dependent and -independent pathways. The resulting stimulation of caspase-1 maturates interleukin (IL)-1β, which leaves the cell, thereby spreading the inflammatory signaling and increasing the synthesis of IL-6 and C-reactive protein (CRP). IL-1β amplifies the NLRP3 inflammatory signaling and promotes sarcoplasmic reticulum (SR) Ca2+ leak and action potential duration (APD) changes in cardiomyocytes (CMs), creating a feedforward signaling network. IL-1β also exerts paracrine effects on cardiac fibroblasts (CFs) and immune cells causing hypertrophy, apoptosis and fibrosis. Activation and perpetuation of this feedforward Ca2+/CaMKII/NLRP3-inflammasome signaling network promotes triggered activity and reentry and increases AF susceptibility. Abbreviations: DADs, delayed afterdepolarizations; EADs, early afterdepolarizations; INa,late, Persistent/late Na+ current; INCX, Na+-Ca2+-exchanger current; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; RyR2, ryanodine receptor type-2; SCaEs, spontaneous SR Ca2+-release events.
Oxidative stress is another nodal point in AF-promoting remodeling.152 Oxidative stress and mitochondrial dysfunction are caused by numerous AF risk factors and amplified by Ca2+ overload-inducing atrial tachycardia.153–155 Besides promoting ectopic activity through Ca2+-handling abnormalities, oxidative stress promotes reentry via ion-channel and gap-junction remodeling, as well as pro-inflammatory and pro-fibrotic signaling (e.g., via nuclear factor κB).152–154 Oxidative stress-related remodeling also increases thrombogenesis156 and causes microvascular flow abnormalities in pig ventricles, coupling electrical consequences to potential thromboembolic and cardiac-functional complications of AF.157 NLRP3-mediated caspase-1 derived IL-1β also causes atrial electrical, Ca2+-handling and structural remodeling in mouse and rabbit atria,73,74,77,158 representing another therapeutically tractable nodal point of AF-related remodeling. Chronic IL-1β increases promote CaMKII-autophosphorylation at Thr287 and oxidation at Met281/282, and increase Ca2+-spark frequency and APD in rodents.78,79,159 These findings support a model in which chronic cardiomyocyte-derived IL-1β creates a self-amplifying feed-forward loop of NLRP3-inflammasome activation at the expense of CaMKII-dependent electrical, Ca2+-handling and structural abnormalities, mechanistically linking IL-1β- and CaMKII-signaling pathways with NLRP3-inflammasome activation (Figure 6). Novel strategies to inhibit CaMKII-, oxidative and/or NLRP3-inflammasome-related signaling might provide new therapeutic approaches for AF prevention and management. The development of novel, multispecific drug approaches provides a potentially innovative way to specifically target signaling systems of interest.160
Gene-regulatory Networks
Many gene-regulatory systems contribute to AF-related atrial remodeling in canine models and AF-patients.161,162 Epigenetic and transcriptional networks underlying AF are reviewed in another review article of this compendium.163 Here, we provide for completeness a short summary of key findings, which are schematically depicted in Figure 7.
Figure 7. Atrial fibrillation-promoting gene-regulatory networks.
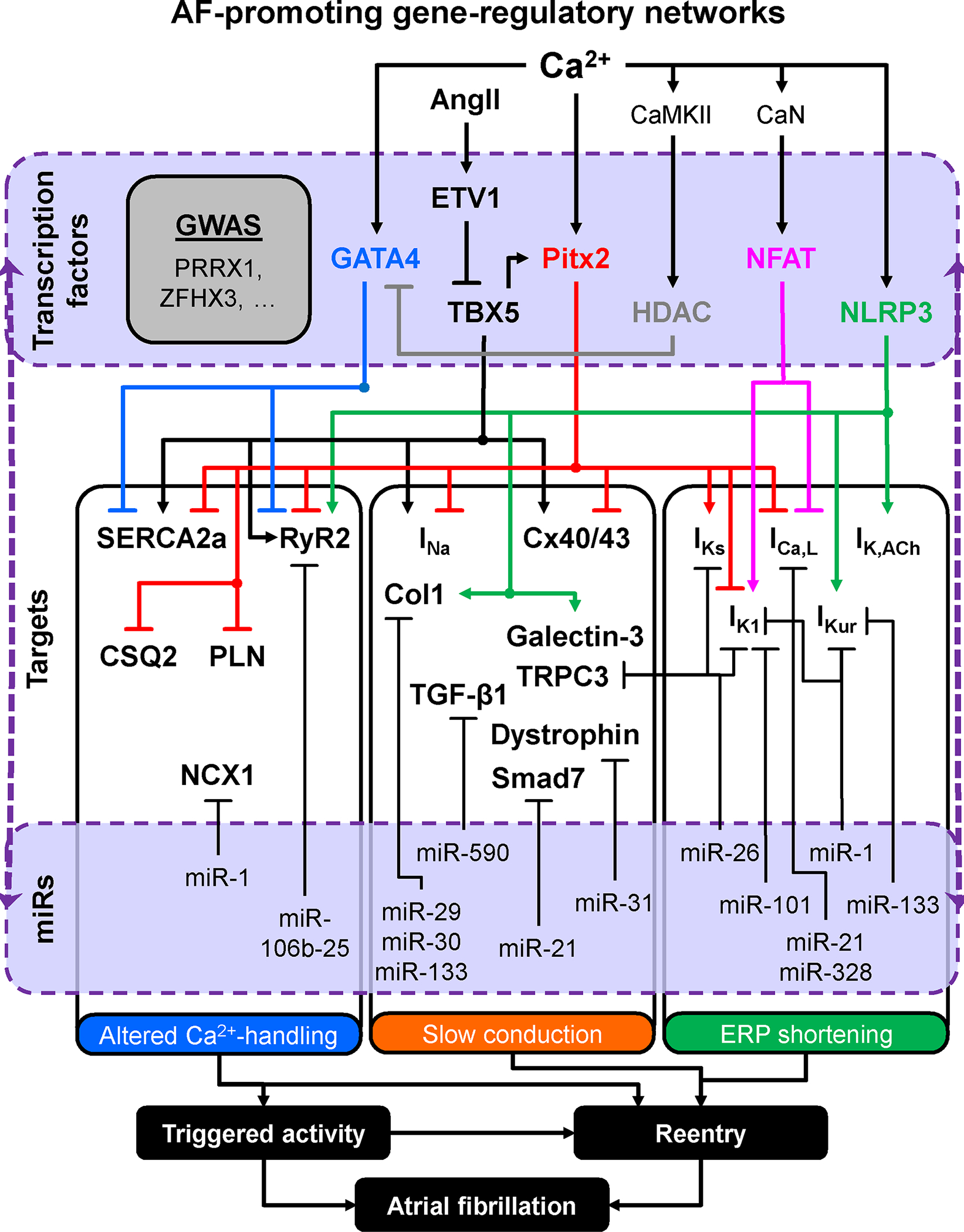
Multiple gene-regulatory networks interact to fine-tune the expression levels of key proteins shaping the effective refractory period (ERP), enabling proper impulse conduction and governing Ca2+-handling processes. Transcriptional alterations in the level of key regulator genes disrupt the network balance, increasing the likelihood of ERP, conduction and Ca2+-handling abnormalities, along with superimposed post-transcriptional changes due to abnormal microRNA (miR) function, leading to the formation of AF triggers and substrates. Abbreviations: Ang-II, angiotensin-II; CaMKII, Ca2+/calmodulin-dependent protein kinase-II; CaN, calcineurin; Col1, collagen-1; CSQ2, calsequestrin-2; Cx40/43, connexin 40/43; ETV1, ETS translocation variant 1 transcription factor; HDAC, histone deacetylase; ICa,L, L-type Ca2+-current; IK1, inward-rectifier K+-current; IK,ACh, acetylcholine-activated inward-rectifier K+-current; IKs, slow delayed-rectifier K+-current; IKur, ultra-rapid delayed-rectifier K+-current; INa, Na+-current; NCX1, Na+-Ca2+-exchanger type-1; NFAT, nuclear factor of activated T-cells; NLRP3, NACHT, LRR and PYD domains-containing protein-3; PITX2, paired Like Homeodomain 2; PLN, phopsholamban; RyR2, ryanodine receptor channel type-2; SERCA2a, SR Ca2+-ATPase type-2a; Smad7, mothers against decapentaplegic homolog 7; TBX5, T-box transcription factor 5; TGF-β1, transforming growth factor β1; TRPC3 transient-receptor potential channel canonical type-3.
Increased nuclear Ca2+-load during AF enhances nuclear CaMKII-phosphorylation and associated HDAC4 export in dogs,164 which is expected to upregulate TFs like Nkx2–5, GATA4 and MEF2c. TFs such as PITX2c and TBX5 are recognized contributors to AF susceptibility via gene-regulatory networks (Figure 7).165–168 TBX5-deficient mice show DAD- and EAD-mediated triggered activity, spontaneous diastolic depolarizations, prolonged APD and slowed atrial conduction, providing both arrhythmogenic triggers and a reentry-maintaining substrate.166 Reduced PITX2c rescues the arrhythmic phenotype.166 Conversely, low PITX2c levels in human atria are associated with Ca2+-handling abnormalities and cellular triggered activity,169 resembling those present in atrial cardiomyocytes of pAF patients,29 suggesting that low levels of either TBX5 or PITX2c could both lead to AF. GATA4 represses the transcription of SERCA2a and RyR2, while reductions in GATA4 eliminate the proarrhythmic phenotype of TBX5-deficient mice.170 ETV1, a TF negatively regulating TBX5, is upregulated in atria of cAF patients.171 Ang-II-driven increases in ETV1 in mice are associated with TBX5 reductions and AF development, mechanistically linking increased Ang-II signaling, abnormal gene-regulatory networks and AF.171 NLRP3 also acts as a transcriptional regulator in T-helper type-2 cells.172 Atrial mRNA levels of RyR2, Kv1.5 (IKur), Kir3.1/3.4 (IK,ACh), collagen-1a, and galectin-3 are all upregulated in mice with cardiomyocyte-restricted constitutive NLRP3 activation.74 Thus, NLRP3 may act as a transcription factor in cardiomyocytes, a concept to be investigated in future work. Finally, miRs control the expression of proteins involved in altered Ca2+-handling, APD-changes, and conduction slowing (reviewed in173,174), in some cases likely by modulating TF-expression and gene-regulatory networks.
The causative role of the putative interactions summarized in Figure 7 in AF pathophysiology remains unproven and for some genes both a decrease and an increase in function relate to AF.167,168 Contemporary approaches using large datasets, extensive genetic information and computational analyses offer new opportunities to identify novel risk variants and mechanisms for AF. A recent study highlighted novel systems and candidate-genes mediating effects on atrial electrophysiology, Ca2+-signaling and structure.175 Pathway and functional enrichment analyses highlighted pathways related to cardiac development; experiments in rabbits with left-atrial enlargement identified the role of a molecular switch from adult to fetal myosin heavy chain, causing contractile and functional heterogeneity that might predispose to AF.175
Clinical Perspectives
A number of molecular paradigms come up repeatedly as central nexuses in various types of AF-substrate development. Examples include CaMKII-activation, oxidative stress and inflammatory signaling, particularly related to the NLRP3-inflammasome. These might represent nodal points worth prioritizing as potential therapeutic targets. Several key gene-regulatory networks in AF have also been identified and subjected to intensive study, as discussed in detail elsewhere in this Compendium. The computational exploration of large genetic datasets coupled to clinical databases has the potential to reveal previously unsuspected novel mechanisms and targets.
Therapeutic Implications, Knowledge Gaps and Future Directions
Despite significant advances in AF-management with respect to antithrombotic therapy and catheter ablation, there remains an important need for improved treatment options.174,176 In particular, catheter ablation has limited efficacy in persistent AF (particularly when long-standing), cannot readily be applied to the millions of symptomatic AF patients worldwide and does not target the underlying AF-promoting atrial cardiomyopathy.107 Risk factor management may indirectly target the AF-promoting atrial cardiomyopathy and can reduce AF burden while increasing the success of rhythm control therapy,4 but intense risk-factor management is challenging to apply and is limited to specific subgroups of AF patients.
Recent advances in our understanding of AF pathophysiology and the identification of nodal players involved in AF-promoting atrial remodeling highlights potential opportunities for new therapeutic approaches. To be practical, any such approach must include absence of non-cardiac toxicity and lack of proarrhythmic liability, feasibility of chronic administration and the absence of negative influences on key homeostatic processes. The latter aspect may be particularly challenging for the key nodal regulators described above because of their key functions in numerous cell types and organ systems. For example, although CaMKII inhibition could be a promising anti-AF strategy, systemic inhibition of CaMKII may impair memory or fertility.177,178 The development of inhibitors that specifically target complex pathways via protein-protein interactions has been proposed for a long time, but this strategy is recently inching closer to practical clinical application.179 This may prove to be a feasible approach to targeting key nodal regulators while leaving their actions on other essential processes intact.180 Biologics have greatly improved the opportunities for specific targeting and are being developed to block inflammatory signaling, which plays a major role in AF.181 They may also prove to be of value in targeting other nodal actors. The major challenges here would be to design a delivery system that can produce stable expression and target it specifically to the atria,182 or at least the heart. Finally, multispecific drugs that combine targeting and effector moieties are increasingly being developed for the therapy of cancer and dyslipidemia, transplant-rejection prevention and management of hemophilia.160 Such compounds might allow for the development of cardiac-specific, possibly even atrial-specific, targeting of key molecular pathways.
Several other elements need to be considered when developing new AF therapies. Since AF is not immediately life-threatening, successful therapies need to be relatively conservative and safe, favoring small molecules or biologicals over complex genetic approaches. Furthermore, market realities including the size of clinical trials required to demonstrate improved clinical outcomes with a new therapeutic approach challenge the willingness to pursue new concepts. The repurposing of existing drugs might help to overcome these limitations. For example, low-dose colchicine significantly reduced the risk of ischemic cardiovascular events after myocardial infarction, possibly (at least partly) through inhibition of the NLRP3-inflammasome.183 Thus, targeting inflammation with low-dose colchicine or other already approved agents might be a viable approach to test the inflammatory hypothesis of AF in general and the putative involvement of the NLRP3 inflammasome in particular. Failure of resolution of inflammation is an emerging concept to explain persisting inflammation that underlies chronic illnesses and a variety of compounds have been identified that promote inflammation resolution. Resolvins are a group of non-toxic omega-3 fatty acid derivatives that promote inflammation-resolution, and one of these (resolvin D1) has shown efficacy in an animal model of AF associated with right heart disease.184 A related substance, icosapent ethyl, has been demonstrated to reduce cardiovascular risk in hypertriglyceridemic patients,185 and might be interesting to study in AF.
Thus, translational research has produced several promising avenues for improved AF therapy. Although it is at present not clear if any of these targets will ultimately be successful in clinical practice, one success story has the capacity to dramatically change AF management and spark substantial additional innovations. Further work is needed to clarify the details of AF-controlling molecular mechanisms and to develop practical approaches to intervening clinically to prevent the occurrence and progression of this important arrhythmia. Our discussion in this section is intended to illustrate the translational potential of the molecular mechanisms we discuss. For a more detailed discussion of challenges and trends in therapeutic development for AF, we refer readers to 2 detailed review articles in this Compendium.186,187
Conclusions
Our understanding of the molecular pathophysiology of AF has increased considerably over the past 10–15 years. Effective translation of this knowledge into practical approaches to taming the arrhythmia has the potential to lead to important advances in patient management, but major challenges in fully understanding and actively applying it need to be overcome for this potential to be realized.
Sources of Funding
The authors’ work is supported by the Netherlands Organization for Scientific Research (ZonMW Veni 91616057 to J.H.), the National Institutes of Health (R01-HL131517, R01-HL136389, and R01-HL089598 to D.D.), the German Research Foundation (DFG, Do 769/4-1 to D.D.), the Canadian Institutes of Health Research (Foundation Grant 148401 to S.N.) and the Heart and Stroke Foundation of Canada (16-12708 to S.N.).
Non-standard Abbreviations and Acronyms
- AF
Atrial fibrillation
- Ang-II
Angiotensin-II
- AP
Action potential
- APD
Action potential duration
- cAF
Persistent (chronic) atrial fibrillation
- CaMKII
Ca2+/calmodulin-dependent protein kinase-II
- cAMP
Cyclic adenosine monophosphate
- Cx
Connexin
- DAD
Delayed afterdepolarization
- EAD
Early afterdepolarization
- HCN channel
Hyperpolarization-activated cyclic nucleotide-gated channel
- HDAC
Histone deacetylases
- HF
Heart failure
- ICa,L
L-type Ca2+-current
- IK1
Basal inward-rectifier K+-current
- IK,ACh
Acetylcholine-activated inward-rectifier K+-current
- IKur
Ultra-rapid delayed-rectifier K+-current
- IL
Interleukin
- INa
Fast Na+-current
- INa,late
Persistent/late Na+-current
- miR
MicroRNA
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NCX1
Na+-Ca2+-exchanger type-1
- NFAT
Nuclear factor of activated T-cells
- NLRP3
NACHT, LRR and PYD domains-containing protein-3
- pAF
Paroxysmal AF
- PKA
Protein kinase-A
- PP1
Protein phosphatase type-1
- PVs
Pulmonary veins
- RyR2
Ryanodine receptor channel type-2
- ROS
Reactive oxygen species
- SAN
Sinoatrial node
- SCaEs
Spontaneous Ca2+-release events
- SERCA2a
SR Ca2+-ATPase type-2a
- SR
Sarcoplasmic reticulum
- TCW
Triggered Ca2+ waves
- TFs
Transcription factors
- TRP
Transient receptor potential
Footnotes
Disclosures
Dobromir Dobrev is a member of Scientific Advisory Boards of OMEICOS Therapeutics, Acesion Pharma and Sanofi and received speaker’s fees for educational lectures from Boston Scientific, Novartis and Bristol-Myers Squibb. His laboratory executed research contracts for OMEICOS. Stanley Nattel served on the Global Advisory Board of Amgen between 2015 and 2019 and is a consultant for Novint Technologies and LQT Therapeutics.
References
- 1.Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. 2014;114:1453–1468 [DOI] [PubMed] [Google Scholar]
- 2.Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S. The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovasc Res. 2016;109:467–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nattel S, Guasch E, Savelieva I, et al. Early management of atrial fibrillation to prevent cardiovascular complications. Eur Heart J. 2014;35:1448–1456 [DOI] [PubMed] [Google Scholar]
- 4.Lau DH, Nattel S, Kalman JM, Sanders P. Modifiable Risk Factors and Atrial Fibrillation. Circulation. 2017;136:583–596 [DOI] [PubMed] [Google Scholar]
- 5.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968 [DOI] [PubMed] [Google Scholar]
- 6.Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. 2014;63:2335–2345 [DOI] [PubMed] [Google Scholar]
- 7.Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666 [DOI] [PubMed] [Google Scholar]
- 8.Ehrlich JR, Cha TJ, Zhang L, Chartier D, Melnyk P, Hohnloser SH, Nattel S. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol. 2003;551:801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hocini M, Ho SY, Kawara T, Linnenbank AC, Potse M, Shah D, Jais P, Janse MJ, Haissaguerre M, De Bakker JM. Electrical conduction in canine pulmonary veins: electrophysiological and anatomic correlation. Circulation. 2002;105:2442–2448 [DOI] [PubMed] [Google Scholar]
- 10.Teh AW, Kistler PM, Lee G, Medi C, Heck PM, Spence S, Morton JB, Sanders P, Kalman JM. Electroanatomic properties of the pulmonary veins: slowed conduction, low voltage and altered refractoriness in AF patients. J Cardiovasc Electrophysiol. 2011;22:1083–1091 [DOI] [PubMed] [Google Scholar]
- 11.Lemola K, Chartier D, Yeh YH, Dubuc M, Cartier R, Armour A, Ting M, Sakabe M, Shiroshita-Takeshita A, Comtois P, Nattel S. Pulmonary vein region ablation in experimental vagal atrial fibrillation: role of pulmonary veins versus autonomic ganglia. Circulation. 2008;117:470–477 [DOI] [PubMed] [Google Scholar]
- 12.Saburkina I, Rysevaite K, Pauziene N, Mischke K, Schauerte P, Jalife J, Pauza DH. Epicardial neural ganglionated plexus of ovine heart: anatomic basis for experimental cardiac electrophysiology and nerve protective cardiac surgery. Heart Rhythm. 2010;7:942–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol. 2016;13:575–590 [DOI] [PubMed] [Google Scholar]
- 14.Malcolme-Lawes LC, Juli C, Karim R, et al. Automated analysis of atrial late gadolinium enhancement imaging that correlates with endocardial voltage and clinical outcomes: a 2-center study. Heart Rhythm. 2013;10:1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vincenti A, Brambilla R, Fumagalli MG, Merola R, Pedretti S. Onset mechanism of paroxysmal atrial fibrillation detected by ambulatory Holter monitoring. Europace. 2006;8:204–210 [DOI] [PubMed] [Google Scholar]
- 16.Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, Nishio Y, Sasaki K, Itoh H, Yokode M, Kita T, Horie M, Kimura T. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. 2012;93:666–673 [DOI] [PubMed] [Google Scholar]
- 17.Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunso S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. 2012;13:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, Cerrone M, Noujaim SF, Denegri M, Jalife J, Priori SG. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci U S A. 2013;110:4291–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:394–396 [DOI] [PubMed] [Google Scholar]
- 20.Lubkemeier I, Andrie R, Lickfett L, Bosen F, Stockigt F, Dobrowolski R, Draffehn AM, Fregeac J, Schultze JL, Bukauskas FF, Schrickel JW, Willecke K. The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J Mol Cell Cardiol. 2013;65:19–32 [DOI] [PubMed] [Google Scholar]
- 21.Li D, Fareh S, Leung TK, Nattel S. Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation. 1999;100:87–95 [DOI] [PubMed] [Google Scholar]
- 22.Mohanty S, Mohanty P, Di Biase L, et al. Long-term follow-up of patients with paroxysmal atrial fibrillation and severe left atrial scarring: comparison between pulmonary vein antrum isolation only or pulmonary vein isolation combined with either scar homogenization or trigger ablation. Europace. 2017;19:1790–1797 [DOI] [PubMed] [Google Scholar]
- 23.Kazemian P, Gollob MH, Pantano A, Oudit GY. A novel mutation in the RYR2 gene leading to catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation: dose-dependent arrhythmia-event suppression by beta-blocker therapy. Can J Cardiol. 2011;27:870 e877–810 [DOI] [PubMed] [Google Scholar]
- 24.Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ, Wehrens XHT. Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. J Am Coll Cardiol. 2013;62:2010–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N. Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol. 2014;7:1214–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benito B, Brugada R, Perich RM, Lizotte E, Cinca J, Mont L, Berruezo A, Tolosana JM, Freixa X, Brugada P, Brugada J. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm. 2008;5:1434–1440 [DOI] [PubMed] [Google Scholar]
- 27.Lemoine MD, Duverger JE, Naud P, Chartier D, Qi XY, Comtois P, Fabritz L, Kirchhof P, Nattel S. Arrhythmogenic left atrial cellular electrophysiology in a murine genetic long QT syndrome model. Cardiovasc Res. 2011;92:67–74 [DOI] [PubMed] [Google Scholar]
- 28.Li YD, Hong YF, Yusufuaji Y, Tang BP, Zhou XH, Xu GJ, Li JX, Sun L, Zhang JH, Xin Q, Xiong J, Ji YT, Zhang Y. Altered expression of hyperpolarization-activated cyclic nucleotide-gated channels and microRNA-1 and -133 in patients with age-associated atrial fibrillation. Mol Med Rep. 2015;12:3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129:145–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voigt N, Trausch A, Knaut M, Matschke K, Varro A, Van Wagoner DR, Nattel S, Ravens U, Dobrev D. Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:472–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayan SM, Franz MR, Clopton P, Pruvot EJ, Krummen DE. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation. 2011;123:2922–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearman CM, Madders GWP, Radcliffe EJ, Kirkwood GJ, Lawless M, Watkins A, Smith CER, Trafford AW, Eisner DA, Dibb KM. Increased Vulnerability to Atrial Fibrillation Is Associated With Increased Susceptibility to Alternans in Old Sheep. J Am Heart Assoc. 2018;7:e009972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu T, Xiong F, Qi XY, Xiao J, Villeneuve L, Abu-Taha I, Dobrev D, Huang C, Nattel S. Altered calcium handling produces reentry-promoting action potential alternans in atrial fibrillation-remodeled hearts. JCI Insight. 2020;5:133754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol Heart Circ Physiol. 2011;301:H1996–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brandenburg S, Kohl T, Williams GS, et al. Axial tubule junctions control rapid calcium signaling in atria. J Clin Invest. 2016;126:3999–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandenburg S, Pawlowitz J, Fakuade FE, et al. Axial Tubule Junctions Activate Atrial Ca2+ Release Across Species. Front Physiol. 2018;9:1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu MB, de Lange E, Garfinkel A, Weiss JN, Qu Z. Delayed afterdepolarizations generate both triggers and a vulnerable substrate promoting reentry in cardiac tissue. Heart Rhythm. 2015;12:2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiferaw Y, Aistrup GL, Louch WE, Wasserstrom JA. Remodeling Promotes Proarrhythmic Disruption of Calcium Homeostasis in Failing Atrial Myocytes. Biophys J. 2020;118:476–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aistrup GL, Arora R, Grubb S, et al. Triggered intracellular calcium waves in dog and human left atrial myocytes from normal and failing hearts. Cardiovasc Res. 2017;113:1688–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Potenza DM, Janicek R, Fernandez-Tenorio M, Niggli E. Activation of endogenous protein phosphatase 1 enhances the calcium sensitivity of the ryanodine receptor type 2 in murine ventricular cardiomyocytes. J Physiol. 2020;598:1131–1150 [DOI] [PubMed] [Google Scholar]
- 41.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Guo T, Oda T, Chakraborty A, Chen L, Uchinoumi H, Knowlton AA, Fruen BR, Cornea RL, Meissner G, Bers DM. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ Res. 2014;114:295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D, Wehrens XH. Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circ Res. 2012;110:465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032 [DOI] [PubMed] [Google Scholar]
- 45.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144 [DOI] [PubMed] [Google Scholar]
- 47.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landstrom AP, Dobrev D, Wehrens XHT. Calcium Signaling and Cardiac Arrhythmias. Circ Res. 2017;120:1969–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoo S, Aistrup G, Shiferaw Y, Ng J, Mohler PJ, Hund TJ, Waugh T, Browne S, Gussak G, Gilani M, Knight BP, Passman R, Goldberger JJ, Wasserstrom JA, Arora R. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010;48:322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiang DY, Li N, Wang Q, Alsina KM, Quick AP, Reynolds JO, Wang G, Skapura D, Voigt N, Dobrev D, Wehrens XH. Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovasc Res. 2014;103:178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alsina KM, Hulsurkar M, Brandenburg S, et al. Loss of Protein Phosphatase 1 Regulatory Subunit PPP1R3A Promotes Atrial Fibrillation. Circulation. 2019;140:681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–680 [DOI] [PubMed] [Google Scholar]
- 55.Chiang DY, Lebesgue N, Beavers DL, Alsina KM, Damen JM, Voigt N, Dobrev D, Wehrens XH, Scholten A. Alterations in the interactome of serine/threonine protein phosphatase type-1 in atrial fibrillation patients. J Am Coll Cardiol. 2015;65:163–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer-Roxlau S, Lammle S, Opitz A, Kunzel S, Joos JP, Neef S, Sekeres K, Sossalla S, Schondube F, Alexiou K, Maier LS, Dobrev D, Guan K, Weber S, El-Armouche A. Differential regulation of protein phosphatase 1 (PP1) isoforms in human heart failure and atrial fibrillation. Basic Res Cardiol. 2017;112:43. [DOI] [PubMed] [Google Scholar]
- 57.Grandi E, Pandit SV, Voigt N, Workman AJ, Dobrev D, Jalife J, Bers DM. Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ Res. 2011;109:1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Song Y, Shryock JC, Belardinelli L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H2031–2039 [DOI] [PubMed] [Google Scholar]
- 59.Greer-Short A, Musa H, Alsina KM, et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm. 2020;17:503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lebek S, Pichler K, Reuthner K, Trum M, Tafelmeier M, Mustroph J, Camboni D, Rupprecht L, Schmid C, Maier LS, Arzt M, Wagner S. Enhanced CaMKII-Dependent Late INa Induces Atrial Proarrhythmic Activity in Patients With Sleep-Disordered Breathing. Circ Res. 2020;126:603–615 [DOI] [PubMed] [Google Scholar]
- 61.Poulet C, Wettwer E, Grunnet M, Jespersen T, Fabritz L, Matschke K, Knaut M, Ravens U. Late Sodium Current in Human Atrial Cardiomyocytes from Patients in Sinus Rhythm and Atrial Fibrillation. PLoS One. 2015;10:e0131432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363 [DOI] [PubMed] [Google Scholar]
- 63.Molina CE, Abu-Taha IH, Wang Q, Rosello-Diez E, Kamler M, Nattel S, Ravens U, Wehrens XHT, Hove-Madsen L, Heijman J, Dobrev D. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients With and Without Atrial Fibrillation. Front Physiol. 2018;9:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen PS, Chen LS, Fishbein MC, Lin SF, Nattel S. Role of the autonomic nervous system in atrial fibrillation: pathophysiology and therapy. Circ Res. 2014;114:1500–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pereira L, Bare DJ, Galice S, Shannon TR, Bers DM. beta-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J Mol Cell Cardiol. 2017;108:8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neef S, Heijman J, Otte K, Dewenter M, Saadatmand AR, Meyer-Roxlau S, Antos CL, Backs J, Dobrev D, Wagner M, Maier LS, El-Armouche A. Chronic loss of inhibitor-1 diminishes cardiac RyR2 phosphorylation despite exaggerated CaMKII activity. Naunyn Schmiedebergs Arch Pharmacol. 2017;390:857–862 [DOI] [PubMed] [Google Scholar]
- 67.Chen WJ, Chang SH, Chan YH, Lee JL, Lai YJ, Chang GJ, Tsai FC, Yeh YH. Tachycardia-induced CD44/NOX4 signaling is involved in the development of atrial remodeling. J Mol Cell Cardiol. 2019;135:67–78 [DOI] [PubMed] [Google Scholar]
- 68.Purohit A, Rokita AG, Guan X, et al. Oxidized Ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation. 2013;128:1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang X, An N, Zhong C, Guan M, Jiang Y, Li X, Zhang H, Wang L, Ruan Y, Gao Y, Liu N, Shang H, Xing Y. Enhanced cardiomyocyte reactive oxygen species signaling promotes ibrutinib-induced atrial fibrillation. Redox Biol. 2020;30:101432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, Chen B, Wu X, Ginsburg KS, Bakhos M, Bers DM, Anderson ME, Song LS, Fill M, Ai X. Stress Signaling JNK2 Crosstalk With CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ Res. 2018;122:821–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gao X, Wu X, Yan J, Zhang J, Zhao W, DeMarco D, Zhang Y, Bakhos M, Mignery G, Sun J, Li Z, Fill M, Ai X. Transcriptional regulation of stress kinase JNK2 in proarrhythmic CaMKIIdelta expression in the aged atrium. Cardiovasc Res. 2018;114:737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan J, Thomson JK, Zhao W, Gao X, Huang F, Chen B, Liang Q, Song LS, Fill M, Ai X. Role of Stress Kinase JNK in Binge Alcohol-Evoked Atrial Arrhythmia. J Am Coll Cardiol. 2018;71:1459–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scott L Jr., Li N, Dobrev D Role of inflammatory signaling in atrial fibrillation. Int J Cardiol 2019;287:195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yao C, Veleva T, Scott L Jr., et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138:2227–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heijman J, Muna AP, Veleva T, et al. Cardiomyocyte NLRP3-inflammatory and Calcium/Calmodulin-dependent Kinase II Signaling Create a Cellular Substrate for Post-operative Atrial Fibrillation. Circ Res. 2020;Under Revision [Google Scholar]
- 76.Fender AC, Kleeschulte S, Stolte S, Leineweber K, Kamler M, Bode J, Li N, Dobrev D. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res Cardiol. 2020;115:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu X, Liu Y, Tu D, Liu X, Niu S, Suo Y, Liu T, Li G, Liu C. Role of NLRP3-Inflammasome/Caspase-1/Galectin-3 Pathway on Atrial Remodeling in Diabetic Rabbits. J Cardiovasc Transl Res. 2020 [DOI] [PubMed] [Google Scholar]
- 78.Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM. TNF-a and IL-1b increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium. 2010;47:378–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Monnerat G, Alarcon ML, Vasconcellos LR, et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun. 2016;7:13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018;138:2530–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIId-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight. 2018;3:e97054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao H, Li H, Wang JJ, et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J. 2018;39:60–69 [DOI] [PubMed] [Google Scholar]
- 83.Qiu Z, He Y, Ming H, Lei S, Leng Y, Xia ZY. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J Diabetes Res. 2019;2019:8151836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Killeen MJ, Sabir IN, Grace AA, Huang CL. Dispersions of repolarization and ventricular arrhythmogenesis: lessons from animal models. Prog Biophys Mol Biol. 2008;98:219–229 [DOI] [PubMed] [Google Scholar]
- 85.Avula UMR, Abrams J, Katchman A, et al. Heterogeneity of the action potential duration is required for sustained atrial fibrillation. JCI Insight. 2019;5:e128765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, Hedley PL, Christiansen M, Olesen SP, Haunso S, Schmitt N, Jespersen T, Svendsen JH. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ Cardiovasc Genet. 2012;5:450–459 [DOI] [PubMed] [Google Scholar]
- 87.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest. 2016;126:112–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pezhouman A, Cao H, Fishbein MC, Belardinelli L, Weiss JN, Karagueuzian HS. Atrial Fibrillation Initiated by Early Afterdepolarization-Mediated Triggered Activity during Acute Oxidative Stress: Efficacy of Late Sodium Current Blockade. J Heart Health. 2018;4: 10.16966/12379-16769X.16146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yaniv Y, Lakatta EG, Maltsev VA. From two competing oscillators to one coupled-clock pacemaker cell system. Front Physiol. 2015;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li YD, Hong YF, Zhang Y, Zhou XH, Ji YT, Li HL, Hu GJ, Li JX, Sun L, Zhang JH, Xin Q, Yusufuaji Y, Xiong J, Tang BP. Association between reversal in the expression of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel and age-related atrial fibrillation. Med Sci Monit. 2014;20:2292–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stillitano F, Lonardo G, Zicha S, Varro A, Cerbai E, Mugelli A, Nattel S. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299 [DOI] [PubMed] [Google Scholar]
- 92.Wang TM, Luk HN, Sheu JR, Wu HP, Chiang CE. Inducibility of abnormal automaticity and triggered activity in myocardial sleeves of canine pulmonary veins. Int J Cardiol. 2005;104:59–66 [DOI] [PubMed] [Google Scholar]
- 93.Jadidi A, Muller-Edenborn B, Chen J, Keyl C, Weber R, Allgeier J, Moreno-Weidmann Z, Trenk D, Neumann FJ, Lehrmann H, Arentz T. The Duration of the Amplified Sinus-P-Wave Identifies Presence of Left Atrial Low Voltage Substrate and Predicts Outcome After Pulmonary Vein Isolation in Patients With Persistent Atrial Fibrillation. JACC Clin Electrophysiol. 2018;4:531–543 [DOI] [PubMed] [Google Scholar]
- 94.Muller-Edenborn B, Minners J, Kocher S, Chen J, Zeh W, Lehrmann H, Allgeier J, Neumann FJ, Arentz T, Jadidi A. Amplified P-wave duration predicts new-onset atrial fibrillation in patients with heart failure with preserved ejection fraction. Clin Res Cardiol. 2019; in press [DOI] [PubMed] [Google Scholar]
- 95.Marrouche NF, Wilber D, Hindricks G, et al. Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA. 2014;311:498–506 [DOI] [PubMed] [Google Scholar]
- 96.Nattel S Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol. 2017;3:425–435 [DOI] [PubMed] [Google Scholar]
- 97.Simon JN, Ziberna K, Casadei B. Compromised redox homeostasis, altered nitroso-redox balance, and therapeutic possibilities in atrial fibrillation. Cardiovasc Res. 2016;109:510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation. 2001;104:2608–2614 [DOI] [PubMed] [Google Scholar]
- 99.Qiu H, Ji C, Liu W, Wu Y, Lu Z, Lin Q, Xue Z, Liu X, Wu H, Jiang W, Zou C. Chronic Kidney Disease Increases Atrial Fibrillation Inducibility: Involvement of Inflammation, Atrial Fibrosis, and Connexins. Front Physiol. 2018;9:1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Qiu H, Liu W, Lan T, Pan W, Chen X, Wu H, Xu D. Salvianolate reduces atrial fibrillation through suppressing atrial interstitial fibrosis by inhibiting TGF-beta1/Smad2/3 and TXNIP/NLRP3 inflammasome signaling pathways in post-MI rats. Phytomedicine. 2018;51:255–265 [DOI] [PubMed] [Google Scholar]
- 101.Harada M, Luo X, Qi XY, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qi XY, Huang H, Ordog B, Luo X, Naud P, Sun Y, Wu CT, Dawson K, Tadevosyan A, Chen Y, Harada M, Dobrev D, Nattel S. Fibroblast inward-rectifier potassium current upregulation in profibrillatory atrial remodeling. Circ Res. 2015;116:836–845 [DOI] [PubMed] [Google Scholar]
- 103.Chung CC, Lin YK, Chen YC, Kao YH, Lee TI, Chen YJ. Vascular endothelial growth factor enhances profibrotic activities through modulation of calcium homeostasis in human atrial fibroblasts. Lab Invest. 2020;100:285–296 [DOI] [PubMed] [Google Scholar]
- 104.Rocken C, Peters B, Juenemann G, Saeger W, Klein HU, Huth C, Roessner A, Goette A. Atrial amyloidosis: an arrhythmogenic substrate for persistent atrial fibrillation. Circulation. 2002;106:2091–2097 [DOI] [PubMed] [Google Scholar]
- 105.Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, Amour J, Leprince P, Dutour A, Clement K, Hatem SN. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur Heart J. 2015;36:795–805a [DOI] [PubMed] [Google Scholar]
- 106.Haemers P, Hamdi H, Guedj K, Suffee N, Farahmand P, Popovic N, Claus P, LePrince P, Nicoletti A, Jalife J, Wolke C, Lendeckel U, Jais P, Willems R, Hatem SN. Atrial fibrillation is associated with the fibrotic remodelling of adipose tissue in the subepicardium of human and sheep atria. Eur Heart J. 2017;38:53–61 [DOI] [PubMed] [Google Scholar]
- 107.Goette A, Kalman JM, Aguinaga L, et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Heart Rhythm. 2017;14:e3–e40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Herum KM, Lunde IG, McCulloch AD, Christensen G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J Clin Med. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kurtenbach S, Kurtenbach S, Zoidl G. Gap junction modulation and its implications for heart function. Front Physiol. 2014;5:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wirka RC, Gore S, Van Wagoner DR, Arking DE, Lubitz SA, Lunetta KL, Benjamin EJ, Alonso A, Ellinor PT, Barnard J, Chung MK, Smith JD. A common connexin-40 gene promoter variant affects connexin-40 expression in human atria and is associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2011;4:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bai D Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. 2014;588:1238–1243 [DOI] [PubMed] [Google Scholar]
- 112.Kanthan A, Fahmy P, Rao R, Pouliopoulos J, Alexander IE, Thomas SP, Kizana E. Human Connexin40 Mutations Slow Conduction and Increase Propensity for Atrial Fibrillation. Heart Lung Circ. 2018;27:114–121 [DOI] [PubMed] [Google Scholar]
- 113.Gemel J, Levy AE, Simon AR, Bennett KB, Ai X, Akhter S, Beyer EC. Connexin40 abnormalities and atrial fibrillation in the human heart. J Mol Cell Cardiol. 2014;76:159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.van der Velden HM, van Kempen MJ, Wijffels MC, van Zijverden M, Groenewegen WA, Allessie MA, Jongsma HJ. Altered pattern of connexin40 distribution in persistent atrial fibrillation in the goat. J Cardiovasc Electrophysiol. 1998;9:596–607 [DOI] [PubMed] [Google Scholar]
- 115.Iwasaki YK, Kato T, Xiong F, Shi YF, Naud P, Maguy A, Mizuno K, Tardif JC, Comtois P, Nattel S. Atrial fibrillation promotion with long-term repetitive obstructive sleep apnea in a rat model. J Am Coll Cardiol. 2014;64:2013–2023 [DOI] [PubMed] [Google Scholar]
- 116.Lazzerini PE, Laghi-Pasini F, Acampa M, et al. Systemic Inflammation Rapidly Induces Reversible Atrial Electrical Remodeling: The Role of Interleukin-6-Mediated Changes in Connexin Expression. J Am Heart Assoc 2019;8:e011006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, Fast VG, Ai X. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res. 2013;97:589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, Nattel S. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res. 2009;105:1213–1222 [DOI] [PubMed] [Google Scholar]
- 119.Jansen JA, van Veen TA, de Jong S, van der Nagel R, van Stuijvenberg L, Driessen H, Labzowski R, Oefner CM, Bosch AA, Nguyen TQ, Goldschmeding R, Vos MA, de Bakker JM, van Rijen HV. Reduced Cx43 expression triggers increased fibrosis due to enhanced fibroblast activity. Circ Arrhythm Electrophysiol. 2012;5:380–390 [DOI] [PubMed] [Google Scholar]
- 120.Pogoda K, Kameritsch P, Retamal MA, Vega JL. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: a revision. BMC Cell Biol. 2016;17 Suppl 1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bikou O, Thomas D, Trappe K, Lugenbiel P, Kelemen K, Koch M, Soucek R, Voss F, Becker R, Katus HA, Bauer A. Connexin 43 gene therapy prevents persistent atrial fibrillation in a porcine model. Cardiovasc Res. 2011;92:218–225 [DOI] [PubMed] [Google Scholar]
- 122.Igarashi T, Finet JE, Takeuchi A, Fujino Y, Strom M, Greener ID, Rosenbaum DS, Donahue JK. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation. 2012;125:216–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shiroshita-Takeshita A, Sakabe M, Haugan K, Hennan JK, Nattel S. Model-dependent effects of the gap junction conduction-enhancing antiarrhythmic peptide rotigaptide (ZP123) on experimental atrial fibrillation in dogs. Circulation. 2007;115:310–318 [DOI] [PubMed] [Google Scholar]
- 124.Gaspo R, Bosch RF, Bou-Abboud E, Nattel S. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ Res. 1997;81:1045–1052 [DOI] [PubMed] [Google Scholar]
- 125.Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131 [DOI] [PubMed] [Google Scholar]
- 126.Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schondube FA, Hasenfuss G, Belardinelli L, Maier LS. Altered Na+ currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–2342 [DOI] [PubMed] [Google Scholar]
- 127.Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114:1483–1499 [DOI] [PubMed] [Google Scholar]
- 128.Blum S, Aeschbacher S, Meyre P, et al. Incidence and Predictors of Atrial Fibrillation Progression. J Am Heart Assoc. 2019;8:e012554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sun H, Chartier D, Leblanc N, Nattel S. Intracellular calcium changes and tachycardia-induced contractile dysfunction in canine atrial myocytes. Cardiovasc Res. 2001;49:751–761 [DOI] [PubMed] [Google Scholar]
- 130.Guichard JB, Xiong F, Qi XY, L’Heureux N, Hiram R, Xiao J, Naud P, Tardif JC, Costa AD, Nattel S. Role of atrial arrhythmia and ventricular response in atrial fibrillation induced atrial remodeling. Cardiovasc Res. 2020 [DOI] [PubMed] [Google Scholar]
- 131.Wakili R, Yeh YH, Yan Qi X, et al. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol. 2010;3:530–541 [DOI] [PubMed] [Google Scholar]
- 132.Greiser M, Kerfant BG, Williams GS, et al. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest. 2014;124:4759–4772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lenski M, Schleider G, Kohlhaas M, Adrian L, Adam O, Tian Q, Kaestner L, Lipp P, Lehrke M, Maack C, Bohm M, Laufs U. Arrhythmia causes lipid accumulation and reduced glucose uptake. Basic Res Cardiol. 2015;110:40. [DOI] [PubMed] [Google Scholar]
- 134.Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ Res. 2008;103:845–854 [DOI] [PubMed] [Google Scholar]
- 135.Luo X, Pan Z, Shan H, et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest. 2013;123:1939–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706 [DOI] [PubMed] [Google Scholar]
- 137.Cha TJ, Ehrlich JR, Chartier D, Qi XY, Xiao L, Nattel S. Kir3-based inward rectifier potassium current: potential role in atrial tachycardia remodeling effects on atrial repolarization and arrhythmias. Circulation. 2006;113:1730–1737 [DOI] [PubMed] [Google Scholar]
- 138.Pandit SV, Berenfeld O, Anumonwo JM, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Zhang Y, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–2387 [DOI] [PubMed] [Google Scholar]
- 140.Makary S, Voigt N, Maguy A, Wakili R, Nishida K, Harada M, Dobrev D, Nattel S. Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ Res. 2011;109:1031–1043 [DOI] [PubMed] [Google Scholar]
- 141.Schmidt C, Wiedmann F, Voigt N, et al. Upregulation of K2P3.1 K+ Current Causes Action Potential Shortening in Patients With Chronic Atrial Fibrillation. Circulation. 2015;132:82–92 [DOI] [PubMed] [Google Scholar]
- 142.Schmidt C, Wiedmann F, Zhou XB, et al. Inverse remodelling of K2P3.1 K+ channel expression and action potential duration in left ventricular dysfunction and atrial fibrillation: implications for patient-specific antiarrhythmic drug therapy. Eur Heart J. 2017;38:1764–1774 [DOI] [PubMed] [Google Scholar]
- 143.Burstein B, Qi XY, Yeh YH, Calderone A, Nattel S. Atrial cardiomyocyte tachycardia alters cardiac fibroblast function: a novel consideration in atrial remodeling. Cardiovasc Res. 2007;76:442–452 [DOI] [PubMed] [Google Scholar]
- 144.Tsai CT, Tseng CD, Hwang JJ, Wu CK, Yu CC, Wang YC, Chen WP, Lai LP, Chiang FT, Lin JL. Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor b1. Cardiovasc Res. 2011;89:805–815 [DOI] [PubMed] [Google Scholar]
- 145.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nattel S, Dobrev D. The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J. 2012;33:1870–1877 [DOI] [PubMed] [Google Scholar]
- 148.Li N, Chiang DY, Wang S, et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. 2014;129:1276–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dewenter M, von der Lieth A, Katus HA, Backs J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ Res. 2017;121:1000–1020 [DOI] [PubMed] [Google Scholar]
- 150.Mesubi OO, Anderson ME. Atrial remodelling in atrial fibrillation: CaMKII as a nodal proarrhythmic signal. Cardiovasc Res. 2016;109:542–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102 [DOI] [PubMed] [Google Scholar]
- 152.Korantzopoulos P, Letsas K, Fragakis N, Tse G, Liu T. Oxidative stress and atrial fibrillation: an update. Free Radic Res. 2018;52:1199–1209 [DOI] [PubMed] [Google Scholar]
- 153.Sovari AA, Dudley SC Jr. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front Physiol 2012;3:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bukowska A, Schild L, Keilhoff G, Hirte D, Neumann M, Gardemann A, Neumann KH, Rohl FW, Huth C, Goette A, Lendeckel U. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp Biol Med (Maywood). 2008;233:558–574 [DOI] [PubMed] [Google Scholar]
- 155.Dudley SC Jr., Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, Harrison DG, Dikalov SI, Langberg J Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation. 2005;112:1266–1273 [DOI] [PubMed] [Google Scholar]
- 156.Cai H, Li Z, Goette A, Mera F, Honeycutt C, Feterik K, Wilcox JN, Dudley SC Jr., Harrison DG, Langberg JJ Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation. 2002;106:2854–2858 [DOI] [PubMed] [Google Scholar]
- 157.Goette A, Bukowska A, Dobrev D, Pfeiffenberger J, Morawietz H, Strugala D, Wiswedel I, Rohl FW, Wolke C, Bergmann S, Bramlage P, Ravens U, Lendeckel U. Acute atrial tachyarrhythmia induces angiotensin II type 1 receptor-mediated oxidative stress and microvascular flow abnormalities in the ventricles. Eur Heart J. 2009;30:1411–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Matsushita N, Ishida N, Ibi M, Saito M, Takahashi M, Taniguchi S, Iwakura Y, Morino Y, Taira E, Sawa Y, Hirose M. IL-1beta Plays an Important Role in Pressure Overload-Induced Atrial Fibrillation in Mice. Biol Pharm Bull. 2019;42:543–546 [DOI] [PubMed] [Google Scholar]
- 159.Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Deshaies RJ. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature. 2020;580:329–338 [DOI] [PubMed] [Google Scholar]
- 161.Gaborit N, Steenman M, Lamirault G, Le Meur N, Le Bouter S, Lande G, Leger J, Charpentier F, Christ T, Dobrev D, Escande D, Nattel S, Demolombe S. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation. 2005;112:471–481 [DOI] [PubMed] [Google Scholar]
- 162.Cardin S, Libby E, Pelletier P, Le Bouter S, Shiroshita-Takeshita A, Le Meur N, Leger J, Demolombe S, Ponton A, Glass L, Nattel S. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res. 2007;100:425–433 [DOI] [PubMed] [Google Scholar]
- 163.Christoffels V, Moskowitz IP, Martin JF, Kirchhof P, de Laat W, Ellinor P. Epigenetic and transcriptional networks underlying AF. Circ Res. 2020;[AF Compendium] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Qi X, Xiao J, Xiong F, Sobue Y, Villeneuve LR, Ljubojevic S, Bers DM, Dobrev D, Nattel S. Abstract 12029: Nuclear IP3 Receptor Knockdown Reveals a Key Role of IP3 Receptor Upregulation in Disrupted Nuclear Calcium-Dependent Signaling in Atrial Fibrillation. Circulation. 2018;138:A12029–A12029 [Google Scholar]
- 165.Roselli C, Chaffin MD, Weng LC, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nadadur RD, Broman MT, Boukens B, et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci Transl Med. 2016;8:354ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lozano-Velasco E, Hernandez-Torres F, Daimi H, Serra SA, Herraiz A, Hove-Madsen L, Aranega A, Franco D. Pitx2 impairs calcium handling in a dose-dependent manner by modulating Wnt signalling. Cardiovasc Res. 2016;109:55–66 [DOI] [PubMed] [Google Scholar]
- 168.Perez-Hernandez M, Matamoros M, Barana A, Amoros I, Gomez R, Nunez M, Sacristan S, Pinto A, Fernandez-Aviles F, Tamargo J, Delpon E, Caballero R. Pitx2c increases in atrial myocytes from chronic atrial fibrillation patients enhancing IKs and decreasing ICa,L. Cardiovasc Res. 2016;109:431–441 [DOI] [PubMed] [Google Scholar]
- 169.Herraiz-Martinez A, Llach A, Tarifa C, Gandia J, Jimenez-Sabado V, Lozano-Velasco E, Serra SA, Vallmitjana A, Vazquez Ruiz de Castroviejo E, Benitez R, Aranega A, Munoz-Guijosa C, Franco D, Cinca J, Hove-Madsen L The 4q25 variant rs13143308T links risk of atrial fibrillation to defective calcium homoeostasis. Cardiovasc Res. 2019;115:578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Laforest B, Dai W, Tyan L, Lazarevic S, Shen KM, Gadek M, Broman MT, Weber CR, Moskowitz IP. Atrial fibrillation risk loci interact to modulate Ca2+-dependent atrial rhythm homeostasis. J Clin Invest. 2019;129:4937–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rommel C, Rosner S, Lother A, et al. The Transcription Factor ETV1 Induces Atrial Remodeling and Arrhythmia. Circ Res. 2018;123:550–563 [DOI] [PubMed] [Google Scholar]
- 172.Bruchard M, Rebe C, Derangere V, Togbe D, Ryffel B, Boidot R, Humblin E, Hamman A, Chalmin F, Berger H, Chevriaux A, Limagne E, Apetoh L, Vegran F, Ghiringhelli F. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol. 2015;16:859–870 [DOI] [PubMed] [Google Scholar]
- 173.Santulli G, Iaccarino G, De Luca N, Trimarco B, Condorelli G. Atrial fibrillation and microRNAs. Front Physiol. 2014;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Heijman J, Guichard JB, Dobrev D, Nattel S. Translational Challenges in Atrial Fibrillation. Circ Res. 2018;122:752–773 [DOI] [PubMed] [Google Scholar]
- 175.Nielsen JB, Thorolfsdottir RB, Fritsche LG, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Goette A, Auricchio A, Boriani G, et al. EHRA White Paper: knowledge gaps in arrhythmia management-status 2019. Europace. 2019;21:993–994 [DOI] [PubMed] [Google Scholar]
- 177.Backs J, Stein P, Backs T, Duncan FE, Grueter CE, McAnally J, Qi X, Schultz RM, Olson EN. The gamma isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proc Natl Acad Sci U S A. 2010;107:81–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ran X, Gestwicki JE. Inhibitors of protein-protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr Opin Chem Biol. 2018;44:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tomasovic A, Brand T, Schanbacher C, et al. Interference with ERK-dimerization at the nucleocytosolic interface targets pathological ERK1/2 signaling without cardiotoxic side-effects. Nat Commun. 2020;11:1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Van Taunay JS, Albelda MT, Frias JC, Lipinski MJ. Biologics and Cardiovascular Disease. J Cardiovasc Pharmacol. 2018;72:77–85 [DOI] [PubMed] [Google Scholar]
- 182.Ni L, Scott L Jr., Campbell HM, Pan X, Alsina KM, Reynolds J, Philippen LE, Hulsurkar M, Lagor WR, Li N, Wehrens XHT. Atrial-Specific Gene Delivery Using an Adeno-Associated Viral Vector. Circ Res. 2019;124:256–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tardif JC, Kouz S, Waters DD, et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381:2497–2505 [DOI] [PubMed] [Google Scholar]
- 184.Hiram R, Xiong F, Naud P, Sirois MG, Tanguay JF, Tardif JC, Nattel S. The inflammation-resolution promoting molecule resolvin-D1 prevents atrial arrhythmogenic remodeling in experimental right heart disease. Cardiovasc Res. 2020;In revision [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr., Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM, Investigators R-I. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med. 2019;380:11–22 [DOI] [PubMed] [Google Scholar]
- 186.Jais P, Reddy VY. Ablating AF in 30 minutes: new technologies for safer and more efficient pulmonary vein isolation. Circ Res. 2020;[AF Compendium] [DOI] [PubMed] [Google Scholar]
- 187.Haldar S, Hueser J. Targeting new therapies for AF. Circ Res. 2020;[AF Compendium] [Google Scholar]