

Abstract
Toll-Like Receptor (TLR) 9 stimulation is required for induction of potent immune responses against pathogen invasion. The use of unmethylated CpG as adjuvants in vaccines provides an excellent means of stimulating adaptive immunity. Our data demonstrate that CpG-C provided prolonged immune responses in the mouse model of tuberculosis when formulated with liposomes and the Mycobacterium tuberculosis antigen ESAT-6. A reduction in the mycobacterial burden was best achieved when administered as an intranasal vaccine and was dependent on type I interferon (IFN). There was a significant difference between CpG-C inoculated wild type and IFN-αR1−/− mice, indicating that type I IFN plays a role in the immune response following CpG-C inoculation. Further analysis showed that early NK cell presence was not an absolute requirement, although elevated IFN-γ levels were detected in the lungs of mice within 48 hours. The reduction in mycobacterial burden was MyD88-independent as CpG-C inoculated MyD88−/− mice showed comparable mycobacterial burdens to wild type mice with no detriment due to the lack of MyD88. Together our data show that pulmonary stimulation of TLR9 bearing antigen presenting cells resulted in the induction of protective immunity against M. tuberculosis infection that was dependent on type I IFN signaling.
Keywords: CpG-C, TLR9, Type I IFN, pulmonary immunity, tuberculosis, vaccine
Introduction
Toll-Like Receptors (TLR) are an integral component of the innate immune response network, interacting with pathogen-associated molecular patterns (PAMPs). Infectious pathogens carry multiple PAMPs that stimulate immune response development during the infection process that contribute to the eventual disease outcome. Interestingly, subunit vaccines have been designed to replicate pathogen-mediated immunity, but without the disease-causing effects, and tend to target, in the majority of cases one TLR. TLR activation through vaccination in the form of molecular adjuvants has been successful, and examples include the TLR4 agonist, monophosphoryl lipid A, and the TLR9 agonists, oligonucleotides that contain unmethylated CpG dinucleotides. Thus, investigating the ability to activate a single innate pathway resulting in adaptive immunity that can result in protective immunity against an infectious pathogen is essential. For the new anti-tuberculosis vaccines currently in development six are subunit vaccines that are formulated with an adjuvant designed to target one TLR1, such as the M72 antigen formulated with AS01 that stimulates innate immunity through TLR4 activation2, while another, H4 formulated with IC31 targets TLR93, 4. Thus, can the activation of one TLR provide sufficient signaling to induce protection against a pathogen that can stimulate multiple TLRs?
Mechanisms associated with killing Mycobacterium tuberculosis remain to be fully elucidated, and a specific host pathway(s) has yet to be identified as the absolute one that will achieve the goal. The current set of experiments were designed to determine the mechanism of immunity generated through stimulation of TLR9 when administered as a component of an intranasal vaccine. We hypothesized that TLR9 stimulation, of pulmonary antigen presenting cells (APCs) was sufficient to induce protective immunity against pulmonary infection. Furthermore, due to the ability of TLR9 to induce type I interferons (IFN), we hypothesized that IFN-α was required to generate protective immunity. Stimulation of TLR9 in APCs and in particular, plasmacytoid dendritic cells (pDC) strongly enhanced DC activation and differentiation, which facilitated T cell activity through multiple mechanisms5–9. Mammalian TLR9, expressed in the endosome of cells, recognizes unmethylated CpG motifs from bacterial DNA10 and interacts with the intracellular adaptor molecule MyD88 to mediate its function11, although MyD88-independent mechanisms have been identified12, 13. The CpG motifs have since been utilized in the form of oligodeoxynucleotides (ODN) that stimulate pDC to potentiate immunity by activation, differentiation and cytokine production14. The role of TLR9 in the host response to M. tuberculosis infection has been controversial: one study demonstrated a requirement for protective immunity15, while another suggested that triggering of MyD88 is of greater importance for immunity16. In humans, genetic variations in TLR9 have resulted in an increased risk of developing tuberculosis1–20. While these previous studies focused on susceptibility to infection, we hypothesized that targeting TLR9, using CpG ODN as a vaccine adjuvant produces a robust pro-inflammatory response that can effectively reduce the mycobacterial burden and that this response required IFN-α.
Immune activation through TLR9 has been utilized in several tuberculosis vaccines formulated with CpG, each with varying results, which may be related to the sub-type of CpG ODN used21–23. As a first step, three CpG subtypes were examined to determine which efficiently induced a robust pro-inflammatory cytokine response. Each was also formulated with the cationic liposome N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP), and then administered intranasally, and the type of pulmonary response examined. To assess T cell responses, the 6-kDa, early secretory antigenic target of M. tuberculosis (ESAT-6; Rv3875), was used to inoculate mice via the intranasal route. In the current studies, ESAT-6 was used as a model antigen to assess the immune response induced by CpG-C stimulation, given that it has been characterized previously in the C57BL/6 mouse24. Our data showed that stimulation by CpG-C of TLR9 on pulmonary mucosal cells induced a robust pro-inflammatory response resulting in antigen-specific T cell-mediated immunity that was dependent on IFN-α. Although the TLR9 adaptor molecule MyD88 molecule is known to be required for downstream signaling, CpG-C mediated protection against M. tuberculosis infection was through a MyD88-independent mechanism.
Materials and Methods
Mice:
Female C57BL/6 mice, aged 6–8 weeks were purchased from Jackson Laboratories (Bar Harbor, MA). B6.129P2(SJL)-Myd88tm1.1Defr/J (MyD88−/−) and B6.129S2-Ifnar1tm1Agt/Mmjax (IFN-αR1−/−) mice bred onto a C57BL/6 background were also obtained from Jackson Laboratories (Bar Harbor, MA). Wild-type C57BL/6 mice were used as controls for these experiments. Mice were housed in a specific temperature and humidity controlled pathogen-free environment. All mice had unlimited access to sterile mouse chow and water. The Colorado State University Institutional Animal Care and Use Committee approved experimental procedures, before commencing experiments.
CpG Oligodeoxynucleotides (ODN):
CpG ODN subtypes A (ODN 2216; 5’-gggggacgatcgtcgggggg-3’), B (ODN 2006; 5’-tcgtcgttttgtcgttttgtcgtt-3’), and C (ODN 2395; 5’-tcgtcgttttcggcgcgcgccg-3’) were purchased from Hycult Biotech Inc. (Plymouth Meeting, PA). The manufacturer’s recommended concentration for in vitro stimulation was between 0.01 to 10 μM after reconstitution in sterile distilled or deionized water. CpG ODN was stored at 4°C for short-term storage, and −80°C for long-term storage.
In vitro bone marrow-derived dendritic cell (BMDC) stimulation assays:
Bone marrow cells were harvested from the femurs of C57BL/6 mice and added to complete medium containing 20 ng/mL GM-CSF (Peprotech, Rocky Hill, NJ) to drive DC differentiation (cRPMI (RPMI-1640 with essential and non-essential amino acids, penicillin, streptomycin (Invitrogen, Carlsbad, CA), and 10% fetal bovine serum (FBS; Atlas, Fort Collins, CO)). Media was changed every 72 hours until the eighth day, after which media was changed to exclude GM-CSF and antibiotics. On day nine, cells were harvested and brought to a concentration of 2.0 × 105 cells per well in a 96 well plate for stimulation for 48 hours with CpG ODN at varying concentrations based on the manufacturer’s recommendation. Positive control wells were cells stimulated with BCG at an MOI of 1:5. Negative control wells included the addition of non-CpG DNA or cRPMI alone.
Inoculation of mice:
Mice were inoculated intranasally (i.n.) with 10μg CpG ODN and cationic liposomes (Avanti, Alabaster, AL) with 20μL of the formulation containing 2μg of ESAT-6 protein. Mice inoculated intragastrically (i.g.), intraperitoneally (i.p.), or subcutaneously (s.c.) were given 100μL containing 50μg CpG ODN with 10μg of ESAT-6 protein. Control groups were mice, inoculated with ESAT-6, cationic liposomes without antigen and non-CpG ODN. A group in which mice received pyrogen-free saline was included for each experiment. Cationic liposomes used were N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP, Avanti Polar, Alabaster, AL) or 1-[2-(oleoyloxy)ethyl]-2-oleyl-3-(2-hydroxyethyl)imidazolinium chloride (DOTIM, kindly provided by Dr. S. Dow, CSU). DOTIM and DOTAP liposomes were prepared in lyophilized cholesterol (Avanti) dissolved in chloroform and diluted in sterile 10% sucrose. Inoculations were administered three times, at two-week intervals. Intra-nasal inoculations were performed while the mouse was anesthetized by ketamine.
Broncho-alveolar lavage (BAL):
To obtain BAL fluid, mice were injected with 150U Heparin (Sodium Salt, Sigma-Aldrich, St. Louis, MO) via the intraperitoneal route, rested for 10 minutes and then humanely euthanized and a catheter inserted into the exposed trachea. Lungs were flushed twice, each with 1mL of PBS containing 5% FBS, and the lavage fluid was stored at −80°C until analyzed for cytokine concentrations. Cells were processed for phenotype analysis.
Mycobacteria:
M. tuberculosis H37Rv (TMCC #102) strain was used for infection studies and was grown as described previously25. Aliquots were stored at −80C and were thawed then sonicated before use.
Aerosol infection:
Mice were infected with approximately 100 CFU of aerosolized M. tuberculosis H37Rv using a Glas-Col inhalation exposure system (Glas-Col, Terre Haute, IN), thirty days after the third and final inoculation, and in prolonged time studies, mice were challenged 120 days later. Thirty days after infection mice were sacrificed and the number of viable organisms quantified in lung and spleen by plating 10-fold serial dilutions of organ homogenates on Difco™ Mycobacteria 7H11 Agar (Becton, Dickinson and Company, Sparks, MD). Colonies were counted after incubating plates at 37°C for 14–21 days.
NK cell depletion:
Each mouse was administered 250 μg via the i.p. route, of either anti-Asialo-GM1 antibody (Accurate Chemical & Scientific Corporation, Westbury, NY) or an isotype control antibody (Polyclonal Rabbit IgG, InVivo MAb, BioXcell, West Lebanon, NH) at two days before receiving intranasal CpG-C/liposome/ESAT-6 and day seven after for each inoculation. Mice were bled on the day of the first inoculation and the day of infection to assess NK cell depletion, using antibodies to CD3 (APC, clone 17A2, Tonbo Biosciences, San Diego, CA), N.K1.1 (PE, clone PK136, Tonbo Biosciences) and NKp46 (FITC, clone 29A1.4, BD Biosciences). For BAL analysis, mice were treated with anti-Asialo-GM1 antibody at days 4 and two before intranasal inoculation.
Cell preparation:
Single-cell suspensions from the lungs and spleens of mice were prepared by mechanical disruption through a 70μm nylon mesh screen and further purified with ACK Lysing Buffer (Life Technologies Corporation, Carlsbad, CA] for lysis of red blood cells. Cells were resuspended in cRPMI and counted using 1% Trypan blue solution to determine the number of viable cells and then diluted accordingly, depending on the analysis.
ELISpot assay:
T cell activation after CpG-C/Liposome/ESAT-6 inoculation was determined by ELISpot analysis for IFN-γ, TNF-α, and IL-2 (eBiosciences/ThermoFisher Scientific, San Diego, CA). Single cell suspensions were incubated overnight at of 5×106 cells/mL in cRPMI in the presence or absence M. tuberculosis H37Rv derived culture filtrate protein (CFP; BEI Resources, Manassas, VA) or ESAT-6 (BEI Resources) depending on the inoculation. Negative controls included cells from non-immunized mice and cells incubated without antigen, while positive control cells were stimulated with Concanavalin A (ConA, Sigma-Aldrich, Saint Louis, MO). Plates were then analyzed by quantifying the number of spots produced by cytokine producing cells using the Series 5 UV-Immunospot Analyzer (C.T.L. Shaker Heights, Ohio).
Flow cytometric analysis:
Antibodies directly conjugated with fluorochromes were used for flow cytometry (purchased from eBiosciences, BD Biosciences, Tonbo Biosciences, San Diego CA, Miltenyi Biotec, San Diego CA). The following antibodies were used: anti-CD11c (APC; clone N418), anti-CD8α (FITC; clone 53–6.7), anti-mPDCA-1 (FITC; Clone: JF05–1C2.4.1), anti-Ly-6C (APC Clone: 1G7.G10). Fluorochrome-conjugated isotype antibodies were used to control for each cell marker specific isotype antibody used. Cells were treated with FcγR (CD16)-blocking antibody (clone 2.4G2 supernatant) and washed. Cell marker-specific antibodies were diluted in fluorescence-activated cell sorting (FACS) buffer (PBS with 0.05% sodium azide) for staining and 25μL of fluorochrome was incubated with cells for 20 mins at 4°C followed by washing in FACS buffer. Cells were then fixed with 4% paraformaldehyde for 25 minutes at 4°C and were stored in FACS buffer before analysis. Flow cytometry was performed using a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA) for eight-color analysis. Cellular responses were analyzed by focusing dot-plot gates to include live lymphoid and myeloid cells, based on forward- and side-scatter characteristics of lung and spleen cells. Approximately 100,000 total events were collected, and data analysis was performed using FlowJo software (FLowJo, Ashland, OR).
ELISA:
IFN-γ, IFN-α, TNF-α, IL-10, IL-12, MCP-1 and IL-6 quantification in culture supernatants was performed by enzyme-linked immunosorbent assay (ELISA; eBiosciences/ThermoFisher Scientific). Plates containing cultured cells were centrifuged at 200xg for 5 minutes; supernatants were aspirated and stored at −80°C until used. ELISA was performed following the manufacturer’s protocol. Color intensity was then read using an Ultramark Microplate Imaging System (BioRad, Hercules, CA). A standard curve was also used with each assay to determine cytokine concentration in pg/mL.
Cytometric Bead Array (CBA):
Multiplex cytokine analysis in culture supernatants was performed by using BD™ CBA mouse inflammatory cytokines kit (BD Biosciences) following the manufacturer’s protocol for the following cytokines: IL-6, IL-10, MCP-1, IFN-γ, and TNF-α. Plates containing cultured cells were centrifuged at 200xg for 5 minutes; supernatants were aspirated and stored at −80°C until used. Supernatants were incubated with CBA beads and samples were analyzed by the FACS Canto II flow cytometer on FCAP Array™ CBA analyzing software (BD Biosciences). A standard curve was also used with each assay to determine cytokine concentration in pg/mL.
Statistical Analyses:
For multivariate analysis, statistical differences between treatment groups were compared using one-way ANOVA analyses with Tukey test post analysis, and non-parametric testing when performed if normality test failed. For comparisons between two treatment groups, Student’s t-test was used based on a 2-tailed hypothesis. Statistical analyses were done using SigmaStat® and Prism 8® software. A p-value <0.05 was considered statistically significant.
Results
Effect of CpG subtypes and concentrations in BMDC.
CpG sub-types were compared in vitro by assessing cytokine secretion in BMDC. The concentrations evaluated were 4, 2, and 1μM (Figure 1). In general, each concentration tested, CpG-B and -C induced higher concentrations of pro-inflammatory cytokines than CpG-A that, at the lower CpG concentrations, were significantly different (One-way ANOVA with Tukey post analysis for each CpG concentration). There was no significant difference in TNF-α and IL-6 concentrations produced by BMDC, for CpG-B and CpG-C at the 4 and 2 μM concentrations, but only at 1μM, CpG-C was better than CpG-B at increasing these cytokine concentrations. IL-10 production displayed a similar trend, except at 4mM there was no difference between stimuli. IL-17 was not produced at any of the CpG concentrations (data not shown). Non-CpG ODN did not have a stimulatory effect on BMDC (data not shown). These data suggested that CpG-C induced increased cytokine concentrations at lower ODN concentrations, significantly better than CpG-A and CpG-B.
Figure 1:
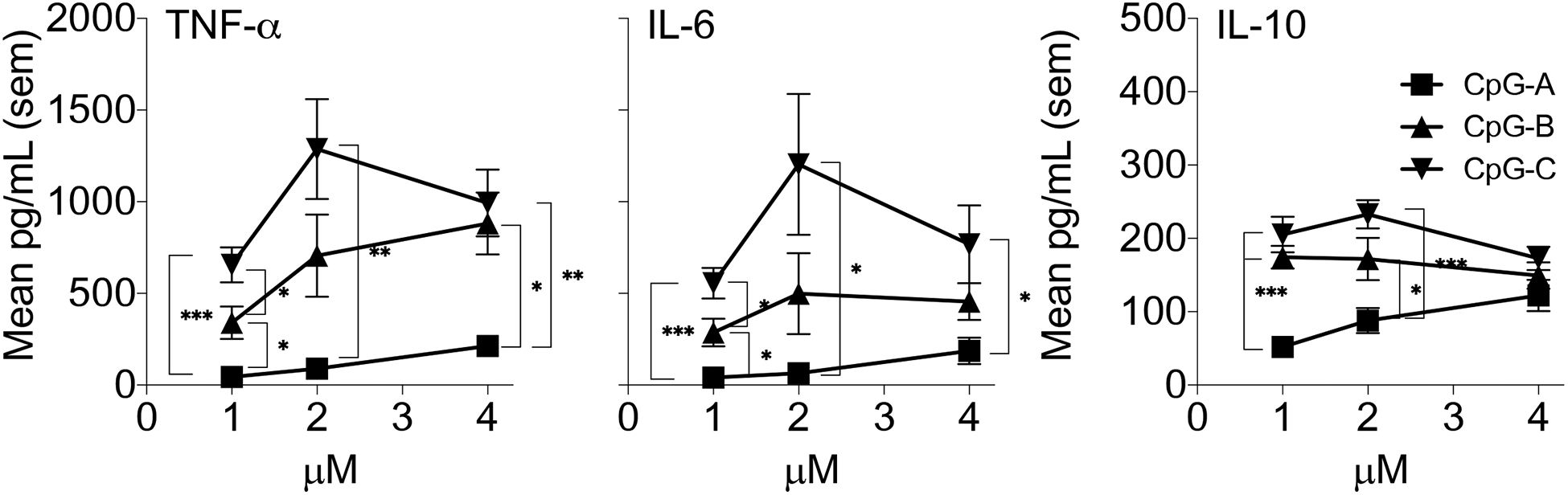
BMDC from C57BL/6 mice were treated with 1μM, 2μM and 4μM of CpG-A, -B, -C ODN for 48 hours and the concentration of TNF-α, IL-6, and IL-10 was assessed in supernatants using the CBA assay. Data are represented as the mean pg/ml and was determined from 7 replicate wells for each treatment. Data were analyzed initially using a One-Way ANOVA for each concentration between the CpG subtypes and then using the Tukey post test to determine differences between groups at each CpG concentration. “ * “ represents results of ANOVA analysis where * = p<0.5, ** = p<0.01, **** = p<0.001
Intranasal inoculation with CpG-A, CpG-B, CpG-C subtypes in C57BL/6 mice.
To determine if significant differences in cytokines induced by each CpG sub-type was related to the induction of protective immunity, each sub-type was formulated with liposomes and ESAT-6 and used to inoculate C57BL/6 mice via the intranasal route. Mice were infected with M. tuberculosis after 30 days and CFU determined (Figure 2A). ESAT-6 was chosen as the antigen because it is known to induce CD4+ and CD8+ T cell-mediated immunity resulting in a significant CFU reduction after pulmonary infection26–28. CpG-B and CpG-C formulations caused a significant decrease in mycobacterial lung burden compared to the saline-treated control group. The spleen bacterial burden in these groups while decreased, was not statistically significant. The bacterial burden in the lungs of the CpG-A inoculated group showed no significant difference, compared to the saline control, and the CpG-C vaccinated group resulted in lower CFU counts than the CpG-A vaccinated group, which was significantly different. Controls included mice inoculated with liposomes/ESAT-6, liposomes in combination with CpG-A, -B, and -C, non-CpG ODN, CpG-C/ESAT-6, and liposomes alone, none of which showed a statistically significant difference in CFU when compared to the saline control (data not shown). The frequency of activated T cells generated by intranasal vaccination with CpG-C/liposomes/ESAT-6 was significantly elevated at day 30 post-vaccination, as determined by the numbers of IFN-γ, TNF-α, and IL-2 expressing splenocytes (Figure 2B). CpG-C/liposomes/ESAT-6 induced a significant increase in the number of spot forming units for all three cytokines at 30 days (Figure 2B), suggesting the ability of the CpG-C to activate T cells and significantly reduce the mycobacterial burden.
Figure 2:
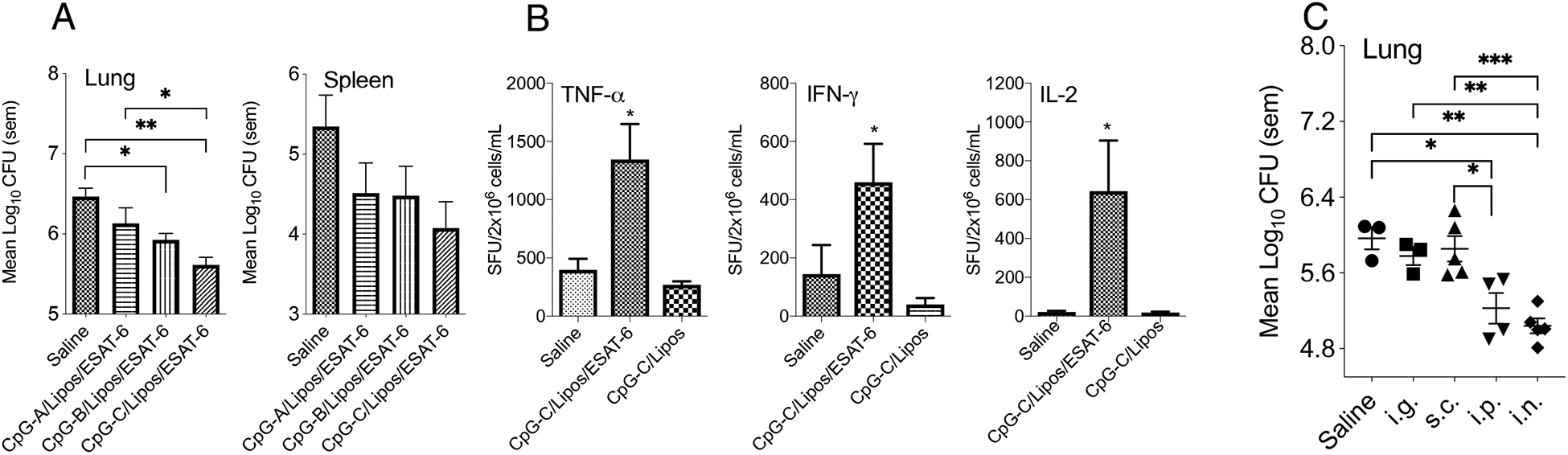
(A) The mycobacterial burden at day 30-post low dose aerosol infection with M. tuberculosis H37Rv in the lungs and spleens of C57BL/6 mice inoculated via the intranasal route with CpG-A, -B, and -C subtypes formulated with liposome and ESAT-6 three times at two-week intervals (N = 5 mice per group). (B) TNF-α, IFN-γ and IL-2 spot forming units (SFU) in spleen cells generated at day 30 post-vaccination with CpG-C based vaccine as shown by ELISpot assay. (C) Mice were inoculated with CpG-C formulated with liposomes and ESAT-6 and inoculated via the intragastric (i.g.), subcutaneous (s.c.), intraperitoneal (i.p.), and intranasal (i.n.) route as described above, rested for 30 days and then infected with M. tuberculosis H37Rv. Log10 CFU was determined 30 days post-infection (N = 3–5 mice per group). The Log10-transformed CFU data was analyzed using the One-way ANOVA for independent measures with post-hoc Tukey test, and SFU data were analyzed using the Student t-test with each treatment group being compared to the Saline-treated group. * = p<0.05, ** = p<0.01, *** = p<0.001
To determine if intranasal inoculation provided a superior reduction in CFU, mice were also inoculated via the intragastric, subcutaneous, and intraperitoneal routes. The routes were chosen as previous reports had indicated that CpG induced potent immune responses against tumors29. Mice inoculated with CpG-C/liposomes/ESAT-6 via the i.n. and s.c. routes produced a significant reduction in Log10 CFU counts compared to the saline-treated group (Figure 2C). Mice inoculated via the i.p. route also had a reduction in mycobacterial counts, but the i.n. route was chosen for the remainder of the studies, because it has been considered to be a possible inoculation route for tuberculosis vaccines currently in clinical trials and targets the organ that is most likely to be infected.30–32. Subsequently, intranasal delivery of CpG-C, liposomes, and ESAT-6 consistently provided a significant decrease in CFU when compared to saline-treated mice in five separate experiments (data not shown).
CpG-C induced early pulmonary responses.
Intranasal inoculation of liposome encapsulated CpG-C induced protective immunity to pulmonary M. tuberculosis infection (Figure 2). We hypothesized that the pulmonary environment became activated soon after CpG-C/liposome inoculation with the induction of pro-inflammatory cytokines and recruitment of early effector myeloid cells that drove mucosal immunity. To test the hypothesis, mice were inoculated with CpG-C/liposomes and the alveolar sac environment sampled by taking broncho-alveolar lavage (BAL) fluid, at 6, 24, and 48 hours to characterize cytokine production and DC recruitment. CpG-C is known to stimulate plasmacytoid DC (pDC) to produce IFN-α, and the BAL IFN-α concentration was elevated at 24 hours, although not significantly, and then decreased at 48 hours. IL-12 was significantly increased at 48 hours after inoculation. TNF-α and MCP-1 concentrations were significantly increased at 24 and 48 hours, while IFN-γ and IL-17 were not significantly increased compared to liposome only (Figure 3A). BAL cells were analyzed for CD11c and CD8α expression and mPDCA and Ly6C expression at the 6 hour time point (Figure 3B and C). CD11c+CD8α+ DC have been shown to play a role in lung adaptive immunity against some pathogens33, 34, and mPDCA+Ly6C+ DC were examined as these were shown to be a subset of DCs present on mucosal tissue35. Thus we asked if the early recruitment of these cells was induced by CpG-C instillation. Significantly elevated levels of CD11cintCD8α+, CD11c+CD8α+, and CD11cintCD8α− DCs were observed within 6 hours of CpG-C intranasal instillation. In addition, percentages of mPDCA+Ly6C+ and mPDCA+Ly6C− DCs were significantly up-regulated within the same time. Taken together these data suggest that CpG-C formulated with liposome induced the early recruitment of diverse subsets of DCs, which preceded the induction of cytokines within the same space.
Figure 3:
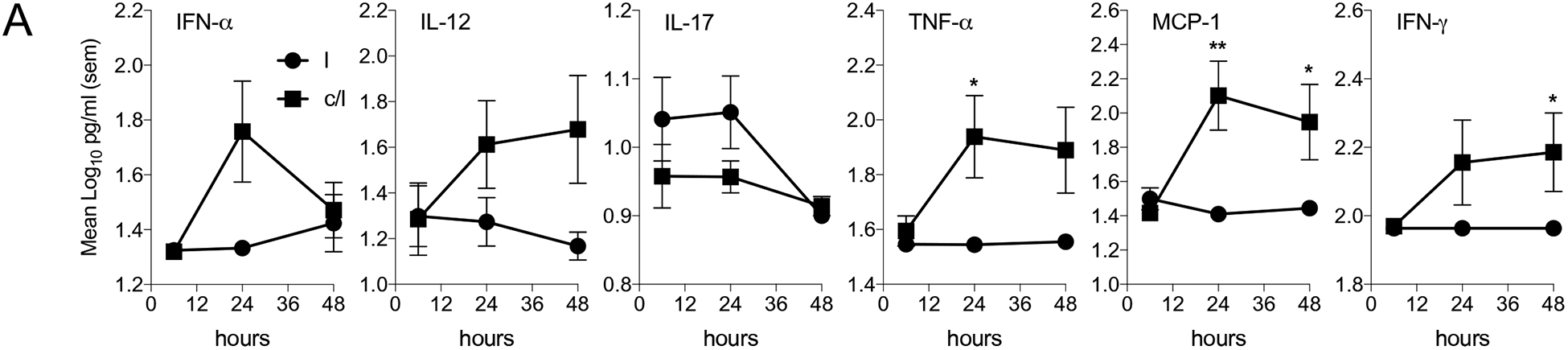
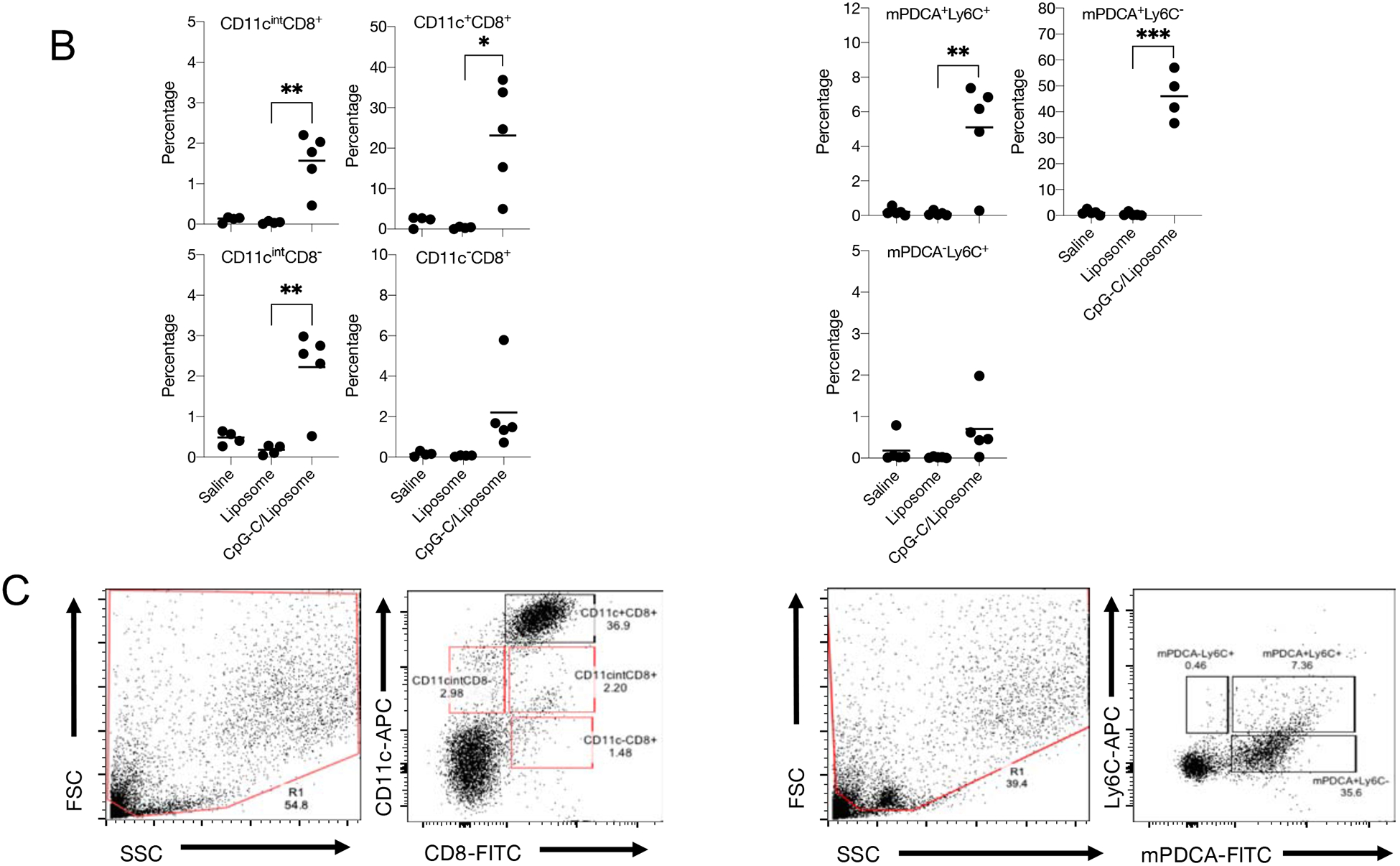
(A) Broncho-alveolar lavage (BAL) fluid from C57BL/6 mice inoculated intranasally, with CpG-C/liposome (c/l (■)) or liposome (l, (●)) alone was assayed for IFN-α, IL-12, IL-17, TNF-α, MCP-1, and IFN-γ. Mice were inoculated via the intranasal route with each formulation and BAL fluid was taken at 6, 24, and 48 hours post-inoculation. (B and C) BAL cells were analyzed at 6 hours after intranasal instillation for CD11c and CD8α expression and mPDCA and Ly6C expression. N = 5 mice per group per time point. Data are representative of 3 experiments. * = p<0.05, ** = p<0.01
To determine if NK cells were responsible for the IFN-γ in the BAL of CpG-C/liposome inoculated mice, these cells were depleted with anti-Asialo-GM1 (Supplemental data), before intranasal inoculation. We also wanted to determine if NK cells played an important role in CpG-C induced IFN-α. At 24-hours the concentrations of IFN-γ in the BAL of anti-Asialo-GM-1 treated mice was significantly reduced compared to isotype control treated mice, suggesting that these cells function during the early innate response (Figure 4A). Although NK cells were reduced, there was no major effect on IFN-α, but the IFN-γ concentration was significantly reduced compared to isotype antibody treated mice (Figure 4A). To determine if this lack of impact on IFN-α was correlated with infection outcome, mice were treated with anti-Asialo-GM1 during vaccination and then infected 30 days later. CFU analysis demonstrated that NK cell depletion during the vaccination period did not alter the ability of mice to reduce CFU in the lung (Figure 4B) and spleen (data not shown). IFN-γ was produced during the early innate mucosal response to CpG-C, and it is unlikely that NK cells played a role in regulating the induction of IFN-α, or the immune response to M. tuberculosis infection. Interestingly, the TNF-α concentrations were significantly reduced in the vaccinated group treated with anti-Asialo-GM1.
Figure 4:
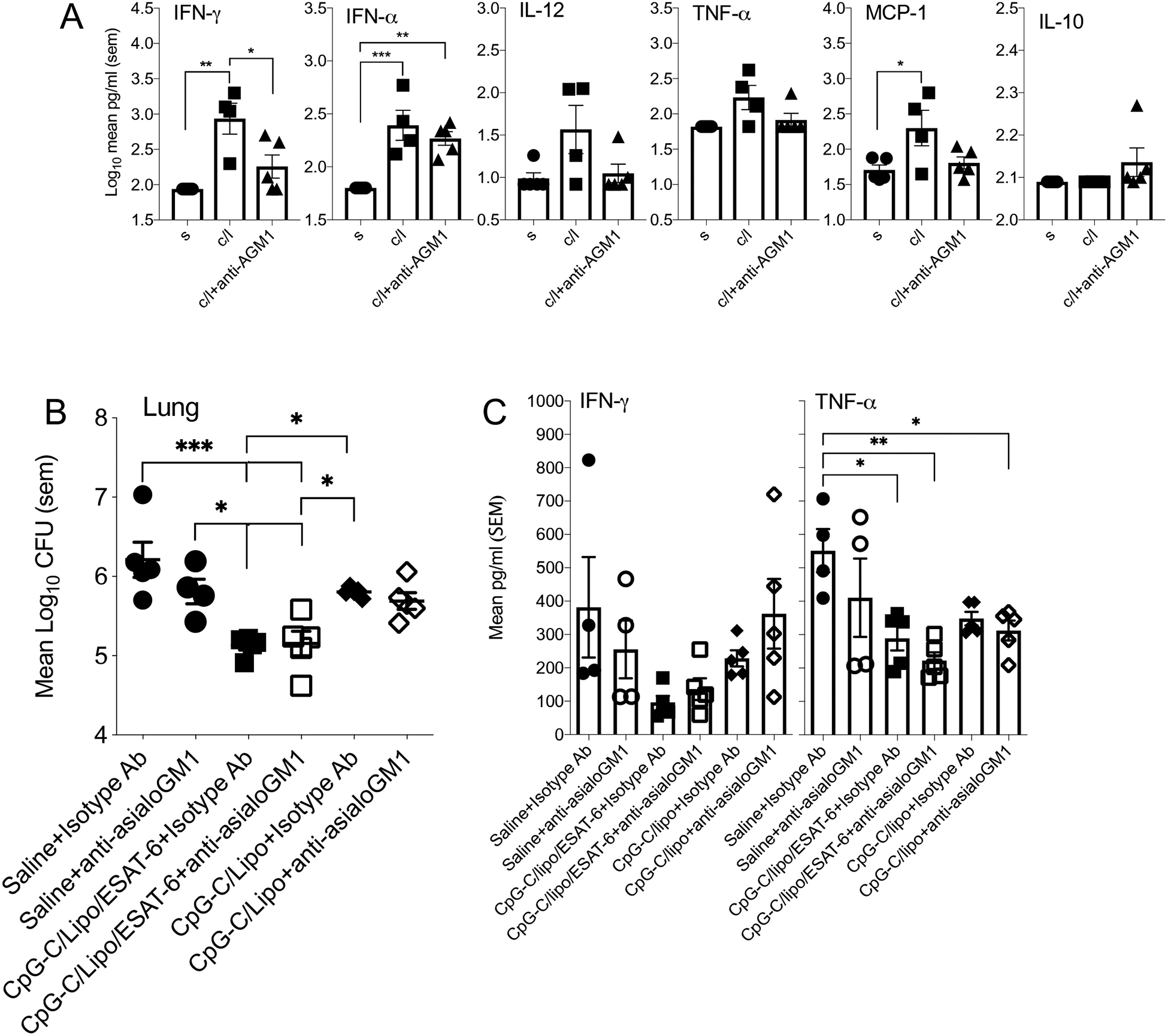
(A) Broncho-alveolar lavage (BAL) fluid from C57BL/6 mice treated with anti-Asialo-GM1 at days 4 and 2 before intranasal inoculation of CpG-C/liposomes/ESAT-6 assayed for IFN-γ, IFN-α, IL-12, TNF-α, MCP-1, and IL-17. Data is from 24 hours post-inoculation. (B) The mycobacterial burden of mice treated with anti-Asialo-GM1 during intranasal inoculation of CpG-C/liposomes/ESAT-6 and infected at day 30 after the last dose with a low dose aerosol of M. tuberculosis H37Rv. Log10 CFU was determined in the lungs and spleens of mice at day 30 post-infection. (C) Cytokine concentration was determined in the lung homogenates from infected mice. Data were analyzed using the One-way ANOVA for independent measures with post-hoc Tukey test. N = 5 mice per group per time point. * = p<0.05, ** = p<0.01
A role for IFN-α in CpG-mediated mucosal immunity.
Given that CpG-C induced elevated levels of pulmonary IFN-α soon after intranasal inoculation we wished to determine if it played a role in the development of adaptive immunity against M. tuberculosis infection. IFN-αR1−/− mice were inoculated with CpG-C/liposome/ESAT-6 and then infected as described previously. At day 30 after infection, the mycobacterial burden in inoculated C57BL/6 mice was significantly reduced in lungs (Figure 5A) compared to saline-treated mice. The CFU in the lungs of IFN-αR1−/− mice inoculated with CpG-C/liposome/ESAT-6 was not significantly different from saline-treated IFN-αR1−/− mice, suggesting the need for type I IFN in the mucosal immunity by this formulation. Thus, type I IFN dependent responses were required for the reduction of the mycobacterial burden. BCG vaccinated IFN-αR1−/− mice had significantly reduced CFU compared to saline-treated IFN-αR1−/− mice, suggesting an alternative mechanism of induction of immunity by BCG.
Figure 5:
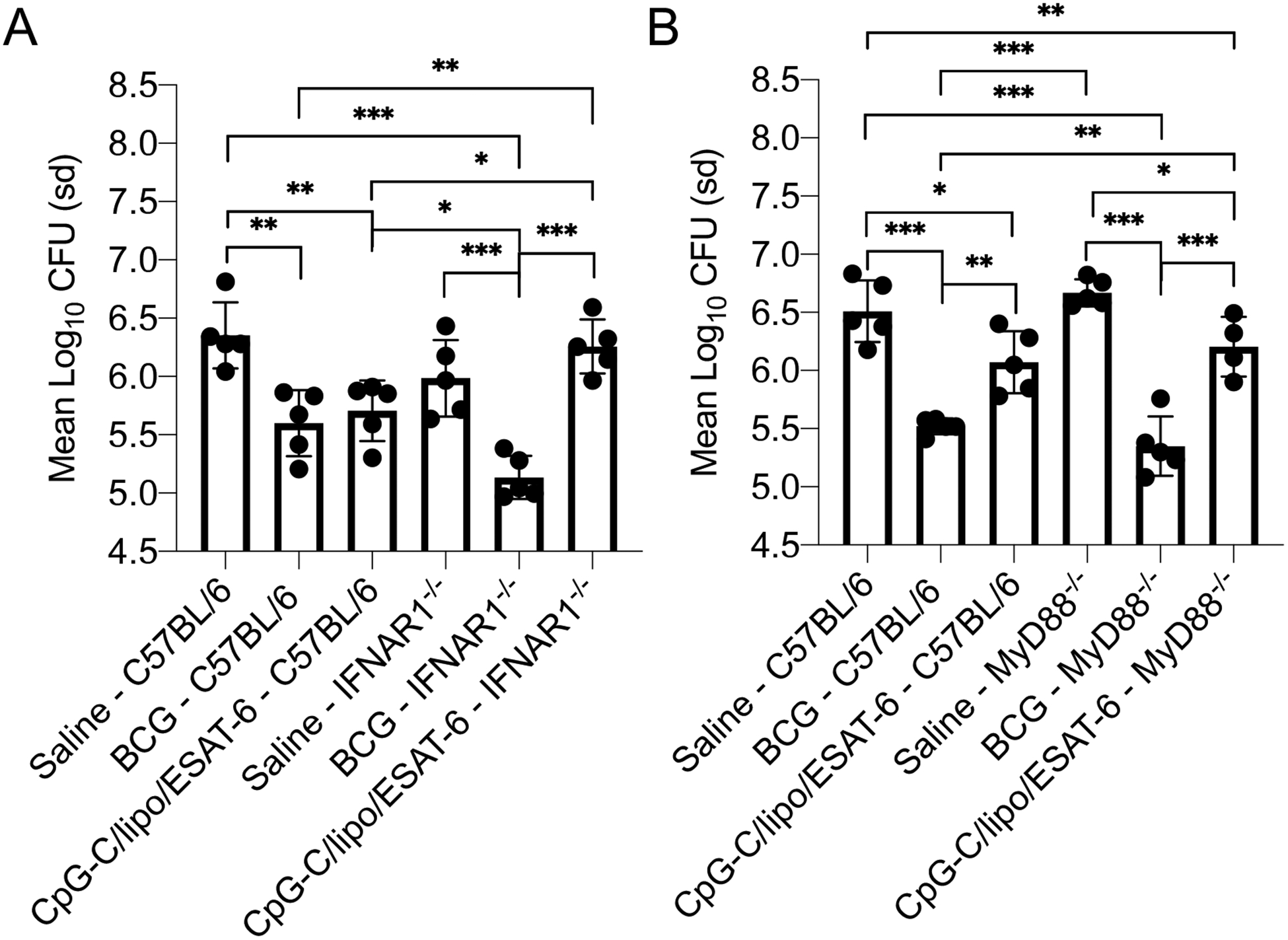
(A) The mycobacterial burden in the lungs of IFNAR1−/− mice and C57BL/6 mice inoculated with either BCG via the subcutaneous route or CpG-C/liposome/ESAT-6 formulation via the intranasal route at day 30 after the final inoculation. Mice were vaccinated with BCG via the subcutaneous route. The mycobacterial burden was determined at day 30 post pulmonary infection with M. tuberculosis H37Rv. (B) The mycobacterial burden in the lungs of MyD88−/− mice and C57BL/6 mice inoculated with either BCG via the subcutaneous route or CpG-C/liposome/ESAT-6 via the intranasal route at day 30 after the final inoculation. Data were analyzed using the One-way ANOVA for independent measures with post-hoc Tukey test. Data are representative of two experiments. N = 4–5 mice per group. * = p<0.05, ** = p<0.01, *** = p<0.001
Role of MyD88 in intra-nasal vaccination with CpG-C based vaccine.
Myeloid differentiation primary response gene 88 (MyD88) is a major intracellular adaptor protein that interacts with the cytoplasmic domain of the majority of TLRs. CpG-C interaction with TLR9 and MyD88 has been demonstrated to be essential for intracellular signaling and cytokine production. To determine the role of MyD88 in the immune mechanism induced by CpG-C/liposome/ESAT-6, MyD88−/− mice were vaccinated and then infected as described above. ANOVA analysis of the Log10 CFU reveal differences between groups of infected mice. Of note, the bacterial burden in the lungs of CpG-C/Liposomes/ESAT-6 inoculated MyD88−/− mice was significantly reduced compared to the saline control MyD88−/− mice at day 30 of infection (Figure 5B). There was no significant difference in the bacterial burden in the CpG-C/liposomes/ESAT-6 vaccinated groups between MyD88−/− mice and wild-type C57BL/6 mice indicating equal levels of protection in the presence or absence of MyD88. Although a trend was observed in the reduction of the bacterial burden in the spleen, there was no significant difference between groups. Similar results were observed with BCG vaccinated mice, which supports a previous report36. Thus MyD88 did not play an essential role in the induction CpG-C based mucosal immunity to M. tuberculosis infection
Protective immunity induced at 120 days post-intranasal vaccination with CpG-C.
An essential requirement for vaccines is the ability to generate long-term immunity. To determine if CpG-C/liposomes/ESAT-6 was capable of inducing prolonged protective immunity, mice were inoculated, rested, then infected 120 days later, and sacrificed 30 days following infection to evaluate the bacterial burden in the lung and spleen. At the end of 120 days, spleen cells were analyzed for the frequency of IFN-γ, TNF-α and IL-2 secreting cells (Figure 6A). There were significantly elevated numbers of IFN-γ and IL-2 secreting cells in the CpG-C/liposomes/ESAT-6 vaccinated group compared to control groups, but not in TNF-α secretion.
Figure 6:
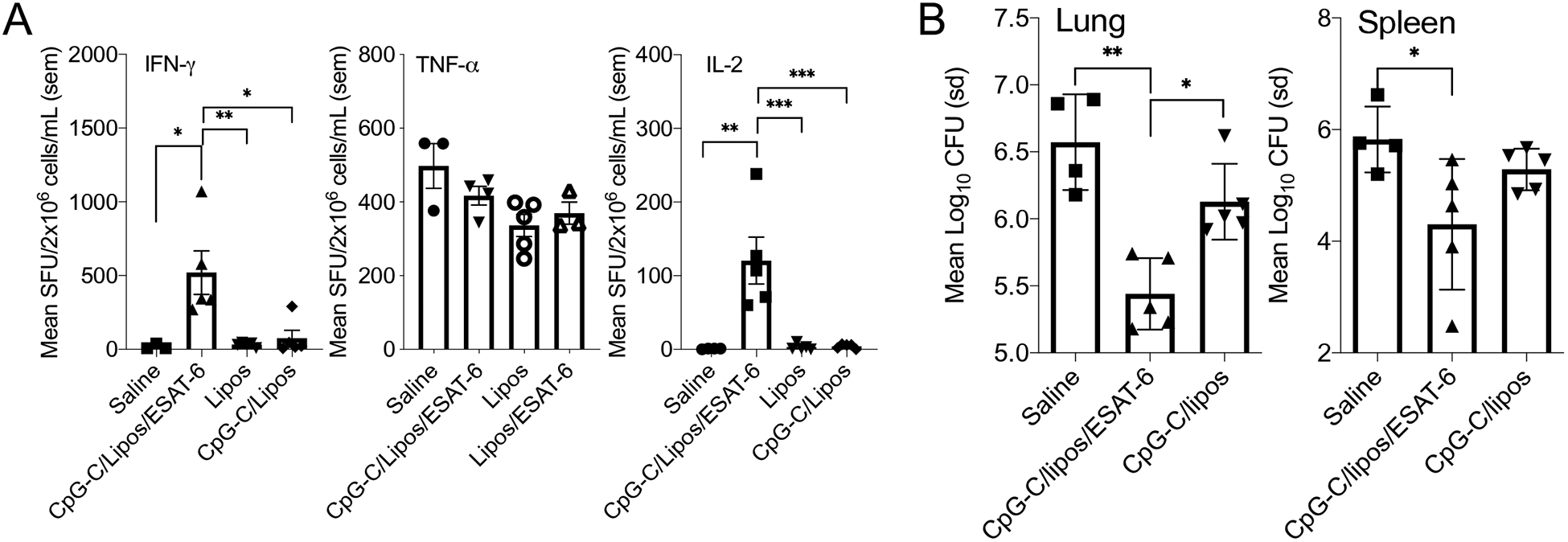
(A) The frequency of IFN-γ, TNF-α, and IL-2 producing cells in the lungs of C57BL/6 mice inoculated intranasally with CpG-C/liposomes/ESAT-6, 120 days earlier. (B) The mycobacterial burden in the lung and spleen of mice were infected with a low dose aerosol of M. tuberculosis H37Rv, 120 days after inoculation. Data are representative of two experiments. N = 4–5 mice per group. * = p<0.05, ** = p<0.01, *** = p<0.001.
Given that the cells were stimulated with M. tuberculosis-derived CFP, it is possible that multiple PAMPs stimulated TNF-α production, resulting in high levels of cytokine secreting cells. The bacterial burden in the lung following infection was significantly reduced in the CpG-C/liposomes/ESAT-6 vaccine group compared to saline control mice (Figure 6B). In the spleen, a similar trend in bacterial burden was observed, with a significant reduction in mycobacterial burden observed in the CpG-C/liposomes/ESAT-6 vaccinated group. These data suggest that CpG-C/liposomes/ESAT-6 induced prolonged immunity.
Discussion
Signaling through TLR9 induces strong adaptive immune responses and thus targeting the receptor in vaccine formulations has been beneficial for promoting protective immunity against intracellular pathogens such as M. tuberculosis. Several anti-tuberculosis protein-based vaccines employ TLR9 agonists to stimulate innate immunity1. The current study showed that type C CpG, more so than types A or B, when administered as an intranasal vaccine caused a significant reduction in the mycobacterial burden in the C57BL/6 mouse model of tuberculosis. Mucosal protective immunity was mediated through type I IFN, that did not require the presence of NK cells during the early innate response in the lungs, for CpG-C to induce anti-tuberculosis immunity. CpG ODN is used in multiple vaccine applications37–39 and is a component of several new tuberculosis vaccines1, one of which, H4 plus IC31 is in clinical trials40–42. However, based on our current studies, it seems as though not all CpG induce protective immunity and our studies suggest that only CpG type C was able to achieve protective immunity in the mouse model. Our findings are supported by reports that have shown that CpG7909, a B-type CpG was not able to induce protective anti-tuberculosis immunity43.
Type I IFN have previously been shown in the mouse model to exacerbate M. tuberculosis infection as it altered immunity44–46. In humans, tuberculosis and influenza co-infection were associated with increased mortality, and influenza co-infection is associated with increased mortality in tuberculosis patients47. In addition, a genetic variant in the human IFNAR1 gene was shown to decrease type I IFN signaling, reducing the risk of tuberculosis48. Our data suggest that type I IFN were required for the development of CpG-C mucosal vaccination-mediated immunity and may describe a different mechanism, representing a scenario in which a cytokine that may be detrimental during infection may be beneficial during induction of vaccine-mediated immunity. The benefit of type I IFN may be related to multiple factors such as duration of exposure of antigens, where it has been reported that during chronic exposure, type I IFN is less effective than during acute exposure. Type I IFN has been associated with proinflammatory responses when expressed acutely, but become immunosuppressive during prolonged expression49. This scenario may be related to vaccination in that antigen and adjuvant are present for short periods, resulting in induction of immunity, while during infection, the excessive amount of type I IFN results in immunosuppressive activity. Indeed, type I IFN signaling in DC has been shown to cause maturation and antigen presentation by DCs to promote adaptive immunity50. Kutchey et al.51 showed that IFNα/β was partially required for controlling pulmonary BCG infection, suggesting a possible role of type I IFN in the clearance of attenuated mycobacteria, and involvement in mucosal vaccination. The recent development of IC31, an adjuvant made of a synthetic anti-microbial cationic peptide and TLR9 agonist d(IC)13 anionic ODN1a52–54, and its use in combination with H4 polypeptide vaccine have shown the importance of inducing type I IFN in mediating anti-tuberculosis immunity through vaccination3. The data presented in the current studies demonstrated that type I IFN was required to cause a significant reduction in mycobacterial burden after intranasal inoculation. In addition, activation of TLR9 resulted in prolonged protective immunity.
CpG ODN has been shown to enhance host defense in a high dose M. tuberculosis infection model through an IFN-γ-dependent mechanism55, and the inclusion of CpG increased the magnitude of the Th1 response induced by BCG vaccination56. In general, mechanisms of CpG induced immunity include up-regulation of co-stimulatory molecules and IL-12 production by dendritic cells57. Global gene analysis of CpG stimulated RAW cells also demonstrated the up-regulation of IL-12 and IL-6 genes within 12 hours, showing the selective induction of specific networks by CpG12. Our data support the previous observations and provide evidence that CpG can stimulate immune responses in the lungs within 24 to 48 hours after inoculation resulting in pro-inflammatory cytokine increases and DC recruitment. Our data also showed that TLR9 signaling was MyD88-independent, indicating that alternative mechanisms were responsible for mediating protective immunity. Previously, others had shown that poly(I:C) (TLR3 agonist) induced NF-κB activation and mitogen-activated protein (MAP) kinases independently of MyD8858 and that TIR domain-containing adaptor inducing IFN-β (TRIF), but not MyD88 was essential for TLR3- and TLR4-mediated signaling pathways59. In regards to M. tuberculosis, although no report has examined the role of MyD88 in sub-unit vaccine-induced immunity, others have demonstrated that MyD88 was essential for controlling M. tuberculosis growth16, 36. Our data supports previous reports that showed that BCG vaccination was able to significantly reduce the mycobacterial burden and prolong the life of MyD88−/− mice36. We now show here that a sub-unit vaccine formulated with CpG-C resulted in the induction of anti-mycobacterial immunity in the absence of MyD88. A TLR9/MyD88-independent, activation mechanism in mouse and human CD4+ T cells, using CpG, was identified, and suggests the presence of other possible receptors for the agonist60. In addition, resveratrol, which suppresses NF-κB activation and cyclooxygenase-2 expression upon TLR3 and TLR4 stimulation, did not affect TLR2 or TLR9 stimulation61. Our data support the idea that MyD88 is not an absolute requirement for induction of CpG-C mediated mucosal immunity against M. tuberculosis infection. Thus, using multiple TLR agonists in vaccine formulations may be a better strategy for future vaccine development, such that multiple pathways can be stimulated so that more potent immune responses can be produced.
Elevated BAL IFN-γ concentrations were associated with NK cells, as anti-asialoGM1-treated mice had lower concentrations than isotype controls, but in the absence of these cells, there was not a significant effect on the eventual outcome of infection. Aerosolized CpG was shown to polarize naive alveolar macrophages toward an M1-like phenotype, which then activate NK cells62, thus indicating an inter-relationship between lung cells at the critically important early stages of immune development. Concerning the development of pulmonary immunity by a vaccine against tuberculosis our data would suggest that NK cells were not significant.
In summary, CpG-C was a superior activator of pro-inflammatory cytokines when compared head-to-head with CpG types A and B and thus was used to induce mucosal immunity. The current studies have identified an essential role for type I IFN in the induction of mucosal immunity generated by CpG-C, that was NK cell and MyD88-independent. These data are novel in that they are the first to identify non-traditional pathways of stimulating anti-tuberculosis immunity. The pathways utilized by CpG-C to induce mucosal immunity are currently under investigation.
Supplementary Material
Acknowledgements
This work was supported by the NIH/NIAID program Advanced Small Animal Models for the Testing of Candidate Therapeutic and Preventative Interventions against Mycobacteria (HHSN272201000009I-003, task order 12 and task order A80) at Colorado State University (AAI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mendez-Samperio P Development of tuberculosis vaccines in clinical trials: Current status. Scand J Immunol 2018;88:e12710. doi: 10.1111/sji.12710 [DOI] [PubMed] [Google Scholar]
- 2.Del Giudice G, Rappuoli R, Didierlaurent AM. Correlates of adjuvanticity: A review on adjuvants in licensed vaccines. Semin Immunol 2018;39:14–21. doi: 10.1016/j.smim.2018.05.001 [DOI] [PubMed] [Google Scholar]
- 3.Aboutorabian S, Hakimi J, Boudet F, Montano S, Dookie A, Roque C, Ausar SF, Rahman N, Brookes RH. A high ratio of IC31((R)) adjuvant to antigen is necessary for H4 TB vaccine immunomodulation. Hum Vaccin Immunother 2015;11:1449–1455. doi: 10.1080/21645515.2015.1023970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lingnau K, Riedl K, von Gabain A. IC31 and IC30, novel types of vaccine adjuvant based on peptide delivery systems. Expert Rev Vaccines 2007;6:741–746. doi: 10.1586/14760584.6.5.741 [DOI] [PubMed] [Google Scholar]
- 5.Sepulveda-Toepfer JA, Pichler J, Fink K, Sevo M, Wildburger S, Mudde-Boer LC, Taus C, Mudde GC. TLR9-mediated activation of dendritic cells by CD32 targeting for the generation of highly immunostimulatory vaccines. Hum Vaccin Immunother 2019;15:179–188. doi: 10.1080/21645515.2018.1514223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fekete T, Suto MI, Bencze D, Mazlo A, Szabo A, Biro T, Bacsi A, Pazmandi K. Human Plasmacytoid and Monocyte-Derived Dendritic Cells Display Distinct Metabolic Profile Upon RIG-I Activation. Front Immunol 2018;9:3070. doi: 10.3389/fimmu.2018.03070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko YA, Chan YH, Liu CH, Liang JJ, Chuang TH, Hsueh YP, Lin YL, Lin KI. Blimp-1-Mediated Pathway Promotes Type I IFN Production in Plasmacytoid Dendritic Cells by Targeting to Interleukin-1 Receptor-Associated Kinase M. Front Immunol 2018;9:1828. doi: 10.3389/fimmu.2018.01828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laustsen A, Bak RO, Krapp C, Kjaer L, Egedahl JH, Petersen CC, Pillai S, Tang HQ, Uldbjerg N, Porteus M, Roan NR, Nyegaard M, Denton PW, Jakobsen MR. Interferon priming is essential for human CD34+ cell-derived plasmacytoid dendritic cell maturation and function. Nat Commun 2018;9:3525. doi: 10.1038/s41467-018-05816-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masuta Y, Yamamoto T, Natsume-Kitatani Y, Kanuma T, Moriishi E, Kobiyama K, Mizuguchi K, Yasutomi Y, Ishii KJ. An Antigen-Free, Plasmacytoid Dendritic Cell-Targeting Immunotherapy To Bolster Memory CD8(+) T Cells in Nonhuman Primates. J Immunol 2018;200:2067–2075. doi: 10.4049/jimmunol.1701183 [DOI] [PubMed] [Google Scholar]
- 10.Pohar J, Yamamoto C, Fukui R, Cajnko MM, Miyake K, Jerala R, Bencina M. Selectivity of Human TLR9 for Double CpG Motifs and Implications for the Recognition of Genomic DNA. J Immunol 2017;198:2093–2104. doi: 10.4049/jimmunol.1600757 [DOI] [PubMed] [Google Scholar]
- 11.Hemmi H, Kaisho T, Takeda K, Akira S. The roles of Toll-like receptor 9, MyD88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J Immunol 2003;170:3059–3064. [DOI] [PubMed] [Google Scholar]
- 12.Tross D, Petrenko L, Klaschik S, Zhu Q, Klinman DM. Global changes in gene expression and synergistic interactions induced by TLR9 and TLR3. Mol Immunol 2009;46:2557–2564. doi: 10.1016/j.molimm.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abe T, Hemmi H, Miyamoto H, Moriishi K, Tamura S, Takaku H, Akira S, Matsuura Y. Involvement of the Toll-like receptor 9 signaling pathway in the induction of innate immunity by baculovirus. J Virol 2005;79:2847–2858. doi: 10.1128/JVI.79.5.2847-2858.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurk M, Schulte B, Kritzler A, Noll B, Uhlmann E, Wader T, Schetter C, Krieg AM, Vollmer J. C-Class CpG ODN: sequence requirements and characterization of immunostimulatory activities on mRNA level. Immunobiology 2004;209:141–154. doi: 10.1016/j.imbio.2004.02.006 [DOI] [PubMed] [Google Scholar]
- 15.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med 2005;202:1715–1724. doi: 10.1084/jem.20051782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, Sonntag T, Kroger S, Akira S, Mossmann H, Kirschning CJ, Wagner H, Freudenberg M, Ehlers S. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, −4 and −9. Eur J Immunol 2008;38:680–694. doi: 10.1002/eji.200736458 [DOI] [PubMed] [Google Scholar]
- 17.Torres-Garcia D, Cruz-Lagunas A, Garcia-Sancho Figueroa MC, Fernandez-Plata R, Baez-Saldana R, Mendoza-Milla C, Barquera R, Carrera-Eusebio A, Ramirez-Bravo S, Campos L, Angeles J, Vargas-Alarcon G, Granados J, Gopal R, Khader SA, Yunis EJ, Zuniga J. Variants in toll-like receptor 9 gene influence susceptibility to tuberculosis in a Mexican population. J Transl Med 2013;11:220. doi: 10.1186/1479-5876-11-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bharti D, Kumar A, Mahla RS, Kumar S, Ingle H, Shankar H, Joshi B, Raut AA, Kumar H. The role of TLR9 polymorphism in susceptibility to pulmonary tuberculosis. Immunogenetics 2014;66:675–681. doi: 10.1007/s00251-014-0806-1 [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Li X, Cui W, Guan L, Shen F, Xu J, Zhou F, Li M, Gao C, Jin Q, Liu J, Gao L. Potential association of pulmonary tuberculosis with genetic polymorphisms of toll-like receptor 9 and interferon-gamma in a Chinese population. BMC Infect Dis 2013;13:511. doi: 10.1186/1471-2334-13-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jahantigh D, Salimi S, Alavi-Naini R, Emamdadi A, Owaysee Osquee H, Farajian Mashhadi F. Association between TLR4 and TLR9 gene polymorphisms with development of pulmonary tuberculosis in Zahedan, southeastern Iran. ScientificWorldJournal 2013;2013:534053. doi: 10.1155/2013/534053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agger EM, Rosenkrands I, Olsen AW, Hatch G, Williams A, Kritsch C, Lingnau K, von Gabain A, Andersen CS, Korsholm KS, Andersen P. Protective immunity to tuberculosis with Ag85B-ESAT-6 in a synthetic cationic adjuvant system IC31. Vaccine 2006;24:5452–5460. doi: 10.1016/j.vaccine.2006.03.072 [DOI] [PubMed] [Google Scholar]
- 22.Zaks K, Jordan M, Guth A, Sellins K, Kedl R, Izzo A, Bosio C, Dow S. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol 2006;176:7335–7345. [DOI] [PubMed] [Google Scholar]
- 23.Verwaerde C, Debrie AS, Dombu C, Legrand D, Raze D, Lecher S, Betbeder D, Locht C. HBHA vaccination may require both Th1 and Th17 immune responses to protect mice against tuberculosis. Vaccine 2014;32:6240–6250. doi: 10.1016/j.vaccine.2014.09.024 [DOI] [PubMed] [Google Scholar]
- 24.Brandt L, Oettinger T, Holm A, Andersen AB, Andersen P. Key epitopes on the ESAT-6 antigen recognized in mice during the recall of protective immunity to Mycobacterium tuberculosis. J Immunol 1996;157:3527–3533. [PubMed] [Google Scholar]
- 25.Grover A, Taylor J, Troudt J, Keyser A, Arnett K, Izzo L, Rholl D, Izzo A. Kinetics of the immune response profile in guinea pigs after vaccination with Mycobacterium bovis BCG and infection with Mycobacterium tuberculosis. Infect Immun 2009;77:4837–4846. doi: 10.1128/IAI.00704-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciabattini A, Pettini E, Andersen P, Pozzi G, Medaglini D. Primary activation of antigen-specific naive CD4+ and CD8+ T cells following intranasal vaccination with recombinant bacteria. Infect Immun 2008;76:5817–5825. doi: 10.1128/IAI.00793-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grover A, Troy A, Rowe J, Troudt JM, Creissen E, McLean J, Banerjee P, Feuer G, Izzo AA. Humanized NOG mice as a model for tuberculosis vaccine-induced immunity: a comparative analysis with the mouse and guinea pig models of tuberculosis. Immunology 2017;152:150–162. doi: 10.1111/imm.12756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.West NP, Thomson SA, Triccas JA, Medveczky CJ, Ramshaw IA, Britton WJ. Delivery of a multivalent scrambled antigen vaccine induces broad spectrum immunity and protection against tuberculosis. Vaccine 2011;29:7759–7765. doi: 10.1016/j.vaccine.2011.07.109 [DOI] [PubMed] [Google Scholar]
- 29.Westwood JA, Potdevin Hunnam TC, Pegram HJ, Hicks RJ, Darcy PK, Kershaw MH. Routes of delivery for CpG and anti-CD137 for the treatment of orthotopic kidney tumors in mice. PLoS One 2014;9:e95847. doi: 10.1371/journal.pone.0095847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dietrich J, Andersen C, Rappuoli R, Doherty TM, Jensen CG, Andersen P. Mucosal administration of Ag85B-ESAT-6 protects against infection with Mycobacterium tuberculosis and boosts prior bacillus Calmette-Guerin immunity. J Immunol 2006;177:6353–6360. doi: 10.4049/jimmunol.177.9.6353 [DOI] [PubMed] [Google Scholar]
- 31.Andersen CS, Dietrich J, Agger EM, Lycke NY, Lovgren K, Andersen P. The combined CTA1-DD/ISCOMs vector is an effective intranasal adjuvant for boosting prior Mycobacterium bovis BCG immunity to Mycobacterium tuberculosis. Infect Immun 2007;75:408–416. doi: 10.1128/IAI.01290-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giri PK, Khuller GK. Is intranasal vaccination a feasible solution for tuberculosis? Expert Rev Vaccines 2008;7:1341–1356. doi: 10.1586/14760584.7.9.1341 [DOI] [PubMed] [Google Scholar]
- 33.Liao Y, Liu X, Huang Y, Huang H, Lu Y, Zhang Y, Shu S, Fang F. Expression pattern of CD11c on lung immune cells after disseminated murine cytomegalovirus infection. Virol J 2017;14:132. doi: 10.1186/s12985-017-0801-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunne PJ, Moran B, Cummins RC, Mills KH. CD11c+CD8alpha+ dendritic cells promote protective immunity to respiratory infection with Bordetella pertussis. J Immunol 2009;183:400–410. doi: 10.4049/jimmunol.0900169 [DOI] [PubMed] [Google Scholar]
- 35.GeurtsvanKessel CH, Lambrecht BN. Division of labor between dendritic cell subsets of the lung. Mucosal Immunol 2008;1:442–450. doi: 10.1038/mi.2008.39 [DOI] [PubMed] [Google Scholar]
- 36.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest 2004;114:1790–1799. doi: 10.1172/JCI21027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi T, Momota M, Kuroda E, Kusakabe T, Kobari S, Makisaka K, Ohno Y, Suzuki Y, Nakagawa F, Lee MSJ, Coban C, Onodera R, Higashi T, Motoyama K, Ishii KJ, Arima H. DAMP-Inducing Adjuvant and PAMP Adjuvants Parallelly Enhance Protective Type-2 and Type-1 Immune Responses to Influenza Split Vaccination. Front Immunol 2018;9:2619. doi: 10.3389/fimmu.2018.02619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golshani M, Amani M, Siadat SD, Nejati-Moheimani M, Arsang A, Bouzari S. Comparison of the protective immunity elicited by a Brucella cocktail protein vaccine (rL7/L12+rTOmp31+rSOmp2b) in two different adjuvant formulations in BALB/c mice. Mol Immunol 2018;103:306–311. doi: 10.1016/j.molimm.2018.10.002 [DOI] [PubMed] [Google Scholar]
- 39.He J, Huang F, Zhang J, Chen H, Chen Q, Zhang J, Li J, Zheng Z, Chen D, Chen J. DNA prime-protein boost vaccine encoding HLA-A2, HLA-A24 and HLA-DR1 restricted epitopes of CaNA2 against visceral leishmaniasis. Immunology 2019;156:94–108. doi: 10.1111/imm.13007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Bree C, van Crevel R, Netea MG. H4:IC31 Vaccine or BCG Revaccination for Tuberculosis. N Engl J Med 2018;379:1969. doi: 10.1056/NEJMc1811046 [DOI] [PubMed] [Google Scholar]
- 41.Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, Mabwe S, Makhethe L, Erasmus M, Toefy A, Mulenga H, Hanekom WA, Self SG, Bekker LG, Ryall R, Gurunathan S, DiazGranados CA, Andersen P, Kromann I, Evans T, Ellis RD, Landry B, Hokey DA, Hopkins R, Ginsberg AM, Scriba TJ, Hatherill M, Team CS. Prevention of M. tuberculosis Infection with H4:IC31 Vaccine or BCG Revaccination. N Engl J Med 2018;379:138–149. doi: 10.1056/NEJMoa1714021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deshmukh SS, Magcalas FW, Kalbfleisch KN, Carpick BW, Kirkitadze MD. Tuberculosis vaccine candidate: Characterization of H4-IC31 formulation and H4 antigen conformation. J Pharm Biomed Anal 2018;157:235–243. doi: 10.1016/j.jpba.2018.05.048 [DOI] [PubMed] [Google Scholar]
- 43.Hu S, Chen H, Ma J, Chen Q, Deng H, Gong F, Huang H, Shi C. CpG7909 adjuvant enhanced immunogenicity efficacy in mice immunized with ESAT6-Ag85A fusion protein, but does not confer significant protection against Mycobacterium tuberculosis infection. J Appl Microbiol 2013;115:1203–1211. doi: 10.1111/jam.12315 [DOI] [PubMed] [Google Scholar]
- 44.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE 3rd, Freedman VH, Kaplan G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc Natl Acad Sci U S A 2001;98:5752–5757. doi: 10.1073/pnas.091096998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redford PS, Mayer-Barber KD, McNab FW, Stavropoulos E, Wack A, Sher A, O’Garra A. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis 2014;209:270–274. doi: 10.1093/infdis/jit424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, Vance RE. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat Microbiol 2019;4:2128–2135. doi: 10.1038/s41564-019-0578-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walaza S, Tempia S, Dawood H, Variava E, Wolter N, Dreyer A, Moyes J, Von Mollendorf C, McMorrow M, Von Gottberg A, Haffejee S, Venter M, Treurnicht FK, Hellferscee O, Martinson NA, Ismail N, Cohen C. The Impact of Influenza and Tuberculosis Interaction on Mortality Among Individuals Aged >/=15 Years Hospitalized With Severe Respiratory Illness in South Africa, 2010–2016. Open Forum Infect Dis 2019;6:ofz020. doi: 10.1093/ofid/ofz020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang G, deWeerd NA, Stifter SA, Liu L, Zhou B, Wang W, Zhou Y, Ying B, Hu X, Matthews AY, Ellis M, Triccas JA, Hertzog PJ, Britton WJ, Chen X, Feng CG. A proline deletion in IFNAR1 impairs IFN-signaling and underlies increased resistance to tuberculosis in humans. Nat Commun 2018;9:85. doi: 10.1038/s41467-017-02611-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srivastava S, Koch LK, Campbell DJ. IFNalphaR signaling in effector but not regulatory T cells is required for immune dysregulation during type I IFN-dependent inflammatory disease. J Immunol 2014;193:2733–2742. doi: 10.4049/jimmunol.1401039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar AM, Colonna M, Steinman RM. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 2009;206:1589–1602. doi: 10.1084/jem.20090247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuchtey J, Fulton SA, Reba SM, Harding CV, Boom WH. Interferon-alphabeta mediates partial control of early pulmonary Mycobacterium bovis bacillus Calmette-Guerin infection. Immunology 2006;118:39–49. doi: 10.1111/j.1365-2567.2006.02337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aichinger MC, Ginzler M, Weghuber J, Zimmermann L, Riedl K, Schutz G, Nagy E, von Gabain A, Schweyen R, Henics T. Adjuvating the adjuvant: facilitated delivery of an immunomodulatory oligonucleotide to TLR9 by a cationic antimicrobial peptide in dendritic cells. Vaccine 2011;29:426–436. doi: 10.1016/j.vaccine.2010.11.003 [DOI] [PubMed] [Google Scholar]
- 53.Olafsdottir TA, Lingnau K, Nagy E, Jonsdottir I. IC31, a two-component novel adjuvant mixed with a conjugate vaccine enhances protective immunity against pneumococcal disease in neonatal mice. Scand J Immunol 2009;69:194–202. doi: 10.1111/j.1365-3083.2008.02225.x [DOI] [PubMed] [Google Scholar]
- 54.Olafsdottir TA, Lingnau K, Nagy E, Jonsdottir I. Novel protein-based pneumococcal vaccines administered with the Th1-promoting adjuvant IC31 induce protective immunity against pneumococcal disease in neonatal mice. Infect Immun 2012;80:461–468. doi: 10.1128/IAI.05801-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Juffermans NP, Leemans JC, Florquin S, Verbon A, Kolk AH, Speelman P, van Deventer SJ, van der Poll T. CpG oligodeoxynucleotides enhance host defense during murine tuberculosis. Infect Immun 2002;70:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freidag BL, Melton GB, Collins F, Klinman DM, Cheever A, Stobie L, Suen W, Seder RA. CpG oligodeoxynucleotides and interleukin-12 improve the efficacy of Mycobacterium bovis BCG vaccination in mice challenged with M. tuberculosis. Infect Immun 2000;68:2948–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sparwasser T, Vabulas RM, Villmow B, Lipford GB, Wagner H. Bacterial CpGDNA activates dendritic cells in vivo: T helper cell-independent cytotoxic T cell responses to soluble proteins. Eur J Immunol 2000;30:3591–3597. doi: 10.1002/1521-4141(200012)30:12<3591::AID-IMMU3591>3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- 58.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001;413:732–738. doi: 10.1038/35099560 [DOI] [PubMed] [Google Scholar]
- 59.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003;301:640–643. doi: 10.1126/science.1087262 [DOI] [PubMed] [Google Scholar]
- 60.Landrigan A, Wong MT, Utz PJ. CpG and non-CpG oligodeoxynucleotides directly costimulate mouse and human CD4+ T cells through a TLR9- and MyD88-independent mechanism. J Immunol 2011;187:3033–3043. doi: 10.4049/jimmunol.1003414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J Immunol 2005;175:3339–3346. [DOI] [PubMed] [Google Scholar]
- 62.Sommariva M, Le Noci V, Storti C, Bianchi F, Tagliabue E, Balsari A, Sfondrini L. Activation of NK cell cytotoxicity by aerosolized CpG-ODN/poly(I:C) against lung melanoma metastases is mediated by alveolar macrophages. Cell Immunol 2017;313:52–58. doi: 10.1016/j.cellimm.2017.01.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.