

Summary
The relationship between heme metabolism and angiogenesis is poorly understood. The final synthesis of heme occurs in mitochondria, where ferrochelatase (FECH) inserts Fe2+ into protoporphyrin IX to produce proto-heme IX. We previously showed that FECH inhibition is antiangiogenic in human retinal microvascular endothelial cells (HRECs) and in animal models of ocular neovascularization. In the present study, we sought to understand the mechanism of how FECH and thus heme is involved in endothelial cell function. Mitochondria in endothelial cells had several defects in function after heme inhibition. FECH loss changed the shape and mass of mitochondria and led to significant oxidative stress. Oxidative phosphorylation and mitochondrial Complex IV were decreased in HRECs and in murine retina ex vivo after heme depletion. Supplementation with heme partially rescued phenotypes of FECH blockade. These findings provide an unexpected link between mitochondrial heme metabolism and angiogenesis.
Subject Areas: Physiology, Developmental Genetics, Cell Biology
Graphical Abstract
Highlights
-
•
Heme synthesis inhibition changes mitochondrial morphology in endothelial cells
-
•
Loss of heme causes buildup of mitochondrial ROS and depolarized membrane potential
-
•
Endothelial cells have damaged oxidative phosphorylation and glycolysis on heme loss
-
•
Damage is due to loss of heme-containing Complex IV, restored by exogenous heme
Physiology; Developmental Genetics; Cell Biology
Introduction
An imbalance in mitochondrial metabolism has been implicated in the development of neovascular diseases catalyzed by aberrant angiogenesis. However, the role of heme synthesis in the development of mitochondrial dysfunction in neovascular diseases is unclear.
Neovascularization is a common phenomenon seen in vascular diseases like cancer, type 2 diabetes mellitus, proliferative diabetic retinopathy (PDR), wet age-related macular degeneration (AMD), and retinopathy of prematurity (ROP) (Friedman et al., 2004; Hellstrom et al., 2013; Kempen et al., 2004; Kizhakekuttu et al., 2012). Neovascularization is the disease process where new blood vessels grow from pre-existing ones, and in the eye this can contribute to ocular complications like hemorrhages, retinal detachment, and loss of central vision (Campochiaro, 2013). Endothelial cells (ECs) are key to this process, mediating different cellular functions essential for angiogenesis like proliferation, migration, and vascular permeability (Vandekeere et al., 2015).
We previously reported that heme synthesis inhibition is anti-angiogenic in retinal ECs in vitro and in animal models of ocular neovascularization. Blocking heme production in human retinal microvascular ECs (HRECs) decreased proliferation, migration, and endothelial tube formation and caused reduced protein expression of total and phosphorylated VEGF receptor 2 (Basavarajappa et al., 2017). This antiangiogenic effect was only seen in ocular ECs, whereas ocular non-endothelial cells were not similarly affected. Heme synthesis inhibition was associated with smaller ocular neovascular lesions in choroidal and retinal neovascularization mouse models (Basavarajappa et al., 2017; Pran Babu et al., 2020). However, the mechanism underlying this effect remains unknown.
The final synthesis of heme takes place in the mitochondria, when ferrochelatase (FECH) inserts ferrous ion into a precursor protoporphyrin IX (PPIX) to form protoheme (iron-protoporphyrin IX) (Dailey et al., 2017; Nilsson et al., 2009; Poulos, 2014). By directly targeting FECH, cells can be depleted of heme and heme-containing proteins, with a concomitant buildup of PPIX (Atamna et al., 2001; Vijayasarathy et al., 1999). Apart from the role of heme in oxygen transport and storage, heme acts as a prosthetic group in many hemoprotein enzymes involved in oxidative phosphorylation, plus cytochrome P450s, catalases, and nitric oxide synthase (Chiabrando et al., 2014).
In the present study, we aimed to assess the relationship between mitochondrial physiology in human ECs and heme inhibition. We hypothesized that heme inhibition would lead to defects in heme-containing enzymes of the mitochondrial electron transport chain (ETC) and thus affect mitochondrial function of ECs. We show that loss of heme altered mitochondrial morphology and dynamics, causing increased reactive oxygen species (ROS) levels and depolarized membrane potential. Our studies reveal that heme synthesis is required for EC respiration and complex IV (cytochrome c oxidase; CcO) function specifically and can negatively impact glycolytic capacity of ECs. Thus, we characterize a previously unknown role of heme in cellular metabolism that facilitates EC function in angiogenesis.
Results
Heme Inhibition Caused Changes to Mitochondrial Morphology and Increased Oxidative Stress
To block heme production, we used a competitive inhibitor of FECH, the terminal enzyme responsible for catalyzing heme synthesis (Shi and Ferreira, 2006) (see Transparent Methods). Blockade of FECH with active site inhibitor N-methylprotoporphyrin (NMPP) causes accumulation of precursor PPIX (Figure S1A). HRECs treated with NMPP showed changes in mitochondrial fragmentation and shape (Figure 1A). NMPP-treated cells had decreased form factor values, indicating reduced mitochondrial branching (Figures 1B and S1B–S1G), owing to more highly fragmented mitochondria (Figure 1A, inset image). Mitochondria appeared less elongated and elliptical in NMPP-treated HRECs as seen by lower aspect ratio values (Figure 1C). To determine mitochondrial mass, we used flow cytometry and quantified median fluorescence intensity (MFI) of NMPP-treated HRECs (Doherty and Perl, 2017). NMPP treatment led to reduced MFI, indicating decreased mass in mitochondria (Figure 1D).
Figure 1.

FECH Blockade Altered Mitochondrial Morphology and Increased Oxidative Stress
(A–C) (A) HRECs treated with DMSO or FECH inhibitor NMPP stained with MitoTracker green (MTG). Inset images indicate magnified region marked in red boxes. Form factor (B) and aspect ratio (C) as quantified using ImageJ. Individual data points indicate mean of mitochondria analyzed from each of 12 individual cells per treatment group.
(D–F) (D) Quantification of MTG fluorescence using flow cytometry and calculated median fluorescence intensity (MFI). qPCR analysis of mRNA expression under FECH knockdown (E) or NMPP treatment (F).
(G) HRECs stained with mitoSox ROS in red and Hoechst staining in blue.
(H and I) (H) Representative fluorescence peaks as measured by flow cytometry followed by (I) quantification of cells positive for red fluorescence.
Bar graphs indicate mean ± SEM, n = 3. Representative results from three independent experiments. ns, non-significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, two-tailed unpaired Student's t test. Scale bars, 20 μm. See also Figures S1 and S4.
We tested mRNA levels of key mitochondrial fusion proteins involved in mitochondrial dynamics, MFN2 and OPA1, and found significantly lower levels under FECH blockade conditions (Figures 1E and 1F). Fission regulator DNM1L (encoding Drp1) showed no change. We measured mitochondrial-specific ROS using MitoSox ROS, and cells treated with NMPP showed a marked increase in ROS levels (Figure 1G). We quantified this increase using flow cytometry and found a significant increase in MitoSox ROS-labeled HRECs treated with NMPP (Figures 1H and 1I). This effect of elevated ROS levels was also seen in RF/6A cells treated with NMPP (Figures S4A and S4B). This primate cell line has properties of chorioretinal ECs (Lou and Hu, 1987) and thus provides corroboration of the primary HREC data.
Loss of FECH Depolarized Mitochondrial Membrane Potential
To assess mitochondrial health, we next measured membrane potential using JC-1, a polychromatic dye that on excitation forms red aggregates and green monomers depending on the energized or deenergized state of mitochondria. Both siRNA-mediated knockdown of FECH and chemical inhibition using NMPP induced loss of healthy red aggregates and an increase in monomers as seen by the green fluorescence (Figures 2A and 2D). This JC-1 excitation was quantified using flow cytometry on HRECs labeled with JC-1 dye and showed reduced red to green fluorescence ratio, indicative of mitochondrial depolarization (Figures 2B, 2C, 2E, and 2F). RF/6A cells also showed depolarized membrane potential after NMPP treatment (Figures S4C–S4E).
Figure 2.

Loss of FECH Reduced Mitochondrial Membrane Potential in HRECs
(A–F) HRECs stained with JC-1 dye showing green monomers and red aggregates under FECH knockdown condition (A) and NMPP treatment (D). Representative dot plots of FL1 versus FL2 channel from three individual experiments, measuring red and green fluorescence using flow cytometry after FECH knockdown (B) and NMPP treatment (E). Red and green arrows indicate quadrants expressing FL1-red and FL2-green fluorescent cells. (C and F) Quantification of red:green fluorescence from flow experiment. Bar graphs indicate mean ± SEM, n = 3; ns, non-significant, ∗p < 0.05, ∗∗∗p < 0.001, one-way ANOVA with Tukey's post hoc tests. Scale bars, 20 μm. See also Figure S4.
Reduced CcO Expression and Activity Rescued by Hemin
To determine where heme depletion was influencing mitochondrial function, we evaluated protein complexes of the ETC after heme inhibition, as complexes II, III, and IV contain heme in their prosthetic groups (Kim et al., 2012). FECH knockdown resulted in a significant decrease of only CcO (Figures 3A and 3B). NMPP-treated cells showed a similar decrease in CcO, and we also found increased expression of Complex V (ATP synthase) upon treatment with this small molecule (Figures 3A and 3C). Additionally, we also assessed mRNA expression of ETC complex genes, NDUFS1 (complex I), SDHA (complex II), UQCRQ (complex III), MT-CO1 (mitochondrially encoded CcO subunit 1), and COX4I1 (nuclear encoded CcO subunit 1). Only the nuclear-encoded CcO gene showed a significant decrease in mRNA expression (Figure S5A). We then examined the protein levels of heme-containing subunit 1 of CcO (COX4I1) and found a significant decrease under both knockdown and NMPP treatment (Figures 3D–3G). Enzyme activity of CcO was also reduced, and total levels of the complex were significantly reduced after FECH blockade (Figures 3H–3K). Moreover, enzyme activities of complexes I–III remained unaffected (Figures S5B–S5D). To confirm if our results were dependent on heme, we sought to rescue the phenotype of CcO reduction by external supplementation of heme to the cells. Hemin, a stable form of heme, was able to alleviate CcO protein expression depletion and partially rescue CcO enzyme activity (Figures 3L–3N). RF/6A cells showed a similar rescue phenotype of CcO enzyme after heme addition (Figures S6A–S6D). We also used another heme synthesis inhibitor, succinylacetone, which blocks the second committed enzyme in heme synthesis, 5-aminolevulinic acid dehydratase (ALAD), upstream of the PPIX-FECH step (Dailey et al., 2017; Vandekeere et al., 2018). HRECs treated with succinylacetone showed reduction in protein expression of CcO and also Complex III (Figures S7A and S7B).
Figure 3.

FECH Inhibition Caused Reduced CcO Expression, Rescued by Hemin
(A) HRECs under FECH siRNA or NMPP-treated conditions were immunoblotted for Complexes I–V as indicated.
(B and C) Quantification of the blots was graphed as shown relative to appropriate control.
(D) FECH siRNA-treated HRECs were probed for CcO nuclear encoded subunit 1 (COX4I1) and FECH along with housekeeping control and (E) quantified. Similarly, HRECs treated with NMPP were blotted for CcO subunit 1 (F) with (G) quantification as shown. CcO enzyme activity was measured under FECH knockdown condition (H) and NMPP treatment (J) and total CcO levels were quantified by ELISA for both the conditions (I and K).
(L and M) CcO protein expression and quantification under defined conditions.
(N) CcO enzyme activity was partially rescued in NMPP-treated cells exposed to hemin. Immunoblot images representative from three independent experiments.
Bar graphs indicate mean ± SEM, n = 3–4; ns, non-significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. (B, C, E, G, H–K) unpaired Student's t test (M and N) one-way ANOVA with Tukey's post hoc tests. See also Figures S5–S7.
FECH Blockade Reduced Mitochondrial Function of Retinal ECs
To understand whether loss of function of an important complex in the ETC resulted in defects in mitochondrial respiration, we assessed maximal oxygen consumption rate (OCR) induced by the potential gradient uncoupler FCCP (Figures S2A and S2B). FCCP induces uninhibited flow of electrons across the ETC, causing the enzymes of the respiratory chain to use metabolic substrates at full potential and, in turn, revealing the maximal cellular capacity that can meet energy demands under metabolically stressed conditions (Dranka et al., 2011). Under metabolic stress, both HRECs and RF/6A cells underwent increased glycolysis to meet energy demands (Figures S2C–S2F).
Based on these optimized parameters (Figure S3), we measured OCR of HRECs after FECH knockdown and observed reduced basal respiration (Figures 4A and 4B). Uncoupler-induced maximal respiration was significantly decreased, with a marked reduction also found in OCR-linked ATP production and spare respiratory capacity (Figures 4C–4E). Similarly, NMPP treated cells showed a dose-dependent decrease in basal (Figures 4F and 4G) and maximal respiration (Figure 4H) along with a decline in OCR-linked ATP production and spare respiratory capacity (Figures 4I and 4J). We saw a similar decrease in mitochondrial respiratory activity in RF/6A cells (Figures S6E–S6I). Succinylacetone-treated HRECs also showed a decrease in basal and maximal respiration along with reduced OCR-linked ATP production and spare respiratory capacity (Figures S7C–S7G).
Figure 4.

Loss of FECH Reduced Mitochondrial Respiration
(A and F) OCR kinetic traces for HRECs under FECH knockdown or NMPP chemical inhibition.
(B and G) Basal respiration.
(C and H) Maximal respiration.
(D and I) OCR-linked ATP production, and (E and J) spare respiratory capacity were calculated based on OCR curves for the respective treatment group.
(A and F) Representative OCR curve of three individual experiments. Bar graphs indicate mean ± SEM, n = 3; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (B, C, D, and E) unpaired Student's t test (G, H, I, and J) one-way ANOVA with Tukey's post hoc tests. See also Figures S6 and S7.
Inhibition of FECH Led to Decreased Glycolytic Function
Microvascular ECs are highly glycolytic compared with other cell types (De Bock et al., 2013). Under mitochondrial stress, both HREC and RF/6A cells had increased glycolysis over mitochondrial respiration in our system (Figures S2C–SF). Hence, we next investigated this key cellular energetic pathway in HRECs by measuring changes in the pH of the extracellular medium. Cells were briefly starved in glucose-deficient medium, followed by induction of glycolysis using a saturating dose of glucose. Interestingly, HRECs under siRNA-mediated FECH knockdown and NMPP treatment each had decreased glycolytic capacity and glycolysis (Figures 5A–5C and 5E–5G). Both conditions depleted glycolytic reserve, as seen by a marked decrease in ECAR after 2-DG injection (Figures 5D and 5H).
Figure 5.

FECH Inhibition Caused a Decrease in Glycolytic Function
(A–H) (A and E) ECAR kinetic traces for HRECs under FECH knockdown or NMPP chemical inhibition. (B and F) Glycolysis, (C and G) glycolytic capacity, and (D and H) glycolytic reserve were calculated based on ECAR curves for the respective treatment group.
(A and E) Representative ECAR curve of three individual experiments. Bar graphs indicate mean ± SEM, n = 3; ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, unpaired Student's t test. 2-DG, 2-deoxyglucose.
FECH Inhibition In Vivo Caused Impaired Mitochondrial Energetics in Retina
We further determined if the effect of NMPP had the same phenotype in vivo in an intact eye. For this, we administered NMPP intravitreally to mice and measured OCR of retina in an ex vivo assay (Figure 6A). We found that retinas treated with NMPP had a significant decrease in basal respiration and a similar decline in maximal respiration (Figures 6B and 6C). Spare respiratory capacity was severely reduced in the retinas of NMPP-treated animals compared with their vehicle-treated counterparts (Figure 6D). We also observed about a 30% reduction in protein expression of subunit 1 of CcO in the NMPP-treated retinas (Figures 6E and 6F).
Figure 6.

FECH Inhibition In Vivo Decreased Mitochondrial Respiration in Retina
(A) Representative OCR kinetic traces for retina from animals treated with NMPP.
(B–D) (B) Basal respiration, (C) maximal respiration, and (D) spare respiratory capacity were calculated based on OCR curves for the respective treatment groups.
(E) Immunoblot showing CcO nuclear encoded subunit 1 (COX4I1) protein expression from three pooled retinal tissue lysates from NMPP treated eyes and (F) quantification of immunoblots. Graphs indicate mean ± SEM for two tissue punches from each retina per treatment group, n = 6–7 per treatment condition. ∗p < 0.05, ∗∗∗p < 0.001, unpaired Student's t test.
Discussion
Heme synthesis blockade suppresses pathological angiogenesis by poorly understood mechanisms. In the present study, we sought to investigate the effects of heme inhibition on the mitochondrial function of retinal ECs by directly studying heme-containing complexes of the ETC and documenting mitochondrial physiology under heme depletion.
The mitochondrial ETC is a major source of ROS induced by vascular endothelial growth factor (VEGF) in hyperglycemic and hypoxic cellular environments (Cheng et al., 2011; Pearlstein et al., 2002; Wang et al., 2018). In PDR, increased mitochondrial ROS and impaired Ca2+ signaling can cause an increase in oxidative stress (Kowluru and Mishra, 2015; Pangare and Makino, 2012; Tang et al., 2014; Wang et al., 2018). Complex III of the ETC was recently shown to be important for umbilical vein EC respiration and thus proliferation during angiogenesis (Diebold et al., 2019). Mitochondrial dysfunction in the retinal pigment epithelium and photoreceptors has been reported for wet AMD (Barot et al., 2011; Lefevere et al., 2017), but evidence in retinal ECs is limited. Metabolic factors like succinate and adenosine, generated from the Krebs cycle and ATP metabolism, respectively, are proangiogenic for hypoxia-driven neovascularization (Sapieha et al., 2010). However, the exact mechanism of how metabolites disrupt mitochondrial energetics in ischemic retinal ECs remains unclear (Grant et al., 1999; Sapieha et al., 2008).
Mitochondrial dysfunction in ECs leads to pathological angiogenesis. Heme and heme-containing enzymes play a significant role in this process, but the linkage between these phenomena has not been extensively explored. Serine synthesis deficiency induced heme depletion in ECs and caused decreased mitochondrial respiration and multiorgan angiogenic defects in animals (Vandekeere et al., 2018), whereas heme accumulation in ECs due to an altered heme exporter affects angiogenesis and causes endoplasmic reticulum stress (Petrillo et al., 2018). Apart from ECs, non-small cell lung carcinoma cells with elevated heme synthesis levels had increases in enzyme activities of the ETC. Increased production of heme was associated with increased migratory and invasive properties of these cells and xenograft tumors in mice (Sohoni et al., 2019). Here, we demonstrated how heme depletion by blockade of the terminal synthesis enzyme FECH led to defects in CcO and ETC disruption (Figure 7). Retinal and choroidal ECs alike have increased mitochondrial-specific oxidative stress, dysfunctional mitochondrial physiology, and disruption of glycolysis as a result of these defects.
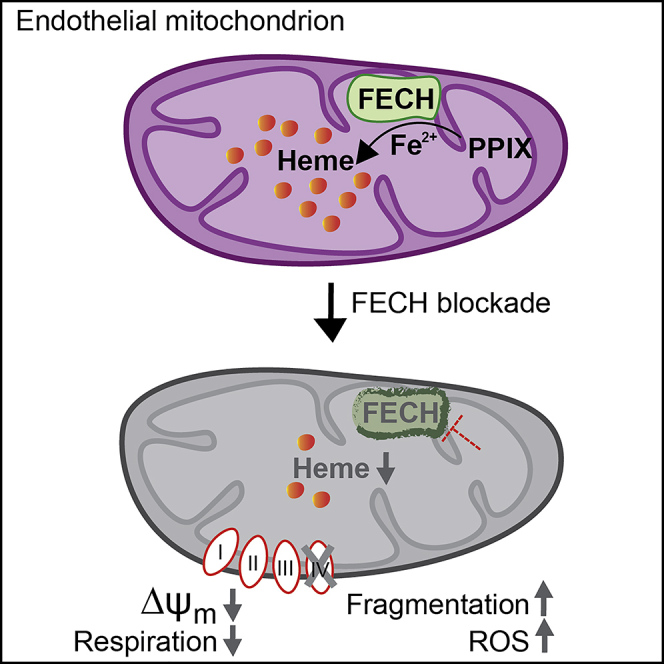
Figure 7.

Schematic Model of Mitochondrial Dysfunction on Heme Loss
Heme synthesis begins with the condensation of glycine and succinyl CoA in the mitochondrial matrix. The final step is the insertion of ferrous ion into protoporphyrin IX (PPIX), catalyzed by ferrochelatase (FECH) to produce protoheme (also known as heme b). Protoheme and its derivatives are available for different cellular enzymes, including complexes of the ETC (e, electrons). Heme a is synthesized by sub-hemylation steps and utilized by CcO (Complex IV) for composition of the holoenzyme. Heme synthesis blockade by inhibiting the terminal synthesis enzyme FECH leads to CcO defects and disrupts cellular energetics. Mitochondrial dynamics is altered with reduced fusion and mass, depolarized membrane potential (ΔΨm) and elevated reactive oxygen species (ROS).
Changes to mitochondrial morphology can indicate alterations in mitochondrial dynamics and cellular function. Our findings of reduced form factor and aspect ratio after FECH blockade indicate greater fragmentation and decreased length (Duraisamy et al., 2019; Picard et al., 2013). Furthermore, reduced mitochondrial mass was evident by our flow cytometric analysis. HRECs after heme depletion also had reduced MFN2 and OPA1 transcript levels. Reduction of these mitochondrial fusion protein marker genes confirms a change in mitochondrial dynamics. An increase in fragmented mitochondria also leads to increased ROS levels (Jezek et al., 2018), as confirmed by our results. Moreover, disruption of fusion proteins can also lead to an increase in ROS levels (Kim et al., 2018), as also evident in our cell model. Although we did not observe any change in fission protein expression, decreased MFN2 is also consistent with the smaller, fragmented mitochondria we observed, similar to those seen in diabetic mouse coronary ECs (Makino et al., 2010).
Inner mitochondrial membrane potential (ΔΨm) was considerably decreased after FECH knockdown or NMPP treatment. This finding was explained by our result of reduced CcO expression and activity, which might have collapsed the potential gradient and caused a reduced membrane potential. Our mitochondrial energetics findings further support this result. A significant reduction in FCCP-induced maximal respiration indicates defective CcO, which is unable to perform the role of electron transfer. Uncoupling brought on by FCCP causes the ETC to rely on CcO to carry out oxidative phosphorylation by the free-flowing electrons (Dranka et al., 2011). Under FECH inhibition conditions, FCCP-induced maximal respiration was affected, indicating CcO dysfunction due to heme loss.
Heme depletion by directly blocking FECH primarily affected levels and activity of CcO of all the other heme-containing complexes of the ETC in ECs of the retina and choroid. CcO subunit 1 contains heme a and a3 (not found in complexes I–III and V). Heme a is made in a series of sub-synthesis hemylation steps carried out by heme a synthase and is essential for proper folding and stability of CcO (Kim et al., 2012) (Figure 7). CcO is particularly sensitive to FECH inhibition-induced heme depletion, possibly affecting the hemylation process downstream of protoheme synthesis, leading to a smaller pool of heme a (Atamna et al., 2001; Sinkler et al., 2017). COX10 and COX15, proteins responsible for conversion of heme b (synthesized by FECH) to heme a were among the top-scoring genes that caused ETC disruption after heme depletion in acute myeloid leukemia cells (Lin et al., 2019), further suggesting the significance of heme a supply as a mediator of mitochondrial function, since it is a prosthetic group specifically in CcO holoenzyme. Furthermore, blocking the skeletal muscle-specific subunit 7a of CcO decreased mitochondrial ATP production in mouse hindlimb muscles. Interestingly, this was associated with reduced muscle angiogenesis and decreased capillarity (Lee et al., 2012), further supporting a link between CcO and EC function. Our findings on the mRNA and protein levels plus enzyme activities of all ETC complexes are consistent with this hypothesis that FECH inhibition preferentially disrupts CcO expression and function.
Loss of functional CcO was restored at least partially by extracellular heme supplementation. Additionally, blocking heme synthesis upstream of FECH by using succinylacetone also led to a similar mitochondrial phenotype as FECH blockade. This suggests that the EC phenotype after FECH inhibition is heme dependent and not due to an off-target effect of NMPP or due to PPIX buildup/toxicity (Wyld et al., 1997). NMPP caused significant overexpression of complex V (not seen under FECH knockdown), which could be a secondary effect of damage to CcO, causing compensatory upregulation of ATP synthase (Havlickova Karbanova et al., 2012; Rolland et al., 2013). Notably, succinylacetone produced a decrease in both Complex III and CcO, perhaps due to a more profound heme synthesis blockade than that achieved by FECH inhibition or knockdown. Our studies previously reported effects of NMPP and FECH knockdown specific to ECs of retina, choroid, brain, and umbilical vein, and we did not observe a similar anti-proliferative phenotype on FECH blockade in other ocular cell types of non-endothelial origin (Basavarajappa et al., 2017). However, the phenotype of FECH deficiency on ETC complexes in other cell types beyond ECs would be an interesting aspect for future exploration.
Perhaps non-endothelial cell types can meet their energy demands from the glycolysis pathway in the event of heme depletion-induced ETC dysfunction (Rafikov et al., 2015). Conversely, loss of mitochondrial respiration in ECs due to lack of functional oxidative phosphorylation did not result in compensation by the glycolysis pathway in ECs (Zielinski et al., 2016). ECs rely upon aerobic glycolysis for nearly 85% of their energy, a feature that is highly active during aberrant angiogenesis (De Bock et al., 2013). Severe heme depletion could possibly disrupt NADH/H+ and redox homeostasis, as already evident from ETC dysfunction. Moreover, increased ROS levels in ECs can shift their metabolic needs to accommodate cellular damage and dysfunction, in which case both cellular energetic systems are collapsed (Vandekeere et al., 2018; Warren et al., 2014; Wellen and Thompson, 2010). Interestingly, mitochondrial transcription of CcO subunit 1 (MT-CO1) mRNA is not affected after heme loss (although protein levels were decreased), but the nuclear encoded CcO gene COX4I1 was significantly decreased at both the mRNA and protein levels after FECH inhibition. This is in keeping with heme acting at the post-transcriptional level to stabilize the assembly of the CcO subunits with minimal effect on CcO mitochondrial transcription itself.
In summary, our findings provide a previously unidentified link between heme synthesis, angiogenesis, and mitochondrial energetics. Specifically, heme synthesis blockade via FECH leads to a reduction in heme availability for hemoprotein components of the ETC, notably CcO, likely due to a shortage of heme a and a3. This is then associated with reduced oxidative phosphorylation, with concomitant loss of mitochondrial membrane potential and changes in mitochondrial shape and dynamics, as well as increased ROS production (Figure 7). Blockade of heme during the early stages of the pathway (via ALAD) disrupts heme b synthesis, affecting the ETC proteins beyond just Complex IV. This potentially explains how ECs rely on a constant supply of heme during increased energy demands, essential for the formation of neovessels. The role of heme synthesis in mitochondrial function and pathological angiogenesis has been overlooked. Our observations bridge this gap in knowledge by characterizing the metabolic phenotype of ECs under heme deficiency. Loss of heme provokes prominent anti-angiogenic effects that might be exploited therapeutically for neovascular eye disease. Our findings invite future studies to further our understanding of metabolic dysfunction in neovascularization.
Limitations of the Study
Retinal ECs are technically laborious for experimental manipulations. Apart from being difficult to transfect, low absolute heme levels precluded spectral determination of hemes a, b, and c. NMPP as a FECH inhibitor could be deleterious to other hemoproteins in ECs and could have off-target effects, although we titrated NMPP to well below toxic levels in these studies and complemented NMPP findings with a knockdown approach and succinylacetone treatment. Similarly, using hemin to rescue FECH inhibition-induced heme depletion has limitations. Hemin itself can introduce mitochondrial toxicity (Higdon et al., 2012), and complete rescue of ETC damage through hemin supplementation is challenging. Additionally, damage to CcO due to insufficient heme could impair formation of ETC supercomplexes (Acin-Perez et al., 2008), which was not assessed in our study. Resolving mitochondrial lysates isolated from NMPP-treated ECs in their endogenous form, for example, by a native gel electrophoresis method, could identify disruption to the assembly of these complexes (Jha et al., 2016).
We previously tested a genetic mouse model with a M98K point mutation in the Fech gene (Fechm1Pas) leading to deficiency in FECH activity and found reduced neovascular lesions in the eye (Basavarajappa et al., 2017; Pran Babu et al., 2020). This Fechm1Pas mouse model shows pronounced PPIX buildup and increased mitochondrial respiratory activity, specifically CcO (Navarro et al., 2005), likely a compensatory response to constitutive heme depletion. This renders them problematic for corroborating our findings in vivo. Moreover, complete loss of function of FECH is incompatible with survival (Magness et al., 2002). For these reasons, for in vivo validation we employed here an acute model of FECH blockade using local administration of NMPP intravitreally. Nonetheless, this FECH inhibition in vivo had energetic effects in the retina similar to those seen on retinal and choroidal ECs cultured in vitro. However, an important limitation is that this technique cannot distinguish the energetic profiles of ECs from other cell types present in the retina.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Timothy W. Corson (tcorson@iu.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by NIH/NEI R01EY025641 (T.W.C.). The authors thank members of the Corson laboratory for comments on the manuscript; the Indiana University School of Medicine Melvin and Bren Simon Cancer Center Angio BioCore, which is supported by NIH/NCI P30CA082790; the Iron and Heme Core facility at the University of Utah, supported in part by NIH/NIDDK U54DK110858; and the Indiana Center for Biomedical Innovation, which is supported in part by the Indiana Clinical and Translational Sciences Institute funded, in part by NIH/NCATS UL1TR002529. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contributions
Conceptualization, T.S. and T.W.C.; Investigation, T.S., K.S., B.P., and M.J.R.; Formal Analysis and Visualization, T.S.; Writing – Original Draft, T.S.; Writing – Review & Editing, T.S., K.S., B.P., M.J.R., and T.W.C.; Funding Acquisition, T.W.C.
Declaration of Interests
T.W.C. is a named inventor on patent applications related to this work. The other authors declare no competing interests.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101391.
Supplemental Information
References
- Acin-Perez R., Fernandez-Silva P., Peleato M.L., Perez-Martos A., Enriquez J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Atamna H., Liu J., Ames B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: revelance to aging. J. Biol. Chem. 2001;276:48410–48416. doi: 10.1074/jbc.M108362200. [DOI] [PubMed] [Google Scholar]
- Barot M., Gokulgandhi M.R., Mitra A.K. Mitochondrial dysfunction in retinal diseases. Curr. Eye Res. 2011;36:1069–1077. doi: 10.3109/02713683.2011.607536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa H.D., Sulaiman R.S., Qi X., Shetty T., Sheik Pran Babu S., Sishtla K.L., Lee B., Quigley J., Alkhairy S., Briggs C.M. Ferrochelatase is a therapeutic target for ocular neovascularization. EMBO Mol. Med. 2017;9:786–801. doi: 10.15252/emmm.201606561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R., Quaegebeur A., Ghesquiere B., Cauwenberghs S., Eelen G. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- Campochiaro P.A. Ocular neovascularization. J. Mol. Med. (Berl) 2013;91:311–321. doi: 10.1007/s00109-013-0993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Siow R.C., Mann G.E. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid. Redox Signal. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- Chiabrando D., Vinchi F., Fiorito V., Mercurio S., Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014;5:61. doi: 10.3389/fphar.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey H.A., Dailey T.A., Gerdes S., Jahn D., Jahn M., O’Brian M.R., Warren M.J. Prokaryotic heme biosynthesis: multiple pathways to a common essential product. Microbiol. Mol. Biol. Rev. 2017;81 doi: 10.1128/MMBR.00048-16. e00048–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold L.P., Gil H.J., Gao P., Martinez C.A., Weinberg S.E., Chandel N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat. Metab. 2019;1:158–171. doi: 10.1038/s42255-018-0011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty E., Perl A. Measurement of mitochondrial mass by flow cytometry during oxidative stress. React. Oxyg Species (Apex) 2017;4:275–283. doi: 10.20455/ros.2017.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranka B.P., Benavides G.A., Diers A.R., Giordano S., Zelickson B.R., Reily C., Zou L., Chatham J.C., Hill B.G., Zhang J. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraisamy A.J., Mohammad G., Kowluru R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:1617–1626. doi: 10.1016/j.bbadis.2019.03.013. [DOI] [PubMed] [Google Scholar]
- Friedman D.S., O’Colmain B.J., Munoz B., Tomany S.C., McCarty C., de Jong P.T., Nemesure B., Mitchell P., Kempen J., Eye Diseases Prevalence Research Group Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Grant M.B., Tarnuzzer R.W., Caballero S., Ozeck M.J., Davis M.I., Spoerri P.E., Feoktistov I., Biaggioni I., Shryock J.C., Belardinelli L. Adenosine receptor activation induces vascular endothelial growth factor in human retinal endothelial cells. Circ. Res. 1999;85:699–706. doi: 10.1161/01.res.85.8.699. [DOI] [PubMed] [Google Scholar]
- Havlickova Karbanova V., Cizkova Vrbacka A., Hejzlarova K., Nuskova H., Stranecky V., Potocka A., Kmoch S., Houstek J. Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation. Biochim. Biophys. Acta. 2012;1817:1037–1043. doi: 10.1016/j.bbabio.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Hellstrom A., Smith L.E., Dammann O. Retinopathy of prematurity. Lancet. 2013;382:1445–1457. doi: 10.1016/S0140-6736(13)60178-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon A.N., Benavides G.A., Chacko B.K., Ouyang X., Johnson M.S., Landar A., Zhang J., Darley-Usmar V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H1394–H1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek J., Cooper K.F., Strich R. Reactive oxygen species and mitochondrial dynamics: the yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants (Basel) 2018;7:13. doi: 10.3390/antiox7010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha P., Wang X., Auwerx J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE) Curr. Protoc. Mouse Biol. 2016;6:1–14. doi: 10.1002/9780470942390.mo150182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempen J.H., O’Colmain B.J., Leske M.C., Haffner S.M., Klein R., Moss S.E., Taylor H.R., Hamman R.F., Eye Diseases Prevalence Research Group The prevalence of diabetic retinopathy among adults in the United States. Arch. Ophthalmol. 2004;122:552–563. doi: 10.1001/archopht.122.4.552. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Khalimonchuk O., Smith P.M., Winge D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta. 2012;1823:1604–1616. doi: 10.1016/j.bbamcr.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.M., Youn S.W., Sudhahar V., Das A., Chandhri R., Cuervo Grajal H., Kweon J., Leanhart S., He L., Toth P.T. Redox regulation of mitochondrial fission protein drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. 2018;23:3565–3578. doi: 10.1016/j.celrep.2018.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizhakekuttu T.J., Wang J., Dharmashankar K., Ying R., Gutterman D.D., Vita J.A., Widlansky M.E. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler Thromb. Vasc. Biol. 2012;32:2531–2539. doi: 10.1161/ATVBAHA.112.256024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru R.A., Mishra M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta. 2015;1852:2474–2483. doi: 10.1016/j.bbadis.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Lee I., Huttemann M., Liu J., Grossman L.I., Malek M.H. Deletion of heart-type cytochrome c oxidase subunit 7a1 impairs skeletal muscle angiogenesis and oxidative phosphorylation. J. Physiol. 2012;590:5231–5243. doi: 10.1113/jphysiol.2012.239707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevere E., Toft-Kehler A.K., Vohra R., Kolko M., Moons L., Van Hove I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion. 2017;36:66–76. doi: 10.1016/j.mito.2017.03.006. [DOI] [PubMed] [Google Scholar]
- Lin K.H., Xie A., Rutter J.C., Ahn Y.R., Lloyd-Cowden J.M., Nichols A.G., Soderquist R.S., Koves T.R., Muoio D.M., MacIver N.J. Systematic dissection of the metabolic-apoptotic interface in AML reveals heme biosynthesis to be a regulator of drug sensitivity. Cell Metab. 2019;29:1217–1231. doi: 10.1016/j.cmet.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou D.A., Hu F.N. Specific antigen and organelle expression of a long-term rhesus endothelial cell line. In Vitro Cell Dev Biol. 1987;23:75–85. doi: 10.1007/BF02623586. [DOI] [PubMed] [Google Scholar]
- Magness S.T., Maeda N., Brenner D.A. An exon 10 deletion in the mouse ferrochelatase gene has a dominant-negative effect and causes mild protoporphyria. Blood. 2002;100:1470–1477. doi: 10.1182/blood-2001-12-0283. [DOI] [PubMed] [Google Scholar]
- Makino A., Scott B.T., Dillmann W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53:1783–1794. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro S., Del Hoyo P., Campos Y., Abitbol M., Moran-Jimenez M.J., Garcia-Bravo M., Ochoa P., Grau M., Montagutelli X., Frank J. Increased mitochondrial respiratory chain enzyme activities correlate with minor extent of liver damage in mice suffering from erythropoietic protoporphyria. Exp. Dermatol. 2005;14:26–33. doi: 10.1111/j.0906-6705.2005.00248.x. [DOI] [PubMed] [Google Scholar]
- Nilsson R., Schultz I.J., Pierce E.L., Soltis K.A., Naranuntarat A., Ward D.M., Baughman J.M., Paradkar P.N., Kingsley P.D., Culotta V.C. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009;10:119–130. doi: 10.1016/j.cmet.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangare M., Makino A. Mitochondrial function in vascular endothelial cell in diabetes. J. Smooth Muscle Res. 2012;48:1–26. doi: 10.1540/jsmr.48.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlstein D.P., Ali M.H., Mungai P.T., Hynes K.L., Gewertz B.L., Schumacker P.T. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb. Vasc. Biol. 2002;22:566–573. doi: 10.1161/01.atv.0000012262.76205.6a. [DOI] [PubMed] [Google Scholar]
- Petrillo S., Chiabrando D., Genova T., Fiorito V., Ingoglia G., Vinchi F., Mussano F., Carossa S., Silengo L., Altruda F. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018;25:573–588. doi: 10.1038/s41418-017-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M., White K., Turnbull D.M. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: a quantitative three-dimensional electron microscopy study. J. Appl. Physiol. 2013;114:161–171. doi: 10.1152/japplphysiol.01096.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos T.L. Heme enzyme structure and function. Chem. Rev. 2014;114:3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pran Babu S.P.S., White D., Corson T.W. Ferrochelatase regulates retinal neovascularization. FASEB J. 2020 doi: 10.1096/fj.202000964R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafikov R., Sun X., Rafikova O., Meadows M.L., Desai A.A., Khalpey Z., Yuan J.X., Fineman J.R., Black S.M. Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol. 2015;6:278–286. doi: 10.1016/j.redox.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland S.G., Motori E., Memar N., Hench J., Frank S., Winklhofer K.F., Conradt B. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc. Natl. Acad. Sci. U S A. 2013;110:E2967–E2976. doi: 10.1073/pnas.1303872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapieha P., Sirinyan M., Hamel D., Zaniolo K., Joyal J.S., Cho J.H., Honore J.C., Kermorvant-Duchemin E., Varma D.R., Tremblay S. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 2008;14:1067–1076. doi: 10.1038/nm.1873. [DOI] [PubMed] [Google Scholar]
- Sapieha P., Joyal J.S., Rivera J.C., Kermorvant-Duchemin E., Sennlaub F., Hardy P., Lachapelle P., Chemtob S. Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 2010;120:3022–3032. doi: 10.1172/JCI42142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z., Ferreira G.C. Modulation of inhibition of ferrochelatase by N-methylprotoporphyrin. Biochem. J. 2006;399:21–28. doi: 10.1042/BJ20060753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkler C.A., Kalpage H., Shay J., Lee I., Malek M.H., Grossman L.I., Huttemann M. Tissue- and condition-specific isoforms of mammalian cytochrome c oxidase subunits: from function to human disease. Oxid Med. Cell Longev. 2017;2017:1534056. doi: 10.1155/2017/1534056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohoni S., Ghosh P., Wang T., Kalainayakan S.P., Vidal C., Dey S., Konduri P.C., Zhang L. Elevated heme synthesis and uptake underpin intensified oxidative metabolism and tumorigenic functions in non-small cell lung cancer cells. Cancer Res. 2019;79:2511–2525. doi: 10.1158/0008-5472.CAN-18-2156. [DOI] [PubMed] [Google Scholar]
- Tang X., Luo Y.X., Chen H.Z., Liu D.P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014;5:175. doi: 10.3389/fphys.2014.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekeere S., Dewerchin M., Carmeliet P. Angiogenesis revisited: an overlooked role of endothelial cell metabolism in vessel sprouting. Microcirculation. 2015;22:509–517. doi: 10.1111/micc.12229. [DOI] [PubMed] [Google Scholar]
- Vandekeere S., Dubois C., Kalucka J., Sullivan M.R., Garcia-Caballero M., Goveia J., Chen R., Diehl F.F., Bar-Lev L., Souffreau J. Serine synthesis via PHGDH is essential for heme production in endothelial cells. Cell Metab. 2018;28:573–587.e13. doi: 10.1016/j.cmet.2018.06.009. [DOI] [PubMed] [Google Scholar]
- Vijayasarathy C., Damle S., Lenka N., Avadhani N.G. Tissue variant effects of heme inhibitors on the mouse cytochrome c oxidase gene expression and catalytic activity of the enzyme complex. Eur. J. Biochem. 1999;266:191–200. doi: 10.1046/j.1432-1327.1999.00843.x. [DOI] [PubMed] [Google Scholar]
- Wang Z., Zhao H., Guan W., Kang X., Tai X., Shen Y. Metabolic memory in mitochondrial oxidative damage triggers diabetic retinopathy. BMC Ophthalmol. 2018;18:258. doi: 10.1186/s12886-018-0921-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren C.M., Ziyad S., Briot A., Der A., Iruela-Arispe M.L. A ligand-independent VEGFR2 signaling pathway limits angiogenic responses in diabetes. Sci. Signal. 2014;7:ra1. doi: 10.1126/scisignal.2004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen K.E., Thompson C.B. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyld L., Burn J.L., Reed M.W., Brown N.J. Factors affecting aminolaevulinic acid-induced generation of protoporphyrin IX. Br. J. Cancer. 1997;76:705–712. doi: 10.1038/bjc.1997.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski L.P., Smith A.C., Smith A.G., Robinson A.J. Metabolic flexibility of mitochondrial respiratory chain disorders predicted by computer modelling. Mitochondrion. 2016;31:45–55. doi: 10.1016/j.mito.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.