

Abstract
Volume-regulated anion channels (VRACs) are critically involved in regulating cell volume, and leucine-rich repeat-containing protein 8A (LRRC8A, SWELL1) is an obligatory subunit of VRACs. Cell swelling occurs early after brain ischemia, but it is unclear whether neuronal LRRC8a contributes to ischemia-induced glutamate release and brain injury. We found that Lrrc8a conditional knockout (Lrrc8a-cKO) mice produced by crossing NestinCre+/− with Lrrc8aflox+/+ mice died 7–8 weeks of age, indicating an essential role of brain LRRC8A for survival. Middle cerebral artery occlusion (MCAO) caused an early increase in LRRC8A protein levels in the hippocampus in wild-type (WT) mice. Whole-cell patch-clamp recording in brain slices revealed that oxygen-glucose deprivation significantly increased the amplitude of VRAC currents in hippocampal CA1 neurons in WT but not in Lrrc8a-cKO mice. Hypotonicity increased the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in hippocampal CA1 neurons in WT mice, and this was abolished by DCPIB, a VRAC blocker. But in Lrrc8a-cKO mice, hypotonic solution had no effect on the frequency of sEPSCs in these neurons. Furthermore, the brain infarct volume and neurological severity score induced by MCAO were significantly lower in Lrrc8a-cKO mice than in WT mice. In addition, MCAO-induced increases in cleaved caspase-3 and calpain activity, two biochemical markers of neuronal apoptosis and death, in brain tissues were significantly attenuated in Lrrc8a-cKO mice compared with WT mice. These new findings indicate that cerebral ischemia increases neuronal LRRC8A-dependent VRAC activity and that VRACs contribute to increased glutamatergic input to hippocampal neurons and brain injury caused by ischemic stroke.
Keywords: electrophysiology, excitotoxicity, ion channel, reperfusion, presynaptic, SWELL1 channel, synaptic plasticity, NMDA receptor
Graphical Abstract
Introduction
Brain ischemia initially causes oxygen and glucose deprivation and relatively acute (from hours to the first few days) cell death in the region where blood flow is severely restricted, ultimately leading to an infarct core. The peri-infarct tissue, known as ischemic penumbra, is partially and chronically (days to weeks) injured after ischemia (Bandera et al., 2006). Because of the slow time course of cell death in the penumbra, it is considered salvageable brain tissue and a target for neuroprotective interventions. Excessive Na+ and Ca2+ influx and cell swelling mediated by multiple receptors and ion channels are the earliest evidence of excitotoxicity in neurons and non-neuronal cells (Rungta et al., 2015; Song and Yu, 2014). It is thus important to determine how cell swelling is involved in ischemic brain injury.
Cell swelling usually initiates the regulatory volume decrease process mediated mainly by volume-regulated anion channels (VRACs), which are formed by multiple different leucine-rich repeat-containing protein 8 (LRRC8) family members. LRRC8A (SWELL1) is the obligatory subunit and can form homo- or heterohexameric channels with its paralogs LRRC8B, C, D, and E (Bao et al., 2018; Qiu et al., 2014; Voss et al., 2014). Pharmacologically blocking VRACs reduces cerebral infarction in animal models (Kimelberg et al., 2000; Zhang et al., 2008). However, all VRAC blockers, including 4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB), have off-target effects (Bowens et al., 2013). VRACs are widely expressed in astrocytes and neurons in the brain, although recent work has largely focused on the role of VRACs expressed on astrocytes in ischemic brain injury. For example, in cultured astrocytes, LRRC8A knockdown attenuates ischemia-induced glutamate release (Wilson et al., 2019). Furthermore, conditional knockout of Lrrc8a in astrocytes generated with GfapCre/+ mice reduces ischemic injury and glutamate release (Yang et al., 2019). It remains unclear how brain ischemia affects the activity of neuronal VRACs in the penumbra and whether LRRC8A-dependent VRACs play a role in ischemic neuronal death.
Glutamate excitotoxicity is a key mechanism underlying ischemic brain injury (Luo et al., 2018; Rossi et al., 2000; Simon et al., 1984). Glutamate is classically released from presynaptic terminals via Ca2+-dependent vesicular exocytosis involving voltage-activated Ca2+ channels and N-methyl-D-aspartate (NMDA) receptors (Corlew et al., 2008; Katayama et al., 1991). Swelling-activated VRACs typically drive intracellular Cl− out and are permeable to small osmolytes, including glutamate, aspartate, and taurine, to decrease intracellular osmotic water in many cell types (Osei-Owusu et al., 2018). During brain ischemia, VRACs may be a major source of glutamate release because blocking VRACs with tamoxifen attenuates glutamate release in the penumbra (Feustel et al., 2004). Although neuronal swelling can increase excitability in hippocampal CA1 neurons via NMDA receptors (Lauderdale et al., 2015), the roles of LRRC8A-dependent VRACs in increased glutamatergic input to neurons and ischemic brain injury are not fully understood.
In the present study, we tested the hypothesis that cerebral ischemia potentiates the activity of neuronal LRRC8A-dependent VRACs, which contribute to increased glutamatergic input to hippocampal neurons and ischemic brain injury. By using Lrrc8a conditional knockout (Lrrc8a-cKO) mice, we provide new evidence that ischemia increases the activity of LRRC8A-dependent VRACs in hippocampal neurons, which potentiates glutamate release and contributes to ischemic brain injury. Our study, using a different mouse model of Lrrc8a KO, provides an independent validation about the role of VRACs in brain ischemia suggested by a recent study focusing on astrocytic VRACs (Yang et al., 2019). Our findings offer additional new insight beyond astrocytic VRACs in the pathophysiology of ischemic stroke.
Methods and Materials
Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center and were performed in accordance with the National Institutes of Health guidelines for the ethical use of animals. NestinCre+/− mice were obtained from Jackson Laboratory (C57BL/6 genetic background; #003771, Bar Harbor, ME). Lrrc8aflox+/+ mice (genetic background C57BL/6) were generated as described previously (Zhang et al., 2017), and the LoxP sites flanked the protein coding region of exon3 in the Lrrc8a gene. Lrrc8a-cKO (NestinCre/+::Lrrc8aflox/flox) mice were produced by crossing male NestinCre+/− mice with female Lrrc8aflox+/+ mice. Age-matched littermates (NestinCre/-::Lrrc8aflox/flox) were used as wild-type (WT) controls. Blood pressure in mice was measured using a tail-cuff method (CODA Monitor, Kent Scientific, Torrington, CT) (Li et al., 2015; Ma et al., 2019). All mice were housed in a pathogen-free environment (24 ± 2 °C, 12 h light/dark cycle) with free access to standard laboratory food and water ad libitum.
Open-field test
The open-field test was performed as we described previously (Zhou et al., 2018). The test apparatus was purchased from Noldus (Boston, MA) with a size of 40 cm (length) × 40 cm (width) × 35 cm (height). Within the open-field testing area, a square of 24 cm (length) × 24 cm (width) in the center was defined as the inner zone, and the remaining space was defined as the outer zone. Mice were placed in a quiet procedure room for 30 min before starting the test. Prior to each test, 95% ethanol was used to remove any scent clues left by the previous mouse. Mice were placed at the center of the testing area and were allowed to freely move for 10 min. The travel path of each mouse was monitored by the Color GigE Camera (Noldus). Total travel distance and total time spent in inner and outer zones were analyzed using the Autotyping software (Patel et al., 2014).
Rotarod test
An accelerating rotarod apparatus was used to conduct the rotarod test (IITC Life Science, Woodland Hills, CA). Each mouse was placed on a rotarod opposite to the rotating direction. In each trial, rotating speed started at 4 rotations/min and progressively increased to a maximum of 40 rotations/min over 300 s (Cai et al., 2013; Zhou et al., 2018). Each animal was given three trials at 20-min intervals. Latency to fall from the accelerating rotating rod in each mouse was automatically registered.
Ischemic stroke model
The middle cerebral artery occlusion (MCAO) method was used to induce brain ischemia, as described previously (Luo et al., 2018). Briefly, mice were anesthetized with isoflurane (1.5% in a mixture of 70% NO2 and 30% O2). A nylon monofilament with a diameter of 0.16 mm was inserted to the right common carotid artery and advanced to the origin of the right middle cerebral artery. The monofilament was kept in place for 90 min and then removed to allow for reperfusion. Sham control mice were subjected to similar surgical procedures without MCAO. During the surgery, the body temperature of mice was kept at 37 ± 0.5°C using a heating pad. After recovery from anesthesia, mice were returned to the animal housing facility. After surgery, buprenorphine (0.1 mg/kg, intraperitoneally) was administrated immediately to minimize postoperative pain.
Neurological deficit was determined on a scale of 0 to 5 in mice, as reported previously (Longa et al., 1989; Luo et al., 2018). The scoring was defined as follows: 0, no observable neurological deficits; 1, failure to extend left forepaw completely (a mild focal neurological deficit); 2, circling to the left (a moderate focal neurological deficit); 3, falling to the left (a severe focal neurological deficit); 4, not walking spontaneously with a depressed level of consciousness; 5, death due to brain ischemia/reperfusion. Mice with a neurological deficit score of 4 or 5 after MCAO were excluded from analysis.
The infarct area in brain coronal sections was identified by staining with 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich, St Louis, MO). Digital images were captured, and infarct volume was quantified using Image-J software and further analyzed according to the following formula: (contralateral hemispheric volume – ipsilateral hemispheric non infarcted volume)/(contralateral hemispheric volume × 2) (Luo et al., 2018; Swanson et al., 1990).
Immunoblotting
While the mice were under deep anesthesia with 2–3% isoflurane, the forebrains of sham control and MCAO mice were removed quickly. The cerebral cortex, hippocampus, and striatum were dissected separately and homogenated in cold RIPA buffer with a 1% protease inhibitor cocktail to extract proteins. Fifty μg proteins from each sample were subjected to polyacrylamide gel electrophoresis (Invitrogen, Carlsbad, CA) and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% milk in tris-buffered saline buffer at room temperature for 1 hour, and then precipitated with mouse anti-LRRC8A (1:1000, #SAB1412855, Sigma-Aldrich), mouse anti-Spectrin alpha chain (1:500, #MAB1622, Millipore Corporation), rabbit anti-cleaved caspase-3 (Asp175) (1:1000, #9661, Cell Signaling Technology, Danvers, MA), or rabbit anti-GAPDH (1:2000, #5174, Cell Signaling Technology). The LRRC8A antibody targeted the amino acid sequence from 711 to 810 at the C-terminus of the mouse LCRR8A protein. Immunoblots were detected with an enhanced chemiluminescence kit (Thermo Fisher Scientific, Waltham, MA), quantified using an Odyssey Fc Imager (LICOR, Lincoln, NE), and normalized using the GAPDH band on the same blot (Luo et al., 2018).
Brain slice preparation and whole-cell recordings
Brain slices were prepared as described previously (Luo et al., 2018). Mice were decapitated under deep anesthesia with 2–3% isoflurane. The brain was quickly removed and placed to an ice-cold artificial cerebrospinal fluid (aCSF) pre-saturated with 95% O2 and 5% CO2. The aCSF contained (in mM): 126.0 NaCl, 3.0 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 10.0 glucose, and 26.0 NaHCO3 (315 ± 5 mOsm). Transversal hippocampus brain slices of 300-μm thickness were sectioned using a microtome (Leica Microsystems Inc.) and then incubated in aCSF for 1 hour at 34°C before further experiments. Hippocampal CA1 pyramidal neurons were visualized under an upright microscope equipped with infrared and differential interference contrast optics (BX51W1, Olympus). Recording pipette resistance was 4–6 MΩ when filled with internal solutions. All external solutions were continually saturated with 95% O2 and 5% CO2 and perfused at 3 ml/min.
VRAC-mediated chloride current, IVRAC, in brain slices was recorded as reported previously (Inoue and Okada, 2007). Cell membrane potential was held at −40 mV, and step voltage was applied from −100 mV to +100 mV in 20-mV increments. The IVRAC was isolated using DCPIB, a specific VRAC blocker (Decher et al., 2001). DCPIB was bath applied via a syringe pump with a final concentration of 30 μM (Wang et al., 2017). The internal solution contained (in mM) 120 NMDG, 120 gluconic acid, 4 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, and 0.3 Na-GTP (pH adjusted to 7.25–7.27 with NMDG; 296 ± 5 mOsm). The external solution contained (in mM) 80 choline-Cl, 20 tetraethyl ammonium chloride, 2.5 KCl, 1.25 NaH2PO4, 2 4-aminopyridine, 4 MgCl2, 26 NaHCO3, 11 glucose, 0.0005 tetrodotoxin (TTX), and 50 mannitol (310 ± 5 mOsm). To record IVRAC in oxygen-glucose deprivation (OGD) conditions, we treated brain slices with an OGD solution for 5 min followed by a 10-min recovery in normal aCSF. The OGD external solution contained (in mM) 126.0 NaCl, 3.0 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 10.0 sucrose, and 26.0 NaHCO3 (315 ± 5 mOsm) continuously gassed with 95% N2/5% CO2 (Luo et al., 2018). The recording signal was processed through a Multiclamp 700B amplifier, filtered at 1 kHz, and digitized by DigiData 1440 at 20 kHz (Molecular Devices). Series resistance was monitored continuously throughout recording processes. Data were not included for analysis if series resistance changed >20%.
Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded using whole-cell voltage-clamp mode. Cell membrane potential was held at −60 mV (Chen and Pan, 2006; Huang et al., 2019; Li et al., 2008). The pipette internal solution contained the following (in mM) 135.0 K-gluconate, 5.0 tetraethyl ammonium, 2.0 MgCl2, 0.5 CaCl2, 5.0 HEPES, 5.0 EGTA, 5.0 Mg-ATP, 0.5 Na-GTP, and 10 lidocaine N-ethyl bromide (adjusted to pH 7.2–7.4 with 1 mol/L KOH; 290–300 mOsm). QX314 was included to block voltage-gated sodium channels. The sEPSCs were recorded in isotonic and hypotonic external solutions (Fiacco et al., 2007; Lauderdale et al., 2015). Isotonic external solution contained (in mM) 80 NaCl, 2.5 KCl, 2.4 CaCl2, 1.3 MgCl2, 1.3 NaH2PO4, 26 NaHCO3, 10 glucose, and 90 mannitol (315 ± 5 mOsm). Hypotonic external solution contained (in mM) 80 NaCl, 2.5 KCl, 2.4 CaCl2, 1.3 MgCl2, 1.3 NaH2PO4, 26 NaHCO3, and 10 glucose (260 ± 5 mOsm).
TTX and 6,7-dinitro-2,3-dihydroxyquinoxaline (DNQX) were obtained from Sigma Aldrich. QX-314 and DCPIB were purchased from Tocris. DCPIB was dissolved with dimethyl sulfoxide (DMSO), and the final concentration of DMSO was 0.05% in the bath solution. DNQX and TTX were dissolved with deionized water to make stock solutions and then diluted with the perfusion solution in electrophysiological experiments.
Data analysis
All data were expressed as mean ± SEM. The frequency and amplitude of sEPSCs were analyzed offline using a peak detection program (MiniAnalysis; Synaptosoft, Leonia, NJ) (Guo et al., 2013; Zhu et al., 2016). Whole-cell VRAC currents were analyzed with Clampfit 9.2 software (Molecular Devices, San Jose, CA), and DCPIB-sensitive VRAC currents were obtained by digitally subtracting VRAC currents recorded in the presence of DCPIB from VRAC currents recorded without DCPIB. A two-tailed Student t test was used for comparison between two groups, and one-way or two-way analysis of variance (ANOVA) with the Bonferroni post-hoc test was used to determine differences among three or more groups. Repeated measures ANOVA with the Dunnett’s post-hoc test was performed to determine differences at different time points within the same group. All statistical analyses were performed using Prism software (version 7; GraphPad Software Inc., La Jolla, CA). P < 0.05 was considered statistically significant.
Results
Phenotypic assessment of Lrrc8a–cKO mice
LRRC8A is an obligatory subunit of VRACs (Qiu et al., 2014; Voss et al., 2014). Nestin is highly expressed in stem and progenitor cells of the neural lineage, and NestinCre+/− mice are commonly used to generate neuronal gene knockout in the entire nervous system (Mignone et al., 2004; Swaminathan et al., 2016; Wang et al., 2015). Compared with NestinCre+/− mice, other available neuronal Cre mice, such as NexCre+/− or CaMKIIαCre+/− mice, remove target genes only in a small population of neurons in limited brain regions. To genetically remove functional VRACs in the brain, we conditionally knocked out Lrrc8a by crossing NestinCre+/− mice with Lrrc8aflox/flox mice. Lrrc8a-cKO mice appeared normal at birth, but all male and female Lrrc8a-cKO mice died unexpectedly at the age of 7–8 weeks old. The cause of mortality was unclear, although some seizure activity was observed in Lrrc8a-cKO mice before death. We thus used 6-week-old Lrrc8a-cKO mice and age- and sex-matched WT control mice for all the experiments described below.
Immunoblotting showed that the protein level of LRRC8A was diminished in various brain regions, including the hippocampus, frontal cortex, and striatum, in 6-week-old Lrrc8a-cKO mice compared with that in wild-type (WT) mice (n = 8–9 mice per group; Fig. 1A). Body weight was significantly lower in Lrrc8a-cKO mice than in WT mice (P < 0.0001, t(22) = 9.427, n = 12 mice per group; Fig. 1B). Systolic blood pressure, measured using a tail-cuff method, was similar in Lrrc8a-cKO and WT mice (n = 6 mice per group; Fig. 1B).
Figure 1. Confirmation and phenotypic assessment of Lrrc8a-cKO mice.
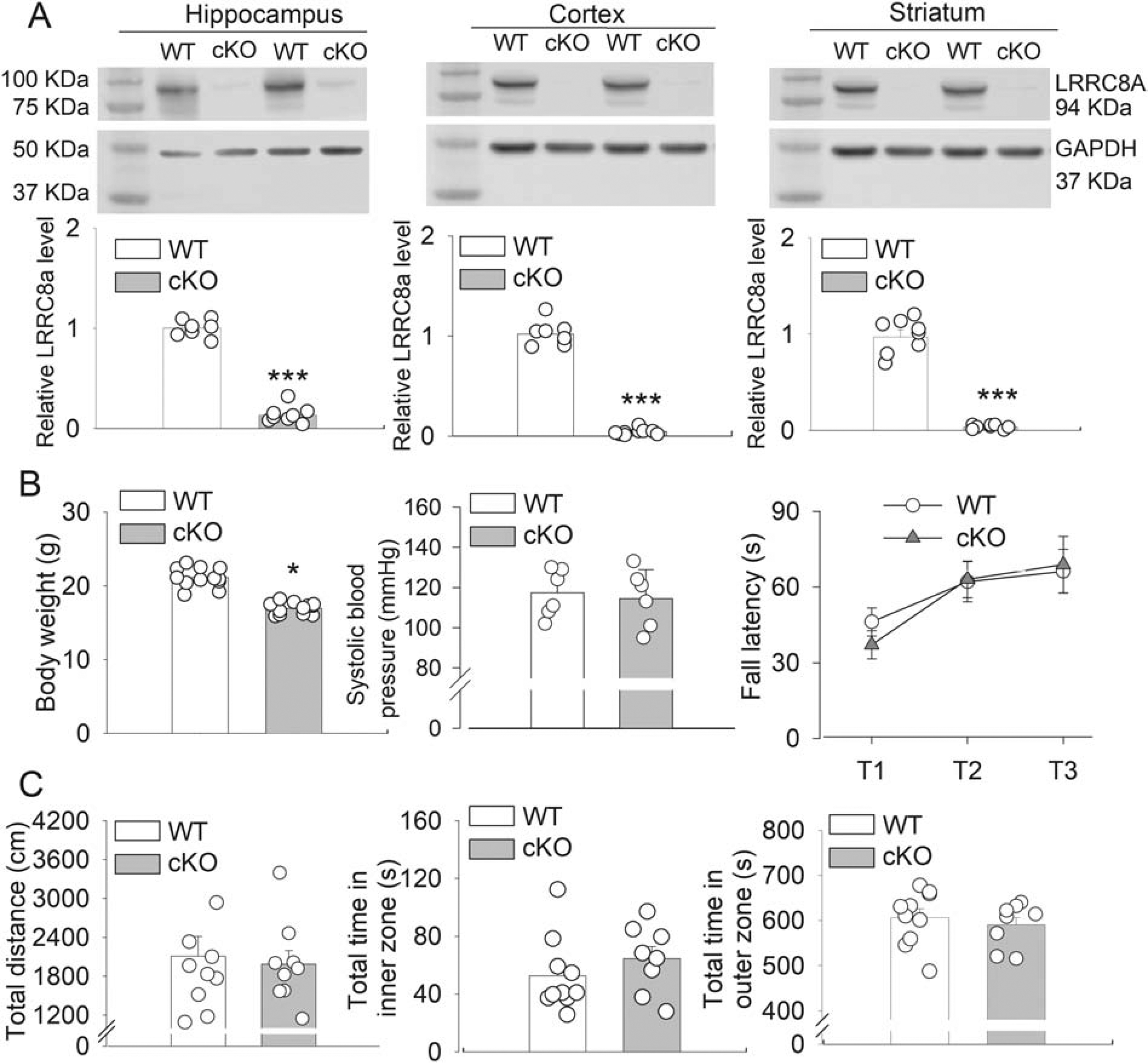
(A) Representative Western blot images and group data show the LRRC8A protein levels in the hippocampus, cerebral cortex, and striatum of 6-week-old WT and Lrrc8a-cKO mice (hippocampus and cortex, n = 8 mice per group; striatum, n = 8 WT mice and 9 Lrrc8a-cKO mice). (B) Summary data show body weight (n = 12 mice per group), systolic blood pressure (n = 6 mice per group), and fall latency in the rotarod test (n = 11 WT mice and 8 Lrrc8a-cKO mice) in 6-week-old WT and Lrrc8a-cKO mice. (C) Group data show the total distance and total time spent in inner and outer zones in the open-field test in 6-week-old WT and Lrrc8a-cKO mice (n = 9 mice per group). *P < 0.05, ***P < 0.001 compared with WT mice.
We used a rotarod test to evaluate motor coordination and learning in Lrrc8a-cKO and WT mice. The falling latencies were similar in these two groups (n = 11 WT mice and 8 Lrrc8a-cKO mice; Fig. 1B). We also used an open-field test to assess general locomotor activity in Lrrc8a-cKO and WT mice. The total travel distance and the total time spent in inner and outer zones did not differ significantly between Lrrc8a-cKO and WT mice (n = 9 mice per group; Fig. 1C).
Brain ischemia transiently increases LRRC8A protein levels in the hippocampus
To determine the effect of cerebral ischemia on LRRC8A protein levels, we used immunoblotting to quantify LRRC8A protein amounts in brain regions of WT mice subjected to middle cerebral artery occlusion (MCAO) for 90 min. LRRC8A protein levels were measured in brain tissues obtained 6 and 24 hours after reperfusion. In the hippocampus, LRRC8A protein levels (~94 kDa) were significantly increased 6 hours after MCAO-reperfusion compared with the sham control group (P < 0.0001, t(14) = 24.87, n = 8 mice per group; Fig. 2A). However, at 24 hours after MCAO-reperfusion, LRRC8A protein levels in the hippocampus did not differ significantly from that of the sham control group (Fig. 2A).
Figure 2. Effect of brain ischemia on LRRC8A protein levels in brain tissues of WT mice.
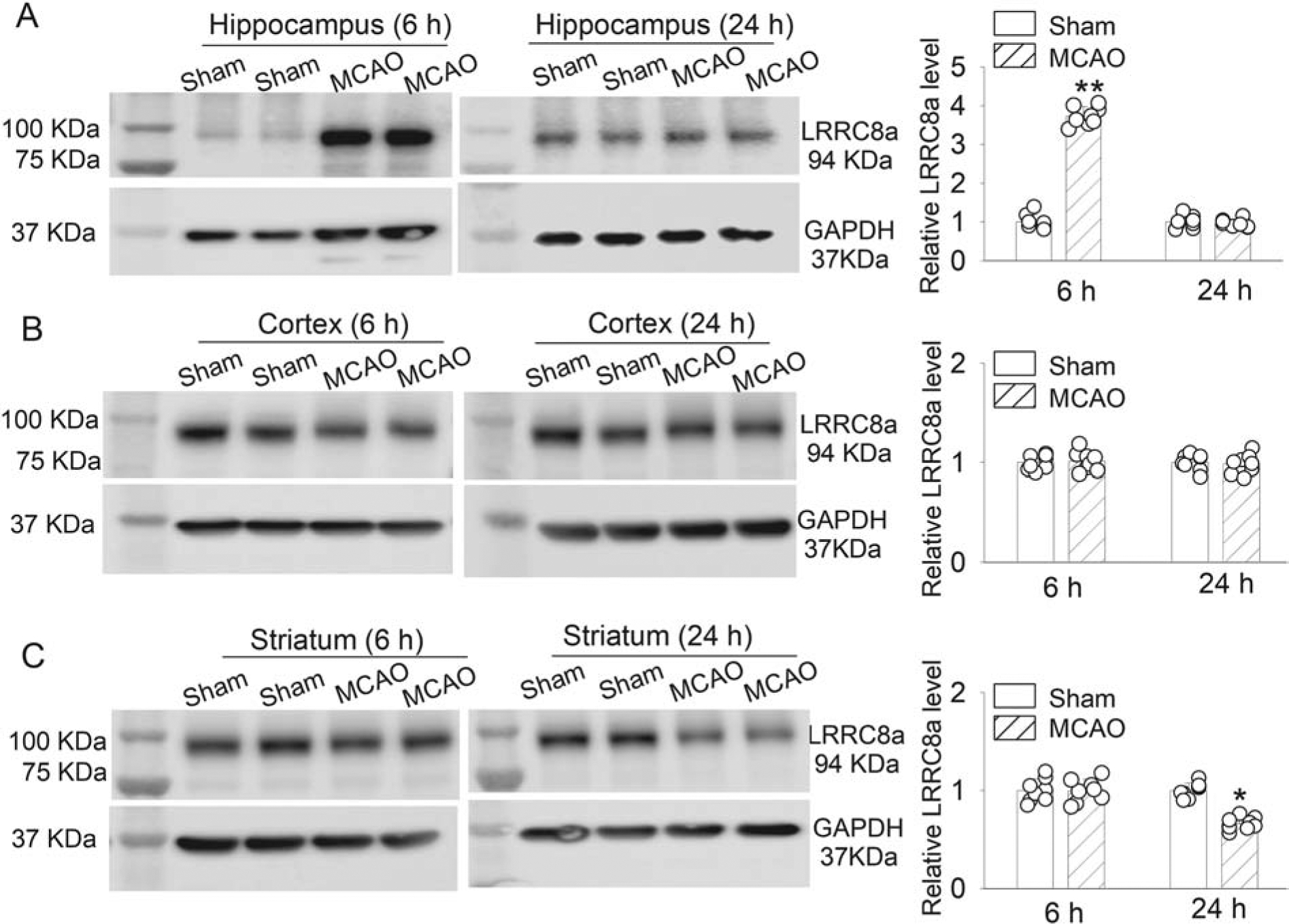
(A-C) Representative blotting images and mean data show LRRC8A protein levels in the hippocampus (A), cerebral cortex (B), and striatum (C) at 6 and 24 hours after MCAO induction in 6-week-old WT mice (n = 8 mice per group). Ctrl: sham control. *P < 0.05, **P < 0.01 compared with the sham control group.
In the cerebral cortex, LRRC8A protein levels were not significantly altered at either 6 or 24 hours after MCAO-reperfusion (Fig. 2B). Moreover, LRRC8A protein levels in the striatum were not altered significantly 6 hour after reperfusion but were significantly reduced 24 hours after reperfusion, compared with levels in sham control mice (P < 0.0001, t(14) = 9.553, n = 8 WT mice per group; Fig. 2C). These results indicate that the LRRC8A protein levels undergo dynamic time-dependent changes in various brain regions after cerebral ischemia and reperfusion.
Mimicking ischemia with oxygen-glucose deprivation increases LRRC8A-dependent VRAC activity in hippocampal neurons
The hippocampus is an ischemic penumbra region in the MCAO model (Kiewert et al., 2010). Functional VRACs are present in hippocampal CA1 pyramidal neurons (Zhang et al., 2011). We next recorded whole-cell VRAC currents (IVRAC) in hippocampal CA1 pyramidal neurons to determine whether ischemia affects VRAC activity in WT and Lrrc8a-cKO mice. Because brain ischemia leads to severe damage to cell membranes, it is impossible to record whole-cell VRAC activity in neurons obtained from ischemic brain tissues. We thus used oxygen-glucose deprivation (OGD) to simulate ischemia in brain slices. OGD was induced for 5 min followed by 10 min of recovery in normal aCSF (Luo et al., 2018). VRAC currents were pharmacologically isolated using 30 μM DCPIB, a specific VRAC blocker, via bath application (Wang et al., 2017). Under the non-OGD condition, current-voltage (I-V) analysis showed that DCPIB-sensitive VRAC currents in hippocampal CA1 neurons from WT mice displayed typical outward rectification at positive holding potentials. The reversal potential of IVRAC was close to −40 mV, which is similar to what was reported previously (Fig. 3A–D) (Inoue and Okada, 2007; Zhang et al., 2011). In Lrrc8a-cKO mice, DCPIB-sensitive IVRAC was largely diminished in hippocampal CA1 pyramidal neurons (P < 0.0001, F(1,10) = 125.3; n = 15 neurons from 4 WT mice, n = 17 neurons from 4 Lrrc8a-cKO mice; Fig. 3A–D), indicating that LRRC8A is essential for VRAC function in hippocampal CA1 neurons.
Figure 3. Effect of oxygen-glucose deprivation (OGD) on VRAC activity in hippocampal CA1 pyramidal neurons of WT and Lrrc8a-cKO mice.
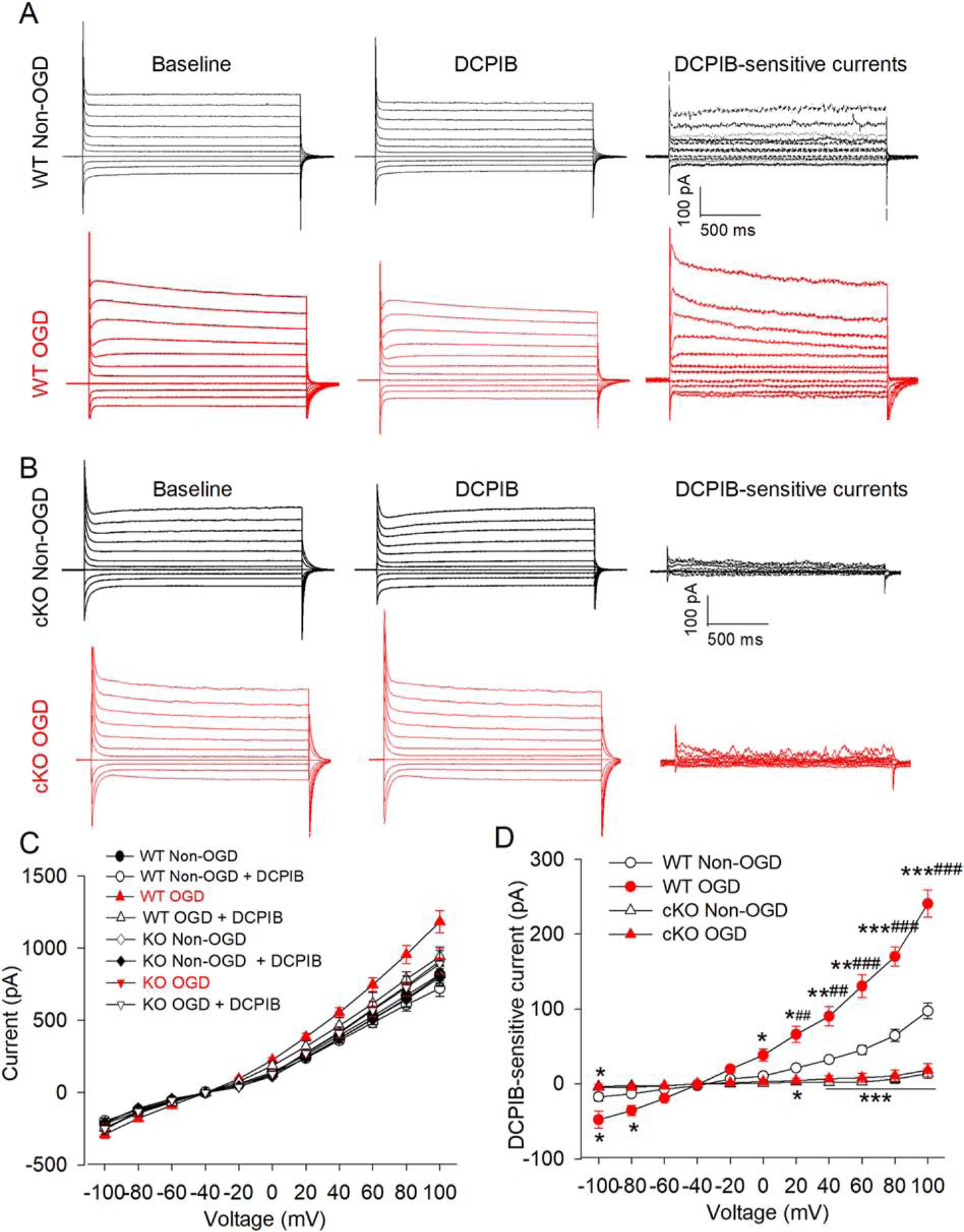
(A and B) Representative current traces of DCPIB-sensitive VRAC currents in hippocampal CA1 pyramidal neurons with and without OGD in WT (A) and Lrrc8a-cKO mice (B). VRAC currents were recorded using a step voltage protocol from −100 mV to +100 mV in 20-mV increments. DCPIB-sensitive VRAC currents were obtained by digitally subtracting currents recorded during DCPIB application from currents recorded before DCPIB application (baseline). (C and D) Current-voltage (I-V) curve shows the effect of OGD on total chloride currents (C) and DCPIB-sensitive VRAC currents (D) in hippocampal CA1 pyramidal neurons from 6-week-old WT and Lrrc8a-cKO mice (n = 15 neurons from 4 WT mice; n = 17 neurons from 4 Lrrc8a-cKO mice). *P < 0.05, **P < 0.01, ***P < 0.001 compared with WT non-OGD group at the same holding potential. ##P < 0.01, ###P < 0.001 compared with the Lrrc8a-cKO OGD group.
OGD caused a large increase in the amplitude of DCPIB-sensitive IVRAC in hippocampal CA1 neurons from WT mice (P < 0.0001, F(1,10) = 108.5, n = 15 neurons from 4 WT mice per group, Fig. 3A and D). In contrast, in Lrrc8a-cKO mice, OGD did not significantly increase DCPIB-sensitive IVRAC in hippocampal CA1 neurons (n = 10 neurons from 4 mice in WT and WT+DCPIB groups, n = 14 neurons from 5 mice in the Lrrc8acKO group; Fig. 3B and D). OGD had no effect on the reversal potential of DCPIB-sensitive IVRAC in hippocampal CA1 neurons from WT and Lrrc8a-cKO mice. These results indicate that ischemia potentiates neuronal VRAC activity, which is critically dependent on LRRC8A.
LRRC8A-dependent VRAC activation induced by hypotonicity potentiates glutamatergic input to hippocampal neurons
Cerebral ischemia causes excessive glutamate release, which can induce excitotoxicity and neuronal death via glutamate NMDA receptor activation (Lipton, 2006; Luo et al., 2018; Simon et al., 1984). To determine whether increased VRAC activity after ischemia contributes to increased glutamate release, we used whole-cell voltage-clamp recordings to measure glutamate-mediated spontaneous excitatory postsynaptic currents (sEPSCs) in hippocampal CA1 pyramidal neurons in brain slices from WT and Lrrc8a-cKO mice. Because brain edema and cell swelling are an early feature of brain ischemia (Simard et al., 2007), we used hypotonicity-induced osmotic swelling to induce VRAC activation. The glutamatergic sEPSCs are a commonly used measure of action potential–dependent and –independent glutamatergic input to CA1 neurons in brain slices (Li et al., 2008). We first measured baseline sEPSCs in hippocampal CA1 neurons in the presence of isotonic solution and then switched to hypotonic solution via bath perfusion for 6 min. In WT mice, hypotonic challenge significantly increased the frequency, but not amplitude, of sEPSCs in CA1 neurons (P = 0.0001, t(9) =3.39, n = 10 neurons from 4 mice; Fig. 4A–D). The sEPSCs were completely blocked by 20 μM of DNQX, a specific glutamate AMPA receptor antagonist (Fig. 4A).
Figure 4. Effect of hypotonic challenge on spontaneous excitatory postsynaptic currents (sEPSCs) in hippocampal CA1 pyramidal neurons of WT and Lrrc8a-cKO mice.
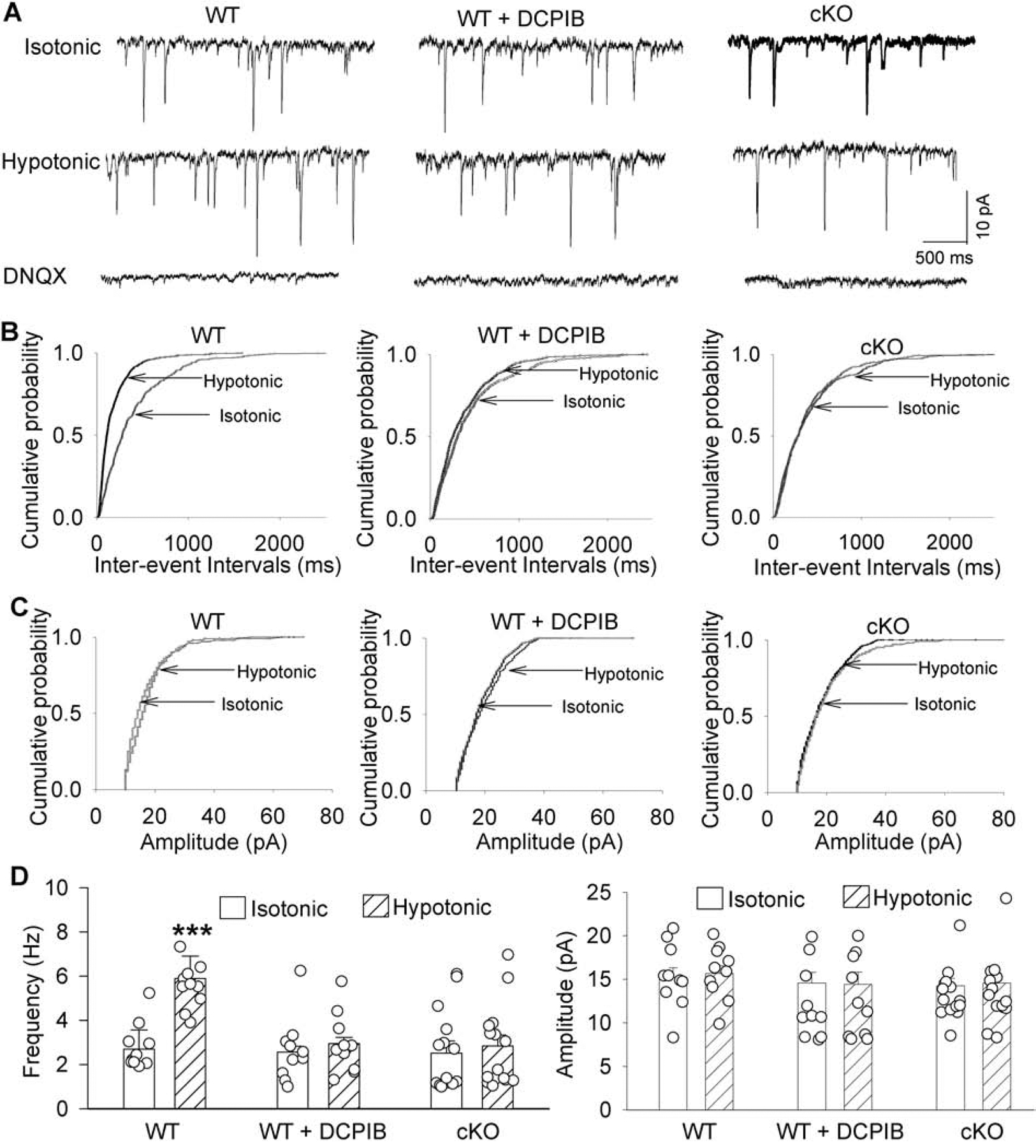
(A-C) Representative recording traces (A) and cumulative plots (B and C) show the effects of DCPIB and hypotonic solution on the frequency and amplitude of glutamatergic sEPSCs in hippocampal CA1 pyramidal neurons from 6-week-old WT and Lrrc8acKO mice. Note that blocking glutamate AMPA receptors with DNQX abolished sEPSCs. (D) Group data show the effects of DCPIB and hypotonic solution on the frequency and amplitude of glutamatergic sEPSCs in hippocampal CA1 pyramidal neurons from 6-week-old WT and Lrrc8a-cKO mice (n = 10 neurons from 4 mice in WT and WT+DCPIB groups; n = 14 neurons from 5 mice in the Lrrc8a-cKO group). ***P < 0.001 compared with the isotonic condition within the same group.
To determine whether VRACs contribute to cell swelling–induced glutamate release, we bath-applied DCPIB (30 μM) during sEPSC recordings in hippocampal CA1 pyramidal neurons from WT mice. Application of DCPIB had no effect on baseline sEPSCs of CA1 neurons in WT mice with isotonic solution (Fig. 4A–D). However, in the presence of DCPIB, hypotonic challenge failed to significantly increase the frequency of sEPSCs in CA1 neurons from WT mice (P = 0.62, t(9) = 0.51, n = 10 neurons from 4 mice; Fig. 4A–D).
The baseline frequency and amplitude of sEPSCs in CA1 neurons did not differ significantly between Lrrc8a-cKO and WT mice, suggesting that VRACs are not actively involved in baseline glutamate release. In contrast, hypotonic challenge had no effect on the frequency or amplitude of sEPSCs in CA1 neurons from Lrrc8a-cKO mice (P = 0.098, t(13) = 1.78, n = 14 neurons from 5 mice, Fig. 4A–D). Together, these data suggest that hypotonicity-induced osmotic swelling augments synaptic glutamatergic input to CA1 pyramidal neurons through LRRC8A-dependent VRACs.
LRRC8A contributes to cerebral ischemia-induced brain injury
To determine whether LRRC8A-dependent VRACs play a role in ischemic brain injury, we measured brain infarct volume in WT and Lrrc8a-cKO mice subjected to MCAO for 90 min followed by 24 hours of reperfusion. The brain infarct regions contain a substantial proportion of hemisphere, including most of the cortex, striatum, hippocampus, and thalamus, in both WT and Lrrc8a-cKO mice. The infarct volume in coronal brain sections was significantly smaller in Lrrc8a-cKO mice than in WT mice (P = 0.0045, t(18) =3.24; n = 9 WT mice, n = 11 Lrrc8a-cKO mice; Fig. 5A and B). Moreover, the neurological severity score was significantly lower in Lrrc8a-cKO mice than in WT mice (P = 0.0251, t(18) = 2.45; Fig. 5C). These findings suggest that LRRC8A-dependent VRACs contribute to ischemic brain damage and associated neurological deficit.
Figure 5. Middle cerebral artery occlusion (MCAO)-induced brain infarct volume and neurological dysfunction in WT and Lrrc8a-cKO mice.
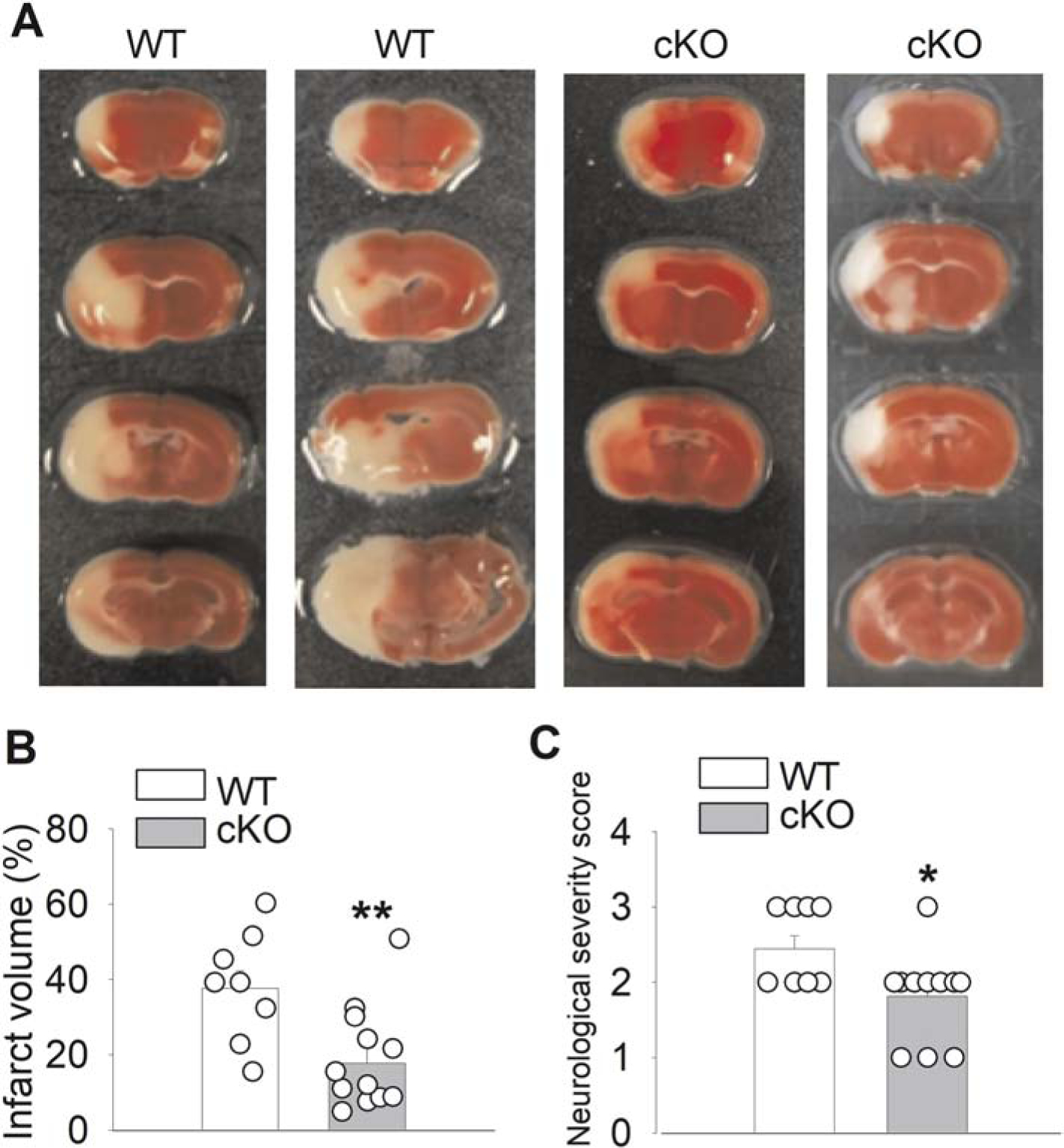
(A and B) Representative 2,3,5-triphenyltetrazolium chloride staining (A) and quantification (B) of brain infarct volume in 6-week-old WT and Lrrc8a-cKO mice subjected to MCAO followed by 24 hours of reperfusion (n = 9 WT mice, n = 11 Lrrc8a-cKO mice). (C) Summary data show the neurological severity score in 6-week-old WT and Lrrc8a-cKO mice subjected to MCAO followed by 24 hours of reperfusion (n = 9 WT mice, n = 11 Lrrc8a-cKO mice). *P < 0.05, **P < 0.01 compared with WT mice.
LRRC8A is involved in cell death caused by cerebral ischemia
Cleaved caspase-3 (Le et al., 2002) and spectrin breakdown product (spectrin BD) resulting from excessive calpain activation (Markgraf et al., 1998) are well-characterized neuronal death markers with cerebral ischemia. Brain ischemia-induced activation of calpain cleaves alpha II spectrin, a cytoskeleton protein, to a breakdown product with a molecular weight of 150 kDa (Pike et al., 2004). The protein level of cleaved caspase-3 (~17 kDa) and spectrin BD in the hippocampus, cerebral cortex, and striatum did not differ significantly between WT sham and Lrrc8a-cKO sham mice (n = 8 mice per group; Fig. 6A–C). Although MCAO significantly increased the protein level of cleaved caspase-3 and spectrin BD in the hippocampus, cortex, and striatum in both WT and Lrrc8a-cKO mice, the increased level of cleaved caspase-3 and spectrin BD in these three ischemic regions was significantly less in Lrrc8a-cKO than in WT mice (n = 8 mice in each group; Fig. 6A–C). These results suggest that LRRC8A-dependent VRACs mediates neuronal injury and death caused by cerebral ischemia.
Figure 6. Effect of MCAO on cleaved caspase-3 and spectrin BD levels in brain tissues of WT and Lrrc8a-cKO mice.
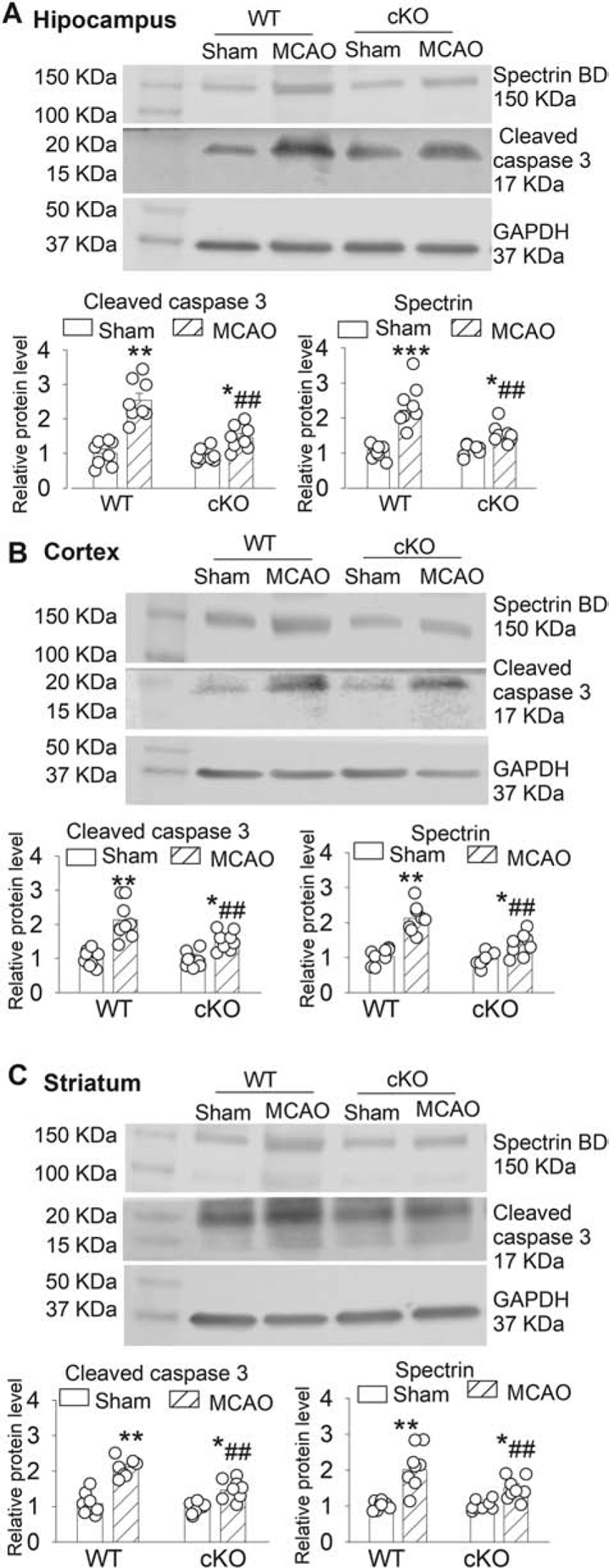
(A-C) Representative Western blot images and summary data show the effect of MCAO on cleaved caspase-3 and spectrin breakdown product (spectrin BD) in the hippocampus (A), cerebral cortex (B), and striatum (C) from 6-week-old WT and Lrrc8a-cKO mice (n = 8 mice per group). Ctrl: sham control. *P < 0.05, **P < 0.01, ***P < 0.001 compared with respective values in the sham control group. ##P < 0.01 compared with the WT MCAO group.
Discussion
Neurons undergo rapid swelling in the soma and dendrites during excitotoxic insults associated with stroke and brain trauma. Focal swellings along the dendrites called varicosities are considered to be a hallmark of acute excitotoxic neuronal injury (Hori and Carpenter, 1994; Hsu and Buzsaki, 1993). In the present study, we found that LRRC8A, an essential subunit of functional VRACs, was transiently increased in the hippocampus, a penumbra region, 6 hours after MCAO-reperfusion. We also found that the VRAC activity in hippocampal CA1 neurons was potentiated by mimicking ischemia with OGD. In our study, blocking with DCPIB or genetic knockout of LRRC8A diminished VRAC currents in hippocampal neurons increased by OGD, suggesting that LRRC8A-dependent VRACs play a major role in Cl− conductance in ischemic neurons. Interestingly, LRRC8A protein levels in the striatum were significantly decreased 24 hours after MCAO. Because the striatum is an infarct core region and is very susceptible to ischemia in the MCAO model (Kiewert et al., 2010), massive cell death and associated protein degradation may account for MCAO-induced reduction in LRRC8A protein levels in the striatum 24 hours after MCAO-reperfusion.
Our study demonstrated that ablating LRRC8A attenuated MCAO-induced brain infarct volume and neurological dysfunction, indicating that LRRC8A-dependent VRACs contribute to ischemic brain injury. Blocking VRACs with DCPIB has been shown to attenuate brain damage induced by MCAO (Zhang et al., 2008). IAA-94, another VRAC blocker, also reduces glutamate-induced necrosis in cultured cortical neurons (Inoue and Okada, 2007). Blocking VRACs with DCPIB or conditional knockout of LRRC8A in astrocytes decreases ischemic brain injury (Han et al., 2014; Yang et al., 2019). Increased calpain activity plays an important role in neuronal death after brain ischemia (Curcio et al., 2016; Neumar et al., 2001). We found that ablating brain LRRC8A reduced calpain activity and cleaved caspase-3 in various ischemic brain tissues, further supporting the role of LRRC8A-dependent VRACs in ischemia-induced neuronal injury. It should be noted that the LRRC8A protein was almost undetectable in brain tissues of Lrrc8a-cKO mice, indicating that Lrrc8a-cKO generated using NestinCre+/− mice probably ablated Lrrc8a in both neurons and astrocytes. VRACs expressed in both astrocytes and neurons are likely involved in ischemic brain damage. There are currently no Cre mice that allow the deletion of genes specifically in all brain neurons. Our Lrrc8a-cKO mice all died 7–8 weeks after birth, demonstrating an essential role of brain LRRC8a for the survival. We did not use NexCre+/− or CaMKIIαCre+/− mice in our study since they ablate genes only in a subpopulation of neurons in limited brain regions. Because of the unexpected premature fatality of Lrrc8a-cKO mice, we did not assess the long-term effect of Lrrc8a-cKO on neurological function after MCAO, which is a limitation of this study. Also, we did not measure cerebral blood flow in Lrrc8acKO and WT mice before or during MCAO. Notably, it has been shown that blocking VRACs with tamoxifen does not alter cerebral blood flow at the baseline and during MCAO (Feustel et al., 2004).
It remains uncertain exactly how ischemia-induced VRAC activation is involved in neuronal injury. Although initial VRAC activation during ischemia may help to restore cell volume, sustained activation of VRAC can cause necrotic neuronal death (Barros et al., 2001). Excitotoxic insult–induced swelling of neuronal soma and dendrites seems to be dependent on extracellular Cl− (Dessi et al., 1994; Inglefield and Schwartz-Bloom, 1998; Rothman, 1985). In neurons, even a transitory volume increase can cause bursting firing (Azouz et al., 1997; Chebabo et al., 1995; Lauderdale et al., 2015) and spreading depression (Dudek et al., 1990). Under a marked depolarizing condition by glutamate receptor activation, VRACs likely serve as a pathway for swelling-aggravating Cl− influx in cortical neurons (Inoue and Okada, 2007). Consistent with this notion, excitotoxic Cl− influx is associated with neuronal death and can be reduced by putative VRAC blockers (Babot et al., 2005; Van Damme et al., 2003). Thus, VRACs likely play a major role in Cl− influx that is involved in neuronal excitotoxic death by ischemia (Fig. 7). Other Cl− influx pathways in neurons during ischemia may include voltage-gated chloride channels and Na+-K+−2Cl− cotransporters (Pond et al., 2006; Wang et al., 2006). Although cell swelling can activate VRACs, we did not specifically assess whether Lrrc8a-cKO affects brain edema or cell swelling induced by MACO. Nevertheless, it has been shown that pharmacological blocking of VRACs attenuates NMDA-induced focal cell swelling in cultured cortical neurons (Inoue and Okada, 2007).
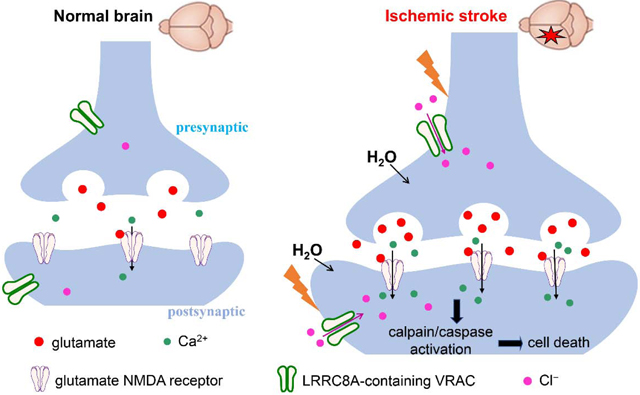
Figure 7. Schematic drawing outlines the potential role of neuronal LRRC8A-containing volume-regulated anion channels (VRACs) in ischemic brain damage.
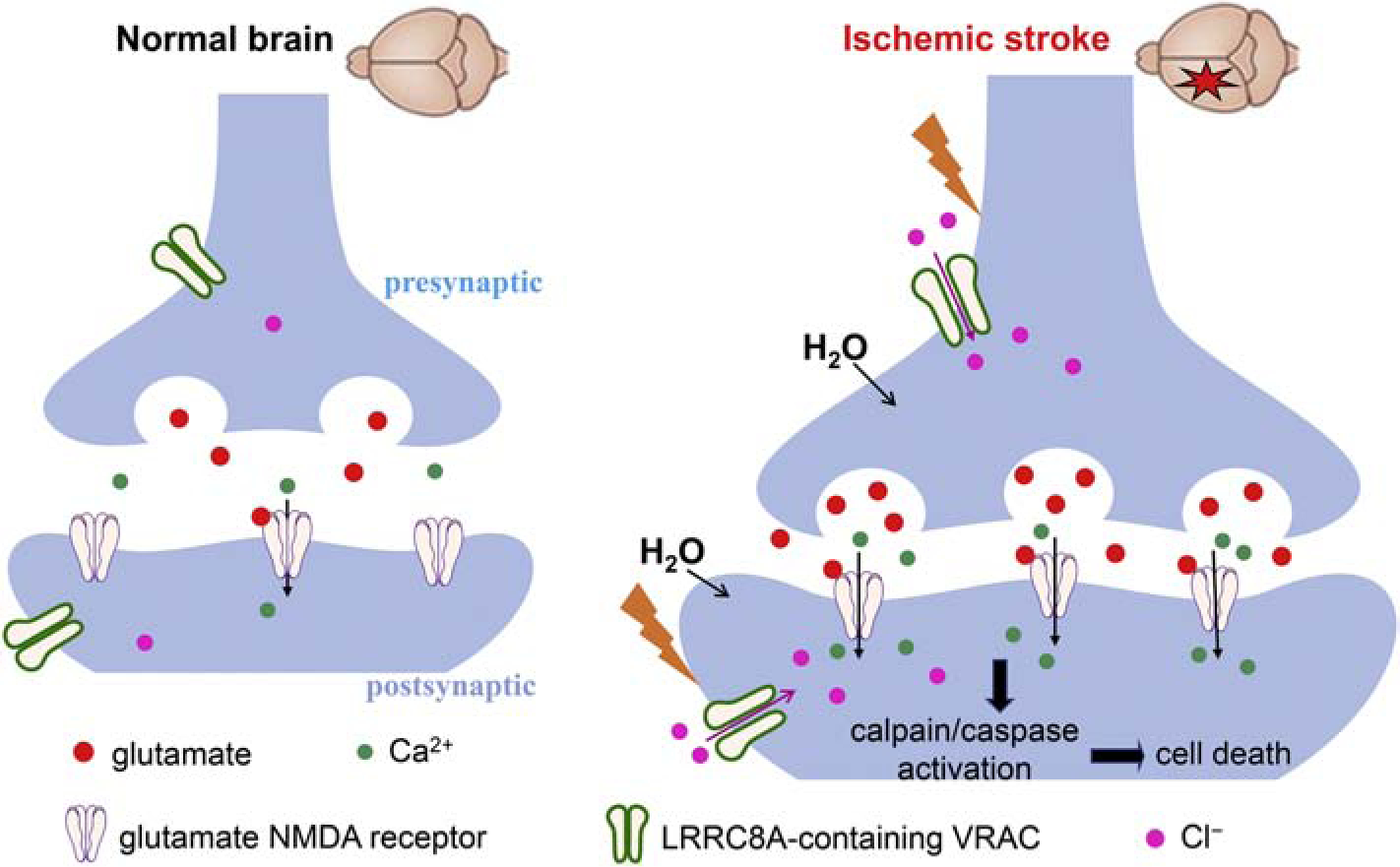
Neuronal VRACs are not activated or involved in glutamate release in the normal brain. Cerebral ischemia causes rapid cell swelling and neuronal depolarization to increase the activity of VRACs expressed in presynaptic and postsynaptic neurons. Augmented VRAC activity in ischemic neurons leads to chloride influx and glutamate release and subsequent activation of glutamate NMDA receptors, which can induce excitotoxicity and neuronal death through calplain/caspase-mediated signaling pathways.
Our study showed that ablating LRRC8A attenuated swelling-augmented glutamatergic input to hippocampal CA1 neurons, suggesting that neuronal LRRC8A-dependent VRACs are involved in swelling-induced glutamate release during ischemia. VRAC-dependent glutamate release likely occurs in the penumbra, because VRAC activation requires sufficient intracellular ATP and oxygen levels. Although previous studies reported that swelling-induced glutamate release is mediated by VRACs in astrocytes (Kimelberg, 2004; Liu et al., 2006), VRACs are also located at presynaptic terminals and dendritic varicosities in neurons (Inoue and Okada, 2007; Zhang et al., 2011). Therefore, swelling-induced VRAC activation in neurons may directly release glutamate to the postsynaptic neurons during ischemia. However, neuronal swelling induced by activation of the glutamate receptors can lead to activation of VRACs (Inoue and Okada, 2007). Glutamate released via NMDA receptor-activated VRACs might in turn activate additional glutamate NMDA and calcium-permeable AMPA receptors. Thus, VRACs may promote the influx of not only Cl− but also Ca2+ and Na+, thereby aggravating ischemic neuronal injury.
In conclusion, our study provides new information showing that cerebral ischemia increases neuronal LRRC8A-dependent VRAC activity and that these VRACs play an important role in ischemic brain damage. VRAC activation by cerebral ischemia can promote glutamate release, which in turn can stimulate VRACs, aggravating neuronal excitotoxic death (Fig. 7). Our study not only independently validates the role of VRACs in brain ischemia but also provides additional new insight about dynamic changes in neuronal VRAC activity caused by ischemia. Therefore, inhibiting neuronal VRAC activity could be a rational strategy for treating ischemic stroke. DCPIB is perhaps the most selective VRAC blocker at the present time. However, DCPIB cannot cross the blood-brain barrier upon systemic administration (Zhang et al., 2008). Because VRACs are widely expressed in brain neurons and astrocytes, inhibiting VRACs in the brain would be expected to produce certain side effects in humans. Future studies are needed to design appropriate strategies to attenuate brain ischemia-induced VRAC hyperactivity without blocking the normal VRAC function.
Highlights:
Cerebral ischemia transiently increases LRRC8A protein levels in the hippocampus.
LRRC8A is essential for hippocampal neuronal VRAC activity increased by ischemia.
LRRC8A is required for hypotonicity-potentiated glutamatergic input to neurons.
LRRC8A-dependent VRACs contribute to brain injury caused by cerebral ischemia.
Acknowledgments
This study was supported by the National Institutes of Health (Grants HL131161 and NS101880) and the N.G. and Helen T. Hawkins Endowment (to H.-L.P.).
List of Abbreviations:
- VRAC
volume-regulated anion channel
- LRRC8A
leucine-rich repeat-containing protein 8A
- NMDA
N-methyl-D-aspartate
- Lrrc8a-cKO
Lrrc8a conditional knockout
- WT
wild-type
- MCAO
middle cerebral artery occlusion
- DCPIB
4-[(2-butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid
- OGD
oxygen-glucose deprivation
- sEPSC
spontaneous excitatory postsynaptic current
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure
The authors declare no conflict of interest with the contents of this article.
References
- Azouz R, Alroy G, Yaari Y, 1997. Modulation of endogenous firing patterns by osmolarity in rat hippocampal neurones. J Physiol 502 (Pt 1), 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babot Z, Cristofol R, Sunol C, 2005. Excitotoxic death induced by released glutamate in depolarized primary cultures of mouse cerebellar granule cells is dependent on GABAA receptors and niflumic acid-sensitive chloride channels. Eur J Neurosci 21, 103–112. [DOI] [PubMed] [Google Scholar]
- Bandera E, Botteri M, Minelli C, Sutton A, Abrams KR, Latronico N, 2006. Cerebral blood flow threshold of ischemic penumbra and infarct core in acute ischemic stroke: a systematic review. Stroke 37, 1334–1339. [DOI] [PubMed] [Google Scholar]
- Bao J, Perez CJ, Kim J, Zhang H, Murphy CJ, Hamidi T, Jaubert J, Platt CD, Chou J, Deng M, Zhou MH, Huang Y, Gaitan-Penas H, Guenet JL, Lin K, Lu Y, Chen T, Bedford MT, Dent SY, Richburg JH, Estevez R, Pan HL, Geha RS, Shi Q, Benavides F, 2018. Deficient LRRC8A-dependent volume-regulated anion channel activity is associated with male infertility in mice. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Barros LF, Hermosilla T, Castro J, 2001. Necrotic volume increase and the early physiology of necrosis. Comp Biochem Physiol A Mol Integr Physiol 130, 401–409. [DOI] [PubMed] [Google Scholar]
- Bowens NH, Dohare P, Kuo YH, Mongin AA, 2013. DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol Pharmacol 83, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai YQ, Chen SR, Pan HL, 2013. Upregulation of nuclear factor of activated T-cells by nerve injury contributes to development of neuropathic pain. J Pharmacol Exp Ther 345, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Aitken PG, Somjen GG, 1995. Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res 695, 203–216. [DOI] [PubMed] [Google Scholar]
- Chen Q, Pan HL, 2006. Regulation of synaptic input to hypothalamic presympathetic neurons by GABA(B) receptors. Neuroscience 142, 595–606. [DOI] [PubMed] [Google Scholar]
- Corlew R, Brasier DJ, Feldman DE, Philpot BD, 2008. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio M, Salazar IL, Mele M, Canzoniero LM, Duarte CB, 2016. Calpains and neuronal damage in the ischemic brain: The swiss knife in synaptic injury. Prog Neurobiol 143, 1–35. [DOI] [PubMed] [Google Scholar]
- Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K, 2001. DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol 134, 1467–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessi F, Charriaut-Marlangue C, Ben-Ari Y, 1994. Glutamate-induced neuronal death in cerebellar culture is mediated by two distinct components: a sodium-chloride component and a calcium component. Brain Res 650, 49–55. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Tasker JG, 1990. Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett 120, 267–270. [DOI] [PubMed] [Google Scholar]
- Feustel PJ, Jin Y, Kimelberg HK, 2004. Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 35, 1164–1168. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD, 2007. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron 54, 611–626. [DOI] [PubMed] [Google Scholar]
- Guo YX, Li DP, Chen SR, Pan HL, 2013. Distinct intrinsic and synaptic properties of pre-sympathetic and pre-parasympathetic output neurons in Barrington’s nucleus. J Neurochem 126, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q, Liu S, Li Z, Hu F, Zhang Q, Zhou M, Chen J, Lei T, Zhang H, 2014. DCPIB, a potent volume - regulated anion channel antagonist, attenuates microglia-mediated inflammatory response and neuronal injury following focal cerebral ischemia. Brain Res 1542, 176–185. [DOI] [PubMed] [Google Scholar]
- Hori N, Carpenter DO, 1994. Functional and morphological changes induced by transient in vivo ischemia. Exp Neurol 129, 279–289. [DOI] [PubMed] [Google Scholar]
- Hsu M, Buzsaki G, 1993. Vulnerability of mossy fiber targets in the rat hippocampus to forebrain ischemia. J Neurosci 13, 3964–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Chen SR, Chen H, Pan HL, 2019. Endogenous transient receptor potential ankyrin 1 and vanilloid 1 activity potentiates glutamatergic input to spinal lamina I neurons in inflammatory pain. J Neurochem 149, 381–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglefield JR, Schwartz-Bloom RD, 1998. Activation of excitatory amino acid receptors in the rat hippocampal slice increases intracellular Cl- and cell volume. J Neurochem 71, 1396–1404. [DOI] [PubMed] [Google Scholar]
- Inoue H, Okada Y, 2007. Roles of volume-sensitive chloride channel in excitotoxic neuronal injury. J Neurosci 27, 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Kawamata T, Tamura T, Hovda DA, Becker DP, Tsubokawa T, 1991. Calcium-dependent glutamate release concomitant with massive potassium flux during cerebral ischemia in vivo. Brain Res 558, 136–140. [DOI] [PubMed] [Google Scholar]
- Kiewert C, Mdzinarishvili A, Hartmann J, Bickel U, Klein J, 2010. Metabolic and transmitter changes in core and penumbra after middle cerebral artery occlusion in mice. Brain Res 1312, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, 2004. Increased release of excitatory amino acids by the actions of ATP and peroxynitrite on volume-regulated anion channels (VRACs) in astrocytes. Neurochem Int 45, 511–519. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Feustel PJ, Jin Y, Paquette J, Boulos A, Keller RW Jr., Tranmer BI, 2000. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 11, 2675–2679. [DOI] [PubMed] [Google Scholar]
- Lauderdale K, Murphy T, Tung T, Davila D, Binder DK, Fiacco TA, 2015. Osmotic Edema Rapidly Increases Neuronal Excitability Through Activation of NMDA Receptor-Dependent Slow Inward Currents in Juvenile and Adult Hippocampus. ASN Neuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA, 2002. Caspase activation and neuroprotection in caspase-3- deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A 99, 15188–15193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM, Pan HL, 2008. Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. J Physiol 586, 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhou JJ, Pan HL, 2015. Endogenous casein kinase-1 modulates NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. J Physiol 593, 4439–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, 2006. NMDA receptors, glial cells, and clinical medicine. Neuron 50, 9–11. [DOI] [PubMed] [Google Scholar]
- Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ, 2006. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54, 343–357. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R, 1989. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20, 84–91. [DOI] [PubMed] [Google Scholar]
- Luo Y, Ma H, Zhou JJ, Li L, Chen SR, Zhang J, Chen L, Pan HL, 2018. Focal Cerebral Ischemia and Reperfusion Induce Brain Injury Through alpha2delta-1-Bound NMDA Receptors. Stroke 49, 2464–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Chen SR, Chen H, Pan HL, 2019. Endogenous AT1 receptor-protein kinase C activity in the hypothalamus augments glutamatergic input and sympathetic outflow in hypertension. J Physiol 597, 4325–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markgraf CG, Velayo NL, Johnson MP, McCarty DR, Medhi S, Koehl JR, Chmielewski PA, Linnik MD, 1998. Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke 29, 152–158. [DOI] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G, 2004. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol 469, 311–324. [DOI] [PubMed] [Google Scholar]
- Neumar RW, Meng FH, Mills AM, Xu YA, Zhang C, Welsh FA, Siman R, 2001. Calpain activity in the rat brain after transient forebrain ischemia. Exp Neurol 170, 27–35. [DOI] [PubMed] [Google Scholar]
- Osei-Owusu J, Yang J, Vitery MDC, Qiu Z, 2018. Molecular Biology and Physiology of Volume-Regulated Anion Channel (VRAC). Curr Top Membr 81, 177–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Gullotti DM, Hernandez P, O’Brien WT, Capehart BP, Morrison B 3rd, Bass C, Eberwine JE, Abel T, Meaney DF, 2014. An open-source toolbox for automated phenotyping of mice in behavioral tasks. Front Behav Neurosci 8, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike BR, Flint J, Dave JR, Lu XC, Wang KK, Tortella FC, Hayes RL, 2004. Accumulation of calpain and caspase-3 proteolytic fragments of brain-derived alphaII-spectrin in cerebral spinal fluid after middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab 24, 98–106. [DOI] [PubMed] [Google Scholar]
- Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD, 2006. The chloride transporter Na(+)-K(+)-Cl- cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci 26, 1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP, Patapoutian A, 2014. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D, 2000. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. [DOI] [PubMed] [Google Scholar]
- Rothman SM, 1985. The neurotoxicity of excitatory amino acids is produced by passive chloride influx. J Neurosci 5, 1483–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJC, Cain SM, Cullis PR, Snutch TP, MacVicar BA, 2015. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell 161, 610–621. [DOI] [PubMed] [Google Scholar]
- Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V, 2007. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon RP, Swan JH, Griffiths T, Meldrum BS, 1984. Blockade of N-methyl-D-aspartate receptors may protect against ischemic damage in the brain. Science 226, 850–852. [DOI] [PubMed] [Google Scholar]
- Song M, Yu SP, 2014. Ionic regulation of cell volume changes and cell death after ischemic stroke. Transl Stroke Res 5, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan A, Delage H, Chatterjee S, Belgarbi-Dutron L, Cassel R, Martinez N, Cosquer B, Kumari S, Mongelard F, Lannes B, Cassel JC, Boutillier AL, Bouvet P, Kundu TK, 2016. Transcriptional Coactivator and Chromatin Protein PC4 Is Involved in Hippocampal Neurogenesis and Spatial Memory Extinction. J Biol Chem 291, 20303–20314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR, 1990. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 10, 290–293. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Callewaert G, Eggermont J, Robberecht W, Van Den Bosch L, 2003. Chloride influx aggravates Ca2+-dependent AMPA receptor-mediated motoneuron death. J Neurosci 23, 4942–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T, Jentsch TJ, 2014. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638. [DOI] [PubMed] [Google Scholar]
- Wang L, Chang X, She L, Xu D, Huang W, Poo MM, 2015. Autocrine action of BDNF on dendrite development of adult-born hippocampal neurons. J Neurosci 35, 8384–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Lu Y, Gunasekar S, Zhang Y, Benson CJ, Chapleau MW, Sah R, Abboud FM, 2017. The volume-regulated anion channel (LRRC8) in nodose neurons is sensitive to acidic pH. JCI Insight 2, e90632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Deriy LV, Foss S, Huang P, Lamb FS, Kaetzel MA, Bindokas V, Marks JD, Nelson DJ, 2006. CLC-3 channels modulate excitatory synaptic transmission in hippocampal neurons. Neuron 52, 321–333. [DOI] [PubMed] [Google Scholar]
- Wilson CS, Bach MD, Ashkavand Z, Norman KR, Martino N, Adam AP, Mongin AA, 2019. Metabolic constraints of swelling-activated glutamate release in astrocytes and their implication for ischemic tissue damage. J Neurochem 151, 255–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z, 2019. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 102, 813–827.e816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cao HJ, Kimelberg HK, Zhou M, 2011. Volume regulated anion channel currents of rat hippocampal neurons and their contribution to oxygen-and-glucose deprivation induced neuronal death. PLoS One 6, e16803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xie L, Gunasekar SK, Tong D, Mishra A, Gibson WJ, Wang C, Fidler T, Marthaler B, Klingelhutz A, Abel ED, Samuel I, Smith JK, Cao L, Sah R, 2017. SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol 19, 504–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ, Kimelberg HK, 2008. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp Neurol 210, 514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JJ, Li DP, Chen SR, Luo Y, Pan HL, 2018. The alpha2delta-1-NMDA receptor coupling is essential for corticostriatal long-term potentiation and is involved in learning and memory. J Biol Chem 293, 19354–19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Chen SR, Pan HL, 2016. Muscarinic receptor subtypes differentially control synaptic input and excitability of cerebellum-projecting medial vestibular nucleus neurons. J Neurochem 137, 226–239. [DOI] [PMC free article] [PubMed] [Google Scholar]