

Abstract
The etiological spectrum of ultra-rare developmental disorders remains to be fully defined. Chromatin regulatory mechanisms maintain cellular identity and function, where misregulation may lead to developmental defects. Here, we report pathogenic variations in MSL3, which encodes a member of the chromatin-associated male-specific lethal (MSL) complex responsible for bulk histone H4 lysine 16 acetylation (H4K16ac) in flies and mammals. These variants cause an X-linked syndrome affecting both sexes. Clinical features of the syndrome include global developmental delay, progressive gait disturbance, and recognizable facial dysmorphism. MSL3 mutations affect MSL complex assembly and activity, accompanied by a pronounced loss of H4K16ac levels in vivo. Patient-derived cells display global transcriptome alterations of pathways involved in morphogenesis and cell migration. Finally, we use histone deacetylase inhibitors to rebalance acetylation levels, alleviating some of the molecular and cellular phenotypes of patient cells. Taken together, we characterize a syndrome that allowed us to decipher the developmental importance of MSL3 in humans.
Introduction
Recent advances in whole-exome sequencing have led to the discovery of numerous causative genes for human diseases. However, gathering a large number of patients represents a significant bottleneck for gene identification and appropriate molecular analysis of rare developmental diseases. Mutations of epigenetic regulators were found to cause neurodevelopmental disorders through mechanisms potentially altering the chromatin landscape1. To date, over 40 Mendelian disorders have been identified that are caused by mutations of the epigenetic machinery and associated with neurological dysfunction, 80% of which are autosomally encoded2,3,4. Fluctuations in the histone acetylation balance, due to the interplay between acetylase and deacetylase functions during development, contributes to the final shape and performance of the adult brain and the whole organism5,6,7,8. While several modifications within the histone core were found to alter the chromatin architecture9, histone H4 lysine 16 acetylation (H4K16ac) appears uncommon among the tail modifications as it directly affects higher-order chromatin structure10. The male-specific lethal (MSL) complex-associated histone acetyltransferase KAT8, also known as MOF (males-absent on the first protein), is responsible for bulk H4K16ac in flies and mammals11. Given its crucial importance, we know surprisingly little about the role of MSL-mediated H4K16ac during development and whether its misregulation is associated with any disease in humans.
Results
Mutations in MSL3 cause a neurodevelopmental disorder
Through the international Matchmaker Exchange initiative12, we have identified de novo variants in MSL homolog 3 (MSL3; previously known as MSL3L1) RefSeq gene NG_012564.1) in 15 unrelated families from Europe, the USA, and Australia (Fig. 1a, b and Supplementary Fig. 1a). In the matchmaking cohort, we identified 11 truncating variants (p1–3, p5–11, and p13), one exon deletion (p12), one missense variant (p4), two gene deletions (p14–15) and one chromosomal inversion (p16) (Fig. 1c and Table 1). Patients 1–9 were gathered from a cohort of approximately 10,000 individuals with intellectual disability, demonstrating a significant enrichment for MSL3 de novo truncating variants in this cohort compared with random occurrence (uncorrected P = 8.98 × 10−16; false discovery rate (FDR) P = 1.64 × 10−11) (Supplementary Fig. 1b,c and Table 1). The enrichment for truncating variants in this cohort of patients is consistent with the data from the Exome Aggregation Consortium (ExAC) database, suggesting that MSL3 is extremely intolerant to loss-of-function variations (probability of being loss-of-function intolerant (pLI) = 0.95)13. For patients 5 and 6 (monozygotic twins), deep sequencing of leucocyte parental DNA did not detect an alternative allele. Patient 16 inherited the X-inversion, causing a putative in-frame transcript between MSL3 and GAB3 from his mildly affected mother (Table 1 and Supplementary Fig. 1d).
Figure 1. Mutations in MSL3 cause a novel neurodevelopmental disorder.
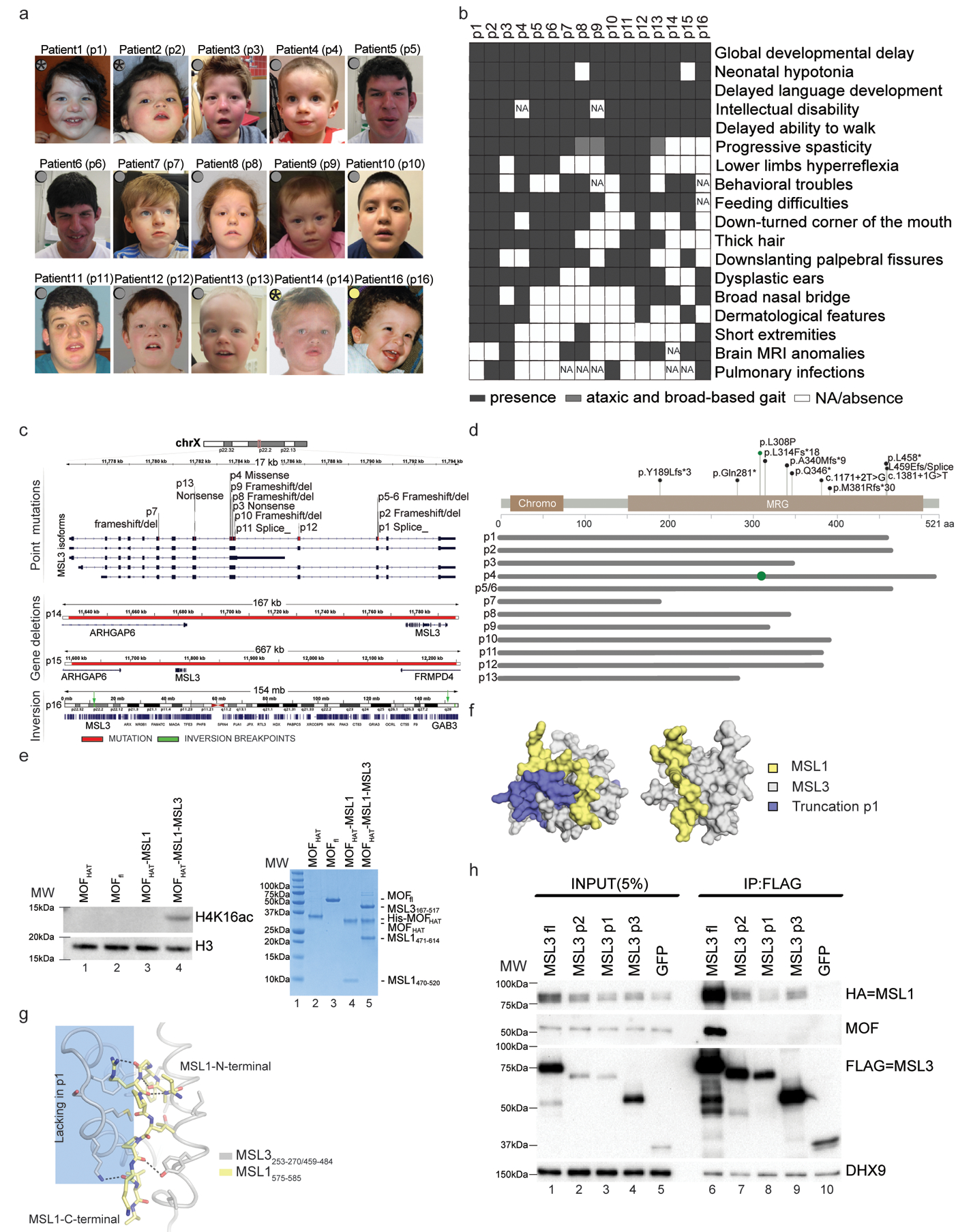
(a) Photographs of patients with MSL3 mutations. Gray circles indicate intragenic variants. Yellow circles indicate whole-gene deletions. Stars show the patients who donated skin biopsies. Written consent for the use of photographs was obtained from the parents of affected individuals. (b) Human Phenotype Ontology heat map of patients’ common clinical features. Blank boxes represent either absent or unreported symptoms. MRI, magnetic resonance imaging; NA, not analyzed. (c) The MSL3 locus and identified variants are highlighted in red. p1–13 variants occur within MSL3 (top). p13/p14 deletions span ARHGAP6, MSL3, and FRMD4 (middle). p16 chromosomal inversion and breakpoints (green) are between Xp22.2 and Xq28 (bottom). (d) Annotation of the pathogenic variants on the primary protein structure. Gray lines represent predicted proteins, where most SNVs (except for p.Leu308Pro; shown as green circle) result in MRG domain truncation. aa, amino acid. fs, frameshift. (e) Left, cropped H4K16ac and H3 immunoblot of a HAT assay performed on nucleosomal substrate with the indicated proteins (full-length (fl; 2–458) and HAT domain (174–458)). Right (lanes 2–5), Coomassie-stained gel of the assayed proteins. The experiment was repeated twice with similar results. MW, molecular weight (lane 1). (f) Modeled surface representation of the MSL1–MSL3 complex (Protein Data Bank: 2Y0N; MSL1, yellow; MSL3, gray). Missing parts in p1 are highlighted in purple (left) or removed (right). (g) Schematic representing the MSL1 (575–585, yellow) and MSL3 (253–270/459–484, gray) interaction interface. Hydrogen bonds are shown in black. α-helices 5/6, which are missing in p1, are shaded blue. (h) FLAG co-immunoprecipitations of FLAG-tagged full-length MSL3, MSL3 variants (p2, p1, and p3) and a GFP control with cropped immunoblots. MSL3-FLAG-tag, MSL3(p2,p1,p3)-FLAG-tag, GFP-FLAG-tag expression vectors were transiently co-transfected with MSL1-HA-tag expression vector into Flp-In T-Rex 293 cells. DHX9 serves as input loading control. The experiment was repeated 4 times with similar results.
Table 1.
MSL3 variants identified in the 16 reported individuals of this study.
| Genomic position hg19 |
cDNA NM_078629.3 |
Protein Q8N5Y2–1 |
Inheritance | |
|---|---|---|---|---|
| Patient 1 | chrX:g.11790375G>T | c.1381+1G>T | NA | de novo |
| Patient 2 | chrX:g.11790367_11790374del | c.1374_1381del | p.(Leu459Glufs*13) | de novo |
| Patient 3 | chrX:g.11783713C>T | c.1036C>T | p.(Gln346*) | de novo |
| Patient 4 | chrX:g.11783600T>C | c.923T>C | p.(Leu308Pro) | de novo |
| Patient 5 | chrX:g.11790365C>T | c.1372C>T | p.(Arg458*) | de novo – Parental mosaicism |
| Patient 6 | chrX:g.11790365C>T | c.1372C>T | p.(Arg458*) | de novo – Parental mosaicism |
| Patient 7 | chrX:g.11780349_11780350delAC | c.566_567delAC | p.(Tyr189Leufs*3) | de novo |
| Patient 8 | chrX:g.11783695delG | c.1018delG | p.(Ala340Leufs*9) | de novo |
| Patient 9 | chrX:g.11783615dupC | c.938dupC | p.(Leu314Phefs*18) | de novo |
| Patient 10 | chrX:g.11783802_11783818dup17 | c.1125_1141dup17 | p.(Met381Argfs*30) | de novo |
| Patient 11 | chrX:g.11783850T>G | c.1171+2T>G | N/A | de novo |
| Patient 12 | Del exon 10 | . | . | de novo |
| Patient 13 | chrX:g.11781990C>T | c.841C>T | p.(Gln281*) | de novo |
| Patient 14 | chrX:g.11633731_11797224del | . | . | de novo |
| Patient 15 | chrX:g.11600773_12249902del | . | . | NA – Father not tested, absent in the Mother |
| Patient 16 | Inv(X)(p22.2q28) | . | . | Inherited from the mother – de novo in the mother |
NA: Not available.
MSL3 is known for its role during X chromosome dosage compensation via H4K16ac in Drosophila. Its absence affects male flies and manifests in lethality in the last larval stage of development11,14,15. The human MSL3 gene resides on the X chromosome and its mutations presented here affect both hemizygous male and heterozygous female individuals (Fig. 1a and Supplementary Table 1). Common features across the 16 patients include hypotonia, feeding difficulties in early infancy, and global delay in the acquisition of major milestones including walking and speech. Intellectual disability was also scored. The phenotypic spectrum is more homogeneous across patients of both sexes with single nucleotide variants (SNVs) compared with patients with chromosomal rearrangements. The patients with SNVs display progressive spasticity or an ataxic gait (9/13), resulting in the inability to walk (5/13). The facial morphology strongly overlaps across patients with telecanthus (8/13), epicanthal folds (9/13), down-slanting palpebral fissures (9/13), downturned corners of the mouth (8/13), and dysplastic ears (11/13) (Fig. 1b and Supplementary Table 1; see also the detailed clinical descriptions in Supplementary Note 1). Together, these clinical observations delineate a recognizable MSL3 syndrome. In line with these phenotypes, MSL3 is ubiquitously expressed in the developing human brain until 17–20 weeks post-conception. Other MSL complex members display a similar expression pattern, although their RNA levels seem slightly higher (Supplementary Fig. 1e)16.
Most of the pathogenic MSL3 variants affect the carboxy (C)-terminal MRG domain (Fig. 1d and Supplementary Fig. 1f), which is responsible for MSL complex formation while enhancing the activity of MOF towards H4K16ac (Fig. 1e)17,18.
Using in silico modeling of the crystal structure of the human MSL3–MSL1 complex17, we predicted that the truncations present in the patients impair the integration of mutant MSL3 into the MSL complex (Fig. 1f, g). To test this, we transiently transfected expression vectors encoding wild-type (full-length) MSL3 and three different MSL3 mutants, representing patients 1, 2, and 3 (p1, p2, and p3), into Flp-In T-REx 293 cells. We observed that in contrast with wild-type MSL3, which showed robust interaction with MSL1 and MOF, all three mutants lost the interaction with MOF and, to a lesser extent, with MSL1 (Fig. 1h, lanes 6–9). Two of the mutations also decreased MSL3 protein stability (Fig. 1h, lanes 2 and 3). Taken together, we find that the mutations present in the patients with MSL3 syndrome lead to severely compromised MSL complex integrity.
Bulk reduction of H4K16ac in cells from patients with MSL3 mutations
To characterize the molecular consequences of MSL3 mutations in patients, we obtained skin biopsies from two female patients harboring frameshift mutations (p1 and p2), one female patient with a locus deletion (p14), and one control tissue from p1’s healthy mother (referred to as Control). In addition, we analyzed two more skin biopsies from healthy donors as controls (referred to as Control 2 and Control 3, see Methods). These samples were used to perform histological analyses and establish primary human dermal fibroblast (HDF) cell culture models for all molecular analyses presented below.
Hematoxylin and eosin staining confirmed that the overall tissue architecture was preserved in both Control and patient specimens (Supplementary Fig. 2a). The MSL complex is responsible for bulk H4K16ac in flies19,20 and mammalian cell culture models14,15,21. Based on this, we performed immunostaining of H4K16ac in patient samples compared with skin punches of Controls. While the H4K16ac staining had homogeneous intensity in dermal and epidermal layers of all three control samples, MSL3 patient cells showed a heterogeneous pattern with a marked and variable reduction in H4K16ac (Fig. 2a, asterisks). This striking reduction of H4K16ac could result from decreased levels of MOF in MSL3 patient cells. However, MOF immunostaining displayed a homogenous distribution within all cells of the tissue section (Fig. 2b). This indicates that the loss of H4K16ac is not caused by the reduction in MOF levels per se. Rather, it is due to a misregulation of its enzymatic activity.
Figure 2. Bulk reduction of H4K16 acetylation in MSL3 patient cells.
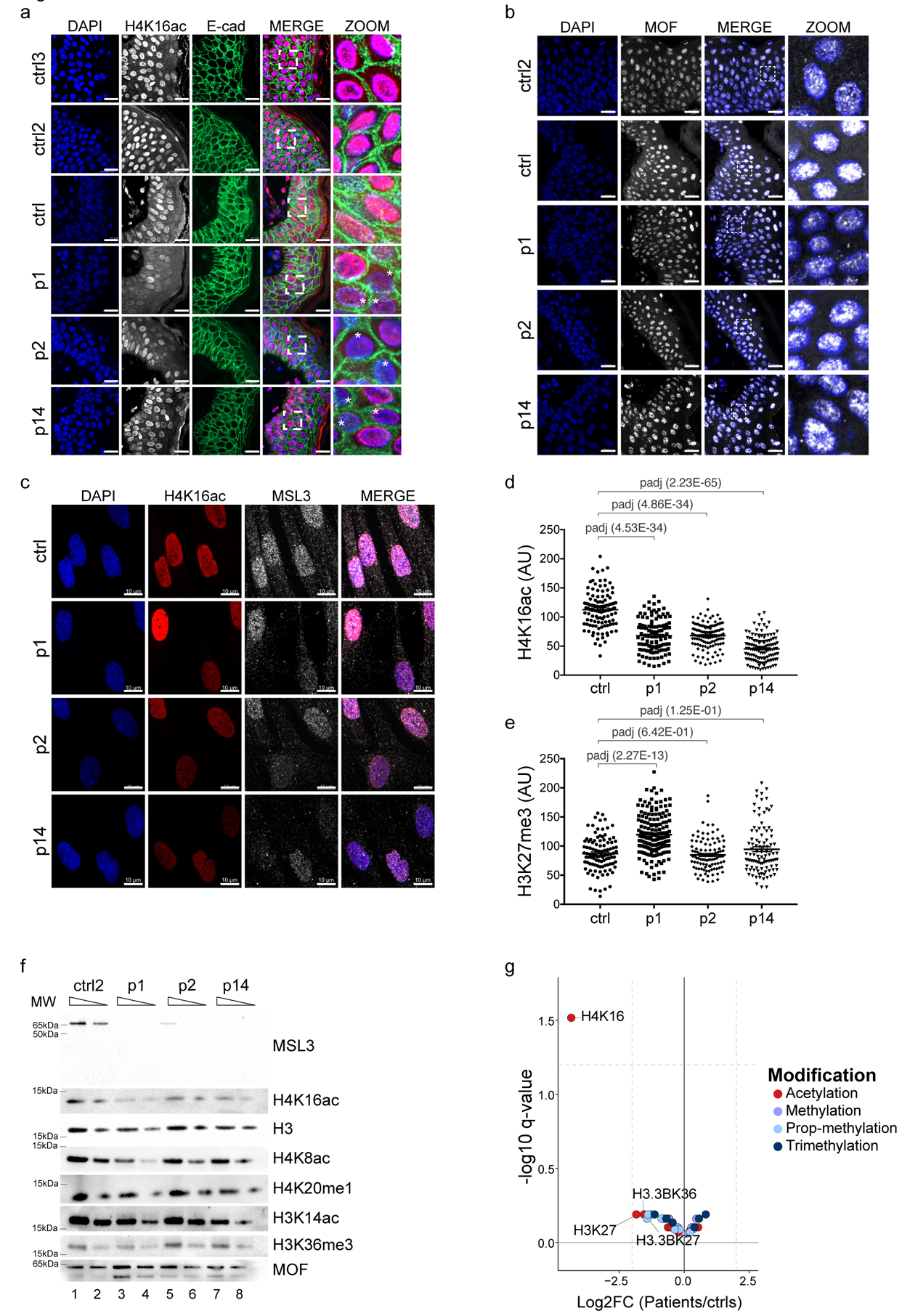
(a) Immunohistochemistry of FFPE skin sections for H4K16ac (white, but pink in the merged images) and E-cadherin (E-cad, green). DAPI staining is shown in blue. Asterisks highlight cells with reduced H4K16ac staining. Scale bars = 20 μm, the pictures in the final column (Magnified) represent a 4.4× magnification of the merged column pictures. Staining was repeated twice with similar results. (b) As in a, but for MOF (white). The pictures in the final column (Magnified) represent a 5.1× magnification of the merged column pictures. c, Immunostaining of the Control and p1-, p2-, and p14-derived primary HDFs for H4K16ac (red) and MSL3 (white). Scale bar = 10 μm. The staining was repeated twice (n = 2 sections per slide). (d,e) Quantification of staining intensities for H4K16ac (d) and H3K27me3 (e) from one representative experiment for the Control and p1, p2, and p14 HDFs. Each data point corresponds to the intensity of a single cell with the center line representing the mean ± s.e.m. P values were determined by ordinary one-way analysis of variance followed by Bonferroni correction. (f) Cropped immunoblot of HDF nuclear extracts. 100 and 50% dilutions were loaded. The experiment was reproduced three times. MW, molecular weight. (g)Volcano plot of the indicated histone modification abundance displayed as log2[fold-change] (x axis, p1, p2, and p14 HDFs (n = 6; 2 for each patient) compared with the Control HDFs (n = 3)) versus the statistical significance (y axis, two-sided, moderated t-test) as determined by LC-MS. Vertical dashed lines represent fourfold changes, and the horizontal dashed line shows a −log10[q value] of 1.2, Benjamini–Hochberg-corrected FDR of 0.05. Red circles show acetylated sites, violet circles show monomethylated sites, light blue circles show propionyl methylated sites, and dark blue circles show trimethylated sites. Detailed results of the statistical analyses related to d and e are provided in Supplementary Table 5, and those for g are presented in Supplementary Table 2.
We confirmed these observations in primary fibroblast cells established from skin punches, where cells lacking functional MSL3 also displayed a severe decrease in H4K16ac (Fig. 2c, d), but not in other histone modifications tested (for example, H3K27me3; Fig. 2e and Supplementary Fig. 2b). Remarkably, quantification of these stainings showed a global reduction of H4K16ac in these heterozygous XMSL3-WT/XMSL3-MUT female patient samples (Fig. 2d). Such a staining pattern is unusual for an X-linked gene in females, where conventionally, heterozygous mutations would be expected to result in a 50:50 population of wild-type and mutant cells. Targeted sequencing of MSL3 complementary DNA (cDNA) for patients 1 and 2 demonstrated mild co-expression of the mutated and wild-type allele at the RNA level (Supplementary Fig. 2d). While the nature of MSL3 mutation in p14 (whole gene deletion) precludes the expression of any mutant MSL3 protein, we note that P1/2 proteins were highly unstable (Fig. 1h), making it unlikely that the MSL3-linked syndrome is generally caused by a dominant negative effect. However, human MSL3 was recently found to be a facultative escapee22 that is stochastically and variably expressed from both female X chromosomes. We therefore propose that MSL3 haploinsufficiency might cause the variable but consistent reduction of H4K16ac in female patients.
Since there could be potential crosstalk between H4K16ac and other residues on the histone tails23, and given that MSL3 has been linked to H3K36me3 and H4K20me124,25, we validated the above findings by bulk western blot analyses and quantitative liquid chromatography–mass spectrometry (LC-MS). In line with the immunofluorescence, we exclusively scored a reduction of MSL3 and H4K16ac levels, while MOF and other histone modifications remained unaffected in western blots (Fig. 2f, and Supplementary Figs. 2b and 5). Affirming these results, when comparing the three patients with two different controls by comprehensive histone LC-MS analysis, the modification that was by far the most significantly affected—both by P value and fold-change—was H4K16ac (Fig. 2g Supplementary Fig. 2e, f and Supplementary Table 2). Hence, loss of MSL3 in patients appears to primarily affect H4K16ac.
H4K16ac has previously been linked to cell-cycle regulation21,26,27,28. We noticed that patient-derived fibroblasts grew significantly slower than Control cells (Supplementary Fig. 2g). Nevertheless, no clear difference of any specific cell-cycle phase could be scored (Supplementary Fig. 2h). Cells did not show signs of G1/S arrest either by reverse transcription quantitative PCR (RT-qPCR) for senescence markers, or by X-gal staining (Supplementary Fig. 2i,j). Furthermore, no differences were scored for γH2AX signal, indicating no apparent increase in DNA damage (data not shown).
To further dissect the possible cause for the slow growth of female MSL3 mutant cells, we next challenged them by the removal of serum and tested their capacity to respond to changes in environmental conditions. Interestingly, on performing these starvation experiments, we observed that patient-derived fibroblasts showed a delay in exiting the G0 phase (Supplementary Fig. 2k). This suggests that MSL3 mutant cells may fail to timely respond to growth factor signals—a property that is crucial during development.
Transcriptome deregulation in cells from patients with MSL3 mutations
Since MSL3 is part of a chromatin-modifying complex involved in transcription regulation21,29,30, we next performed RNA sequencing (RNA-Seq) experiments on three patient-derived primary fibroblast cell lines. For this purpose, we analyzed two different passages compared with a passage-matched Control in synchronized conditions. Differential expression analysis was performed using DESeq2, and genes with an FDR < 0.05 were considered differentially expressed (Fig. 3a). As gene expression differences can reflect genetic background or environmental influences between individuals31, we decided to consider only transcripts that were consistently deregulated in all three patients. This identified a set of 519 commonly misregulated genes (Fig. 3a,b and Supplementary Fig. 3a).
Figure 3. Loss of MSL3 leads to transcriptome misregulation of developmental pathways.
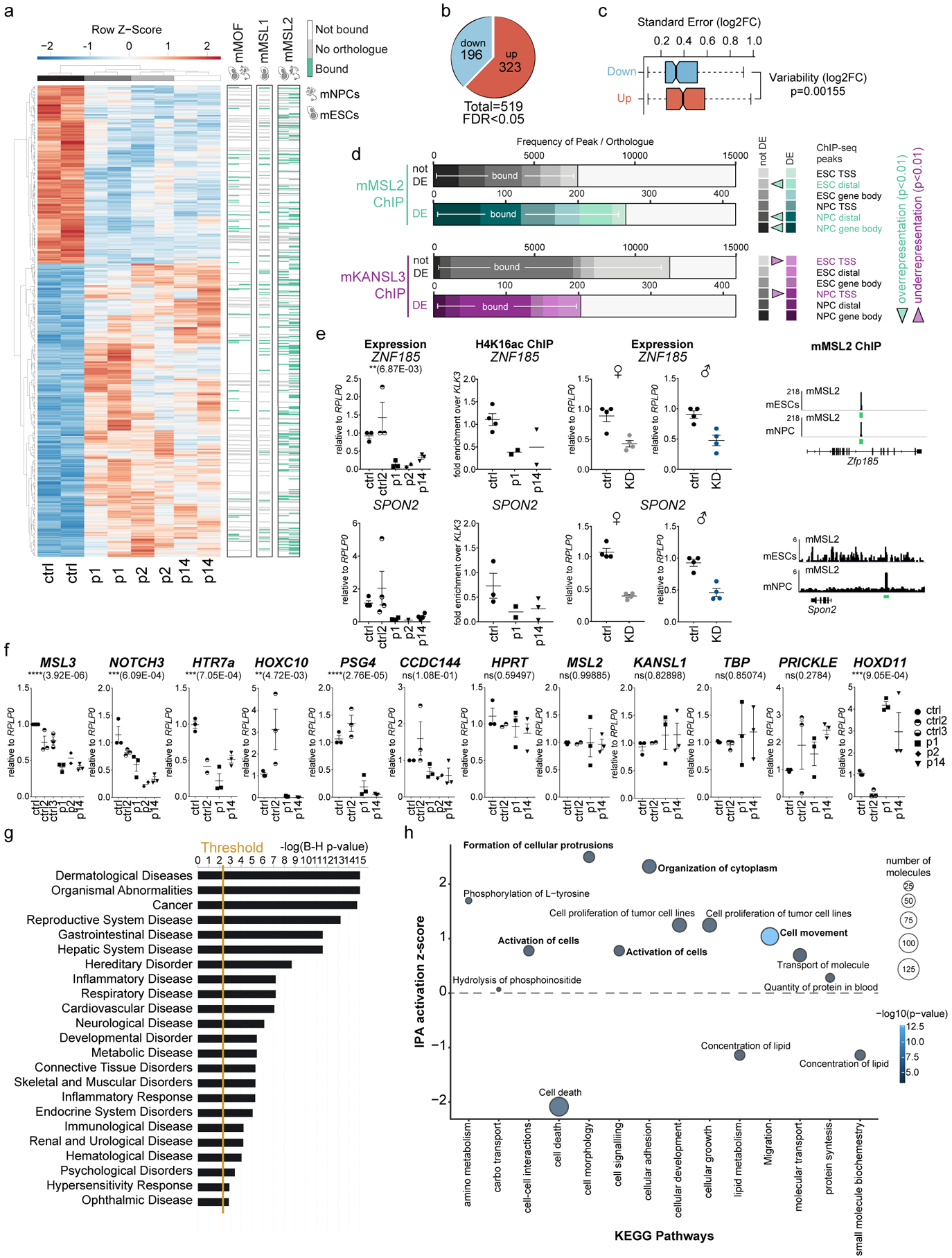
(a) (Left) Heat map showing the z-scores of differentially expressed (DE) genes (p1, p2, and p14 versus Control) obtained by RNA-Seq (FDR 0.05). Right, MACS2 ChIP-Seq21 peaks for mouse MSL1 (mMSL1), mouse MSL2 (mMSL2) and mouse MOF (mMOF) at mouse orthologs of MSL3 patient DE genes (green, bound; white no peak; gray, no ortholog) in mouse embryonic stem cells (mESCs) and mouse neural progenitor cells (mNPCs). (b) Pie chart of DE genes. (c) Box plot displaying DESeq2 log2[fold-change] s.e. for the DE downregulated (n = 196) versus DE upregulated genes (n = 323). The P value was calculated using a two-sided Welch two-sample t-test. (d) ChIP-Seq peak frequency plot for mouse mMSL2 or mouse KANSL3 (mKANSL3) at unaffected/not DE versus misregulated/MSL3 DE genes in patients. P values for ChIP peak over-representation or under-representation within the groups were calculated using a one-sided Fisher’s test. TSS, transcription start site. (e) Dot plots (center line representing the mean± s.e.m.) of expression and H4K16ac ChIP-qPCR analyses. Expression levels were determined in controls and patients and on MSL3 knockdown (KD) in male (♂) and female (♀) control HDFs. H4K16ac ChIP-qPCR enrichment values were calculated relative to input and expressed as the fold-change over KLK3. Right, genome browser snapshots of mMSL2 ChIP-Seq enrichment at corresponding mouse orthologs. The y-axis represents 1× sequencing depth normalized read densities (number of reads per bin/(total number of mapped reads * fragment length / effective genome size). (f) RT-qPCR expression analysis displayed as dot plots with the center line representing the mean ± s.e.m. Expression levels were normalized to RPLP0 and expressed relative to the Control. NS, not significant. P values were determined by ordinary one-way analysis of variance followed by Bonferroni correction. (g) IPA z-score plot for the disease and function signature of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Padj < 0.005 cut-off). Benjamini–Hochberg multiple testing correction was applied (n = 519 DEgenes). The vertical gold line shows the threshold. (h) Bubble plot representing perturbation pathway analysis for molecular and cellular functions. The bubble size shows the molecule abundance, while the color represents the IPA z-score (see scale bar). The most representative sub-pathway by number of molecules was selected to represent the full signature (n = 477 transcripts). For the dot plots in (e,f), each dot represents an independent experiment in HDFs. Further details and statistical test values for each plot are provided in Supplementary Table 5.
We first explored whether altered expression of these genes could be directly related to MSL-mediated H4K16ac. We noticed that the extent of misregulation (log2[fold-change]) was much more consistent between different patients in the downregulated subset compared with the upregulated group of genes (Fig. 3a,c). This supports the idea that the upregulated group of genes arise from secondary responses within each individual, which may be more variable. We also found that the orthologs of the differentially expressed genes in patient cells were significantly enriched for mMSL2 chromatin immunoprecipitation (ChIP) peaks in mouse cells21. In contrast, mKANSL3 peaks were significantly under-represented, corroborating the hypothesis that the observed effects are caused by H4K16ac deposited by the MSL complex, but not the KANSL complex (Fig. 3a,d). Therefore, we performed H4K16ac ChIP experiments in patient cells and focused on conserved mammalian MSL targets that are mMSL2 bound in mouse cells and very consistently downregulated in both patient cells, as well as on MSL3 knockdown in male and female fibroblasts (Fig. 3e). These results substantiated our conclusion, as H4K16ac was lost from ZNF185 and SPON2, but not H3F3B (Fig. 3e and Supplementary Fig. 3b).
We next focused on analyzing the biological functions and pathways orchestrated by the differentially expressed genes in the MSL3-linked syndrome. Intriguingly, we found differentially expressed Notch, Hox and Wnt family members, which are key morphogenetic regulators of development from Drosophila to mammals. We confirmed these findings by RT-qPCR, for which we included several additional control cell lines (Control 2 and Control 3). Remarkably, patient cell lines showed very consistent behavior in the selected set of downregulated as well as unaffected control genes (Fig. 3f, Supplementary Fig. 3c, and Supplementary Table 5). We could also confirm the reduction at the protein level for HTR7 (5-hydroxytryptamine receptor 7) and UNC5B (netrin receptor UNC5B) in the patient skin specimens (Supplementary Fig. 3d).
Pathway perturbation analysis highlighted ‘dermatological diseases’, ‘organismal abnormalities’, ‘gastrointestinal disease’, and ‘neurological disease’ as top-scoring disease signatures (Fig. 3g). More specifically, the genes commonly affected on MSL3 mutation are overall involved in the activation of cellular movement, organization of the cytoskeleton, or the formation of cell protrusions, while mechanisms to reduce cell death are repressed (Fig. 3h). These are all characteristic symptoms of the MSL3-linked syndrome underscoring the relevance of our RNA-Seq dataset (see Supplementary Table 3 for differentially expressed and clinically relevant genes based on patient phenotypes).
Next, we were interested in dissecting the primary gene expression perturbations caused by acute loss of MSL3 versus secondary effects that may accumulate in patients during development. Therefore, we performed MSL3 knockdown experiments in control primary fibroblast cell lines, as well as MSL3 rescue experiments in patient cells, to determine the extent to which we could phenocopy the gene expression changes detected by RNA-Seq. We observed an acute downregulation of multiple target genes (for example, ZNF185, SPON2, NOTCH3, and FAM157) on MSL3 knockdown in both male and female cells (Supplementary Fig. 3e). Genes found to be upregulated in patient cells typically did not respond (for example, PRICKLE) or were sometimes downregulated (WNT2). When we performed the inverse experiment by providing MSL3 in patient cells, we noted a similar trend (Fig. 4a). We could rescue several downregulated targets (ZNF185, SPON2, FOSB, and EGR2) by re-expressing MSL3. Interestingly, these genes were also upregulated in Control cells, indicating that they might be more generally susceptible to alteration of MSL3 and H4K16ac levels. Taken together, our gene expression analysis suggests that a lack of the MSL complex results in acute downregulation of several biologically relevant genes, including key developmental regulators. This misregulation may then gradually trigger secondary perturbances, collectively resulting in the global disease state of the MSL3 syndrome.
Figure 4. HDAC inhibition alleviates cellular and molecular phenotypes of MSL3 patient cells.
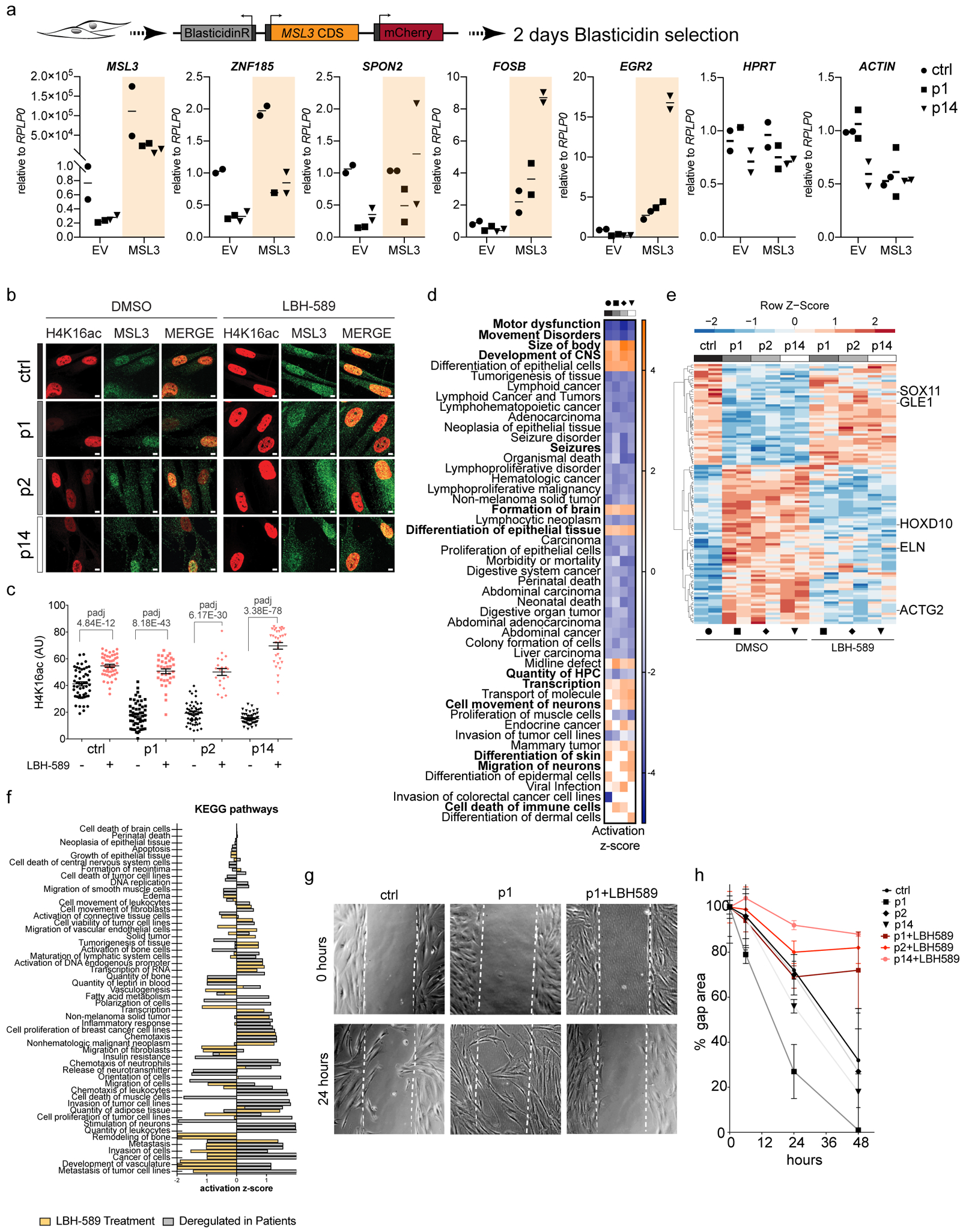
(a) MSL3 and empty expression vector (EV) were transfected in Control and p1/p14 HDFs. Dot plots depict RNA levels from each independent experiment normalized to RPLP0 relative to the EV. Center lines represent the mean (n = 2). CDS, protein coding sequence; BlasticidinR, blasticidin S resistance gene. (b) Immunofluorescence for H4K16ac (red) and MSL3 (green) on treatment with 4 μM LBH-589. Scale bar = 5 μm. The staining was reproduced twice. (c) Quantification of b. Data points represent H4K16ac intensities of individual cells from one representative experiment. Center lines represent the mean± s.e.m. P values were determined by ordinary one-way analysis of variance followed by Bonferroni correction. Detailed results of the statistical analyses are shown in Supplementary Table 5. (d) Heat map of activation z-scores obtained from IPA (comparison analysis; disease and function signature (n = 3,391 for the Control; n = 2,740 for p1; n = 2,487 for p2; n = 2,737 for p14; Padj < 1E−4 for differentially expressed genes on LBH-589 treatment (2 μM)). Activated pathways were plotted for |±z| > 2.5 and a log10[P value] cut-off of 1.5. Bold type shows the pathways impacting on patient cell physiology. CNS, central nervous system. Black circle, Control; square, p1; rhombus, p2 and triangle, p14. (e) Heat map representing z-scores of differentially expressed genes reverting to not differentially expressed on LBH-589 treatment (n = 109). Five disease-relevant transcripts are highlighted. Black circle, Control; square, p1; rhombus, p2 and triangle, p14. (f) IPA comparison analysis of KEGG pathway z-scores in genes rescued on LBH-589 (n = 109 with an expression cut-off of log ratio > |±0.2|). (g) Representative differential interference contrast images of HDFs at 0 and 24 h after creating a ‘scratch’ (gap area). (h) Quantification of the percentage gap area at 0, 6, 24, and 48 h for Control and patient HDFs grown with or without LBH-589 (2 μM). The center line represents the average ± s.e.m. (n = 3).
Histone deacetylase inhibitor (HDACi)-mediated alleviation of phenotypes in patient cells
Given the clear link between H4K16ac and MSL3 mutation, we considered whether it would be possible to rescue the phenotypes of patient cells not only by reintroducing MSL3 (Fig. 4a) but also by readjusting acetylation levels. We therefore tested three different classes of HDACi that are currently approved for clinical trial phase II or III (SAHA (suberoylanilide hydroxamic acid; also known as vorinostat)32,33, LBH-589 (panobinostat)34, and MGCD0103 (mocetinostat)35, as well as trichostatin A as a positive control36,37. These inhibitors show strong effects on histone H4 acetylation and, in particular, H4K16ac in human transformed cells37 (Supplementary Fig. 4a). Hence, we referred to the aforementioned study for the use of HDACi to modulate H4K16ac in our primary untransformed cells.
Treatment of Control and patient cells with HDACi resulted in bulk histone hyperacetylation and triggered upregulation of acetylation-sensitive genes (Supplementary Fig. 4b–d). We first verified that the response to HDACi is effective in all mutant cells, irrespective of the MSL3/H4K16ac levels. We therefore performed immunostainings on HDACi-treated patient cells and observed an overall increase of H4K16ac at the single-cell level from both high and low MSL3 populations (Fig. 4b). Quantification of these stainings in patient-derived cells confirmed the rescue of H4K16ac, which was now comparable to the healthy Control (Fig. 4c). Next, we tested whether this has a positive influence on gene expression. Remarkably, treatment with HDACi specifically rescued the expression level of selected downregulated targets, while control genes or upregulated targets remained largely unchanged (Supplementary Fig. 4e; other data not shown). Unlike p1, p2 and p14 patient cells responded more selectively to LBH-589, while the other two HDACi (SAHA and MGCD0103) showed only a modest rescue of gene expression defects (Supplementary Fig. 4e). To characterize the HDACi response in MSL3-deficient cells in a more comprehensive manner, we performed RNA-Seq on treatment with LBH-589. As expected, HDACi treatment caused major transcriptional changes (Supplementary Fig. 4f and Supplementary Table 4). However, we successfully observed previously detected pathways, as well as potential side effects of this drug in Control and patient cells38 (Fig. 4d and Supplementary Table 4). This supports the notion that the primary HDF cell lines used in our analysis represent a relevant model for studying the MSL3-linked syndrome.
Interestingly, a large set of the differentially expressed genes reversed their expression toward normal levels on HDACi treatment, although the extent varied between different genes and patients (Supplementary Fig. 4g and Supplementary Table 3). We focused our attention on the 116 genes that were fully rescued to control levels and hence did not score as differentially expressed after panobinostat treatment (Fig. 4e). Among them, clinically relevant genes based on the patient phenotypes were scrutinized. One would expect a positive clinical impact by restoring a basal expression of, for example, SOX11 (OMIM 615866), HOXD10 (OMIM 142984), LEP (OMIM 164160) or ACTG2 (OMIM 102545) based on their role in human disease and patient symptoms, including cognitive performance, gait disturbance, being overweight, and feeding difficulties/constipation, respectively. Accordingly, KEGG pathway analysis indicated that HDACi may generally rewire deregulated processes relevant to the disease state of these patients (Fig. 4f and Supplementary Table 3).
Hence, we wondered whether we would be able to rescue the previously observed cell-cycle phenotype and migratory signature using HDACi. Indeed, genes involved in G0 arrest (for example, GAS1), were rescued toward the expression levels in Control cells on HDACi treatment (Supplementary Fig. 4h), while displaying a slight increase in G1/S arrest. Moreover, in agreement with the activation of the respective cellular pathways (Fig. 3h), we had observed that MSL3 patient-derived fibroblasts migrate faster than Control cells. Ergo, we assessed whether the positive influence of HDACi on gene expression would also lead to a beneficial outcome on a cellular level. Strikingly, on HDACi treatment, we could restore the phenotype of MSL3 mutant cells, as they now adopted the migratory behavior of the Control cells (Fig. 4g, h and Supplementary Fig. 4i). Therefore, HDACi have the potential to both molecularly as well phenotypically alleviate at least some of the defects in MSL3 mutant cells, thus providing a promising therapeutic avenue for these patients.
Discussion
In summary, we report a syndrome where loss of MSL3 is associated with a pronounced loss of bulk H4K16ac. The patients presented in our study display a complex phenotype including neurodevelopmental delay, with a recurrent facial dysmorphism and a progressive neurological disorder. This suggests that MSL-mediated H4K16ac in humans may be involved in the regulation of complex developmental processes; for example, those occurring in the brain5,39,40. This is intriguing as it is in contrast with the sex-specific function of MSL3 during dosage compensation in flies, suggesting a more context-specific role in mammals. Given the conserved role of H4K16ac in chromatin decompaction from yeast to humans, it remains to be demonstrated why its physiological roles evolved to be so organism specific. Our study also raises a general question about the role of H4K16ac, as MSL3 patients survive to adulthood compared with the early embryonic lethality of the Mof mutant in mice41,42,43,44. An important point to consider in this context is that although MSL3 mutations lead to a severe loss of H4K16 acetylation, remaining H4K16ac may be sufficient to allow developmental progress to a certain extent, since MOF is still expressed in these cells. While no MOF variants have been reported in any developmental disorder yet, de novo heterozygous loss-of-function variants of KANSL1 were reported to cause Koolen–de Vries syndrome (KdVS)45,46,47. KANSL1 encodes a scaffold component of the MOF–KANSL complex. However, the disease characteristics of KdVS are symptomatically and molecularly distinct from patients with MSL3 mutations with regard to features of facial dysmorphism—specifically, palpebral fissure orientation, nose tip and mouth shape, and visceral and brain abnormalities, which are absent in patients with MSL3 mutations. Although KdVS patients also display neuromuscular disorders, the progressive gait disturbance observed in the MSL3-linked syndrome may be a distinctive feature. Given the multifaceted and widespread roles of MOF in processes such as metabolism44, autophagy48, and stress response43, we speculate that the MSL3 mutations presented here may provide separation-of-function alleles, allowing us to dissect the role of altered H4K16ac deposited by the MOF–MSL complex versus the MOF–KANSL complex during human development.
The MSL3-linked syndrome affects both hemizygous males and heterozygous females with comparable symptoms, which is difficult to reconcile with classical models of X-chromosomal inheritance49. More recently, it has been appreciated that X-linked disorders can display intermediate penetrance; for example, due to varying degrees of X chromosome inactivation in females50. This includes variations in escape genes, such as IQSEC251, or examples like XLH or E1α, where similar phenotypes can be observed in both heterozygous females and hemizygous males. For MSL3, this can be presumably linked to its property of being a facultative escapee, where various degrees of X chromosome inactivation and escape might provide an explanation for the phenotypic and molecular differences among female patients. This is notable for the mother of p16, who is a carrier of the disease-causing allele, but is only mildly affected. The nature of the MSL3 variants, as well as environmental effects modulating epigenome plasticity, might additionally contribute to the phenotypic outcome of the syndrome. A comprehensive assessment of the allele-specific expression of MSL3 in different types of tissues, and the establishment of alternative cellular and animal models, will be required to dissect the apparently very complex regulation and phenotypic outcome in the MSL3-linked syndrome in both sexes.
Lastly, the positive outcome of HDACi partially alleviating migratory and cell-cycle defects provides a preliminary, yet promising ground for future studies, especially in light of the degenerative nature of the MSL3-linked syndrome. More efforts will need to be undertaken to potentially ameliorate the symptomatology and delay the probable myelopathy-caused hyperreflexia, for which HDACi have already shown positive results52. The possibility of alleviating at least some of the symptoms of these patients through repurposing of anticancer drugs provides yet another case where epigenetic therapy using HDACi may pave ways for treatments of other diseases affecting epigenetic modifiers. Taken together, our characterization of this syndrome not only provides insights into the developmental roles of MSL-associated H4K16ac in humans, but also underscores the general relevance of studying sex-specific aspects related to X-linked diseases.
Methods
Subjects, whole-exome sequencing, and variant validation
Sixteen individuals from 15 unrelated families with a disease-causing variant in MSL3 were included in this study after we obtained informed consent. The local ethics committee of the Dijon University Hospital approved this study (2016-A01347–44). The patients were followed up by clinical geneticists from France, the United Kingdom, the Netherlands, Belgium, Denmark, Germany, the US, Estonia, and Australia. Thirteen variants were identified by whole-exome sequencing for the diagnosis of an undiagnosed rare disorder associated with multiple congenital anomalies and syndromic intellectual disability. Two deletions were identified by array comparative genomic hybridization and SNP array, and the X chromosome inversion breakpoint in MSL3 was mapped by fluorescent in-situ hybridization and cloning of the junction fragment. The clinical observations were gathered through the Matchmaker Exchange initiative.
Blood or DNA samples from affected individuals and their parents, as well as informed consent, were obtained from all participants in accordance with the site-specific institutional boards of each country. Dedicated consent for the collection of skin biopsies was obtained.
De novo variants were analyzed by PCR and direct Sanger sequencing using DNA from patients and their parents. Primer sequences and detailed methods are available on request. In the family with two affected siblings (p5 and p6) and suspected parental somatic mosaicism, confirmation and estimation of the percentage of cells bearing the variant was performed using DNA extracted from the peripheral blood and saliva of all members of the family (the two patients and parents). Targeted sequencing for the variant position was performed on a MiSeq instrument (Illumina) at depths of 1,706 and 1,398 folds for the variant position in the father and mother, respectively. Genotyping identified 12 and 6 variant alleles, resulting in allelic balances of 0.007 and 0.004, respectively. In p1, the splice variant analysis was conducted on total RNA extracted from peripheral blood with TRIzol (Life Technologies). Sanger sequencing confirmed the splicing defect. Written consent was obtained from the parents and/or guardians of the affected individuals both for participation in the study and for use of the clinical photographs in this report.
Enrichment of de novo MSL3 variants in the matchmaker cohort
The statistical significance after Benjamini–Hochberg correction for enrichment of functional and/or loss-of-function de novo mutations in the matchmaker cohort was estimated as previously described53. The gene-specific mutation rates were published54. Because MSL3 is X linked, we assumed that the matchmaker cohort had an equivalent proportion of males and females (5,000 each; 15,000 alleles). The uncorrected P value was corrected for multiple testing, taking 18,280 genes to apply the Benjamini–Hochberg FDR correction.
Exome analysis
Each collaborating team followed a published methodology for sample and library preparation, sequencing data production, analysis, and interpretation55,56,57,58,59.
Parental segregation analysis
Genomic DNA was amplified by PCR using a HotStarTaq PCR kit (Qiagen). PCR products were purified using an Agencourt CleanSEQ system (Beckman Coulter) and sequenced with a BigDye Terminator Cycle Sequencing kit (version 3.1; Applied Biosystems) on an ABI 3730 sequencer (Applied Biosystems). Sequence data were analyzed using Mutation Surveyor version 4.0.9 (SoftGenetics).
MiSeq cDNA analysis
Total RNA was extracted from peripheral blood with TRIzol (Life Technologies). The integrity of the RNA was assessed on 1.2% agarose gel, and the concentration and purity were determined by optical densitometry. cDNA was generated with a QuantiTect Reverse Transcription kit (Qiagen) and PCR amplified (Supplementary Table 5) with PrimeStar GXL (Takara). A Nextera XT DNA Prep Kit (Illumina) was used to create a cDNA library and sequenced on a MiSeq instrument (Illumina). Resulting data were aligned to the human reference genome (GRCh37/hg19) using STAR2 (version 2.5.3)60. Aligned reads were sorted using Picard (version 2.17). Splice junctions and transcript organization were visualized using the sashimi plot of the Integrative Genomics Viewer.
Haplotype
The MSL3 locus was amplified from genomic DNA by long-range PCR using PrimeSTAR GXL (Takara). PCR products were pooled for each case. The Nextera XT DNA Prep kit (Illumina) was used to create a tagged and fragmented DNA library, which was sequenced on the MiSeq (Illumina). Resulting data were aligned to the reference genome (GRCh37/hg19) using the Burrows–Wheeler Aligner (version 0.7.6). GATK (version 2.1–10) enabled indel realignment and base quality score recalibration. Variants with a quality score of >30 and an alignment quality score of >20 were annotated with SeattleSeq Annotation (version 137). The haplotype was determined using HaplotypeCaller from VCF files.
Protein modeling
The complex structure of the MSL1–MSL3 complex (Protein Data Bank: 2Y0N) was analyzed and visualized with Pymol (version 1.7.4)
Cloning for protein expression and purification
The human MOF174–458 and MOF2–458 (UniProt Q9H7Z6–1) CDS were cloned in a modified pET15b vector (His10-tag). MSL1470–520 or MSL1471–616 (UniProt Q68DK7–1) CDS were cloned into the multiple cloning site (MCS) 1 of a pACYC coexpression vector (Novagen) with MOF174–458 cloned in MCS2. The coding sequence of MSL3167–517 (UniProt Q8N5Y2–1) was cloned in MCS1 of a modified pRSFDuet vector. Proteins were expressed in lysogeny broth (LB) using Rosetta2 pLysS (MOF174–458 and MOF2–458) or BL21(DE3) cells (MOF174–458–MSL1470–520 and MOF174–458–MSL1471–616–MSL3167–517). Expression was induced with isopropyl β-D-1-thiogalactopyranoside (0.1–1 mM, OD600 0.5–0.7, 18 °C overnight). After lysis by sonication (MOF174–458, MOF174–458–MSL1470–520, and MOF174–458–MSL1471–616–MSL3167–517: 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 500 mM NaCl, 20 mM imidazole, 5 mM β-mercaptoethanol, 0.01% (w/v) taurodeoxycholate, 1 ethylenediaminetetraacetic acid (EDTA)-free cOmplete (Roche), pH 7.5; MOF2–458: 50 mM HEPES, 100 mM NaCl, 20 mM imidazole, 5 mM β-mercaptoethanol, 0.01% (w/v) taurodeoxycholate, 1 cOmplete (Roche), pH 7.5) and clarification by centrifugation, the supernatant was applied to a HisTrap HP column (GE Healthcare). This was washed with lysis buffer (for MOF174–458 and MOF174–458–MSL1471–616–MSL3167–517 containing 2,000 mM NaCl) before elution with an imidazole gradient (20–300 mM). Protein-containing fractions were pooled.
After buffer exchange (50 mM HEPES, 100 mM NaCl, 20 mM imidazole, 5 mM β-mercaptoethanol, 0.01% (w/v) taurodeoxycholate, pH 7.5), the solutions containing MOF2–458 and the trimeric MOF complex were applied to a Heparin HP column (GE Healthcare) and eluted with a NaCl gradient (100–2,000 mM NaCl). Protein-containing fractions were pooled and concentrated. Finally, MOF174–458, MOF2–458, MOF174–458–MSL1470–520, and MOF174–458–MSL1471–616–MSL3167–517 were applied to a Superdex S75 (MOF174–458, MOF2–458 and MOF174–458–MSL1470–520) or S200 (MOF174–458–MSL1471–616–MSL3167–517) gel filtration column (25 mM HEPES, 500 mM NaCl, pH 7.5). Protein-containing fractions were pooled, concentrated, flash-frozen, and stored at −80 °C. Chromatography was performed on Äkta purifier systems. The purity of all assayed proteins and complexes was >90%. The protein concentration was determined by Bradford assay.
Nucleosome reconstitution and HAT assays
Nucleosomal core particles were reconstituted using a PCR-amplified 147-base pair DNA fragment61 with 5′ Cy5 and 3′ Biotin and Xenopus laevis histone octamers applying salt dialysis62. The nucleosomal core particle quality was assessed using agarose gel electrophoresis. For HAT assays, 100 nM MOF174–458, MOF2–458, MOF174–458–MSL1470–520, or MOF174–458–MSL1471–616–MSL3167–517 were mixed with AcCoA (Sigma; final concentration 33 μM) and nucleosomes (0.54 μg) in assay buffer (10 mM Tris, 200 mM NaCl, 1 mM EDTA, pH 8.0), and incubated for 1 h (37 °C, 600 r.p.m.). The reaction was stopped by the addition of a 4× sodium dodecyl sulfate (SDS) sample buffer followed by SDS polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblot.
Cell culture, drug treatment, and transfections
Primary HDFs were isolated from 3 mm skin biopsies using 0.25% collagenase type I, 0.05% DNase I, 20% fetal calf serum high glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco) overnight at 37 °C. HDFs were allowed to attach and cultured in DMEM supplemented with 10% fetal calf serum, penicillin-streptomycin. All experiments were performed at early passages (2–8). LBH-589 (Selleck Chemicals), MGCD0103 (Toronto Research Chemicals), SAHA (Adipogen) and trichostatin A were dissolved in dimethyl sulfoxide (DMSO). For most experiments, before experimental treatment, cells were starved overnight in Hank’s Balanced Salt Solution (24020133; Thermo Fisher). Flp-In T-Rex 293 cells (R78007; Thermo Fisher) were maintained in DMEM-GlutaMAX supplemented with sodium pyruvate, glucose and 10% fetal bovine serum. They were also maintained in zeocin- and blasticidin-containing medium according to the manufacturer’s protocol. All transgenes used in immunoprecipitation experiments were cloned into pcDNA5/FRT/TO (V6520–20; Thermo Fisher) with a C-terminal 3×FLAG –HBH-tag (HBH, His6-biotinylation sequence-His6). Entire tag sequence in aa: LEDYKDHDGDYKDHDIDYKDDDDKRGSHHHHHHAGKAGEGEIPAPLAGTVSKILVKEGDTVKAGQTVLVLEAMKMETEINAPTDGKVEKVLVKERDAVQGGQGLIKIGVHHHHHH. The MSL1 coding sequence was cloned with a C-terminal 3×HA-Avi tag. This vector was co-transfected with the MSL3 wild type (full length), truncating mutants or a green fluorescent protein (GFP) coding sequence (as an immunoprecipitation negative control) in a 1:1 ratio (500 ng each on 6-well plates for nuclear FLAG-tag immunoprecipitation) with a 3:1 lipid (Lipofectamine 2000 (μl)) to DNA (μg) ratio. Some 6 h after transfection, transgenes were induced with 0.1 μg ml−1 doxycyline for 16 h before lysate preparation. The hsMSL3–3×FLAG –HBH coding sequence was subcloned into a custom pCAG–MCS–BGH(polyA), pSV40–BlasticidinR–SV40(polyA), pSV40–mCherry–SV40(polyA) vector created in multiple steps from an SV40 promoter containing backbone pMB1610 (Addgene 65853). The vector and map can be provided on request. For HDF transfections, 0.1 μg plasmid was used with Lipofectamine 3000 on 1 × 105 cells in Opti-MEM followed by selection with 30 μg ml−1 blasticidin-HCl (Invitrogen; A111139–03) overnight. Cells were then grown in fresh media for 24 h before RNA extraction.
FLAG immunoprecipitation
Nuclei were isolated with 0.5% NP-40 in isotonic lysis buffer (10 mM Tris HCl, pH 7.5, with 2 mM MgCl2, 3 mM CaCl2, and 0.3 M sucrose). Nuclear pellet was extracted using HMGT buffer (25 mM HEPES pH 7.5, 10 mM MgCl2, 10% glycerol, 0.2% Tween-20) containing 420 mM KCl. The lysate was clarified by centrifugation before slowly reducing KCl to 150 mM by dilution. FLAG-M2 beads (Sigma; 1804) were used for overnight immunoprecipitation followed by HMGT buffer (150 mM KCl) washes. Proteins were eluted by incubating for 10 min at 70 °C in 2× SDS sample buffer.
MSL3 knockdown in HDFs
Silencer Select (Ambion–Thermo Fisher) small interfering RNA ID s21530 and control small interfering RNA were used at a final concentration of 50 nM for 32 h before RNA extraction.
Expression analysis in HDFs and RT–qPCR
Total RNA was prepared from 12-well-plate-cultured cells using TRIzol (Thermo Fisher) and purified with Direct-zol RNA MiniPrep and MicroPrep kits (Zymo Research). cDNA samples were synthesized using Maxima Reverse Transcriptase (K1671; Thermo Fisher) from 20–50 ng of total RNA. RT–qPCR reactions were performed on a LightCycler 480 Multiwell Plate 384 (Roche) using SYBR Green Master Mix (Roche). The primer efficiency was tested using serial dilutions.
Immunofluorescence and immunohistochemistry
HDFs were seeded as required for the experimental setup and fixed in 4% formaldehyde in a μ-Slide 8-Well on ice. Formalin-fixed, paraffin-embedded (FFPE) section and HDF immunostainings were performed according to published procedures63 using MSL3 antibody (USBI129933; MaxPab; 1:200) as primary, together with H4K16ac (07–329; Millipore; 1:500), H3K27me3 ((C36B11) 9733; Cell Signaling Technology; 1:250), E-cadherin (610181; BD Bioscience; 1:200), and KAT8 (EPR15803) (ab200660; Abcam; 1:500) antibodies.
Multi-stacked and tailed images were acquired on a Spinning disk confocal microscope (Zeiss). The fluorescence intensity was measured on orthogonal projections from the same stack number using ImageJ (1.47 v) and corrected for background. Fluorescently labeled secondary antibodies (Invitrogen) were used at a 1:300 dilution: Goat-anti-Mouse-Alexa Fluor 594 (A-11032), Goat-anti-Rabbit-Alexa Fluor 594 (A-11037), Goat-anti-Rabbit-Alexa Fluor 488 (A-11034), and Goat-anti-Mouse-Alexa Fluor 488 (A-11001).
Immunoblotting
Cells were fractionated using a subcellular fractionation kit (78840; Thermo Fisher), and the buffers were supplemented with cOmplete (Roche) and sodium butyrate (19–137; Merck Millipore). The protein concentration was determined using Qubit protein assay reagent. Samples were denatured at 95 °C for 10 min in Pierce Lane Marker Reducing Sample Buffer, resolved by NuPAGE Bis-Tris denaturing conditions, and transferred to 0.22 μm polyvinylidene difluoride membranes followed by blocking in 5% non-fat milk in phosphate buffered saline (PBS) (0.3% Tween) before incubation with antibodies: MSL3 (USBI129933, MaxPab, USBiological, 1:1000 in bovine serum albumin), H3 (61475; Active Motif; 1:5000), H4K16ac (39167; Active Motif; 1:200 and 07–329; Millipore; 1:2000), Flag (M2; Sigma; 1:1000), HA (MMS-101P; Covance; 1:1000), MOF (A300–992A; Bethyl; 1:1000), DHX9 (NDH-II (E10) sc-137183; 1:2000–1:5000), H3K36me3 (ab9050; Abcam; 1:1000), acH3 (rabbit; Millipore; 1:1000), H4K20me1 (ab9051; Abcam; 1:1000), H4K8ac (61103; Active Motif; 1:1000), H3K14ac (39599; Active Motif; 1:1000), acH4 (06–598; Millipore; 1:1000), and H3K27me3 (9733; Cell Signaling Technology, 1:1000).
Mass spectrometry sample preparation
After histone extraction (78840; Thermo Fisher), proteins were separated on a 16% Tris-Glycine gel (Novex) and stained with colloidal Coomassie. Gel bands containing histones were cut out. After de-staining, reduction, and in-gel alkylation, histones were propionylated in a 79:1 mixture of isopropanol and propionic anhydride (240311; Sigma) for 2 h. Gel pieces were washed with Milli-Q water, dried in a SpeedVac centrifuge and rehydrated in hydroxylamine followed by incubation with ammonium hydroxide solution to reestablish a neutral pH. After further washes with ammonium bicarbonate-based buffer and acetonitrile, gel pieces were again dehydrated. Subsequent tryptic digestion was performed as described previously64. Recovered peptides were desalted on STAGE tips according to ref.65.
LC-MS bottom-up analysis of histone peptides
Digested and desalted peptides were analyzed by tandem LC-MS (LC-MS/MS) using nanoflow high-performance liquid chromatography (Easy nLC1200; Thermo Fisher) coupled with a Q Exactive hybrid mass spectrometer (Thermo Fisher) through a nanoelectrospray ion source (Proxeon). Peptides were loaded on a 20-cm-long, 75-μm-wide analytical column filled in-house with 1.9 μm C18 beads (Reprosil C18; Dr. Maisch) at a flow rate of 500 nl min−1. The analytical column was then equilibrated for 5 min (5% buffer A),followed by 60 min of peptide elution at a flow rate of 300 nl min−1 with a linearly increasing concentration of organic solvent (80% acetonitrile, 0.1% formic acid). Q Exactive was operated in positive mode, employing data-dependent acquisition of fragmentation spectra of multiple-charged peptides for their identification.
MaxQuant software (version 1.6.0.16) was used for raw file peak extraction and protein identification. Peptide and protein FDRs were set to 0.01 using a decoy database. Additionally, ‘match between runs’ was used with a matching time window of 1 min and an alignment time window of 20 min.
LC-MS data analysis
A list of the ratios of the modified lysine or arginine sites derived from MaxQuant (‘…sites.txt’) was imported to Perseus66. The data matrix was modified by applying the ‘expand site table’ step, removing empty rows, as well as contaminants and entries from the decoy database. All site intensities were log2 transformed and normalized by subtracting the mean of the whole sample from the particular histone site intensities67. A moderated t-test was used to test for significant differences between control and patient samples. Differences with a q value < 0.05 (Benjamini–Hochberg corrected) were considered significant.
Paraffin embedding and sectioning
Human skin specimens (2 mm) were fixed overnight in PBS containing 4% formaldehyde. After dehydration in ethanol/PBS and Histolemon series, they were incubated in paraffin at 58 °C overnight and cast into molds covered with paraffin. Blocks were sectioned using an RM2155 microtome (Leica) at 5–10 μm. Additional FFPE skin sections age matched to the control were purchased from BioCat.
Hematoxylin and eosin staining
Paraffin sections were dewaxed and rehydrated step-wise. The samples were stained in hematoxylin solution (0.1% hematoxylin, 5% KAl(SO4)2, 0.02% KIO3). Counterstaining was performed by incubating the slides in eosin solution (1% eosin). Slides were mounted in Permount Mounting Medium (Thermo Fisher) and analyzed using brightfield microscopy.
Cell-cycle analysis by fluorescence-activated cell sorting (FACS)
HDFs were synchronized by mitotic shake followed by serum starvation overnight. Cells were induced to cycle for 6 h by the addition of complete media. 1 × 106 HDFs were fixed in suspension in 70% cold ethanol while gently shaking for 20 min at 4 °C. After washing in FACS buffer (0.2% bovine serum albumin in PBS), H4K16ac staining was performed at room temperature for 90 min (39167; Active Motif; 1:500 and 07–329; Millipore; 1:500 in FACS buffer). This was followed by 30 min incubation with Ki67-FITC as indicated (BD Biosciences; 556026) and allophycocyanin-labeled Anti-Rabbit secondary antibodies. Propidium iodide (1 μg ml−1) was used to stain the nuclei. After additional washes, cells were analysed in FACS buffer using LSR II (BD Biosciences).
Alive singlets, based on forward scatter (FSC)/side scatter (SSC), were gated for further analysis. Three channel analyses were performed using antibodies/dyes with no fluorescence absorbance overlap. Machine laser intensities were set up using beads. For downstream analysis in FlowJo (version 10.4.1), alive cells were gated, followed by re-gating of only single cells. For cell-cycle phase analyses, the ‘cell cycle’ automatic function was run using propidium iodide.
Colorimetric detection of senescence-associated β-galactosidase
This was performed as described previously68. Pictures were taken with a conventional charge-coupled device camera on a regular inverted cell culture microscope with a 10× objective.
RNA-Seq
Approximately 50 ng RNA was isolated from the synchronized control, p1, p2, and p14 HDFs at passage 4 and passage 7, and subjected to poly(A) RNA-Seq (TruSeq Stranded mRNA Sample Prep kit; Illumina). Reads were mapped to the GRCh37/hg19 reference genome with HISAT2 (version 2.2.0-beta)69 using default parameters. Primary alignments were assigned to features with featureCounts (version 1.4.6-p2)70 using the GRCh37 Gencode annotation (release 19). Differential expression analysis was performed using DESeq2 (version 1.18.1)71 with default settings. We identified differentially expressed genes between the control and all patients with patients treated as biological replicates at FDR-adjusted P < 0.05 (Control versus p1/p2/p14). When LBH-589-treated patients were compared with DMSO-treated controls (FDR < 0.05), we considered previously identified differentially expressed genes as ‘rescued’ if they were not differentially expressed anymore. To assess the global effect of LBH-589, genes were identified as differentially expressed in DMSO versus the LBH-589 treatment, irrespective of the cell line, at FDR-adjusted Padj < 0.001. Network maps and pathways/cell components were generated in ingenuity pathway analysis (IPA; Qiagen; version 01–07) using curated lists from the Ingenuity Pathways Knowledge Base. Activation z-scores were used to infer the activation states (‘increased’ or ‘decreased’) of implicated biological functions. Differentially expressed genes were annotated using the OMIM morbid map (version November 2017).
ChIP sequencing (ChIP-Seq) analysis
MACS2 ChIP-Seq peaks of mMSL2, mMOF and mKANSL321 were split in TSS (gene overlapping within ±1 kb of the TSS), gene body (gene overlapping from TSS + 1 kb until the gene end) or distal (closest gene to any peak) groups based on the NCBIM37 Gencode annotation (version 1) and compared with the differentially expressed genes in MSL3 patients. Mouse orthologues of differentially expressed MSL3 genes were identified on Ensembl BioMart. Overlaps, frequency calculations, statistical analyses, and plots were generated in R (version 1.1.383).
Scratch assay of HDFs
An equal number of cells were seeded in Culture Insert-2 (IBIDI) in 12 wells and allowed to attach. Cells were starved overnight before adding fresh media with LBH-589 (2 μM) or DMSO. The cell-free gaps in which the cell migration could be visualized were created by removal of the silicon comb. Pictures were taken with a conventional charge-coupled device camera on a regular inverted cell culture microscope with a 10× objective at 0, 6, 24, and 48 h after removal of the comb and drug administration. Images were analysed using ImageJ (1.47 v); the cell-free gap was measured and plotted as ratio over time 0 for each corresponding cell line.
H4K16ac Chromatin Immunoprecipitation (ChIP)
HDFs were starved overnight and collected by centrifugation after cell counting. Cells were crosslinked using 1% formaldehyde for 10 min. Sodium butyrate was added to all buffers. Cells were fractionated (78840; Thermo Fisher) and nuclei dissolved in lysis buffer (20 mM Tris pH 8, 5 mM MgCl2, 1 mM CaCl2, 0.25 M sucrose, 1% Triton-X100, 10 μM EDTA, and 0.5% SDS). Some 50,000 lysed nuclei were diluted with 5× dilution buffer (10 mM Tris-HCl pH 8, 140 mM NaCl, 0.1% Sodium Deoxycholate, 1% Triton-X100, 1 mM EDTA, 1× protease, and 0.1% SDS) and chromatin sheared using a Diagenode Bioruptor Pico (7 cycles with 30 s ON/OFF. Immunoprecipitation timing and antibody concentration were tightly set up using flow cytometry (50,000 nuclei, 2 h, room temperature, 1:750 dilution). Dynabeads-bound chromatin was washed using 2× immunoprecipitation dilution buffer, 1× High Salt wash buffer (400 mM NaCl), 1× LiCl buffer, and 1× TE (10 mM Tris, 1 mM EDTA, pH 8.0). Chromatin was reverse cross-linked at 65 °C overnight followed by RNAse A and Proteinase K digestion. DNA was purified using phenol–chloroform extraction followed by ethanol precipitation.
Statistics
All statistics were calculated with GraphPad Prism 6/7 or R (version 1.1.383). One-way ANOVA was performed for multiple comparisons followed by Bonferroni corrections for individual pairing. Dot plots are presented with the center line as the average± SEM. In the box plots in Fig. 3c and Supplementary Fig. 2e, the line that divides the box into two parts represents the median, while the ends are the upper and lower quartiles (first and third quartile). The extreme lines shows the highest and lowest value excluding outliers. Bar plots represent the average±SEM., with overlaid data points representing independent experiments. Results were considered significant at an FDR (P value) below 5%. Additional information and statistical analysis results are provided in the figure legends and Supplementary Table 5.
Web Resources
NHLBI Exome Sequencing Project Exome Variant Server: http://evs.gs.washington.edu/EVS/
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/
Picard: www.picard.sourceforge.net
University of Burgundy Centre de Calcul, https://haydn2005.u-bourgogne.fr/dsi-ccub/
Seattle Seq Annotation tool: snp.gs.washington.edu/SeattleSeqAnnotation137/
ExAC browser: http://exac.broadinstitute.org/
GATK, HaplotypeCaller: https://www.broadinstitute.org/gatk/gatkdocs/org_broadinstitute_gatk_tools_walkers_haplotypecaller_HaplotypeCaller.php
EBI Expression Atlas: https://www.ebi.ac.uk/gxa/home/
Ensembl Biomart: https://www.ensembl.org/biomart/
Integrative Genome Viewer: https://software.broadinstitute.org/software/igv/
DECIPHER, http://decipher.sanger.ac.uk;
ProteomeXchange Consortium, http://proteomecentral.proteomexchange.org;
National Center for Biotechnology Information (NCBI) Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/; NCBI
ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/.
Reporting Summary
Further information on experimental design and reagents can be found in the Life Sciences Reporting Summary.
Data availability
RNA-seq data have been deposited to the Gene Expression Omnibus under the accession number GSE102250.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository72 with the dataset identifier PXD009317.
The damaging variants reported as disease-causing in this article were deposited in ClinVar database under study accession SUB2871008.
Supplementary Material
Acknowledgments
We are grateful to the patients and their families for their cooperation and interest in the study. We thank N. Iovino, B. Sheikh, and I. Ilik for critical reading of the manuscript. We also thank C. Pessoa Rodrigues and A. Karoutas for technical help, insightful discussion, and advice. We thank V. Bhardwaj for advice and consulting on RNA-Seq analysis, and S. Kübart, A. Schröer, J. Wirth, and H.-G. Nothwang for help with inversion breakpoint mapping. We thank L. Wells for patient recruitment and clinical data collection. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009–003), a parallel funding partnership between the Wellcome Trust, Department of Health, and Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the authors and not necessarily those of the Wellcome Trust or Department of Health. This study has UK Research Ethics Committee (REC) approval (10/H0305/83, granted by the Cambridge South REC; and GEN/284/12, granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research through the Comprehensive Clinical Research Network. This study makes use of DECIPHER (see URLs), which is funded by Wellcome. Sequencing for patient 12 was provided by the Center for Mendelian Genomics at the Broad Institute of MIT and Harvard, and was funded by the National Human Genome Research Institute, National Eye Institute, and National Heart, Lung and Blood Institute grant UM1 HG008900 to D. MacArthur and H. Rehm. This work was supported by CRC992, CRC1140, and CRC746 (awarded to A.A.). It was also supported by the council of Burgundy, German Human Genome Program (grant number 01KW99087) and National Genome Research Network (project numbers 01GR0105 and 01GS08160), awarded to V.M.K. and A.R., respectively. C.I.K.V. was supported by a Human Frontier Science Program long-term fellowship (000233/2014-L).
Footnotes
Competing Financial Interests
The authors declare no competing financial interests
References
- 1.Ronan JL, Wu W & Crabtree GR From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet 14, 347–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ropers H-H & Hamel BCJ X-linked mental retardation. Nat. Rev. Genet 6, 46–57 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Lubs HA, Stevenson RE & Schwartz CE Fragile X and X-linked intellectual disability: four decades of discovery. Am. J. Hum. Genet 90, 579–590 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjornsson HT The Mendelian disorders of the epigenetic machinery. Genome Res. 25, 1473–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tapias A & Wang Z-Q Lysine Acetylation and Deacetylation in Brain Development and Neuropathies. Genomics Proteomics Bioinformatics 15, 19–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J-H, Hart SRL & Skalnik DG Histone deacetylase activity is required for embryonic stem cell differentiation. Genesis 38, 32–38 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Hsieh J, Nakashima K, Kuwabara T, Mejia E & Gage FH Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc. Natl. Acad. Sci. U. S. A 101, 16659–16664 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mews P et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tessarz P & Kouzarides T Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol 15, 703–708 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Shogren-Knaak M et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Keller CI & Akhtar A The MSL complex: juggling RNA-protein interactions for dosage compensation and beyond. Curr. Opin. Genet. Dev 31, 1–11 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Philippakis AA et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum. Mutat 36, 915–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith ER et al. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol 25, 9175–9188 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taipale M et al. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol. Cell. Biol 25, 6798–6810 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Consortium GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kadlec J et al. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat. Struct. Mol. Biol 18, 142–149 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Zhao X et al. Crosstalk between NSL histone acetyltransferase and MLL/SET complexes: NSL complex functions in promoting histone H3K4 di-methylation activity by MLL/SET complexes. PLoS Genet. 9, e1003940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelbart ME, Larschan E, Peng S, Park PJ & Kuroda MI Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat. Struct. Mol. Biol 16, 825–832 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akhtar A & Becker PB Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 5, 367–375 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Chelmicki T et al. MOF-associated complexes ensure stem cell identity and Xist repression. Elife 3, e02024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tukiainen T et al. Landscape of X chromosome inactivation across human tissues. Nature 550, 244–248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Y et al. Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem 285, 4268–4272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morales V, Regnard C, Izzo A, Vetter I & Becker PB The MRG domain mediates the functional integration of MSL3 into the dosage compensation complex. Mol. Cell. Biol 25, 5947–5954 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D et al. Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat. Struct. Mol. Biol 17, 1027–1029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaquero A et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 20, 1256–1261 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonenfant D et al. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol. Cell. Proteomics 6, 1917–1932 (2007). [DOI] [PubMed] [Google Scholar]
- 28.McManus KJ & Hendzel MJ The relationship between histone H3 phosphorylation and acetylation throughout the mammalian cell cycle This paper is one of a selection of papers published in this Special Issue, entitled 27th International West Coast Chromatin and Chromosome Conference, and has undergone the Journal’s usual peer review process. Biochem. Cell Biol 84, 640–657 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Conrad T & Akhtar A Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nat. Rev. Genet 13, 123–134 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Conrad T et al. The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Dev. Cell 22, 610–624 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Kasowski M et al. Extensive variation in chromatin states across humans. Science 342, 750–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Ruijter AJM, Van Gennip AH, Caron HN, Stephan K & Van Kuilenburg ABP Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J 370, 737–749 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marks PA & Breslow R Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol 25, 84–90 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Bradner JE et al. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol 6, 238–243 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coni S et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep 7, 44079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lillico R, Sobral MG, Stesco N & Lakowski TM HDAC inhibitors induce global changes in histone lysine and arginine methylation and alter expression of lysine demethylases. J. Proteomics 133, 125–133 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Schölz C et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol 33, 415–423 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Tzogani K et al. The European Medicines Agency Review of Panobinostat (Farydak) for the Treatment of Adult Patients with Relapsed and/or Refractory Multiple Myeloma. Oncologist (2017). doi: 10.1634/theoncologist.2017-0301 [DOI] [Google Scholar]
- 39.Volmar C-H & Wahlestedt C Histone deacetylases (HDACs) and brain function. Neuroepigenetics 1, 20–27 (2015). [Google Scholar]
- 40.Dulac C Brain function and chromatin plasticity. Nature 465, 728 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas T, Dixon MP, Kueh AJ & Voss AK Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol. Cell. Biol 28, 5093–5105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta A et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol 28, 397–409 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheikh BN et al. MOF maintains transcriptional programs regulating cellular stress response. Oncogene 35, 2698–2710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee A et al. MOF Acetyl Transferase Regulates Transcription and Respiration in Mitochondria. Cell 167, 722–738.e23 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Koolen DA et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet 38, 999–1001 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Sharp AJ et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet 38, 1038–1042 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Shaw-Smith C et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet 38, 1032–1037 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Füllgrabe J et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500, 468–471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan TH SEX LIMITED INHERITANCE IN DROSOPHILA. Science 32, 120–122 (1910). [DOI] [PubMed] [Google Scholar]
- 50.Dobyns WB et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am. J. Med. Genet. A 129A, 136–143 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Zerem A et al. The molecular and phenotypic spectrum of IQSEC2-related epilepsy. Epilepsia 57, 1858–1869 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Lezin A et al. Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1 associated myelopathy/tropical spastic paraparesis patients. Blood 110, 3722–3728 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Lelieveld SH et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci 19, 1194–1196 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilissen C et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Retterer K et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med 18, 696–704 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Thevenon J et al. Diagnostic odyssey in severe neurodevelopmental disorders: toward clinical whole-exome sequencing as a first-line diagnostic test. Clin. Genet 89, 700–707 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Pajusalu S, Reimand T & Õunap K Novel homozygous mutation in KPTN gene causing a familial intellectual disability-macrocephaly syndrome. Am. J. Med. Genet. A 167, 1913–1915 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thåström A et al. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol 288, 213–229 (1999). [DOI] [PubMed] [Google Scholar]
- 62.Dyer PN et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Basilicata MF, Frank M, Solter D, Brabletz T & Stemmler MP Inappropriate cadherin switching in the mouse epiblast compromises proper signaling between the epiblast and the extraembryonic ectoderm during gastrulation. Sci. Rep 6, 26562 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shevchenko A, Tomas H, Havlis J, Olsen JV & Mann M In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Rappsilber J, Mann M & Ishihama Y Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Tyanova S et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Kuhn M Contributions from Jed Wing and Steve Weston and Andre Williams and Chris Keefer and Allan Engelhardt and Tony Cooper and Zachary Mayer and Brenton Kenkel and the R Core Team and Michael Benesty and Reynald Lescarbeau and Andrew Ziem and Luca Scrucca., caret: Classification and Regression Training. in R Package Version 6.0−−47 (2015). [Google Scholar]
- 68.Stemmler MP, Hecht A & Kemler R E-cadherin intron 2 contains cis-regulatory elements essential for gene expression. Development 132, 965–976 (2005). [DOI] [PubMed] [Google Scholar]
- 69.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaudel M et al. Exploring the potential of public proteomics data. Proteomics 16, 214–225 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited to the Gene Expression Omnibus under the accession number GSE102250.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository72 with the dataset identifier PXD009317.
The damaging variants reported as disease-causing in this article were deposited in ClinVar database under study accession SUB2871008.