

Abstract
Bispecific antibodies (BsAbs) that engage T cells bind to tumor cells via a tumor-associated antigen and to T cells through surface CD3. BsAbs have promising antitumor properties in vivo. Here we describe the effects of Fc silencing on BsAbs-driven T-cell trafficking to solid tumors. We used BsAbs driven by disialoganglioside GD2 or oncoprotein ErbB2 (HER2) and built on the IgG(L)-scFv platform with or without Fc silencing. We studied the kinetics of T-cell infiltration from blood into solid tumor masses when driven by these BsAbs. We also investigated the therapeutic efficacy of these BsAbs in two mouse models: immunodeficient mice xenografted with patient-derived GD2+ neuroblastoma or HER2+ breast cancer, and human CD3ε transgenic mice implanted with a GD2+ murine tumor. BsAbs built with intact Fc domain failed to drive T cells to tumor thereby failing to achieve an antitumor effect in mice. T cells became sequestered in lungs by myeloid cells or depleted in circulation. In contrast, when Fc function was silenced by N297A±K322A mutations, T cells were able to infiltrate into subcutaneous solid tumors, a prerequisite for successful therapy outcome.
Keywords: Fc silencing, N297A, T cell–engaging bispecific antibody, T-cell trafficking, antitumor, GD2, HER2
Introduction
T cell–engaging bispecific antibody (BsAb) binds to both CD3 on T cells and antigens on tumor cells simultaneously to facilitate T cell–mediated cytotoxicity. A number of BsAbs, across a variety of formats, have been shown to successfully redirect T cells to kill tumor cells in vivo (1-7). T-cell infiltration is essential for BsAbs to successfully mediate antitumor responses. The extent of T-cell infiltration is a useful indicator for the potential of successful immunotherapy against solid tumors (8,9). Strong T-cell infiltration has been associated with beneficial clinical outcome for various solid tumor diagnoses (10). Various methods have been developed to study T-cell trafficking into solid tumors in vivo, including use of radioactive isotopes, such as 111In (11) and 89Zr (12). Luciferase has proven to be a nontoxic reporter for imaging gene modified T cells noninvasively and monitoring their persistence and distribution in various organs over time in preclinical models (13,14).
For BsAbs, Fc-mediated immune functions are not necessary and are at times unwanted (15). In order to avoid or reduce antigen-independent cytokine release syndrome (CRS) due to crosslinking of CD3 and Fcγ receptors followed by nonspecific activation of immune cells, mutations have been introduced into Fc domain to eliminate FcγR binding for T cell–engaging bispecific antibodies (1,2,4,16,17).
Human IgG contains a single conserved site in the CH2 domain of Fc, Asn297, for N-linked glycosylation. Glycans at this site are the bridge between two CH2 domains that determine the interaction between Fc domain and Fcγ receptors. The N297A mutation removes N-glycosylation (18) thereby preventing binding to Fcγ receptors. The K322A mutation removes binding to C1q and prevents activation of the complement cascade (19). The K322A mutation has been used to reduce complement activation and in attempts to reduce IgG related pain side effects (20). However, it has not been determined whether these mutations could affect T-cell trafficking and antitumor activity of BsAbs.
Here, we focused on the N297A and K322A mutations to study the effects of loss of FcγR and C1q binding on T-cell trafficking and anti-tumor efficacy in two mouse models. We also investigated the fate of T cells when they are exposed to BsAbs with intact Fc. Our results confirmed the necessity of Fc silencing, proved the association between T-cell infiltration and antitumor effect for immunotherapy using BsAbs, and clarified the effect of deglycosylation and/or silencing of C1q binding on BsAb-driven T-cell trafficking to solid tumors and antitumor activity in vivo.
Materials and Methods
Construction and expression of BsAbs
The constructions of GD2 and HER2 series of BsAbs were as described previously (1,2). For each BsAb, scFv of huOKT3 was fused to the C-terminus of the light chain of human IgG1 via a C-terminal (G4S)3 linker (21). Both mutations on Fc, N297A and K322A, were generated with site-directed mutagenesis via primer extension in polymerase chain reactions (22). The nucleotide sequence encoding each BsAb was synthesized by GenScript and was subcloned into a mammalian expression vector. Each BsAb was produced using the Expi293™ expression system (Thermo Fisher Scientific) separately. Antibodies were purified with protein A affinity column chromatography. The purity of these antibodies was evaluated by size-exclusion high-performance liquid chromatography (SE-HPLC). The stabilities of these antibodies were monitored by SEC-HPLC weekly for four weeks at 37°C. Purity was quantified by the % monomeric peak eluting at the correct retention time.
Cell lines
M14 (melanoma) was obtained from University of California, Los Angeles; Neuroblastoma IMR-32 (CCL-127) and breast carcinoma AU565 (CRL-2351) were obtained from American Type Culture Collection (ATCC). All the cell lines used were authenticated by short tandem repeat profiling with PowerPlex 1.2 System (Promega), and routinely tested for mycoplasma using MycoAlert™ PLUS Mycoplasma Detection Kit (Lonza). The cells were cultured in RPMI1640 (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Life Technologies) at 37°C in a 5% CO2 humidified incubator. The cells were passaged three times after thaw.
Cell cytotoxicity (51Cr release assay)
Cell cytotoxicity was assayed using 51Cr release as described in (23). Effector T cells were isolated from human PBMCs using Pan T cell isolation kit (Miltenyi Biotec), and then activated and expanded using Dynabeads™ Human T-Activator CD3/CD28 (Thermo Fisher Scientific) for 14 days. Effector to target ratio was 10:1 for T cell–mediated cytotoxicity assays. NK92MI cells, which were transfected with human FcγRIIIa-158V, were used for ADCC assays with effector-to-target ratio of 20:1.
In vitro binding kinetics studies by surface plasmon resonance (SPR)
The interactions between BsAbs and Fcγ receptors were measured using Biacore T-100 Biosensor system (GE healthcare). The protein ligands were immobilized on the sensor chip using the Amino Coupling Kit (GE Healthcare). A blank flow cell was used as negative control. All the BsAbs were diluted in HBS-EP buffer (0.01 mol/L HEPES, pH 7.4, 0.15 mol/L NaCl, 3 mmol/L EDTA, and 0.05% v/v Surfactant P20), and injected over the sensor surface. The results were analyzed using the Biacore T-100 evaluation software.
Antitumor effect in human tumor xenografts
All animal procedures were performed in compliance with Memorial Sloan Kettering Cancer Center’s institutional Animal Care and Use Committee (IACUC) guidelines. BALB/c Rag2−/−IL-2Rγc−/− (double knockout, DKO) mice and heterozygous human CD3ε transgenic mice [B6.Cg-Tg(CD3E)600Cpt/J mice were bred with wildtype C57BL/6 mice to generate huCD3ε transgenic F1 heterozygotes] were used in this study. Patient-derived xenografts (PDXs) were established from fresh surgical specimens with MSKCC IRB approval. Tumor cells in Matrigel (Corning Corp) were implanted subcutaneously on the right flank of each mouse. Tumor size was measured using handheld Peira TM900 imaging device (Peira bvba). T cells isolated from peripheral blood were stimulated with Dynabeads™ Human T-Activator CD3/CD28 and expanded for eight days before injection with the presence of IL-2 (30 IU/ml). BsAbs and activated human T cells were injected intravenously at the same time and 1000 IU IL-2 given subcutaneously. IVIG (50 mg/dose), and 2.4G2 (mAb to Ly6G from Bioxcell, 200 μg/dose) were given three times per week intraperitoneally. The first doses were injected 48 hours before human T cells. The weights of the mice were monitored and no weight loss >15% was observed.
T-cell transduction
T cells isolated from peripheral blood were stimulated with Dynabeads™ Human T-Activator CD3/CD28 for 24 hours. T cells were transduced with retroviral constructs containing tdTomato and click beetle red luciferase in RetroNectin-coated 6-well plates in the presence of IL-2 (100 IU/ml) and protamine sulfate (4 μg/mL). Transduced T cells were cultured for 8 days before being used in animal experiments.
Bioluminescence imaging (BLI)
To monitor homing of T cells to tumor, BsAbs and luciferase transduced T cells were injected intravenously to mice at the same time, and 1000 IU IL-2 was given subcutaneously. Mice were then anesthetized and imaged after intravenous injection of 3 mg D-luciferin (Gold Biotechnology) at different days post T-cell injection. Images were acquired using IVIS SpectrumCT In Vivo Imaging System (Caliper Life Sciences). Bioluminescence images were overlaid with photographs, and regions of interest (ROI) were drawn based on the location and contour of tumor using Living image 2.60 (Xenogen). The total counts of photons (photon/s) were obtained. Bioluminescence signals before T-cell injection were used as baselines.
Immunohistochemistry and immunofluorescence staining
Immunohistochemistry (IHC) and immunofluorescence (IF) were performed at the MSK Molecular Cytology Core Facility using Discovery XT processor (Ventana Medical Systems) as described in (2). Tumor samples were fixed and embedded in paraffin. Anti-human CD45, anti-Myeloperoxidase, anti-mouse CD31 and anti-mouse CD68 were used, which was followed by biotinylated secondary antibody. The detection was performed using a DAB detection kit (Ventana Medical Systems) or Alexa Fluor™ 488 or 568 Tyramide Reagent (Invitrogen). IHC images were captured from tumor sections using a Nikon ECLIPSE Ni-U microscope and NIS-Elements 4.0 imaging software. IF images were captured with Leica Inverted Confocal SP8 and processed with Imaris (Bitplane). Cells were counted with Qupath 0.1.2.
Flow cytometry analysis and cytokine assay
For cell lines, goat anti-human IgG-PE was purchased from SouthernBiotech. For blood samples from mice, the following antibodies were purchased from Biolegend: anti-human CD45-APC (HI30), anti-human CD3-Percp/Cy5.5 (SK7), anti-mouse CD45-Brilliant Violet 711™ (30-F11), anti-mouse CD11b-Brilliant Violet 570™ (M1/70), and anti-mouse Ly6G-PE/Dazzle™ 594 (1A8). Cytokine amounts were measured with flow cytometry using LEGENDplex™ Human and mouse Th1 Panel (5-plex) (Biolegend) following manufacturer’s protocol. The experiments were conducted using a BD LSRFortessa flow cytometer and analyzed with FlowJo v10.
ELISA
Plasma amounts of GD2-BsAb, GD2-BsAb (N297A) and GD2-BsAb (N297A+K322A) were assayed by ELISA. The rat anti-hu3F8 anti-idiotypic antibody A1G4 (24) was adsorbed onto 96-well microtiter plates to capture the serum BsAb, which was detected with peroxidase-conjugated mouse anti-human IgG1 (Jackson ImmunoResearch) using o-Phenylenediamine dihydrochloride (OPD) as substrate. Pharmacokinetics (PK) analysis was performed by non-compartmental modelling of the BsAb concentration over time using the Phoenix WinNonlin software (v7.0, Certara, Princeton, NJ).
Mouse anti-human antibody response (MAHA) was determined by ELISA. Diluted mouse plasma was allowed to react with GD2-BsAb (N297A+K322A) coated on 96-well microtiter plates, and the bound MAHA was detected by peroxidase-conjugated goat anti-mouse IgG Fc (Southern Biotech) using OPD substrate.
Plasma amount of human IgG was determined by the Human ELISA Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. The plasma amount of mIgG1 in huCD3ε transgenic mice and DKO mice was measured using the Mouse IgG ELISA Kit (Bethyl Laboratories) according to manufacturer’s instructions.
Statistical analysis
Differences between samples indicated in the figures were tested for statistical significance by the Student’s t-test with GraphPad Prism 8, and p < 0.05 was considered statistically significant.
Area under the curve (AUC) was calculated using GraphPad Prism 8 (25).
Results
Mutations on Fc domain have no effect on in vitro antigen binding or T-cell cytotoxicity
We analyzed six purified BsAbs of interest: three specific to GD2 including GD2-BsAb, GD2-BsAb (N297A) and GD2-BsAb (N297A+K322A); and three specific to HER2, including HER2-BsAb, HER2-BsAb (N297A) and HER2-BsAb (N297A+K322A). The control BsAbs (Ctrl-BsAbs) used in this study bound to CD3 but not the tumors of interest based on immunohistochemistry (negative staining).
To confirm that the mutations on Fc domain did not affect binding to antigen, we used the GD2+ melanoma cell line M14 and the HER2+ breast cancer cell line AU565 for flow cytometry analysis. In the GD2-binding series, GD2-BsAb, GD2-BsAb (N297A) and GD2-BsAb (N297A+K322A) showed comparable binding to M14 (Fig. 1A). In the HER2-binding series, we demonstrated equivalent binding to AU565 regardless of Fc mutations on HER2 series of BsAbs (Fig. 1B).
Figure 1.
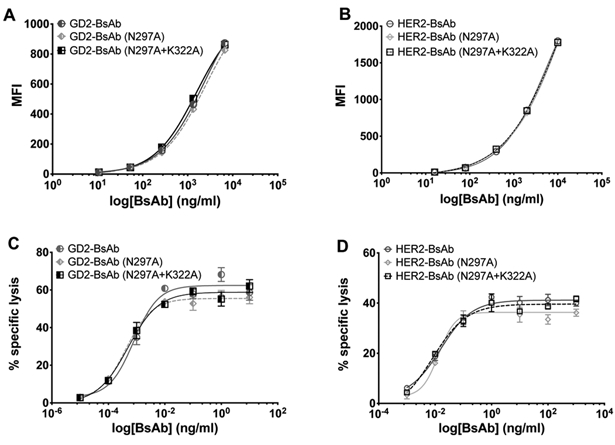
Comparisons of binding to cell lines and antibody dependent T cell–mediated cytotoxicity assays. A, GD2-BsAb and its Fc mutants binding to GD2+ melanoma cell line M14. B, HER2-BsAb and its Fc mutants binding to HER2+ breast cancer cell line AU565. C, GD2-BsAb and its Fc mutants redirected T-cell killing in 51Cr release assay of GD2+ neuroblastoma cell line IMR-32. Effector to target cell ratio was 10:1. D, HER2-BsAb and its Fc mutants redirected T-cell killing in 51Cr release assay of HER2+ breast cancer cell line AU565. Effector to target cell ratio was 10:1. The results shown are representative results from at least three independent repeats. No significant differences were observed between binding or cytotoxicity.
In order to verify that Fc mutations would not affect BsAb-dependent T cell–mediated cytotoxicity (ADTC) of BsAbs, we performed 51Cr release assays on GD2+ neuroblastoma cell line IMR-32 (Fig. 1C) and HER2+ breast cancer cell line AU565 (Fig. 1D). For each antigen-specific series, all three BsAbs showed comparable T cell–killing efficiency, confirming the preservation of in vitro ADTC potency despite Fc modifications.
Mutations on Fc domain do not impact purity, stability or pharmacokinetics
To confirm that these Fc mutations did not differentially impact stability, we examined the purity of six BsAbs by SEC-HPLC, right after purification and over four weeks of continuous incubation at 37°C. For both GD2-specific BsAb (Supplementary Fig. S1A) and HER2-specific BsAb sets (Supplementary Fig. S1B), wild type (WT) Fc, N297A, and N297A+K322A mutants, the percentage of monomer peaks were comparable and remained comparable over the course of the entire 37°C incubation. The HER2 series of BsAbs all started out with lower purity than that of the GD2 series but remained comparable over time. When pharmacokinetics of GD2-BsAb and its two Fc mutants were studied in C57BL/6 and BALB/c Rag2−/−IL-2Rγc−/− (DKO) mice, no differences were found among any of the Fc mutants (Supplementary Fig. S2).
N297A on Fc domain abolishes binding to human and mouse Fcγ receptors
To confirm that N297A eliminates binding to human FcγRIIIa, we evaluated antibody-dependent cell-mediated cytotoxicity (ADCC) for the two sets of BsAbs using NK92MI effector cells that were stably transfected with human FcγRIIIa-158V and either GD2+ IMR-32 or HER2+ AU565 cells (Fig. 2A and 2B). As expected, all BsAbs containing N297A mutations showed no ADCC, whereas BsAbs with intact Fc, GD2-BsAb and HER2-BsAb, mediated lysis of target cells.
Figure 2.
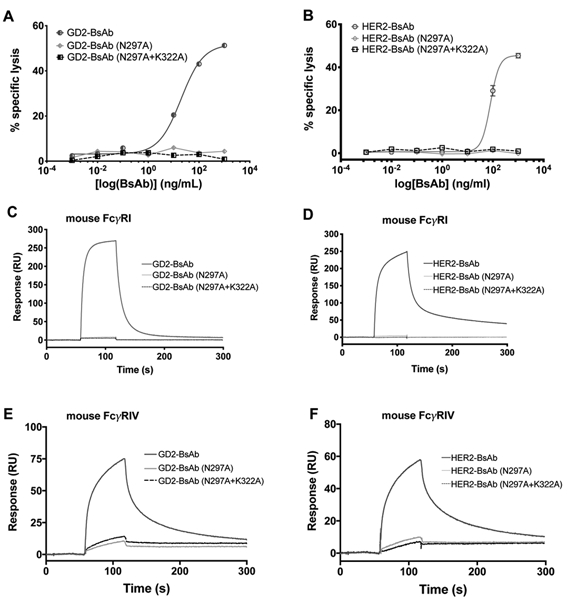
Comparisons of antibody-dependent cellular cytotoxicity and surface plasmon resonance analysis of binding to mouse Fcγ receptors. A, Antibody-dependent cellular cytotoxicity of GD2-BsAb and its Fc mutants against GD2+ IMR-32 cells. Effector to target ratio was 20:1. B, Antibody-dependent cellular cytotoxicity of HER2-BsAb and its Fc mutants against HER2+ AU565 Cells. Effector to target ratio was 20:1. Binding kinetics of BsAb and Fc mutants to mouse FcγRI (C, GD2-bind series, and D, HER2-binding series) and FcγRIV (E, GD2-bind series, and F, HER2-binding series). Protein binding to the SPR chip surface was measured in resonance units (RU). The results shown are representative results from at least three independent repeats.
We also investigated binding of BsAbs to mouse FcγRI and FcγRIV using surface plasmon resonance (SPR). Although GD2-BsAb and HER2-BsAb with WT Fc showed affinity for FcγRI and FcγRIV, BsAbs with Fc mutations N297A or N297A+K322A lost binding to mouse FcγRI and retained only minimal binding to mouse FcγRIV (Fig. 2C, 2D, 2E and 2F).
K322A on Fc domain eliminates complement-dependent cytotoxicity
To evaluate the role of N297A and K322A mutations in reducing binding to C1q and complement activation, we compared antibody-dependent complement-dependent cytotoxicity (CDC) of the GD2 series of BsAbs using GD2+ IMR-32 cell lines (Supplementary Fig. S3A). Removal of glycosylation reduced CDC; double mutations on Fc domain eliminated CDC. SPR showed that GD2-BsAb and HER2-BsAb bound human C1q strongly. The single mutation of N297A reduced binding and the double mutations of N297A + K322A abolished C1q binding (Supplementary Fig. S3B and C).
Fc silencing led to differences in antitumor effect in huCD3ε transgenic mice
To determine the role of Fc mutations on BsAb function in vivo, we first studied the effect of a single dose (5 μg) of GD2-BsAb and GD2-BsAb (N297A+K322A) on peripheral T cells in tumor-free huCD3ε transgenic mice. Mice treated with GD2-BsAb displayed significantly lower frequencies of huCD3+ T cells in the peripheral blood (24 hours post BsAb) and in the spleen (72 hours post BsAb) than mice treated with BsAbs carrying the double mutations (Supplementary Fig. S4A and S4B). In addition, after four doses of BsAbs, GD2-BsAb treated mice had significantly lower percentages of huCD3+ T cells in the peripheral blood when compared with the other three groups (Supplementary Fig. S4C), possibly contributing to these differences in tumor response. This indicates a possible depletion effect on circulating T cells from BsAbs when Fc domain was not silenced.
Next, we sought to identify how these mutations impacted each BsAb’s in vivo antitumor efficacy. Here we used human CD3ε transgenic mice, subcutaneously implanted with 5 × 104 EL4, a GD2+ mouse lymphoma (Fig. 3A). Mice (n=10 per group) were treated with 5 μg of each BsAb intravenously, twice per week, starting on four days after tumor implantation. All three GD2+ specific BsAbs delayed tumor growth compared with the control group. The two Fc mutants (N297A and N297A+K322A) showed stronger antitumor responses than that with WT Fc domain (Fig. 3B). To compare the treatment effects of different BsAbs, the area under the curve was calculated for each mouse on the last day of follow-up (day 31 post treatment). BsAb treatment groups were compared using unpaired t tests (Welch’s correction, unequal variance), showing significant differences between GD2-BsAb and GD2-BsAb (N297A+K322A) groups (p = 0.04) and between GD2-BsAb and GD2-BsAb (N297A) groups (p = 0.006). Mice in the control group had to be sacrificed before day 31 because of tumor size exceeding 10% of the body weight (~2 g, or 2000 mm3). Three out of the ten mice in the GD2-BsAb group showed no treatment effect, whereas all 10 out of 10 mice in both the GD2-BsAb(N297A+K322A) group and the GD2-BsAb (N297A) groups showed tumor suppression (Supplementary Fig. S5). One of the limitations of using human antibodies in huCD3ε transgenic mice is the mouse anti-drug antibody (ADA) response after two weeks of treatment (341±188 ng/ml, n=5), which confounded interpretation of antitumor response after the initial weeks of treatment. For these reasons, instead of using the immunocompetent mouse model, we focused our studies in the immunodeficient BALB/c Rag2−/−IL-2Rγc−/− DKO mouse model.
Figure 3.
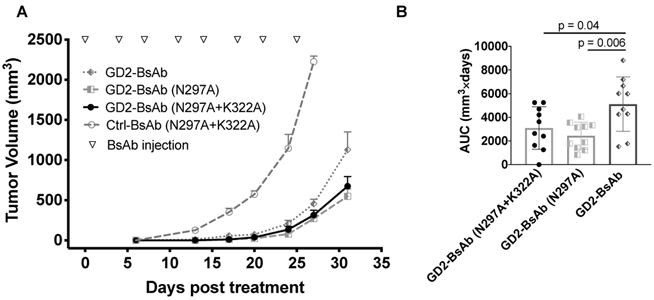
Comparisons of efficacy of GD2-BsAb and its mutants in huCD3ε transgenic mice (n =10 mice/group). A, Tumor growth curves of GD2+mouse lymphoma following treatment with Ctrl-BsAb, GD2-BsAb and its Fc mutants. B, Individual EL4 tumor AUC in huCD3ε transgenic mice following treatment with GD2-binding BsAbs after 31 days of treatment; values calculated by Prism GraphPad 8. BsAb treatment groups were compared using unpaired t tests (Welch’s correction, unequal variance), showing significant differences between GD2-BsAb and GD2-BsAb (N297A+K322A) groups (p = 0.04) and between GD2-BsAb and GD2-BsAb (N297A) groups (p = 0.006). The results shown are representative results from at least three independent repeats.
BsAbs with intact Fc domain fail to mediate effective T cell infiltration into tumor
To evaluate the role of BsAb Fc mutations in T cell infiltration, we examined the GD2 series of BsAbs in the xenograft immunodeficient mouse model. Here we implanted GD2+ patient-derived xenograft (PDX) neuroblastoma (Piro 20) tumors subcutaneously into DKO mice and treated the mice intravenously with BsAb (1ug) and activated human T cells (10 million). T cells were transduced with luciferase-expressing retrovirus (54% transduction), after activation for trafficking in vivo. BsAbs were repeated twice per week for a total of two weeks. To improve the survival of the human T cells, mice were also supplemented with subcutaneous doses of human IL-2 (1000U) twice per week. When imaged by bioluminescence, T cells first appeared to localize to the lung and we used this to confirm successful T-cell injection for all T cell–trafficking experiments. (Supplementary Fig. S6). By day 3, GD2-BsAb failed to draw T cells into the tumors, which showed no more infiltration than those treated with a control antibody. In contrast, both GD2-BsAb (N297A) and GD2-BsAb (N297A+K322A) groups showed accumulation of T cells in the tumor (>50-fold greater than GD2BsAb), peaking around day 6, and decreasing thereafter (Fig. 4A and B). These differences in infiltration were confirmed by IHC staining with human CD45, where only mice treated with Fc-silenced BsAbs showed high numbers of T cells in tumor. Mice treated with BsAb with WT Fc displayed minimal T-cell numbers inside tumor (Fig. 4C). AUC analysis also showed significant differences in T-cell accumulation caused by Fc silencing (Supplementary Fig. S7A).
Figure 4.

T-cell trafficking into GD2+ PDX in DKO mice to exert antitumor effects. A, Representative bioluminescence images of T-cell trafficking over days of BsAb treatment. B, Quantitation of T-cell infiltration into tumors over time as measured by bioluminescence (n = 5 mice/group) expressed as total flux or radiance (photons/sec) in each pixel integrated over the entire tumor contour (ROI). C, Representative immunohistochemistry images of huCD45+ T-cell infiltration in tumor sections on day 6 (100X magnifications). D, Tumor response among GD2+ neuroblastoma PDX (Piro 20) (n = 5 mice/group) after treatment with Ctrl-BsAb, GD2-BsAb and its Fc mutants. The results shown are representative results from at least three independent repeats. The significant differences were calculated based on AUC, shown in Supplementary Fig. S7.
We next used a second tumor model, implanted with a HER2+ breast cancer PDX (M37) subcutaneously into DKO mice and treated with a single injection of 10 million T cells transduced with luciferase. BsAbs and IL-2 were also administered as described above. Based on bioluminescence, T cells driven by HER2-BsAb showed minimal tumor infiltration (Fig 5A and 5B). In contrast, T cells driven by HER2-BsAb Fc-silenced by N297A+K322A or N297A mutations showed >400-fold greater tumor infiltration on day 6, showing similar trends with GD2-BsAb (N297A+K322A) and (N297A), which is statistically significant by AUC analysis for the cumulative effect (Supplementary Fig. S7B). The presence and absence of tumor infiltrating T cells were confirmed by immunohistochemistry (Fig. 5C).
Figure 5.

T-cell trafficking into HER2+ human breast cancer PDX in DKO mice to exert antitumor effect. A, Representative bioluminescence images of T-cell trafficking over days of BsAb treatment. B, Quantitation of T-cell trafficking into tumors over time by bioluminescence (n = 5 mice/group) expressed as total flux or radiance (photons/sec) in each pixel integrated over the entire tumor contour (ROI). C, Representative immunohistochemistry images of huCD45+ T-cell infiltration in tumor sections on day 6 (100X magnifications). D, Tumor response among HER2+ breast cancer PDX (M37) (n = 5 mice/group) after treatment with Ctrl-BsAb, Her2-BsAb and its Fc mutants. The results shown are representative results from at least three independent repeats. The significant differences were calculated based on AUC, shown in Supplementary Fig. S8.
Irrespective of target or tumor models, BsAbs (N297A) seem comparable to BsAbs (N297A+K322A) in driving T-cell infiltration into tumor. BsAbs with intact Fc domain failed to demonstrate T-cell trafficking into tumors, instead being sequestered in the lungs as late as day 3 post T cells injection (Figs. 4A and 5A).
BsAbs with intact Fc domains have no antitumor effects in xenograft mouse model
To investigate the therapeutic impact of T-cell trafficking, we evaluated T cell–antitumor function using two PDX models: GD2+ Neuroblastoma (Piro 20) or HER2+ Breast Cancer (M37). Tumor-bearing mice were treated with three doses of T cells (20 M, i.v.) and five doses of BsAb (1 μg per dose, i.v.) given twice per week with IL-2 (1000U, s.c.). Similar to what we saw in the huCD3ε transgenic mice, GD2-BsAb (N297A) and GD2-BsAb (N297A+K322A) significantly suppressed GD2+ tumor growth. GD2-BsAb with intact Fc domain failed to show any differences in tumor growth over Ctrl-BsAb (Fig. 4D). AUC analysis also showed significant differences caused by Fc silencing (Supplementary Fig. S8A).
Results were similar for the HER2 series of BsAbs, where tumor growth was suppressed by HER2-BsAb (N297A) and HER2-BsAb (N297A+K322A), whereas mice treated with HER2-BsAb with WT Fc domain failed to show any antitumor effect (Fig. 5D). Again, AUC analysis showed significant differences caused by Fc silencing (Supplementary Fig. S8B). These results confirmed that Fc domain controls the antitumor effects of T cell–engaging BsAbs and can drive the failure of T cell trafficking to tumors.
BsAbs with intact Fc domain sequester T cells in lungs in xenograft mouse model
To understand how BsAbs with intact Fc domain failed to direct T cells into tumor, we went back to our bioluminescence model to evaluate the distribution of T cells in BsAb treated xenografted mice. 30 minutes after the infusion of T cells and BsAb, T cells were visible in the lungs for all treatment groups regardless of Fc status. By 24 hours, mice treated with BsAbs carrying WT Fc domain displayed lower human CD45+ T-cell percentages in peripheral blood compared with the other groups (Fig. 6A). Similar to Ctrl-BsAb, the WT Fc GD2-BsAb group also had significantly more circulating human IFN-γ when compared with the other groups (Supplementary Fig. S9A), and more circulating mouse IL-6 (Supplementary Fig. S9B). We observed significantly higher percentages of mouse polymorphonucler myeloid-derived suppressor cells (PMN-MDSC, CD11b+Ly6G+) in peripheral blood among mice treated with WT GD2-BsAb or Ctrl-BsAb when compared with mice treated with the Fc-silenced GD2BsAb variants (Supplementary Fig. S10). By 72 hours, IHC (n=5 mice/group) revealed a difference in T-cell distribution in lungs versus spleens (Fig. 6B). With intact Fc, the majority of T cells were sequestered in the lung around the perivascular space, confirmed by mouse CD31 immunofluorescence staining (Fig. 6C); few or no T cells homed to the spleen or the tumor. In contrast, when Fc domain was silenced, T-cell distribution in the lung was scant and scattered, accompanied by robust homing to the spleen and migration into the tumor. By confocal microscopy, mouse macrophages and neutrophils were concentrated around T cells in the perivascular regions when Fc-intact BsAbs were used (Fig. 6D and E). The results suggest and active process in which human T cells are trapped through an Fc-dependent process mediated by mouse myeloid cells.
Figure 6.

Enumeration of T cells in blood, lung, spleen, and tumor in mice xenografted with GD2+ neuroblastoma PDX (Piro 20). A, Percentages of T cells in blood 24 hours after T-cell injection (n = 10 mice/group). B, Representative IHC images of huCD45+ T-cell infiltration in lung and spleen sections 72 hours after T-cell injection (n = 5 mice/group, 100× magnifications), with comparison among Ctrl-BsAb, GD2-BsAb, and their Fc-mutants. C, Representative IF images of huCD45+ T cells (magenta), CD31+ mouse endothelial cells (green), and nuclei (DAPI, blue; 200× magnifications) in mouse lungs treated with different BsAbs. D, Representative IF images of huCD45+ T cells (red), CD68+ mouse macrophages (green), and nuclei (DAPI, blue; 200× magnifications). Two images on the bottom were recorded at 400× magnification (left, Ctrl-BsAb; right, GD2-BsAb). E, Representative IF images of huCD45+ T cells (red), myeloperoxidase+ mouse neutrophils (green), and nuclei (DAPI, blue; 200× magnifications). Two images on the bottom were recorded at 400× magnification (left, Ctrl-BsAb; right, GD2-BsAb). The results shown are representative results from at least three independent repeats. Statistical significance of all results was determined by t test. If it is not labeled, it is not significant.
Since DKO mice do not contain B cells, their amount of circulating immunoglobulin should be lower than that of immunocompetent mice. To test if the occupation of Fcγ receptors by circulating IgG could play a role in sequestration of T cells, we treated DKO mice bearing GD2+ melanoma M14 cells with either intravenous immunoglobulin (IVIG) or an anti-CD16/CD32 (2.4G2) in addition to GD2-BsAbs, to block the mouse Fc receptors. By day 5, more tumor infiltration was evident in both IVIG and 2.4G2 treated groups, although rescue of the defects evident in WT Fc BsAb was partial (Supplementary Fig. S11).
We next studied the effect of IVIG on T-cell sequestration in lung and engraftment in spleen. Tumor-free DKO mice were injected with T cells, together with either GD2-BsAb ± IVIG, or GD2-BsAb (N297A+K322A) ± IVIG. Immunodeficient DKO mice had no endogenous IgG as measured by ELISA. After administration of human IVIG to saturate the FcRs in these mice, serum human IgG amount was measurable (9.4±1.6 mg/ml, n=5) when the BsAb treatment injections were initiated. These IgG amounts were higher than the endogenous mouse IgG amount in immunocompetent huCD3ε transgenic mice (1.56±0.43 mg/ml, n=5). IVIG had no effect on T-cell distribution in the lung for Fc-silenced GD2-BsAb (N297A+K322A). In contrast, IVIG allowed T cells in the lungs of GD2-BsAb with WT Fc domain treated mice to redistribute to the spleen (Fig. 7). These results suggested that Fc receptor blockade could alleviate the sequestration mediated by intact Fc domain in GD2-BsAb, and also indicated that additional mechanisms might be responsible for the failure of T cells to infiltrate to tumor.
Figure 7.

Effect of IVIG on T-cell trafficking. A, Representative IHC images of huCD45+ T cells in lungs and spleens of mice treated with and without IVIG (100× magnifications). B, Enumeration of T cells per pixel2 (area) of IHC images of huCD45+ T cells in mouse spleens. Cells were counted from two randomly selected fields for each mouse (n = 10). The results shown are representative results from at least three independent repeats. Statistical significance of all results was determined by t test. If it is not labeled, it is not significant.
Relation of Fc silencing and T-cell infiltration into kidney
We next examined kidney infiltration by T cells at 72 hours after injection (Supplementary Fig. S12A) using IHC. Mice treated with GD2-BsAb and GD2-BsAb (N297A) showed significantly higher ratios of T cellkidney/T cellspleen (Supplementary Fig. S12B), suggesting that complement activation was associated with renal T-cell infiltration. We did not observe any morphologic evidence of acute renal damage associated with T-cell infiltration during this 2-week observation.
Discussion
In this study we showed that BsAb-driven T-cell infiltration into solid tumors in vivo was necessary for tumor response. We used a luciferase reporter for non-invasive dynamic, bioluminescence monitoring of T-cell trafficking in individual mice and delineated T-cell trafficking patterns in vivo. Our results showed that Fc silencing could play a role in permitting T-cell trafficking into tumors and that differences in Fc functions could affect the overall antitumor response, sequestration of T cells into the lung, splenic engraftment and renal infiltration.
In this study, we investigated six BsAbs, with and without Fc mutations, directed at two targets, GD2 and HER2. The N297A mutation silenced Fc function by removing N-glycosylation (18) eliminating most ADCC and CDC functions. The K322A mutation removed binding to C1q and prevented activation of the complement cascade. The two mutations (N297A and N297A+K322A) did not affect antigen binding or efficiency in T cell–mediated cytotoxicity in vitro, stability in vitro, or pharmacokinetics in vivo. In addition, N297A did not eliminate binding to FcγRI or C1q.
For both antigen systems, N297A and the double mutant N297A+K322A were most efficient in driving T cells into solid tumors and maintaining them for the longest duration inside solid masses. In contrast, when the Fc domain was fully functional, T-cell infiltration was minimal to non-existent. These homing capabilities translated into large differences in antitumor efficacy of these antibodies. In both antigen systems, near complete tumor ablation was observed when BsAbs with N297A+K322A or N297A mutations were administered to mice after adoptive transfer of activated human T cells. In contrast, BsAbs with intact Fc domain had minimal therapeutic benefit in immunodeficient mice engrafted with activated human T cells, or in transgenic mice carrying huCD3ε+ naïve T cells. In addition, BsAbs with intact Fc functions depleted T-cell numbers in the peripheral blood and spleens of huCD3ε transgenic mice.
We also found that T cells could not escape from lungs of mice injected with BsAbs with intact Fc, which led to failure of T-cell trafficking to tumor. Injected human T cells mostly stayed in the perivascular region of lung. We further showed that mouse neutrophils and macrophages, cells that expressed Fcγ receptors, also tended to gather in the same regions. In contrast, in mice treated with Fc-silenced BsAbs, T cells and mouse myeloid cells did not congregate in the same distributions. After Fcγ receptor blockade using IVIG, T-cell infiltration into tumors improved. Following blockade with anti-FcγR (2.4G2), T-cell infiltration into tumor improved. Additional mechanisms might be involved.
Catumaxomab, a T cell–engaging bispecific antibody (anti-EpCAM × anti-CD3), has functional Fc. Catumaxomab was designed to bind and activate Fcγ receptor bearing cells for ADCC (26). A dose-limiting toxicity was the cytokine-release-syndrome (CRS) via T-cell activation induced by Fc domain (27). In our study, we observed elevated cytokine expression induced by BsAbs with WT Fc, IFN-γ from injected human T cells and IL-6 from host myeloid cells. IL-6 could be released when neutrophils are cross-linked with FcγRI or FcγRII (28), and with the high PMN-MDSC/neutrophil count, this elevated cytokine was expected. In addition, when T cells were sequestered and destroyed in the lungs by myeloid cells, an “inflammatory” response would lead to neutrophilia. However, after Fc silencing, cytokine release in DKO mice could be substantially reduced. These data support the role of Fc silencing in reducing CRS that is otherwise expected to accompany BsAb therapy.
The single N297A mutation didn’t show any significant differences when compared to double mutant N297A+ K322A in Fcγ receptor binding, or in T-cell infiltration into solid tumor, or in antitumor effect in the mouse models. However, we observed a preferential T-cell infiltration into kidney if complement activation was not removed. Although we did not observe morphologic evidence of acute renal damage associated with a single injection of T cells, longer observation, especially after multiple doses of both T cells and BsAb, will be needed to study the effects of kidney tropism. Other studies have also used Fc silencing to avoid complement inactivation so as to avoid infusion reactions (29-31). Before we can extrapolate these results from mouse models to the clinic, further studies may be needed, given species differences in complement systems, efficiency of Fc, and complement activation by human IgG1-Fc (32). Some renal toxicities have emerged as late effects in response to treatment with immune checkpoint (e.g. CTLA-4 and PD-1) inhibitor (ICI) antibodies (33). Additionally, residual Fc functions of the IgG4 subclass ICI has been associated with trogocytosis and paralysis of NK cells (34). Whether residual complement activation could cause renal damage by BsAb will have to await results from clinical trials.
Bispecific antibodies (BsAbs) have emerged as a strategy for the treatment of hemophilia (35) and cancer (36). To date, two BsAb formats have been approved by the FDA for clinical use (35,36); over 60 are in preclinical and clinical development (37-39). The majority utilize human IgG-Fc, built either on the IgG framework or using Fc fusion systems to increase serum half-lives of small recombinant fragments. Our study suggests that WT Fc domain in BsAbs could decrease their efficacy with the potential for undesirable cytokine storm and organ infiltration by T cells. None of these adverse properties were predicted by in vitro assays. Many mutations have been described to silence Fc domain (e.g. N297G, D265A, L234A+L235A±P329G) (40-44), but little is known about their in vivo immunological properties other than pharmacokinetics (45). Attempts to maintain or enhance FcR competence have been utilized to provide additional antitumor activity without considering the negative impact beyond cytokine release and non-selective T-cell activation. It is possible that BsAbs with silenced Fc domain will be more able to prevent sequestration of T cells by BsAb in the lung or the reticuloendothelial system.
Reduction in circulating T cells could be the result of T-cell redistribution and/or T-cell destruction. In our studies, all injected T cells were labeled with luciferase and their trafficking followed and quantified by bioluminescence in real time, as well as by immunohistochemistry at necropsy. These orthogonal methods of live imaging and immunohistology were the basis of our conclusions. Besides Fc-mediated sequestration, T-cell trafficking or redistribution is likely an orchestrated interplay of adhesion ligands and receptors on both T cells and endothelial cells, as well the network of chemokines and receptors, that require investigations beyond the scope of our studies. Both humanized SCID or huCD3ε transgenic mouse models have limitations in predicting clinical efficacy or toxicity in human. We have mainly focused on the immunodeficient DKO mice model because it allowed us to study human T-cell trafficking utilizing the luciferase reporter system without the confounding issues of neutralizing antibodies. Despite their limitations, immunodeficient mice mimic the clinical setting in that patients who come to phase I/II studies with high risk metastatic disease (e.g. neuroblastoma) usually have undergone myelo- and lympho- suppressive chemoradiotherapy and have deficient T-cell functions. Our data suggest that with an intact Fc, BsAb can bind to the Fcγ receptors on immune cells, such as neutrophils, macrophages and even NK cells while activating the complement cascade, and in that process activate and damage T cells, sequestering them to the reticuloendothelial system, releasing cytokine and preventing T cells from tumor infiltration. A deeper understanding of the role of residual Fc functions, whether in ADCC or in CDC, should avoid unexpected acute or late effects in the clinical development of BsAbs, a requisite consideration since the lesson of TGN1412 (46,47).
Supplementary Material
Acknowledgement
The authors would like to thank Dr. Mamoru Ito of Central Institute for Experimental Animals, Kawasaki, Japan, for providing the DKO mice, Dr. John H. Sampson of Duke University, Durham North Carolina, for providing the huCD3ε transgenic mice, Dr Piro Lito of MSK for providing the Piro 20 neuroblastoma PDX, and Dr Maurizio Scaltriti of MSK for providing M37 breast cancer PDX. We thank Dr. Irene Cheung for help with PK study and ADA assays, Dr. Alessandra Piersigilli and Dr. Sebastien Monette for evaluation of H&E and IHC slides and Dr. Brian Santich for the helpful discussions and suggestions.
Financial support:
This work was supported in part by funds from Enid A. Haupt Endowed Chair, the Robert Steel Foundation, and Kids Walk for Kids with Cancer. Technical service provided by the MSK Animal Imaging Core Facility, Antitumor Assessment Core Facility, and Molecular Cytology Core Facility were supported in part by the NCI Cancer Center Support Grant P30 CA008748.
Footnotes
Conflicts of Interest
NKC reports receiving commercial research grants from Y-mabs Therapeutics and Abpro-Labs Inc.; holding ownership interest/equity/options in Y-mabs Therapeutics Inc., and in Abpro-Labs, and owning stock options in Eureka Therapeutics. NKC is the inventor of pending and issued patents filed by MSK, including hu3F8 and 8H9 licensed to Y-mabs Therapeutics, beta-glucan to Biotec Pharmacon, and HER2 bispecific antibody to Abpro-labs. NKC is an advisory board member for Abpro-Labs and Eureka Therapeutics. LW, SSH, HX, and VP were named as inventors of pending and/or issued patents filed by MSK.
References:
- 1.Lopez-Albaitero A, Xu H, Guo H, Wang L, Wu Z, Tran H, et al. Overcoming resistance to HER2-targeted therapy with a novel HER2/CD3 bispecific antibody. Oncoimmunology 2017;6:e1267891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol Res 2015;3:266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A 2010;107:12605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishiguro T, Sano Y, Komatsu SI, Kamata-Sakurai M, Kaneko A, Kinoshita Y, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med 2017;9. [DOI] [PubMed] [Google Scholar]
- 5.Iizuka A, Nonomura C, Ashizawa T, Kondou R, Ohshima K, Sugino T, et al. A T-cell-engaging B7-H4/CD3 bispecific Fab-scFv antibody targets human breast cancer. Clin Cancer Res 2019. [DOI] [PubMed] [Google Scholar]
- 6.Wu Z, Guo HF, Xu H, Cheung NV. Development of a Tetravalent Anti-GPA33/Anti-CD3 Bispecific Antibody for Colorectal Cancers. Mol Cancer Ther 2018;17:2164–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoseini SS, Guo H, Wu Z, Hatano MN, Cheung NV. A potent tetravalent T-cell-engaging bispecific antibody against CD33 in acute myeloid leukemia. Blood Adv 2018;2:1250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann B, Biburger M, Bruckner C, Ipsen-Escobedo A, Gordan S, Lehmann C, et al. Tumor location determines tissue-specific recruitment of tumor-associated macrophages and antibody-dependent immunotherapy response. Sci Immunol 2017;2. [DOI] [PubMed] [Google Scholar]
- 9.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J 2010;16:399–403. [DOI] [PubMed] [Google Scholar]
- 10.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298–306. [DOI] [PubMed] [Google Scholar]
- 11.Pittet MJ, Grimm J, Berger CR, Tamura T, Wojtkiewicz G, Nahrendorf M, et al. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proc Natl Acad Sci U S A 2007;104:12457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mall S, Yusufi N, Wagner R, Klar R, Bianchi H, Steiger K, et al. Immuno-PET Imaging of Engineered Human T Cells in Tumors. Cancer Res 2016;76:4113–23. [DOI] [PubMed] [Google Scholar]
- 13.Santos EB, Yeh R, Lee J, Nikhamin Y, Punzalan B, Punzalan B, et al. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nat Med 2009;15:338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szyska M, Herda S, Althoff S, Heimann A, Russ J, D'Abundo D, et al. A Transgenic Dual-Luciferase Reporter Mouse for Longitudinal and Functional Monitoring of T Cells In Vivo. Cancer Immunol Res 2018;6:110–20. [DOI] [PubMed] [Google Scholar]
- 15.Hristodorov D, Fischer R, Linden L. With or without sugar? (A)glycosylation of therapeutic antibodies. Mol Biotechnol 2013;54:1056–68. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Lam CK, Long V, Widjaja L, Yang Y, Li H, et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clin Cancer Res 2017;23:1506–18. [DOI] [PubMed] [Google Scholar]
- 17.Klupsch K, Baeriswyl V, Scholz R, Dannenberg J, Santimaria R, Senn D, et al. COVA4231, a potent CD3/CD33 bispecific FynomAb with IgG-like pharmacokinetics for the treatment of acute myeloid leukemia. Leukemia 2018. [DOI] [PubMed] [Google Scholar]
- 18.Chao DT, Ma X, Li O, Park H, Law D. Functional characterization of N297A, a murine surrogate for low-Fc binding anti-human CD3 antibodies. Immunol Invest 2009;38:76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isaacs JD, Greenwood J, Waldmann H. Therapy with monoclonal antibodies. II. The contribution of Fc gamma receptor binding and the influence of C(H)1 and C(H)3 domains on in vivo effector function. J Immunol 1998;161:3862–9. [PubMed] [Google Scholar]
- 20.Sorkin LS, Otto M, Baldwin WM 3rd, Vail E, Gillies SD, Handgretinger R, et al. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 2010;149:135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orcutt KD, Ackerman ME, Cieslewicz M, Quiroz E, Slusarczyk AL, Frangioni JV, et al. A modular IgG-scFv bispecific antibody topology. Protein Eng Des Sel 2010;23:221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reikofski J, Tao BY. Polymerase chain reaction (PCR) techniques for site-directed mutagenesis. Biotechnol Adv 1992;10:535–47. [DOI] [PubMed] [Google Scholar]
- 23.Cheng M, Ahmed M, Xu H, Cheung NK. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer 2015;136:476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung NK, Cheung IY, Canete A, Yeh SJ, Kushner B, Bonilla MA, et al. Antibody response to murine anti-GD2 monoclonal antibodies: correlation with patient survival. Cancer Res 1994;54:2228–33. [PubMed] [Google Scholar]
- 25.<https://www.graphpad.com/support/faq/area-under-dose-response-data/>.
- 26.Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. MAbs 2010;2:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jager M, Schoberth A, Ruf P, Hess J, Hennig M, Schmalfeldt B, et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res 2012;72:24–32. [DOI] [PubMed] [Google Scholar]
- 28.Ericson SG, Zhao Y, Gao H, Miller KL, Gibson LF, Lynch JP, et al. Interleukin-6 production by human neutrophils after Fc-receptor cross-linking or exposure to granulocyte colony-stimulating factor. Blood 1998;91:2099–107. [PubMed] [Google Scholar]
- 29.Seckinger A, Delgado JA, Moser S, Moreno L, Neuber B, Grab A, et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017;31:396–410. [DOI] [PubMed] [Google Scholar]
- 30.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, et al. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel 2016;29:457–66. [DOI] [PubMed] [Google Scholar]
- 31.Bacac M, Klein C, Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. OncoImmunology 2016;5:e1203498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, et al. Crosstalk between human IgG isotypes and murine effector cells. J Immunol 2012;189:3430–8. [DOI] [PubMed] [Google Scholar]
- 33.Wanchoo R, Karam S, Uppal NN, Barta VS, Deray G, Devoe C, et al. Adverse Renal Effects of Immune Checkpoint Inhibitors: A Narrative Review. American journal of nephrology 2017;45:160–9. [DOI] [PubMed] [Google Scholar]
- 34.Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, et al. Checkpoint Inhibition of KIR2D with the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK-cells in Patients with Myeloma. Clin Cancer Res 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, et al. Factor VIII-Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. N Engl J Med 2016;374:2044–53. [DOI] [PubMed] [Google Scholar]
- 36.Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 2015;16:57–66. [DOI] [PubMed] [Google Scholar]
- 37.Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs 2017;9:182–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spiess C, Zhai QT, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Molecular Immunology 2015;67:95–106. [DOI] [PubMed] [Google Scholar]
- 39.Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacology & therapeutics 2018;182:161–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell 2018;9:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, et al. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol 2008;181:6664–9. [DOI] [PubMed] [Google Scholar]
- 42.Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin Cancer Res 2016;22:3286–97. [DOI] [PubMed] [Google Scholar]
- 43.Harwood SL, Alvarez-Cienfuegos A, Nunez-Prado N, Compte M, Hernandez-Perez S, Merino N, et al. ATTACK, a novel bispecific T cell-recruiting antibody with trivalent EGFR binding and monovalent CD3 binding for cancer immunotherapy. Oncoimmunology 2017;7:e1377874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J Biol Chem 2017;292:3900–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leabman MK, Meng YG, Kelley RF, DeForge LE, Cowan KJ, Iyer S. Effects of altered FcgammaR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs 2013;5:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunig T The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nat Rev Immunol 2012;12:317–8. [DOI] [PubMed] [Google Scholar]
- 47.Bartholomaeus P, Semmler LY, Bukur T, Boisguerin V, Romer PS, Tabares P, et al. Cell Contact-Dependent Priming and Fc Interaction with CD32+ Immune Cells Contribute to the TGN1412-Triggered Cytokine Response. J Immunol 2014;192:2091–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.