

Abstract
Gulf War Illness (GWI) is a chronic multi-symptom disorder, characterized by symptoms such as fatigue, pain, cognitive and memory impairment, respiratory, skin and gastrointestinal problems, that is experienced by approximately one-third of 1991 Gulf War veterans. Over the nearly three decades since the end of the war, investigators have worked to elucidate the initiating factors and underlying causes of GWI. A significant portion of this research has indicated a strong correlation between GWI and exposure to a number of different acetycholinesterase inhibitors (AChEIs) in theater, such as sarin and cyclosarin nerve agents, chlorpyrifos and dichlorvos pesticides, and the anti-nerve agent prophylactic pyridostigmine bromide. Through studying these exposures and their relationship to the symptoms presented by ill veterans, it has become increasingly apparent that GWI is the likely result of an underlying neuroimmune disorder. While evidence indicates that AChEIs are a key exposure in the development of GWI, particularly organophosphate AChEIs, the mechanism(s) by which these chemicals instigate illness appears to be related to “off-target”, non-cholinergic effects. In this review, we will discuss the role of AChEI exposure in the development and persistence of GWI; in particular, how these chemicals, combined with other exposures, have led to a chronic neuroimmune disorder.
This article is part of the special issue entitled ‘Acetylcholinesterase Inhibitors: From Bench to Bedside to Battlefield’.
Keywords: Gulf war illness, Acetylcholinesterase inhibitor, Neuroimmune, Neuroinflammation
1. Introduction
Acetylcholinesterase inhibitors (AChEIs) comprise a class of chemicals that can be generally categorized as therapeutics, pesticides, or nerve agents. During the 1991 Gulf War, soldiers had the potential to be exposed to all three of these categories of AChEIs: the pesticides chlorpyrifos (CPF) and dichlorvos (DDVP) were regularly and pervasively used for pest control, pyridostigmine bromide (PB) was prescribed to be taken every 8 h as a prophylactic against potential nerve agent exposure, and the demolition of ammunition storage facilities released sarin and cyclosarin nerve agents and potentially exposed veterans to these chemical weapons (Tuovinen et al., 1999; Institute of Medicine (US) Committee on Health Effects Associated with Exposures During the Gulf War, 2000; Research Advisory Committee on Gulf War Veteran’ Illnesses, 2008; White et al., 2016). Exposure to AChEIs has been repeatedly implicated as the potential cause of Gulf War Illness (GWI), a chronic multi-symptom disorder affecting nearly one-third of the veterans that returned from the 1991 conflict, by both epidemiological (Sullivan et al., 2003; Research Advisory Committee on Gulf War Veteran’ Illnesses, 2008; Steele et al., 2012; Kerr, 2015; White et al., 2016; Sullivan et al., 2018) and preclinical studies (Henderson et al., 2001; Amourette et al., 2009; Lamproglou et al., 2009; Abdullah et al., 2012; Abdullah et al., 2013; Parihar et al., 2013; Hattiangady et al., 2014; Ojo et al., 2014; Nutter et al., 2015; O’Callaghan et al., 2015; Zakirova et al., 2015; Abdullah et al., 2016; Cooper et al., 2016; Phillips and Deshpande, 2016; Pierce et al., 2016; Alhasson et al., 2017; Emmerich et al., 2017; Flunker et al., 2017; Locker et al., 2017; Shetty et al., 2017; Zakirova et al., 2017; Ashbrook et al., 2018; Carreras et al., 2018; Cooper et al., 2018; Koo et al., 2018; Macht et al., 2018; Miller et al., 2018; Phillips and Deshpande, 2018; Seth et al., 2018; Macht et al., 2019; Michalovicz et al., 2019). Acute exposure to toxic levels of AChEIs, particularly the organophosphate (OP) compounds like the pesticides and nerve agents, has a number of systemic effects (i.e. salivation, lacrimation, urination, defecation, gastrointestinal upset, emesis, miosis [SLUDGEM]) and can cause seizures. However, it is the long-term neurological symptoms associated with exposure to these compounds that harmonize with many of the long-lasting symptoms of GWI: fatigue, pain, mood disorders, cognitive and memory impairment (Fukuda et al., 1998; Steele, 2000; Haley et al., 2001; Golomb, 2008; Sullivan et al., 2003; Maule et al., 2018). Furthermore, the similarity of these symptoms to those of sickness behavior, the adaptive behavioral response elicited by illnessor infection-induced neuroinflammation (Dantzer and Kelley, 2007; Dantzer et al., 2008), has suggested that GWI is the result of an underlying chronic neuroimmune disorder; a hypothesis that has been supported by both preclinical animal (O’Callaghan et al., 2015; Zakirova et al., 2016; Alhasson et al., 2017; Locker et al., 2017; Shetty et al., 2017; Ashbrook et al., 2018; Carreras et al., 2018; Kodali et al., 2018; Koo et al., 2018; Macht et al., 2018, 2019; Miller et al., 2018; Hernandez et al., 2019; Joshi et al., 2019; Madhu et al., 2019; Michalovicz et al., 2019) and clinical studies (Broderick et al., 2013; Parkitny et al., 2015; White et al., 2016; Coughlin, 2017; Abou-Donia et al., 2017; Georgopoulous et al., 2017; Alshelh et al., 2020). In spite of the fact that the ACh signaling facilitated by AChE inhibition is typically anti-inflammatory (Pavlov et al., 2003; Pavlov and Tracey, 2005), several preclinical studies have directly shown a connection between GWI-relevant AChEI exposures and neuroinflammation (Ojo et al., 2014; O’Callaghan et al., 2015; Locker et al., 2017; Ashbrook et al., 2018; Koo et al., 2018; Miller et al., 2018). Moreover, while there are many hurdles to directly evaluate neuroinflammation in living humans, a recent clinical Positron Emission Tomography (PET) study found strong signals for the neuroinflammatory marker, 18 kDa transducer protein (TSPO), throughout the cortex of veterans with GWI compared to controls, providing the first direct, in vivo data to show neuroinflammation in the illness (Alshelh et al., 2020).
In this review, we will discuss the three major groups of AChEIs that soldiers were exposed to during the 1991 Gulf War that have been associated with GWI: nerve agents, pesticides, and pyridostigmine bromide. In particular, we will focus on discussing the preclinical body of data that has supported the hypothesis that GWI is the result of an underlying neuroinflammatory/neuroimmune disorder. Lastly, we will discuss emerging evidence that suggests that this neuroimmune dysfunction is the result of persistent, non-cholinergic effects of AChEI exposure.
1.1. Acetylcholinesterase inhibitor exposures in GWI
The emergence of a chronic multi-symptom disorder in veterans immediately following the 1991 Gulf War raised questions regarding the possibility that harmful exposures occurred during the war (Pennisi, 1996). Soldiers that were deployed during the 1991 Gulf War were exposed to a number of conditions that could have increased their risk for negative health impacts, including pesticides, nerve agent, oil well fire smoke, depleted uranium munitions, prophylactic drugs and vaccinations, paints, and psychological and/or physiological stress (White et al., 2016). However, the prevalence of neurological and neuropsychological symptoms among veterans with GWI has fueled an interest in the neurotoxicant exposures that may have been experienced in theater (White et al., 2016). Among these neurotoxicants, there was the potential for and, in some cases, documentation of, the exposure of soldiers to both irreversible and reversible AChEIs during deployment. The delineation between these two subcategories of AChEIs is important because the perceived risks associated with irreversible OP AChEIs, i.e. those pesticides and nerve agents that permanently bind and inactivate the enzyme, is greater than the reversible AChEIs (including pharmaceuticals) that only modulate enzyme activity through on-off binding dynamics. In this section, we will discuss both subclasses of AChEIs that have been investigated in GWI and how exposure to these compounds may contribute to the associated chronic neuroinflammatory dysfunction.
1.2. Sarin, cyclosarin, and nerve agent surrogates
Exposure to sarin and/or cyclosarin nerve agent, both OP AChEIs, was a very real risk to soldiers deployed during the 1991 Gulf War. Interviews with veterans of the 1991 Gulf War have indicated that many of them recall hearing chemical alarms in camp during deployment (Haley and Kurt, 1997; Kurt, 1998; Haley and Tuite, 2013; Chao et al., 2016). Additionally, several investigations have determined that the destruction of multiple munitions storage sites by U.S. troops released a plume containing both sarin and cyclosarin that is predicted to have exposed upwards of 100,000 soldiers (Institute of Medicine (US) Committee on Health Effects Associated with Exposures During the Gulf War, 2000; Research Advisory Committee on Gulf War Veteran’ Illnesses, 2008). Interestingly, a survey of soldiers that were in proximity to one of these detonation sites, Khamisiyah, found that the majority of respondents did not report any acute cholinergic symptoms (Institute of Medicine (US) Committee on Health Effects Associated with Exposures During the Gulf War, 2000). However, many studies of sick veterans have uncovered an association between potential nerve agent exposures and GWI presentation (see review in Golomb, 2008; White et al., 2016). While nerve agent exposure has been associated with incidence of high blood pressure, diabetes, arthritis and chronic bronchitis in GWI (Zundel et al., 2019), the observations of MRI-assessed brain structural abnormalities like decreased gray and white matter volumes and increased ventricle size (Heaton et al., 2007; Chao et al., 2010, 2011, 2014, 2015; Chao and Zhang, 2018), as well as poor neurobehavioral performance (White et al., 2001; Proctor et al., 2006; Toomey et al., 2009) and an increased risk for brain cancer (Bullman et al., 2005) are more indicative of the long-term brain effects of nerve agent exposure. These studies combined with the result of the Department of Defense survey suggest that GWI may be the result of low-level exposures to sarin and/or cyclosarin that went undetected due to a lack of immediate/acute neurotoxic effects.
Though posing an obvious neurotoxicological risk, only a handful of preclinical studies have investigated the role of potential sarin exposure in the development of GWI. A few of these studies have investigated the potential outcomes of low-dose, sub-lethal exposure(s) to sarin or sarin surrogates alone in relationship to GWI symptomology. While subclinical inhalation of sarin has been demonstrated to be capable of producing signs of immunosuppression, such as reduced T-cell responses and reduced corticosterone levels (Henderson et al., 2001; Kalra et al., 2002), as well as autonomic imbalance and cardiomyopathy (Shewale et al., 2012), these studies do not directly correlate with the neuroimmune dysfunction hypothesis for GWI. However, repeated, low-dose exposure to the sarin surrogate diisopropyl fluorophosphate (DFP) has been shown to instigate depression, cognitive impairment, and neuronal damage (Phillips and Deshpande, 2016, 2018). In these studies, rats were injected with 0.4–0.5 mg/kg, s.c. DFP once a day for 5 days and, when assessed three to six months after exposure, demonstrated memory impairments in novel object recognition and object location tasks, signs of anxiety and depression as indicated by increased immobility time in the forced swim test, reduced sucrose preference, and reduced time in the open arms of the elevated plus maze. These behavioral observations were concurrent with increased levels of neuronal calcium and neuronal cell damage in the hippocampus. In addition, 14-day exposure to the same dose of DFP was found to result in impaired axonal transport (Naughton et al., 2018). While there were no direct evaluations of neuroinflammation or neuroimmune dysfunction in these studies, an expanding body of research has suggested an association between neuroinflammation and depression (Walker et al., 2013), as well as axonal transport deficits (Errea et al., 2015). However, these results not only support a role for nerve agent exposure in the development of GWI symptoms, but also demonstrate that these low-dose exposures are capable of producing long-lasting effects that can translate to the decades of persistent illness experienced by veterans with GWI.
Deployed soldiers were exposed to significant physiological stressors such as exercise and extreme temperatures (Young et al., 1992; Sapolsky, 1998; Sullivan et al., 2003) that had the potential to interact with or modulate the responses to chemical exposures. As such, several studies investigating the contribution of nerve agent exposure to GWI have found positive correlations to illness symptomology when sarin surrogates have been combined with a stressor (O’Callaghan et al., 2015; Locker et al., 2017; Ashbrook et al., 2018; Koo et al., 2018; Miller et al., 2018; Craddock et al., 2018; Michalovicz et al., 2019; Belgrad et al., 2019). In this GWI rodent model, a single dose of DFP is preceded by a chronic (4–7 day) exposure to exogenous corticosterone (CORT) provided in the drinking water (200 mg/L) to mimic the high physiological stress experienced by soldiers during deployment. Though currently acute in scope, the extensive evaluation of this model has strongly supported the role of neuroimmune dysfunction as the underlying cause of GWI showing that this combination of exposures results in CORT priming of the DFP neuroinflammatory response leading to significantly increased inflammatory cytokine mRNA throughout the brain, including cortex, hippocampus, and striatum (O’Callaghan et al., 2015; Locker et al., 2017; Koo et al., 2018; Miller et al., 2018), along with alterations in neuroimmune signaling via histone modification and DNA methylation changes (Ashbrook et al., 2018) shortly after the exposures. Furthermore, evaluation of this paradigm using a literature-derived, logic model of neuron-glia interactions indicated the potential for GWI to derive from an aberrant homeostatic neuroinflammatory profile (Craddock et al., 2018). Notably, these neuroinflammatory effects occur in the absence of significant peripheral inflammation (Michalovicz et al., 2019). While DFP alone has some minor proinflammatory effects in the liver and serum, these effects are largely suppressed by the prior CORT exposure, findings that differentiate them from the CORT priming of the DFP response in the brain and highlight the role of neuroinflammation in GWI. Interestingly, similar results were found in a clinical evaluation of neuroinflammation where veterans with GWI presented with an increased neuroinflammatory PET signal in the cortex in spite of there being no difference in plasma cytokine levels in comparison to healthy controls (Alshelh et al., 2020).
While no changes have been observed in astrocytes or microglia, the brain’s primary immune cells, in the acute time points following exposure in this model (O’Callaghan et al., 2015), Belgrad et al. (2019) found that DFP alone had effects on oligodendrocytes, another glial cell type with immune function (Peferoen et al., 2014). In this study, DFP exposure decreased the number of mature and proliferating oligodendrocytes in the rat cortex and corpus callosum out to 21 days post-exposure while combined CORT and DFP exposure ameliorated these effects. However, this combined exposure resulted in an increase in myelin basic protein, a protein crucial to proper myelin structure, that may be indicative of demyelination or injury (Kristensson et al., 1986; Bartholdi and Schwab, 1998). As such, Naughton et al. (2018) found that their chronic DFP exposure paradigm resulted in disordered, de-compacted myelin sheaths. These exposures have also been translated into neuronal cell culture and shown to affect microtubule acetylation and axonal transport of mitochondria (Rao et al., 2017). In addition to these cellular and molecular level changes, high-order diffusion MRI of GWI rat brains demonstrated alterations in brain structure and connectivity concurrent with neuroinflammation that may be indicative of subtle structural changes in dendrites or glial processes (Koo et al., 2018). Taken at early time points following GWI exposure, these diffusion changes may capture the initiating conditions that have led to the more significant changes in brain structure reported by traditional MRI in veterans with GWI (Heaton et al., 2007; Chao et al., 2010, 2011, 2014, 2015; Chao and Zhang, 2018), and suggests neuroinflammation as an underlying cause.
While both sarin and DFP can significantly increase brain ACh levels by inhibiting AChE activity, the exacerbated neuroinflammation instigated by corticosterone priming of DFP exposure was associated with a reduction in ACh levels and mitigation of AChE inhibition in the brain (Locker et al., 2017; Miller et al., 2018). These studies provide strong evidence for a causative association between nerve agent and wartime stress exposure and GWI, suggesting that these exposures have resulted in a persistent shift in how the neuroimmune system functions; ultimately, allowing for a chronic neuroinflammatory state that underlies the neurological and systemic issues experienced by the ill veterans. These results also propose a mechanism by which a condition of high physiological stress facilitated the circumvention of the anti-inflammatory cholinergic signaling pathway. Furthermore, the other studies discussed here (Phillips and Deshpande, 2016, 2018; Naughton et al., 2018) used low doses of DFP that did not produce acute cholinergic crisis. Though the potential for veterans with GWI to have been exposed to chemical weapons containing nerve agent has been highly controversial in the past, studies that have investigated these exposures at low doses, alone or in combination with the high physiological stress experienced in theater, have found compelling evidence for the involvement of nerve agent in the development of a chronic neuroimmune disorder underlying GWI. Moreover, the recent evidence indicating that wartime stress may have reduced the anticholinergic effects of these agents (Locker et al., 2017; Miller et al., 2018) suggests that other, non-cholinergic mechanisms are likely responsible for GWI (Terry, 2012; Naughton and Terry, 2018).
1.3. Chlorpyrifos (CPF) and dichlorvos (DDVP)
In addition to potential low-dose nerve agent exposure, frequent and pervasive pesticide usage also constituted a repeated, daily exposure in theater that was employed to help prevent vector-borne illnesses. According to the Environmental Exposure Report on Pesticides, soldiers were exposed to pesticides via treated uniforms and tents, flea collars, pest strips, fogging, and personal application with an estimated 41,000 having been overexposed (Winkenwerder, 2003); these exposures have been associated with GWI (Haley and Kurt, 1997; Golier et al., 2007; Golomb, 2008; Steele et al., 2012; White et al., 2016; Sullivan et al., 2018). Among these pesticides, veterans with GWI were exposed to the OP AChEIs, CPF and DDVP. Highlighting these exposures, a recent study of GW military pesticide applicators indicated a strong association between cognitive impairments and higher levels of exposure to DDVP pest strips (Sullivan et al., 2018). Unfortunately, to our knowledge, there has been no direct evaluation of GWI-related DDVP exposure in animal models, but DDVP exposure has the potential to produce neuroimmune responses such as microglial activation, increased inflammatory cytokine expression and neurodegeneration (Kaur et al., 2007; Binukumar et al., 2011). However, several studies have investigated the potential role of CPF exposure in GWI and found significant neurological effects (Ojo et al., 2014; Hernandez et al., 2015; Nutter et al., 2015; Cooper et al., 2016, 2018; Flunker et al., 2017; Locker et al., 2017; Miller et al., 2018). Among these studies, investigators have demonstrated that repeated exposure to CPF can cause persistent impairment of axonal transport (Hernandez et al., 2015), loss of synaptic integrity and neurogenesis in the hippocampus (Ojo et al., 2014), and decreased pain threshold when combined with PB, PER and DEET (Nutter et al., 2015; Cooper et al., 2018; Flunker et al., 2017; Cooper et al., 2018, 2018). While these results do not directly support the neuroimmune dysfunction hypothesis of GWI, conditions like neuropathic pain, impaired hippocampal neurogenesis, and axonal transport deficits have been associated with neuroinflammation (Ellis and Bennett, 2013; Walker et al., 2013; Errea et al., 2015; Valero et al., 2017). However, a few studies have directly evaluated neuroimmune-related consequences of CPF exposure. Specifically, it has been shown that exposure to the active metabolite of CPF, chlorpyrifos oxon (CPO; 8 mg/kg, i.p.), results in exacerbated neuroinflammation in mice when combined with prior exogenous exposure to the stress hormone CORT as indicated by an increase of brain inflammatory cytokine mRNA across different brain areas (Locker et al., 2017; Miller et al., 2018). Chronic exposure to CPF alone or in combination with PB and the pesticide permethrin (PER) in mice for 10 days was found to cause an increase in GFAP, indicative of neuroinflammation-associated astrocyte activation; these results were brain region specific, with CPF alone increasing GFAP in motor cortex and hippocampus and CPF + PB + PER combined exposure causing astrogliosis in the piriform cortex and basolateral amygdala (Ojo et al., 2014). Furthermore, similar to the results seen with the sarin surrogate DFP, prior stressor exposure mitigated brain AChE inhibition and decreased ACh levels instigated by CPF exposure (Locker et al., 2017; Miller et al., 2018). While these studies highlight a role for OP AChEI pesticides in the neuroimmune dysfunction associated with GWI, more investigation is needed to understand the mechanisms by which they instigate illness (i.e. non-cholinergic pathways).
1.4. Pyridostigmine bromide (PB)
The requirement for soldiers deployed during the 1991 Gulf War to take prophylactic doses of PB in hopes of preventing serious complications from potential nerve agent exposures has made the drug a prime target for investigation in GWI. As a reversible AChEI with minimal permeability across the blood brain barrier (BBB), little risk was expected from prophylactic treatment with PB particularly considering the dire consequences of nerve agent exposure. Substantiating this notion is the prevalence of reversible AChEIs as pharmacological agents for the treatment of several illnesses, including myasthenia gravis, Alzheimer’s disease, glaucoma, and others (Čolović et al., 2013). However, as early as 1997, epidemiological studies began to uncover an association between chemical exposures, including PB, and the emerging illness in a large population of veterans (Haley et al., 1997a; Haley et al., 1997b; Haley and Kurt, 1997; The Iowa Persian Gulf Study Group, 1997). Over the years, these initial studies have been expanded to reveal strong correlations between PB exposure alone or in combination with other chemicals and the various symptoms of GWI (Kurt, 1998; Nisenbaum et al., 2000; Schumm et al., 2001; Wolfe et al., 2002; Sullivan et al., 2003; Golomb, 2008; Research Advisory Committee on Gulf War Illnesses, 2008; Steele et al., 2012; Steele et al., 2015; White et al., 2016; Sullivan et al., 2018; Zundel et al., 2019). In particular, a few studies found a positive correlation between the number of PB pills taken and the severity of individual GWI cases (Wolfe et al., 2002; Lucas et al., 2007; Golomb, 2008). While most studies focused on evaluating GWI as the collection of its symptoms, a few studies have found specific associations between GWI, PB use, and neuromuscular dysfunction and suppressed cortisol responses (Golier et al., 2006, 2007), as well as gene-exposure interactions with butyrylcholinesterase genotypes (Steele et al., 2015) and increased risk of heart attack and diabetes (Zundel et al., 2019).
While these clinical findings have driven numerous preclinical studies to investigate the role of PB exposures in GWI, there has been minimal evidence to support its involvement in the underlying neuroimmune dysfunction associated with the illness. As such, when PB has been evaluated alone without exposure to any other mediating factors, the drug has minimal deleterious effects on mice or rats (Abou-Donia et al., 1996; Amourette et al., 2009; Lamproglou et al., 2009; Barbier et al., 2009; Locker et al., 2017; Macht et al., 2018; Hernandez et al., 2019). While very few studies have looked directly at neuroinflammation as a result of PB exposure, those that have investigated this directly, found very minimal proinflammatory effects of the drug with an inclination towards anti-inflammatory outcomes (Locker et al., 2017; Hernandez et al., 2019). Moreover, while it has been suggested that stress may increase the permeability of the BBB allowing for PB to gain access to the brain (Friedman et al., 1996; Hanin, 1996; Shen, 1998; Shaikh et al., 2003), several studies investigating the possibility that wartime stress affected the brain accessibility of PB have found that multiple stressor methods do not increase BBB permeability, affect PB’s reduction of brain ChE activity, nor elicit the elaboration of inflammatory markers in the brain or blood in animal models of GWI (Sinton et al., 2000; Song et al., 2002; Tian et al., 2002; Amourette et al., 2009; Locker et al., 2017; Macht et al., 2018). These results suggest that the relationship between PB exposure and GWI that has been supported by epidemiological studies is not straightforward but does not seem to support a role for PB alone in the neuroimmune dysfunction hypothesis.
1.4.1. PB in combination with other GW-relevant chemical exposures
As stated previously, veterans suffering with GWI had the potential to be exposed to many chemicals in theater. As such, a recent study of GW military pesticide applicators found a strong association between combined high pesticide and PB exposure and greater cognitive impairment along with higher rates of GWI (Sullivan et al., 2018). Other studies involving co-exposures to PB and sarin had mixed results. Abou-Donia et al. (2002) found that while PB offered some peripheral ChE protection it did not mitigate sarin-reduced brain AChE activity. Furthermore, while all exposure combinations (PB alone, sarin alone, PB + sarin) caused worsened sensorimotor impairments compared to controls, as assessed by grip strength and beam- and inclined plane-walking (Abou-Donia et al., 2002), the results were largely dependent on the dosage of sarin given and the amount of time following exposure. Specifically, combined exposure with PB was detrimental with higher dosages of sarin for the beam-walk score and degree at which slipping occurred on the inclined plane, but protective for the same tasks when combined with lower dosages of sarin. A separate study found that PB protected against short-term sarin-induced neurobehavioral impairment, as indicated by enhanced acoustic startle response and anxiety/decreased habituation in the open field test, while PB + sarin increased pain threshold at 16 weeks after exposure (Scremin et al., 2003). However, it is difficult to compare the results between these two studies as they used drastically different exposure models.
In addition, a significant number of exposure models have been developed that combine PB with other, non-AChEI, GW-relevant chemicals including: permethrin (PER) with or without stress (Abdullah et al., 2011; Zakirova et al., 2015; Alhasson et al., 2017; Nizamutdinov et al., 2018; Seth et al., 2018; Joshi et al., 2019); PER and DEET with or without stress (Abou-Donia et al., 1996; Abdel-Rahman et al., 2002; Abdullah et al., 2012; Parihar et al., 2013; Hattiangady et al., 2014; Megahed et al., 2015; Pierce et al., 2016; Shetty et al., 2017; Carerras et al., 2018; Petrescu et al., 2018; Madhu et al., 2019); PER and CPF with or without DEET (Ojo et al., 2014; Nutter et al., 2015; Cooper et al., 2016, 2018; Flunker et al., 2017). The prominence of these mixtures in GWI models, which largely revolve around combined exposures with PER and DEET, stem from a recommendation in the report from the Institute of Medicine (US) Committee to Review the Health Consequences of Service During the Persian Gulf War (1995) and an initial study of these chemicals in a hen model (Abou-Donia et al., 1996). While some of these models have demonstrated neuroimmune-related effects, these models generally evaluate conditions only when all chemical exposures are combined and, therefore, the results are likely due to the other chemicals employed in these models rather than PB. However, one study presented by Cooper et al. (2018) found that inclusion of PB in their combination exposure with permethrin, chlorpyrifos, and DEET was requisite for the development of chronic pain and aberrant nociceptor signaling. Overall, the disparity in positive results between models that more directly investigate the role of PB in GWI and those that use combinations with other chemicals like sarin, PER, and/or CPF further highlights the likelihood that PB had a modulatory effect on the response to these other exposures in the initiation of GWI.
2. How did acetylcholinesterase inhibitor exposures cause the neuroimmune dysfunction associated with GWI?
While it has been suggested that there is a strong connection between AChEI exposures and the development of GWI and these chemicals have been demonstrated to have numerous cellular, biochemical, physiological, and neuropsychological effects, the direct mechanism by which exposure has resulted in this chronic illness has remained obscure. As summarized in this review, we suggest that the literature indicates that OPs that irreversibly inhibit AChE, like sarin and its surrogates, chlorpyrifos, and dichlorvos, seem to be the key exposure for the development of neuroimmune dysfunction in GWI. Under normal conditions, OP AChEIs exert their toxicological effects by organophosphorylating and inactivating the cholinesterase enzymes which produces acute toxicity. However, it is unclear whether exposed veterans with GWI exhibited any signs of acute AChEI intoxication. As far as potential sarin or cyclosarin exposure is concerned, the survey conducted of soldiers within the vicinity of the Kamisiyah detonation suggests that they did not experience signs of acute toxicity (Institute of Medicine (US) Committee on Health Effects Associated with Exposures During the Gulf War, 2000), suggestive of low-level exposure. As discussed in this review, a number of studies have recapitulated GWI in animal models using low doses of sarin, sarin surrogates, or pesticides. Similarly, it has been shown that under conditions of high physiological stress, the cholinergic toxicity of agents like DFP and CPO is suppressed as evidenced by a reduction in the percentage of enzyme inhibition and amelioration of the increase in brain ACh (Locker et al., 2017; Miller et al., 2018). We hypothesize that these conditions, e.g. low-level exposures alone or combined with high physiological stress, avoid the acute cholinergic toxicity of the OP AChEIs due to a dose-dependent interaction with the enzyme itself. In the case of low level exposure, the doses of AChEIs encountered may be below threshold for significant AChE inhibition. When considering the interaction of AChEI exposures and high physiological stress, studies have shown that stress or CORT exposures can increase AChE activity (Oriaku and Soliman, 1986; Fatranska et al., 1987; Tsakiris and Kontopoulos, 1992); thus, altering the activity of AChE in the tissue has the potential to compensate for the inhibition instigated by AChEI exposure by normalizing ACh levels. Therefore, in both of these conditions, exposure to OP AChEIs would have minimal functional impact on cholinergic signaling and not trigger the prototypical cholinergic anti-inflammatory signaling pathway (Pavlov et al., 2003; Pavlov and Tracey, 2005); facilitating the significant neurological results demonstrated following OP AChEI exposure, including signs of neuroimmune dysfunction, such as increased expression of neuroinflammatory cytokines and astrogliosis (Ojo et al., 2014; Locker et al., 2017; Miller et al., 2018). Thus, we suggest that GWI may be the result of aberrant neuroimmune signaling that is instigated by the organophosphorylation of non-cholinesterase targets by the OP AChEIs, a hypothesis that has been previously suggested (Grigoryan et al., 2008, 2009; Terry et al., 2012; Naughton and Terry, 2018) and warrants further investigation in relationship to neuroinflammatory signaling.
3. Conclusions
As stated throughout this review, GWI is believed to be a result of a combination of exposures/conditions that were experienced by soldiers during deployment. In the years that have followed the 1991 Gulf War, research has suggested that AChEIs are chief among these exposures as culprits in the development of GWI, particularly when combined with stressors to mimic the extreme conditions experienced by soldiers during deployment. Overall, the irreversible, OP AChEIs, both nerve agents and pesticides, are more likely to have played a primary role in the development of GWI as they show a strong correlation with the neuroimmune dysfunction suspected to be the underlying cause of illness among these veterans. While the administration of PB pills has long been suspect in the development of GWI, our review of the literature has suggested that the correlation between PB and GWI is not strongly supported by preclinical investigations using animal models of exposure; thus, the role of PB in GWI remains unclear. In spite of their common cholinergic functions, we strongly suspect that GWI is the result of the actions of OP AChEI exposures on non-cholinergic targets (Grigoryan et al., 2008, 2009; Terry et al., 2012; Locker et al., 2017; Miller et al., 2018; Naughton and Terry, 2018), namely organophosphorylation of proteins that are crucial to neuroinflammatory signaling. Long-term low dose inhibition of AChE by these compounds and other contributing factors, such as physiological stress and other chemical exposures, may have played a role as well to facilitate the development of a chronic neuroimmune disorder (Fig. 1). By the continued investigation into potential mechanisms underlying GWI pathobiology, we may be able to uncover therapeutic targets that can be modulated to more successfully treat GWI symptoms and/or reverse the underlying aberrant neuroimmune function associated with this disorder.
Fig. 1. Mechanism of acetylcholinesterase inhibitor-induced neuroimmune dysfunction in Gulf War Illness.
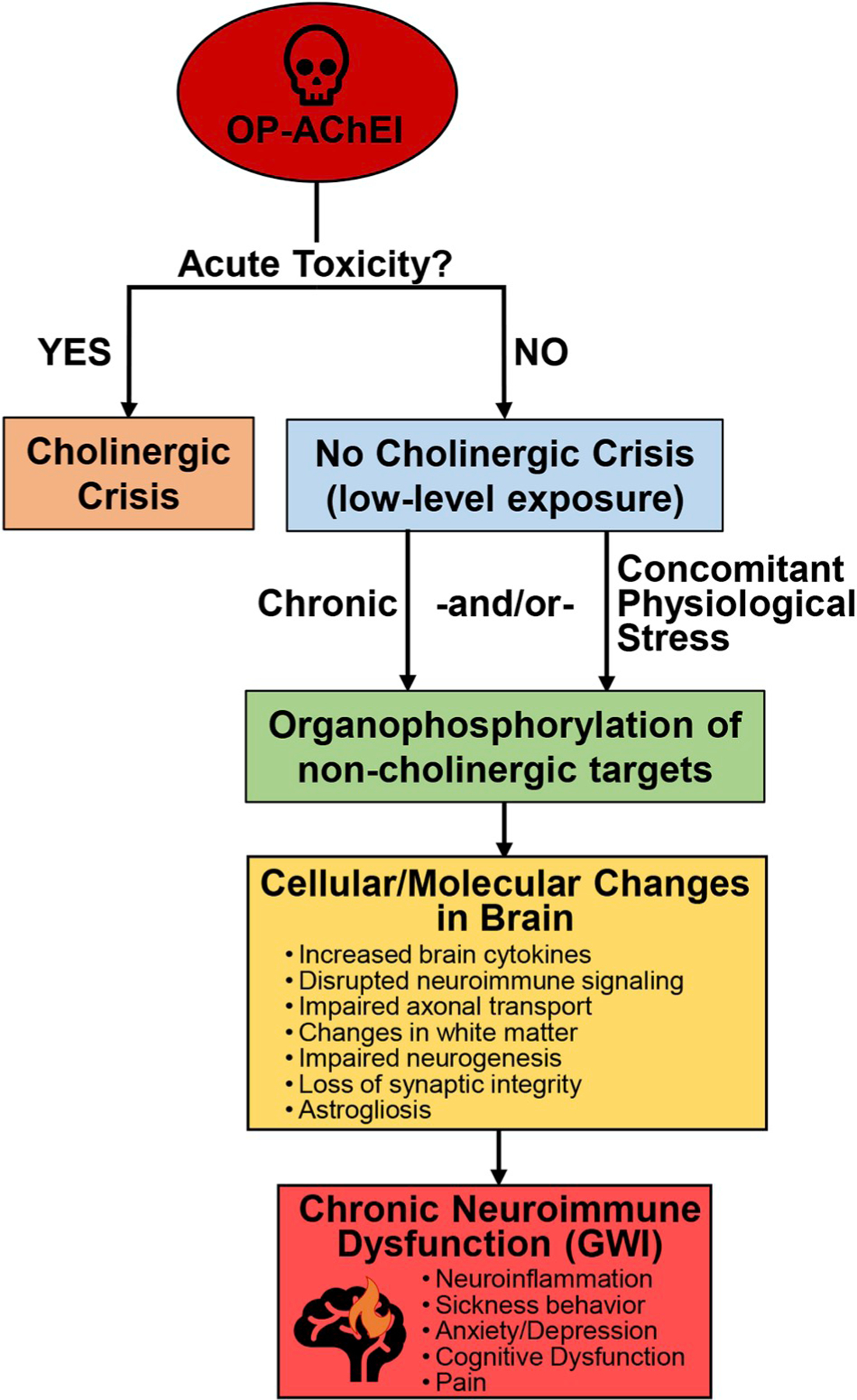
Acetylcholinesterase inhibitors (AChEIs), in particular the organophosphate (OP) chemicals, can instigate illness in two ways: (1) acute toxicity that results in cholinergic crisis (salivation, lacrimation, urination, defecation, gastrointestinal upset, emesis, miosis [SLUDGEM]; seizures) and carries a higher risk of mortality; (2) long-term illness in the absence of an acute cholinergic crisis. The latter condition is proposed to be the result of chronic low-level AChEI exposure with or without concurrent exposure to physiological stress. The myriad of cellular and molecular effects that have been demonstrated in the brain as a result of these exposures are hypothesized to be the consequences of organophosphorylation of non-cholinergic targets, e.g. neuroinflammatory signaling mediators. Ultimately, these effects culminate into a state of chronic neuroimmune dysfunction, the underlying cause of Gulf War Illness (GWI).
HIGHLIGHTS.
Gulf War Illness (GWI) is associated with underlying neuroimmune dysfunction.
Veterans with GWI were exposed to several acetylcholinesterase inhibitors (AChEIs).
Organophosphate AChEIs, nerve agent and pesticides, can instigate neuroinflammation.
While associated with GWI, pyridostigmine does not cause neuroinflammation.
Lack of acute toxicity suggests GWI results from non-cholinergic actions of AChEIs.
Acknowledgements
This work was supported by a Department of Defense, Congressionally Directed Medical Research Program: Gulf War Illness award (W81XWH-13-2-0072). The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention. This work was supported by the Assistant Secretary of Defense for Health Affairs, through the Gulf War Illness Research Program. Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
References
- Abdel-Rahman A, Shetty AK, Abou-Donia MB, 2002. Disruption of the blood-brain barrier and neuronal cell death in cingulate cortex, dentate gyrus, thalamus, and hypothalamus in a rat model of Gulf-War syndrome. Neurobiol. Dis 10 (3), 306–326. [DOI] [PubMed] [Google Scholar]
- Abdullah L, Crynen G, Reed J, Bishop A, Phillips J, Ferguson S, Mouzon B, Mullan M, Mathura V, Mullan M, Ait-Ghezala G, Crawford F, 2011. Proteomic CNS profile of delayed cognitive impairment in mice exposed to Gulf War agents. NeuroMolecular Med. 13 (4), 275–288. [DOI] [PubMed] [Google Scholar]
- Abdullah L, Evans JE, Bishop A, Reed JM, Crynen G, Phillips J, Pelot R, Mullan MA, Ferro A, Mullan CM, Ait-Ghezala G, Crawford FC, 2012. Lipidomic profiling of phosphocholine-containing brain lipids in mice with sensorimotor deficits and anxiety-like features after exposure to Gulf War agents. NeuroMolecular Med. 14 (4), 349–361. [DOI] [PubMed] [Google Scholar]
- Abdullah L, Evans JE, Montague H, Reed JM, Moser A, Crynen G, Gonzalez A, Zakirova Z, Ross I, Mullan C, Mullan M, Ait-Ghezala G, Crawford F, 2013. Chronic elevation of phosphocholine containing lipids in mice exposed to Gulf War agents pyridostigmine bromide and permethrin. Neurotoxicol. Teratol 40, 74–84. [DOI] [PubMed] [Google Scholar]
- Abdullah L, Evans JE, Joshi U, Crynen G, Reed J, Mouzon B, Baumann S, Montague H, Zakirova Z, Emmerich T, Bachmeier C, Klimas N, Sullivan K, Mullan M, Ait-Ghezala G, Crawford F, 2016. Translational potential of long-term decreases in mitochondrial lipids in a mouse model of Gulf War Illness. Toxicology 372, 22–33. [DOI] [PubMed] [Google Scholar]
- Abou-Donia MB, Conboy LA, Kokkotou E, Jacobson E, Elmasry EM, Elkafrawy P, Neely M, Bass CR, Sullivan K, 2017. Screening for novel central nervous system biomarkers in veterans with Gulf War Illness. Neurtoxicol. Teratol 61, 36–46. [DOI] [PubMed] [Google Scholar]
- Abou-Donia MB, Dechkovskaia AM, Goldstein LB, Bullman SL, Khan WA, 2002. Sensorimotor deficit and cholinergic changes following coexposure with pyridostigmine bromide and sarin in rats. Toxicol. Sci 66 (1), 148–158. [DOI] [PubMed] [Google Scholar]
- Abou-Donia MB, Wilmarth KR, Jensen KF, Oehme FW, Kurt TL, 1996. Neurotoxicity resulting from coexposure to pyridostigmine bromide, deet, and permethrin: implication of Gulf War chemical exposures. J. Toxicol. Environ. Health 48 (1), 35–56. [DOI] [PubMed] [Google Scholar]
- Alhasson F, Das S, Seth R, Dattaroy D, Chandrashekaran V, Ryan CN, Chan LS, Testerman T, Curch J, Hofseth LJ, Horner R, Nagarkatti M, Nagarkatti P, Lasley SM, Chatterjee S, 2017. Altered gut microbiome in a mouse model of Gulf War Illness causes neuroinflammation and intestinal injury via leaky gut and TLR4 activation. PloS One 12 (3), e0172914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshelh Z, Albrech DS, Bergan C, Akeju O, Clauw DJ, Conboy L, Edwards RR, Kim M, Lee YC, Protsenko E, Napadow V, Sullivan K, Loggia ML, 2020. In-vivo imaging of neuroinflammation in veterans with Gulf War illness. Brain Behav. Immun 10.1016/j.bbi.2020.01.020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amourette C, Lamproglou I, Barbier L, Fauquette W, Zoppe A, Viret R, Diserbo M, 2009. Gulf War illness: effects of repeated stress and pyridostigmine treatment on blood-brain barrier permeability and cholinesterase activity in rat brain. Behav. Brain Res 203, 207–214. [DOI] [PubMed] [Google Scholar]
- Ashbrook DG, Hing B, Michalovicz LT, Kelly KA, Miller JV, de Vega WC, Miller DB, Broderick G, O’Callaghan JP, McGowan PO, 2018. Epigenetic impacts of stress priming of the neuroinflammatory response to sarin surrogate in mice: a model of Gulf War illness. J. Neuroinflammation 15 (1), 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier L, Diserbo M, Lamproglou I, Amourette C, Peinnequin A, Fauquette W, 2009. Repeated stress in combination with pyridostigmine Part II: changes in cerebral gene expression. Behav. Brain Res 197 (2), 292–300. [DOI] [PubMed] [Google Scholar]
- Bartholdi D, Schwab ME, 1998. Oligodendroglial reaction following spinal cord injury in rat: transient upregulation of MBP mRNA. Glia 23 (3), 278–284. [DOI] [PubMed] [Google Scholar]
- Belgrad J, Dutta DJ, Bromley-Coolidge S, Kelly KA, Michalovicz LT, Sullivan KA, O’Callaghan JP, Fields RD, 2019. Oligodendrocyte involvement in gulf war illness. Glia 67 (11), 2107–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binukumar BK, Bal A, Gill KD, 2011. Chronic dichlorvos exposure: microglial activation, proinflammatory cytokines and damage to nigrostriatal dopaminergic system. NeuroMolecular Med. 13 (4), 251–265. [DOI] [PubMed] [Google Scholar]
- Broderick G, Ben-Hamo R, Vashishtha S, Efroni S, Nathanson L, Barnes Z, Fletcher MA, Klimas N, 2013. Altered immune pathway activity under exercise challenge in Gulf War Illness: an exploratory analysis. Brain Behav. Immun 28, 159–169. [DOI] [PubMed] [Google Scholar]
- Bullman TA, Mahan CM, Kang HK, Page WF, 2005. Mortality in US Army Gulf War veterans exposed to 1991 Khamisiyah chemical munitions destruction. Am. J. Publ. Health 95 (8), 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras I, Aytan N, Mellott T, Choi JK, Lehar M, Crabtress L, Leite-Morris K, Jenkins BG, Blusztajn JK, Dedeoglu A, 2018. Anxiety, neuroinflammation, cholinergic and GABAergic abnormalities are early markers of Gulf War illness in a mouse model of the disease. Brain Res. 1681, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LL, Rothlind JC, Cardenas VA, Meyerhoff DJ, Weiner MW, 2010. Effects of low-evel exposure to sarin and cyclosarin during the 1991 Gulf War on brain function and brain structure in US veterans. Neurotoxicology 31 (5), 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LL, Abadjian L, Hlavin J, Meyerhoff DJ, Weiner MW, 2011. Effects of low-level sarin and cyclosarin exposure and Gulf War Illness on brain structure and function: a study at 4T. Neurotoxicology 32 (6), 814–822. [DOI] [PubMed] [Google Scholar]
- Chao LL, Kriger S, Buckley S, Ng P, Mueller SG, 2014. Effects of low-level sarin and cyclosarin exposure on hippocampal subfields in Gulf War Veterans. Neurotoxicology 44, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LL, Zhang Y, Buckley S, 2015. Effects of low-level sarin and cyclosarin exposure on white matter integrity in Gulf War Veterans. Neurotoxicology 48, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LL, Reeb R, Esparza IL, Abadjian LR, 2016. Associations between the self-reported frequency of hearing chemical alarms in theater and regional brain volume in Gulf War Veterans. Neurotoxicology 53, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LL, Zhang Y, 2018. Effects of low-level sarin and cyclosarin exposure on hippocampal microstructure in Gulf War Veterans. Neurtoxicol. Teratol 68, 36–46. [DOI] [PubMed] [Google Scholar]
- Colović MB, Krstić DZ, Lazarević-Pašti TD, Bondžić AM, Vasić VM, 2013. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr. Neuropharmacol 11 (3), 315–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper BY, Johnson RD, Nutter TJ, 2016. Exposure to Gulf War Illness chemicals induces functional muscarinic receptor maladaptations in muscle nociceptors. Neurotoxicology 54, 99–110. [DOI] [PubMed] [Google Scholar]
- Cooper BY, Flunker LD, Johnson RD, Nutter TJ, 2018. Behavioral, cellular and molecular maladaptations covary with exposure to pyridostigmine bromide in a rat model of gulf war illness pain. Toxicol. Appl. Pharamcol 352, 119–131. [DOI] [PubMed] [Google Scholar]
- Coughlin SS, 2017. A neuroimmune model of gulf war illness. J. Environ. Health Sci 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock TJA, Michalovicz LT, Kelly KA, Rice MA Jr., Miller DB, Klimas NG, Morris M, O’Callaghan JP, Broderick G, 2018. A logic model of neuronal-glial interaction suggests altered homeostatic regulation in the perpetuation of neuroinflammation. Front. Cell. Neurosci 12, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Kelley KW, 2007. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun 21 (2), 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW, 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci 9, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis A, Bennett DL, 2013. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth 111 (1), 26–37. [DOI] [PubMed] [Google Scholar]
- Emmerich T, Zakirova Z, Klimas N, Sullivan K, Shetty AK, Evans JE, Ait-Ghezala G, Laco GS, Hattiangady B, Shetty GA, Mullan M, Crynen G, Abdullah L, Crawford F, 2017. Phospholipid profiling of plasma from GW veterans and rodent models to identify potential biomarkers of Gulf War Illness. PloS One 12 (4), e0176634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errea O, Moreno B, Gonzalez-Franquesa A, Garcia-Roves PM, Villoslada P, 2015. The disruption of mitochondrial axonal transport is an early event in neuroinflammation. J. Neuroinflammation 12, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatranska M, Budai D, Oprsalova Z, Kvetnansky R, 1987. Acetycholine and its enzymes in some brain areas of the rat under stress. Brain Res. 424, 109–114. [DOI] [PubMed] [Google Scholar]
- Flunker LK, Nutter TJ, Johnson RD, Cooper BY, 2017. DEET potentiates the development and persistence of anticholinesterase dependent chronic pain signs in a rat model of Gulf War Illness pain. Toxicol. Appl. Pharmacol 316, 48–62. [DOI] [PubMed] [Google Scholar]
- Friedman A, Kaufer D, Shemer J, Hendler I, Soreq H, Tur-Kaspa I, 1996. Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat. Med 2 (12), 1382–1385. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Nisenbaum R, Stewart G, Thompson WW, Robin L, Washko RM, Noah DL, Barrett DH, Randall B, Herwaldt BL, Mawle AC, Reeves WC, 1998. Chronic multisymptom illness affecting air force veterans of the gulf war. J. Am. Med. Assoc 280, 981–988. [DOI] [PubMed] [Google Scholar]
- Georgopoulous AP, James LM, Carpenter AF, Engdahl BE, Leuthold AC, Lewis SM, 2017. Gulf War illness (GWI) as a neuroimmune disease. Exp. Brain Res 235 (10), 3217–3225. [DOI] [PubMed] [Google Scholar]
- Golier JA, Schmeidler J, Legge J, Yehuda R, 2006. Enhanced cortisol suppression to dexamethasone associated with Gulf War deployment. Psychoneuroendocrinology 31 (10), 1181–1189. [DOI] [PubMed] [Google Scholar]
- Golier JA, Schmeidler J, Legge J, Yehuda R, 2007. Twenty-four hour plasma cortisol and adrenocorticotropic hormone in Gulf War veterans: relationships to posttraumatic stress disorder and health symptoms. Biol. Psychiatr 62 (10), 1175–1178. [DOI] [PubMed] [Google Scholar]
- Golomb BA, 2008. Acetylcholinesterase inhibitors and gulf war illnesses. Proc. Natl. Acad. Sci. U. S. A 105, 4295–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Schopfer LM, Thompson CM, Terry AV, Masson P, Lockridge O, 2008. Mass spectrometry identifies covalent binding of soman, sarin, chlorpyrifos oxon, diisopropyl fluorophosphate, and FP-biotin to tyrosines on tubulin: a potential mechanism of long term toxicity by organophosphorus agents. Chem. Biol. Interact 175 (1–3), 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan H, Li B, Anderson EK, Xue W, Nachon F, Lockridge O, et al. , 2009. Covalent binding of the organophosphorus agent FP-biotin to tyrosine in eight proteins that have no active site serine. Chem. Biol. Interact 180 (3), 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley RW, Hom J, Roland PS, Bryan WW, Van Ness PC, Bonte FJ, Devous MD Sr, Mathews D, Fleckenstein JL, Wians FH Jr., Wolfe GI, Kurt TL, 1997a. Evaluation of neurologic function in Gulf War veterans. A blinded case-control study. J. Am. Med. Assoc 277 (3), 223–230. [PubMed] [Google Scholar]
- Haley RW, Kurt TL, Hom J, 1997b. Is there a Gulf War Syndrome? Searching for syndromes by factor analysis of symptoms. J. Am. Med. Assoc 277 (3), 215–222. [PubMed] [Google Scholar]
- Haley RW, Kurt TL, 1997. Self-reported exposure to neurotoxic chemical combinations in the Gulf War. A cross-sectional epidemiologic study. J. Am. Med. Assoc 277 (3), 231–237. [PubMed] [Google Scholar]
- Haley RW, Tuite JJ, 2013. Epidemiologic evidence of health effects from long-distance transit of chemical weapons fallout from bombing early in the 1991 Persian Gulf War. Neuroepidemiology 40 (3), 178–189. [DOI] [PubMed] [Google Scholar]
- Haley RW, Luk GD, Petty F, 2001. Use of structural equation modeling to test the construct validity of a case definition of Gulf War syndrome: invariance over developmental and validation samples, service branches and publicity. Psychiatr. Res 102 (2), 175–200. [DOI] [PubMed] [Google Scholar]
- Hanin I, 1996. The Gulf War, stress and a leaky blood-brain barrier. Nat. Med 2 (12), 1307–1308. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Mishra V, Kodali M, Shuai B, Rao X, Shetty AK, 2014. Object location and object recognition memory impairments, motivation deficits and depression in a model of Gulf War illness. Front. Behav. Neurosci 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton KJ, Palumbo CL, Proctor SP, Killiany RJ, Yurgelun-Todd DA, White RF, 2007. Quantitative magnetic resonance brain imaging in US army veterans of the 1991 Gulf War potentially exposed to sarin and cyclosarin. Neurotoxicology 28 (4), 761–769. [DOI] [PubMed] [Google Scholar]
- Henderson RF, Barr EB, Blackwell WB, Clark CR, Conn CA, Kalra R, March TH, Sopori ML, Tesfaigzi Y, Menache MG, Mash DC, Dokladny K, Kozak W, Kozak A, Wachulec M, Rudolph K, Kluger MJ, Singh SP, Razani-Boroujerdi S, Langley RJ, 2001. Response of F344 rats to inhalation of subclinical levels of sarin: exploring potential causes of Gulf War illness. Toxicol. Ind. Health 17 (5–10), 294–297. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Beck WD, Naughton SX, Poddar I, Adam BL, Yanasak N, Middleton C, Terry AV Jr., 2015. Repeated exposure to chlorpyrifos leads to prolonged impairments of axonal transport in the living rodent brain. Neurotoxicology 47, 17–26. [DOI] [PubMed] [Google Scholar]
- Hernandez S, Fried DE, Grubišić V, McClain JL, Gulbransen BD, 2019. Gastrointestinal neuroimmune disruption in a mouse model of Gulf War illness. Faseb. J 33 (5), 6168–6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute of Medicine (Us) Committee on Health Effects Associated with Exposures During the Gulf War, 2000. In: In: Vaccines Fulco CE, Liverman CT, Sox HC (Eds.), Gulf War and Health: Volume 1. Depleted Uranium, Sarin, Pyridostigmine Bromide National Academies Press (US); 2000, Washington (DC). [PubMed] [Google Scholar]
- Institute of Medicine (Us) Committee to Review the Health Consequences of Service During the Persian Gulf War, 1995. Health Consequences of Service during the Persian Gulf War: Initial Findings and Recommendations for Immediate Action. National Academies Press (US); 1995, Washington (DC). [PubMed] [Google Scholar]
- Joshi U, Pearson A, Evans JE, Langlois H, Saltiel N, Ojo J, Klimas N, Sullivan K, Keegan AP, Oberlin S, Darcey T, Cseresznye A, Raya B, Paris D, Hammock B, Vasylieva N, Hongsibsong S, Stern LJ, Crawford F, Mullan M, Abdullah L, 2019. A permethrin metabolite is associated with adaptive immune responses in Gulf War Illness. Brain Behav. Immun 81, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra R, Singh SP, Razani-Boroujerdi S, Langley RJ, Blackwell WB, Henderson RF, Sopori ML, 2002. Subclinical doses of the nerve gas sarin impair T cell responses through the autonomic nervous system. Toxicol. Appl. Pharmacol 184 (2), 82–87. [PubMed] [Google Scholar]
- Kaur P, Radotra B, Minz RW, Gill KD, 2007. Impaired mitochondrial energy metabolism and neuronal apoptotic cell death after chronic dichlorvos (OP) exposure in rat brain. Neurotoxicology 28 (6), 1208–1219. [DOI] [PubMed] [Google Scholar]
- Kerr KJ, 2015. Gulf War illness: an overview of events, most prevalent health outcomes, exposures, and clues as to pathogenesis. Rev. Environ. Health 30 (4), 273–286. [DOI] [PubMed] [Google Scholar]
- Kodali M, Hattiangady B, Shetty GA, Bates A, Shuai B, Shetty AK, 2018. Curcumin treatment leads to better cognitive and mood function in a model of Gulf War Illness with enhanced neurogenesis, and alleviation of inflammation and mitochondrial dysfunction in the hippocampus. Brain Behav. Immun 69, 499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BB, Michalovicz LT, Calderazzo S, Kelly KA, Sullivan K, Killiany RJ, O’Callaghan JP, 2018. Corticosterone potentiates DFP-induced neuroinflammation and affects high-order imaging in a rat model of Gulf War Illness. Brain Behav. Immun 67, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensson K, Homes KV, Duchala CS, Zeller NK, Lazzarini RA, Dubois-Dalcq M, 1986. Increased levels of myelin basic protein transcripts gene in virus-induced demyelination. Nature 322 (6079), 544–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurt TL, 1998. Epidemiological association in US veterans between Gulf War illness and exposures to anticholinesterases. Toxicol. Lett 102–103, 523–526. [DOI] [PubMed] [Google Scholar]
- Lamproglou I, Barbier L, Diserbo M, Fauvelle F, Fauquette W, Amourette C, 2009. Repeated stress in combination with pyridostigmine Part I: long-term behavioural consequences. Behav. Brain Res 197 (2), 301–310. [DOI] [PubMed] [Google Scholar]
- Locker AR, Michalovicz LT, Kelly KA, Miller JV, Miller DB, O’Callaghan JP, 2017. Corticosterone primes the neuroinflammatory response to Gulf War Illness-relevant organophosphates independently of acetylcholinesterase inhibition. J. Neurochem 142, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas KE, Rowe PC, Armenian HK, 2007. Latency and exposure-health association in Gulf War veterans with early fatigue onsets: a case-control study. Ann. Epidemiol 17 (10), 799–806. [DOI] [PubMed] [Google Scholar]
- Macht VA, Woodruff JL, Grillo CA, Wood CS, Wilson MA, Reagan LP, 2018. Pathophysiology in a model of gulf war illness: contributions of pyridostigmine bromide and stress. Psychoneuroendocrinology 96, 195–202. [DOI] [PubMed] [Google Scholar]
- Macht VA, Woodruff JL, Maissy ES, Grillo CA, Wilson MA, Fadel JR, Reagan LP, 2019. Pyridostigmine bromide and stress interact to impact immune function, cholinergic neurochemistry and behavior in a rat model of Gulf War Illness. Brain Behav. Immun 80, 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhu LN, Attaluri S, Kodali M, Shuai B, Upadhya R, Gitai D, Shetty AK, 2019. Neuroinflammation in Gulf War Illness is linked with HMGB1 and complement activation, which can be discerned from brain-derived extracellular vesicles in the blood. Brain Behav. Immun 81, 430–443. [DOI] [PubMed] [Google Scholar]
- Maule AL, Janulewicz PA, Sullivan KA, Krengle MH, Yee MK, McClean M, White RF, 2018. Meta-analysis of self-reported health symptoms in 1990–1991 gulf war and gulf war-era veterans. BMJ Open 8 (2), e016086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megahed T, Hattiangady B, Shuai B, Shetty AK, 2015. Parvalbumin and neuropeptide Y expressing hippocampal GABA-ergic inhibitory interneuron numbers decline in a model of Gulf War illness. Front. Cell. Neurosci 8, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovicz LT, Locker AR, Kelly KA, Miller JV, Barnes Z, Fletcher MA, Miller DB, Klimas NG, Morris M, Lasley SM, O’Callaghan JP, 2019. Corticosterone and pyridostigmine/DEET exposure attenuate peripheral cytokine expression: supporting a dominant role for neuroinflammation in a mouse model of Gulf War Illness. Neurotoxicology 70, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JV, LeBouf RF, Kelly KA, Michalovicz LT, Ranpara A, Locker AR, Miller DB, O’Callaghan JP, 2018. The neuroinflammatory phenotype in a mouse model of Gulf War Illness is unrelated to brain regional levels of acetylcholine as measured by quantitative HILIC-UPLC-MS/MS. Toxicol. Sci 165 (2), 302–313. [DOI] [PubMed] [Google Scholar]
- Naughton SX, Hernandez CM, Beck WD, Poddar I, Yanasak N, Lin PC, Terry AV Jr., 2018. Repeated exposures to diisopropylfluorophosphate result in structural disruptions of myelinated axons and persistent impairments of axonal transport in the brains of rats. Toxicology 92–103 406–407. [DOI] [PubMed] [Google Scholar]
- Naughton SX, Terry AV Jr., 2018. Neurotoxicity in acute and repeated organophosphate exposure. Toxicology 408, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum R, Barrett DH, Reyes M, Reeves WC, 2000. Deployment stressors and a chronic multisymptom illness among Gulf War veterans. J. Nerv. Ment. Dis 188 (5), 259–266. [DOI] [PubMed] [Google Scholar]
- Nizamutdinov D, Mukherjee S, Deng C, Strauss HM, Shapiro LA, 2018. Gulf War agents pyridostigmine bromide and permethrin cause hypersensitive nociception that is restored after vagus nerve stimulation. Neurotoxicology 69, 93–96. [DOI] [PubMed] [Google Scholar]
- Nutter TJ, Johnson RD, Cooper BY, 2015. A delayed chronic pain like condition with decreased Kv channel activity in a rat model of Gulf War Illness pain syndrome. Neurotoxicology 51, 67–79. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Kelly KA, Locker AR, Miller DB, Lasley SM, 2015. Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J. Neurochem 133 (5), 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo JO, Abdullah L, Evans J, Reed JM, Montague H, Mullan MJ, Crawford FC, 2014. Exposure to an organophosphate pesticide, individually or in combination with other Gulf War agents, impairs synaptic integrity and neuronal differentiation, and is accompanied by subtle microvascular injury in a mouse model of Gulf War agent exposure. Neuropathology 34 (2), 109–127. [DOI] [PubMed] [Google Scholar]
- Oriaku ET, Soliman KF, 1986. Effect of stress and glucocorticoids on the gastrointestinal cholinergic enzymes. Arch. Int. Pharmacodyn. Ther 280 (1), 136–144. [PubMed] [Google Scholar]
- Parihar VK, Hattiangady B, Shuai B, Shetty AK, 2013. Mood and memory deficits in a model of Gulf War illness are linked with reduced neurogenesis, partial neuron loss, and mild inflammation in the hippocampus. Neuropsychopharmacology 38 (12), 2348–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkitny L, Middleton S, Baker K, Younger J, 2015. Evidence for abnormal cytokine expression in Gulf War Illness: a preliminary analysis of daily immune monitoring data. BMC Immunol. 16, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov VA, Tracey KJ, 2005. The cholinergic anti-inflammatory pathway. Brain Behav. Immun 19, 493–499. [DOI] [PubMed] [Google Scholar]
- Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ, 2003. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol. Med 9, 125–134. [PMC free article] [PubMed] [Google Scholar]
- Peferoen L, Kipp M, Valk P, Noort JM, Amor S, 2014. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141 (3), 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi E, 1996. Chemicals behind gulf war syndrome? Science 272 (5261), 479–480. [DOI] [PubMed] [Google Scholar]
- Petrescu AD, Grant S, Frampton G, McMillin M, Kain J, Kodali M, Shetty AK, DeMorrow S, 2018. Gulf war illness-related chemicals increase CD11b/c+ monocyte infiltration into the liver and aggravate hepatic cholestasis in a rodent model. Sci. Rep 8 (1), 13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KF, Deshpande LS, 2016. Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War Illness. Neurotoxicology 52, 127–133. [DOI] [PubMed] [Google Scholar]
- Phillips KF, Deshpande LS, 2018. Chronic neurological morbidities and elevated hippocampal calcium levels in a DFP-based rat model of gulf war illness. Mil. Med 183 (Suppl. l_1), 552–555. [DOI] [PubMed] [Google Scholar]
- Pierce LM, Kurata WE, Matsumoto KW, Clark ME, Farmer DM, 2016. Long-term epigenetic alterations in a rat model of Gulf War Illness. Neurotoxicology 55, 20–32. [DOI] [PubMed] [Google Scholar]
- Proctor SP, Heaton KJ, Heeren T, White RF, 2006. Effects of sarin and cyclosarin exposure during the 1991 Gulf War on neurobehavioral functioning in US army veterans. Neurotoxicology 27 (6), 931–939. [DOI] [PubMed] [Google Scholar]
- Rao AN, Patil A, Brodnik ZD, Qiang L, Espana RA, Sullivan KA, Black MM, Baas PW, 2017. Pharmacologically increasing microtubule acetylation corrects stress-exacerbated effects of organophosphates on neurons. Traffic 18 (7), 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Research Advisory Committee (Rac) on Gulf War Veterans’ Illnesses, 2008. Gulf War Illness and the Health of Gulf War Veterans: Scientific Findings and Recommendations. U.S. Government Printing Office, Washington, DC. [Google Scholar]
- Sapolsky RM, 1998. The stress of Gulf War syndrome. Nature 393 (6683), 308–309. [DOI] [PubMed] [Google Scholar]
- Schumm WR, Reppert EJ, Jurich AP, Bollman SR, Castelo C, Sanders D, Webb FJ, 2001. Pyridostigmine bromide and the long-term subjective health status of a sample of female reserve component Gulf War veterans: a brief report. Psychol. Rep 88 (1), 306–308. [DOI] [PubMed] [Google Scholar]
- Scremin OU, Shih TM, Huynh L, Roch M, Booth R, Jenden DJ, 2003. Delayed neurologic and behavioral effects of subtoxic doses of cholinesterase inhibitors. J. Pharmacol. Exp. Therapeut 304 (3), 1111–1119. [DOI] [PubMed] [Google Scholar]
- Seth RK, Kimono D, Alhasson F, Sarkar S, Albadrani M, Lasley SK, Horner R, Janulewicz P, Nagarkatti M, Nagarkatti P, Sullivan K, Chatterjee S, 2018. Increased butyrate priming in the gut stalls microbiome associated-gastrointestinal inflammation and hepatic metabolic reprogramming in a mouse model of Gulf War Illness. Toxicol. Appl. Pharmacol 350, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh J, Karanth S, Chakraborty D, Pruett S, Pope CN, 2003. Effects of daily stress or repeated paraoxon exposures on subacute pyridostigmine toxicity in rats. Arch. Toxicol 77 (10), 576–583. [DOI] [PubMed] [Google Scholar]
- Shen ZX, 1998. Pyridostigmine bromide and gulf war syndrome. Med. Hypotheses 51 (3), 235–237. [DOI] [PubMed] [Google Scholar]
- Shetty GA, Hattiangady B, Upadhya D, Bates A, Attaluri S, Shuai B, Kodali M, Shetty AK, 2017. Chronic oxidative stress, mitochondrial dysfunction, Nrf2 activation and inflammation in the Hippocampus accompany heightened systemic inflammation and oxidative stress in an animal model of gulf war illness. Front. Mol. Neurosci 10, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewale SV, Anstadt MP, Horenziak M, Izu B, Morgan EE, Lucot JB, Morris M, 2012. Sarin causes autonomic imbalance and cardiomyopathy: an important issue for military and civilian health. J. Cardiovasc. Pharmacol 60 (1), 76–87. [DOI] [PubMed] [Google Scholar]
- Sinton CM, Fitch TE, Petty F, Haley RW, 2000. Stressful manipulations that elevate corticosterone reduce blood-brain barrier permeability to pyridostigmine in the Rat. Toxicol. Appl. Pharmacol 165 (1), 99–105. [DOI] [PubMed] [Google Scholar]
- Song X, Tian H, Bressler J, Pruett S, Pope C, 2002. Acute and repeated restraint stress have little effect on pyridostigmine toxicity or brain regional cholinesterase inhibition in rats. Toxicol. Sci 69 (1), 157–164. [DOI] [PubMed] [Google Scholar]
- Steele L, 2000. Prevalence and patterns of Gulf War illness in Kansas veterans: association of symptoms with characteristics of person, place, and time of military service. Am. J. Epidemiol 152 (10), 992–1002. [DOI] [PubMed] [Google Scholar]
- Steele L, Sastre A, Gerkovich MM, Cook MR, 2012. Complex factors in the etiology of Gulf War illness: wartime exposures and risk factors in veteran subgroups. Environ. Health Perspect 120, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele L, Lockridge O, Gerkovich MM, Cook MR, Sastre A, 2015. Butyrylcholinesterase genotype and enzyme activity in relation to Gulf War illness: preliminary evidence of gene-exposure interaction from a case-control study of 1991 Gulf War veterans. Environ. Health 14, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K, Krengel M, Proctor SP, Devine S, Heeren T, White RF, 2003. Cognitive functioning in treatment-seeking Gulf War veterans: pyridostigmine bromide use and PTSD. J. Psychopathol. Behav. Assess 25 (2), 95–103. [Google Scholar]
- Sullivan K, Krengel M, Bradford W, Stone C, Thompson TA, Heeren T, White RF, 2018. Neuropsychological functioning in military pesticide applications from the Gulf War: effects on information processing speed, attention, and visual memory. Neurotoxicol. Teratol 65, 1–13. [DOI] [PubMed] [Google Scholar]
- Terry AV Jr., 2012. Functional consequences of repeated organophosphate exposure: potential non-cholinergic mechanisms. Pharmacol. Ther 134 (3), 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Iowa Persian Gulf Study Group, 1997. Self-reported illness and health status among Gulf War veterans. A population-based study. J. Am. Med. Assoc 277 (3), 238–245. [PubMed] [Google Scholar]
- Tian H, Song X, Bressler J, Pruett S, Pope CN, 2002. Neither forced running nor forced swimming affect acute pyridostigmine toxicity or brain-regional cholinesterase inhibition in rats. Toxicology 176 (1–2), 39–50. [DOI] [PubMed] [Google Scholar]
- Toomey R, Alpern R, Vasterlin JJ, Baker DG, Reda DJ, Lyons MJ, Henderson WG, Kang HK, Eisen SA, Murphy FM, 2009. Neuropsychological functioning of U.S. Gulf War veterans 10 years after the war. J. Int. Neuropsychol. Soc 15 (5), 717–729. [DOI] [PubMed] [Google Scholar]
- Tsakiris S, Kontopoulos AN, 1992. Time changes in Na+,K+-ATPase, Mg++-ATPase, and acetylcholinesterase activities in the rat cerebrum and cerebellum caused by stress. Pharamcol. Biochem. Behav 44 (2), 339–342. [DOI] [PubMed] [Google Scholar]
- Tuovinen K, Kaliste-Korhonen E, Raushel FM, Hanninen O, 1999. Success of pyridostigmine, physostigmine, eptastigmine and phosphotriesterase treatments in acute sarin intoxication. Toxicology 134 (2), 169–178. [DOI] [PubMed] [Google Scholar]
- Valero J, Bernardino L, Cardoso FL, Silva AP, Fontes-Ribeiro C, Ambrosio AF, Malva JO, 2017. Impact of neuroinflammation on hippocampal neurogenesis: relevance to aging and Alzheimer’s disease. J. Alzheimers. Dis 60 (s1), S161–S168. [DOI] [PubMed] [Google Scholar]
- Walker AK, Kavelaars A, Heijnen CJ, Dantzer R, 2013. Neuroinflammation and comorbidity of pain and depression. Pharmacol. Rev 66 (1), 80–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RF, Proctor SP, Heeren T, Wolfe J, Krengel M, Vasterling J, Lindem K, Heaton KJ, Sutker P, Ozonoff DM, 2001. Neuropsychological function in Gulf War veterans: relationship to self-reported toxicant exposures. Am. J. Ind. Med 40 (1), 42–54. [DOI] [PubMed] [Google Scholar]
- White RF, Steele L, O’Callaghan JP, Sullivan K, Binns JH, Golomb BA, Bloom FE, Bunker JA, Crawford F, Graves JC, Hardie A, Klimas N, Knox M, Meggs WJ, Melling J, Philbert MA, Grashow R, 2016. Recent research on Gulf War illness and other health problems in veterans of the 1991 Gulf War: effects of toxicant exposures during deployment. Cortex 74, 449–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkenwerder W, 2003. Environmental Exposure Report: Pesticides Final Report. U. S. Department of Defense, Office of the Special Assistant to the the Undersecretary of Defense (Personnel and Readiness) for Gulf War Illnesses Medical Readiness and Military Deployments. (Washington, D. C: ). [Google Scholar]
- Wolfe J, Proctor SP, Erickson DJ, Hu H, 2002. Risk factors for multisymptom illness in US Army veterans of the Gulf War. J. Occup. Envrion. Med 44 (3), 271–281. [DOI] [PubMed] [Google Scholar]
- Young RC Jr., Rachal RE, Huguley JW 3rd, 1992. Environmental health concerns of the Persian gulf war. J. Natl. Med. Assoc 84 (5), 417–424. [PMC free article] [PubMed] [Google Scholar]
- Zakirova Z, Tweed M, Crynen G, Reed J, Abdullah L, Nissanka N, Mullan M, Mullan MJ, Mathura V, Crawford F, Ait-Ghezala G, 2015. Gulf War agent exposure causes impairment of long-term memory formation and neuropathological changes in a mouse model of Gulf War Illness. PloS One 10 (3), e0119579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakirova Z, Crynen G, Hassan S, Abdullah L, Horne L, Mathura V, Crawford F, Ait-Ghezala G, 2016. A chronic longitudinal characterization of neurobehavioral and neuropathological cognitive impairment in a mouse model of gulf war agent exposure. Front. Integr. Neurosci 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakirova Z, Reed J, Crynen G, Horne L, Hassan S, Mathura V, Mullan M, Crawford F, Ait-Ghezala G, 2017. Complementary proteomic approaches reveal mitochondrial dysfunction, immune and inflammatory dysregulation in a mouse model of Gulf War Illness. Proteonomics Clin. Appl 11 (9–10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zundel CG, Krengel MH, Heeren T, Yee MK, Grasso CM, Janulewicz Lloyd PA, Coughlin SS, Sullivan K, 2019. Rates of chronic medical conditions in 1991 gulf war veterans compared to the general population. Int. J. Environ. Res. Publ. Health 16 (6), E949pii. [DOI] [PMC free article] [PubMed] [Google Scholar]