

Abstract
Aim: Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a key regulator of serum low-density lipoprotein (LDL) cholesterol levels, and sortilin is linked to lipoprotein metabolism. Although statin therapy increases PCSK9 levels, effects of this therapy on plasma sortilin levels have not been evaluated. The purpose of the present study was to examine the effects of statins on plasma PCSK9 and sortilin levels, and association of statin-induced increase in PCSK9 levels with sortilin.
Methods: Serum lipid levels and plasma PCSK9 and sortilin levels were measured at baseline and 8 months after statin therapy in 90 statin-naive patients with coronary artery disease (CAD). Pitavastatin 4 mg/day was used to treat 44 patients and pravastatin 20 mg/day to treat the remaining 46 patients.
Results: For both statin groups, significant increases in hetero-dimer PCSK9 levels (pitavastatin: 31%, p < 0.0001; pravastatin: 34%, p = 0.03) and decreases in sortilin levels (pitavastatin: −8%, p = 0.02; pravastatin: −16%, p = 0.002) were observed. Although a reduction in LDL cholesterol was greater in the pitavastatin group than in the pravastatin group, no significant differences were observed in percentage changes in hetero-dimer PCSK9 and sortilin levels. A significant positive correlation was observed between percentage changes in hetero-dimer PCSK9 levels and those in sortilin levels (pitavastatin: r = 0.359, p = 0.02; pravastatin: r = 0.276, p = 0.06).
Conclusions: Use of pitavastatin and pravastatin increased plasma PCSK9 and decreased sortilin levels. Statin-induced increases in PCSK9 were associated with changes in sortilin in statin-naive patients with CAD.
Keywords: Low-density lipoprotein cholesterol, Proprotein convertase subtilisin/kexin type 9, Sortilin, Statin
Introduction
Elevated low-density lipoprotein (LDL) cholesterol levels are key factors associated with the occurrence of atherosclerosis and coronary artery disease (CAD), which are the leading causes of morbidity and mortality worldwide. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a key regulator of serum LDL cholesterol levels1–3) and is secreted by the liver into the circulation and binds the hepatic LDL receptors, causing their subsequent degradation4–7).
Recent genome-wide association studies have identified that a genetic variant at a locus on chromosome 1p13.3 is strongly associated with LDL cholesterol levels and the risk of myocardial infarction8–10). This locus contains the following four genes: SORT1, CELSR2, PSRC1, and MYBPHL, and a subsequent study has shown that SORT1 affects lipid metabolism11). SORT1 encodes sortilin, a type 1 transmembrane protein12, 13), which acts as a multiligand receptor and is linked to systemic lipoprotein metabolism and PCSK9 secretion14–17).
Lowering LDL cholesterol levels is currently considered the most effective strategy for preventing CAD. The use of statins results in the elevation of LDL receptor expression that increases the uptake of LDL particles from the circulation18). However, the use of statins causes an increase in the expression of PCSK919), counteracting the beneficial effects of statins. Thus, PCSK9 is an attractive drug target for hypercholesterolemia, and results from clinical trials are promising20, 21). Although statin therapy increases PCSK9 levels, the effects of statins on plasma sortilin levels have not been evaluated. Therefore, in this observational, longitudinal study, we examined the effects of statin therapy on plasma PCSK9 and sortilin levels and the association of statin-induced increase in PCSK9 with sortilin in statin-naive patients with CAD. Furthermore, we evaluated the effects of two types of statins, intensive lipid-lowering therapy with pitavastatin and moderate lipid-lowering therapy with pravastatin, on plasma PCSK9 and sortilin levels.
Methods
Patients and Study Design
Data were obtained from the Treatment With Statin on Atheroma Regression Evaluated by Intravascular Ultrasound With Virtual Histology (TRUTH) study, which was a prospective, open-labeled, randomized, multicenter trial performed at 11 Japanese centers to compare the effects of 8-month treatment with pitavastatin versus pravastatin on coronary atherosclerosis using virtual histology intravascular ultrasound22). In brief, 164 patients with angina pectoris who were not receiving lipid-lowering therapy were randomly treated with either pitavastatin (4 mg/day, intensive lipid-lowering group) or pravastatin (20 mg/day, moderate lipid-lowering group).
The patients included in the TRUTH study were considered for the present study if they fulfilled the following criteria: the allocated statins were continued during the study period (8 months) and an adequate plasma volume was available in frozen samples for the required measurements. In total, 90 patients met the inclusion criteria. Pitavastatin was used to treat 44 patients and pravastatin to treat the remaining 46 patients.
The TRUTH study was conducted in accordance with the Declaration of Helsinki and with the approval of the ethical committees of the 11 participating institutions. Each patient enrolled in the present study provided written informed consent.
Laboratory Analysis
Serum lipid levels were measured before treatment (at baseline) and at the 8-month follow-up22). Serum total cholesterol, LDL cholesterol, high-density lipoprotein cholesterol, and triglycerides levels were measured using standard enzymatic methods (AU2700; Beckman Coulter, CA, USA) and commercial enzymatic kits (Kyowa Medex, Tokyo, Japan). Plasma PCSK9 and sortilin levels at baseline and at the 8-month follow-up were measured at a central laboratory (BML, Kawagoe, Japan) using sandwich enzyme-linked immunosorbent assays (ELISA)23). For soluble sortilin, sortilin-specific antibodies were used for detection. In brief, mAb A9E [0.3 µg/mL in phosphate-buffered saline (PBS)] was coated onto a microplate (Nunc Maxisorp, Thermo Scientific) by incubation at 4°C overnight. The wells were then blocked with PBS containing 1% bovine serum albumin for 2 h at room temperature. After the plate had been washed with PBS containing 0.1% Tween20 (PBST), the calibrator (recombinant soluble sortilin) and plasma/serum samples (1:100) diluted with PBST containing 0.3% BSA was added and incubated for 2 h at room temperature. After washing the plate, horseradish peroxidase-labeled mAb E12A (0.03 µg/mL in PBST containing 0.3% BSA) was allowed to react for 1 h at room temperature. After washing, 3, 3′, 5, 5′-tetramethylbenzidine substrate solution (Dako) was added and incubated for 0.5 h. The reaction was then stopped by adding 0.5M sulfuric acid. The absorbance was measured at 450 nm using a microplate reader.
Statistical Analysis
Statistical analysis was performed using StatView, version 5.0 (SAS Institute, Cary, North Carolina). The results are expressed as mean ± SD or median (range). Differences in continuous variables between the two groups were compared using the unpaired t-test when the variables had a normal distribution and the Mann-Whitney U-test when they were not normally distributed. Differences in continuous variables within each group were compared using the paired t-test when the variables had a normal distribution and the Wilcoxon signed rank-sum test when they were not normally distributed. Categorical variables between the two groups were compared using the chi-square test or the Fisher's exact test. Univariate regression analyses were performed to assess the relation between percentage changes in sortilin and those in various lipid parameters. Statistical significance was set at p < 0.05.
Results
The baseline characteristics of the subjects are listed in Table 1. No differences in the baseline characteristics were found between the two groups, except for the frequency of calcium channel blocker use.
Table 1. Baseline characteristics of patients.
| All patients (n = 90) | Pitavastatin (n = 44) | Pravastatin (n = 46) | p value | |
|---|---|---|---|---|
| Age (years) | 67 ± 10 | 67 ± 9 | 67 ± 11 | 0.92 |
| Men (%) | 73 (81%) | 38 (86%) | 35 (76%) | 0.33 |
| Body mass index (kg/m2) | 24.2 ± 3.2 | 23.9 ± 3.2 | 24.4 ± 3.3 | 0.52 |
| Status of coronary artery disease | 0.89 | |||
| Stable angina pectoris (%) | 63 (70%) | 30 (68%) | 33 (72%) | |
| Unstable angina pectoris (%) | 27 (30%) | 14 (32%) | 13 (28%) | |
| Hypertension (%) | 59 (66%) | 27 (61%) | 32 (70%) | 0.55 |
| Diabetes mellitus (%) | 39 (43%) | 16 (36%) | 23 (50%) | 0.27 |
| Smoker (%) | 20 (22%) | 11 (25%) | 9 (20%) | 0.66 |
| ACE inhibitors or ARBs (%) | 49 (54%) | 22 (50%) | 27 (59%) | 0.54 |
| Calcium channel blockers (%) | 47 (52%) | 15 (34%) | 32 (70%) | 0.002 |
| β blockers (%) | 9 (10%) | 5 (11%) | 4 (9%) | 0.74 |
Data are expressed as mean ± SD or as number (percentage).
ACE, angiotensin-converting enzyme; ARB, angiotensin-receptor blocker.
Serum lipid levels and plasma PCSK9 and sortilin levels at baseline and at the follow-up are shown in Table 2. Serum LDL cholesterol levels decreased significantly in both statin groups, with a significantly greater reduction in the pitavastatin group (−42% vs. −28%, p = 0.0001). Furthermore, high-density lipoprotein cholesterol levels increased significantly in both statin groups. Significant increases in total and hetero-dimer PCSK9 levels were observed in both statin groups (pitavastatin: 29%, p = 0.0001, and 31%, p < 0.0001; pravastatin: 33%, p = 0.03, and 34%, p = 0.03, respectively). Plasma furin-cleaved PCSK9 levels were not significantly changed from baseline in either group. Significant decreases in sortilin levels from baseline were observed in both statin groups (pitavastatin: −8%, p = 0.02; pravastatin: −16%, p = 0.002). Although a reduction in LDL cholesterol level was greater in the pitavastatin group than in the pravastatin group, percentage changes in hetero-dimer PCSK9 and sortilin levels were not different (Fig. 1).
Table 2. Serum lipid levels and plasma PCSK9 and sortilin levels at baseline and at the 8-month follow-up.
| All patients (n = 90) |
Pitavastatin (n = 44) |
Pravastatin (n = 46) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | p value | Baseline | Follow-up | p value | Baseline | Follow-up | p value | |
| Total cholesterol (mg/dL) | 203 ± 34 | 157 ± 27 | < 0.0001 | 196 ± 31 | 143 ± 24 | < 0.0001 | 210 ± 36 | 172 ± 22 | < 0.0001 |
| % change | −22 ± 13 | −26 ± 13* | −17 ± 12 | ||||||
| LDL cholesterol (mg/dL) | 130 ± 31 | 84 ± 25 | < 0.0001 | 123 ± 24 | 71 ± 20 | < 0.0001 | 137 ± 36 | 96 ± 23 | < 0.0001 |
| % change | −35 ± 17 | −42 ± 15** | −28 ± 16 | ||||||
| Triglycerides (mg/dL) | 114 (36 to 573) | 98 (34 to 396) | 0.07 | 116 (36 to 573) | 91 (34 to 292) | 0.02 | 112 (53 to 316) | 108 (40 to 396) | 0.75 |
| % change | −17 (−75 to 168) | −20 (−76 to 121) | −12 (−75 to 168) | ||||||
| HDL cholesterol (mg/dL) | 47 ± 12 | 51 ± 13 | 0.001 | 48 ± 12 | 51 ± 14 | 0.03 | 47 ± 11 | 51 ± 13 | 0.02 |
| % change | 11 ± 24 | 9 ± 21 | 12 ± 28 | ||||||
| PCSK9 | |||||||||
| Total (ng/mL) | 125 ± 40 | 148 ± 42 | < 0.0001 | 126 ± 40 | 154 ± 43 | 0.0001 | 124 ± 41 | 142 ± 41 | 0.03 |
| % change | 31 ± 66 | 29 ± 40 | 33 ± 83 | ||||||
| Hetero-dimer (ng/mL) | 113 ± 36 | 135 ± 41 | < 0.0001 | 113 ± 35 | 141 ± 41 | < 0.0001 | 112 ± 37 | 130 ± 40 | 0.03 |
| % change | 33 ± 69 | 31 ± 40 | 34 ± 89 | ||||||
| Furin-cleaved (ng/mL) | 12 (4 to 44) | 12 (6 to 31) | 0.58 | 12 (4 to 44) | 12 (6 to 31) | 0.7 | 11 (5 to 23) | 12 (6 to 30) | 0.3 |
| % change | 0 (−63 to 233) | −6 (−57 to 225) | 8 (−63 to 233) | ||||||
| Sortilin (ng/mL) | 8.7 ± 2.3 | 7.4 ± 2.7 | < 0.0001 | 8.2 ± 1.9 | 7.3 ± 2.0 | 0.02 | 9.2 ± 2.6 | 7.5 ± 3.3 | 0.002 |
| % change | −12 ± 27 | −8 ± 28 | −16 ± 26 | ||||||
Data are expressed as mean ± SD or median (range).
LDL, low-density lipoprotein; HDL, high-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin type 9.
p = 0.0003
p = 0.0001 compared with pravastatin group.
Fig. 1.
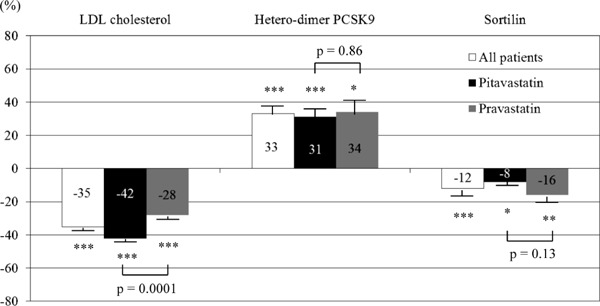
Percentage changes in LDL cholesterol, hetero-dimer PCSK9, and sortilin levels at the 8-month follow-up.
Although the reduction in LDL cholesterol levels was greater in the pitavastatin group than in the pravastatin group, no significant differences in percentage changes in hetero-dimer PCSK9 and sortilin were observed. Data are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.0001 compared with baseline.
Correlations between baseline sortilin levels and lipid parameters are shown in Table 3. Baseline sortilin levels did not correlate with baseline PCSK9 or LDL cholesterol levels or percentage changes in these levels.
Table 3. Correlations between baseline sortilin levels and lipid parameters.
| All patients |
Pitavastatin |
Pravastatin |
||||
|---|---|---|---|---|---|---|
| r | p value | r | p value | r | p value | |
| Baseline level | ||||||
| Total cholesterol | 0.118 | 0.27 | 0.229 | 0.13 | −0.004 | 0.98 |
| LDL cholesterol | 0.104 | 0.33 | 0.133 | 0.39 | 0.026 | 0.87 |
| Triglycerides | −0.032 | 0.76 | 0.220 | 0.15 | −0.267 | 0.07 |
| HDL cholesterol | −0.023 | 0.83 | −0.106 | 0.5 | 0.048 | 0.75 |
| Hetero-dimer PCSK9 | 0.085 | 0.43 | 0.084 | 0.59 | 0.094 | 0.54 |
| Percentage change | ||||||
| Total cholesterol | 0.008 | 0.94 | −0.035 | 0.82 | −0.113 | 0.45 |
| LDL cholesterol | 0.039 | 0.71 | 0.124 | 0.42 | −0.166 | 0.27 |
| Triglycerides | 0.119 | 0.26 | −0.047 | 0.76 | 0.159 | 0.29 |
| HDL cholesterol | 0.052 | 0.63 | −0.026 | 0.87 | 0.074 | 0.63 |
| Hetero-dimer PCSK9 | 0.020 | 0.85 | −0.286 | 0.06 | 0.110 | 0.47 |
LDL, low-density lipoprotein; HDL, high-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin type 9.
We assessed correlations between percentage changes in sortilin and lipid parameters (Table 4) and found weak but significant positive correlations between percentage changes in hetero-dimer PCSK9 and those in sortilin (r = 0.272, p = 0.009) (Fig. 2). These correlations were observed in both statin groups (pitavastatin: r = 0.359, p = 0.02; pravastatin: r = 0.276, p = 0.06) (Table 4).
Table 4. Correlations between percentage changes in sortilin and lipid parameters.
| All patients |
Pitavastatin |
Pravastatin |
||||
|---|---|---|---|---|---|---|
| r | p value | r | p value | r | p value | |
| Total cholesterol (% change) | 0.100 | 0.35 | 0.166 | 0.28 | 0.182 | 0.22 |
| LDL cholesterol (% change) | −0.105 | 0.32 | −0.008 | 0.96 | −0.085 | 0.57 |
| Triglycerides (% change) | 0.141 | 0.19 | 0.020 | 0.9 | 0.308 | 0.04 |
| HDL cholesterol (% change) | 0.108 | 0.31 | 0.061 | 0.69 | 0.169 | 0.26 |
| Hetero-dimer PCSK9 (% change) | 0.272 | 0.009 | 0.359 | 0.02 | 0.276 | 0.06 |
LDL, low-density lipoprotein; HDL, high-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin type 9.
Fig. 2.
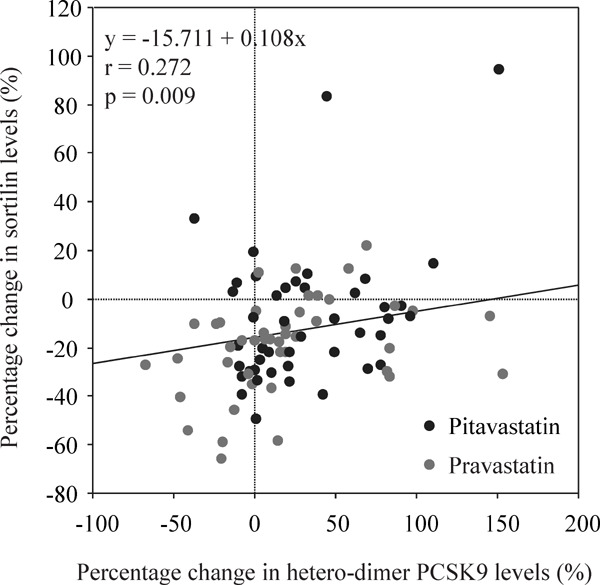
Correlations between percentage changes in hetero-dimer PCSK9 and those in sortilin.
Weak but significant positive correlations were observed between percentage changes in hetero-dimer PCSK9 and those in sortilin.
Discussion
The major findings of the present study are as follows. First, plasma hetero-dimer PCSK9 levels increased significantly 8 months after statin therapy in both the pitavastatin and pravastatin groups. Second, plasma sortilin levels were significantly reduced in both statin groups. Although the reduction of LDL cholesterol was greater in the pitavastatin group than in the pravastatin group, the differences in the increase in hetero-dimer PCSK9 levels and the decrease in sortilin levels were not significant between these two statin groups. Finally, percentage changes of heterodimer PCSK9 levels were positively correlated with those of sortilin levels.
PCSK9, a serine protease produced by the liver, is a newly identified protein that plays a key role in cholesterol homeostasis24). PCSK9 degrades hepatic LDL receptors and subsequently increases LDL cholesterol levels4–7). In addition, plasma PCSK9 is also involved in the inflammatory process24, 25). Thus, PCSK9 may have a broader physiological role in the vascular system. The use of statins is associated with an increase in the expression of PCSK919), counteracting the beneficial effects of statins. The mechanism underlying PCSK9 degradation of LDL receptors is extremely complex. Recently, PCSK9 was found to bind to LDL receptors, subsequently targeting them for intracellular destruction within the hepatocyte26–28). The effect of 4 mg of pitavastatin on the reduction of LDL cholesterol levels was significantly greater than that of 20 mg of pravastatin. However, no significant difference in percentage changes in PCSK9 levels was observed between these two statin groups. The amount of plasma PCSK9 may not reflect the total amount of statin-induced increases in hepatic PCSK9 secretion because with high levels of PCSK9, greater levels are bound to hepatic LDL receptors, removing them from circulation. In addition, increases in circulating PCSK9 levels caused by statin therapy differ over the shortand long-term29). This explains why a significant difference was not observed in the increase of PCSK9 levels from baseline between the two statin groups.
The main functions of sortilin are to transport ligands between the trans-Golgi network and the early endosomes and to bind and internalize various ligands across the cell membrane by receptor-mediated endocytosis13, 16). Although the exact mechanism underlying the effects of sortilin on lipid metabolism has not been fully examined, sortilin binds to LDL on the cell surface30) and increases the amount of LDL internalized18, 31). In addition, sortilin has been shown to bind apolipoprotein A-V and lipoprotein lipase32, 33). The effect of sortilin on very low-density lipoprotein (VLDL) synthesis, as determined by overexpression and knockdown studies, is conflicting11, 34, 35). Musunuru et al.11) reported that sortilin reduced VLDL synthesis and thereby reduced LDL cholesterol levels, whereas Kjolby et al.34) reported that sortilin increased VLDL synthesis and thereby increased LDL cholesterol levels. Strong et al.35) reported that increased hepatic sortilin expression reduced hepatic apolipoprotein B secretion and increased LDL catabolism, providing dual mechanisms underlying the reduction of plasma LDL cholesterol levels. Thus, the effect of sortilin on LDL cholesterol levels is controversial. In the present study, baseline sortilin levels did not correlate with baseline PCSK9 or LDL cholesterol levels or percentage changes in these levels, indicating that sortilin could not predict the LDL cholesterol-lowering effects of statins.
It is still unclear how the hepatic expression and plasma sortilin levels are regulated. Sortilin is constitutively released from the cell surface following shedding by metalloproteinases36) and can therefore be detected in human plasma. Once released from the cell surface, sortilin does not influence PCSK9 activity37). Recently, two proteins were suggested to enhance PCSK9-mediated degradation of the LDL receptor: one is sortilin, which binds to PCSK9 in the trans-Golgi network and possibly facilitates its secretion37), and the other is amyloid precursor-like protein 2 (APLP2), which facilitates trafficking of the PCSK9-LDL receptor complex to endosomes/lysosomes38). However, Butkinaree et al.39) reported that PCSK9 enhanced LDL receptor degradation independent of sortilin or APLP2 ex vivo and in mice. Interestingly, when co-expressed with PCSK9, both sortilin and APLP2 were targeted for lysosomal degradation, and sortilin enhanced the stability of APLP2. Considering these findings, the effect of sortilin on the functions of APLP2 needs to be studied in specific tissues, especially the brain, small intestine, and colon, where the expression of both transcripts is quite high. We could not explain the precise mechanism of why plasma sortilin levels decreased and how circulating sortilin levels were regulated because this is a clinical study. However, consistent with the previous report that circulating PCSK9 and sortilin are positively correlated37), we found significant positive correlations between percentage changes in PCSK9 and those in sortilin.
Although the homozygote for the major allele at chromosome 1p13.3 locus is associated with > 90% reduced expression of sortilin in the human liver and with increased levels of LDL cholesterol11), the heterozygote with missense mutations in the SORT1 gene had no apparent effect on serum cholesterol levels30), suggesting a marginal effect of the SORT1 gene on LDL cholesterol levels. Further studies on the function of sortilin in LDL metabolism will better elucidate the role of genetic variants at the SORT1 gene in the regulation of lipoprotein metabolism and modulation of CAD risk.
The present study has several limitations. First, it was a post hoc analysis of the TRUTH trial and all subjects had CAD. Second, plasma PCSK9 and sortilin levels were measured using frozen samples at only two timepoints. Third, we did not evaluate the amount of LDL receptor, sortilin, or PCSK9 expression in hepatocytes. Finally, the small number of patients included in the study made the statistical power insufficient.
Despite these limitations, to the best of our knowledge, this is the first study that evaluated the effects of statins, particularly two different types of statins, on plasma PCSK9 and sortilin levels at the same time. Thus, both pitavastatin and pravastatin increased plasma PCSK9 levels and decreased sortilin levels. Moreover, changes in PCSK9 levels were positively correlated with those in sortilin levels, but LDL cholesterol levels were not. A prospective, randomized study with a greater number of patients would be required to confirm our conclusions.
Conclusions
Both pitavastatin and pravastatin increased plasma PCSK9 levels and decreased sortilin levels. Statin-induced increases in PCSK9 levels were associated with changes in sortilin levels in statin-naive patients with CAD.
Disclosures
None.
Conflicts of Interest
None.
References
- 1). Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci, 2007; 32: 71-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Seidah NG, Khatib AM, Prat A. The proprotein convertases and their implication in sterol and/or lipid metabolism. Biol Chem, 2006; 387: 871-877 [DOI] [PubMed] [Google Scholar]
- 3). Lambert G, Krempf M, Costet P. PCSK9: a promising therapeutic target for dyslipidemias? Trends Endocrinol Metab, 2006; 17: 79-81 [DOI] [PubMed] [Google Scholar]
- 4). Graham MJ, Lemonidis KM, Whipple CP, Subramaniam A, Monia BP, Crooke ST, Crooke RM. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res, 2007; 48: 763-767 [DOI] [PubMed] [Google Scholar]
- 5). Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest, 2006; 116: 2995-3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Park SW, Moon YA, Horton JD. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem, 2004; 279: 50630-50638 [DOI] [PubMed] [Google Scholar]
- 7). Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chrétien M, Prat A, Seidah NG. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem, 2004; 279: 48865-48875 [DOI] [PubMed] [Google Scholar]
- 8). Myocardial Infarction Genetics Consortium. Genomewide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet, 2009; 41: 334-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, König IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H, WTCCC and the Cardiogenics Consortium Genomewide Association Analysis of Coronary Artery Disease. N Engl J Med, 2007; 357: 443-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10). Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T, Martin NG, Pedersen NL, Kyvik KO, Kaprio J, Hofman A, Freimer NB, Jarvelin MR, Gyllensten U, Campbell H, Rudan I, Johansson A, Marroni F, Hayward C, Vitart V, Jonasson I, Pattaro C, Wright A, Hastie N, Pichler I, Hicks AA, Falchi M, Willemsen G, Hottenga JJ, de Geus EJ, Montgomery GW, Whitfield J, Magnusson P, Saharinen J, Perola M, Silander K, Isaacs A, Sijbrands EJ, Uitterlinden AG, Witteman JC, Oostra BA, Elliott P, Ruokonen A, Sabatti C, Gieger C, Meitinger T, Kronenberg F, Döring A, Wichmann HE, Smit JH, McCarthy MI, van Duijn CM, Peltonen L, ENGAGE Consortium Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet, 2009; 41: 47-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, Rader DJ. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nat Genet, 2010; 466: 714-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Petersen CM, Nielsen MS, Nykjaer A, Jacobsen L, Tommerup N, Rasmussen HH, Roigaard H, Gliemann J, Madsen P, Moestrup SK. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J Biol Chem, 1997; 272: 3599-3605 [DOI] [PubMed] [Google Scholar]
- 13). Quistgaard EM, Madsen P, Grøftehauge MK, Nissen P, Petersen CM, Thirup SS. Ligands bind to Sortilin in the tunnel of a ten-bladed beta-propeller domain. Nat Struct Mol Biol, 2009; 16: 96-98 [DOI] [PubMed] [Google Scholar]
- 14). Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, Wahlstrand B, Hedner T, Corella D, Tai ES, Ordovas JM, Berglund G, Vartiainen E, Jousilahti P, Hedblad B, Taskinen MR, Newton-Cheh C, Salomaa V, Peltonen L, Groop L, Altshuler DM, Orho-Melander M. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet, 2008; 40: 189-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Willnow TE, Petersen CM, Nykjaer A. VPS10P-domain receptors - regulators of neuronal viability and function. Nat Rev Neurosci, 2008; 9: 899-909 [DOI] [PubMed] [Google Scholar]
- 16). Willnow TE, Kjølby M, Nykjaer A. Sortilins: new players in lipoprotein metabolism. Curr Opin Lipidol, 2011; 22: 79-85 [DOI] [PubMed] [Google Scholar]
- 17). Kjølby M, Nielsen MS, Petersen CM. Sortilin, encoded by the cardiovascular risk gene SORT1, and its suggested functions in cardiovascular disease. Curr Atheroscler Rep, 2015; 17: 496. [DOI] [PubMed] [Google Scholar]
- 18). Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol, 2009; 29: 431-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res, 2008; 49: 394-398 [DOI] [PubMed] [Google Scholar]
- 20). Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med, 2012; 367: 1891-1900 [DOI] [PubMed] [Google Scholar]
- 21). Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med, 2012; 366: 1108-1118 [DOI] [PubMed] [Google Scholar]
- 22). Nozue T, Yamamoto S, Tohyama S, Umezawa S, Kunishima T, Sato A, Miyake S, Takeyama Y, Morino Y, Yamauchi T, Muramatsu T, Hibi K, Sozu T, Terashima M, Michishita I. Statin treatment for coronary artery plaque composition based on intravascular ultrasound radiofrequency data analysis. Am Heart J, 2012; 163: 191-199 [DOI] [PubMed] [Google Scholar]
- 23). Hori M, Ishihara M, Yuasa Y, Makino H, Yanagi K, Tamanaha T, Kishimoto I, Kujiraoka T, Hattori H, Harada-Shiba M. Removal of plasma mature and furin-cleaved proprotein convertase subtilisin/kexin 9 by low-density lipoprotein-apheresis in familial hypercholesterolemia: development and application of a new assay for PCSK9. J Clin Endcrinol Metab, 2015; 100: E41-E49 [DOI] [PubMed] [Google Scholar]
- 24). Li S, Li JJ. PCSK9: A key factor modulating atherosclerosis. J Atheroscler Thromb, 2015; 22: 221-230 [DOI] [PubMed] [Google Scholar]
- 25). Li S, Zhu CG, Guo YL, Xu RX, Zhang Y, Sun J, Li JJ. The relationship between the plasma PCSK9 levels and platelet indices in patients with stable coronary artery disease. J Atheroscler Thromb, 2015; 22: 76-84 [DOI] [PubMed] [Google Scholar]
- 26). Li J, Tumanut C, Gavigan JA, Huang WJ, Hampton EN, Tumanut R, Suen KF, Trauger JW, Spraggon G, Lesley SA, Liau G, Yowe D, Harris JL. Secreted PCSK9 promotes LDL receptor degradation independently of proteolytic activity. Biochem J, 2007; 406: 203-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). McNutt MC, Lagace TA, Horton JD. Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J Biol Chem, 2007; 282: 20799-20803 [DOI] [PubMed] [Google Scholar]
- 28). Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem, 2007; 282: 18602-18612 [DOI] [PubMed] [Google Scholar]
- 29). Nozue T, Hattori H, Ishihara M, Iwasaki T, Hirano T, Kawashiri MA, Yamagishi M, Michishita I. Comparison of effects of pitavastatin versus pravastatin on serum proprotein convertase subtilisin/kexin type 9 levels in statinnaive patients with coronary artery disease. Am J Cardiol, 2013; 111: 1415-1419 [DOI] [PubMed] [Google Scholar]
- 30). Tveten K, Strøm TB, Cameron J, Berge KE, Leren TP. Mutations in the SORT1 gene are unlikely to cause autosomal dominant hypercholesterolemia. Atherosclerosis, 2012; 225: 370-375 [DOI] [PubMed] [Google Scholar]
- 31). Linsel-Nitschke P, Heeren J, Aherrahrou Z, Bruse P, Gieger C, Illig T, Prokisch H, Heim K, Doering A, Peters A, Meitinger T, Wichmann HE, Hinney A, Reinehr T, Roth C, Ortlepp JR, Soufi M, Sattler AM, Schaefer J, Stark K, Hengstenberg C, Schaefer A, Schreiber S, Kronenberg F, Samani NJ, Schunkert H, Erdmann J. Genetic variation at chromosome 1p13.3 affects sortilin mRNA expression, cellular LDL-uptake and serum LDL levels which translates to the risk of coronary artery disease. Atherosclerosis, 2010; 208: 183-189 [DOI] [PubMed] [Google Scholar]
- 32). Nielsen MS, Jacobsen C, Olivecrona G, Gliemann J, Petersen CM. Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem, 1999; 274: 8832-8836 [DOI] [PubMed] [Google Scholar]
- 33). Nilsson SK, Christensen S, Raarup MK, Ryan RO, Nielsen MS, Olivecrona G. Endocytosis of apolipoprotein A-V by members of the low density lipoprotein receptor and the VPS10p domain receptor families. J Biol Chem, 2008; 283: 25920-25927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34). Kjolby M, Andersen OM, Breiderhoff T, Fjorback AW, Pedersen KM, Madsen P, Jansen P, Heeren J, Willnow TE, Nykjaer A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab, 2010; 12: 213-223 [DOI] [PubMed] [Google Scholar]
- 35). Strong A, Ding Q, Edmondson AC, Millar JS, Sachs KV, Li X, Kumaravel A, Wang MY, Ai D, Guo L, Alexander ET, Nguyen D, Lund-Katz S, Phillips MC, Morales CR, Tall AR, Kathiresan S, Fisher EA, Musunuru K, Rader DJ. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Invest, 2012; 122: 2807-2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). Hermey G, Sjøgaard SS, Petersen CM, Nykjaer A, Gliemann J. Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem J, 2006; 395: 285-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Gustafsen C, Kjolby M, Nyegaard M, Mattheisen M, Lundhede J, Buttenschøn H, Mors O, Bentzon JF, Madsen P, Nykjaer A, Glerup S. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab, 2014; 19: 310-318 [DOI] [PubMed] [Google Scholar]
- 38). DeVay RM, Shelton DL, Liang H. Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J Biol Chem, 2013; 288: 10805-10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Butkinaree C, Canuel M, Essalmani R, Poirier S, Benjannet S, Asselin MC, Roubtsova A, Hamelin J, Marcinkiewicz J, Chamberland A, Guillemot J, Mayer G, Sisodia SS, Jacob Y, Prat A, Seidah NG. Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9 convertase-mediated low density lipoprotein receptor degradation but interact with each other. J Biol Chem, 2015; 290: 18609-18620 [DOI] [PMC free article] [PubMed] [Google Scholar]