

Summary
Cas9 is a prokaryotic RNA-guided DNA endonuclease that binds substrates tightly in vitro but turns over rapidly when used to manipulate genomes in eukaryotic cells. Little is known about the factors responsible for dislodging Cas9 or how they influence genome engineering. Unbiased detection through proximity labeling of transient protein interactions in cell-free Xenopus laevis egg extract identified the dimeric histone chaperone FACT as an interactor of substrate-bound Cas9. FACT is both necessary and sufficient to displace dCas9, and FACT immunodepletion converts Cas9’s activity from multi-turnover to single-turnover. In human cells, FACT depletion extends dCas9 residence times, delays genome editing, and alters the balance between indel formation and homology directed repair. FACT knockdown also increases epigenetic marking by dCas9-based transcriptional effectors with a concomitant enhancement of transcriptional modulation. FACT thus shapes the intrinsic cellular response to Cas9-based genome manipulation most likely by determining Cas9 residence times.
Keywords: CRISPR, Cas9, FACT complex, SPT16, SSRP1, histone chaperone
Graphical Abstract
eTOC blurb
S. pyogenes Cas9 binds very tightly to DNA. It has been unclear how cells remove Cas9 from the genome. Wang et al. determine that the histone chaperone complex FACT displaces Cas9 from its substrate in eukaryotic systems. FACT knockdown potentiates CRISPR-based tools and alters Cas9 gene editing outcomes.
Introduction
Cas9 is a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated RNA-guided DNA endonuclease that is directed to a target DNA molecule by forming a ribonucleoprotein (RNP) complex with a guide RNA (gRNA) (Doudna and Charpentier, 2014; Hsu et al., 2014; Jinek et al., 2012; Knott and Doudna, 2018). After unwinding its duplex substrate, Cas9 uses two nuclease domains to generate a double-stranded break (DSB) (Doudna and Charpentier, 2014; Hsu et al., 2014; Jinek et al., 2012; Knott and Doudna, 2018; Sternberg et al., 2015). Subsequent break repair relies on a cell’s endogenous machinery to either incorporate sequences using a DNA template through homology-directed repair (HDR) or introduce insertions or deletions (indels) during non-homologous end joining (NHEJ) (Maggio and Goncalves, 2015). The ease of programming Cas9 to generate targeted DSBs and initiate break repair has enabled its widespread use as a genome editing agent (Cong et al., 2013; Jinek et al., 2012; 2013; Mali et al., 2013).
Mutations that inactivate Cas9’s nuclease activity preserve its capacity to bind a gRNA and target DNA (Qi et al., 2013). Catalytically inactive Cas9 (dCas9) has dramatically expanded the CRISPR toolbox. Fusing various effector proteins to dCas9 has enabled CRISPR-based methods to activate or repress gene expression, manipulate the three-dimensional architecture of nuclei, image genomic loci, track RNA molecules, and identify proteins at specific loci (Chen et al., 2013; Gao et al., 2018; Gilbert et al., 2013; Hilton et al., 2015; Konermann et al., 2015; Liu et al., 2017; Myers et al., 2018; Nelles et al., 2016; Qi et al., 2013; Schmidtmann et al., 2016; Wang et al., 2018).
The relationship between the efficacy of the CRISPR-Cas9 toolbox and Cas9’s lifetime on a genomic target site is unclear. For example, CRISPR transcriptional effectors localize histone acetyltransferases or methyltransferases around a transcription start site (TSS) to manipulate expression of endogenous genes (Gilbert et al., 2013; Hilton et al., 2015). Such transcriptional engineering presumably relies on providing these histone modifiers sufficient time at the TSS to deposit the appropriate epigenetic marks. Conversely, Cas9’s utility as a targeted nuclease may be predicated on its removal from the genome because Cas9 itself masks the DSB from cellular repair enzymes (Clarke et al., 2018; Richardson et al., 2016b). Cas9 residence times and unloading might thus play crucial roles in Cas9-based interventions.
While some Cas9 molecules behave as multi-turnover enzymes (Yourik et al., 2019), the widely used Streptococcus pyogenes Cas9 and dCas9 exhibit extremely stable protein-DNA interactions and possess residence times of over five hours in vitro (Raper et al., 2018; Richardson et al., 2016b; Sternberg et al., 2014). Estimates of residence times in live cells vary (Ma et al., 2016; Shao et al., 2016), but some experiments indicate that S. pyogenes Cas9 stays bound to its target in mammalian cells for as little as five minutes (Knight et al., 2015) and imply that cellular factors promote turnover. Given that DNA repair requires at least 30 minutes to occur (Mao et al., 2008), the ability to detect resolved genomic edits only one hour after electroporation of Cas9 RNPs (Kim et al., 2014) further suggests that cells actively remove Cas9 from the genome either purposefully or as a byproduct of normal genome metabolism.
Prior work has suggested that RNA polymerases can dislodge Cas9 from DNA in vitro when the gRNA anneals to the non-coding DNA strand but not to the coding strand (Clarke et al., 2018). Targeting the non-coding strand with a gRNA roughly correlated with increased editing rates in human cells and an increased ability of a bacterially-encoded CRISPR system to fight phage infection. However, the ability to edit non-transcribed regions of the human genome implies that RNA polymerases are not solely responsible for Cas9 eviction. Moreover, the ability to edit post-mitotic cells such as neurons suggests that replicative DNA polymerases are also not solely responsible for unloading Cas9 (Nishiyama et al., 2017; Suzuki et al., 2016).
Here, we find that metazoan cellular extracts contain a factor responsible for rapid multi-turnover activity of Cas9 on DNA substrates. An unbiased proteomics approach to mark proteins transiently associating with substrate-bound Cas9 and dCas9 identified both components of the heterodimeric Facilitates Chromatin Transcription (FACT) histone chaperone complex, SPT16 and SSRP1. In Xenopus extracts, immunodepletion of FACT subunits prevented dCas9 displacement and converted Cas9’s activity from multi-turnover to single-turnover. In a minimal buffer system, recombinant FACT was sufficient to displace dCas9. In living human cells, FACT promoted dCas9 unbinding, modified Cas9 editing outcomes, and played a strand-independent role in determining the extent of epigenetic marking and transcriptional regulation from dCas9-based effectors. These results reveal an unanticipated functional interaction between Cas9 and the eukaryotic machinery responsible for regulating nucleosome assembly.
Results
Cell-Free X. laevis Egg Extract Promotes Rapid Turnover of Cas9 from DNA Substrates
Xenopus egg extracts have a long track record of dissecting nuclear dynamics and interrogating processes such as DNA replication, chromosome segregation, and DNA repair (Heald et al., 1996; Hoogenboom et al., 2017; Kalab et al., 2006; Knipscheer et al., 2009; Lebofsky et al., 2009). We therefore used high-speed supernatant (HSS) of total Xenopus laevis egg lysate (Lebofsky et al., 2009) to look for cellular factors that promote the dissociation of a pre-formed Cas9 RNP-DNA complex.
We first tested the ability of HSS to promote the dissociation of S. pyogenes Cas9 RNPs from linear and plasmid DNA substrates harboring a single on-target site. We incubated RNPs with twice as many moles of DNA for 45 minutes before adding buffer or HSS. With excess substrate, complete cleavage would occur only if Cas9 possesses multi-turnover activity. Consistent with prior in vitro data (Richardson et al., 2016b; Sternberg et al., 2014), a Cas9 RNP targeting linear double-stranded DNA in buffer failed to cleave the excess substrate and thus behaved as a single-turnover enzyme even under these multi-turnover conditions (Figure 1A). However, incubation of RNP and target DNA in HSS and ATP yielded a steady conversion of substrate to product that was consistent with multi-turnover behavior (Figure 1A). Pre-depleting HSS ATP levels using calf intestinal phosphatase (CIP) and adding an excess of the non-hydrolysable analogue ATPγS reverted Cas9’s behavior to single-turnover (Figure 1A). We observed similar results when targeting Cas9 to a circular plasmid under multi-turnover conditions (Figure 1B).
Figure 1. Energy-Dependent Release of Cas9 and dCas9 from DNA in HSS.

(A) Time course of Cas9 RNPs programmed against a linear DNA substrate in a 1:2 molar ratio in buffer, HSS with ATP, or HSS with CIP and ATPγS.
(B) Time course of Cas9 RNPs programmed against a plasmid substrate in a 1:2 molar ratio in buffer, HSS with ATP, or HSS with CIP and ATPγS.
(C) Schematic of the single-turnover competition assay.
(D) ATP-dependent unloading of dCas9 off a plasmid substrate in X. laevis HSS. Presence of linearized DNA after addition of a 10-fold molar excess of Cas9 indicates removal of dCas9 while persistence of circular DNA indicates stable binding of dCas9. See also Figure S1C.
We next developed a competition assay to determine whether factors in HSS induce dCas9 removal from a circular DNA substrate under single turnover conditions (Figure 1C). We allowed a dCas9 RNP to equilibrate in buffer with a plasmid containing a single on-target site and then incubated this RNP-plasmid complex with buffer alone, HSS containing an ATP-regenerating system (ARS, see STAR Methods), or HSS pre-incubated with CIP and excess ATPγS. Finally, we added a 10-fold molar excess of catalytically active Cas9 programmed with the identical on-target gRNA. Persistent binding of dCas9 should prevent binding of Cas9 and preclude cleavage while dCas9 unloading would grant Cas9 access to the target site. The presence of linearized DNA thus provides a readout of dCas9 unloading. Consistent with our data using multi-turnover conditions, buffer alone did not promote dCas9 dissociation from the plasmid (Figure 1D Lanes 3-5). By contrast, HSS containing the ARS rapidly removed dCas9 to allow the catalytically active Cas9 to cleave the plasmid within 45 minutes (Figure 1D Lanes 6-8). Conversely, ATP-depleted HSS supplemented with ATPγS did not evict the majority of dCas9 molecules, thereby preventing Cas9 from cleaving the plasmid (Figure 1D Lanes 9-11). Overall, our data with linear and circular DNA substrates indicate that HSS contains at least one factor capable of dislodging Cas9 from its target, enabling re-binding to uncleaved molecules and multi-turnover behavior.
Unbiased Cas9 Interaction Marking Identifies the FACT Histone Chaperone as Required for Cas9 Removal and Multi-Turnover Activity in HSS
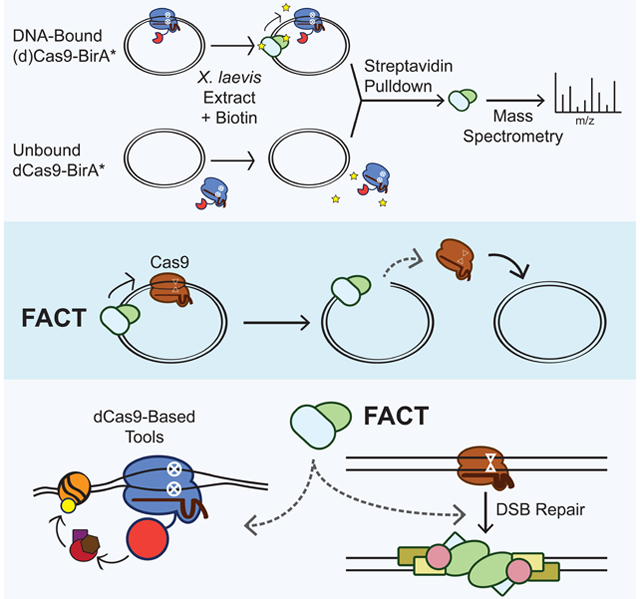
To identify factors that remove Cas9 from DNA in HSS, we fused Cas9 and dCas9 to the promiscuous biotin ligase BirA* (Arg118Gly). BirA* covalently labels nearby proteins, which can then be isolated with Streptavidin-coupled beads and identified through mass spectrometry proteomics (Gao et al., 2018; Roux et al., 2012; Schmidtmann et al., 2016). BirA*’s utility for our purposes is derived from our ability to extend the labeling time beyond a few minutes, thereby allowing the system to accumulate biotinylated versions of transient yet repeated interactors (Roux et al., 2013). BirA*-dCas9 fusions expressed in living cells have helped identify DNA interactors at repetitive genomic regions (Schmidtmann et al., 2016), but excess BirA*-dCas9 unbound to the genomic target has complicated its use at non-repetitive loci. We reasoned that the ability to form a defined Cas9-BirA* RNP-DNA or dCas9-BirA* RNP-DNA species in HSS could enable identification of Cas9 and dCas9 removal factors.
We expressed and purified recombinant Cas9-BirA* and dCas9-BirA* in and from bacterial cells (Data S1 and Figure S1A). The fused biotin ligase neither compromised Cas9’s nuclease activity nor hindered rapid dislodging of dCas9 in HSS (Figures S1B and S1C). We programmed Cas9-BirA* with an on-target gRNA, dCas9-BirA* with the same on-target gRNA, or dCas9-BirA* with a non-targeting (NT) gRNA. We added a ten-fold molar excess of plasmid substrate relative to each RNP in buffer and then added this mixture to HSS containing ARS and biotin (Figure 2A). Streptavidin pulldown and label-free proteomic mass spectrometry (Wuhr et al., 2014) identified biotinylated X. laevis proteins that were specifically enriched by gRNA-mediated binding of Cas9 or dCas9 to the plasmid relative to non-specific biotinylation when dCas9 was complexed with the NT gRNA. Cas9-BirA* and dCas9-BirA* programmed with the on-target gRNA had nearly identical interactors (Figure S1D), consistent with prior in vitro data indicating that Cas9 obscures the DSB so that repair factors are not preferentially enriched around Cas9 (Clarke et al., 2018; Richardson et al., 2016b).
Figure 2. FACT Complex Interacts with DNA-Bound Cas9 and dCas9 to Promote Eviction and Multi-Turnover Behavior.

(A) Schematic of samples prepared for mass spectrometry. dCas9-BirA* programmed with the on-target gRNA, Cas9-BirA* programmed with the on-target guide, and dCas9-BirA* programmed with a NT sgRNA were incubated with a 10-fold molar excess of plasmid and then added to HSS containing the ARS and biotin. Biotinylated proteins were isolated with Streptavidin-coupled beads and identified through mass spectrometry.
(B) Volcano plot of biotinylated proteins in Cas9-BirA*–on-target gRNA samples (N = 3 biological replicates) versus dCas9-BirA*–NT gRNA samples (N = 3 biological replicates). Colored circles correspond to factors that were significantly enriched (p < 0.05) according to a Limma analysis. Red circles correspond to the two components of the FACT complex. See also Figure S1D and Table S1.
(C) Enrichment of biotinylated SSRP1 and SPT16 in HSS containing dCas9-BirA*–on-target gRNA versus dCas9-BirA*–NT gRNA.
(D) FACT immunodepletion inhibits dCas9 eviction in HSS. A 10-fold molar excess of Cas9 was added to dCas9 RNP-plasmid complexes incubated in SSRP1 or SPT16-immunodepleted extract either in the presence or absence of recombinant FACT. See also Figure S1E.
(E) FACT promotes Cas9’s multi-turnover activity. Cas9 RNPs were incubated with a plasmid substrate in a 1:2 molar ratio with either buffer, mock-immunodepleted HSS with ATP, SSRP1-immunodepleted HSS with ATP, or HSS with CIP and ATPγS for 180 min.
(F) Recombinant FACT displaces dCas9 pre-loaded on a plasmid in a minimal buffer system.
Three major sets of DNA-bound Cas9 interactors were apparent by comparing the on-target samples in which Cas9 was bound to the plasmid to the NT background control in which Cas9 was unbound to the target: PIP4K2C; H/ACA-associated proteins DKC1, NHP2, NOP10, and GAR1; and both components of the FACT heterodimer, SPT16 and SSRP1 (Figure 2B and Table S1). PIP4K2C is a lipid kinase that converts phosphatidylinositol-4-phosphate to phosphatidylinositol-4,5-bisphosphate. PIP4K2C is not explicitly linked to DNA metabolism, but it has recently been found that phosphoinositides accumulate at sites of double-stranded DNA damage (Wang et al., 2017). H/ACA RNPs are involved in pseudouridylation of RNA, maintenance of telomere integrity, and ribosome biogenesis (Kiss et al., 2010). The ability of H/ACA-associated proteins to interact with unique RNA secondary structures and preserve genomic integrity could imply roles in the cellular response to Cas9 binding. However, PIP4K2C and H/ACA proteins are not known to destabilize protein-DNA interactions and thus were not top candidates for being the Cas9 release factors in HSS.
The FACT complex is a histone chaperone with established roles in nucleosome assembly and remodeling and thus represented an attractive candidate to mediate Cas9 removal in HSS. FACT is a heterodimer consisting of SSRP1 and SPT16, both of which were strongly enriched by proximity biotinylation and unbiased proteomics. FACT promotes chromosomal transactions by removing the H2A-H2B dimer specifically or generally weakening histone contacts within chromatin (Okuhara et al., 1999; Orphanides et al., 1998; Winkler and Luger, 2011). Individual testing by Streptavidin pulldown and Western blotting confirmed that binding of dCas9-BirA* to a plasmid leads to increased biotinylation of SSRP1 and SPT16 (Figure 2C).
To determine whether FACT is necessary to displace Cas9 in HSS, we immunodepleted either SSRP1 or SPT16 from HSS and ensured that immunodepletion of either FACT subunit did not affect Cas9 degradation (Figure S1E). In the single-turnover competition assay, immunodepletion of either FACT component prevented even a 10-fold molar excess of Cas9 from accessing a DNA target that was pre-bound by dCas9 (Figure 2D). Add-back of recombinant human FACT to SSRP1 or SPT16-immunodepleted extracts rescued the ability of HSS to dislodge dCas9 (Figure 2D). Notably, under multi-turnover conditions, we found that immunodepletion of SSRP1 from HSS was sufficient to convert Cas9’s activity from multi-turnover to apparently single-turnover (Figure 2E). FACT is therefore necessary to displace Cas9 from DNA substrates in a cell-free system. To determine whether FACT is also sufficient to displace Cas9, we incubated recombinant human FACT with a DNA target pre-bound by dCas9 under single-turnover conditions in a minimal buffer system (Figure 2F). Even in buffer alone, FACT was capable of displacing dCas9 to expose the target site to Cas9 (Figure 2F).
FACT Depletion Increases dCas9 Residence Times in Human Cells
To directly monitor FACT’s ability to displace Cas9 in live human cells, we conducted fluorescence recovery after photobleaching (FRAP) microscopy. To generate visible foci of Cas9 bound to the genome, we stably expressed dCas9-HaloTag in a U2OS cell line containing tandem arrays of lacO and tetO sites in a single locus (Janicki et al., 2004) (Figure 3A and Figure S2A). Each array contained roughly 50,000 repeats of lacO and 75,000 repeats of tetO. We then co-transfected a tetO gRNA plasmid (Table S2) and either a NT or SPT16 siRNA. Depletion of SPT16 led to a concomitant reduction in SSRP1 levels (Figure S2B) consistent with prior reports that levels of the two FACT subunits are interdependent (Safina et al., 2013). Forty-eight hours after transfection, we labeled HaloTag with the dye JF549, photobleached dCas9-HaloTag molecules bound to the synthetic arrays, and monitored fluorescence recovery with confocal imaging.
Figure 3. FACT Depletion Enhances dCas9 Binding Times in Human Cells.

(A) Schematic of synthetic arrays bound by dCas9-HaloTag. See also Figure S2A.
(B) Representative FRAP images of dCas9-HaloTag targeted to arrays in U2OS cells treated with either NT (top) or SPT16 (bottom) siRNAs. Scale bars = 10 μm.
(C) Compiled FRAP curves for cells treated with either NT (N = 27) or SPT16 (N = 31) siRNAs. Black lines are the average recovery curves for each condition. See also Figure S2C.
(D) Quantification of fraction recovery 500s after photobleaching for cells transfected with NT or SPT16 siRNAs. Statistical significance was calculated through a Mann-Whitney U Test.
Three-dimensional FRAP imaging at the arrays revealed that dCas9-HaloTag within cells containing wild-type levels of FACT (N = 27) recovered to roughly half of their original fluorescence levels 500 seconds after photobleaching (Figures 3B, 3C, and 3D). dCas9-HaloTag within cells treated with SPT16 siRNAs (N = 31) on average recovered less than a third of their original fluorescence levels after the same time period (Figures 3B, 3C, and 3D), indicating that FACT is likely responsible for displacing Cas9 from the genome in live cells. FACT knockdown extended the mean dCas9-HaloTag residence times at the arrays by approximately 30% from 183.5 to 238.3 seconds (Figure S2C).
Knockdown of FACT Alters Cas9 Genome Editing Outcomes in Human Cells
To determine how FACT knockdown affects phenotypic outcomes of Cas9-based interventions, we first asked whether FACT influences Cas9-based genome editing in intact human cells. We specifically measured editing rates and outcomes via amplicon next generation sequencing (amplicon-NGS) after FACT knockdown (Tables S2-4). Sixty hours after transfecting K562 cells with either NT or SPT16 siRNAs (Figure S3A), we electroporated separate cultures of cells with Cas9 RNPs targeted to eight different loci, including a non-transcribed gene desert. For each locus, we performed editing reactions with and without a matched single-stranded oligodeoxynucleotide (ssODN) HDR donor that programs a PAM mutation at the appropriate locus.
Knockdown of FACT did not significantly alter indel frequencies measured after 48 hours in the absence of an HDR donor (Figure S3B). Consistent with previous reports (Richardson et al., 2016a), inclusion of an ssODN donor increased total editing (indels plus HDR) (Figure S3C). This increase in editing was consistent across all eight gRNAs tested and rescued otherwise relatively ineffective gRNAs.
In the presence of an ssODN, siRNA-mediated depletion of SPT16 did not affect total editing frequencies relative to the NT siRNA control 48 hours after Cas9 RNP electroporation (Figure S3D). However, a time course of editing rates at one site (VEGFA) (Figure S3E) revealed that SPT16 knockdown significantly impeded the rate of HDR (Figure 4A). HDR levels for SPT16-depleted cells was approximately 50% of that in NT siRNA-treated cells 12 hours after electroporation of Cas9 RNPs (Figure 4A). Indels in SPT16-depleted cells were somewhat reduced at early timepoints but caught up to that of NT siRNA-treated cells within 24 hours (Figure 4B). Measurements taken 48 hours after electroporation indicate that SPT16 knockdown ultimately increased indel frequencies and concomitantly reduced HDR frequencies by up to 50% at multiple loci (Figures 4C and 4D). At the eight loci tested, we observed no marked difference in the effect of SPT16 knockdown on gRNAs that targeted the coding or non-coding strand (Figure 4C and 4D). While knockdown of SPT16 did not alter the relative abundance of the five most common edited alleles containing indels for a CD59 locus, depletion of FACT altered their absolute frequencies and the relative abundance of several alleles each comprising less than 1% of all editing outcomes (Figure 4E). We found similar conservation of the most frequent allele but re-ordering of minor alleles after editing the VEGFA locus (Figure S3F). Both CD59 and VEGFA loci possessed a long tail of rare editing outcomes that were mostly deletions (Figure S3G).
Figure 4. FACT Alters Cas9 Genome Editing Outcomes in Human Cells.

(A) HDR rates from amplicon-NGS sequencing of VEGFA 0, 1, 2, 3, 6, 9, 12, and 24 hours after electroporation of Cas9 RNPs in the presence of an HDR donor (N = 3 biological replicates). Data are represented as mean ± standard deviation. See also Figure S3E.
(B) Indel rates from amplicon-NGS sequencing of VEGFA 0, 1, 2, 3, 6, 9, 12, and 24 hours after electroporation of Cas9 RNPs in the presence of an HDR donor (N = 3 biological replicates). Data are represented as mean ± standard deviation. See also Figure S3E.
(C) HDR rates from amplicon-NGS sequencing of eight different loci 48 hours after electroporation of Cas9 RNPs in the presence of an HDR donor (N = 3 biological replicates). Data are represented as mean ± standard deviation. See also Figure S3D.
(D) Indel rates from amplicon-NGS sequencing of eight different loci 48 hours after electroporation of Cas9 RNPs in the presence of an HDR donor (N = 3 biological replicates). Data are represented as mean ± standard deviation. See also Figure S3D.
(E) Representative alleles from cells edited with Cas9 programmed with a CD59 gRNA and a PAM-out ssODN HDR donor. See also Figure S3G.
Knockdown of FACT Increases Epigenetic Marking and Transcriptional Phenotypes from dCas9-Based Effectors in Human Cells
We next interrogated whether increased Cas9 residence times after FACT depletion influences the potency of dCas9-based transcriptional effectors that rely on the recruitment of epigenetic modifying enzymes to a target site. We specifically investigated whether FACT influences the deposition of chromatin marks by two different modifiers that can be fused to dCas9. The histone acetyltransferase p300 directly deposits H3K27ac marks on chromatin to upregulate transcription and has been deployed for CRISPR activation (CRISPRa) (Hilton et al., 2015). The Kruppel-associated box (KRAB) domain is a transcriptional repressor that recruits other factors to methylate histones and has been used for CRISPR interference (CRISPRi) (Gilbert et al., 2013).
We began by interrogating the role of FACT in dCas9-based histone acetylation. We generated HEK293T cells that stably express both dCas9-p300 and a gRNA targeting the CD25 TSS on either the coding or non-coding strand (Figure S4A and Table S2). Also known as IL2RA, CD25 encodes a subunit of the interleukin-2 receptor and is poorly expressed under basal conditions (Uhlen et al., 2015), but its expression can be induced using CRISPRa (Simeonov et al., 2017). We transfected NT or SPT16 siRNAs into dCas9-p300 cells expressing either a coding or non-coding strand gRNA (Figure S4B). Relative to the NT siRNA control, SPT16 depletion induced a significant increase in H3K27ac levels with either targeted gRNA according to qPCR using primers (Table S5) that amplified either a region upstream (Figure 5A) or inclusive (Figure S4C) of the protospacer. Knockdown of SPT16 did not increase basal histone acetylation when dCas9-p300 was paired with a NT gRNA (Figures 5A and S4C). siRNA knockdown of SSRP1 in the dCas9-p300 cell lines yielded similar increases in H3K27ac levels as knockdown of SPT16 (Figure S4D).
Figure 5. FACT Depletion Increases Epigenetic Marking and Transcriptional Phenotypes From dCas9-based Effectors in Human Cells.

(A) Knockdown of SPT16 increases H3K27 acetylation in HEK293T dCas9-p300 cells (N = 3 biological replicates). Fold enrichment is the amount of H3K27ac after SPT16 depletion normalized to the amount of H3K27ac after treatment with a NT siRNA. qPCR primers amplified a region 9 base pairs upstream of the non-coding-strand gRNA protospacer and 46 base pairs upstream of the coding strand gRNA protospacer. Data are represented as mean ± standard deviation. See also Figure S4.
(B) Knockdown of SPT16 increases H3K9 methylation in K562 dCas9-KRAB cells (N = 3 biological replicates). Fold enrichment is the amount of H3K9me2 after SPT16 depletion normalized to the amount of H3K9me2 after treatment with a NT siRNA. qPCR primers amplified a region 66 base pairs upstream of both the non-coding and coding strand gRNA protospacers. Data are represented as mean ± standard deviation. See also Figure S4.
(C) Representative histograms of CD55 levels in CRISPRi cells after treatment with NT or SPT16 siRNAs.
(D) FACT depletion enhances dCas9-KRAB-mediated knockdown of CD55 in K562 cells expressing CD55 TSS gRNAs (N = 3 biological replicates). CRISPRi cells were stained with FITC anti-CD55 after transfection of either NT or SPT16 siRNAs. gRNAs bind to either the coding (C) or non-coding (NC) strand and are labeled according to their distance in base pairs from the TSS. Data are represented as mean ± standard deviation. See also Figure S5.
(E) Representative histograms of CD59 levels in CRISPRi cells after treatment with NT or SPT16 siRNAs.
(F) FACT depletion enhances dCas9-KRAB-mediated knockdown of CD59 in K562 cells expressing CD59 TSS gRNAs (N = 3 biological replicates). CRISPRi cells were stained with FITC anti-CD59 after transfection of either NT or SPT16 siRNAs. gRNAs are labeled as in (D). Data are represented as mean ± standard deviation. See also Figure S5.
We used a similar approach to interrogate the role of FACT in dCas9-based histone methylation with K562 cells stably expressing dCas9-KRAB. We took advantage of conveniently located PAMs to target dCas9-KRAB to either the coding or non-coding strand of an identical location at the CD55 TSS (Figure S4E). CD55 is a ubiquitously and highly expressed gene (Uhlen et al., 2015) that encodes a cell surface glycoprotein involved in the complement system. Transfection of SPT16 siRNAs increased levels of H3K9 methylation when dCas9-KRAB was targeted with a gRNA to either the coding or non-coding strand (Figures 5B and S4F and Table S5). Knockdown of SPT16 did not increase basal histone methylation when dCas9-KRAB was paired with a NT gRNA (Figures 5B and S4F). We found similar increases in H3K9me2 using an siRNA against SSRP1 (Figure S4G).
We asked whether increased epigenetic marking by dCas9-based effectors during knockdown of FACT translates into increased transcriptional phenotypes. Targeting dCas9-p300 to the CD25 TSS with gRNAs at varying distances from the TSS (Table S2) generated cell populations that were between 12% and 95% CD25-positive, but knockdown of SPT16 did not further increase CD25 expression for any gRNA tested (Figures S5A and S6).
Targeting dCas9-KRAB to the CD55 TSS with gRNAs at varying distances from the TSS (Table S2) generated cell populations that were between 8% and 77% CD55-positive (Figures 5C, 5D, and S6). Notably, siRNA knockdown of SPT16 potentiated the degree of CRISPRi as measured by a decrease in CD55-positive cells (Figures 5C, 5D, and S6) with some CRISPRi cell populations exhibiting more than a 60% drop in CD55-positive cells upon SPT16 depletion. This transcriptional phenotype was observed regardless of the strand to which the gRNA bound. We found similar potentiation of CRISPRi transcriptional phenotypes after SPT16 knockdown when targeting CD59 (Figures 5E, 5F, and S6), which is even more highly expressed on the cell surface than CD55 (Uhlen et al., 2015). Increased CRISPRi was still dependent on proper targeting of dCas9-KRAB to the typical CRISPRi window around each gene’s TSS (Gilbert et al., 2013) as targeting dCas9-KRAB several kilobase pairs downstream of the TSS was ineffective even during knockdown of FACT (Figures S5B and S6). Similarly, localizing dCas9 unattached to an effector at various distances from the CD55 or CD59 TSSs did not affect transcription even after SPT16 knockdown (Figures S5C, S5D, and S6).
Discussion
Programmable prokaryotic nucleases such as Cas9 are widely used for eukaryotic genome and transcriptome manipulation, but it is largely unclear how host cells interface with these foreign enzymes. While several studies have uncovered how histones impede Cas9 target search and binding (Hilton et al., 2015; Horlbeck et al., 2016; Kallimasioti-Pazi et al., 2018; Knight et al., 2015; Yarrington et al., 2018), none have described how proteins responsible for restructuring and remodeling nucleosomes might affect Cas9. We have found that the histone chaperone FACT is required for Cas9 unloading and multi-turnover activity in cell-free extract. In live human cells, direct observations of dCas9 turnover at a synthetic array revealed that FACT promoted displacement of dCas9. Knockdown of FACT also inhibited templated repair of Cas9-induced breaks, increased indel formation, and increased the efficacy of dCas9-based transcriptional effectors.
FACT not only maintains nucleosome integrity by tethering the H3-H4 tetramer to DNA and helping deposit H2A-H2B dimers after displacement but also promotes nucleosome disassembly by dislodging H2A-H2B dimers and uncoiling DNA (Chen et al., 2018; Gurova et al., 2018; Hsieh et al., 2013; Kemble et al., 2015; Valieva et al., 2016; Winkler et al., 2011). Cas9 is a prokaryotic enzyme never before seen by eukaryotic histone chaperones, making it unlikely that FACT specifically recognizes Cas9. The simplest model is that FACT’s nucleosome-displacing capabilities allow it to displace genomic roadblocks such as Cas9. Indeed, a recent structure of FACT bound to a nucleosome reveals that FACT primarily contacts the double-stranded DNA and makes only peripheral interactions with histones (Liu et al., 2020). This finding could imply that FACT scans genomic DNA looking for impediments and helps resolve them when encountered. Our data do not rule out the potential importance of other histone chaperones or chromatin remodelers in genome surveillance and Cas9 displacement but highlight a prominent role of FACT in this process.
Modulating the turnover of Cas9 from a eukaryotic genome could inform the extent to which genome editing and transcriptional regulation rely upon repeated rounds of Cas9 binding and eviction. Indeed, we found that knockdown of FACT had no effect on basal epigenetic marking when dCas9-p300 or dCas9-KRAB was paired with a NT gRNA but increased specific epigenetic marking when either effector was guided to multiple target sites. The situation of DNA double-strand break repair is more complex. Kinetic analysis of Cas9-induced break repair has suggested that cells primarily invoke error-prone pathways to slowly repair Cas9 damage in a single round (Brinkman et al., 2018). Conversely, experiments inducing adjacent Cas9 breaks or modulating DNA repair with non-homologous single-stranded DNA implied that cells invoke error-free repair pathways that enable repeated rounds of Cas9 binding and eviction preceding eventual end-point mutation (Guo et al., 2018; Richardson et al., 2016a; Wang et al., 2019). We found that knockdown of FACT reduced Cas9-induced HDR and increased indels, which might imply that the balance between HDR and indels is influenced by multiple rounds of Cas9 cleavage and turnover. However, we note that histone chaperones and chromatin remodelers can also directly influence DNA repair through both direct and indirect mechanisms (Aleksandrov et al., 2018; Ayrapetov et al., 2014; Gospodinov et al., 2011; Lademann et al., 2017; Lans et al., 2012; Piquet et al., 2018; Price and D'Andrea, 2013). For example, FACT helps recruit DNA repair factors such as RNF20 (Oliveira et al., 2014). However, enforced chromatin relaxation and reduced nucleosome occupancy are sufficient to promote RNF20 localization, H2AX deposition, and end resection (Kari et al., 2011; Oliveira et al., 2014; Piquet et al., 2018). It is still unclear whether Cas9 owes its genome editing prowess to single-turnover or multi-turnover kinetics, but it is likely that the same nucleosome remodeling activity that promotes Cas9 displacement also promotes HDR.
While Cas9 unloading within X. laevis egg extracts is ATP-dependent, FACT activity is ATP-independent (Orphanides et al., 1998). Notably, we found that depletion of ATP or immunodepletion of FACT are both sufficient to abrogate the multi-turnover behavior of Cas9 in Xenopus extract. It is possible that egg extracts actively recruit FACT to DNA-bound Cas9 and dCas9 in an ATP-dependent manner. We note that the most enriched factor in our proteomics data set is the lipid kinase PIP4K2C, and recent work has revealed that phosphoinositides localize around DNA lesions to recruit proteins through the lipids’ associations with Pleckstrin Homology (PH) domains (Wang et al., 2017). Intriguingly, both SPT16 and SSRP1 contain PH domains (Kemble et al., 2013; Zhang et al., 2015), but a great deal of additional work is necessary to determine if lipid metabolism around the R-loop generated by Cas9 binding is responsible for recruitment of FACT to Cas9.
While RNA polymerases are capable of dislodging Cas9 in a strand-specific manner in vitro (Clarke et al., 2018), our data argue that FACT plays a prominent role in Cas9 removal within eukaryotic systems. While FACT is often associated with transcription (Mason and Struhl, 2003; Saunders et al., 2003), it possesses nucleosome remodeling activity separate from RNA polymerases. The egg extract we employed is transcriptionally silent and does not initiate DNA replication (Lebofsky et al., 2009), indicating that basal FACT activity decoupled from transcription or replication within egg extract may be sufficient to remove Cas9 from its substrate. While previous studies have reported RNA polymerase-mediated displacement of Cas9 bound to the non-coding strand (Clarke et al., 2018), we and others have found no strand bias in dCas9-based epigenetic reprogramming (Gilbert et al., 2013; Hilton et al., 2015; Konermann et al., 2015; Qi et al., 2013).
Targeting dCas9 downstream of a TSS in E. coli effectively suppresses gene expression presumably because dCas9 acts as a potent transcriptional roadblock to the bacterial RNA polymerase (Qi et al., 2013). Transcriptional reprogramming in human cells is less effective with dCas9 alone and is greatly increased by recruitment of an epigenetic modifier (Gilbert et al., 2013; Qi et al., 2013), suggesting that dCas9 is not a major roadblock to human RNA polymerases. We found that depleting FACT increased epigenetic marking and CRISPRi phenotypes for both coding and non-coding gRNAs across multiple loci. Notably, we also found that depleting FACT was insufficient to turn dCas9 alone into a transcriptional roadblock. These results indicate that either that human RNA polymerases can displace Cas9 or that dCas9 occupancy is so low to begin with that any small increase in dCas9 residence times after FACT depletion is not sufficient to generate a detectable effect on transcription. These results are consistent with Cas9’s utility as a generalized genome editing tool effective at editing both highly transcribed genes and transcriptionally silent regions.
FACT’s Cas9-displacing activity markedly influences epigenetic reprogramming by dCas9-fused effectors. Knockdown of FACT in dCas9-p300 cells induced an up to 7-fold increase in histone acetylation. However, FACT depletion did not increase CRISPRa transcriptional phenotypes in these cells even though recruiting histone acetyltransferases such as p300 has been previously shown to effectively upregulate transcription (Hilton et al., 2015). These results thus suggest that factors beyond histone acetylation are the critical bottlenecks to increased CD25 expression. By contrast, increasing dCas9-KRAB’s residence time by globally downregulating FACT potentiates both dCas9-based histone methylation and transcriptional downregulation. This downregulation was evident only when we localized the KRAB domain around TSSs. Together with prior observations that Cas9-effectors are ineffective as short-lived RNPs but potent when expressed through permanent lentiviral constructs, our data suggest that the effectiveness of CRISPRi depends upon dCas9’s residence time at a TSS. Future approaches might specifically increase residence time without affecting other genome transactions.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Jacob E. Corn (jacob.corn@biol.ethz.ch).
Materials Availability
All plasmids are available upon request.
Data and Code Availability
Amplicon-NGS data were deposited in SRA as PRJNA634106. Code for amplicon sequencing data processing is available at https://github.com/staciawyman/cortado.
Experimental Model and Subject Details
HEK293T dCas9-p300 cells were a generous gift from Dr. Charles Gersbach. Parental K562 cells were acquired from the UC Berkeley Cell Culture Facility. K562 dCas9-KRAB were identical to those previously reported (Richardson et al., 2018). U2OS cells in which a cassette of tetO and lacO sequences had been integrated at high copy number at a single genomic locus (Janicki et al., 2004) were modified to stably express dCas9-HaloTag with a puromycin selection marker, and individual clones were isolated by limiting dilution. All cells were regularly tested for mycoplasma contamination. HEK293T and U2OS cells were maintained in DMEM with glutamax (Gibco) supplemented with 10% fetal bovine serum, 1% sodium pyruvate (Gibco), and 100 U/ml penicillin-streptomycin (Gibco). dCas9-HaloTag expressing cells were also maintained in 0.5 μg/ml puromycin and 50 μg/ml hygromycin. K562 cells were maintained in RPMI (Gibco) supplemented with 10% fetal bovine serum, 1% sodium pyruvate, and 100 U/ml penicillin-streptomycin. All cells were maintained at 37°C with 5% CO2.
Method Details
X. laevis HSS
X. laevis HSS was prepared as previously described (Lebofsky et al., 2009). Aliquots were snap frozen and thawed as necessary. To immunodeplete SSRP1, HSS diluted 1:10 in Unloading Buffer (20 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.01% Tween) was exposed to two rounds of SSRP1 antibody. For each round, 150 μl of Dynabeads™ Protein G (Thermo Fisher Scientific) was washed three times in PBS, resuspended in 35 μg of SSRP1 antibody (Santa Cruz Biotechnology # sc-74536) and 825 μl of PBS, incubated with rotation for 90 min at room temperature, washed three times with PBST, resuspended in 2.4 μl of HSS diluted 1:10 in Unloading Buffer to 24 μl, and mixed for 45 min at room temperature. SPT16 antibody was generously provided by Dr. Hasan Yardimci. To immunodeplete SPT16, HSS was incubated with two rounds of 200 μl SPT16 antibody conjugated to 150 μl of Dynabeads™ Protein G. Mock depletions were conducted with the same amount of IgG antibody (BioLegend #400101).
Cas9, FACT, RNA, and Donor DNA Preparation
Streptococcus pyogenes Cas9 (pMJ915, Addgene #69090) with two nuclear localization signal sequences and an HA tag at the C-terminus (Lin et al., 2014) was expressed in Rosetta2 DE3 (QB3-Berkeley Macrolab) cells. Cell pellets were sonicated, clarified, Ni2+-affinity purified (HisTraps, GE Life Sciences), TEV cleaved, cation-exchanged (HiTrap SP HP, GE Life Sciences), size excluded (Sephacryl S-200, GE Life Sciences) and eluted at 40 μM in 20 mM HEPES KOH pH 7.5, 5% glycerol, 150 mM KCl, and 1 mM DTT. dCas9, Cas9-BirA*, and dCas9-BirA* were similarly expressed and purified. Recombinant human FACT was generously provided by Dr. Danny Reinberg. gRNAs were generated by HiScribe™ (New England Biolabs) T7 in vitro transcription using PCR-generated DNA as a template and purified using RNeasy Mini columns (Qiagen) (dx.doi.org/10.17504/protocols.io.dm749m). ssODN donor DNA was obtained by ordering unmodified ultramer oligonucleotides (Integrated DNA Technologies). For generation of stable cell lines, gRNAs were cloned into the lentiviral pLG1-library vector (Addgene #84832) as previously described (Horlbeck et al., 2016).
Multi-Turnover Cas9 Activity
84 fmol of Cas9 diluted in Unloading Buffer to a volume of 1 μl was added to 504 fmol of gRNA diluted in Unloading Buffer to a volume of 0.5 μl. Cas9 and gRNA were incubated for 15 min at room temperature. 168 fmol of either linear or plasmid substrate was then added to the RNPs, and reaction mixtures were incubated for 45 min at room temperature. Next, either 3 μl of Unloading Buffer and 1 μl of 100 mM ATP, 3 μl of diluted HSS (1:8 in Unloading Buffer) and 1 μl of 100 mM ATP, 3 μl of mock-depleted and diluted HSS (1:8 in Unloading Buffer) and 1 μl of 100 mM ATP, 3 μl of SSRP1-depleted and diluted HSS (1:8 in Unloading Buffer) and 1 μl of 100 mM ATP, or 3 μl of diluted HSS (1:8 in Unloading Buffer) supplemented with CIP, ATPγS, and 1 μl of Unloading Buffer were added. 0.5 μl of CIP (New England Biolabs) and 0.6 μl of 34 mM ATPγS (Sigma-Aldrich) was added to every 18 μl of diluted HSS for the final condition. An additional 3 μl of Unloading Buffer and 1 μl of 100 mM ATP, 3 μl of diluted HSS (1:8 in Unloading Buffer) and 1 μl of 100 mM ATP, 3 μl of mock-depleted and diluted HSS (1:8 in Unloading Buffer) and 1 μl of 100 mM ATP, 3 μl of SSRP1-depleted and diluted HSS (1:8 in Unloading Buffer) and 1 μl of 100 mM ATP, or 3 μl of diluted HSS (1:8 in Unloading Buffer) supplemented with CIP and ATPγS and 1 μl of Unloading Buffer were added to the corresponding samples after 5, 10, 30, 60, 90, 120, and 150 min. Samples were mixed with Proteinase K (Sigma), incubated at 50°C for 30 min, and run on an agarose gel.
Plasmid Protection in HSS
504 fmol of the on-target gRNA diluted in 0.5 μl of Unloading Buffer was added to either 0.5 μl of Unloading Buffer, 420 fmol of dCas9 diluted in 0.5 μl of Unloading Buffer, or 420 fmol of BirA*-dCas9 diluted in 0.5 μl of Unloading Buffer. Samples were incubated at room temperature for 15 min, added to 84 fmol of plasmid DNA diluted in 2.0 μl of Unloading Buffer, and incubated for 45 min at room temperature. Either 18 μl of Unloading Buffer, 18 μl of HSS supplemented with 0.55 μl of ARS, or 18 μl of HSS supplemented with 0.5 μl of CIP (New England Biolabs) and 0.6 μl of 34 mM ATPgS (Sigma-Aldrich) was added to the reaction mixtures. A stock solution of ARS was generated by mixing 10 μl of 100 mM ATP (VWR), 5 μl of 2 M phosphocreatine (Sigma-Aldrich), and 0.5 μl of 5 mg/ml creatine phosphokinase (Sigma-Aldrich). Samples were incubated for 15 min at room temperature. Next, either 1 μl of Unloading Buffer or 4.2 pmol of Cas9 diluted in 0.5 μl of Unloading Buffer pre-complexed with 5.04 pmol of the on-target gRNA diluted in 0.5 μl of Unloading Buffer was added to the reaction mixtures and incubated for 30 min. Samples were incubated with Proteinase K (Sigma-Aldrich) at 50°C for 30 minutes and then run on an agarose gel. The percentage of linear DNA was quantified through ImageJ. For the single-turnover protection assay using immunodepleted extracts, either 18 μl of diluted HSS (1:10 in Unloading Buffer) that was mock-depleted, 18 μl of diluted HSS (1:10 in Unloading Buffer) depleted of SSRP1, or 18 μl of diluted HSS (1:10 in Unloading Buffer) depleted of SPT16 was added to the RNPs after the 45 min incubation with the plasmid.
For FACT add-back experiments, 2 μg of recombinant human FACT was added to 18 μl of diluted HSS (1:10 in Unloading Buffer) immunodepleted of SSRP1 or SPT16 and incubated with pre-formed dCas9 RNP-plasmid complexes for 4 hours at room temperature prior to addition of Cas9 RNPs.
Mass Spectrometry
5.04 pmol of the on-target gRNA diluted in 14 μl of Unloading Buffer was added to 4.2 pmol of either Cas9-BirA* or dCas9-BirA*. As a reference sample, 5.04 pmol of a non-targeting gRNA diluted in 14 μl of Unloading Buffer was added to 4.2 pmol of dCas9-BirA*. RNPs were incubated at room temperature for 15 min. Samples were then added to 52.4 pmol of plasmid DNA, and reaction mixtures were incubated for 45 min at room temperature. 45 μl of HSS supplemented with 1.36 μl of ARS and biotin at a final concentration at 5 μM was added to these samples, and the resulting solutions were incubated for 60 min at room temperature. 405 μl of Unloading Buffer, 2.5 μl of Apyrase (New England Biolabs), and 56.25 μl of Apyrase Buffer was added to samples, which were incubated at 30°C for 15 minutes. 25 μl of DNase I (New England Biolabs) and 56.26 μl of DNase I Buffer were then added to samples, which were incubated at 37°C for 15 minutes. Samples were diluted 1:2 in Unloading Buffer and then mixed with 250 μl of MyOne™ Streptavidin C1 Dynabeads™ (Thermo Fisher Scientific) that had been washed three times in 50 mM Tris pH 7.4, 500 mM NaCl, 0.4% SDS, 5 mM EDTA, and 1 mM DTT. Samples were incubated overnight at 4°C with rotation.
Beads were washed once with 1 ml of 0.1% sodium deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, and 50 mM HEPES pH 7.5, once with 1 ml of 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 10 mM Tris pH 8.1, and twice with 1 ml of 50 mM Tris pH 7.4, 50 mM NaCl. Beads were then washed five times with 1 ml of 50 mM ammonium bicarbonate and then resuspended in 100 μl of 50 mM ammonium bicarbonate containing 0.01% ProteaseMAX (Promega) and 3 μg of sequencing-grade trypsin (Promega). Samples were incubated with mixing at 37°C for 4 hours after which the supernatant was collected and transferred to a new tube. Beads were washed again with 50 μl of 50 mM ammonium bicarbonate, and supernatants were pooled. 2 μl of formic acid (Fisher Scientific) was added to acidify the samples to a pH of ~3.0. Samples were then spun down to dryness in a speedvac and submitted to the University of California, Davis Proteomics Core for Multi-Dimension Protein Identification Technology mass spectrometry. Trypsinized peptides were mapped to the X. laevis proteome using the PHROG database (Wuhr et al., 2014). Protein enrichment levels were analyzed by the Limma Bioconductor package.
Western Blots
For X. laevis HSS, samples of equal volumes were incubated with Laemmli Buffer (Bio-Rad) at 95°C for 5 min. For human tissue culture, cells were washed in PBS and then lysed in 1X RIPA Buffer (Millipore Sigma) supplemented with Halt™ Protease Inhibitors (Thermo Fisher Scientific) at 4°C for 60 min. Samples were spun down at 15,000 g for 15 min, and the protein concentrations of the cleared lysates were measured using a BCA Protein Assay Kit (Thermo Fisher Scientific). 30 ug of lysate was denatured by incubation with Laemmli Buffer at 95°C for 5 min.
Both X. laevis and mammalian protein samples were resolved on Mini-PROTEAN® TGX™ 420% gels (Bio-Rad), and resolved proteins were transferred (TransBlot Turbo, Bio-Rad) to nitrocellulose membranes. Membranes were blocked in 5% milk in TBST for 30 min at room temperature and incubated with primary antibodies in 5% milk in TBST overnight at 4°C. Membranes were washed three times in TBST, incubated with secondary antibodies (LI-COR Biosciences) in 5% milk in TBST for 45 min, and then exposed on an Odyssey® CLx Imaging System (LI-COR Biosciences). X. laevis protein levels were probed using the following antibodies: GAPDH (Cell Signaling Technology #2118 1:5000), SPT16 (Cell Signaling Technology #12191 1:1000), SSRP1 (Santa Cruz Biotechnology #sc-74536 1:1000), and Cas9 (Novus Biologicals #7A9-3A3 1:1000). Human cell protein levels were probed using the following antibodies: GAPDH (Cell Signaling Technology #2118 1:5000), SPT16 (Cell Signaling Technology #12191 1:1000), SSRP1 (BioLegend #609701 1:100), and Cas9 (Novus Biologicals #7A9-3A3 1:1000).
Lentiviral Packaging and Transduction
Lentiviral packaging of all constructs was performed in HEK293T cells. Plasmids were transfected using TransIT®-LT1 Transfection Reagent (Mirus) at a ratio of μg of total DNA to 3 μl of the transfection reagent. The plasmid mixture consisted of 50% lentiviral transfer plasmid, 40% ΔVPR plasmid, and 10% VSVG plasmid. Virus was harvested at 48 and 72 hours after transfection, passed through a 0.45 μM filter, and added to target cells for transduction. 48 hours after transduction, both K562 and HEK293T cells were exposed to puromycin at 1 μg/ml. Cells were maintained in media containing puromycin for at least two passages to ensure complete selection.
siRNA Transfection
For FRAP imaging, 150,000 cells were co-transfected with 1 μg of the tetO gRNA plasmid and 30 pmol of siRNA complexed with 4 μl of Lipofectamine™ 2000 (Thermo Fisher Scientific) in Opti-MEM (Gibco). For Western blots and flow cytometry, 50,000 cells were transfected with 7.5 pmol of siRNA complexed with 2.25 μl of Lipofectamine™ RNAiMAX (Thermo Fisher Scientific) in Opti-MEM (Gibco). For editing experiments, 1,200,000 K562 cells were transfected 180 pmol of siRNA complexed with 54 μl of Lipofectamine™ RNAiMAX in Opti-MEM (Gibco). Cells were transfected in the absence of penicillin-streptomycin. 12 hours after transfection, cells were transferred to fresh media containing penicillin-streptomycin. For ChIP experiments, 5,000,000 HEK293T or K562 cells were transfected with 750 pmol of siRNA complexed with 225 μl of Lipofectamine™ RNAiMAX in Opti-MEM (Gibco). The following siRNAs were used: SMARTpool ON-TARGETplus SUPT16H siRNA (Horizon Discovery #L-009517-00), ON-TARGETplus SSRP1 siRNA (Horizon Discovery #J-011783-07), and ON-TARGETplus Nontargeting Pool (Horizon Discovery #D-001810-10).
Fluorescence Recovery After Photobleaching
dCas9-HaloTag was stably expressed in a previously reported U2OS cell line containing arrays of lacO and tetO (Janicki et al., 2004). Targeting of dCas9-HaloTag to the arrays was confirmed by expressing mOrange-LacI-NLS to mark the array and labeling the HaloTag with the JF646 dye (Chong et al., 2018). For FRAP, forty-eight hours after co-transection of the gRNA plasmid and siRNA, cells were seeded onto glass bottom microwell dishes (MatTek Corporation) and labeled with 100 nm of JF549. Cells were imaged on an inverted Zeiss LSM 710 AxioObserver confocal microscope equipped with a 40x/NA 1.4 oil immersion objective and an incubation chamber at 37°C and 5% CO2. For each experiment, the FRAP spot was bleached with a 561 nm laser set to maximum power. Recovery was monitored by a time-lapse of Z-stacks at 10-20 second intervals with a 0.68 mm step size. Due to the long interval between Z-stacks, the fast mode of recovery immediately after photobleaching cannot be measured.
FRAP images were analyzed as previously described (Hansen et al., 2017). Briefly, for each FRAP movie, the XY position of the array was manually annotated, its Z position was inferred by the maximum intensity pixel in the XY annotated column, and the fluorescence intensity around the spot was integrated. An unbleached control spot was used to normalize for loss of fluorescence due to bleaching during recovery, and the FRAP curves were normalized so that the pre-bleach intensity was 1.
Flow Cytometry
Sixty hours after transfection of siRNAs, cells were washed once in 1% BSA in PBS and then stained on ice for 1 hour. Cells were stained in 50 μl of either a PE CD25 antibody (BioLegend #302606 1:100), FITC CD55 antibody (BioLegend #311306 1:100), or FITC CD59 antibody BioLegend #304706). Samples were also stained with PE Mouse IgG1 κ antibody (BioLegend #400112 1:100), FITC Mouse IgG1, κ antibody (BioLegend #400108 1:100), or FITC Mouse IgG2a, κ antibody (BioLegend #400208 1:100) as an isotype control. Cells were washed three times in 1% BSA in PBS. Fluorescence was measured using the Attune NxT Flow Cytometer (Thermo Fisher Scientific).
Chromatin Immunoprecipitation
Sixty hours after transfections, cells were trypsinized if necessary, washed once in PBS, and then incubated in 10 ml of 1% formaldehyde in PBS for 15 min at room temperature. Reactions were quenched with 1.54 ml of 1.5 M glycine. Samples were spun down, resuspended in 1 ml of ice-cold PBS, spun down again, and snap-frozen in liquid nitrogen.
Cell pellets were lysed after thawing on ice by incubation first in 10 ml of 50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, and Halt™ Protease Inhibitors at 4°C for 10 min. Samples were spun down, the supernatant was aspirated, and the pellet was resuspended in 10 ml of 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM Tris, and Halt™ Protease Inhibitors at 4°C for 10 min. Samples were spun down again, the supernatant was aspirated, and the pellets were resuspended in 900 μl of 10 mM Tris, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl, 0.1% sodium deoxycholate, 0.5% sarcosine, and Halt™ Protease Inhibitors. Resuspended samples were sonicated with a Sartorius probe sonicator with three 1-minute intervals with an amplitude of 70% and a cycle of 0.9. Sonicated samples were added to 5.1 ml of 10 mM Tris, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl, 0.1% sodium deoxycholate, 0.5% sarcosine, and Halt™ Protease Inhibitors, and 600 μl of 10% Triton X-100. Solutions were centrifuged at 4°C at max speed for 20 minutes. 150 μl of the supernatant was retained as input. 6 ml of supernatant was used for immunoprecipitation.
8 μg of H3K9me2 (Abcam ab1220), H3K27ac (Abcam ab4729), or IgG antibody (BioLegend #400101) was incubated with 100 μl of Protein G Dynabeads™ (Thermo Fisher Scientific) at room temperature for 2 hours, washed three times with 0.5% BSA in PBS, resuspended in 100 μl of 0.5% BSA in PBS, and added to 3 ml of cell lysate. Samples were incubated overnight at 4°C with rotation. Beads were then washed four times with 1 ml of 10 mM HEPES, 500 mM LiCl, 1 mM EDTA, 1% NP-40, and 0.7% sodium deoxycholate, washed once in 1 ml of TBS, resuspended in 200 μl of 50 mM Tris, 10 mM EDTA, and 1% SDS, and incubated at 65°C overnight. Supernatants were collected and added to 200 μl of TE. 100 μl of input lysate was added to 300 μl of TE. 1 μl of RNAse A (New England Biolabs) was added to the samples, which were incubated at 37°C for 1 hour. 4 μl of Proteinase K (Thermo Fisher Scientific) was added to the samples, which were incubated at 57°C for 1 hour. 2 ml of Buffer PB (Qiagen) was added to the samples, which were flowed over MinElute columns (Qiagen). Columns were washed with Buffer PE (Qiagen) and eluted in 30 μl of Buffer EB (Qiagen). Immunoprecipitation samples were diluted 1:3 in distilled water while input samples were diluted 1:49 in distilled water.
qPCR reactions were performed in a total volume of 10 μl containing 2 μl of diluted samples, 5 ul of 2X ssoFast™ EvaGreen Supermix with Low ROX (Bio-Rad), and primers (Table S5) each at a final concentration of 500 nM. Samples were run on an Applied Biosystems™ StepOne™ Real-Time PCR System (Fisher Scientific). The thermocycler was set for 95°C for 2 minutes and 40 cycles of 95°C for 5 seconds and 55°C for 10 seconds. Fold enrichment of the assayed genes over a control locus were calculated using the 2−ΔΔCt method.
Cas9 Electroporation
Cells were electroporated with Cas9 RNPs 60 hours after transfection of siRNAs. For each electroporation, 30 pmol of Cas9 was diluted to a final volume of 3.5 μl with Cas9 Buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl2, 10% glycerol, and 1 mM TCEP). Cas9 was incubated with 36 pmol of gRNA diluted to a final volume of 3.5 μl in Cas9 Buffer. The resulting mixture was incubated for 10 min at room temperature. For HDR experiments, 1 μl of 100 μM ssODN donor was then added to the RNPs. 200,000 cells were washed once in PBS, resuspended in 16 μl of Buffer SF (Lonza), added to the RNP complexes, and electroporated using the FF120 program on the 4D-Nucleofector™ (Lonza). Reaction mixtures were incubated at room temperature for 10 min after electroporation and then transferred to pre-warmed media.
Next-Generation Sequencing
Either 0, 1, 2, 3, 6, 9, 12, 24 or 48 hours after electroporation, genomic DNA was harvested cells using QuickExtract™ DNA Extraction Solution (Lucigen). 200 ng of genomic DNA from edited cells was amplified using primer pairs from primer set 1 in a 30-cycle PCR reaction (Table S4). PCR products were SPRI cleaned, and 25 ng of SPRI-cleaned amplicons were amplified again using primer pairs from primer set 2 (Table S4) in a 12-cycle PCR reaction. Amplicons from the second PCR were SPRI cleaned, and 10 ng of SPRI-cleaned amplicons were used in a 9-cycle PCR reaction with Illumina compatible primers. All PCRs were conducted with PrimeSTAR® GXL DNA Polymerase (Takara) according to the manufacturer’s instructions. Libraries were pooled and submitted to the Vincent J. Coates Genomics Sequencing Laboratory at the University of California, Berkeley for 300 bp paired-end cycle processing using an Illumina MiSeq sequencing Kit (Illumina Inc., San Diego, CA).
Samples were deep sequenced to a depth of at least 10,000 reads. Reads were trimmed of adapters and low quality bases. Paired reads were joined into a single read and aligned to the input reference and donor sequences using NEEDLE (Li et al., 2015). Editing outcomes were determined and quantified using a modified version (https://github.com/staciawyman/cortado) of CRISPResso (Pinello et al., 2016). Reads were classified as NHEJ if an insertion or deletion in the alignment overlapped a 6 bp window around the cut site. Reads were classified as HDR if they were not NHEJ and contained the primary edit specified in the donor sequence. Percent NHEJ and HDR are calculated as the number of reads divided by the number of aligned reads. Indel size distributions were visualized through CRISPResso2 (Clement et al., 2019).
Quantification and Statistical Analysis
All analysis was performed using data from three biological replicates. Data are presented as mean ± standard deviation. Statistical analyses were performed in the PRISM software using a Student’s t-test. FRAP data was analyzed using a Mann-Whitney U Test. Statistical details can be found in figures and the corresponding figure legends. Significance was defined as p < 0.05.
Supplementary Material
Table S1. Interactors of Cas9-BirA* and dCas9-BirA* in X. laevis HSS, Related to Figure 2
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SSRP1 | Santa Cruz Biotechnology | Cat#sc-74536; RRID:AB_1129655 |
| SPT16 | Dr. Hasan Yardimci | N/A |
| Purified Mouse IgG1 | BioLegend | Cat#400101 |
| GAPDH | Cell Signaling Technology | Cat#2118; RRID:AB_561053 |
| SPT16 | Cell Signaling Technology | Cat#12191; RRID: AB_2732025 |
| Cas9 | Novus Biologicals | Cat#7A9-3A3 |
| SSRP1 | BioLegend | Cat#609701; RRID:AB_315730 |
| PE anti-human CD25 | BioLegend | Cat#302606; RRID:AB_314276 |
| FITC anti-human CD55 | BioLegend | Cat#311306; RRID:AB_314863 |
| FITC anti-human CD59 | BioLegend | Cat#304706; RRID:AB_2076126 |
| PE Mouse IgG1 κ isotype control | BioLegend | Cat#400112; RRID:AB_2847829 |
| FITC Mouse IgG1, κ Isotype control | BioLegend | Cat#400108 |
| FITC Mouse IgG2a, κ isotype control | BioLegend | Cat#400208 |
| H3K9me2 | Abcam | Cat#ab1220; RRID:AB_449854 |
| H3K27ac | Abcam | Cat#ab4729; AB_2118291 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| S. pyogenes Cas9 | QB3-Berkeley Macrolab | N/A |
| S. pyogenes dCas9 | QB3-Berkeley Macrolab | N/A |
| S. pyogenes Cas9-BirA* | This manuscript | N/A |
| S. pyogenes dCas9-BirA* | This manuscript | N/A |
| Recombinant Human FACT | Dr. Danny Reinberg | N/A |
| Critical Commercial Assays | ||
| HiScribe™ T7 High Yield RNA Synthesis Kit | New England Biolabs | Cat#E2040S |
| Deposited Data | ||
| Amplicon-NGS Raw Sequence Reads | This manuscript | SRA:PRJNA634106 |
| Oligonucleotides | ||
| SMARTpool ON-TARGETplus SUPT16H siRNA | Horizon Discovery | Cat#L-009517-00 |
| ON-TARGETplus SSRP1 siRNA | Horizon Discovery | Cat#J-011783-07 |
| ON-TARGETplus Nontargeting Pool | Horizon Discovery | Cat#D-001810-10 |
| Recombinant DNA | ||
| pMJ915 | Lin et al., 2014 | Addgene Plasmid #69090 |
| pCRISPRia-v2 | Horlbeck et al., 2016 | Addgene Plasmid #84832 |
| Software and Algorithms | ||
| Cortado Amplicon-NGS Analysis | https://github.com/staciawyman/cortado | N/A |
Highlights.
Histone chaperone FACT is necessary & sufficient to remove Cas9 from DNA in vitro
FACT turns Cas9 from single turnover to multi-turnover
FACT depletion in human cells delays Cas9 DSB repair & alters editing outcomes
FACT depletion increases dCas9 residence to increase epigenetic marking & CRISPRi
Acknowledgements
A.S.W. is supported by the National Science Foundation Graduate Research Fellowships Program and the Li Ka Shing Foundation. R.A.W. is supported by postdoctoral fellowship 131415-PF-17-168-01-DMC from the American Cancer Society. Y.H., D.T.W., A.B.H., and X.D. are supported by NIH grants UO1-497 EB021236 and U54-DK107980. A.B.H. and X.D. are supported by California Institute for Regenerative Medicine grant LA1-08013. A.B.H is supported by the NIH predoctoral fellowship T32 GM098218. C.D.R., B.G.G., K.R.K., J.T.V., S.K.W., J.J.S., and J.E.C. are supported by the Li Ka Shing Foundation and Heritage Medical Research Institute. J.T.V. is supported by CIRM TRAN1-09292. J.C.W. is supported by NIH grant HL098316. J.C.W. is a Howard Hughes Medical Institute Investigator and an American Cancer Society Research Professor. This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. We would like to thank Dr. Danny Reinberg for generously providing recombinant FACT and Dr. Hasan Yardimci for generously providing the SPT16 antibody.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
J.E.C. is a co-founder of Spotlight Therapeutics.
References
- Aleksandrov R, Dotchev A, Poser I, Krastev D, Georgiev G, Panova G, Babukov Y, Danovski G, Dyankova T, Hubatsch L, et al. (2018). Protein Dynamics in Complex DNA Lesions. Mol Cell 69, 1046–1061.e5. [DOI] [PubMed] [Google Scholar]
- Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, and Price BD (2014). DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A 111, 9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, de Haas M, Holland HA, Akhtar W, and van Steensel B (2018). Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol Cell 70, 801–813.e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li G-W, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Dong L, Hu M, Wang Y-Z, Xiao X, Zhao Z, Yan J, Wang P-Y, Reinberg D, Li M, et al. (2018). Functions of FACT in Breaking the Nucleosome and Maintaining Its Integrity at the Single-Nucleosome Level. Mol Cell 71, 284–293.e284. [DOI] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, et al. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R, Heler R, MacDougall MS, Yeo NC, Chavez A, Regan M, Hanakahi L, Church GM, Marraffini LA, and Merrill BJ (2018). Enhanced Bacterial Immunity and Mammalian Genome Editing via RNA-Polymerase-Mediated Dislodging of Cas9 from Double-Strand DNA Breaks. Mol Cell 71, 42–55.e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, et al. (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096. [DOI] [PubMed] [Google Scholar]
- Gao XD, Tu L-C, Mir A, Rodriguez T, Ding Y, Leszyk J, Dekker J, Shaffer SA, Zhu LJ, Wolfe SA, et al. (2018). C-BERST: defining subnuclear proteomic landscapes at genomic elements with dCas9-APEX2. Nat Methods 15, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodinov A, Vaissiere T, Krastev DB, Legube G, Anachkova B, and Herceg Z (2011). Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol Cell Biol 31, 4735–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Feng Y-L, Xiao J-J, Liu Q, Sun X-N, Xiang J-F, Kong N, Liu S-C, Chen G-Q, Wang Y, et al. (2018). Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 19, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurova K, Chang H-W, Valieva ME, Sandlesh P, and Studitsky VM (2018). Structure and function of the histone chaperone FACT - Resolving FACTual issues. Biochim Biophys Acta Gene Regul Mech. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, and Darzacq X (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald R, Tournebize R, Blank T, Sandaltzopoulos R, Becker P, Hyman A, and Karsenti E (1996). Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature 382, 420–425. [DOI] [PubMed] [Google Scholar]
- Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom WS, Klein Douwel D, and Knipscheer P (2017). Xenopus egg extract: A powerful tool to study genome maintenance mechanisms. Dev Biol 428, 300–309. [DOI] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh F-K, Kulaeva OI, Patel SS, Dyer PN, Luger K, Reinberg D, and Studitsky VM (2013). Histone chaperone FACT action during transcription through chromatin by RNA polymerase II. Proc Natl Acad Sci U S A 110, 7654–7659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. (2004). From silencing to gene expression: real-time analysis in single cells. Cell 116, 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, and Doudna J (2013). RNA-programmed genome editing in human cells. Elife 2, e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalab P, Pralle A, Isacoff EY, Heald R, and Weis K (2006). Analysis of a RanGTP-regulated gradient in mitotic somatic cells. Nature 440, 697–701. [DOI] [PubMed] [Google Scholar]
- Kallimasioti-Pazi EM, Thelakkad Chathoth K, Taylor GC, Meynert A, Ballinger T, Kelder MJE, Lalevee S, Sanli I, Feil R, and Wood AJ (2018). Heterochromatin delays CRISPR-Cas9 mutagenesis but does not influence the outcome of mutagenic DNA repair. PLoS Biol 16, e2005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari V, Shchebet A, Neumann H, and Johnsen SA (2011). The H2B ubiquitin ligase RNF40 cooperates with SUPT16H to induce dynamic changes in chromatin structure during DNA double-strand break repair. Cell Cycle 10, 3495–3504. [DOI] [PubMed] [Google Scholar]
- Kemble DJ, McCullough LL, Whitby FG, Formosa T, and Hill CP (2015). FACT Disrupts Nucleosome Structure by Binding H2A-H2B with Conserved Peptide Motifs. Mol Cell 60, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble DJ, Whitby FG, Robinson H, McCullough LL, Formosa T, and Hill CP (2013). Structure of the Spt16 middle domain reveals functional features of the histone chaperone FACT. J Biol Chem 288, 10188–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, and Kim J-S (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24, 1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T, Fayet-Lebaron E, and Jady BE (2010). Box H/ACA small ribonucleoproteins. Mol Cell 37, 597–606. [DOI] [PubMed] [Google Scholar]
- Knight SC, Xie L, Deng W, Guglielmi B, Witkowsky LB, Bosanac L, Zhang ET, Beheiry EI,M, Masson J-B, Dahan M, et al. (2015). Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science 350, 823–826. [DOI] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, and Walter JC (2009). The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326, 1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, and Doudna JA (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lademann CA, Renkawitz J, Pfander B, and Jentsch S (2017). The INO80 Complex Removes H2A.Z to Promote Presynaptic Filament Formation during Homologous Recombination. Cell Rep 19, 1294–1303. [DOI] [PubMed] [Google Scholar]
- Lans H, Marteijn JA, and Vermeulen W (2012). ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R, Takahashi T, and Walter JC (2009). DNA replication in nucleus-free Xenopus egg extracts. Methods Mol Biol 521, 229–252. [DOI] [PubMed] [Google Scholar]
- Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, and Lopez R (2015). The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43, W580–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, and Doudna JA (2014). Enhanced homology-directed human genome engineering by controlled timing of CRISpR/Cas9 delivery. Elife 3, e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Y, Chen Y, Li M, Zhou F, Li K, Cao H, Ni M, Liu Y, Gu Z, et al. (2017). In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell 170, 1028–1043.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou K, Zhang N, Wei H, Tan YZ, Zhang Z, Carragher B, Potter CS, D'Arcy S, and Luger K (2020). FACT caught in the act of manipulating the nucleosome. Nature 577, 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Tu L-C, Naseri A, Huisman M, Zhang S, Grunwald D, and Pederson T (2016). CRISPR-Cas9 nuclear dynamics and target recognition in living cells. J Cell Biol 214, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio I, and Goncalves MAFV (2015). Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol 33, 280–291. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 7, 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, and Struhl K (2003). The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol Cell Biol 23, 8323–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SA, Wright J, Peckner R, Kalish BT, Zhang F, and Carr SA (2018). Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat Methods 15, 437–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelles DA, Fang MY, O'Connell MR, Xu JL, Markmiller SJ, Doudna JA, and Yeo GW (2016). Programmable RNA Tracking in Live Cells with CRISPR/Cas9. Cell 165, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama J, Mikuni T, and Yasuda R (2017). Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 96, 755–768.e755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuhara K, Ohta K, Seo H, Shioda M, Yamada T, Tanaka Y, Dohmae N, Seyama Y, Shibata T, and Murofushi H (1999). A DNA unwinding factor involved in DNA replication in cell-free extracts of Xenopus eggs. Curr Biol 9, 341–350. [DOI] [PubMed] [Google Scholar]
- Oliveira DV, Kato A, Nakamura K, Ikura T, Okada M, Kobayashi J, Yanagihara H, Saito Y, Tauchi H, and Komatsu K (2014). Histone chaperone FACT regulates homologous recombination by chromatin remodeling through interaction with RNF20. J Cell Sci 127, 763–772. [DOI] [PubMed] [Google Scholar]
- Orphanides G, LeRoy G, Chang CH, Luse DS, and Reinberg D (1998). FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92, 105–116. [DOI] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, and Yuan G-C (2016). Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquet S, Le Parc F, Bai S-K, Chevallier O, Adam S, and Polo SE (2018). The Histone Chaperone FACT Coordinates H2A.X-Dependent Signaling and Repair of DNA Damage. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BD, and D'Andrea AD (2013). Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper AT, Stephenson AA, and Suo Z (2018). Functional Insights Revealed by the Kinetic Mechanism of CRISPR/Cas9. J. Am. Chem. Soc 140, 2971–2984. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, Bray NL, and Corn JE (2016a). Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nat Commun 7, 12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CD, Kazane KR, Feng SJ, Zelin E, Bray NL, Schafer AJ, Floor SN, and Corn JE (2018). CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat Genet 50, 1132–1139. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, and Corn JE (2016b). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol 34, 339–344. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, and Burke B (2013). BioID: a screen for protein-protein interactions. Curr Protoc Protein Sci 74, Unit19.23. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safina A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, and Gurova KV (2013). Complex mutual regulation of facilitates chromatin transcription (FACT) subunits on both mRNA and protein levels in human cells. Cell Cycle 12, 2423–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, and Lis JT (2003). Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301, 1094–1096. [DOI] [PubMed] [Google Scholar]
- Schmidtmann E, Anton T, Rombaut P, Herzog F, and Leonhardt H (2016). Determination of local chromatin composition by CasID. Nucleus 7, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Zhang W, Hu H, Xue B, Qin J, Sun C, Sun Y, Wei W, and Sun Y (2016). Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system. Nucleic Acids Res 44, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov DR, Gowen BG, Boontanrart M, Roth TL, Gagnon JD, Mumbach MR, Satpathy AT, Lee Y, Bray NL, Chan AY, et al. (2017). Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, LaFrance B, Kaplan M, and Doudna JA (2015). Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, Hatanaka F, Yamamoto M, Araoka T, Li Z, et al. (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Valieva ME, Armeev GA, Kudryashova KS, Gerasimova NS, Shaytan AK, Kulaeva OI, McCullough LL, Formosa T, Georgiev PG, Kirpichnikov MP, et al. (2016). Large-scale ATP-independent nucleosome unfolding by a histone chaperone. Nat Struct Mol Biol 23, 1111–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nakamura M, Abbott TR, Zhao D, Luo K, Yu C, Nguyen CM, Lo A, Daley TP, La Russa M, et al. (2019). CRISPR-mediated live imaging of genome editing and transcription. Science 365, 1301–1305. [DOI] [PubMed] [Google Scholar]
- Wang H, Xu X, Nguyen CM, Liu Y, Gao Y, Lin X, Daley T, Kipniss NH, La Russa M, and Qi LS (2018). CRISPR-Mediated Programmable 3D Genome Positioning and Nuclear Organization. Cell 175, 1405–1417.e1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-H, Hariharan A, Bastianello G, Toyama Y, Shivashankar GV, Foiani M, and Sheetz MP (2017). DNA damage causes rapid accumulation of phosphoinositides for ATR signaling. Nat Commun 8, 2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DD, and Luger K (2011). The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J Biol Chem 286, 18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DD, Muthurajan UM, Hieb AR, and Luger K (2011). Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem 286, 41883–41892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhr M, Freeman RMJ, Presler M, Horb ME, Peshkin L, Gygi S, and Kirschner MW (2014). Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr Biol 24, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarrington RM, Verma S, Schwartz S, Trautman JK, and Carroll D (2018). Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Proc Natl Acad Sci U S A 115, 9351–9358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yourik P, Fuchs RT, Mabuchi M, Curcuru JL, and Robb GB (2019). Staphylococcus aureus Cas9 is a multiple-turnover enzyme. Rna 25, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zeng F, Liu Y, Shao C, Li S, Lv H, Shi Y, Niu L, Teng M, and Li X (2015). Crystal Structure of Human SSRP1 Middle Domain Reveals a Role in DNA Binding. Sci Rep 5 18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Interactors of Cas9-BirA* and dCas9-BirA* in X. laevis HSS, Related to Figure 2
Data Availability Statement
Amplicon-NGS data were deposited in SRA as PRJNA634106. Code for amplicon sequencing data processing is available at https://github.com/staciawyman/cortado.