

Summary
Empirical optimization of stem cell differentiation protocols is time-consuming, labor-intensive, and typically does not comprehensively interrogate all relevant signaling pathways. Here we describe barcodelet single-cell RNAseq (barRNA-seq), which enables systematic exploration of cellular perturbations by tagging individual cells with RNA ‘barcodelets’ to identify them based on the treatments they receive. We apply barRNA-seq to simultaneously manipulate up to seven developmental pathways and study effects on embryonic stem cell (ESC) germ layer specification and mesodermal specification, uncovering combinatorial effects of signaling pathway activation on gene expression. We further develop a data-driven framework for identifying combinatorial signaling perturbations that drive cells toward specific fates, including several annotated in an existing scRNAseq gastrulation atlas, and utilize this approach to guide ESC differentiation into a notochord-like population. We expect barRNAseq will have broad utility for investigating and understanding how cooperative signaling pathways drive cell fate acquisition.
eTOC Blurb
An eTOC blurb should also be included that is no longer than 50 words describing the context and significance of the findings for the broader journal readership. When writing this paragraph, please target it to non-specialists by highlighting the major conceptual point of the paper in plain language, without extensive experimental detail. The blurb must be written in the third person and refer to “Corresponding Author Last Name and colleagues.”
See examples here: https://www.cell.com/cell-stem-cell/current
Deriving stem cell differentiation protocols is time-consuming and labor-intensive. Yeo et al. describe a method for exploring in a single highly multiplexed experiment the combinatorial effects of up to seven signaling pathways on embryonic stem cell differentiation. An analysis framework identifies combinatorial signaling perturbations driving cells toward specific fates.
Graphical abstract
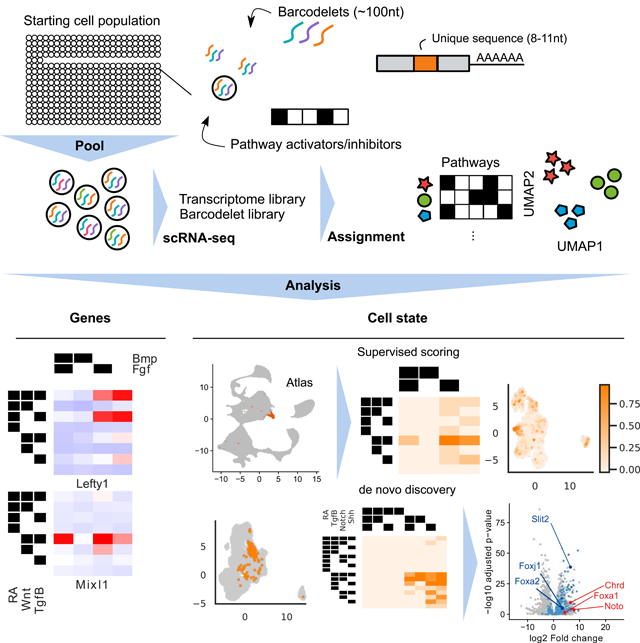
Introduction
Embryonic stem cells (ESCs) can be directed to differentiate into a variety of valuable cell types for disease modeling, drug screening, and regenerative medicine via the precise combinatorial and temporal manipulation of a relatively small set of intercellular signaling pathways (Cohen and Melton, 2011; Shi et al., 2017).While many protocols have been developed to direct the differentiation of ESCs into a variety of cell types using lessons learned from embryonic patterning of those cell types, many cell types of therapeutic value are inaccessible because the appropriate spatiotemporal combination of signaling pathways involved in specifying them has not been elucidated.
Current methods for discovering protocols to differentiate cells into a desired state are highly empirical. If ESC differentiation, like embryonic cell fate determination, is represented as a progressively branching lineage tree, current approaches collect detailed data on specific branching paths while leaving most branches unstudied (Figure 1A).We hypothesize that a systematic cataloging of the gene expression effects of each path through a lineage tree would aid our understanding of what cell types arise from alternative conditions.
Figure 1. barRNA-seq enables combinations of a set of treatments to be observed in a single highly-multiplexed experiment.

(A) Cell fate determination depicted as a decision tree. Circles in dashed lines depict unexplored branches.
(B) 2Schematic of barRNA-seq experiment. Cells are treated with a set of activators and inhibitors targeting pathways of interest, and transfected with a combination of barcodelets associated with the set of treatments they observed. Cells are subsequently pooled for scRNA-seq. Barcodelets are readily observed in scRNA-seq, and can then be used to assign a treatment group to each cell
(C) Schematic of barcodelet. Barcodelets are small RNA molecules containing adapter sequences, a variable barcode, an RT-qPCR primer and a polyA tail for oligo-dT binding
(D) Barcodelet count fraction in germ layer dataset. Cells are grouped based on the whether the top 5 most abundant barcodelets form a valid combination
(E) Distribution of summed barcodelet count fraction of top 5 most abundant barcodelets in germ layer dataset, grouped by whether or not the top 5 most abundant barcodelets form a valid combination
(F) Summary of datasets. Asterisks indicate percentages reported with respect to cells in which >10 barcodelet UMIs were observed
(G) UMAP visualization of cells collected at each starting point (top: mESC, bottom: mesendoderm). Cells are colored according to source dataset (left) and whether they were control cells (right)
See also Figure S1G
To address our hypothesis, we developed barcodelet single-cell RNA-sequencing (barRNA-seq) to systematically examine an entire lineage tree defined by the combinational modulation of signaling pathways. In barRNA-seq, barcodelets are simply added to a treatment condition, become durably associated with individual cells, and appear as unique messages in scRNA-seq data (Figure 1B-C).We apply our assay to study the consequences of the combinatorial modulation of signaling pathways during early mESC differentiation toward germ layer and mesodermal fates.
We chose to study mESC differentiation toward germ layer and mesodermal fates because it faithfully models in vivo germ layer specification and patterning, for which there is a wealth of existing gene expression data available. The emergence of droplet-based single cell RNA sequencing (scRNA-seq) (Klein et al., 2015; Macosko et al., 2015) has allowed developmental biologists to produces atlases of gene expression at different stages of mouse embryonic development (Ibarra-Soria et al., 2018; Pijuan-Sala et al., 2019).
Combinatorial modulation of signaling pathways has been shown to govern developmental transitions (Loh et al., 2014, 2016; Davidson, 2010): Wnt and Tgfβ signals are vital in specification of mesendoderm, which is further specified to endoderm and mesoderm by Tgfβ and Bmp signals. Anterior-posterior axial patterning is accomplished primarily through Wnt, Fgf and retinoic acid (RA) signaling inputs, dorsal-ventral specification is primarily influenced by Bmp and Shh signaling, and left-right axis determination instigated by Notch and Tgfβ signaling (Meno et al., 1998). The complex interplay of these seven signaling pathways have been implicated in development of all germ layers and axes.Nonetheless, a comprehensive view of how signaling inputs combine to modulate cell fate is still lacking.
One reason for the lack of comprehensive understanding of combinatorial signaling input is the lack of suitably high-throughput approaches to measure their transcriptional consequences. Bulk transcriptomic approaches have been hampered by high cost and effort per sample. Droplet-based scRNA-seq has primarily been used to map diversity in unmanipulated samples, but has more recently been used to multiplex a large number of samples together in a single experiment (Adamson et al., 2016; Dixit et al., 2016; Jaitin et al., 2016; Kang et al., 2018). Recently, methods complementary to barRNA-Seq have been developed to barcode cells by treatment condition prior to scRNA-seq (Shin et al., 2019; Stoeckius et al., 2017), but thus far these methods have not been applied to systematic dissection of stem cell differentiation.
We use barRNA-Seq to measure transcriptome-wide expression in 32 to 384 distinct populations per experiment at single cell resolution. We systematically explore the combinatorial effects of activation/inhibition of up to seven signaling pathways during ESC germ layer patterning. We start by showing that the expression of an underappreciated fraction of genes are dependent on the combinatorial activation and inhibition of these signaling pathways. We probe the mechanistic basis of combinatorial signaling control of individual gene expression by the functional evaluation of individual enhancers, identifying individual promoters and promoter/enhancer pairs that carry out combinatorial gene regulation. We develop an analysis framework that identifies treatment combinations associated with distinct expression states via both a supervised approach that leverages a reference atlas to score our in vitro cells, as well as an unsupervised approach that relies only on the assay data. We show how this data-driven framework enables us to propose specific combinatorial signaling logics that give rise to well-defined cell types by then using it to guide differentiation of a Foxa2+ notochord-like cell population. Altogether, this work highlights the promise of highly multiplexed scRNA-seq in elucidating the drivers of stem cell differentiation.
Results
Barcodelet scRNA-seq enables the observation of hundreds of treatment conditions in a single highly-multiplexed experiment
To simultaneously record scRNA-seq measurements for hundreds of populations in a single experiment, we developed a barcoding method compatible with oligo(dT)-based reverse transcription. We performed in vitro transcription to produce ~100 nt RNA molecules we call barcodelets composed of an Illumina sequencing adapter sequence, a variable 8–11 nt condition-specific barcode, a primer sequence for RT-qPCR quantification, and a 28 nt poly-adenine stretch for oligo(dT) binding (Figure 1C). We tested several RNA transfection approaches to introduce barcodelets into cells, identifying TransIT-mRNA to be optimal for high-efficiency RNA integration into cells (Figure S1A). We found the survival and gene expression of mouse ESCs (mESCs) and their derivatives to be unaffected by RNA transfection up to 72 hours post-transfection, with no detectable differential expression (<= 1 gene differentially expressed at p < 0.01, Figure S1B-C). Cellular populations are labeled with a unique combination of 2–5 distinct barcodelet species. The combinatorial complexity of this labeling strategy allows the theoretical disambiguation of hundreds of thousands of populations per experiment.
We perform single-cell RNA-seq using 10X Chromium droplet-based scRNA-seq and produce a cDNA library from all of the cells in a single experiment (Zheng et al., 2017).We divide cDNA molecules that include 10x Chromium cell barcodes (separate from our condition barcodelets) into separate short cDNA (<500-bp) and long cDNA (>500-bp) pools. Barcodelet cDNA is 170–180-bp long, and so we perform barcodelet-specific library preparation on the short pool and transcriptome library preparation on the long pool. This separation is crucial since barcodelets are present at extremely high levels that would swamp out transcriptome reads if prepared together. Cell barcodes allow for the association of barcodelets with their associated transcriptome reads.For our experiments we collected >4*108 transcriptome reads and >1*107 barcodelet reads per experiment.
We first applied barRNA-seq to systematically discover signaling pathway interactions during mESC germ layer specification. Epiblast-stage mESCs were divided into 32 treatment groups comprising every combination of activation or inhibition of five key developmental signaling pathways: Wnt, retinoic acid (RA), Tgfβ, Bmp, and Fgf. Activators comprised a mix of growth factors and small molecule agonists, inhibitors were all small molecule antagonists, and all activators and inhibitors were dosed at concentrations predetermined to be active (Supplementary Table 1). While serum is used during differentiation, which has been shown to enable endogenous activation of signaling pathways (Ying et al., 2003), the use of small molecule inhibitors that function intracellularly should effectively prevent such signaling. The 32 populations were each transfected with a unique combination of 5 of 10 unique barcodelet species, each corresponding to either the activator or inhibitor of a pathway (Figure 1D). Populations were then pooled for 10X Chromium droplet-based scRNA-seq. We refer to this dataset as the germ layer dataset.
To assign each cell a treatment combination, we inspect the top barcodelets in the data. We note that since each of the 32 treatment conditions is specified by a combination of 5 from a pool of 10 possible barcodelet species, only ~12% of combinations would be valid. In this germ layer dataset, we observe that the most abundant barcodelets in 75.6% of cells cells form a valid combination (Figure 1F). Furthermore, cells in which the top barcodelets form a valid combination have a higher summed barcode count fraction over the most abundant barcodelets than cells in which the top barcodelets do not form a valid combination (Figure 1E, Figure S1D-E). Hence, by thresholding on the summed barcode count fraction, we are able to control the false positive rate of our assignments. To ensure the fidelity of our labeling system for downstream analysis, we choose a strict false positive rate of 1%, which allowed us to assign 6369, or 68.2% of cells in which at least 10 barcodelet UMIs were observed (Figure 1F), with a median of 131 cells in each group.
We next hypothesized that we would only be able to observe later signaling events by starting with a population of already differentiated cells. We hence applied barRNA-seq to mesendodermal cells. To produce these anterior primitive streak mesendodermal cells, epiblast-stage cells were treated for 24 hours with agonists of Wnt, Tgfβ, and Fgf and inhibitors of RA and Bmp. As has been shown (Loh et al., 2016), we find that co-activation of Wnt, Tgfβ, and Fgf supports maximal induction of primitive streak mesendoderm genes T, Mixl1, and Mesp1, and inhibition of Bmp biases toward anterior primitive streak with maximal expression of Gsc and Eomes. We then exposed these anterior primitive streak mesendodermal cells to all combinations of activation or inhibition of seven pathways, adding Hedgehog and Notch pathways to the previous five pathways. This setup yielded 128 unique combinatorial treatments, which were performed with 2–4 biological replicates to yield a median of 21 cells for 384 unique populations (Figure 1F, S1G). We refer to this dataset as the mesendoderm dataset.
Finally, to enable us to assess replicate consistency and to boost power for downstream analysis, we repeated the experiment with both starting states using the 5 original pathways assayed in the germ layer dataset, for a median of 64 cells across 144 conditions with 2–4 biological replicates. This dataset also includes cells which received no treatments, which we refer to as control cells. We refer to this dataset as the mixed dataset.
Expression data for each dataset was first processed individually (see methods), and then cells corresponding to each starting state (mESC, mesendoderm) were combined for downstream analysis and visualization (Figure 1F-G, Figure S1I,K). In addition, we further filtered transcriptomic outgroups of extraembryonic endoderm (ExEn) using a semi-supervised, smoothed clustering approach initialized with known gene markers. ExEn cells are known to arise from aberrant differentiation of ESCs, and they comprise 4–5% of cells in all treatment groups, largely independent of treatment. The ExEN outgroup is highly transcriptionally distinct (Figure S1H,J), and hence removing them reduces noise in downstream analyses. We note that in bulk transcriptomic approaches, confounding populations cannot be accounted for in analysis.
Combinatorial treatments induce specific and consistent expression profiles
We first performed two internal validation checks to assess if barcodelets have correctly labeled the cells with the treatments they observed. First, we hypothesized that cells assigned the same treatment group should be closer in expression space than cells assigned different treatment groups. We observe this to be the case both when comparing the expression distance in PCA space between pairs of cells receiving the same/different sets of treatments across datasets (Figure 2A, Mann-Whitney p-value << 0.01), and by visualizing the cells from each dataset using UMAP (Figure 2C-D, S2). Cells that received the same treatment combination are also more likely to be closer in expression space even if they are from different datasets, and there is strong correlation of aggregated expression profiles across datasets, suggesting good replicate consistency (Figure 2B, S1L).
Figure 2. Combinatorial treatments induce specific and consistent expression profiles.

(A) Distribution of distances between pairs of cells across conditions and datasets. Distance is computed as Euclidean distance in PCA space. Distances between cells within the same condition are significantly smaller than distances between cells from different conditions, and between treatment and control cells (Mann-Whitney p-value << 0.01)
(B) Heatmap depicting correlation of mean expression profiles for cells assigned to each treatment combination across datasets. The last column (indicated by grey circles) corresponds to control cells
(C, D) Visualization of cells assigned to a few treatment groups for both starting states (mESC: RA+Wnt-Tgfβ+Bmp-Fgf+, RA+Wnt-Tgfβ-Bmp+Fgf-, RA-Wnt-Tgfβ-Bmp-Fgf-; Mesendoderm: RA-Wnt+Tgfβ+Bmp+Fgf-, RA+Wnt+Tgfβ-Bmp+Fgf+, RA-Wnt-Tgfβ-Bmp-Fgf-). UMAP visualization of cells assigned to that treatment condition, colored by dataset
(E) Table showing results on pathway status classification task. In addition to AUROC, top 10 genes ranked by coefficient are shown.
See also Figure S1L, S2
Secondly, we hypothesized that gene expression should be strongly predictive of the activation/inhibition status of each individual signaling pathway. To test this, we trained logistic regression models to predict the signaling status of each pathway given gene expression. Our classifiers mostly achieved excellent classification performance in a held-out test set (median AUROC = 0.957 for mESC, 0.955 for mesendoderm) (Figure 2E). The lower performance of Notch and Shh models may be because their effects on gene expression are not as readily observed except in combination with other pathways. We examined the genes with the highest positive coefficients for each classifier, and found that genes were consistent between the two starting states, and that many have previously been described to be downstream of these pathways (Figure 2E). This includes several well-known targets of Wnt signaling such as Cdx1, Cdx2, Lef1, Wnt6, and Axin2 (Sherwood et al., 2011), as well as Id1, Id2, and Id3, which have previously been shown to be downstream effectors of Bmp signaling (Hollnagel et al., 1999). We note that even for Notch and Shh, the genes with highest positive coefficients are Nrarp and Ptch1, which are known feedback inhibitors of the respective pathways (Lamar et al., 2001;Stone et al., 1996). Altogether, these results indicate that barRNA-seq allows accurate labeling of multiplexed populations derived from combinatorial signaling treatment of germ-layer mESC.
A regression analysis framework identifies combinatorial interactions in signaling pathways
Inspection of the mean gene expression within each treatment condition highlights complex interactions between pathways in regulating gene expression (Figure 3A). To assess in a principled manner the extent to which genes are under combinatorial control of these signaling pathways, we developed a Bayesian regression analysis framework to identify the effects of these pathways on the top 2000 most variable genes within the barRNA-seq dataset. Scaled normalized expression of each gene was modeled as arising from a normal distribution, with the mean dependent on the treatments that each cell had received. For each gene, we compared the fit of models that included varying orders of interaction terms to determine optimal model complexity. We defined the optimal model to be the model with the lowest complexity that has a model score within the 95% confidence interval of the best model score (See STAR methods). Of these top 2000 most variable genes, we found that the optimal model for ~60% of genes include second-order and higher terms, suggesting that the majority of differentially expressed embryonic genes are under combinatorial signaling control. (Figure 3C)
Figure 3. Gene expression at the germ layer stage requires highly complex interactions between signaling pathways.

(A) Mean scaled gene expression of genes chosen for validation of cells that observed different treatment combinations
(B) Correlation of average scRNA-seq expression with GFP expression for reporter cell lines. Spearman rank correlation coefficient is reported for each gene and is significant at p < 0.05 for all genes except Trh. 95% confidence intervals are estimated via bootstrap (n = 1000).
(C) Schematic of Bayesian regression analysis framework. Model fitting: X is a covariate matrix with order of each term annotated above. Black indicates 1. Models are fit with varying orders of terms included. Model comparison: Model comparison plot for an example gene, with model order on the y-axis and estimate of model fit on the x-axis. The vertical grey-dashed line indicates the score of the best model fit. Empty circles indicate the mean score of that model, black horizontal lines indicate the standard deviation of the score, and grey horizontal lines indicates the standard deviation of the difference between the score of that model and the best model. (See methods) Lower scores imply a better model fit.
(D, E) Hierarchical clustering of top 2000 most variable genes. Clusters are annotated beneath with alternating dark/light grey. The Lefty1 cluster (red, *) and T/Mixl1 cluster (blue, **) are magnified and relevant genes indicated See also Figure S3
Focusing first on cells starting in the mESC state, our results not only recapitulate known linear and combinatorial pathway effects, but also uncover genes whose expression requires previously unknown combinatorial signaling at the germ layer stage. For example, Lefty1 is known to be a Tgfβ target gene (Besser, 2004), and we do find that activation of Tgfβ increases Lefty1 expression. However, we find that Lefty1 expression is greatly increased when RA and Tgfβ are coactivated, and that Bmp activation ablates this effect. Hence, RA+Tgfβ+Bmp- conditions induce highest Lefty1 expression. Examples of other genes include: Hoxa1, a canonical RA-induced gene (Simeone et al., 1990), which we find to be also dependent on Wnt and Fgf input, and Mixl1, a mesendoderm-specific gene (Hart et al., 2002) which we find to depend on the appropriate combinatorial input of four signaling pathways (Figure 3A, S3A-B).
To discover genes sharing similar regulatory logic, we clustered genes by their optimal model coefficients using correlation as the distance metric (see STAR methods), and found that sets of genes share the same combinatorial regulation profile (Figure 3D). For example, Lefty1, Lefty2, and Pycr2, which lie in a contiguous stretch on chromosome 1, all share a combinatorial regulatory pattern that favors high expression in RA+Tgfβ+Bmp- conditions (Figure 3A, 3D). Several other genes that are in distinct chromosomal locations, including Fgf8 and Trh, share a highly similar combinatorial regulatory logic. Genes with similar combinatorial regulatory logic to Mixl1 (high expression in RA-Wnt+Tgfβ+Fgf+ conditions, (Figure 3A, 3E) include additional key mesendodermal genes such as T, Gsc, and Mesp1. (Figure 3E).
To validate the highly combinatorial regulation of some of these developmental genes we monitored the expression of eight genes (Cdx2, Hoxa1, Lefty1, Mest, Mixl1, T, Tdgf1, Trh) by constructing corresponding GFP knock-in reporter cell lines through CRISPR/Cas9-based homology-directed repair (Arbab et al., 2015). We compared average scRNA-seq expression among cells in each combinatorial treatment condition with GFP expression in the GFP cell lines and find strong concordance (Figure 3B, S3G-I). Analysis of these GFP reporter cell lines reveals that, in optimal inductive conditions, the vast majority (64.9–97.6%) of cells display above-control transgene fluorescence for Brachyury, Hoxa1, and Lefty1 (Fig. S3G-I), suggesting that differentiation is relatively uniform. Thus, unbiased analysis of barRNA-seq data allows the discovery of hundreds of genes regulated by highly combinatorial signaling logic.
For cells starting in the mesendoderm state, we again observed complex signaling interactions (Figure S3C-F), and validated the accuracy of the gene expression in barRNA-seq by comparing averaged scRNA-seq expression of Lefty1, Mixl1, Mest, Tdgf1 and Tfrc with flow cytometric GFP expression (Lefty1, Mixl1, Mest, Tdgf1) or antibody staining (Tfrc) in matched conditions. Again, we found that expression in our validation expression correlates with average scRNA-seq expression (Figure S3D), demonstrating again that barRNA-seq yields faithful gene expression information. However, the correlation is weaker than in the previous experiments, likely as a result of the reduced power from multiplexing a larger number of conditions into a single experiment.
Enhancers and promoters can implement signaling based combinatorial control of gene expression
We hypothesized that signaling-based combinatorial control of gene expression could be mechanistically linked to certain regulatory DNA sequences.For 17 genes that were regulated combinatorially at the germ layer stage, we identified 1-kb enhancer regions centered on strong ChIP-Seq peaks for Wnt effector Tcf/Lef members Tcf7l1 (Cole et al., 2008) and Tcf7l2 (Szczesnik et al., 2019) or RA effector Rarg (Mazzoni et al., 2013).We cloned these enhancer regions into a Tol2 transposon (Urasaki et al., 2006) genome-integrated GFP enhancer reporter construct with a minimal promoter (Sherwood et al., 2014).We then derived mESC lines with transposon-mediated random genomic integration of these reporter constructs and measured GFP fluorescence under combinatorial signaling conditions matching the germ layer barRNA-seq experiment.
Of the 17 enhancer reporter constructs, eight showed activation in response to at least one of the pathways that activate the nearby combinatorially regulated gene. For these other nine enhancers, we posit that other enhancers must be required for signal-dependent gene activation in germ layer-stage mESCs, which can partially be attributed to the lack of ChIP-Seq data in the exact starting population. To determine whether enhancer reporter constructs exhibited combinatorial activity, we computed the mean difference of the observed and activity under a linear model where the activity depends additively on contributions of each pathway (Figure 4A).Enhancers with large deviation from this linear model can be inferred to require combinatorial signaling. Of the constructs tested, an Evx1 upstream enhancer containing a strong Tcf7l2 binding site most strongly exhibited combinatorial activity, recapitulating the gene’s pattern of maximal activation in RA-Wnt+ conditions that cannot be explained by linear effects of the two pathways (p < 0.2, Figure S4A, 4B). Another example is Pbx1, which is maximally activated by RA+Wnt- conditions. We observe that an intronic Pbx1 enhancer with a strong Rarg binding site (Figure S4B) recapitulates this combinatorial regulatory pattern (p < 0.1, Figure 4C).
Figure 4. Sequence features implement signaling-based combinatorial control of gene expression.

(A) Expected and observed normalized GFP expression of enhancer reporter assays for genes chosen for validation. Expected expression was computed under a linear model where effects are assumed to be linearly additive. Asterisks denote result of paired t-tests on log expression. * is significant at p < 0.2, ** at p < 0.1.
(B, C) Mean expression of GFP reporters in enhancer reporter assay (left) and scRNA-seq (right) showing combinatorial effects. Expression in enhancer reporter assay is normalized to the −/− condition. (B) Assay results for Evx1, which is most highly expressed in RA-/Wnt+, (C) Assay results for Pbx1, which is most highly expressed in RA+/Wnt-
(D) Expected and observed normalized GFP expression of reporters containing sequence features from the enhancer, the native promoter, or both for Cdx1 and Lefty1. Expected expression was computed under a linear model where effects are assumed to be linearly additive. Asterisks denote result of paired t-tests. * is significant at p < 0.2, *** at p < 0.05.
(E, F) Mean expression of GFP reporters in enhancer reporter assay (left) and scRNA-seq (right) showing combinatorial effects. Expression in enhancer reporter assay is normalized to the −/− condition. (B) Assay results for Cdx1, which is most highly expressed in RA-/Wnt+, (C) Assay results for Lefty1, which is most highly expressed in RA+/Tgfβ+/Bmp- See also Figure S4
We further investigated the regulation of Cdx1 and Lefty1, where the tested enhancers did not recapitulate the observed regulation of the gene. We hypothesized that combinatorial regulation of these genes may require the promoter region instead of or in addition to the enhancer region (Figure S4C-D). We replaced the minimal promoter in the enhancer reporter construct with a 1-kb fragment of each gene’s native promoter. We found for both genes that while the enhancer alone failed to replicate the combinatorial regulation of the native gene, the gene’s native promoter was able to recapitulate combinatorial activity (p < 0.05). In the case of Cdx1, combining the enhancer and native promoter further increased combinatorial activity in the Wnt+RA+ condition that maximally activates the gene, which would not have been predicted from the enhancer alone results (p < 0.05, Figure 4D-F). In sum, individual enhancers can be sufficient to implement combinatorial regulatory logic for some genes, while the native promoters alone or together with enhancers can implement combinatorial regulatory logic for other genes.
Specific differentiation conditions correspond to in vivo embryonic cell types
We next turn from gene-level to cell-state level analysis, developing an analysis framework for identifying specific differentiation conditions under which cells are driven to particular fates. To do so, we employed two complementary approaches: (1) a supervised approach, where we used published labeled in vivo data to score our dataset, and (2) an unsupervised approach, where we identified stable subpopulations from our data de novo (Figure 5A).
Figure 5. Specific differentiation conditions starting from mESC correspond to known embryonic cell types.

(A) Schematic of two approaches for identifying specific different conditions under which cells are driven to particular fates. Top: A supervised approach, using an external atlas to score cells. Bottom: An unsupervised approach that identifies stable subpopulations
(B, C, D) Results of applying supervised approach to cells starting at the mESC stage
(B) Heatmap depicting mean score of each cell type classifier for cells assigned to each treatment combination
(C) Distribution of classifier scores visualized on the UMAP
(D) Expression of gene markers identified for each cell type classifier visualized on the UMAP. Gene markers shown are within the top 20 genes most correlated with the cell type classifier score
For the supervised approach, we hypothesized that many cell types found in vivo during early development would be present amongst our exhaustive examination of 160 in vitro differentiation conditions. To investigate this, we used a published dataset of single cell RNA-seq profiles of 116,312 cells derived from E6.5 - E8.5 mouse embryos (Pijuan-Sala et al., 2019). We first built classifiers for each of the 37 labeled cell types manually annotated in this published dataset. Our classifiers achieved >0.8 accuracy on all subpopulations in held-out test sets (Supplementary Table 3). We then used these classifiers to score cells from our experiments and ranked treatment conditions by the mean score in each treatment condition.
Applying this approach to cells starting at the mESC state, we found that specific combinatorial signaling conditions are associated with particular embryonic subpopulations (Figure 5B-D). We found that induction of the anterior primitive streak was indeed associated with RA-Wnt+Bmp-Fgf+ conditions, as previously shown (Loh et al., 2016). These cells are enriched for mesendodermal T, Mixl1, and Mesp1, and were used as precursors to our mesoderm differentiation experiments.
We also identified a number of additional embryonic populations associated with specific combinatorial differentiation conditions. For example, induction of annotated neural crest cells was associated most strongly with cells receiving inhibition of Wnt, Tgfβ, and Bmp. Inhibition of Smad signaling downstream of Tgfβ and Bmp is known to induce neural fates (Chambers et al., 2009). Furthermore, RA is known to posteriorize ESC-derived neural precursors to promote spinal cord fates over rostral fates (Wichterle et al., 2002). Accordingly, RA+Wnt-Tgfβ-Bmp- cells are enriched in neural tube markers such as Pax6, Sfrp1 and Sfrp5 (Rimini et al., 1999). Another example is surface ectoderm, which we find to be correlated with RA+Tgfβ-Bmp+Fgf-. Bmp signaling is known to promote non-neural ectoderm fate acquisition (Leung et al., 2013), and our data confirms that non-neural ectodermal marker genes Tfap2a, Dlx5, and Bambi (Reichert et al., 2013) are all induced by Bmp signaling. Finally, caudal mesoderm cells were associated with RA+Wnt+Bmp-Fgf+. These cells have posterior neural and mesodermal fate potential that self-renews in the tailbud of embryos to progressively populate posterior embryonic structures and is known to differentiate in response to combined Wnt and Fgf (Turner et al., 2014). Canonical caudal mesoderm genes Cdx1, Cdx4, and Fgfbp3 are enriched in this population. Thus, our barRNA-seq data confirms and refines the signaling combinations involved in germ layer differentiation.
Applying this approach to cells starting at the mesendoderm state identified later signaling-based events in development. Previous work had confirmed that paraxial (also called presomitic) mesoderm is specified from ESC-derived mesendoderm by activation of Wnt and inhibition of Bmp and Tgfβ (Loh et al., 2016). We find that RA-Wnt+Tgfβ-Bmp- cells indeed are scored highest by the paraxial mesoderm classifier. Gene markers associated with this classifier include Tbx6, Dlx1 and Aldh1a2. The combinatorial logic driving cells to lateral plate mesoderm fates also fits with what has been found previously. Loh et al had also previously identified that cells receiving Bmp with inhibition of RA, Wnt and Tgfβ are driven towards lateral plate and extraembryonic mesoderm formation, as marked by the gene Hand1. In support of this signaling logic, we find that the classifier for allantois, an extraembryonic mesoderm structure, assigned the highest scores to cells receiving RA-Wnt-Tgfβ-Bmp+ treatment (Figure 6A-C). Based on gene expression, it is likely that cells receiving this treatment condition are more broadly specified as lateral plate and extraembryonic mesoderm, and the published allantois annotation may be too specific for the annotated cells. It has also been previously reported that ESC-derived mesendoderm cells treated in RA-Wnt-Bmp- conditions give rise to cells resembling the node (Winzi et al., 2011). We identify that cells receiving RA-Tgfβ+Wnt-Bmp- conditions are enriched in expression of node marker genes such as Tdgf1, Lhx1, Gsc, Eomes, and Otx2, and thus we propose that Tgfβ is an additional factor in node fate acquisition from mesendoderm cells. To test the involvement of Tgfβ in node fate acquisition, we tested whether Tdgf1-GFP expression from mesendoderm cells was sensitive to Tgfβ treatment. We found that Tdgf1-GFP expression was maximal in mesendoderm cells treated with RA-Tgfβ+Wnt-Bmp- conditions, and substituting Tgfβ activation with Tgfβ inhibition significantly decreased Tdgf1-GFP expression (Figure S5A-B).
Figure 6. Mesendodermal cells are directed to diverse fates by combinatorial signaling.

(A, B, C) Results of applying supervised approach to cells in the mesendoderm dataset
(A) Heatmap depicting mean score of each cell type classifier for cells assigned to each treatment combination
(B) Distribution of classifier scores visualized on the UMAP
(C) Expression of gene markers identified for each cell type classifier visualized on the UMAP. Gene markers shown are within the top 20 genes most correlated with the cell type classifier score
See also Figure S6
To investigate whether signaling pathway combinations associated with particular cell states by our assay was concordant with the reference atlas, we visualized the mean expression of the top ten genes most predictive of each pathway’s activity (from Figure 2E) in the E8.25 embryonic atlas that were also highly variable in the atlas (Figure S5C-D). We find some concordance with the results resulted in our paper. For example, caudal mesoderm is associated with the RA+Wnt+Bmp-Fgf+ condition in our assay, and indeed genes associated with RA, Wnt and to a lesser extent, Fgf, do appear to be upregulated in cells labeled as caudal mesoderm in the E8.25 atlas. Similarly, spinal cord is associated with RA+Bmp-Tgfβ- conditions as is well supported in the literature, and the populations annotated as allantois, which expresses canonical markers of lateral plate mesoderm, and extraembryonic mesoderm show expected Bmp+ signatures. However, we also found in our assay that anterior primitive streak was associated with RA-Wnt+Bmp-Fgf+. Although this is consistent with previous work (Loh et al. 2016), it cannot be easily inferred from the atlas data (Fig. S5C-D). Hence, this analysis does not consistently provide clear insight into which pathways must be activated or repressed to drive differentiation into each cell type, either because this is a relatively small gene set whose predictive power is limited due to the dynamic nature of embryonic signaling, or the expression profiles of cell states do not necessarily have to reflect pathway modulations that were required to direct cells toward that state. Overall, we believe that our approach of building classifiers based on high-dimensional gene expression profiles of embryonic subpopulations and scoring their similarity with barRNA-seq populations yields more accurate insight into the signaling pathways involved in the formation of these embryonic populations.
Combinatorial treatments drive mesendodermal cells cells toward diverse and specific fates
In addition to confirming previously known differentiation conditions for ESC-derived mesendoderm, we sought to identify stable subpopulations in our dataset that were not annotated by our supervised approach. Hence, we also devised an unsupervised de novo strategy, which not only independently discovered cell subpopulations already identified by the supervised approach described above hence validating the approach (Figure S6), but also allowed us to identify additional cell fates. For each cluster, treatment conditions are then ranked by membership and gene markers were determined via differential expression analysis (Figure 5A).
Using this method, we annotated two additional populations from the mesendoderm dataset: a Runx3/Cdx4+ cell population encompassing primitive hematopoietic cells that favored RA-Wnt+Bmp+ conditions, as well as a Foxa2+/T+ cell subpopulation with similarity in gene expression to embryonic notochord cells that were most enriched for in the RA-Wnt-Bmp-Tgfβ+ condition (Figure 7A-C). To determine if these conditions allow for differentiation of notochord cells from mESCs, we performed mESC-derived mesendoderm differentiation of a Foxa2-GFP reporter cell line in the presence of either this proposed combination of conditions or variants thereof where one factor was substituted for its paired activator/inhibitor. We were able to recapitulate the results of our barRNA-seq assay, finding that in the presence of optimal RA-Wnt-Bmp-Tgfβ+Shh+ conditions, on average 14.9% of cells show Foxa2-GFP expression (Figure 7E, S7B). Inhibition of Tgfβ or activation of Bmp or Wnt reduced Foxa2+ cell numbers and expression dramatically (Figure 7D, S7A).
Figure 7. Cells expressing notochord markers are selected for by activating Tgfβ+.

(A) Stable clusters of cells identified by unsupervised clustering analysis
(B) Expression of gene markers identified for each cell type classifier visualized on the UMAP. Gene markers shown are differentially expressed with respect to the rest of the cell population at q-value < 0.05
(C) Fraction of cells assigned to each treatment combination that belong to the corresponding cluster in (A)
(D) Average and standard deviation of flow cytometric Foxa2-GFP expression normalized to the Tgfβ+Shh+Fgf-Bmp-Notch-Wnt baseline.
(E) Flow cytometry plots showing Foxa2-GFP expression in the presence of RA-Wnt-Bmp-Tgfβ+
(F) Differential expression analysis of bulk RNA-seq expression of Foxa2+ putative notochord cell population vs. control cells. Dots colored in blue are genes that were also found to be significantly differentially expressed with positive fold change in barRNA-seq at corrected p-value < 0.05. Annotated dark blue dots are known notochord markers. Dots colored in red are additional notochord markers found to be significantly differentially expressed in the bulk RNA-seq with adjusted p-value < 0.05.
(G) RT-qPCR of notochord markers in FoxaGFP+ and control cell populations. Up-regulation of all notochord markers is significant by t-test at p-value < 0.05 on log RT-qPCR normalized expression.
(H) Immunofluorescence staining of notochord markers in notochord-like and control differentiation conditions. All panels are 20X-magnified, Hoechst 33342 is included in the merged panels.
To further characterize this cell population, we conducted bulk RNA-sequencing of the Foxa2-GFP+ cells treated with optimal RA-Wnt-Bmp-Tgfβ+Shh+ conditions. When comparing the set of positive markers identified from the notochord-like cluster discovered in barRNA-seq with the positive markers identified in the bulk RNA-sequencing analysis of the sorted Foxa2-GFP+ population, we found the barRNA-seq markers to be significantly over-represented in the bulk RNA-seq markers (hypergeometric p-value << 1e-45), and to have the highest fraction of overlap (55.8%) amongst all barRNA-seq clusters. In this way, the bulk RNA-sequencing recapitulates the scRNA-seq data. These positive markers include T, Slit2, Foxj1, Tmem176a and Tmem176b (Figure 7F, Supplementary Table 4-5). The higher sensitivity of bulk RNA-sequencing additionally allowed us to identify a larger set of differentially expressed genes, including previously described notochord markers Noto, Slit2 and Chrd (Figure 7F) (Peng et al., 2019). We confirmed robust up-regulation of these notochord markers in the sorted population with respect to the control population using RT-qPCR (Figure 7G). Immunofluorescence experiments show that clusters of cells in notochord-like conditions but not control conditions express proteins for all three of the markers assessed-- Slit2, Chrd, and Foxa2 (Figure 7H). barRNA-seq has therefore enabled us to elucidate specific signaling combinations that drive differentiation toward novel cell types of interest.
Discussion
barRNA-seq reveals the combinatorial complexity underlying embryonic cell fate specification by systematically characterizing the response of ESC-derived epiblast-stage and mesendoderm-stage cells to dozens of signaling conditions. We find that a significant fraction of genes are sensitive to specific combinatorial signaling conditions. Furthermore, while in certain cases sets of genes exhibit strong co-regulation (e.g. Lefty1, Lefty2, and Pycr2) we find many more cases in which genes exhibit specific regulatory regimes not shared by other similarly regulated genes (Figure 3E). We also provide evidence that combinatorial signaling pathway response sometimes occurs at the level of individual enhancers or promoters, and can be integrated by multiple regulatory elements. These findings suggest that signaling pathways have evolved a wide variety of interaction modes. Altogether, our findings support a deep network of evolved regulatory relationships governing the exquisitely controlled process of embryo patterning.
Emerging “cell atlas” approaches that characterize native populations at single-cell resolution are a great resource for interpreting in vitro differentiation data (Han et al., 2018; Regev et al., 2017; Tabula Muris Consortium et al., 2018). By comparing in vitro ESC-derived populations to in vivo cells from gastrulation-stage and germ layer-stage embryos, we find that certain ESC-derived cells show striking similarity to embryo-derived cells whereas others do not have obvious embryonic counterparts. For example, ESC germ layer-derived neural crest, neural tube, non-neural ectoderm, and caudal mesoderm cells as well as mesendoderm-derived paraxial mesoderm, somitic mesoderm, lateral plate mesoderm (annotated as allantois in the embryonic dataset), and anterior primitive streak cells exhibit high transcriptome-wide similarity to their embryonic counterparts. On the other hand, several transcriptomically coherent populations emerge from the ESC mesendodermal cells that share expression of known key marker genes but lack strong transcriptome-wide similarity to cells from germ layer stage embryos, including notochord cells and primitive blood precursors. It is possible that these cells would have correlates in embryos from a different stage of development. It is also possible that the embryonic correlates occur so rarely or transiently that they are not sufficiently represented in the embryonic dataset. A third possibility is that some of the combinatorial signaling conditions provided never occur in embryos or only occur in the presence of additional signals not provided in our experiment. Additionally, in vivo pathway activity may not consistently provide clear insight into how pathways should be modulated to control cell fate (Supplementary Information). Hence, complementary perturbation-based technologies such as barRNA-seq are important to further elucidate causes of cellular differentiation and heterogeneity.
Developmental cell fate specification is a highly complex process, and the assay conditions used in this work only begin to approach the possible signaling conditions to which an embryonic cell could be exposed. The ability to perform transcriptomic analysis on hundreds of conditions per experiment opens up possibilities to explore such complexities. Future work could examine additional signaling pathways to assess the full range of inputs capable of impacting transcriptomic state of a single starting population. Examining differential activity among ligands within a signaling pathway will also be key, as different ligands within a pathway have been shown to induce different signaling outcomes (Antebi et al., 2017; Nandagopal et al., 2018). Moreover, developmental fate specification often occurs through graded morphogenetic activity (Slack, 2014), and our highly multiplexed system is ideal to analyze how signaling pathways act dose-dependently in isolation and in combination with other pathways.
Several recent studies have introduced methods to label distinct populations in the context of scRNA-seq (Kang et al., 2018; Shin et al., 2019; Stoeckius et al., 2017). Multiplexing experiments using scRNA-sequencing not only lowers cost by treating each cell as an experiment, but also enables better quality control by filtering of transcriptomic outgroups and reducing batch effects. Importantly, there exists a trade-off between the number of conditions that may be multiplexed into an experiment and the resolution at which we can carry out downstream analysis. Collecting a larger number of cells per condition unlocks gene-level analysis as well as insights into population heterogeneity, while studies focusing on cell states or aggregate gene signatures can lower the number of cells per condition to maximize throughput. Combining any of these multiplexing methods with the experimental and analytical framework we describe should advance our ability to understand cells’ ability to integrate combinatorial inputs into coherent output.
STAR Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard Sherwood (rsherwood@rics.bwh.harvard.edu)
Materials Availability
Cell lines and plasmids are available upon request.
Data and Code Availability
The single-cell and bulk RNA sequencing datasets generated during this study have been deposited to GEO under accession number GSE122009. Custom scripts for computational modeling and statistical analysis are available in: Data S1.
Experimental model and subject details
Culture and differentiation of 129P2/OlaHsd male mouse embryonic stem cells was modified slightly from previously published protocols (Sherwood et al., 2014). mESCs were grown at 37 degrees Celsius in mESC media: Knockout DMEM supplemented with 15% ES-tested fetal bovine serum, 0.1 mM nonessential amino acids, Glutamax, 0.55 mM 2mercaptoethanol (all from Thermo Fisher), 1X ESGRO LIF, 5 μM GSK3 inhibitor XV, and 50 μM UO126 (Millipore). Cells were regularly tested for mycoplasma. C-terminal GFP fusion knock-in lines were constructed using a previously published protocol (Arbab et al., 2015).
To induce differentiation, cells were grown for 48 hours in mESC media without GSK3 inhibitor XV and UO126 and then seeded at 10^4 cells/cm2 in mESC media without GSK3 inhibitor XV and UO126. After 24 hours, media was replaced with serum-free differentiation media: Advanced DMEM supplemented with N-2, B27 Supplement without vitamin A, and Glutamax (Thermo Fisher). After 24 hours, media was replaced with differentiation media: Advanced DMEM with 2% ES-tested FBS and Glutamax. After 24 hours in differentiation media, cells were treated in differentiation media with combinatorial treatment conditions for 24 hours in all germ layer experiments. Small molecules and growth factors along with the concentrations used in the described experiments are listed in Table S1. To induce mesendoderm differentiation, cells were treated for 24 hours in differentiation media with CD 2665, Activin A, GSK3 inhibitor XV, Dorsomorphin, and Fgf2 and then exposed to combinatorial treatment conditions for 24 hours in differentiation media.
Method details
Barcodelet preparation and administration
Barcodelets were prepared by in vitro transcription using the Megashortscript T7 Transcription Kit (Thermo Fisher). Input barcodelet DNA was prepared by PCR amplification with one of 30 specific barcodelet species and common PCR primers (Bcdlt_T7pro_fw and Bcdlt_step1_rv using 35 cycles of PCR with 2X NEBNext Mastermix (NEB) with 65 degree annealing temperature followed by PCR purification (Qiagen). All sequences are listed in Table S2.
Transfection of barcodelets was performed using TransIT-mRNA (Mirus) 2–16 hours before harvest for single cell RNA-seq. Each well of a 96-well plate received 200 ng total barcodelet RNA mixed equally among the 2–5 component barcodelet species. Barcodelets for each well were diluted in 5 μL OptiMEM with 0.1 μL mRNA-Boost reagent. This mixture was mixed well with a mixture of 5 μL OptiMEM + 0.1 μL TransIT-mRNA reagent, incubated at room temperature for 2–5 minutes, and pipetted onto cells in differentiation media with combinatorial treatment conditions.
Single-cell RNA-seq
Cells were prepared by trypsinization, resuspension in PBS + 0.04% BSA, and repeated filtering through a 70 μM cell strainer. Cells were then counted and loaded onto the 10X Genomics Chromium Single Cell Gene Expression chip using the manufacturer’s protocol, aiming for 10,000 cells. Library prep was performed according to manufacturer’s protocol with the following adaptations.
cDNA amplification was performed for 12 cycles. During post cDNA reaction cleanup, after addition of 60 uL SPRIselect reagent and magnetic separation, the supernatant is collected into a new tube. The cleanup protocol is otherwise followed for this bead-bound fraction containing the transcriptome. After this protocol is complete, add 100 uL SPRIselect beads to the supernatant from the 60 uL SPRIselect step. Follow the remaining cleanup steps starting from magnetic separation of this new bead-bound fraction, which contains barcodelets. Run QC on both transcriptome and barcodelet samples, expecting the transcriptome to look as described in the 10X Chromium manual and the barcodelet sample to have a peak around 175 bp. Complete the transcriptome library prep as described in the manual, using 20 uL of input and saving the remaining 20 uL as backup. Complete the barcodelet library prep using the protocol below:
Continue with 20 uL (half) of reserved barcodelet-specific cDNA. Perform qPCR step to establish correct number of PCR cycles.
In a qPCR tube, combine:
| 0.2μL | barcodelet-specific cDNA Library |
| 7.5μL | 2X NEBNext mastermix (NEB) |
| 0.375 μL | 20 uM Bcdltr2_ixN701_fw |
| 0.375 uL | 20 uM PE1 |
| 5.8 μL | H20 |
| 0.75 μL | 20X EvaGreen Dye (Biotium) |
Mix well and perform qPCR using the following thermocycling protocol: 98°C 30s. 30x (98°C 10sec, 67°C 30sec, 72°C 30sec). 72°C, 5 min. 4°C hold. Perform the following PCR for the number of cycles indicated in the qPCR Ct count.
In a PCR tube, combine:
| 20μL | cDNA Library |
| 50μL | 2X NEBNext MasterMix |
| 2.5 μL | 20 uM Bcdltr2_ixX_fw (use different indexed primer for each sample) |
| 2.5 uL | 20 uM PE1 |
| 25 μL | H20 |
Mix well and perform PCR using the following thermocycling protocol: 98°C 30s. X cycles (98°C 10sec, 67°C 30sec, 72°C 30sec) as determined from qPCR. 72°C, 5 min. 4°C hold. Purify product with 1.6X AMPure/SPRISelect (160μL), resuspend in 20μL DS buffer (10 mMTris-HCl pH8.0, 0.1 mM EDTA). Test library quality on TapeStation or Bioanalyzer (Agilent), expecting a single peak at 205–210 bp.
Pool with transcriptome for Illumina Nextseq sequencing at a molar ratio of 9:1 (9 parts transcriptome, 1 part barcodelet). Use no more than 10 uL of either final product. Prepare a 10 uL mix (5 uL each) from 100 uM stocks of the two custom index primers you will need: PE2rc_indexseqprimer and Multiplexr2_indexseqprimer, both ordered HPLC-purified.
Sequence using a NextSeq 75 nt kit. Reads should be split as follows (make sure to use a custom index primer):
Read 1: 26 cycles
i7 index (custom primer): 8 cycles
i5 index: 0 cycles
Read 2: 57 cycles
Flow cytometry and enhancer/promoter reporter experiments
Cells for flow cytometry were trypsinized and resuspended in flow cytometry buffer (Phenol Red-free DMEM + 2% FBS + 2 mM EDTA (Thermo Fisher)). PE anti-CD71 (Tfrc) clone RI7217 (Biolegend) was used at 1:800 and stained in flow cytometry buffer for 20 minutes at 4 degrees. Cells were run on a BD FACSymphony, and median expression values were obtained from FACSDiva software on cells gated for Fsc vs. Ssc for 2–4 replicates per experiment.
For enhancer and promoter reporter experiments, a Tol2 transposon site-flanked base plasmid was subcloned from a previously published plasmid (Sherwood et al., 2014). This plasmid has a minimal promoter derived from the mouse Hspa1a promoter, followed by GFP, a poly-adenylation signal, and a Hygromycin resistance cassette. Cloning of 17 enhancers chosen for their presence of robust ChIP-Seq peaks for the RA effector Rarg or Wnt effector Tcf7l2 was performed by cloning ~1 kb regions from the mouse genome into the MluI site between the GFP ORF and the polyA site using InFusion (Clontech). Promoters were cloned into either the base plasmid or enhancer-containing plasmids by replacing the Hsp1a1 promoter at the SnaBI and EcoRV sites and cloning ~1 kb regions from the mouse genome in their place using InFusion. Primers used in the cloning and resulting enhancer and promoter regions are presented in Table S2. Stable mESC lines were made by Tol2 transposition of these constructs into wild-type mESCs followed by stable Hygromycin selection. Differentiation was performed as described above.
Analysis of notochord-like differentiation
For experiments testing transcriptome differences in the presence or absence of barcodelet transfection, mESC differentiation to mesendoderm was performed as described above in the presence of barcodelet tranfection 24 or 72 hours prior to RNA isolation. For Foxa2-GFP+ notochord-like cell analysis, differentiation of mESCs to mesendoderm was performed as described above in four biological replicates per experiment, then cells were trypsinized and re-plated at 5*10^4 cells/cm2 onto 804G conditioned media-coated wells and treated for 24 hours with the conditions described in the text. Optimal Foxa2-GFP efficiency was seen in cells treated with 10 ng/mL Activin, 500 nM Dorsomorphin, 100 nM IWR1, and 2 uM Sag. Control conditions included no additional reagents. For RT-qPCR and bulk RNA-seq, Foxa2-GFP+ were flow cytometrically isolated prior to RNA isolation. 3’-enriched RNA-sequencing was performed using the Lexogen Quantseq 3’ mRNA-seq kit using manufacturer-suggested protocols and sequenced using Illumina Nextseq at >5*10^6 reads per sample. For RT-qPCR, reverse transcription was performed using M-MuLV first strand synthesis kit (NEB), and qPCR was performed using Onetaq polymerase (NEB) and EvaGreen (Biotium) for detection using a Bio-Rad CFX96 qPCR machine. Expression values were normalized to Actb. Immunofluorescence was performed by fixing cells for 15 minutes with 4% paraformaldehyde. Primary antibody staining was performed overnight in PBS + 0.05% Triton X-100 + 5% donkey serum (Jackson ImmunoResearch) with rabbit anti-Foxa2 (Abcam ab40874, 1:1,000), goat anti-Chordin (R&D Systems AF758, 1:40), and/or sheep anti-Slit2 (R&D Systems AF5444, 1:40) as described in the text. Secondary antibody staining was performed for 1 hour at room temperature using DyLight 594- and DyLight 649-conjugated antibodies (Jackson ImmunoResearch). Hoechst 33342 was added at 1 μg/mL before imaging. Immunofluorescence images were captured using a Leica DMI 6000b inverted fluorescence microscope, and image analysis with the Leica AF6000 software package.
Quantification and statistical analysis
Assignment of treatment combination
Barcodelet reads are mapped using the 10X cellranger pipeline (v1.3.1) modified with a custom mapping step that first aligns and trims the common right flanking sequence, and then maps the remaining sequence to the known pool of unique barcodelet sequences. The alignment allows for 4 mismatches in the right flanking sequence, and 2 mismatches in the unique subsequence. The output of the pipeline is UMI-unique barcodelet counts for each cell.
To assign each cell a treatment combination, we inspect the top k most abundant barcodelet species in the count data, where k is the number of barcodelets each cell was labeled with, and compute the summed count fraction of these top k barcodelets. We expect that invalid combinations observed are due to noise. Since we also know the expected fraction of invalid combination based on the size of the starting pool of barcodelets, we can estimate the false positive rate at any given summed count fraction. To ensure labeling fidelity, we set the threshold on the summed count fraction at a false positive rate of 1%, and assign all cells above that threshold for which the barcodelet combination was valid. We retain the set of cells which have been assigned a treatment combination and that also have a high quality transcriptome (see next section) for downstream analysis.
Transcriptome preprocessing
Transcriptome reads were mapped using the 10X cellranger pipeline (v1.3.1) to mm10 (v.1.2.0). UMI-unique counts were then preprocessed using Seurat (Butler et al., 2018) using standard procedure. Following normalization and log-scaling of UMI-unique counts, 5000 variable genes are identified for each dataset. Then, cells from each starting state are integrated using 2000 anchor features. PCA is then run using the set of anchor features to get the first 30 PCs, which is subsequently used for UMAP visualization. Empirically, visualization does not appear to be largely sensitive to the number of PCs used.
To remove transcriptomic outgroups, we use a smoothed clustering method. Clusters are first initialized using a 2-component Gaussian mixture model fit on a small set of known gene markers. Since this often results in noisy clusters, the clusters are then refined using k-nearest neighbors voting in the embedding space, where distance is defined as the Euclidean distance in PCA space and k is set to 20. The set of gene markers used for the ExEN outgroup are: Col4a1, Lama1, Sparc, Sox17
Internal validation
Expression similarity was computed as the Euclidean distance between cells in PCA space. Distances were computed between pairs of cells which were assigned the same treatment combination, or different treatment combination, across datasets. Distances were also computed for each cell vs. the control cell population as a special case. Expression profiles in each treatment combination were aggregated by taking the mean of the scaled expression across all cells assigned to that treatment combination.
To check that gene expression is predictive of individual pathway activation, logistic regression models were fit using 5-fold cross-validation on a training set consisting of 80% of the data, and tested on the remaining held-out set of 20%. To identify genes predictive of individual pathway activation, genes are ranked by their model coefficient. Models were then also fit to predict combinatorial pathway activation (see supplementary information).
To check if gene expression is also predictive of combinatorial pathway activation, logistic regression models were fit using 5-fold cross-validation on a training set consisting of 80% of the data to predict combinatorial pathway status (multitask) for increasing numbers of pathways. This is in contrast to when each pathway is predicted independently (multilabel). For a given number of pathways, difference in model accuracy was then assessed for the randomly held-out test sets (remaining 20%) via paired t-test. We found that model accuracy is consistently higher when predicting combinatorial pathway status than predicting pathway independently, suggesting that combinatorial gene expression effects are present in our data (Figure S1F).
A Bayesian regression analysis framework for modeling gene expression
Log-scaled normalized gene expression is modeled as:
where X is a matrix typically comprised of linear terms indicating activation or inhibition of each pathway, as well as interaction terms. For each gene, we fit and compare models including only an intercept term (0th order) and up to 4th order terms (For e.g. PathwayA : PathwayB : PathwayC : PathwayD) using pymc3 (Salvatier et al., 2016). For model inference, 1000 samples are drawn for 2 chains after 1000 tuning steps. Model comparison was then performed using leave-one-out (LOO) cross-validation via Pareto-smoothed importance sampling (PSIS). We define the optimal model as the model with the lowest complexity that has a model score within 1.96 * standard error (95% confidence interval (CI)) of the best model score. We define a coefficient to be non-zero if the 95% CI does not contain 0 in the optimal model for that gene. To find gene clusters, we perform hierarchical clustering on the coefficient matrix, using average linkage and correlation as the distance metric. Then, for visualization, flat clusters are formed using the inconsistency criterion, thresholding at 1.15. We fit models for the top 2000 genes used for integration.
Concordance of scRNA-seq and reporter data
Spearman rank correlation coefficient is computed for comparing average scRNA-seq expression with GFP expression for reporter cell lines. 95% confidence intervals are estimated via bootstrap.
Analysis of flow-cytometry data for enhancer/promoter reporter experiments
For each replicate, median fluorescence intensity was normalized with respect to the baseline condition (all inhibitors). To determine if a reporter was combinatorial, we computed the expected expression under a linear model, and then tested if the difference between this expected expression and the actual observed expression was significant using a paired t-test.
Scoring cells using classifiers trained on existing in vivo data
Data from Pijuan-Sala et al. was downloaded from https://content.cruk.cam.ac.uk/jmlab/atlas_data.tar.gz. Cells annotated as doublets or stripped were first removed. To reduce computation time, genes expressed in fewer than 5 cells were also removed. Raw counts were first scaled and normalized in Seurat using a similar workflow as described above. Cells were integrated across stages using the first 30 dimensions of CCA to find integration anchors. Integrated counts were then scaled again for input to downstream classifiers. For visualization, PCA was run to obtain the first 30 PCs, which were then used for UMAP.
Logistic regression models were fit for each cell type on a random split of 80% of the data, using the scaled expression of the intersection of the set of anchor genes in the reference atlas dataset, and the query barRNA-seq dataset. Each model was fit with an elastic net penalty using 5-fold cross-validation and stochastic gradient descent learning, searching over the weight on the regularization parameter (ranging from 1e-4 to 1e0). Then, models were used to score cells from our barRNA-seq assay. For each classifier, treatment groups were ranked by the mean score. To identify genes associated with each classifier, we ranked genes by the correlation of their expression and the classifier score in the query dataset.
The annotations for paraxial and somatic mesoderm in Pijuan-Sala et al are reversed from their previous work (Ibarra-Soria et al., 2018) and established markers of mesoderm subtypes (Loh et al., 2016), and thus we have manually reversed these labels to reflect the canonical markers for these two cell states.
Unsupervised de novo discovery of cell subpopulations
To enable de novo discovery of cell subpopulations not present in reference datasets, we first perform unsupervised graph clustering (Blondel et al., 2008; Traag et al., 2015). We selected the resolution parameter such that clustering resulted in partitions that were relatively invariant to small perturbations in the resolution parameter (Lambiotte, 2010). Stability analysis was conducted using 100 initializations and over a range of 150 resolution parameters ranging from 0.5 to 4, using normalized variation of information to compare partitions. For each cluster, we performed differential expression analysis using Seurat to identify conserved positive gene markers (i.e. with positive fold change) at combined p-value < 0.05. Combined p-values were corrected for multiple testing using Benjamini-Hochberg. Then, for each cluster, treatment groups were ranked by the fraction of cells in each treatment group that had membership in that cluster.
Characterization of Foxa2GFP+ population
Bulk RNA-seq reads were mapped using the Quantseq 3’ mRNA mapping pipeline as described by lexogen. Briefly, reads were first trimmed using bbduk from the bbmap suite (v37.75) trimming for low quality tails, poly-A read-through and adapter contamination using the recommended parameters. Then, reads were mapped using the STAR aligner (v2.5.2b) (Dobin et al., 2013) with the recommended modified-Encode settings. Finally, HT-seq (v0.9.1) count was used to obtain per-gene counts.
All bulk RNA-sequencing analysis was done using DESeq2 (v1.24.0) (Love et al., 2014). One of the samples (Foxa2GFP+ replicate 1) had a very small number of counts and was removed from downstream analysis. For differential expression of control samples that received only barcodelets and no treatments against samples that received barcodelets as well as treatments, samples are grouped by the transfection protocol as well as treatment. For differential expression analysis of the Foxa2GFP+ population, all samples that received only barcodelets and no treatments are grouped together as a control group. In both cases, for visualization, count data is transformed by applying a regularized logarithm.
Supplementary Material
Table S1: Concentrations of small molecules and growth factors used in all experiments. Related to Figure 1F
Table S3: Classifier performance on held-out test set. Related to Figure 5
Table S4: Differentially expressed genes detected from bulk expression data of Foxa2+ notochord-like cell population with respect to control population. Related to Figure 7F
Table S5: Differentially expressed genes detected from bulk expression data of Foxa2+ notochord-like cell population with respect to Foxa2- population. Related to Figure 7F
Data S1: Custom analysis notebooks/scripts for computational modeling and statistical analysis. Related to STAR Methods.
Highlights.
barRNA-seq enables systematic exploration of combinatorial signaling control
Complex interactions between pathways regulate gene expression during development
Data-driven framework identifies combinatorial signaling driving fate acquisition
Stem cell differentiation systematically mapped to embryonic single cell atlas
Acknowledgments
The authors thank Lendy Chu and Chad Araneo for technical assistance and the MIT Biomicro Center and the Harvard Medical School Biopolymers Facility for sequencing. The authors acknowledge funding from 1RO1HG008363 (D.K.G.), 1R01HG008754 (D.K.G.), 1R01NS109217 (D.K.G.), 1K01DK101684 (R.I.S.), 1R21OD025309 (R.I.S), the Human Frontier Science Program, NWO, Brigham Research Institute, Harvard Stem Cell Institute, American Cancer Society (R.I.S.), and the Agency for Science, Technology and Research Graduate Academy (G.Y.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W. (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antebi YE, Linton JM, Klumpe H, Bintu B, Gong M, Su C, McCardell R, and Elowitz MB (2017). Combinatorial Signal Perception in the BMP Pathway. Cell 170, 1184–1196.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbab M, Srinivasan S, Hashimoto T, Geijsen N, and Sherwood RI (2015). Cloning-free CRISPR. Stem Cell Reports 5, 908–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser D. (2004). Expression of nodal, lefty-a, and lefty-B in undifferentiated human embryonic stem cells requires activation of Smad2/3. J. Biol. Chem 279, 45076–45084. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DE, and Melton D. (2011). Turning straw into gold: directing cell fate for regenerative medicine. Nat. Rev. Genet 12, 243–252. [DOI] [PubMed] [Google Scholar]
- Cole MF, Johnstone SE, Newman JJ, Kagey MH, and Young RA (2008). Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 22, 746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH (2010). Emerging properties of animal gene regulatory networks. Nature 468, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadue P, Huber TL, Paddison PJ, and Keller GM (2006). Wnt and TGF-beta signaling are required for the induction of an in vitro model of primitive streak formation using embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A 103, 16806–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1107.e17. [DOI] [PubMed] [Google Scholar]
- Hart AH, Hartley L, Sourris K, Stadler ES, Li R, Stanley EG, Tam PPL, Elefanty AG, and Robb L. (2002). Mixl1 is required for axial mesendoderm morphogenesis and patterning in the murine embryo. Development 129, 3597–3608. [DOI] [PubMed] [Google Scholar]
- Hollnagel A, Oehlmann V, Heymer J, Rüther U, and Nordheim A. (1999). Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J. Biol. Chem 274, 19838–19845. [DOI] [PubMed] [Google Scholar]
- Ibarra-Soria X, Jawaid W, Pijuan-Sala B, Ladopoulos V, Scialdone A, Jörg DJ, Tyser RCV, Calero-Nieto FJ, Mulas C, Nichols J, et al. (2018). Defining murine organogenesis at single-cell resolution reveals a role for the leukotriene pathway in regulating blood progenitor formation. Nat. Cell Biol 20, 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, and Amit I. (2016). Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896.e15. [DOI] [PubMed] [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamar E, Deblandre G, Wettstein D, Gawantka V, Pollet N, Niehrs C, and Kintner C. (2001). Nrarp is a novel intracellular component of the Notch signaling pathway. Genes Dev. 15, 1885–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AW, Kent Morest D, and Li JYH (2013). Differential BMP signaling controls formation and differentiation of multipotent preplacodal ectoderm progenitors from human embryonic stem cells. Dev. Biol 379, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Ang LT, Zhang J, Kumar V, Ang J, Auyeong JQ, Lee KL, Choo SH, Lim CYY, Nichane M, et al. (2014). Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 14, 237–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Chen A, Koh PW, Deng TZ, Sinha R, Tsai JM, Barkal AA, Shen KY, Jain R, Morganti RM, et al. (2016). Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell 166, 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, and Wichterle H. (2013). Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat. Neurosci 16, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meno C, Shimono A, Saijoh Y, Yashiro K, Mochida K, Ohishi S, Noji S, Kondoh H, and Hamada H. (1998). lefty-1 is required for left-right determination as a regulator of lefty-2 and nodal. Cell 94, 287–297. [DOI] [PubMed] [Google Scholar]
- Nandagopal N, Santat LA, LeBon L, Sprinzak D, Bronner ME, and Elowitz MB (2018). Dynamic Ligand Discrimination in the Notch Signaling Pathway. Cell 172, 869–880.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G, Suo S, Cui G, Yu F, Wang R, Chen J, Chen S, Liu Z, Chen G, Qian Y, et al. (2019). Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature 572, 528–532. [DOI] [PubMed] [Google Scholar]
- Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, Mulas C, Ibarra-Soria X, Tyser RCV, Ho DLL, et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. (2017). The Human Cell Atlas. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert S, Randall RA, and Hill CS (2013). A BMP regulatory network controls ectodermal cell fate decisions at the neural plate border. Development 140, 4435–4444. [DOI] [PubMed] [Google Scholar]
- Rimini R, Beltrame M, Argenton F, Szymczak D, Cotelli F, and Bianchi ME (1999). Expression patterns of zebrafish sox11A, sox11B and sox21. Mech. Dev 89, 167–171. [DOI] [PubMed] [Google Scholar]
- Salvatier J, Wiecki TV, and Fonnesbeck C. (2016). Probabilistic programming in Python using PyMC3. PeerJ Comput. Sci 2, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Maehr R, Mazzoni EO, and Melton DA (2011). Wnt signaling specifies and patterns intestinal endoderm. Mech. Dev 128, 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, and Gifford DK (2014). Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol 32, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Inoue H, Wu JC, and Yamanaka S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov 16, 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Lee W, Lee JH, and Bang D. (2019). Multiplexed single-cell RNA-seq via transient barcoding for simultaneous expression profiling of various drug perturbations. Sci Adv 5, eaav2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone A, Acampora D, Arcioni L, Andrews PW, Boncinelli E, and Mavilio F. (1990). Sequential activation of HOX2 homeobox genes by retinoic acid in human embryonal carcinoma cells. Nature 346, 763–766. [DOI] [PubMed] [Google Scholar]
- Slack J. (2014). Establishment of spatial pattern. Wiley Interdiscip. Rev. Dev. Biol 3, 379–388. [DOI] [PubMed] [Google Scholar]
- Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, and Smibert P. (2017). Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, et al. (1996). The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134. [DOI] [PubMed] [Google Scholar]
- Szczesnik T, Ho JWK, and Sherwood R. (2019). Dam mutants provide improved sensitivity and spatial resolution for profiling transcription factor binding. Epigenetics Chromatin 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, and Principal investigators (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DA, Hayward PC, Baillie-Johnson P, Rué P, Broome R, Faunes F, and Martinez Arias A. (2014). Wnt/β-catenin and FGF signalling direct the specification and maintenance of a neuromesodermal axial progenitor in ensembles of mouse embryonic stem cells. Development 141, 4243–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urasaki A, Morvan G, and Kawakami K. (2006). Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 174, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, and Jessell TM (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397. [DOI] [PubMed] [Google Scholar]
- Winzi MK, Hyttel P, Dale JK, and Serup P. (2011). Isolation and characterization of node/notochord-like cells from mouse embryonic stem cells. Stem Cells Dev. 20, 1817–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, and Smith A. (2003). BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Concentrations of small molecules and growth factors used in all experiments. Related to Figure 1F
Table S3: Classifier performance on held-out test set. Related to Figure 5
Table S4: Differentially expressed genes detected from bulk expression data of Foxa2+ notochord-like cell population with respect to control population. Related to Figure 7F
Table S5: Differentially expressed genes detected from bulk expression data of Foxa2+ notochord-like cell population with respect to Foxa2- population. Related to Figure 7F
Data S1: Custom analysis notebooks/scripts for computational modeling and statistical analysis. Related to STAR Methods.
Data Availability Statement
The single-cell and bulk RNA sequencing datasets generated during this study have been deposited to GEO under accession number GSE122009. Custom scripts for computational modeling and statistical analysis are available in: Data S1.