

Abstract
Charcot-Marie-Tooth disease type 2E/1F (CMT2E/1F) is a peripheral neuropathy caused by mutations in neurofilament protein L (NFL), which is one of five neurofilament subunit proteins that co-assemble to form neurofilaments in vivo. Prior studies on cultured cells have shown that CMT2E/1F mutations disrupt neurofilament assembly and lead to protein aggregation, suggesting a possible disease mechanism. However, electron microscopy of axons in peripheral nerve biopsies from patients has revealed accumulations of neurofilament polymers of normal appearance and no evidence of protein aggregates. To reconcile these observations, we re-examined the assembly of seven CMT2E/1F NFL mutants in cultured cells. None of the mutants assembled into homopolymers in SW13vim- cells, but P8R, P22S, L268/269P, and P440/441L mutant NFL assembled into heteropolymers in the presence of neurofilament protein M (NFM) alone, and N98S, Q332/333P, and E396/397K mutant NFL assembled in the presence of NFM and peripherin. P8R, P22S, N98S, L268/269P, E396/397K and P440/441L mutant NFL co-assembled into neurofilaments with endogenous NFL, NFM, and α-internexin in cultured neurons, though the N98S and E396/397K mutants showed reduced filament incorporation, and the Q332/333P mutant showed limited incorporation. We conclude that all the mutants are capable of assembling into neurofilaments but for some of the mutants this was dependent on the identity of the other neurofilament proteins available for co-assembly, and most likely also their relative expression level. Thus, caution should be exercised when drawing conclusions about the assembly capacity of CMT2E/1F mutants based on transient transfections in cultured cells.
Keywords: Charcot-Marie-Tooth disease type 2E, Charcot-Marie-Tooth disease type 1F, CMT2E, CMT1F, neurofilament, axonal transport, assembly
INTRODUCTION
Charcot-Marie-Tooth (CMT) disease, or hereditary motor and sensory neuropathy, is one of the most common inherited neurological disorders with an incidence of 1 in 2,500 individuals (Saporta & Shy, 2013; Skre, 1974). CMT presents with a loss of motor and sensory function in the periphery that slowly progresses proximally over time. The disease is debilitating but not fatal, with an age of onset ranging from infancy to middle-age (Suter & Scherer, 2003). Clinically, CMT is categorized by electromyography as either type 1 (decreased nerve conduction velocities, indicative of demyelination), or type 2 (decreased compound muscle action potentials, indicative of axonal degeneration and denervation) (Suter & Scherer, 2003). Genetically, the disease can be caused by mutations in >80 different genes (Timmerman, Strickland, & Züchner, 2014).
Mutations in the low molecular weight neurofilament triplet protein (NFL) give rise to CMT disease that is classified as type 2E or 1F depending on the clinical presentation (Horga et al., 2017; Yang, Gu, Burnette, & Li, 2016). One explanation for this variable diagnosis is that there is a loss of axon caliber which decreases conduction velocity without demyelination (Lancaster et al., 2018), thereby making what is an axonal (type 2) disease sometimes appear to be demyelinating (type 1). More than 30 disease-causing mutations have been described which span the entire length of the NFL protein. Most are dominant missense mutations, which are the focus of our present study.
Neurofilaments, which are the intermediate filaments of neurons, are abundant, space-filling, cytoskeletal polymers that are transported into and along axons where they contribute to the growth of axonal caliber (Brown, 2014; Perrot, Berges, Bocquet, & Eyer, 2008). NFL is one of five neurofilament subunit proteins (NFL, NFM, NFH, peripherin and α-internexin) that co-assemble to form neurofilament polymers (Lee & Cleveland, 1996; Yuan et al., 2006, 2012). The expression of these proteins varies depending on the neuronal cell type and stage of development, with peripherin expressed only in the peripheral nervous system (Yuan et al., 2012).
Several laboratories have investigated the effects of CMT2E/1F missense mutations on NFL co-assembly with other neurofilament proteins in SW13vim- cells, which are human adrenal adenocarcinoma cells that lack endogenous cytoplasmic intermediate filaments (Sarria, 1990; Yamamichi-Nishina et al., 2003). The P8R and P8Q mutants formed filamentous bundles or non-filamentous aggregates whereas the P8L, P22T, E90K, N98S, and Q332/333P mutants formed only non-filamentous aggregates (Brownlees et al., 2002; Gentil, Mushynski, & Durham, 2013; Leung, Nagan, Graham, & Liem, 2006; Perez-Olle, Jones, & Liem, 2004; Perez-Olle, Leung, & Liem, 2002; Perez-Olle et al., 2005; Sasaki et al., 2006; Yum, Zhang, Mo, Li, & Scherer, 2009). The P22S mutant formed filaments in one study, though they were excessively bundled or fragmented in some cells (Perez-Olle et al., 2005), but formed only non-filamentous aggregates in another (Sasaki et al., 2006). The P8R and Q332/333P mutants also formed aggregates and/or inclusions in cultured neurons (Brownlees et al., 2002; Perez-Olle et al., 2005; Zhai, Lin, Julien, & Schlaepfer, 2007), though some have reported that this was preceded by incorporation into neurofilaments (Gentil et al., 2013; Tradewell, Durham, Mushynski, & Gentil, 2009). The general conclusion from all these reports is that most of the CMT2E/1F NFL mutants examined are unable to assemble into filaments and, instead, the mutant NFL protein has a dominant negative effect on filament assembly creating amorphous aggregations within the cell (Brownlees et al., 2002; Leung et al., 2006; Sasaki et al., 2006; Zhai et al., 2007).
In contrast to the studies in cell culture, electron microscopy has revealed abundant neurofilament polymers in axons of sural nerve biopsies from CMT2E/1F patients (Benedetti et al., 2010; Doppler, Kunstmann, Krüger, & Sommer, 2017; G M Fabrizi et al., 2004; Gian Maria Fabrizi et al., 2007; Züchner, Vorgerd, Sindern, & Schröder, 2004) as well as in cell bodies and proximal axons of a mouse model of CMT2E/1F (Lancaster et al., 2018; Zhao, Brown, & Liem, 2017). Neurofilaments are also abundant in embryonic and adult neurons cultured from these mice, albeit with axonal and/or and somal accumulations (Zhao et al., 2017). To reconcile these apparently conflicting reports, we reexamined the assembly capabilities of CMT2E/1F mutant NFL in cultured cells.
MATERIALS AND METHODS
Molecular cloning
The wild type human NFL cDNA plasmid (phNFL) was created using a cDNA construct provided by Shin-Ichi Hisanaga (Sasaki et al., 2006). The cDNA (GenBank Accession number NM_006158) was excised with BamHI (New England Biolabs, Ipswich, MA, USA) and cloned into the multiple cloning site of vector pEGFP-C1 (Takara Bio USA, Mountain View, CA, USA) in which the GFP sequence had previously been excised (Uchida, Alami, & Brown, 2009). The CMT2E/1F mutant NFL plasmids were created by site-directed mutagenesis (QuikChange Lightning Site-Directed Mutagenesis Kit, Agilent Technologies, Santa Clara, CA, USA) using the wild type construct as a template. The amino acid and nucleotide changes were as follows: Pro8Arg, 23C>G (phNFL-P8R); Pro22Ser, 64C>A, 65C>G (phNFL-P22S); Asn98Ser, 293A>G (phNFL-N98S); Leu268Pro, 803T>C (phNFL-L268P); Gln332Pro, 995A>C (phNFL-Q332P); Glu396Lys, 1186G>A (phNFL-E396K); Pro440Leu, 1319C>T (phNFLP440L). The human NFM cDNA plasmid (phNFM) was created using a cDNA provided by Virginia Lee (Pleasure, Lee, & Nelson, 1990; GenBank Accession number NM_005382.2). A Kozak consensus sequence was added by PCR and then the coding region was excised using EcoRI and KpnI (New England Biolabs) and subcloned into the Takara Bio vector (without GFP) described above. The human peripherin cDNA (PRPH; GenBank Accession number NM_006262.3) in the mammalian-expression vector pCMV-SPORT6 was purchased from Dharmacon (Lafayette, CO, USA, cat# MHS 6278–202802013). The human α-internexin cDNA plasmid (phINA) was created using a cDNA construct purchased from Dharmacon (cat# MHS 6278–202831005). The cDNA (GenBank accession number NM_032727.3) was amplified by PCR to introduce flanking EcoRI and KpnI restriction sites, and then subcloned into the into the Takara Bio vector (without GFP) described above.
The wild type rat NFL EGFP fluorescent fusion plasmid (prNFL-EGFP) was created from a rat pNFL-myc construct provided by Chris Miller (Yates et al., 2009). First, we used PCR to amplify the NFL sequence without the myc tag. Then, we cloned this PCR product into the multiple cloning site of a pEGFP-N3 vector (Takara Bio USA) using EcoRI and BamHI (New England Biolabs) to yield the linker sequence 5’-GGATCCATCGCCACC-3’. The resulting plasmid coded for GFP connected to the C-terminus of rat NFL by a 5 amino acid linker with the sequence -Gly-Ser-Ile-Ala-Thr-. The sequence was confirmed to agree with GenBank reference sequence NM_031783.1 except for a silent mutation at nucleotide 1590G>A. CMT2E/1F mutations homologous to those in the human NFL constructs were introduced into the prNFL-EGFP construct by site-directed mutagenesis as described above. The amino acid and nucleotide changes were as follows: Pro8Arg, 23C>G (prNFL-P8R-EGFP); Pro22Ser, 64C>A, 65C>G (prNFL-P22S-EGFP); Asn98Ser, 293A>G (prNFL-N98S-EGFP); Leu269Pro, 806T>C, 807G>C (prNFL-L269P-EGFP); Gln333Pro, 998A>C, 999G>C (prNFL-Q333P-EGFP); Glu397Lys, 1189G>A (prNFL-E397K-EGFP); Pro441Leu, 1322C>T, 1323A>G (prNFL-P441L-EGFP). Note that there is a serine inserted at position 251 in the rat sequence which shifts all amino acids thereafter by one when compared with the human sequence. Thus, the mutations L268P, Q332P, E396K and P440L in human NFL correspond to L269P, Q333P, E397K and P441L, respectively, in rat NFL. To reflect this, these mutants are referred to as L268/269P, Q332/333P, E396/397K and P440/441L in this manuscript. The untagged rat NFM cDNA plasmid (prNFM) was generated by excising the GFP from the pEGFP-NFM construct previously reported (Wang, Ho, Sun, Liem, & Brown, 2000) using AgeI and BspEI restriction enzymes (New England Biolabs).
The sequence of the entire open reading frame of each construct was confirmed using Sanger sequencing. All plasmids were amplified in DH5α competent cells (Thermo Fisher Scientific, Waltham, MA, USA) and then purified to transfection quality and concentration using a Qiagen endotoxin-free maxi kit (Qiagen USA, Germantown, MD, USA).
Cell culture
Human adrenal adenocarcinoma SW13vim- cells, which lack endogenous vimentin, were obtained from Dr. Robert Evans of the University of Colorado, Denver, Denver, CO (Sarria, 1990) and cultured in DMEM/F12 (Thermo Fisher Scientific) supplemented with 5% fetal bovine serum (FBS) (HyClone, GE Healthcare, Chicago, IL, USA) and 10 μg/ml gentamicin (MilliporeSigma, Burlington, MA, USA) as described previously (Yan, Jensen, & Brown, 2007). Cortical neurons were obtained from newborn (postnatal day 0) rats and cultured using the glial-sandwich method (Goslin, Hannelore, & Banker, 1998; Kaech & Banker, 2006) in NbActiv4™ medium (BrainBits, Springfield, IL, USA) as described previously (Uchida, Monsma, Fenn, & Brown, 2016). Both SW13vim- cells and neurons were cultured in coverslip-bottomed 35 mm dishes coated with poly-D-lysine (MilliporeSigma) and maintained at 37°C in a humidified atmosphere with 5% CO2 as described previously (Uchida et al., 2016).
Transfection
SW13vim- cells were transfected with human neurofilament plasmids 16–20 hours after plating using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions with the following exceptions: the Lipofectamine was diluted in DMEM/F12 media, the mixture was incubated at 37°C for 20 minutes before adding it to the culture dishes, and the dishes were incubated for 3–4 hours at 37°C before the media was replaced. Rat cortical neurons were transfected with rat neurofilament plasmids by electroporation using a Lonza Nucleofector™ II electroporator and Rat Neuron Nucleofector™ kit (Basel, Switzerland, cat# VAPG-1003) with program G-013 according to the manufacturer’s instructions. One hour after plating, the culture medium was replaced with fresh medium in order to remove dead cells and debris.
Fixation
SW13vim- cells were fixed 18–24 hours after transfection and neurons were fixed 9 days after plating. After aspirating the medium, the cells were rinsed twice with PBS and then fixed with 4% formaldehyde in PBS (Thermo Fisher Scientific) at room temperature for 30 minutes. After fixation, the cells were rinsed twice more with PBS and then permeabilized with 1% Triton-X in PBS for 15 minutes at room temperature. After permeabilization, the cells were rinsed three times with PBS and immunostained.
Immunostaining
The fixed and permeabilized cells were blocked with 4% donkey serum (Jackson ImmunoResearch, West Grove, PA, USA) in PBS for 10 minutes at room temperature. Cells were incubated in primary and then secondary antibodies diluted in the same blocking solution, each for 45 minutes at 35°C, rinsing and blocking in between. NFL was visualized with a rabbit polyclonal antibody, provided by Virginia Lee (Trojanowski, Kelsten, & Lee, 1989), at a 1:400 dilution and Alexa 488 donkey anti-rabbit secondary antibody (Thermo Fisher Scientific, cat# A-21206) at a 1:400 dilution. NFM was visualized with phospho-independent mouse monoclonal antibody RMO270 (Thermo Fisher Scientific, cat# 13–0700) at a 1:400 dilution and Cy3 donkey anti-mouse secondary antibody (Jackson ImmunoResearch) at a 1:400 dilution. Vimentin was visualized with chicken polyclonal antibody Poly 29191 (BioLegend, San Diego, CA, USA, cat# 919101) at a 1:1800 dilution and Alexa 647 donkey anti-chicken secondary antibody (Jackson ImmunoResearch) at a 1:400 dilution. Alpha-internexin was visualized with rabbit polyclonal antibody αBB, provided by Ron Liem (Ching, Chien, Flores, & Liem, 1999), at a 1:100 dilution and Alexa 647 donkey anti-rabbit secondary antibody (Thermo Fisher Scientific) at a 1:400 dilution. GFP-tagged NFL was visualized with a chicken polyclonal antibody (Abcam, Cambridge, United Kingdom, cat# ab13970) at a 1:1000 dilution and Alexa 488 donkey anti-chicken secondary antibody (Jackson ImmunoResearch) at a 1:400 dilution. After immunostaining, the cells were mounted in Fluoromount™ (Southern Biotech, Birmingham, AL, USA). For counting, the cells were counterstained with DAPI (Thermo Fisher Scientific, cat# D1306) according to the manufacturer’s instructions and mounted in ProLong Gold™ (Thermo Fisher Scientific, cat# P36930).
Microscopy and Imaging
Immunostained SW13vim- and nerve cells were observed using an Andor Revolution WD spinning disk confocal microscope (Andor Technology Ltd, Belfast, UK) incorporating a Yokogawa CSU-W1 confocal scanning unit (Yokogawa Electric Corporation, Tokyo, Japan) attached to a Nikon TiE inverted microscope (Nikon Instruments, Melville, NY). Excitation of fluorophores was achieved using the following solid state laser lines: a 100 mW 405 nm diode laser, 50 mW 488 nm DPSS laser, 50 mW 561 nm DPSS laser, and a 100 mW 640 nm diode laser. A 405/488/561/640 quadruple multi-band pass dichroic was used in combination with the following emission filters: 452/45 (BFP), 525/50 (GFP), 600/50 (RFP), and 700/75 (Cy5). Images were acquired using a Nikon 100x/1.4 NA Plan Apo VC oil-immersion objective and an Andor Neo 16-bit sCMOS camera. Living nerve cells were observed by wide-field epifluorescence after 9 days in culture. The observation medium was composed of Hibernate A medium (low fluorescence formulation) supplemented with 2% (v/v) B27, 0.5 mM glutamax, and 37.5 mM NaCl. Cells were maintained at 37°C using an Okolab H301 stage-top incubator (Okolab, Ottaviano, Italy) and a Bioptechs objective heater (Butler, PA, USA). Time-lapse movies were acquired with 300 ms exposures and 3 second intervals using an ET-GFP filter set (#49002; Chroma Technology, Bellows Falls, VT, USA), a Nikon 100x/1.4 NA Plan Apo VC oil-immersion objective, and an Andor iXon ULTRA 897BV EMCCD camera.
Quantification
Cell counting was conducted on the wide-field epifluorescence microscope described above using an ET-GFP filter set (see above) and an ET-mCherry filter set (Chroma Technology, #49008). Fields of view were chosen randomly in the upper left, upper right, lower right, and lower left quadrants of the dish. Counting was conducted in at least three fields of view in each quadrant. Cells were verified to have or lack visible filaments using both the green and red filter cubes, or with the green filter cube alone. A tally of filamentous and non-filamentous cells for each dish was maintained and then the percent of all cells observed with filaments when positively transfected with both plasmids was averaged across three replicated experiments.
Experimental Design and Statistical Analysis
All experiments were repeated independently at least three times. Each SW13vim- experiment was performed with a different cell passage. Each cortical neuron experiment was performed with cultures established from a different litter of rat pups. The cell counting was performed on at least three separate experiments with at least two dishes per experimental condition. Approximately 100–300 cells were counted per dish in the double transfection experiment, and 30–150 cells were counted per dish in the triple transfection experiment. The statistical analysis for the cell counting was performed with a two-tailed student’s t-test assuming unequal variances. Error bars indicate standard deviation throughout.
RESULTS
Choice of mutants
There are 34 reported CMT2E/1F NFL mutations in human patients (Fig. 1) (Doppler et al., 2017; Fu & Yuan, 2018; Horga et al., 2017; Lerat et al., 2019; Sainio et al., 2018). These are primarily missense mutations, but there are also frameshift, deletion, and nonsense mutations. The mutations are distributed throughout the NFL protein, but appear more frequently in the head and rod domains than in the tail domain. For the present study, we focused on mutations that have been studied previously in cultured cells (Brownlees et al., 2002; Gentil et al., 2013; Perez-Olle et al., 2004, 2002, 2005; Sasaki et al., 2006; Tradewell et al., 2009; Yum et al., 2009; Zhai et al., 2007), and/or mutations for which there are published electron micrographs of nerve biopsies from CMT2E/1F patients (Benedetti et al., 2010; Elbracht et al., 2014; G M Fabrizi et al., 2004; Gian Maria Fabrizi et al., 2007; Horga et al., 2017; Luigetti et al., 2016; Pisciotta et al., 2015; Züchner et al., 2004). Following these criteria, the following mutations were selected: P8R, P22S, N98S, L268P, Q332P, E396K, and P440L (highlighted in red in Fig. 1). These mutations are also among the most commonly encountered in CMT2E/1F patients (Horga et al., 2017).
Fig. 1. Schematic of the known CMT2E/1F NFL mutations.

The NFL protein is represented as a horizontal black bar, with thickened regions representing the portions of the NFL rod domain that are predicted to form α-helical coiled-coils based on sequence alignment with human vimentin (Chernyatina, Guzenko, & Strelkov, 2015). Missense mutations (labeled below the schematic) are the most common, but there are also frameshift, deletion, and nonsense mutations (labeled above the schematic). Within the rod domain, all the nonsense mutations with known inheritance patterns are recessive, while the nonsense mutation found in the tail domain is dominantly inherited. Data from: Mersiyanova et al., 2000; De Jonghe et al., 2001; Georgiou et al., 2002; Yoshihara et al., 2002; Jordanova et al., 2003; Fabrizi et al., 2004, 2007; Züchner et al., 2004; Choi et al., 2004, 2012; Leung et al., 2006; Miltenberger-Miltenyi et al., 2007; Shin et al., 2008; Butinar et al., 2008; Abe et al., 2009; Yum et al., 2009; Bhagavati et al., 2009; Benedetti et al., 2010; Baets et al., 2011; Lin et al., 2011; Sivera et al., 2013; Agrawal et al., 2014; DiVincenzo et al., 2014; Elbracht et al., 2014; Hashiguchi et al., 2014; Manganelli et al., 2014; Drew et al., 2015; Noto et al., 2015; Pisciotta et al., 2015; Berciano et al., 2015, 2016; Luigetti et al., 2016; Werheid et al., 2016; Yang et al., 2016; Doppler et al., 2017; Horga et al., 2017; Fu and Yuan, 2018; Sainio et al., 2018; Lerat et al., 2019. The mutants used in the present study are marked in red.
None of the mutant proteins can assemble into homopolymers
Rodent NFL, M and H are incapable of forming homopolymers in cells (Ching & Liem, 1993; Lee, Xu, Wong, & Cleveland, 1993). In contrast, human NFL can assemble into homopolymers in cells, though it preferentially co-polymerizes with other neurofilament proteins when those proteins are present (Carter et al., 1998). To test the ability of CMT2E/1F mutant human NFL to form homopolymers, we transfected mutant constructs into SW13vim- human adrenal adenocarcinoma cells and visualized the expressed proteins by immunofluorescence microscopy following immunostaining (Fig. 2). SW13vim- cells lack endogenous cytoplasmic intermediate filaments due to spontaneous silencing of vimentin expression (Sarria, 1990; Yamamichi-Nishina et al., 2003) and are therefore a valuable tool for studying intermediate filament proteins because they allow the expression of one or more exogenous proteins in the absence of any endogenous cytoplasmic intermediate filament protein. Since these cells can revert to a vimentin-expressing state over time (Yamamichi-Nishina et al., 2003) we routinely co-stained with a vimentin antibody to confirm the absence of vimentin in the cells that we imaged. The average rate of reversion (i.e. proportion of vimentin-positive cells) in our cultures was 0.8%.
Fig. 2. SW13vim- cells expressing mutant NFL alone.

Representative images of SW13vim- cells expressing wild type or mutant human NFL and immunostained for NFL. All cells were confirmed to lack vimentin protein by co-staining for vimentin (data not shown). A’ shows an entire SW13vim- cell transfected with wild type human NFL. The yellow line marks the outline of the cell and the white box marks the portion of the cell shown in A. For the other SW13vim- cell images in this figure and the rest of this paper we use a higher magnification but show only a portion of each cell in each panel in order to conserve space. Wild type human NFL formed homopolymers in 64% of the transfected cells (note the short filaments in A), but the human NFL mutants did not form filaments in any of the transfected cells (note the punctate appearance of the NFL protein in B-H). Scale bar = 5 μm.
On average, 64% of the cells transfected with wild type human NFL alone contained filaments but, typically, they were relatively short (Fig. 2A). The remaining cells exhibited a punctate fluorescence (447 cells counted in four dishes from two separate experiments). In contrast, none of the cells expressing human CMT2E/1F mutant NFL contained filaments (124–462 cells counted in two dishes for each mutant from one experiment). Instead, these cells all presented with punctate fluorescence (Fig. 2B–H). The punctate, as opposed to diffuse, appearance suggests that the protein may have had some capacity for self-association, perhaps into short oligomeric assemblies, but the resolution of our images is not sufficient to confirm this. However, these data show that while wild type human NFL can form homopolymers, all of the human CMT2E/1F mutants examined lack this capability.
The P8R, P22S, L268P & P440L mutants can co-assemble with NFM
Mammalian neurons express multiple neurofilament proteins in vivo and these proteins preferentially co-assemble. For example, in rodents the developmental expression of NFL is always accompanied by the co-expression of NFM and peripherin and/or α-internexin, and these proteins form heteropolymers (Nixon & Shea, 1992; Perrot & Eyer, 2009). Also, in cultured neonatal mouse sympathetic neurons which express NFL, NFM, α-internexin and peripherin, all four of these proteins are incorporated along the entire length of every neurofilament (Yan et al., 2007). Similarly, when NFL is co-expressed with other neurofilament proteins in SW13vim- cells, these proteins preferentially co-assemble and are incorporated along the entire length of each neurofilament (Ching & Liem, 1993; Lee et al., 1993). Thus, neurofilaments are heteropolymers in vivo.
To investigate the ability of the CMT2E/1F mutants to form heteropolymers, we co-expressed the wild-type and mutant human NFL with human NFM in SW13vim- cells. Fig. 3 shows representative images and Fig. 4 shows the quantification. Essentially every cell that was positively transfected with both wild type NFL and NFM had filaments (Fig. 3A, Fig. 4). In addition, we also observed filaments in most cells that were co-transfected with the P8R, P22S, L268P and P440L CMT2E/1F NFL mutants. The L268P and P440L NFL mutants formed filaments in 82% and 95% of positively transfected cells, respectively (Fig. 4), and those filaments were indistinguishable from the wild type at the resolution of our images (Fig. 3E & H). The P8R and P22S mutants formed filaments in 84% and 76% of transfected cells, respectively (Fig. 4), though these filaments tended to be somewhat shorter in length (Fig. 3B–C). The other three mutants (N98S, Q332P, and E396K) formed filaments in less than 1% of cells that were co-expressing NFM (Fig. 4). The NFL and NFM in these cells co-localized in non-filamentous puncta (Fig. 3D, F & G), very similar in appearance to the mutant NFL in the single transfection experiments shown in Fig. 2.
Fig. 3. SW13vim- cells expressing mutant NFL and NFM.
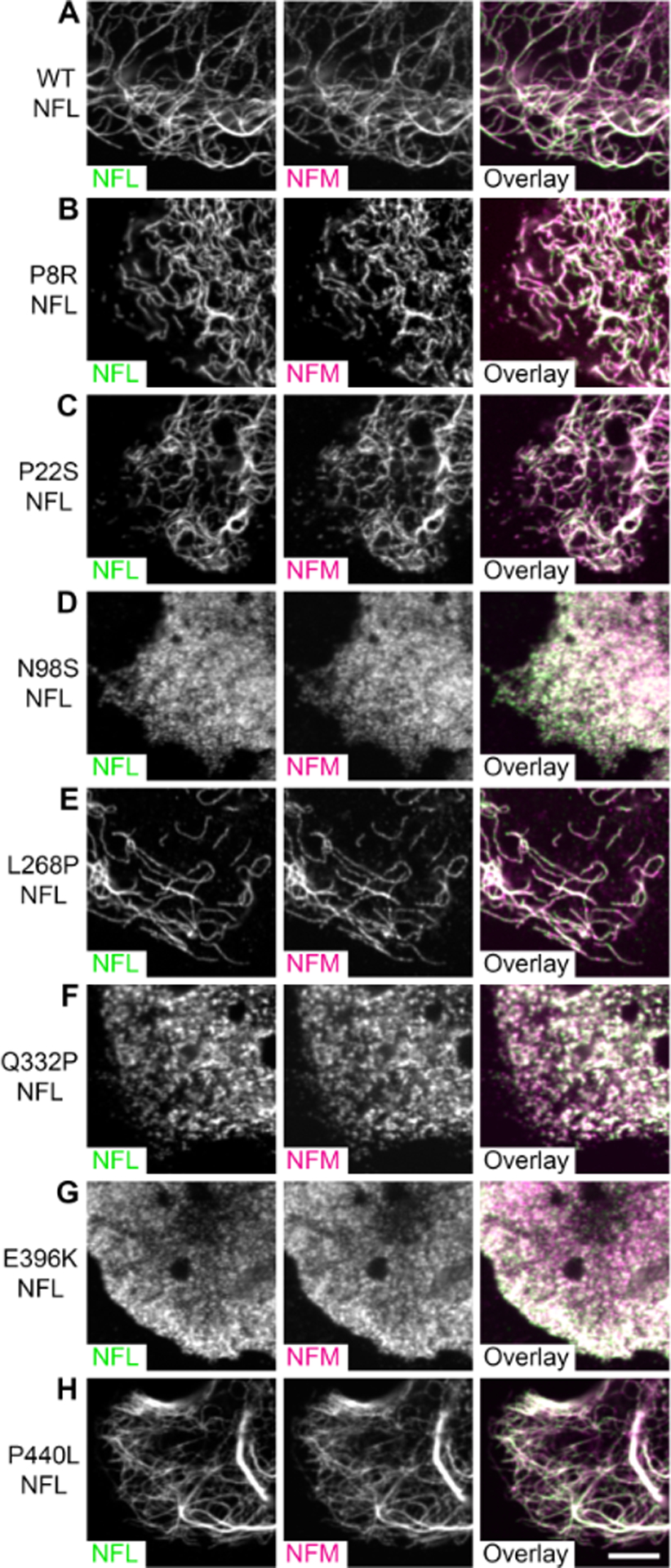
Representative images of SW13vim- cells co-transfected with human NFL and human NFM and immunostained for NFL (green) and NFM (magenta). All cells were confirmed to lack vimentin by co-staining for vimentin (data not shown). (A) WT NFL and NFM readily co-assembled into long filaments. Mutant NFL co-assembled with NFM for 4 of the 7 mutants examined: (B) P8R, (C) P22S, (E) L268P, and (H) P440L. The remaining three mutants did not form filaments: (D) N98S, (F) Q332P and (G) E396K. Scale bar = 5 μm.
Fig. 4. Quantification of neurofilament assembly in SW13vim- cells expressing mutant NFL and NFM.

Quantification of the percentage of cells expressing both NFL and NFM that also contained filaments for the experiment shown in Fig. 3. The data for the wild type was obtained by counting 12 or more areas per dish in 12 dishes from 6 different experiments (2 dishes per experiment). The data for each mutant was obtained by counting 12 or more fields of view per dish in 6 dishes from 3 different experiments (2 dishes per experiment). All the mutants were statistically significantly different from that of wild type (two-tailed Student’s t-test assuming unequal variances). The asterisks represent the p values: p<0.005(**), p<0.0005(***), p<0.00005(****). The mutants had the following degrees of freedom and p-values, respectively: P8R (5, 1.5 × 10−4); P22S (5, 1.6 × 10−5); N98S (11, 1.5 × 10−27); L268P (5, 1.2 × 10−5); Q332P (10, 6.6 × 10−23); E396K (7, 1.2 × 10−15); and P440L (5, 2.6 × 10−3). Error bars represent the standard deviation.
The N98S, Q332P and E396K mutants can assemble with NFM and peripherin
To investigate the potential of additional neurofilament polypeptides to rescue the assembly defect observed for the N98S, Q332P and E396K NFL mutants in the presence of NFM, we co-expressed these mutant proteins with human NFM and peripherin in SW13vim- cells. The presence of peripherin did not interfere with the assembly of wild type NFL (Fig. 5A) which formed filaments in 96% of transfected cells (Fig. 6). In addition, we also observed assembly of the N98S, Q332P and E396K NFL mutants into filaments, which did not occur in the absence of peripherin (Figs. 3 & 4). The appearance of the filaments was comparable to those formed with wild type NFL (Fig. 5B, C & D), though the incorporation of the N98S and Q332P mutants was less continuous and the proportion of transfected cells with filaments was lower for all three mutants (44%, 36%, and 70% for N98S, Q332P and E396K respectively; Fig. 6). We attempted to perform quadruple staining of these cells for vimentin, NFL, NFM and peripherin, but were unable to identify a satisfactory peripherin antibody. We also explored substituting ɑ-internexin for peripherin but did not observe a noticeable improvement in the extent of filament assembly compared to NFL plus NFM alone, so we did not pursue those experiments further (data not shown).
Fig. 5. SW13vim- cells expressing mutant NFL, NFM and peripherin.

Representative images of SW13vim- cells co-transfected with human NFL, NFM and peripherin and immunostained for NFL (green) and NFM (magenta). (A) The introduction of peripherin does not alter the ability for wild type NFL to form filaments with NFM. (B-D) Those NFL mutants that did not assemble into filaments with NFM alone (N98S, Q332P, and E396K) were able to form filaments in the presence of NFM and peripherin. Scale bar = 5 μm.
Fig. 6. Quantification of neurofilament assembly in SW13vim- cells expressing mutant NFL, NFM and peripherin.

Quantification of the percentage of cells expressing NFL and NFM that also contained filaments for wild type NFL and the N98S, Q332P and E396K mutants. The data for each NFL construct was obtained by counting 12 or more fields of view per dish in 6 dishes from 3 different experiments (2 dishes per experiment). All three mutants formed filaments in many cells, though the percentage of cells with filaments was less than for the wild type (two-tailed Student’s t-test assuming unequal variances). The asterisks represent the p values: p<0.005(**), p<0.0005(***), p<0.00005(****). The mutants had the following degrees of freedom and p-values, respectively: N98S (5, 3.2 × 10−4); Q332P (8, 3.5 × 10−9); E396K (6, 9.6 × 10−4). Error bars represent the standard deviation.
CMT2E/1F NFL mutant subunits can assemble into an endogenous filamentous network
The NFL mutations examined here are inherited dominantly, so patients are heterozygous for the mutant allele. Thus, in patients the mutant NFL would be co-expressed with wild type NFL in addition to the other expressed neurofilament proteins. To mimic this, we investigated the assembly properties of CMT2E/1F mutant rat NFL in primary cultures of neonatal rat cortical neurons, which express NFL, NFM and α-internexin but very little NFH and no peripherin (our unpublished observations). To visualize the mutant NFL, we used GFP-tagged rat NFL constructs and then either fixed the cells for immunostaining or visualized the fusion proteins by live imaging. To avoid the elevated diffuse GFP-NFL that can result from GFP-NFL overexpression in these neurons, we co-transfected with untagged rat NFM. Note that rat NFL has a serine insertion at amino acid 251 which shifts all mutations beyond this point by one when compared to the human mutations. Thus, L268P, Q332P, E396K and P440L in human NFL correspond to L269P, Q333P, E397K and P441L in rat NFL.
Fig. 7 shows representative images of neurofilaments in the soma of cortical neurons immunostained for GFP, NFM and α-internexin. The relatively low neurofilament content of these neurons permits single filaments to be resolved in the cell bodies and along the axons (Uchida, Colakoglu, Wang, Monsma, & Brown, 2013). The P8R, P22S, L269P and P441L mutants assembled into filaments that co-stained with NFM and α-internexin along their entire length and that were indistinguishable in length and organization from the filaments in neurons expressing wild-type (Fig. 7B, C, E, H). The E397K, N98S, and Q333P mutants also assembled into filaments with NFM and α-internexin, but with successively decreasing efficacy. The neurofilament content in these cells was lower and the cell soma contained elevated punctate staining for the mutant protein, most prominently for the Q333P mutant (Fig. 7D, F & G). However, in no case did we observe the aggregates and somal sequestration described in previous reports (Brownlees et al., 2002; Perez-Olle et al., 2005; Sasaki et al., 2006; Tradewell et al., 2009; Zhai et al., 2007). These data reveal considerable diversity in the assembly properties of the CMT2E/1F mutants. The P8R, P22S, L269P and P441L mutants all apparently co-assembled fully with endogenous neurofilament proteins in these neurons whereas the N98S, Q333P and E397K mutants apparently caused a partial disruption of the endogenous neurofilament array and exhibited elevated non-filamentous mutant protein.
Fig. 7. Assembly of GFP-tagged mutant NFL in primary rat cortical neurons.

Representative images of cultured primary rat cortical neurons transfected with GFP-tagged rat NFL and immunostained for GFP (green), NFM (magenta) and ɑ-internexin (INA; blue) 9 days after plating. Each panel shows a single neuronal cell body with several branching processes extending outward as well as processes passing through the field of view from other neighboring neurons. The P8R (B), P22S (C), L269P (E) and P441L (H) mutants all formed filaments comparable in appearance to the wild type (A). The N98S (D) and E397K (G) mutants assembled less efficiently, exhibiting elevated punctate GFP fluorescence in the soma (see green fluorescence in Overlay). The Q333P mutant (F) exhibited very limited assembly into filaments and higher levels of punctate fluorescence in the soma. Note that there is a serine insertion at amino acid 251 in the rat NFL gene which shifts all mutation nomenclature by one when compared to the human mutations. Thus, the L268P, Q332P, E396K and P440L mutations in human NFL correspond to L269P, Q333P, E397K and P441L in rat NFL. Scale bar = 10 μm.
While the images in Fig. 7 were selected to be representative on average, it is important to note that there was considerable variation from cell to cell for some of the mutants, particularly N98S but also to a lesser extent E397K (Fig. 8). Most of the cells transfected with these mutants exhibited lower neurofilament densities and weaker incorporation along single neurofilaments than for the wild type protein or the P8R, P22S, L269P and P441L mutants, with elevated punctate staining for the mutant protein in the cell bodies (Fig. 8B & E). However, some cells exhibited abundant neurofilaments with incorporation indistinguishable from the wild type (Fig. 8A & D) and others exhibited very low neurofilament densities and limited incorporation (Fig. 8C & F) similar to the typical appearance of cells expressing for the Q332P mutant (compare to Fig. 7F). Thus, the N98S and E397K mutants retained the capacity for robust assembly into neurofilament polymers in cortical neurons under certain conditions.
Fig. 8. Examples of the cellular variation in assembly for two NFL mutants in primary rat cortical neurons.

Images of cells exhibiting robust assembly (A, D), less assembly (B, E) and limited or no assembly (C, F) for the N98S mutant (A-C) and the E397K mutant (D-F). The transfection and immunostaining conditions were identical to those in Fig. 7. Note that the cells with more limited incorporation also exhibit elevated punctate GFP fluorescence in the soma, indicative of less efficient incorporation into filaments (see green fluorescence in Overlay). Scale bar = 10 μm.
To confirm the incorporation of the mutant proteins into neurofilament polymers in cortical neurons, we examined cells expressing GFP-tagged mutant NFL by live imaging. In experiments using the Q333P mutant, we did not encounter any cells containing green fluorescent filaments. This suggests that any incorporation of this mutant protein into neurofilaments was too low to detect by live-cell imaging and is consistent with the limited incorporation observed by immunostaining (Fig. 7F). However, the other mutants did incorporate into filaments. The P8R, P22S, L269P, and P441L mutants all exhibited levels of incorporation comparable to wild type NFL, with bright and continuous GFP fluorescence along the entire length of each filament (Fig. 9A, B, C, E, G). The N98S and E397K mutants exhibited noticeably weaker fluorescence, resulting in a lower signal-to-noise ratio, though these proteins were still distributed continuously throughout the filaments (Fig. 9D, F).
Fig. 9. Live imaging of GFP-tagged mutant NFL incorporation along single filaments.

Primary rat cortical neurons were transfected with GFP-tagged wild type (A) and mutant (B-G) rat NFL and then imaged live 9 days after plating. We show three representative examples (1, 2, 3) of single axonal neurofilaments for each mutant. For each mutant, the three examples were taken from three different dishes and from at least two independent experiments. The Q333P mutant is not shown because we did not observe any cells with filaments containing detectable GFP fluorescence using this mutant. Note that the signal-to-noise ratio of the filaments is comparable to wild type for the P8R, P22S, L269P and P440L mutants (B, C, E, G), but lower for the N98S and E397K mutants (D, F). Scale bar = 3 μm.
Neurofilaments containing CMT2E/1F mutant NFL move rapidly and intermittently in cultured axons
There have been reports that P8R, P22S, P22T and Q333P mutations in NFL disrupt the axonal transport of neurofilaments in cultured neurons (Brownlees et al., 2002; Perez-Olle et al., 2005). However, this conclusion was based on a sequestration of neurofilament proteins in cell bodies and depletion from axons, which we did not observe in our experiments. To address this, we performed live imaging of neurons expressing GFP-tagged mutant NFL. As stated previously, we did not detect GFP-tagged neurofilaments in neurons expressing the Q333P NFL mutant protein so were unable to record neurofilament movement for this mutant. Cells expressing GFP-tagged P8R, P22S, N98S, L269P, E397K and P441L mutant NFL all contained bright fluorescent filaments that moved in rapid, intermittent and bidirectional manner through gaps in the neurofilament array (Fig. 10). The behavior of the filaments was qualitatively indistinguishable from filaments containing GFP-tagged wild type NFL (Fig. 10) though as noted above, the filaments containing the N98S and E397K mutants were fainter, resulting in a lower signal-to-noise ratio (Fig. 10D & F). Thus, neurofilaments containing P8R, P22S, N98S, L269P, E397K, or P441L mutant NFL were all capable of movement in axons of these neurons.
Fig. 10. Movement of neurofilaments containing GFP-tagged mutant NFL.

Primary rat cortical neurons were transfected with GFP-tagged wild type (A) and mutant (B-G) NFL and then imaged live 9 days after plating. The images are stills from representative time-lapse movies showing the movement of single neurofilament polymers through gaps in the axonal neurofilament array (regions that lack other neurofilament polymers). The time-lapse interval was 3 seconds. The GFP-tagged proteins assembled efficiently into filaments resulting in a low level of diffuse fluorescence in the gaps for all the mutants shown here. The yellow lines trace the paths of the moving neurofilaments, obtained by tracing on a maximum projection of the corresponding time-lapse movie (path). The leading end of the moving filament is indicated by a green arrow, and the trailing end by a magenta arrow. The time point of each representative still image is indicated in the top left corner of each frame. The P8R, P22S, N98S, L269P, E397 mutants all incorporated into filaments that moved in a rapid, intermittent and bidirectional manner qualitatively indistinguishable from the wild type protein, though the images for the N98S and E397K mutants were grainier due to the lower level of incorporation of these mutant proteins (Fig. 9). The Q333P mutant is not shown because we did not observe any cells with filaments containing detectable GFP fluorescence using this mutant. Scale bar = 10 μm.
DISCUSSION
In contrast to wild type NFL, none of the CMT2E/1F mutants examined in this study were able to assemble into homopolymers in SW13vim- cells. However, the P8R, P22S, L268P, and P440L mutants all co-assembled into neurofilaments with NFM alone, and the N98S, Q332P, and E396K mutants co-assembled with NFM and peripherin. Thus, the CMT2E/1F mutations examined here all caused some impairment of the assembly properties of NFL, but all were capable of co-assembly into neurofilaments depending on the context.
In cortical neurons, the P8R, P22S, L269P and P441L mutants all co-assembled with endogenous NFM and ɑ-internexin. These cells also express endogenous NFL, as do CMT2E/1F patients heterozygous for a missense mutation, though we were unable to confirm the presence of endogenous NFL in the assembled filaments because there are no antibodies that distinguish the wild type and mutant NFL proteins. The N98S and E397K mutants also co-assembled into filaments in these cells, but there was considerable variation from cell to cell, with some cells exhibiting a normal neurofilament array with extensive incorporation, and others exhibiting reduced neurofilament density, weaker incorporation, and elevated punctate staining for the mutant protein. The explanation for the variability is not clear, but most likely it reflects variations in expression level that are common in transient transfection experiments, suggesting that the ability of these mutants to assemble into neurofilaments may be sensitive to the stoichiometry of the available neurofilament subunit proteins. The Q333P mutant exhibited very limited incorporation into neurofilaments in the cortical neurons. The ability of this mutant to co-assemble with NFM in SW13vim- cells in the presence of peripherin, and the fact that cortical neurons express no peripherin, suggests that peripherin may be more able to rescue the defective assembly of this mutant than NFM or ɑ-internexin. A caveat to these experiments is that they were performed in cortical neurons. We used these neurons because of their low neurofilament content in primary cell culture, permitting us to readily resolve neurofilament polymers by fluorescence microscopy. Moving forward, it will be important to confirm the assembly properties of the Q332/333P mutants in lower motor and sensory neurons, which are the neuronal cell types primarily affected in CMT2E/1F in vivo, and particularly in different subclasses of those neurons that differ in their peripherin expression (Ferri et al., 1990; Goldstein, Grant, House, Henken, & Gainer, 1996; Holford, Case, & Lawson, 1994). Specifically, our data suggest that neurons which express NFL and peripherin may be less vulnerable to the disruptive effects of the mutant NFL than those which express NFL alone.
Fig. 11 summarizes our observations. Collectively, these data suggest a hierarchy of assembly capability among the CMT2E/1F mutants from unimpaired to most impaired as follows: WT > P440/441L > L268/269P > P8R > P22S > E396/397K > N98S > Q332/333P. The lower neurofilament density in many of the neurons expressing the E397K, N98S and Q333P mutants suggests that these mutants, at least at certain expression levels, are capable of disrupting the stability or assembly of neurofilaments. One possibility is that there is a certain maximum “load” that a filament can tolerate for each mutant and that at levels that exceed this load, filaments may be unstable or fail to form (Fig. 12). According to this hypothesis, the mutants towards the top of the hierarchy (e.g. P440/441L) would be considering “high load” mutants, meaning that filaments can tolerate a high proportion of these mutant subunits, whereas mutants towards the bottom of the hierarchy (e.g. Q332/333P) would be considered “low load” mutants, meaning that the filaments can tolerate only a low proportion of these mutants. Consistent with this idea, we noted that live imaging of GFP-tagged mutants revealed lower incorporation of the N98S and E397K mutants in neurofilaments compared to the other mutants higher up the hierarchy.
Fig. 11. Table summarizing the data for the NFL mutants.

The assembly capability of each of the CMT2E/1F mutants is summarized by experiment. The cell type and the exogenous (transfected) and endogenous neurofilament subunits in those cells are indicated for each experimental condition. The figures containing representative images for each experiment are also indicated. The grey cells denote conditions in which there was no discernible filament formation and the green cells denote conditions in which there was filament formation, with the intensity of the green color and number of “+” symbols denoting the extent of similarity in filament length and density to wild type neurofilaments. Blank cells in the table indicate mutants that were not examined under that particular experimental condition. Abbreviations: NFL (neurofilament protein L), NFM (neurofilament protein M), PRPH (peripherin), INA (ɑ-internexin).
Fig. 12. Proposed effect of mutant load on filament integrity.

Neurofilaments have no fixed subunit stoichiometry so the relative proportion of the various neurofilament polypeptides in the filament can vary depending on their relative level of expression. We propose that some CMT2E/1F mutations in NFL are more disruptive to neurofilament integrity than others, resulting in differences in the maximum mutant subunit “load” that is compatible with neurofilament assembly and stability. “High load” mutants are those such as P440/441L that can incorporate into filaments at relatively high concentration without compromising filament assembly or integrity. In contrast “low load” mutants are those such as Q332/333P that can disrupt filament assembly or integrity even at relatively low concentrations. Whether or not CMT2E/1F mutant protein expression reaches levels sufficient to disrupt filament formation in patients remains to be established.
Several of the mutants that we examined (P8R, P22S, N98S and Q332/333P) have been studied previously in cultured cells, sometimes with conflicting results. Human and rat P8R NFL mutants were reported to form non-filamentous puncta, aggregates, and/or filamentous bundles with NFM and/or NFH in SW13vim- cells and also to form aggregates in neuronal CAD cells (Brownlees et al., 2002; Perez-Olle et al., 2004, 2002, 2005). Similar results were reported for P8Q and P8L NFL mutants in SW13 vim- cells (Perez-Olle et al., 2002). Human P22S mutant NFL was reported to form filaments and filamentous bundles with NFM in SW13vim- cells in one study (Perez-Olle et al., 2004), but non-filamentous aggregates with NFM or NFH in the same cells in another study (Sasaki et al., 2006). Human N98S mutant NFL was reported to form non-filamentous aggregates with NFM in SW13vim- cells (Perez-Olle et al., 2004), and both human and rodent P8R and Q332/333P mutants as well as human P22S and P22T mutants have also been reported to form non-filamentous aggregates in primary neurons (Brownlees et al., 2002; Sasaki et al., 2006; Zhai et al., 2007). Tradewell et al. (2009) reported that rodent P8R and Q333P mutant NFL incorporated into neurofilaments in cultured sensory neurons prior to disrupting those filaments, and Gentil et al. (2013) reported that the rodent P8R mutant caused neurofilament bundling in those neurons. Lastly, human or rodent Q332/333P mutant NFL was reported to form non-filamentous aggregates with NFM and/or NFH in SW13vim- cells and in neuronal CAD cells (Perez-Olle et al., 2005) and primary neurons (Gentil et al., 2013; Sasaki et al., 2006; Tradewell et al., 2009; Zhai et al., 2007). In contrast to these reports, we found that P8R and P22S mutant NFL assembled readily into neurofilaments of normal appearance in both SW13vim- cells and in cortical neurons, and both the N98S and Q332P mutants were capable of assembling into neurofilaments in SW13vim- cells when co-transfected with NFM and peripherin. While the Q333P mutant exhibited very limited incorporation into filaments in cortical neurons, the N98S mutant could assemble into filaments in these neurons.
It should be noted that several of the above studies also reported an impairment of neurofilament entry into axons of CAD cells and primary neurons expressing P8R, P8L, P8Q, E89K, N98S or Q333P mutant NFL, which implied that there was an impairment of neurofilament transport (Brownlees et al., 2002; Perez-Olle et al., 2004, 2005). Importantly, however, those studies also reported that the same mutants disrupted neurofilament assembly. Thus, since neurofilament polymers are the cargoes of neurofilament protein transport, it seems most likely that the transport impairment was due to the effect of the mutants on neurofilament polymer integrity. In our hands, GFP fusions of the P8R, P22S, N98S, L269P, E397K and P441L NFL mutants all co-assembled readily into neurofilaments in cortical neurons and direct observation of the filaments by time-lapse imaging revealed that they moved in a rapid, intermittent, and bidirectional manner that was qualitatively indistinguishable from wild type NFL. We did not perform a kinetic analysis so we cannot exclude the possibility that these mutants caused subtle impairments of neurofilament transport that could have significance for the disease pathogenesis, but our data do demonstrate that neurofilaments containing these mutant proteins are capable of rapid intermittent movement.
The explanation for the differences between our findings and the published reports discussed above is unclear, but we note that all of these studies, including our own, involved transient transfection experiments which afforded no control over expression level. Anecdotally, we have noted that neurofilament proteins aggregate when expressed at high levels. Thus, we suggest that the capacity of the mutant proteins to assemble and be transported may be sensitive to their expression level, and that the filamentous or non-filamentous aggregates that have been reported by others may have been due to elevated levels of expression. Overall, these considerations suggest that caution should be exercised when interpreting the assembly and transport properties of mutant neurofilament proteins in cultured cells using transient transfection.
Our observation on the capacity of the CMT2E/1F mutants to assemble into neurofilaments without disruption of the endogenous neurofilament array is consistent with observations of assembled neurofilaments in electron microscopic studies of nerve biopsies from CMT2E/1F patients. Fabrizi et al. (2004,2007) described a mix of atrophic and swollen axons in patients with the P22S, C322-N326del or L268P mutations and the swollen axons were packed with neurofilament polymers that were disorganized but otherwise normal in appearance. Both Zϋchner et al. (2004) and Fabrizi et al. (2007) described similar neurofilament accumulations in patients with the E396K mutation, as did Benedetti et al. (2010) for a patient with the P440L mutation. Unfortunately, no patient biopsy data exists for the Q332P mutation. Also, the magnification of published electron micrographs of sural nerve biopsies from patients with the P8R and N98S mutations are too low to discern if neurofilament polymers were present (Horga et al., 2017; Luigetti et al., 2016). However, accumulations of neurofilament polymers of normal appearance have been reported in proximal axons and cell bodies of neurons in knock-in mice heterozygous for the N98S mutant allele (Lancaster et al., 2018; Zhao et al., 2017), in the cell bodies and axons of sensory neurons cultured from these mice (Zhao et al., 2017), and in the cell bodies and axons of motor neurons differentiated from a patient-derived iPSC line heterozygous for the N98S mutation (Saporta et al., 2015). In light of our present data, these studies suggest that CMT2E/1F may not be a disorder caused by a failure of neurofilaments to assemble, but rather a disorder caused by disorganization of the neurofilament polymers along axons, resulting in focal accumulations and swellings in some axonal locations and depletion and atrophy in others.
While it is clear that the mutants examined here differ in their capacity for assembly, the clinical literature reveals no clear correlation with disease severity. For example, while the Q332/333P mutant was most refractory to assembly in our experiments, the nerve conduction velocities and age of onset of patients with this mutation do not indicate any greater disease severity than for the P8R mutant, for instance, which assembled readily into neurofilaments in the presence of NFM alone (Horga et al., 2017). One possible explanation is that in vivo the mutations result in subtle changes to the filaments that impair neuronal function without inducing wholesale neurofilament disassembly (Fig. 13). For example, while the mutant NFL incorporates into neurofilaments, it may do so less efficiently resulting in a pool of unincorporated protein that may aggregate or produce damaging off-target interactions, or the mutant NFL may incorporate into filaments, but this incorporation may alter filament interactions and/or transport in ways that lead to neurofilament disorganization and disease. Our present study suggests that the best way to resolve the disease mechanism is to examine the CMT2E/1F mutant NFL protein in a model system expressing all relevant neurofilament subunit proteins, including the mutant, at endogenous levels. A particularly pressing question is whether the mutant protein is stably expressed in patients and whether it incorporates fully into filaments. The NFLN98S/+ CMT2E/1F mouse model engineered by the Liem lab displays a disease phenotype and is a promising model system to accomplish this moving forward (Adebola et al., 2015; Lancaster et al., 2018; Zhao et al., 2017), as are neurons differentiated from patient-derived iPSCs (Saporta et al., 2015).
Fig. 13. Possible pathogenic mechanisms of CMT2E/1F.

We propose three possible mechanisms of pathogenesis for CMT2E/1F which are not mutually exclusive. It is also possible that different mutants could act through different mechanisms or combinations of mechanisms. (A) Healthy neurons. Wild type NFL is fully assembled into filaments resulting in polymers that interact with other proteins and move both bi-directionally along microtubules. (B) Diseased neurons. We consider three possible modes of toxicity: (i) mutant NFL may assemble less efficiently, resulting in an increased pool of unassembled NFL protein that may aggregate or engage in promiscuous interactions; (ii) neurofilaments containing mutant NFL may have altered protein interactions which interfere with their normal function; (iii) neurofilaments containing mutant NFL may have transport defects in either direction.
ACKNOWLEDGEMENTS
We thank Robert Evans for the SW13 human adrenal adenocarcinoma cell line, Virginia Lee for the rabbit polyclonal anti-NFL antibody, Virginia Lee and Don Cleveland for human NFM cDNA, Ron Liem and Shin-Ichi Hisanaga for human NFL cDNA, and Chris Miller and Ron Liem for rat NFL cDNA. This project was funded by National Institutes of Health Grant R01 NS038526 to A.B., with additional support provided by National Institutes of Health Grants P30 NS104177, P30 NS045758, and S10 OD010383. The authors declare that they have no competing financial interests.
DATA SHARING
The data that support the findings of this study are available from the corresponding author upon reasonable request. The plasmids described in this study are available in the Anthony Brown Lab collection on Addgene.
Abbreviations:
- CMT
Charcot-Marie-Tooth
- CMT2E/1F
Charcot-Marie-Tooth type 2E/1F
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- INA ɑ
internexin
- NFL
neurofilament protein L
- NFM
neurofilament protein M
- NFH
neurofilament protein H
- PRPH
peripherin
REFERENCES
- Abe A, Numakura C, Saito K, Koide H, Oka N, Honma A, … Hayasaka K (2009). Neurofilament light chain polypeptide gene mutations in Charcot–Marie–Tooth disease: nonsense mutation probably causes a recessive phenotype. Journal of Human Genetics, 54(2), 94–97. 10.1038/jhg.2008.13 [DOI] [PubMed] [Google Scholar]
- Adebola AA, Di Castri T, He C-Z, Salvatierra LA, Zhao J, Brown K, … Liem RKH (2015). Neurofilament light polypeptide gene N98S mutation in mice leads to neurofilament network abnormalities and a Charcot-Marie-Tooth Type 2E phenotype. Human Molecular Genetics, 24(8), 2163–2174. 10.1093/hmg/ddu736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal PB, Joshi M, Marinakis NS, Schmitz-Abe K, Ciarlini PDSC, Sargent JC, … Beggs AH (2014). Expanding the phenotype associated with the NEFL mutation: neuromuscular disease in a family with overlapping myopathic and neurogenic findings. JAMA Neurology, 71(11), 1413–1420. 10.1001/jamaneurol.2014.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baets J, Deconinck T, De Vriendt E, Zimon M, Yperzeele L, Van Hoorenbeeck K, … De Jonghe P (2011). Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain, 134(9), 2664–2676. 10.1093/brain/awr184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti S, Previtali SC, Coviello S, Scarlato M, Cerri F, Di Pierri E, … Bolino A (2010). Analyzing Histopathological Features of Rare Charcot-Marie-Tooth Neuropathies to Unravel Their Pathogenesis. Archives of Neurology, 67(12), 1498–1505. 10.1001/archneurol.2010.303 [DOI] [PubMed] [Google Scholar]
- Berciano J, García A, Peeters K, Gallardo E, De Vriendt E, Pelayo-Negro AL, … Jordanova A (2015). NEFL E396K mutation is associated with a novel dominant intermediate Charcot–Marie–Tooth disease phenotype. Journal of Neurology, 262(5), 1289–1300. 10.1007/s00415-015-7709-4 [DOI] [PubMed] [Google Scholar]
- Berciano J, Peeters K, García A, López-Alburquerque T, Gallardo E, Hernández-Fabián A, … Jordanova A (2016). NEFL N98S mutation: another cause of dominant intermediate Charcot–Marie–Tooth disease with heterogeneous early-onset phenotype. Journal of Neurology, 263(2), 361–369. 10.1007/s00415-015-7985-z [DOI] [PubMed] [Google Scholar]
- Bhagavati S, Maccabee PJ, & Xu W (2009). The neurofilament light chain gene (NEFL) mutation Pro22Ser can be associated with mixed axonal and demyelinating neuropathy. Journal of Clinical Neuroscience, 16(6), 830–831. 10.1016/j.jocn.2008.08.030 [DOI] [PubMed] [Google Scholar]
- Brown A (2014). Slow Axonal Transport. In Reference Module in Biomedical Sciences (pp. 1–9). 10.1016/B978-008045046-9.02000-3 [DOI] [Google Scholar]
- Brownlees J, Ackerley S, Grierson AJ, Jacobsen NJO, Shea K, Anderton BH, … Miller CCJ (2002). Charcot-Marie-Tooth disease neurofilament mutations disrupt neurofilament assembly and axonal transport. Human Molecular Genetics, 11(23), 2837–2844. 10.1093/hmg/11.23.2837 [DOI] [PubMed] [Google Scholar]
- Butinar D, Starr A, Zidar J, Koutsou P, & Christodoulou K (2008). Auditory nerve is affected in one of two different point mutations of the neurofilament light gene. Clinical Neurophysiology, 119(2), 367–375. 10.1016/j.clinph.2007.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter J, Gragerov A, Konvicka K, Elder G, Weinstein H, & Lazzarini RA (1998). Neurofilament (NF) Assembly; Divergent Characteristics of Human and Rodent NF-L Subunits. Journal of Biological Chemistry, 273(9), 5101–5108. 10.1074/jbc.273.9.5101 [DOI] [PubMed] [Google Scholar]
- Chernyatina AA, Guzenko D, & Strelkov SV (2015). Intermediate filament structure: the bottom-up approach. Current Opinion in Cell Biology, 32(Box 1), 65–72. 10.1016/j.ceb.2014.12.007 [DOI] [PubMed] [Google Scholar]
- Ching GY, Chien CL, Flores R, & Liem RK (1999). Overexpression of alpha-internexin causes abnormal neurofilamentous accumulations and motor coordination deficits in transgenic mice. The Journal of Neuroscience, 19(8), 2974–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching GY, & Liem RK (1993). Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. The Journal of Cell Biology, 122(6), 1323–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B-O, Koo SK, Park M-H, Rhee H, Yang S-J, Choi K-G, … Chung KW (2012). Exome sequencing is an efficient tool for genetic screening of Charcot-Marie-Tooth Disease. Human Mutation, 33(11), 1610–1615. 10.1002/humu.22143 [DOI] [PubMed] [Google Scholar]
- Choi B-O, Lee MS, Shin SH, Hwang JH, Choi K-G, Kim W-K, … Chung KW (2004). Mutational analysis ofPMP22, MPZ, GJB1, EGR2 andNEFL in Korean Charcot-Marie-Tooth neuropathy patients. Human Mutation, 24(2), 185–186. 10.1002/humu.9261 [DOI] [PubMed] [Google Scholar]
- De Jonghe P, Mersivanova I, Nelis E, Del Favero J, Martin JJ, Van Broeckhoven C, … Timmerman V (2001). Further evidence that neurofilament light chain gene mutations can cause Charcot-Marie-Tooth disease type 2E. Annals of Neurology, 49(2), 245–249. [DOI] [PubMed] [Google Scholar]
- DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, … Higgins JJ (2014). The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Molecular Genetics & Genomic Medicine, 2(6), 522–529. 10.1002/mgg3.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler K, Kunstmann E, Krüger S, & Sommer C (2017). Painful Charcot-Marie-Tooth neuropathy type 2E/1F due to a novel NEFL mutation. Muscle & Nerve, 55(5), 752–755. 10.1002/mus.25410 [DOI] [PubMed] [Google Scholar]
- Drew AP, Zhu D, Kidambi A, Ly C, Tey S, Brewer MH, … Kennerson ML (2015). Improved inherited peripheral neuropathy genetic diagnosis by whole-exome sequencing. Molecular Genetics & Genomic Medicine, 3(2), 143–154. 10.1002/mgg3.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbracht M, Senderek J, Schara U, Nolte K, Klopstock T, Roos A, … Rudnik-Schöneborn S (2014). Clinical and morphological variability of the E396K mutation in the neurofilament light chain gene in patients with Charcot-Marie- Tooth disease type 2E. Clinical Neuropathology, 33(09), 335–343. 10.5414/NP300742 [DOI] [PubMed] [Google Scholar]
- Fabrizi GM, Cavallaro T, Angiari C, Bertolasi L, Cabrini I, Ferrarini M, & Rizzuto N (2004). Giant axon and neurofilament accumulation in Charcot-Marie-Tooth disease type 2E. Neurology, 62(8), 1429–1431. 10.1212/01.WNL.0000120664.07186.3C [DOI] [PubMed] [Google Scholar]
- Fabrizi Gian Maria, Cavallaro T, Angiari C, Cabrini I, Taioli F, Malerba G, … Rizzuto N (2007). Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton. Brain, 130(2), 394–403. 10.1093/brain/awl284 [DOI] [PubMed] [Google Scholar]
- Ferri G, Sabani A, Abelli L, Polak JM, Dahl D, & Portier M (1990). Distribution of Peripherin and Neurofilament Protein Immunoreactivity and Effect of Capsaicin. Brain Research, 515, 331–335. [DOI] [PubMed] [Google Scholar]
- Fu J, & Yuan Y (2018). A novel homozygous nonsense mutation in NEFL causes autosomal recessive Charcot–Marie–Tooth disease. Neuromuscular Disorders, 28(1), 44–47. 10.1016/j.nmd.2017.09.018 [DOI] [PubMed] [Google Scholar]
- Gentil BJ, Mushynski WE, & Durham HD (2013). Heterogeneity in the properties of NEFL mutants causing Charcot–Marie–Tooth disease results in differential effects on neurofilament assembly and susceptibility to intervention by the chaperone-inducer, celastrol. The International Journal of Biochemistry & Cell Biology, 45(7), 1499–1508. 10.1016/j.biocel.2013.04.009 [DOI] [PubMed] [Google Scholar]
- Georgiou D-M, Zidar J, Korošec M, Middleton L, Kyriakides T, & Christodoulou K (2002). A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics, 4(2), 93–96. 10.1007/s10048-002-0138-4 [DOI] [PubMed] [Google Scholar]
- Goldstein ME, Grant P, House SB, Henken DB, & Gainer H (1996). Developmental regulation of two distinct neuronal phenotypes in rat dorsal root ganglia. Neuroscience, 71(1), 243–258. 10.1016/0306-4522(95)00404-1 [DOI] [PubMed] [Google Scholar]
- Goslin K, Hannelore A, & Banker G (1998). Rat hippocampal neurons in low-density culture In Banker G & Goslin K (Eds.), Culturing Nerve Cells (pp. 339–370). Cambridge: MIT Press. [Google Scholar]
- Hashiguchi A, Higuchi Y, Nomura M, Nakamura T, Arata H, Yuan J, … Takashima H (2014). Neurofilament light mutation causes hereditary motor and sensory neuropathy with pyramidal signs. Journal of the Peripheral Nervous System, 19(4), 311–316. 10.1111/jns.12102 [DOI] [PubMed] [Google Scholar]
- Holford LC, Case P, & Lawson SN (1994). Substance P, neurofilament, peripherin and SSEA4 immunocytochemistry of human dorsal root ganglion neurons obtained from post-mortem tissue: a quantitative morphometric analysis. Journal of Neurocytology, 23(9), 577–589. 10.1007/BF01262058 [DOI] [PubMed] [Google Scholar]
- Horga A, Laurà M, Jaunmuktane Z, Jerath NU, Gonzalez MA, Polke JM, … Reilly MM (2017). Genetic and clinical characteristics of NEFL -related Charcot-Marie-Tooth disease. Journal of Neurology, Neurosurgery & Psychiatry, 88(7), 575–585. 10.1136/jnnp-2016-315077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, … Timmerman V (2003). Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain : A Journal of Neurology, 126(Pt 3), 590–597. 10.1093/brain/awg059 [DOI] [PubMed] [Google Scholar]
- Kaech S, & Banker G (2006). Culturing hippocampal neurons. Nature Protocols, 1(5), 2406–2415. 10.1038/nprot.2006.356 [DOI] [PubMed] [Google Scholar]
- Lancaster E, Li J, Hanania T, Liem R, Scheideler MA, & Scherer SS (2018). Myelinated axons fail to develop properly in a genetically authentic mouse model of Charcot-Marie-Tooth disease type 2E. Experimental Neurology, 308(April), 13–25. 10.1016/j.expneurol.2018.06.010 [DOI] [PubMed] [Google Scholar]
- Lee MK, & Cleveland DW (1996). Neuronal intermediate filaments. Annual Review of Neuroscience, 19(1), 187–217. 10.1016/S0955-0674(05)80036-5 [DOI] [PubMed] [Google Scholar]
- Lee MK, Xu Z, Wong PC, & Cleveland DW (1993). Neurofilaments are obligate heteropolymers in vivo. The Journal of Cell Biology, 122(6), 1337–1350. 10.1083/jcb.122.6.1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat J, Magdelaine C, Beauvais‐Dzugan H, Espil C, Ghorab K, Latour P, … Lia A-S (2019). A novel pathogenic variant of NEFL responsible for deafness associated with peripheral neuropathy discovered through next‐generation sequencing and review of the literature. Journal of the Peripheral Nervous System, 24(1), 139–144. 10.1111/jns.12310 [DOI] [PubMed] [Google Scholar]
- Leung CL, Nagan N, Graham TH, & Liem RKH (2006). A novel duplication/insertion mutation of NEFL in a patient with Charcot-Marie-Tooth disease. American Journal of Medical Genetics Part A, 140A(9), 1021–1025. 10.1002/ajmg.a.31242 [DOI] [PubMed] [Google Scholar]
- Lin K-P, Soong B-W, Yang C-C, Huang L-W, Chang M-H, Lee I-H, … Lee Y-C (2011). The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PloS One, 6(12), e29393 10.1371/journal.pone.0029393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luigetti M, Padua L, Coraci D, Fabrizi GM, Romano A, & Sabatelli M (2016). Nerve ultrasound in CMT2E/CMT1F due to NEFL mutation: Confirmation of an axonal pathology. Clinical Neurophysiology, 127(9), 2990–2991. 10.1016/j.clinph.2016.06.024 [DOI] [PubMed] [Google Scholar]
- Manganelli F, Tozza S, Pisciotta C, Bellone E, Iodice R, Nolano M, … Santoro L (2014). Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. Journal of the Peripheral Nervous System, 19(4), 292–298. 10.1111/jns.12092 [DOI] [PubMed] [Google Scholar]
- Mersiyanova IV, Perepelov AV, Polyakov AV, Sitnikov VF, Dadali EL, Oparin RB, … Evgrafov OV (2000). A New Variant of Charcot-Marie-Tooth Disease Type 2 Is Probably the Result of a Mutation in the Neurofilament-Light Gene. The American Journal of Human Genetics, 67(1), 37–46. 10.1086/302962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miltenberger-Miltenyi G, Janecke AR, Wanschitz JV, Timmerman V, Windpassinger C, Auer-Grumbach M, & Löscher WN (2007). Clinical and electrophysiological features in Charcot-Marie-Tooth disease with mutations in the NEFL gene. Archives of Neurology, 64(7), 966–970. 10.1001/archneur.64.7.966 [DOI] [PubMed] [Google Scholar]
- Nixon RA, & Shea TB (1992). Dynamics of neuronal intermediate filaments: A developmental perspective. Cell Motility and the Cytoskeleton, 22(2), 81–91. 10.1002/cm.970220202 [DOI] [PubMed] [Google Scholar]
- Noto Y-I, Shiga K, Tsuji Y, Mizuta I, Higuchi Y, Hashiguchi A, … Mizuno T (2015). Nerve ultrasound depicts peripheral nerve enlargement in patients with genetically distinct Charcot-Marie-Tooth disease. Journal of Neurology, Neurosurgery & Psychiatry, 86(4), 378–384. 10.1136/jnnp-2014-308211 [DOI] [PubMed] [Google Scholar]
- Perez-Olle R, Jones ST, & Liem RKH (2004). Phenotypic analysis of neurofilament light gene mutations linked to Charcot-Marie-Tooth disease in cell culture models. Human Molecular Genetics, 13(19), 2207–2220. 10.1093/hmg/ddh236 [DOI] [PubMed] [Google Scholar]
- Perez-Olle R, Leung CL, & Liem RKH (2002). Effects of Charcot-Marie-Tooth-linked mutations of the neurofilament light subunit on intermediate filament formation. Journal of Cell Science, 115(24), 4937–4946. 10.1242/jcs.00148 [DOI] [PubMed] [Google Scholar]
- Perez-Olle R, Lopez-Toledano MA, Goryunov D, Cabrera-Poch N, Stefanis L, Brown K, & Liem RKH (2005). Mutations in the neurofilament light gene linked to Charcot-Marie-Tooth disease cause defects in transport. Journal of Neurochemistry, 93(4), 861–874. 10.1111/j.1471-4159.2005.03095.x [DOI] [PubMed] [Google Scholar]
- Perrot R, Berges R, Bocquet A, & Eyer J (2008). Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Molecular Neurobiology, 38(1), 27–65. 10.1007/s12035-008-8033-0 [DOI] [PubMed] [Google Scholar]
- Perrot R, & Eyer J (2009). Neuronal intermediate filaments and neurodegenerative disorders. Brain Research Bulletin, 80(4–5), 282–295. 10.1016/j.brainresbull.2009.06.004 [DOI] [PubMed] [Google Scholar]
- Pisciotta C, Bai Y, Brennan KM, Wu X, Grider T, Feely S, … Shy ME (2015). Reduced neurofilament expression in cutaneous nerve fibers of patients with CMT2E. Neurology, 85(3), 228–234. 10.1212/WNL.0000000000001773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasure S, Lee V, & Nelson D (1990). Site-specific phosphorylation of the middle molecular weight human neurofilament protein in transfected non-neuronal cells. The Journal of Neuroscience, 10(7), 2428–2437. 10.1523/JNEUROSCI.10-07-02428.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainio MT, Ylikallio E, Mäenpää L, Lahtela J, Mattila P, Auranen M, … Tyynismaa H (2018). Absence of NEFL in patient-specific neurons in early-onset Charcot-Marie-Tooth neuropathy. Neurology Genetics, 4(3), e244 10.1212/NXG.0000000000000244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta MA, Dang V, Volfson D, Zou B, Xie XS, Adebola A, … Dimos JT (2015). Axonal Charcot–Marie–Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Experimental Neurology, 263, 190–199. 10.1016/j.expneurol.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta MA, & Shy ME (2013). Inherited Peripheral Neuropathies. Neurologic Clinics, 31(2), 597–619. 10.1016/j.ncl.2013.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarria a J. (1990). Regulated expression of vimentin cDNA in cells in the presence and absence of a preexisting vimentin filament network. The Journal of Cell Biology, 111(2), 553–565. 10.1083/jcb.111.2.553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Gotow T, Shiozaki M, Sakaue F, Saito T, Julien J-P, … Hisanaga S-I (2006). Aggregate formation and phosphorylation of neurofilament-L Pro22 Charcot–Marie–Tooth disease mutants. Human Molecular Genetics, 15(6), 943–952. 10.1093/hmg/ddl011 [DOI] [PubMed] [Google Scholar]
- Shin JS, Chung KW, Cho SY, Yun J, Hwang SJ, Kang SH, … Choi B-O (2008). NEFL Pro22Arg mutation in Charcot-Marie-Tooth disease type 1. Journal of Human Genetics, 53(10), 936–940. 10.1007/s10038-008-0333-8 [DOI] [PubMed] [Google Scholar]
- Sivera R, Sevilla T, Vilchez JJ, Martinez-Rubio D, Chumillas MJ, Vazquez JF, … Espinos C (2013). Charcot-Marie-Tooth disease: Genetic and clinical spectrum in a Spanish clinical series. Neurology, 81(18), 1617–1625. 10.1212/WNL.0b013e3182a9f56a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skre H (1974). Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clinical Genetics, 6(2), 98–118. 10.1111/j.1399-0004.1974.tb00638.x [DOI] [PubMed] [Google Scholar]
- Suter U, & Scherer SS (2003). Disease mechanisms in inherited neuropathies. Nature Reviews Neuroscience, 4(9), 714–726. 10.1038/nrn1196 [DOI] [PubMed] [Google Scholar]
- Timmerman V, Strickland A, & Züchner S (2014). Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes, 5(1), 13–32. 10.3390/genes5010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tradewell ML, Durham HD, Mushynski WE, & Gentil BJ (2009). Mitochondrial and Axonal Abnormalities Precede Disruption of the Neurofilament Network in a Model of Charcot-Marie-Tooth Disease Type 2E and Are Prevented by Heat Shock Proteins in a Mutant-Specific Fashion. Journal of Neuropathology and Experimental Neurology, 68(6), 642–652. 10.1097/NEN.0b013e3181a5deeb [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Kelsten ML, & Lee VM (1989). Phosphate-dependent and independent neurofilament protein epitopes are expressed throughout the cell cycle in human medulloblastoma (D283 MED) cells. The American Journal of Pathology, 135(4), 747–758. [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Alami NH, & Brown A (2009). Tight Functional Coupling of Kinesin-1A and Dynein Motors in the Bidirectional Transport of Neurofilaments. Molecular Biology of the Cell, 20(23), 4997–5006. 10.1091/mbc.e09-04-0304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Colakoglu G, Wang L, Monsma PC, & Brown A (2013). Severing and end-to-end annealing of neurofilaments in neurons. Proceedings of the National Academy of Sciences, 110(29), E2696–E2705. 10.1073/pnas.1221835110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Monsma PC, Fenn JD, & Brown A (2016). Live-cell imaging of neurofilament transport in cultured neurons. In Methods in Cell Biology (Vol. 131, pp. 21–90). 10.1016/bs.mcb.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ho C, Sun D, Liem RKH, & Brown A (2000). Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nature Cell Biology, 2(3), 137–141. 10.1038/35004008 [DOI] [PubMed] [Google Scholar]
- Werheid F, Azzedine H, Zwerenz E, Bozkurt A, Moeller MJ, Lin L, … Claeys KG (2016). Underestimated associated features in CMT neuropathies: clinical indicators for the causative gene? Brain and Behavior, 6(4), 1–14. 10.1002/brb3.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamichi-Nishina M, Ito T, Mizutani T, Yamamichi N, Watanabe H, & Iba H (2003). SW13 Cells Can Transition between Two Distinct Subtypes by Switching Expression of BRG1 and Brm Genes at the Post-transcriptional Level. Journal of Biological Chemistry, 278(9), 7422–7430. 10.1074/jbc.M208458200 [DOI] [PubMed] [Google Scholar]
- Yan Y, Jensen K, & Brown A (2007). The polypeptide composition of moving and stationary neurofilaments in cultured sympathetic neurons. Cell Motility and the Cytoskeleton, 64(4), 299–309. 10.1002/cm.20184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gu L-Q, Burnette WB, & Li J (2016). N98S mutation in NEFL gene is dominantly inherited with a phenotype of polyneuropathy and cerebellar atrophy. Journal of the Neurological Sciences, 365, 46–47. 10.1016/j.jns.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates DM, Manser C, De Vos KJ, Shaw CE, McLoughlin DM, & Miller CCJ (2009). Neurofilament subunit (NFL) head domain phosphorylation regulates axonal transport of neurofilaments. European Journal of Cell Biology, 88(4), 193–202. 10.1016/j.ejcb.2008.11.004 [DOI] [PubMed] [Google Scholar]
- Yoshihara T, Yamamoto M, Hattori N, Misu K, Mori K, Koike H, & Sobue G (2002). Identification of novel sequence variants in the neurofilament-light gene in a Japanese population: analysis of Charcot-Marie-Tooth disease patients and normal individuals. Journal of the Peripheral Nervous System, 7(4), 221–224. 10.1046/j.1529-8027.2002.02028.x [DOI] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Sasaki T, Chen Y, Kumar A, Veeranna, … Nixon RA (2006). Alpha-Internexin Is Structurally and Functionally Associated with the Neurofilament Triplet Proteins in the Mature CNS. Journal of Neuroscience, 26(39), 10006–10019. 10.1523/JNEUROSCI.2580-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Sasaki T, Kumar A, Peterhoff CM, Rao MV, Liem RK, … Nixon RA (2012). Peripherin Is a Subunit of Peripheral Nerve Neurofilaments: Implications for Differential Vulnerability of CNS and Peripheral Nervous System Axons. Journal of Neuroscience, 32(25), 8501–8508. 10.1523/JNEUROSCI.1081-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum SW, Zhang J, Mo K, Li J, & Scherer SS (2009). A novel recessive Nefl mutation causes a severe, early-onset axonal neuropathy. Annals of Neurology, 66(6), 759–770. 10.1002/ana.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai J, Lin H, Julien J-P, & Schlaepfer WW (2007). Disruption of neurofilament network with aggregation of light neurofilament protein: a common pathway leading to motor neuron degeneration due to Charcot–Marie–Tooth disease-linked mutations in NFL and HSPB1. Human Molecular Genetics, 16(24), 3103–3116. 10.1093/hmg/ddm272 [DOI] [PubMed] [Google Scholar]
- Zhao J, Brown K, & Liem RKH (2017). Abnormal neurofilament inclusions and segregations in dorsal root ganglia of a Charcot-Marie-Tooth type 2E mouse model. PLOS ONE, 12(6), e0180038 10.1371/journal.pone.0180038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S, Vorgerd M, Sindern E, & Schröder JM (2004). The novel neurofilament light (NEFL) mutation Glu397Lys is associated with a clinically and morphologically heterogeneous type of Charcot-Marie-Tooth neuropathy. Neuromuscular Disorders, 14(2), 147–157. 10.1016/j.nmd.2003.10.003 [DOI] [PubMed] [Google Scholar]