

Abstract
Soluble secreted proteins and membrane proteins are subjected to protein quality control pathways during their synthesis in the endoplasmic reticulum (ER) and delivery to other destinations. Foremost among these quality control pathways is the selection of misfolded proteins for ER-associated degradation (ERAD). A growing number of diseases, including Cystic Fibrosis, are linked to the ERAD pathway. In most cases, a membrane protein known as the Cystic Fibrosis Transmembrane Conductance Regulator, or CFTR, is prematurely degraded by ERAD. Cell-based assays and in vitro studies have elucidated factors required for the recognition and degradation of CFTR, yet mechanistic details on how these factors target specific disease-causing variants is limited. Given the possibility that variants might exhibit unique susceptibilities to ubiquitin modification, which is required for proteasome-mediated degradation, we devised an assay that recapitulates this event. Here, we demonstrate that ER-enriched membranes from transfected human cells support CFTR ubiquitination when combined with radiolabeled ubiquitin and isolated enzymes in the ubiquitination cascade. We also show that select disease-causing variants are ubiquitinated more extensively than wild-type channels and to varying degrees. Our system provides a platform to examine how other purified factors impact CFTR ubiquitination and the ubiquitination of additional disease-associated membrane proteins.
Keywords: cystic fibrosis, F508del CFTR, protein quality control, ubiquitination, ERAD
Subject Category: Enzymatic assays and analysis
Graphical Abstract

1. Introduction
Eukaryotic cells utilize a combination of distinct pathways to ensure that misfolded proteins are eliminated before they accumulate and become toxic. These pathways, collectively referred to as protein quality control pathways, act upon proteins localized to a variety of cellular compartments [1, 2]. Because an estimated 30% of all proteins in eukaryotes are soluble proteins or integral membrane proteins that access the secretory pathway [3, 4], protein quality control is particularly active in the endoplasmic reticulum (ER), where these proteins are synthesized, fold, and become post-translationally modified before being delivered to the Golgi for subsequent trafficking to other locations. However, when secretory proteins misfold, they are instead retained and targeted for ER associated degradation (ERAD). During ERAD, ubiquitin is conjugated most commonly onto a lysine side chain in a misfolded protein. Repeated cycles in which an appended ubiquitin is modified by additional ubiquitin molecules leads to the formation of a polyubiquitin chain that acts as potent signal for proteasome-dependent degradation [5, 6]. Once marked, substrates are retrotranslocated from the ER to the cytosol by an AAA+ ATPase, which is known as p97 in humans or Cdc48 in yeast, before being targeted to the 26S proteasome [7–9].
While protein quality control pathways, such as ERAD, protect cells from proteotoxic stress arising from the accumulation of misfolded proteins, these same pathways can cause human diseases when substrates containing subtle defects are targeted too aggressively for premature degradation [8, 10–12]. Solute conducting channels and transporter proteins are especially vulnerable to overzealous protein quality control because their folding is particularly complex. Specifically, nearly all integral membrane solute channels and transporters contain several transmembrane segments [13, 14], which must be translated and properly inserted into the ER membrane before the entire protein can attain its native conformation [15–18]. Compared to other integral membrane proteins, the assembly of these proteins is additionally problematic because the insertion of individual transmembrane helices is unfavorable and requires the synthesis of other helices before membrane integration is complete [14, 19, 20]. Channels also have cytosolic domains that may fold and assemble independently of membrane-spanning domains, as well as domains that ultimately reside in the extracellular space but initially fold within the ER. The coordinated assembly of a protein that resides in these three unique chemical environments is essential to avoid selection by the protein quality control machinery [21–27]. And finally, ion channels and transporters contain pore-associated hydrophilic amino acid residues, which further compromise membrane integration. Due to these challenges, a significant proportion of wild-type ion channels and transporters are targeted for ERAD, and mutations that disrupt folding dramatically shift the precarious balance between folding and degradation in favor of the latter.
The cystic fibrosis transmembrane conductance regulator (CFTR) is an epithelial chloride channel that epitomizes this balance. CFTR is a large (~140 kDa) protein that requires ~10 min to translate and ~20 min to reach a trafficking-competent state [28, 29]. During that time, it is vulnerable to ERAD targeting due to its intrinsically delayed folding kinetics and thermodynamic instability [30–33]. As a result, ~70% of otherwise functional wild-type channels are ubiquitinated and destroyed via ERAD [34]. Mutations that further destabilize CFTR, such as the predominant cystic fibrosis (CF)-causing allele F508del—which compromises the folding of the cytosolic first nucleotide binding domain (NBD1) and attenuates its interaction with the second nucleotide binding domain (NBD2) and first membrane spanning domain (MSD1) [35–37]—cause virtually all channels to be eliminated by ERAD, precluding function at the plasma membrane [29, 38]. Furthermore, F508del CFTR channels which manage to escape ERAD, whether through hypothermic incubation in cell culture or by treatment with small molecule “corrector” compounds, are rapidly endocytosed and targeted to the lysosome for degradation [38–41]. Remarkably, corrected F508del CFTR retains ~50% of the function of the wild-type channel [42, 43]. The activity of corrected F508del CFTR can be augmented further by “potentiator” compounds that restore near wild-type conductance [44, 45]. Because F508del CFTR can be coerced to attain wild-type activity, considerable effort has been made to identify corrector compounds that augment folding [46–49]. However, >300 disease-causing variants in CFTR have been identified, many of which are relatively common but remain refractory to therapeutic correction [50].
To date, approximately two dozen factors have been identified that either promote the maturation of CFTR in the ER or catalyze ERAD [51, 52]. Folding is initially supported by molecular chaperones, such as Hsc70 and Hsp90, which hydrolyze ATP to facilitate CFTR folding [53–56]. The action of these chaperones is modulated by co-chaperones, such as DNAJA1 and DNAJA2, which support productive CFTR folding by enhancing Hsc70 activity [57–59] and bind “hotspots” on CFTR to coordinate folding [60]. Other factors recruit E3 ubiquitin ligases, which attach polyubiquitin onto incompletely folded CFTR channels [61, 62]. The integral membrane ubiquitin ligases RMA1 and its close homologue RNF185 are the first to act upon CFTR in the ER, ubiquitinating channels after MSD1 and NBD1 translation [63–65]. A related ligase, gp78, also ubiquitinates CFTR co-translationally, though it primarily extends chains initiated by RMA1 or RNF185 [66]. In contrast, a cytosolic ubiquitin ligase, CHIP, interacts with CFTR through Hsc70, effectively converting this pro-folding chaperone into an element of the degradative machinery [67, 68]. Because CHIP also targets CFTR for lysosomal degradation after the protein has been endocytosed [63, 69, 70], this enzyme is a major driver of channel degradation [63, 68, 71].
Here, we present an assay that combines isolated enzymes required for polyubiquitination with ER-enriched microsomes prepared from CFTR-expressing human cell cultures. We hypothesized that the ubiquitination of distinct disease-causing CFTR variants would differ based on the degree to which they are misfolded. Indeed, some variants, such as F508del, are ubiquitinated to a greater extent than others that exhibit subtler folding defects. We also found that purified CHIP modestly enhanced the ubiquitination of all variants. Our assay thus recapitulates a key step in CFTR quality control, reflects how aggressively variants are targeted for degradation, and provides a means to better define the effects of contributing factors during CFTR biogenesis.
2. Materials and Methods
2.1. CFTR expression plasmids
pcDNA5/FRT human cell expression vectors for uncommon disease-causing CFTR variants were initially obtained from the Sorscher lab at Emory University [72] and sequence verified through Sanger sequencing (GENEWIZ). In order to enhance the expression of select disease-causing CFTR variants (P67L, G85E, E92K, W1282X, N1303K) with various levels of misfolding [51], inserts containing the CFTR reading frames were removed by restriction digest with ApaI, NotI, and SacII restriction endonucleases (Thermo Fisher #FD1414, New England Biolabs #R3189, New England Biolabs #R0157) and resolved by 0.5% TBE/agarose gel electrophoresis. The excised ~4.5 kb CFTR gene inserts were collected by gel extraction (Thermo Scientific #K0692). In parallel, the pcDNA3.1(+) was obtained from the Kleyman lab at the University of Pittsburgh, digested with the ApaI and NotI restriction endonucleases, resolved by gel electrophoresis, and ~5.4 kb linear vectors were collected by gel extraction. Plasmids encoding wild-type and F508del CFTR variants were previously obtained from the Frizzell lab at the University of Pittsburgh [59].
Ligations were conducted in a 3:1 molar ratio using the Quick Ligation™ Kit (New England Biolabs #M2200) and transformed into NEB5α High Efficiency competent E. coli cells (New England Biolabs #C2987U). Clones containing the ligation products were isolated, and the plasmids were extracted (Thermo Scientific #K0503) and then screened by digestion with EcoRI restriction endonuclease (Thermo Fisher #FD0274). Plasmids matching the digestion pattern of pcDNA3.1(+) wild-type CFTR control (see above) were verified by GENEWIZ Sanger sequencing.
2.2. Isolation and enrichment of the Ube1 ubiquitin activating enzyme
The pET21d His-Ube1 expression vector was a gift from Dr. Cynthia Wolberger at Johns Hopkins University (Addgene plasmid #34965). After transformation into a BL21(DE3) E. coli strain for bacterial expression, the cells were cultured overnight in Luria Broth (LB) containing 100 μg/mL Ampicillin (LB + Amp) at 37°C. The saturated overnight culture was used to inoculate a 0.5 L LB + Amp culture to initial optical density at 600 nm (OD600) of 0.05, which was then incubated at 37°C with shaking at 200 rpm. After reaching an OD600 of 0.3, IPTG was added to a final concentration of 500 μM to induce expression of His-Ube1, and the culture was incubated at 18°C at 200 rpm overnight, as induction at low temperature is required for efficient expression of Ube1 [73]. The culture was centrifuged at 5000 rpm (~3600×g) for 10 min at 4 °C, the supernatant was removed, and cell pellets were stored at −80°C.
Pellets were subsequently thawed on ice with 25 mL of Lysis Buffer (2.6 mM NaH2PO4, 47.4 mM Na2H2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole, 1% PMSF, 0.2% Leupeptin, 0.1% Pepstatin A, 5 mM 2-mercaptoethanol, 0.25% Triton X-100, 2 mg/mL lysozyme) supplemented with 2 Roche cOmplete™ EDTA-free protease inhibitor cocktail tablets (hereafter referred as “PIC”; Millipore Sigma #11873580001) and incubated for at least 30 min with occasional agitation by hand. After clumps were absent and the lysate appeared mucoid, the solution was transferred to an SS34 Sorvall™ centrifuge tube and sonicated in 30 sec bursts followed by 30 sec on ice until it reverted to a thin fluid. The lysate was then centrifuged at 10000 rpm (~12000×g) for 10 min at 4°C. A 15 μL aliquot of cleared lysate was collected in 15 μL 2X SDS Sample Buffer (125 mM Tris, pH 6.8, 125 mM NaCl, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue) as a diagnostic sample, while a small scoop of the lysate pellet was also collected and emulsified in 2X SDS Sample Buffer. The remainder of the cleared lysate was then applied to a chromatography column that had previously been packed with 3 mL of Ni-NTA resin (Qiagen #30210), washed with ddH2O, and equilibrated with 10 mM Imidazole Buffer. Lysate was flowed across the resin at the slowest possible drip rate and a diagnostic sample of the flow-through was collected. The column was washed with 25 mL of 10 mM Imidazole Wash Buffer (2.6 mM NaH2PO4, 47.4 mM Na2H2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole, 1% PMSF, 0.2% Leupeptin, 0.1% Pepstatin A) and then with 25 mL 30 mM Imidazole Wash Buffer (2.6 mM NaH2PO4, 47.4 mM Na2H2PO4 pH 8.0, 300 mM NaCl, 30 mM imidazole, 1% PMSF, 0.2% Leupeptin, 0.1% Pepstatin A). His-Ube1 was eluted with a 30 mL 200 mM Imidazole Elution Buffer (2.6 mM NaH2PO4, 47.4 mM Na2H2PO4 pH 8.0, 300 mM NaCl, 200 mM imidazole, 1% PMSF, 0.2% Leupeptin, 0.1% Pepstatin A) + 1 PIC, which was collected in six 5 mL fractions. Washes and elutions were stored on ice at 4°C overnight, and diagnostic samples were collected in 2X SDS Sample Buffer and stored at −20°C.
Next, the diagnostic samples were thawed and resolved across a 10% polyacrylamide/SDS gel by SDS-PAGE, stained with Coomassie Brilliant Blue stain, and destained with Coomassie Destain (50% methanol, 10% acetic acid) to identify peak fractions containing His-Ube1. These fractions were pooled, dialyzed against 20 mM HEPES, pH 7.4, 20 mM NaCl for 4 hr at 4°C, and His-Ube1 was applied to a previously prepared 3 mL column packed with DEAE-Sepharose® Fast Flow resin (GE Healthcare #17–0709-01) that had been equilibrated in 20 mM HEPES, pH 7.4, 20 mM NaCl. The column was then washed with 20 mL of 20 mM HEPES, pH 7.4, 50 mM NaCl, and His-Ube1 was eluted with 18 mL of 20 mM HEPES, pH 7.4, 150 mM NaCl, followed by 9 ml of 20 mM HEPES, pH 7.4, 200 mM NaCl, followed by 9 ml of 20 mM HEPES, pH 7.4, 300 mM NaCl, all of which were collected in 3 mL fractions. All washes and elutions were stored on ice at 4°C overnight, and diagnostic samples in 2X SDS Sample Buffer were collected for each wash and the elution and stored at 20°C.
The diagnostic samples from this column were subsequently resolved by SDS-PAGE and Coomassie stained as described above, and fractions enriched for intact His-Ube1 were again identified. These fractions were pooled, applied to an Amcon® Ultra-15 30000 MWCO centrifugal filter that had been equilibrated with 50 mM HEPES, pH 7.5, 0.2% Tween 20, and centrifuged at 3333 rpm (2000×g) 4°C until less than 1 mL remained. His-Ube1 was then diluted into 20 mL of 50 mM HEPES, pH 7.5, and recentrifuged until less than 1 mL remained. Concentrated His-Ube1 was next transferred to a new microcentrifuge tube and stored on ice. Diagnostic samples in 2X SDS Sample Buffer taken before and after protein concentration were examined by SDS-PAGE and Coomassie stained, as described above, to determine which proportion of concentrated protein represented intact His-Ube1. Finally, 10 μL aliquots were flash frozen in liquid nitrogen and stored at −80°C.
2.3. Isolation and enrichment of CHIP
The pET151/D-TOPO CHIP and pET151/D-TOPO CHIP134–303 vectors [74] were transformed into Rosetta2(DE3) and BL21(DE3) E. coli, respectively. Transformed E. coli cultures were grown and induced with IPTG, as above, incubated overnight at 18°C with shaking at 200 rpm, and harvested. The lysis and purification of His-CHIP from cell pellets generally followed the procedure used to purify His-Ube1. In brief, cell pellets were thawed, lysed with buffer containing 10 mM imidazole and protease inhibitors, and after the lysates were cleared by centrifugation, they were applied to a chromatography column packed with Ni-NTA resin previously washed and equilibrated with 10 mM imidazole. Bound His-CHIP was washed with 10 mM and 30 mM imidazole, though protease inhibitors were not required for efficient purification of full-length His-CHIP, which was more protease-resistant (data not shown). His-CHIP was eluted in fractions with 200 mM Imidazole Elution Buffer and stored on ice at 4°C. Diagnostic samples were collected at all steps, resolved by SDS-PAGE, and Coomassie stained to indicate peak fractions. These fractions were pooled and dialyzed into 50 mM HEPES, pH 7.0, 50 mM NaCl at 4° overnight.
After dialysis, solutions containing both full length His-CHIP and His-CHIP134–303 appeared cloudy, indicating precipitation of CHIP due to reduced solubility. The supersaturated CHIP solution was transferred to microcentrifuge tubes and centrifuged at 13000 rpm (~15900 ×g) for 5 min at 4°C to remove insoluble CHIP, and the supernatant was transferred to fresh tubes on ice and monitored for further precipitation. After none was observed, full length His-CHIP was diluted 1:1 in 50 mM HEPES, pH 7.0, 50 mM NaCl, 20% glycerol to maintain solubility. A subsequent BCA protein concentration assay indicated that prior to any dilution, saturated solutions of His-CHIP and His-CHIP134–303 were 5.0 mg/mL (128 μM) and 7.5 mg/mL (313 μM), respectively. Fractions were aliquoted, flash frozen in liquid nitrogen, and stored at −80°C.
2.4. Protein concentration determination
Aliquots of isolated, enriched protein were dispensed into wells of a clear, flat-bottomed 96 well plate alongside 10 μL BSA standards (ranging from 0–2000 μg/mL) in 50 mM HEPES, pH 7.5 in 50 mM HEPES, pH 7.5 and quantified by Pierce™ BCA protein concentration assay (Thermo Fisher Scientific #23225). Molarities of these proteins were calculated by using the obtained concentration and the theoretical molecular weight of each protein. Where indicated, the effective molarity of an enriched protein was determined by first calculating the fractional purity of protein appearing at the anticipated molecular mass by gel densitometry, as observed by Coomassie staining, and then multiplying this proportion by the total protein concentration as determined by the BCA protein concentration assay.
2.5. Human cell culture protocols
HEK293 cell cultures were maintained in Falcon® 10 cm sterilized polystyrene dishes (Corning #353003) with 10 mL DMEM (Sigma-Aldrich #D6429) containing 10% FBS (Hyclone #SH30071) at 37°C in a 5% CO2 humidified atmosphere. Upon reaching confluency, cells were passaged by aspirating the media and detached by addition of 2 mL Gibco® TrypLE™ per plate (Thermo Fisher Scientific #12604–021), followed by incubation at ambient temperature for 3 min. Trypsinized cells were then transferred to a sterile conical tube containing 8 mL DMEM and centrifuged at 1500 rpm (405×g) in a clinical centrifuge for 3 min. After centrifugation, the supernatant was aspirated and the cells were resuspended in at least 3 mL DMEM + 10% FBS, of which 100 μL was dispensed into a 10 cm plate along with 10 mL DMEM + 10% FBS and returned to 37°C. For procedures requiring a precise number of cells (e.g., for transfection), 20 μL of the cell suspension was diluted 1:3 in Trypan Blue Stain (Life Technologies #15250–061), of which 10 μL was examined in a hemocytometer to calculate cell density. Cell cultures were passaged at most five times before they were either discarded or stored in glycerol at −80°C (i.e., resuspended instead in 1–3 mL of 90% FBS, 10% DMSO, aliquoted into 1 mL cryovials, and stored in polystyrene foam racks at −80°C). Frozen cultures were revived by thawing and dispensing into 10 cm plates containing 20 mL DMEM + 10% FBS, which was replaced with 10 mL of fresh DMEM + 10% FBS after overnight incubation, after which cells were passaged normally. Only cultures passaged at least once were used for experiments, and all procedures were conducted within a 1300 Series A2 biosafety cabinet (Thermo Fisher Scientific).
2.6. Plasmid transfection into HEK293 cells
Plasmids encoding CFTR were purified from transformed, saturated DH5α or NEB5α E. coli cultures grown in LB + 100 μg/mL ampicillin using either a QIAGEN® Plasmid Maxi Kit (Qiagen #12163) for initial experiments or a higher-yield ZymoPURE™ II Plasmid Maxiprep Kit (Zymo Research #D4203) for subsequent experiments.
Prior to transfection, 0.6×106 HEK293 cells were plated either into wells of a 6 well plate with 2 mL DMEM + 10% FBS per well to test transfection conditions, or into 10 cm plates with 10 mL DMEM + 10% FBS to prepare microsomes. The cultures were incubated for at least 24 hr at 37°C 5% CO2 before transfection. When cultures reached 50% or higher confluency (ideally 70%), cells were transfected using either Lipofectamine™ 2000 or PEI transfection reagents. For Lipofectamine™ 2000 (Thermo Fisher Scientific #11668) transfection, 6 well plate cultures were transfected with 4 μg plasmid and 10 μL Lipofectamine™ 2000 per well by incubating each reagent alone in Gibco® Opti-MEM® (Thermo Fisher Scientific #31985) for 5 min at ambient temperature before the plasmid and reagent were combined for 20 min and added to cells. Cultures were returned to 37°C for 6 hr, after which the media was aspirated and replaced with 2 mL of new DMEM + 10% FBS. For transfection of 10 cm plate cultures, plasmids and transfection reagents were scaled up 6.6-fold to account for the increased surface area. Transfected cultures were incubated overnight at 37°C 5% CO2 prior to any subsequent experiment.
For PEI transfection (Polysciences Inc. #23966–1), a 1 mg/mL solution of MW 25000 PEI in double distilled water (ddH2O) was prepared immediately before transfection, which was dissolved after a 5 min incubation at 95°C. Six well plate cultures were transfected with 2 ng plasmid and 20 μL of 1 mg/mL PEI per well. Each reagent was incubated alone in 1X DPBS (Thermo Fisher # 14190–144) for 5 min at ambient temperature before they were combined for 20 min, and the mixture was diluted 1:2 with Opti-MEM® and added to cells. The cultures were then incubated at 37° for 4 hr after which the media was replaced with 2 mL DMEM + 10% FBS per well. As above, reagents were scaled-up when 10 cm plates were used, and cultures were incubated overnight at 37°C prior to subsequent experiments. Due to its lower cost and similar performance to Lipofectamine™ 2000, transfection by PEI was the preferred method for procedures requiring large numbers of transfected cells (e.g., for the preparation of microsomes).
2.7. Cell lysis and CFTR detection
After transfection to transiently express CFTR, transfection efficiency was assessed by preparing cell lysates for western blots. First, the media was aspirated and replaced with 300 μL (for a 6 well dish) of TNT Buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 1% Triton X-100) + 1 PIC. Plates were incubated on ice for 30 min with occasional agitation by hand before centrifugation at 13000 rpm (~16000×g) for 15 min at 4°C. The total protein concentration of the cleared lysates was determined as described above. Each cleared lysate was diluted into SDS Sample Buffer and volumes corresponding to 30 μg total protein were resolved by 5% polyacrylamide/SDS gels and SDS-PAGE. To assess CFTR expression, blots were treated with 1:2500 mouse anti-CFTR (antibody 217 from the Cystic Fibrosis Foundation Therapeutics antibody distribution program, University of North Carolina at Chapel Hill). All blots were washed with TBST as described above and then treated with 1:5000 horse anti-mouse-HRP secondary antibody in TBST (Cell Signaling Technology #7076) for 4 hr with ambient rocking. After secondary antibody treatment, the blots were again washed, developed with SuperSignal™ West Pico Chemiluminescent Substrate, and imaged using a Bio-Rad ChemiDoc™ XRS+Imager.
2.8. Immunoprecipitation and detection of ubiquitinated CFTR
To isolate ubiquitinated CFTR proteins from HEK293 cells, cells in 10 cm plates were transfected with either pcDNA3.1(+) encoding wild-type CFTR or F508del CFTR using PEI as described above. The cells were simultaneously transfected with an equal quantity of pcDNA3.1(+) encoding HA-tagged ubiquitin. After overnight incubation, the media was aspirated and replaced with 10 mL DMEM + 10% FBS +10 μM MG132 (Selleck Chemicals #S2619). Cultures were returned to 37°C for 1 hr before the media was aspirated, the cells were trypsinized with TrypLE™ for 3 min at ambient temperature, and then transferred to conical tubes containing 6 mL DMEM + 10% FBS. The mixture was centrifuged at 1500 rpm for 3 min, and the cell pellets were lysed with 500 μL TNT Buffer + PIC + 25 μM MG132 + 5 mM NEM on ice for 30 min with occasional agitation by hand. After centrifugation at 13000 rpm (~1600×g) for 15 min at 4°C, 750 μg of total protein, 850 μL of TBS + 0.14% Triton X-100 + 25 μM MG132 + 5 mM NEM + 1 PIC, 60 μL of a 1:1 slurry of ProteinA Sepharose™ CL-4B (GE Healthcare #71–7090-00 AF) in TBS, and 3 μL of mouse anti-CFTR antibody (1:1 mix of CFFT antibodies 217 and 596) were combined and incubated on a rotator at 4°C overnight to immunoprecipitate CFTR. The next morning, the tubes were retrieved and centrifuged at 2000 rpm (380×g) for 2 min at 4°C, the pelleted beads were resuspended in IP Wash Buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.2% SDS, 5 mM EDTA), and then washed three time before the beads were finally resuspended in SDS Sample Buffer. The samples were next incubated at 37°C for 15 min and resolved through duplicate 5% polyacrylamide/SDS gels. After proteins were transferred to nitrocellulose, one set of blots was probed for CFTR as described (see section 2.7, above), while the other set was treated with 1:2000 mouse anti-HA-HRP (3F10) (Millipore Sigma #12013819001) and 1:2000 mouse anti-ubiquitin (P4D1) (Santa Cruz #sc-8017) antibodies, before subsequently being probed with 1:2500 horse anti-mouse-HRP secondary antibody in TBST. Both sets of blots were washed, treated with chemilumenescent substrate, and imaged as described.
2.9. Isolation of ER-enriched microsomes from human tissue culture cells
Human cell cultures were at least 50% confluent at the time of microsome preparation and had been incubated for ~18 hr after transfection with CFTR-encoding plasmids (section 2.6). Microsomes were prepared using no fewer than three 10 cm plates of cells.
To prepare microsomes, the media was aspirated and cells were suspended by treatment with TrypLE™ for 3 min at ambient temperature. After centrifugation, the supernatant was aspirated from the cell pellet, and the cells were resuspended in 1X DPBS that had been chilled on ice. After re-centrifugation and aspiration of the supernatant, the cells were resuspended in 10 mL of Homogenization Buffer (0.5 M sorbitol, 20 mM HEPES, pH 7.4, + 2 PIC) and placed on ice. The cell suspension was then applied to a stainless steel syringe homogenizer fitted with a tungsten carbide ball bearing (Isobiotec), which was 7.976 mm in diameter and allowed for 24 μm clearance, and two 10 mL Luer-Lok™ disposable syringes (Becton Dickinson #309604). After homogenization, lysed cells were diluted 1:2 with Homogenization Buffer, then centrifuged at 720 rpm (93×g) for 5 min at 4°C in a clinical centrifuge to remove nuclei. The supernatant was then collected into a new SS34 Sorvall™ tube to which 10 mL of KOAc Buffer (20 mM HEPES, pH 7.4, 210 mM potassium acetate, 3 mM magnesium acetate, + 1 PIC) was added before centrifugation at 10000 rpm (~12000×g) for 10 min at 4°C. The supernatant was removed and the pelleted microsomes were resuspended in 20 mL of Transport Buffer (20 mM HEPES, pH 7.4, 250 mM sorbitol, 70 mM potassium acetate, 1 mM magnesium acetate, + 1 PIC) and re-centrifuged. Microsomes were then resuspended in 150–350 μL Transport Buffer as judged by the relative size of the microsomal pellet. To more accurately quantify the relative amounts of microsomes, 1:200 dilutions of each microsome preparation in 2% SDS were prepared and the mean absorbance of each preparation at 280 nm (A280) was calculated. After resuspension, the microsomes typically possessed an A280 of 0.10–0.25. Resuspended microsomes were aliquoted and stored at −80°C.
Before using microsomes for in vitro ubiquitination assays, expression of CFTR was confirmed by treating microsomes with 1 U RQ1 RNase-free DNase for 20 min on ice (Promega #M6101). The microsomes were then transferred to fresh microcentrifuge tubes, pelleted at 13000 rpm for 5 min at 4°C, and resuspended with 2X SDS Sample Buffer such that each sample would have a final microsomal A280 of 0.125 in a 20 μL volume. A total of 5 μL of each sample was resolved by 10% polyacrylamide/SDS gel electrophoresis and CFTR was detected by western blot as described above (see section 2.7).
2.10. Radiolabeling of ubiquitin with iodine-125
One day prior to labeling, two 2 mL 7K MWCO Zeba™ Spin Desalting Columns (Thermo Fisher Scientific #89889) were vertically clamped, uncapped, filled with freshly mixed 1X DPBS + 1% BSA, and allowed to drain by gravity flow at ambient temperature. Columns were pre-equilibrated with DPBS/BSA over three washes. After the third wash, the columns were capped, sealed at the ends with Parafilm®, and stored at 4°C overnight.
On the following day, the columns were washed a fourth time with DPBS + 1% BSA and transferred to a chemical fume hood (Thermo Scientific) equipped with an air pump and air filters for radiological monitoring. Tubes stored at −80°C containing 1 M Tris, pH 8.0, 2 M NaCl, 5 mg/mL NaI in 1% BSA, and 33 mM ICl in ddH2O were thawed. ICl was a gift from Dr. Gerry Apodaca at the University of Pittsburgh. One 1.5 mL microfuge tube was set up for each of two separate iodination reactions, and 100 μl of 1 M Tris, pH 8.0, was added to each reaction tube, followed by 20 μL of 1:50 ICl (prepared by diluting 2 μL ICl into 98 μL 2 M NaCl). Next, 4 μL of 125I radionuclide (Perkin Elmer #NEZ033L005MC) was added to each reaction tube on ice and incubated for 1 min, after which 10 μL of thawed 10 mg/mL recombinant human ubiquitin (Boston Biochem #U-100H) in DPBS was added to reactions and incubated for an additional 10 min. The reactions were quenched by the addition of 100 μL 33 mM NaI, transferred to pre-equilibrated desalting columns (1 column per reaction), and allowed to drip by gravity flow into microcentrifuge tubes. After the columns drained, 300 μL of DPBS + 1% BSA was added to each column, which was also collected. This flow-through was discarded and 600 μL of additional DPBS + 1% BSA was added to the column. Eluted 125I-ubiquitin was collected in collection tubes and the product (2×105 – 6×105 cpm per μL) was subsequently aliquoted and stored at −80°C.
2.11. Microsomal CFTR ubiquitination assay
In vitro ubiquitination assays with human cell-derived ER-enriched microsomes containing CFTR were conducted by preparing 30 μL reactions on ice with Buffer 88 (20 mM HEPES, pH 6.8, 250 mM sorbitol, 150 mM potassium acetate, 5 mM magnesium acetate) + PIC, diluted His-Ube1 (1:2 in Buffer 88 + PIC), diluted UbcH5b (Boston Biochem #E2–622; 1:5 in Buffer 88 + PIC), the indicated concentration of His-CHIP, a 10X ATP Regenerating System (10 mM ATP, 400 μM creatine phosphate, 2 mg/mL creatine phosphokinase in Buffer 88), and 1 U RQ1 RNase-free DNase. The microsomes (prepared as described in section 2.9) were thawed and either a fixed quantity of microsomes was added to reactions, regardless of the relative levels of CFTR (such that all reactions contained microsomes with a corresponding A280 of 0.02, i.e., 6 μL of microsomes with an A280 of 0.15 added to a 30 μL reaction), or the quantities of microsomes added to assays were adjusted such that approximately equal amounts of CFTR were added to all reactions. Microsomes were thus diluted or concentrated to prepare working stocks in Buffer 88 + PIC with an A280 of 0.1, 0.3, or 0.5, depending on the amount of CFTR. Equal volumes of these working stocks were then added to reactions. After the addition, the reactions were removed from ice and 5 μL of 125I-ubiquitin was added and mixed briefly by hand. The reactions were then incubated for 45 min at ambient temperature and typically contained 0.15 μM His-Ube1, 0.25 μM UbcH5b, and 2 μM CHIP (where indicated).
After incubation, the reactions were quenched by the addition of 70 μL TBS/SDS (50 mM Tris, pH 7.4, 150 mM NaCl, 14% SDS, 1 PIC) and incubated at 37°C for 15 min. Quenched reactions were supplemented with 900 μL TBS/Triton (50 mM Tris, pH 7.4, 150 mM NaCl, 0.24% Triton X-100, 25 μM MG132, 5 mM NEM, 1 PIC), 45 μL of a 1:1 slurry of ProteinA Sepharose™ CL-4B in 1X TBS, and 1.5 μL Mouse anti-CFTR antibody (1:1 mix of CFFT antibodies 217 and 596), which was then incubated on a rotator at 4°C for 3 hr to immunoprecipitate CFTR. The mixture was centrifuged at 2000 rpm (376×g) for 2 min at ambient temperature to pellet the beads, which were washed two more times in 500 μL IP Wash Buffer + PIC. Finally, the immunoprecipitated CFTR was resuspended in 50 μL 2X SDS Sample Buffer, incubated at 37°C for 10 min, and stored at −20°C.
To quantify the amount of precipitated CFTR and ubiquitinated protein, the precipitates were incubated at 37°C for 10 min and 20 μL was loaded onto duplicate sets of 5% polyacrylamide/SDS gels and resolved by SDS-PAGE. One set of gels were treated with SDS-PAGE Fixing Buffer (25% isopropanol, 10% acetic acid) for 30 min with agitation, then dried onto Whatman® filter paper with a dry ice-cooled vacuum pump gel drier for 1.75 hr. Dried gels were next exposed to a blanked phosphorimager screen overnight. After a minimum of three days, the screen was scanned using either a Typhoon™ FLA 7000 or Amersham™ Typhoon™ phosphorimager (GE Healthcare). Sixteen-bit depth phosphorimages were collected at a 50 μm pixel size using an upper PMT voltage of 800 V. The other set of gels was transferred overnight onto nitrocellulose membranes, which were blocked for 30 min in milk/TBST and treated with primary antibodies. Initial experiments were subjected to western blotting with 1:2500 mouse anti-CFTR (CFFT antibody 217) and 1:2500 horse anti-mouse-HRP as described in section 2.7. Later experiments were treated similarly, but used 1:5000 rabbit anti-CFTR (D6W6L) (Cell Signaling Technology #78335) and 1:5000 goat anti-rabbit-HRP (Cell Signaling Technology #7074) to maximize the signal. In both cases, the blots were washed with TBST, developed with either SuperSignal™ West Pico Chemiluminescent Substrate or ProSignal® Pico ECL Reagent (Genesee Scientific #20–300B), and visualized on a Bio-Rad ChemiDoc™ XRS+ Imager.
2.12. Ubiquitination of enriched CFTR
Highly enriched wild-type CFTR was isolated from stably expressing HEK293 cell lines and provided in a buffer containing HEPES, pH 7.5, 150 mM NaCl, 10 mM magnesium ATP, 0.2 mM TSEP, 0.06% digitonin, and 10% glycerol by Dr. Zhengrong Yang and Dr. John Kappes of the Cystic Fibrosis Foundation Protein Production Core at the University of Alabama [75]. Ubiquitination reactions containing purified CFTR generally followed the same procedure described for the ubiquitination of microsomal CFTR. In brief, reactions contained Buffer 88 + PIC, the ATP Regenerating System, His-Ube1, UbcH5b, and CHIP, where indicated. DNase was omitted. A total of 3 μL (~0.1 μM) purified CFTR was added to reactions along with 3 μL of 125I-ubiquitin. After 30 min at ambient temperature, reactions were quenched and incubated with TBS/Triton, ProteinA Sepharose™, and a 1:1 mix of CFTR antibodies 217 and 596. The solution was incubated at 4°C for 3 hr, and the beads were washed and resuspended in 2X SDS Sample Buffer before incubation at 37°C for 10 min. Samples were then stored at −20°C. Once thawed, they were again incubated at 37°C for 10 min and resolved on duplicate sets of 5% polyacrylamide/SDS gels by SDS-PAGE. One set was fixed, dried, placed on a blanked phosphorimaging screen, and imaged using an Amersham™ Typhoon™ phosphorimager as described in section 2.11. The other set was transferred to nitrocellulose, blocked, and probed with 1:5000 rabbit anti-CFTR (D6W6L), which was followed by addition of 1:5000 goat anti-rabbit-HRP. The signal was developed with the chemiluminescent substrate and imaged as described.
2.13. Image capture and statistical analysis
All western blots and radiographs were collected as unsaturated grayscale images and quantified as either 8-bit or 16-bit TIFFs, respectively. All blot and radiograph densitometry analyses were performed in ImageJ (NIH). Raw integrated densities were exported to Microsoft Excel for quantification. Fold-changes in CFTR ubiquitination between reactions (e.g., with or without added CHIP) were determined by measuring integrated densities for ubiquitinated CFTR at molecular masses greater than ~168 kDa (or at the native mass of truncated CFTR, as appropriate for W1282X CFTR) and calculating the mean ratio between these integrated densities across a set of independently conducted replicates. For further statistical analysis, Student’s two-tailed T-tests were performed using Minitab 19 Statistical Software. Comparison of mean ubiquitination between CFTR variants assumed equal variance. Separately conducted in vitro ubiquitination assays were treated as independent biological replicates.
3. Results
Using a protocol similar to those described previously, in which yeast lysate is incubated with either yeast [76] or human [77] ER-derived microsomes, we designed an in vitro ubiquitination assay that combined ER-enriched microsomes prepared from HEK293 cells transiently expressing F508del CFTR with isolated enzymes required for substrate ubiquitination, an ATP regenerating system, and I-125 radiolabeled ubiquitin (Figure 1A). When I-125 ubiquitin is appended onto CFTR by these enzymes, the protein is observed as a high molecular mass smear after SDS-PAGE, where addition of each ubiquitin (~9 kDa) shifts the native molecular mass of CFTR (168 kDa) correspondingly higher on the gel. Just as observed in control reactions containing yeast cytosol [77], reactions with enriched E1 and E2 enzymes efficiently ubiquitinated F508del CFTR in membranes prepared from HEK293 cells (Figure 1B).
Figure 1. CHIP enhances the in vitro ubiquitination of F508del CFTR.

(A) Schematic depicting the procedure used to ubiquitinate microsomal CFTR in vitro. (B) I-125 radiograph (top) and anti-CFTR western blot (bottom) depicting in vitro ubiquitination of F508del-CFTR. Microsomes were prepared from transiently transfected HEK293 cells and assayed with either yeast cytosol, or purified Ube1 (E1), UbcH5b (E2), and/or CHIP (E3). Either full length CHIP (CHIP1–303) or C-terminally truncated CHIP (CHIP1–297) was added to reactions as indicated. “B” denotes immaturely glycosylated F508del CFTR. “*” denotes a non-specific antibody band observed by CFTR western blot. Molecular masses are indicated in kDa. (C) I-125 radiograph (top) and anti-CFTR western blot (bottom) depicting a time course of in vitro ubiquitination with F508del CFTR HEK293 microsomes, and the E1 and E2 enzymes, conducted either in the absence or presence of full length His-CHIP.
Ubiquitination was not observed in reactions lacking cytosol but in the presence of the enriched enzymes. Addition of a purified E3 ubiquitin ligase, such as CHIP, was unnecessary to catalyze F508del CFTR ubiquitination, most likely due to the presence of endogenous ubiquitin ligases in or on the microsomal membranes [51]. However, addition of His-CHIP enhanced F508del CFTR ubiquitination two-fold (2.00±0.03; n=2) relative to reactions lacking added CHIP (Figure 1B; compare lanes 4 and 5). In contrast, addition of an inactive CHIP mutant, CHIP1–297 [78], failed to enhance ubiquitination (Figure 1B; compare lanes 4 and 6). Other reactions examining reduced combinations of components revealed that addition of only the E2 ubiquitin conjugating enzyme UbcH5b was sufficient to support F508del CFTR ubiquitination, suggesting that E2s are limiting on the surface of the HEK293 cell-derived microsomes (Figure 1B; compare lanes 4 and 9). This result also implies that microsomes contain endogenous E1 ubiquitin activating enzymes, as the addition of only UbcH5b provided near maximal ubiquitination activity. Next, we performed a ubiquitination time course with reactions containing F508del CFTR microsomes. Our results indicated that ubiquitinated CFTR accumulates at a linear rate, even after 90 min of incubation (Figure 1C). Consistent with the data in Figure 1B, CHIP further increased the rate of CFTR ubiquitination by approximately two-fold when added to the reaction. As the difference in signal between reactions with CHIP and those without CHIP was evident after a 45 min incubation time, we elected to conduct all subsequent microsomal in vitro ubiquitination assays at this time point. We also reasoned that a 45 min incubation would allowed for magnified effects should the addition of any reagents inhibit the reaction.
We next coopted this newly developed assay to compare ubiquitination profiles between different CFTR variants. While ~70% of CF patients encode at least one copy of F508del CFTR, >300 annotated variants have been implicated in causing CF (https://cftr2.org). These variants sort into five non-mutually exclusive classes, a significant portion of which are categorized at least in part as class II mutations, which is characterized by impaired folding and maturation in the ER [79, 80]. As a result of these mutations, the pool of functional CFTR is lost due to ER retention and subsequent degradation. While the disease-causing mutation in F508del CFTR resides within NBD1, several disease-causing variants display similar processing defects due to mutations in other domains of the channel, such as MSD1 and NBD2 [81]. Mutations in these other domains could cause distinct misfolded conformations, which may be ubiquitinated to different levels and/or detected and ubiquitinated by different enzymes in the ubiquitin cascade. To test these hypotheses, we selected a subset of disease-causing CFTR variants that are known to misfold (Table 1). These variants include three variants with mutations in MSD1 (P67L, G85E, and E92K), and two variants with mutations in NBD2 (W1282X and N1303K). Wild-type and F508del CFTR were included for a comparison. These variants also differ widely in their ability to be corrected by small molecules or low temperature incubation. While P67L CFTR causes a relatively mild form of disease and is easily correctable [82, 83], G85E is intractable to correction by either hypothermic incubation or corrector compounds [84], despite the remarkably close residence of these mutations in the protein’s linear sequence. By our estimate, approximately three-quarters of patients globally encode at least one of the disease-causing variants examined in this analysis.
Table 1. Select CFTR variants representing a range of CF phenotypes and mutated domains.
CFTR variant information from The Clinical and Functional Translation of CFTR (CFTR2) database, available at https://cftr2.org. Data retrieved from the March 11, 2019 version of CFTR2. “Occurrences” indicates the number of times a particular disease-causing allele has been sequenced in CF patients and recorded in the database (e.g. F508del CFTR homozygotes contribute two distinct occurrences). “Frequency” reflects the proportion that a particular variant appears among all patient occurrences recorded in CFTR2.
| Variant | % of Patients (U.S.) | CFTR2 Allele | Mutant Domain | Disease Severity | Pharmacological Correction | |
|---|---|---|---|---|---|---|
| Occurrences | Frequency | |||||
| WT | — | — | — | — | None | — |
| F508del | 86.4 | 99061 | 0.69700 | NBD1 | Severe | Moderate |
| N1303K | 2.4 | 2246 | 0.01580 | NBD2 | Severe | Moderate |
| W1282X | 2.3 | 1726 | 0.01220 | NBD2 | Severe | Moderate |
| G85E | 0.6 | 616 | 0.00434 | MSD1 | Severe | Low |
| E92K | <0.5 | 49 | 0.00027 | MSD1 | Severe | Moderate |
| P67L | <0.5 | 239 | 0.00168 | MSD1 | Mild | High |
To compare these variants, we initially sought to test if differences would emerge between a narrower subset of variants. To this end, we prepared microsomes from HEK293 cells either lacking CFTR or transiently expressing wild-type CFTR or F508del CFTR and conducted in vitro ubiquitination reactions as described above. We discovered that F508del CFTR was ubiquitinated to a greater degree than wild-type CFTR, despite being expressed at a lower level (Figure 2A). Meanwhile, ubiquitination was absent in reactions containing microsomes from untransfected (control) HEK293 cells, demonstrating that ubiquitinated CFTR is specifically immunoprecipitated in this assay. Taking into account the relative amount of CFTR precipitated after each reaction, we concluded that in reactions containing Ube1, UbcH5b, and CHIP, F508del CFTR was ubiquitinated approximately two-fold more than the wild-type protein (1.84±0.13; n=2). These data indicate, as anticipated, that the PQC machinery preferentially selects F508del CFTR over wild-type CFTR for degradation. No ubiquitination was observed in reactions with microsomes lacking CFTR. Interestingly addition of purified Hsp70 to reactions failed to enhance ubiquitination in the presence of CHIP, suggesting that abundant Hsc70 and perhaps Hsp70 reside on the microsome surface. Indeed, subsequent western blotting of microsomal membranes with an antibody that detects both Hsp70 isoforms confirmed that HEK293-derived membranes contain these molecular chaperones; attempts to remove endogenous chaperones with stringent washing conditions were unsuccessful (data not shown).
Figure 2. CHIP enhances ubiquitination of F508del CFTR to a greater extent than wild-type CFTR.
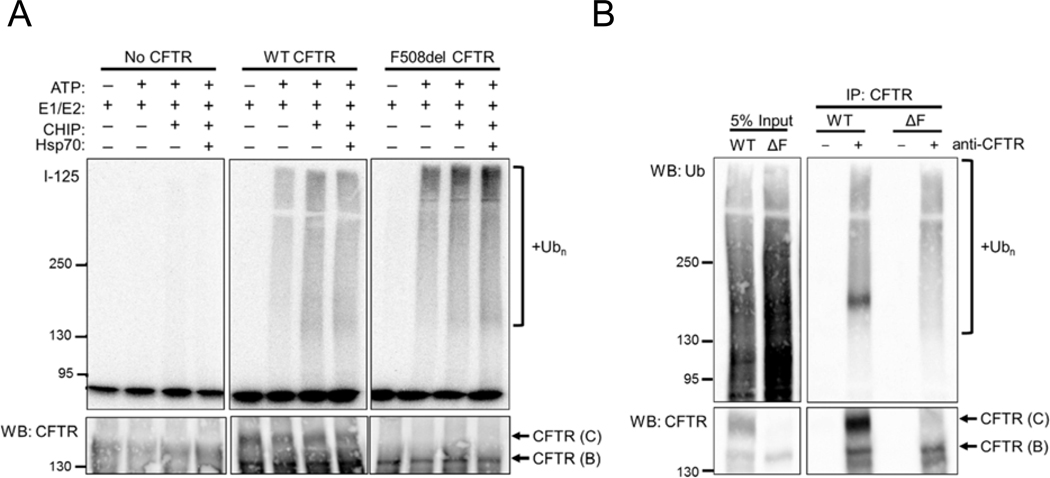
(A) Representative I-125 radiograph (top) and anti-CFTR western blot (bottom) comparing ubiquitination of wild-type (WT) CFTR and F508del CFTR in microsomes prepared from HEK293 cells. Cell were mock transfected or transfected with CDNA3.1 wild-type (WT) CFTR or pCDNA3.1 F508del CFTR prior to microsome preparation. Reactions contained ATP, Ube1 (E1), UbcH5b (E2), CHIP, and Hsp70 as indicated. While ubiquitination was absent in reactions lacking CFTR, polyubiquitination of CFTR was observed in reactions with either wild-type or F508del CFTR. Molecular masses are indicated in kDa. (B) Anti-ubiquitin/anti-HA western blot (top) and anti-CFTR western blot of lysates from HEK293 cells transfected with wild-type (WT) or F508del (ΔF) CFTR expression plasmids as in (A) prior to treatment with MG132 for 1 hr, followed by cell lysis and CFTR immunoprecipitation. CFTR polyubiquitination was observed in immunoprecipitations of each variant with anti-CFTR antibody, as indicated.
To verify that the increased susceptibility of F508del CFTR to ubiquitination recapitulated the situation in live cells, we next expressed wild-type and F508del CFTR in HEK293 cells, treated the cells with MG132 for 1 hr to inhibit the proteasome and accumulate polyubiquitinated CFTR, and immunoprecipitated the protein from cell lysates. As shown in Figure 2B, similar quantities of ubiquitinated wild-type and F508del CFTR were observed, but steady-state expression of the wild-type channel was substantially greater than that of the misfolded variant (~4.5 fold). Therefore, the F508del CFTR channels are ubiquitination to a greater extent per protein than wild-type CFTR, as anticipated based on their relative folding status. Since this in vitro assay faithfully recapitulated the increased selection of a misfolded form CFTR by the ubiquitination machinery, we next sought to compare ubiquitination efficiencies between the subset of CFTR variants outlined in Table 1.
To this end, we expressed each of the CFTR variants along with the wild-type protein in HEK293 cells, collected microsomes, and assayed the membranes either in the presence or absence of added CHIP. Consistent with the results in Figure 2A, F508del CFTR was again ubiquitinated greater than two-fold higher than wild-type CFTR (2.42±0.07; n=6) (Figure 3A, B). In contrast, the other variants were ubiquitinated to intermediate degrees, even when CHIP was added. Although these proteins are differentially misfolded and retained in the ER [83–86], CHIP uniformly enhanced ubiquitination of all variants, but displayed a modest effect that was restricted to a relatively small range of molecular weights (Figure 3C). We also found that the CHIP-dependent enhancement of ubiquitination required binding to Hsc70 or Hsp70, since addition of a CHIP mutant lacking its N-terminal TPR domain (CHIP134–303), which is required for Hsp70/Hsc70 binding [67], failed to enhance CFTR ubiquitination (data not shown). Based on the relative magnitude of the cellular defects of the CFTR variants examined [36, 37, 83–86] (also see Discussion), these results suggest that the protein quality control/ubiquitination machinery is faithfully reconstituted in the in vitro assay.
Figure 3. Misfolded CFTR variants exhibit variable levels of ubiquitination in vitro.

(A) Representative I-125 radiograph (top) and anti-CFTR western blot (bottom) of reactions with CFTR-containing microsomes prepared from HEK293 cells. The variants were expressed from pcDNA3.1 prior to microsome isolation. All reactions contained His-Ube1 (E1), UbcH5b (E2), and ATP. Where indicated, reactions were supplemented with CHIP. Molecular masses are indicated in kDa. (B) Quantification of all reactions performed as in (A), indicating average ubiquitination adjusted for relative CFTR expression levels, normalized to wild-type (WT) CFTR. All variants, except for N1303K CFTR, were ubiquitinated to a significantly greater degree per protein than the wild-type channel (p<0.05), and F508del CFTR was ubiquitinated to a significantly greater degree than all other disease-causing variants tested. n=6; error bars depict standard errors of the mean. Mutated domain and relative pharmacological correction is indicated for each variant. Green arrow: highly corrected; yellow arrow: modestly corrected; red arrows: poorly corrected. (C) Quantification of all reactions performed as in (A), indicating the average fold enhancement of CFTR ubiquitination between reactions containing the designated variant with CHIP (+ CHIP) compared to reactions without added CHIP (- CHIP). Only ubiquitinated species between 150 and 200 kDa were quantified (or between 100 and 150 kDa for the truncated W1282X CFTR variant). Fold enhancement in the presence of CHIP does not differ significantly between the variants tested (p>0.05), however CHIP significantly enhances ubiquitination of all variants compared to reactions lacking CHIP, as quantified in (B). n=6; error bars depict standard errors of the mean.
In addition to the known E3 ubiquitin ligases that have been reported to modify CFTR [51], numerous others might contribute to this critical event in the selection of ERAD substrates [9, 87]. Since many of these ligases are integral membrane proteins or are ER-associated, their presence would have obscured the magnitude of CHIP-dependent ubiquitination. We therefore sought to assess CHIP-supported ubiquitination independent of the contribution of any other E3 ubiquitin ligase. To examine this possibility, we assayed solubilized wild-type CFTR isolated from transfected human cells [75] in place of microsomal CFTR (Figure 4A). As anticipated, CHIP ubiquitinated wild-type CFTR in the presence of ATP (Figures 4B). That CHIP ubiquitinated wild-type CFTR without the addition of purified Hsp70 suggests that the chaperone may co-purify with CFTR, or that CHIP has an inherent affinity for solubilized CFTR, which might be partially unfolded. Notably, CHIP has been found to exhibit endogenous chaperone-like activity [88]. These data support the results of assays with microsomal CFTR on the contribution of CHIP during CFTR quality control. Moreover, because the combination of CHIP and highly enriched CFTR produced ubiquitinated species ranging from 150–250 kDa (Figure 4B), species greater than 250 kDa produced during reactions using microsomes (e.g., Figure 1B) likely result from the activities of other E3 ubiquitin ligases. Therefore, our assay sets the stage to compare the relative contribution of each ubiquitin ligase during the targeting of CFTR for ERAD.
Figure 4. CHIP ubiquitinates highly enriched solubilized wild-type CFTR in vitro.

(A) Schematic depicting the procedure used to ubiquitinate enriched wild-type CFTR. (B) I-125 radiograph (top) and anti-CFTR western blot (bottom) depicting in vitro ubiquitination of wild-type (WT) CFTR that was highly enriched from a stably expressing HEK293 cell line, either in the absence or presence of CHIP. Molecular masses are indicated in kDa.
4. Discussion
While numerous methods exist to identify and characterize the contributions of E3 ubiquitin ligases on the turnover of substrates in the secretory pathway in cells, in vitro reconstitutions can dissect the mechanism of how specific enzymes catalyze this process. Here, we have presented an in vitro ubiquitination assay to study ubiquitin ligases that contribute to the degradation of CFTR during its selection for ERAD.
We show that disease-causing, class II CFTR variants —which exhibit folding defects that trigger degradation by the ERAD pathway [80]— differ in their susceptibilities to the ubiquitination cascade. F508del CFTR was the most susceptible of all variants tested, suggesting that this common allele is the most catastrophically misfolded amongst those tested and is thus especially predisposed to ubiquitination and degradation. This result is in-line with other studies examining a large number of CFTR alleles, which similarly suggested that F508del CFTR is one of the most poorly folded variants [50]. Although we tested a smaller range of variants, we deliberately focused on those containing amino acid substitutions in distinct domains and exhibiting unique susceptibilities to pharmacological correction (Table 1).
We next found that the level of ubiquitination failed to correlate with either the residence of the mutation or its ability to be corrected. Even so, it is notable that P67L, which is easily corrected [83], was ubiquitinated to an intermediate degree relative to wild-type and F508del CFTR. Other variants were similarly ubiquitinated, such as G85E and E92K CFTR. In contrast to P67L, G85E and E92K CFTR are poorly corrected and cause severe symptoms in CF patients [84, 89–91]. Earlier studies with G85E, which introduces a charged glutamate residue into the first transmembrane span of MSD1, proposed that interactions between the transmembrane spans that comprise MSD1 are weakened [92, 93]. An inherent difficulty in establishing stable contacts between these transmembrane spans when this residue is introduced is thought to be why G85E CFTR remains so intractable to correction and potentiation, even when drug cocktails that synergistically augment the function of other misfolding variants are used [94, 95]. On the other hand, P67L CFTR confers a less obtrusive substitution, one that is both chemically conservative and occurs near— but not within —the first transmembrane span of MSD1, as suggested by a multitude of predictive algorithms [92]. Therefore, P67L CFTR likely exhibits more stable folding between the MSD1 transmembrane spans than either G85E or E92K CFTR. Therefore, P67L CFTR is more amenable to correction. It remains surprising that these three variants are ubiquitinated to a similar degree, as we have observed, but potentially any defect in MSD1 assembly, however minor, causes channels to be ubiquitinated more aggressively than wild type CFTR. Upon correction, ubiquitination of these channels may then revert to levels more similar to the wild-type conformation. Additional testing with CFTR variants treated with corrector compounds prior to the isolation of membranes and in vitro assays will be necessary to determine if this is indeed the case (see below).
Another variant, W1282X, was also ubiquitinated to an intermediate degree compared to the wild-type and F508del CFTR variants. The truncation, which lacks the multiple folding defects evident for F508del [96], truncates NBD2, thereby aborting assembly with NBD1. NBD2 is thought to play a key role in the posttranslational assembly of individual domains in the fully folded CFTR protein [97]. Thus, it might be surprising that N1303K CFTR, which also alters NBD2, was ubiquitinated to the same degree as the wild-type protein. Nonetheless, data suggest that this variant is primarily handled by autophagy, rather than ERAD [98]. Together, the testing of a larger subset of class II variants will be necessary to further test these and other hypotheses.
That purified CHIP enhanced ubiquitination of all assayed variants to a similar degree may also appear surprising (Figure 3C). Given that the ligase is thought to have the greatest activity upon fully translated CFTR —i.e., after translation of NBD2 [63]— one might expect that CFTR would be more extensively ubiquitinated due to severe mutations within this domain, yet W1282X CFTR displayed a similar degree of CHIP-dependent ubiquitination as the other variants. As noted above, this truncation likely affects numerous inter-domain interactions, as do each of the other variants. Even wild-type CFTR displayed a similar relative degree of CHIP-dependent enhanced ubiquitination. We suggest that this is a consequence of the diminished basal ubiquitination of wild-type CFTR by the membrane-resident E3 ubiquitin ligases, providing a greater opportunity for purified CHIP to polyubiquitinate it. Regardless, the overall modest effect of CHIP on the ubiquitination of all of the variants strongly suggests that maximal ubiquitination also requires the activity of membrane-inserted, ER-localized E3 ubiquitin ligases that also act on CFTR, i.e., RMA1, RNF185, and gp78 [63, 65, 66]. In the future, selectively silencing the genes encoding each of these ligases will allow for an analysis of the contribution of each one on the ubiquitination of CFTR in microsomes. In principle any integral membrane ERAD substrate that can be immunoprecipitated could be assayed similarly, such as the alpha subunit of the epithelial sodium channel (ENaC), which we previously showed is targeted for ubiquitination in vitro when incubated with yeast cytosol [99]. However, one must note the likelihood that ubiquitination of integral membrane ERAD substrates in this assay reflects not only that of proteins residing in ER membranes, but also that of some subset of proteins localized to post-ER compartments, as evidenced by the presence of maturely glycosylated CFTR (i.e., “band C” CFTR). In fact, the protocol for the microsome preparation used in our studies was originally developed to elucidate Golgi-associated processes [100]. Therefore, membranes derived from the Golgi network, endosomes, or possibly even the plasma membrane are also expected to be present to some degree. While the majority of class II CFTR variants are anticipated to be retained in the ER, other ERAD substrates may be distributed differently between cellular compartments, so care must be taken in determining the suitability of our assay for any substrate examined.
Finally, it was gratifying that purified CHIP ubiquitinated solubilized CFTR. Although the lack of a requirement for an Hsp70 chaperone in this reaction may arise from the innate chaperone-like activity of the ligase [88], this assay is ideally suited to elucidate the contribution of any protein quality factor on CFTR ubiquitination in a minimal system. If a subset of misfolded CFTR variants could be purified and examined, this assay would also provide a direct way to test the susceptibility of variants to CHIP independent of the contributions of any other E3 ubiquitin ligase. This system will also allow for direct tests of chemical chaperones and CFTR correctors/potentiators that are thought to bind directly to CFTR and alter its conformation and perhaps susceptibility to the ERAD pathway. In parallel, CFTR-containing microsomes can now be prepared from cells treated with these same compounds and ubiquitination monitored, as described here. Given that these microsomes are also likely to contain rough ER, as implied by western blotting detection of ribosomal subunits (data not shown), this assay could also be adapted for studies in which in vitro translation is needed. Together, our new tools significantly expand the scope of assays that can be used to probe the molecular mechanisms underlying an incurable and devastating disease.
Highlights.
Mutant forms of CFTR, which cause Cystic Fibrosis, are ubiquitinated and degraded
Assays that reconstitute CFTR ubiquitination will help identify therapeutics
ER membranes with wild-type and diseased CFTR variants can be isolated
CFTR ubiquitination can then be recapitulated with purified components
Misfolded disease-causing CFTR variants exhibit enhanced ubiquitination
Acknowledgments
The authors wish to thank Jennifer Goeckeler-Fried, Dr. Eric Sorscher, Dr. Christopher Guerriero, Dr. George Michael Preston, Dr. Patrick Thibodeau, Dr. Saurav Misra, Dr. Zhengrong Yang, and Dr. John Kappes for reagents, advice, and valuable discussions during the development of these assays. This work was supported by grant R35GM131732 from the National Institutes of Health and by grants BRODSK18G0 and ESTABR17H0 from the Cystic Fibrosis Foundation. Additional support was provided by grant KAPPES18XX0 from the Cystic Fibrosis Foundation.
Abbreviations:
- ERAD
endoplasmic reticulum-associated degradation
- CFTR
cystic fibrosis transmembrane conductance regulator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sun Z and Brodsky JL, Protein quality control in the secretory pathway. J Cell Biol, 2019. 218(10): p. 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fregno I and Molinari M, Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit Rev Biochem Mol Biol, 2019. 54(2): p. 153–163. [DOI] [PubMed] [Google Scholar]
- 3.Ghaemmaghami S, et al. , Global analysis of protein expression in yeast. Nature, 2003. 425(6959): p. 737–41. [DOI] [PubMed] [Google Scholar]
- 4.Venter JC, et al. , The sequence of the human genome. Science, 2001. 291(5507): p. 1304–51. [DOI] [PubMed] [Google Scholar]
- 5.Komander D and Rape M, The Ubiquitin Code. Annu Rev Biochem, 2012. 81: p. 203–229. [DOI] [PubMed] [Google Scholar]
- 6.Varshavsky A, The ubiquitin system, an immense realm. Annu Rev Biochem, 2012. 81: p. 167–176. [DOI] [PubMed] [Google Scholar]
- 7.Richly H, et al. , A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell, 2005. 120(1): p. 73–84. [DOI] [PubMed] [Google Scholar]
- 8.Ruggiano A, Foresti O, and Carvalho P, Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol, 2014. 204(6): p. 869–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christianson JC and Ye Y, Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol, 2014. 21(4): p. 325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Needham PG, Guerriero CJ, and Brodsky JL, Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb Perspect Biol, 2019. 11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houck SA and Cyr DM, Mechanisms for quality control of misfolded transmembrane proteins. Biochim Biophys Acta, 2012. 1818(4): p. 1108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welch WJ, Role of quality control pathways in human diseases involving protein misfolding. Semin Cell Dev Biol, 2004. 15(1): p. 31–8. [DOI] [PubMed] [Google Scholar]
- 13.Hessa T, et al. , Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature, 2005. 433(7024): p. 377–81. [DOI] [PubMed] [Google Scholar]
- 14.Hessa T, et al. , Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature, 2007. 450(7172): p. 1026–30. [DOI] [PubMed] [Google Scholar]
- 15.Chen M and Zhang JT, Topogenesis of cystic fibrosis transmembrane conductance regulator (CFTR): regulation by the amino terminal transmembrane sequences. Biochemistry, 1999. 38(17): p. 5471–7. [DOI] [PubMed] [Google Scholar]
- 16.Hessa T, White SH, and von Heijne G, Membrane insertion of a potassium-channel voltage sensor. Science, 2005. 307(5714): p. 1427. [DOI] [PubMed] [Google Scholar]
- 17.Buck TM, et al. , A novel tripartite motif involved in aquaporin topogenesis, monomer folding and tetramerization. Nat Struct Mol Biol, 2007. 14(8): p. 762–9. [DOI] [PubMed] [Google Scholar]
- 18.Shurtleff MJ, et al. , The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ojemalm K, et al. , Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol Cell, 2012. 45(4): p. 529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, et al. , Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain. Proc Natl Acad Sci U S A, 2007. 104(20): p. 8263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher EA, et al. , The AAA-ATPase p97 facilitates degradation of apolipoprotein B by the ubiquitin-proteasome pathway. J Lipid Res, 2008. 49(10): p. 2149–60. [DOI] [PubMed] [Google Scholar]
- 22.O’Donnell BM, et al. , Endoplasmic reticulum-associated degradation of the renal potassium channel, ROMK, leads to type II Bartter syndrome. J Biol Chem, 2017. 292(31): p. 12813–12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu BH, Strickland EH, and Thomas PJ, Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem, 1997. 272(25): p. 15739–15744. [DOI] [PubMed] [Google Scholar]
- 24.Buck TM and Skach WR, Differential stability of biogenesis intermediates reveals a common pathway for aquaporin-1 topological maturation. J Biol Chem, 2005. 280(1): p. 261–9. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Z, et al. , HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem, 1998. 273(33): p. 21061–6. [DOI] [PubMed] [Google Scholar]
- 26.Staub O, et al. , Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J, 1997. 16(21): p. 6325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu D, et al. , Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J Biol Chem, 1996. 271(37): p. 22791–5. [DOI] [PubMed] [Google Scholar]
- 28.Ward CL and Kopito RR, Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem, 1994. 269(41): p. 25710–25718. [PubMed] [Google Scholar]
- 29.Cheng SH, et al. , Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell, 1990. 63(4): p. 827–834. [DOI] [PubMed] [Google Scholar]
- 30.Ward CL, Omura S, and Kopito RR, Degradation of CFTR by the ubiquitin-proteasome pathway. Cell, 1995. 83(1): p. 121–7. [DOI] [PubMed] [Google Scholar]
- 31.Jensen TJ, et al. , Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell, 1995. 83(1): p. 129–35. [DOI] [PubMed] [Google Scholar]
- 32.Sato S, Ward CL, and Kopito RR, Cotranslational ubiquitination of cystic fibrosis transmembrane conductance regulator in vitro. J Biol Chem, 1998. 273(13): p. 7189–7192. [DOI] [PubMed] [Google Scholar]
- 33.Lukacs GL and Verkman AS, CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med, 2012. 18(2): p. 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lukacs GL, et al. , Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J, 1994. 13(24): p. 6076–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoelen H, et al. , The primary folding defect and rescue of DeltaF508 CFTR emerge during translation of the mutant domain. PLoS One, 2010. 5(11): p. e15458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendoza JL, et al. , Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell, 2012. 148(1–2): p. 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabeh WM, et al. , Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell, 2012. 148(1–2): p. 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukacs GL, et al. , The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem, 1993. 268(29): p. 21592–8. [PubMed] [Google Scholar]
- 39.Okiyoneda T, et al. , Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science, 2010. 329(5993): p. 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukacs GL, et al. , Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem J, 1997. 328 ( Pt 2): p. 353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma M, et al. , Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem, 2001. 276(12): p. 8942–8950. [DOI] [PubMed] [Google Scholar]
- 42.Dalemans W, et al. , Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature, 1991. 354(6354): p. 526–528. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, et al. , Thermally unstable gating of the most common cystic fibrosis mutant channel (DeltaF508): “rescue” by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. J Biol Chem, 2011. 286(49): p. 41937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang H, et al. , Nanomolar affinity small molecule correctors of defective Delta F508-CFTR chloride channel gating. J Biol Chem, 2003. 278(37): p. 35079–35085. [DOI] [PubMed] [Google Scholar]
- 45.Van Goor F, et al. , Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A, 2009. 106(44): p. 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pedemonte N, et al. , Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest, 2005. 115(9): p. 2564–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okiyoneda T, et al. , Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol, 2013. 9(7): p. 444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loo TW, Bartlett MC, and Clarke DM, Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem Pharmacol, 2013. 86(5): p. 612–619. [DOI] [PubMed] [Google Scholar]
- 49.Lopes-Pacheco M, et al. , Correctors rescue CFTR mutations in nucleotide-binding domain 1 (NBD1) by modulating proteostasis. Chembiochem, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sosnay PR, et al. , Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet, 2013. 45(10): p. 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Estabrooks S and Brodsky JL, Regulation of CFTR Biogenesis by the Proteostatic Network and Pharmacological Modulators. Int J Mol Sci, 2020. 21(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukuda R and Okiyoneda T, Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Ubiquitylation as a Novel Pharmaceutical Target for Cystic Fibrosis. Pharmaceuticals (Basel), 2020. 13(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strickland E, et al. , The molecular chaperone Hsc70 assists the in vitro folding of the N-terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem, 1997. 272(41): p. 25421–25424. [DOI] [PubMed] [Google Scholar]
- 54.Loo MA, et al. , Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J, 1998. 17(23): p. 6879–6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bagdany M, et al. , Chaperones rescue the energetic landscape of mutant CFTR at single molecule and in cell. Nat Commun, 2017. 8(1): p. 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Youker RT, et al. , Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell, 2004. 15(11): p. 4787–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meacham GC, et al. , The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J, 1999. 18(6): p. 1492–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farinha CM, et al. , The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem J, 2002. 366(Pt 3): p. 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H, et al. , Cysteine string protein monitors late steps in cystic fibrosis transmembrane conductance regulator biogenesis. J Biol Chem, 2006. 281(16): p. 11312–11321. [DOI] [PubMed] [Google Scholar]
- 60.Baaklini I, et al. , Selective Binding of HSC70 and its Co-Chaperones to Structural Hotspots on CFTR. Sci Rep, 2020. 10(1): p. 4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmidt BZ, et al. , Cysteine string protein promotes proteasomal degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) by increasing its interaction with the C terminus of Hsp70-interacting protein and promoting CFTR ubiquitylation. J Biol Chem, 2009. 284(7): p. 4168–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grove DE, et al. , The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol Biol Cell, 2011. 22(3): p. 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Younger JM, et al. , Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell, 2006. 126(3): p. 571–582. [DOI] [PubMed] [Google Scholar]
- 64.Tomati V, et al. , Genetic Inhibition Of The Ubiquitin Ligase Rnf5 Attenuates Phenotypes Associated To F508del Cystic Fibrosis Mutation. Sci Rep, 2015. 5: p. 12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.El Khouri E, et al. , RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J Biol Chem, 2013. 288(43): p. 31177–31191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morito D, et al. , Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol Biol Cell, 2008. 19(4): p. 1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ballinger CA, et al. , Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol, 1999. 19(6): p. 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meacham GC, et al. , The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol, 2001. 3(1): p. 100–105. [DOI] [PubMed] [Google Scholar]
- 69.Fu L, et al. , Dab2 is a key regulator of endocytosis and post-endocytic trafficking of the cystic fibrosis transmembrane conductance regulator. Biochem J, 2012. 441(2): p. 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fu L, et al. , DeltaF508 CFTR surface stability is regulated by DAB2 and CHIP-mediated ubiquitination in post-endocytic compartments. PLoS One, 2015. 10(4): p. e0123131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Younger JM, et al. , A foldable CFTRΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J Cell Biol, 2004. 167(6): p. 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Han ST, et al. , Residual function of cystic fibrosis mutants predicts response to small molecule CFTR modulators. JCI Insight, 2018. 3(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carvalho AF, et al. , High-yield expression in Escherichia coli and purification of mouse ubiquitin-activating enzyme E1. Mol Biotechnol, 2012. 51(3): p. 254–61. [DOI] [PubMed] [Google Scholar]
- 74.Zhang H, et al. , A bipartite interaction between Hsp70 and CHIP regulates ubiquitination of chaperoned client proteins. Structure, 2015. 23(3): p. 472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hildebrandt E, et al. , A stable human-cell system overexpressing cystic fibrosis transmembrane conductance regulator recombinant protein at the cell surface. Mol Biotechnol, 2015. 57(5): p. 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakatsukasa K and Brodsky JL, in vitro reconstitution of the selection, ubiquitination, and membrane extraction of a polytopic ERAD substrate. Methods Mol Biol, 2010. 619: p. 365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakasone MA, et al. , Structural Basis for the Inhibitory Effects of Ubistatins in the Ubiquitin-Proteasome Pathway. Structure, 2017. 25(12): p. 1839–1855 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ye Z, et al. , Symmetry breaking during homodimeric assembly activates an E3 ubiquitin ligase. Sci Rep, 2017. 7(1): p. 1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Welsh MJ and Smith AE, Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell, 1993. 73(7): p. 1251–1254. [DOI] [PubMed] [Google Scholar]
- 80.Veit G, et al. , From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell, 2016. 27(3): p. 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Goor F, et al. , Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros, 2014. 13(1): p. 29–36. [DOI] [PubMed] [Google Scholar]
- 82.Gilfillan A, et al. , P67L: a cystic fibrosis allele with mild effects found at high frequency in the Scottish population. J Med Genet, 1998. 35(2): p. 122–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sabusap CM, et al. , Analysis of cystic fibrosis-associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases. JCI Insight, 2016. 1(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lopes-Pacheco M, et al. , Combination of Correctors Rescues CFTR Transmembrane-Domain Mutants by Mitigating their Interactions with Proteostasis. Cell Physiol Biochem, 2017. 41(6): p. 2194–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Haggie PM, et al. , Correctors and Potentiators Rescue Function of the Truncated W1282X-CFTR Translation Product. J Biol Chem, 2016. 292(3): p. 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rapino D, et al. , Rescue of NBD2 mutants N1303K and S1235R of CFTR by small-molecule correctors and transcomplementation. PLoS One, 2015. 10(3): p. e0119796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Preston GM and Brodsky JL, The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem J, 2017. 474(4): p. 445–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kopp Y, et al. , CHIP as a membrane-shuttling proteostasis sensor. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Decaestecker K, et al. , Genotype/phenotype correlation of the G85E mutation in a large cohort of cystic fibrosis patients. Eur Respir J, 2004. 23(5): p. 679–84. [DOI] [PubMed] [Google Scholar]
- 90.Gené GG, et al. , N-terminal CFTR missense variants severely affect the behavior of the CFTR chloride channel. Hum Mutat, 2008. 29(5): p. 738–49. [DOI] [PubMed] [Google Scholar]
- 91.Stanke F, et al. , Diversity of the basic defect of homozygous CFTR mutation genotypes in humans. J Med Genet, 2008. 45(1): p. 47–54. [DOI] [PubMed] [Google Scholar]
- 92.Patrick AE, et al. , Alteration of CFTR transmembrane span integration by disease-causing mutations. Mol Biol Cell, 2011. 22(23): p. 4461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xiong X, et al. , Structural cues involved in endoplasmic reticulum degradation of G85E and G91R mutant cystic fibrosis transmembrane conductance regulator. J Clin Invest, 1997. 100(5): p. 1079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Phuan PW, et al. , Nanomolar-potency ‘co-potentiator’ therapy for cystic fibrosis caused by a defined subset of minimal function CFTR mutants. Sci Rep, 2019. 9(1): p. 17640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ensinck M, et al. , Phenotyping of Rare CFTR Mutations Reveals Distinct Trafficking and Functional Defects. Cells, 2020. 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mutyam V, et al. , Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Du K and Lukacs GL, Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell, 2009. 20(7): p. 1903–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu Q, et al. , Rescue of CFTR NBD2 mutants N1303K and S1235R is influenced by the functioning of the autophagosome. J Cyst Fibros, 2018. 17(5): p. 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Buck TM, et al. , The endoplasmic reticulum-associated degradation of the epithelial sodium channel requires a unique complement of molecular chaperones. Mol Biol Cell, 2010. 21(6): p. 1047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Balch WE and Rothman JE, Characterization of protein transport between successive compartments of the Golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch Biochem Biophys, 1985. 240(1): p. 413–25. [DOI] [PubMed] [Google Scholar]