

Abstract
Aim
The aryl hydrocarbon receptor (AhR), a ligand-inducible transcription factor mediating toxic effects of dioxins and uremic toxins, has recently emerged as a pathophysiological regulator of immune-inflammatory conditions. Indoxyl sulfate, a uremic toxin, is associated with cardiovascular disease in patients with chronic kidney disease and has been shown to be a ligand for AhR. The aim of this study was to investigate the potential role of AhR in indoxyl sulfate-induced leukocyte–endothelial interactions.
Methods
Endothelial cell-specific AhR knockout (eAhR KO) mice were produced by crossing AhR floxed mice with Tie2 Cre mice. Indoxyl sulfate was administered for 2 weeks, followed by injection of TNF-α. Leukocyte recruitment to the femoral artery was assessed by intravital microscopy. Vascular endothelial cells were transfected with siRNA specific to AhR (siAhR) and treated with indoxyl sulfate, followed by stimulation with TNF-α.
Results
Indoxyl sulfate dramatically enhanced TNF-α-induced leukocyte recruitment to the vascular wall in control animals but not in eAhR KO mice. In endothelial cells, siAhR significantly reduced indoxyl sulfate-enhanced leukocyte adhesion as well as E-selectin expression, whereas the activation of JNK and nuclear factor-κB was not affected. A luciferase assay revealed that the region between −153 and −146 bps in the E-selectin promoter was responsible for indoxyl sulfate activity via AhR. Mutational analysis of this region revealed that activator protein-1 (AP-1) is responsible for indoxyl sulfate-triggered E-selectin expression via AhR.
Conclusion
AhR mediates indoxyl sulfate-enhanced leukocyte–endothelial interactions through AP-1 transcriptional activity, which may constitute a new mechanism of vascular inflammation in patients with renal disease.
Keywords: Activator protein-1 transcription factor, Aryl hydrocarbon receptor, Chronic kidney disease, Inflammation, Indoxyl sulfate
Introduction
The prevalence of cardiovascular disease in dialysis patients is higher than that in healthy individuals1) because of the high rate of coronary artery diseases in this population2). Even in patients with mild to moderate chronic kidney disease, the estimated glomerular filtration rate is inversely correlated with the risk of cardiovascular disease and mortality3). Furthermore, the incidence of death is far more common than the administration of renal replacement therapy at all stages4). Therefore, the relationship between cardiovascular dysfunction and renal disease, described as “cardiorenal syndrome,” is a well-recognized phenomenon5, 6). Several factors such as uremic toxin exposure, immune system dysfunction, oxidative stress, anemia, and the renin–angiotensin–aldosterone system have been shown to affect the cardiovascular system and the kidney7).
Endothelial dysfunction, particularly that involving leukocyte–endothelial interactions, plays a central role in the initiation and development of atherosclerosis8). Proinflammatory cytokines such as TNF-α and IL-1β induce the expression of several cell adhesion molecules, including E-selectin from the selectin protein family, intercellular cell adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1), which promotes monocyte/macrophage infiltration into the activated endothelium8, 9). The migration of monocytes/macrophages leads to plaque rupture via the formation of a fibrous cap, proliferation of vascular smooth muscle cells, and induction of angiogenesis.
Indoxyl sulfate is a protein-bound uremic toxin synthesized in the liver from indole, a metabolite of tryptophan produced by intestinal flora from dietary protein10), and it is excreted in urine through the kidneys. Therefore, serum levels of indoxyl sulfate increase when kidney function deteriorates11, 12). In dialysis patients, serum concentrations of indoxyl sulfate are approximately 54 times higher than those in healthy subjects13). Moreover, Barreto et al. showed that serum indoxyl sulfate progressively increases with stages of chronic kidney disease and is associated with the incidence of cardiovascular disease and overall mortality in patients with chronic kidney disease12). Several studies have suggested that indoxyl sulfate aggravates cardiovascular disease by inducing cardiac cell fibrosis, endothelial senescence, proliferation of vascular smooth muscle cells, and activation of monocytes/macrophages via an oxidative stress-dependent path way14–22). In addition, we previously showed that indoxyl sulfate enhances cytokine-induced leukocyte–endothelial interactions through E-selectin in vivo and in vitro, and this process is mediated by activation of oxidative stress-dependent intracellular signaling such as JNK and NF-κB in human umbilical vein endothelial cells (HUVEC)23). Interestingly, angiotensin II or hydrogen peroxide-related oxidative stress failed to enhance the expression of E-selectin, suggesting that the inflammatory effect of indoxyl sulfate is mediated in an oxidative stress-independent as well as -dependent manner.
In 2010, Schroeder et al. demonstrated that the aryl hydrocarbon receptor (AhR) is an endogenous receptor for indoxyl sulfate24). AhR is a member of the basic helix–loop–helix Per–Arnt–Sim regulatory protein superfamily. This ligand-inducible transcription factor can be activated by environmental contaminants such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). AhR, which is present in the cytoplasm without stimulation, translocates to the nucleus upon ligand binding and induces the expression of proteins such as cytochrome p450 (Cyp) 1a1 and Cyp1b1 via xenobiotic response elements (XREs). AhR agonists such as TCDD are known to have carcinogenic and teratogenic effects25) and have been shown to exacerbate atherosclerosis via the accumulation of macro-phages in apolipoprotein E-null mice26). We therefore hypothesized that indoxyl sulfate-induced vascular inflammation was regulated by AhR.
Materials and Methods
Reagents
Recombinant mouse TNF-α was obtained from Biolegend (San Diego, CA, USA). Antibodies against E-selectin (A-10: sc-137203), ICAM-1 (G-5: sc-8439), VCAM-1 (H276: sc-8304), and AhR (A-3: sc-133088) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The HRP-conjugated anti-tubulin antibody (PM054-7) was obtained from MBL (Nagoya, Japan). Anti-lamin A/C (SAB4200236) was obtained from Sigma-Aldrich. Normal rabbit IgG antibodies were obtained from Cell Signaling Technology (Beverly, MA). AhR-targeting siRNA (siAhR: J-004990-070005) and non-targeting siRNA (siCont: D-00181001-05) were obtained from Thermo Scientific (Dharmacon Division, Lafayette, CO, USA). The other reagents were prepared as previously described23).
Laboratory Animals
All animal experiments were approved by the Ethical Committee for Animal Experimentation of Tokyo Medical and Dental University (Tokyo, Japan). C57BL/6J-background B6.129(FVB)-Ahr tm3.1Bra/J (AhRfx/fx) mice, harboring the DBA/2 Ahr allele (Ahrd) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The Ahrfx/fx mice were crossed with the Tie2-cre transgenic mice, kindly provided by Dr. Sawada (the University of Chicago)27) to generate offspring that were hemizygous for the Cre transgene and heterozygous for the Ahrfx allele (Cre+Ahrfx/+). Cre+ Ahrfx/fx mice (endothelial cell-specific AhR-knockout mice: eAhr KO) were produced by crossing male Cre+ Ahrfx/+ mice with female Ahrfx/fx mice to prevent Cremediated deletion of the floxed alleles in the female germline28). Male Cre+Ahrfx/fx mice were routinely mated with female Ahrfx/fx mice to obtain eAhR KO mice and Ahrfx/fx littermates, which served as experimental controls that did not express Cre.
Male mice aged 8–10 weeks were anesthetized by intraperitoneal injection of sodium pentobarbital (Schering-Plough Corporation; Kenilworth, NJ, USA) at a dose of 65 mg/kg. A micro-osmotic pump (model 2002; Alzet; Alza, Palo Alto, CA, USA) was implanted subcutaneously that released a solution containing 20% (w/v) dimethyl sulfoxide in 100 mmol/L phosphate buffer (pH 8.0) with or without 0.79 mmol/L indoxyl sulfate at a rate of 0.5 µL/h. Two weeks later, 2 µg recombinant mouse TNF-α was injected intraperitoneally. Leukocyte adhesion in the femoral artery was assessed 3 h after this treatment by intravital microscopy (IVM) as previously described23, 29–33). In brief, mice were injected via the left femoral vein with rhodamine 6G chloride (Molecular Probes, Eugene, OR) to label leukocytes in vivo. The femoral artery was visualized with a microscope (model BX51WI; Olympus, Tokyo, Japan) equipped with a water immersion objective. Labeled leukocytes were clearly visualized in the anterior half of the vessels facing the objective, and platelets less than 5 µm in diameter were excluded. All images were recorded using a personal computer with an image analysis program (Meta-Morph; Molecular Devices, Sunnyvale, CA, USA). The number of adherent and rolling leukocytes (i.e., those that did not move for ≥ 3 s during the 1-min recording period and those that passed a reference line perpendicular to the vessel axis, respectively) was counted along a region of interest, a 100 × 100 µm segment of the vessel, and expressed as the number of interacting cells per 104 µm2 of the vessel surface33).
Endothelial Cell Isolation
Mice were euthanized and perfused via the left ventricle with ice-cold PBS, the lungs were dissected and rinsed with PBS. The organs were minced and incubated in HEPES buffer (10 mmol/L HEPES pH 7.4, 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2) containing 2 mg/ml collagenase D and 40 U/ml DNase I for 30 min. The pulmonary cells were collected via a 70-µm cell strainer, and endothelial cells were isolated using CD31 micro-beads (Miltenyi Biotec Inc., Auburn, CA) according to the manufacturer's protocol. Collected cells were analyzed for purity using the phycoerythrin-conjugated anti-CD31 antibody (Miltenyi Biotec) by flow cytometry as previously described18).
Cell Cultures
HUVEC were purchased and cultured as previously described18, 23). HL-60, a human leukemia cell line, was obtained from American Type Culture Collection, and the cells were maintained in RPMI 1640 medium supplemented with 10% FCS, 100 IU/ml penicillin, 0.17 mmol/L streptomycin, and 2 mmol/L L-glutamine.
Real-Time Quantitative PCR
Total RNA was extracted, and cDNA was prepared as previously reported23). Real-time quantitative reverse-transcriptase PCR was performed using a Thermal Cycler Dice Real Time System Single (Takara)34) to quantitate the expression of Cyp1a1, Cyp1b1, PTGS2, CSF-2, and GAPDH mRNA using the following primers: Cyp1a1, forward (F): 5′-TCCAAGAGTCCACCCTTCC-3′, reverse (R): 5′-AAGCATGATCAGTGTAGGGATCT-3′; Cyp1b1, F: 5′-GGCATTAGAGTCAACTACACAAAGC-3′, R: 5′-GAATGGCAAGTGCCAAAAA-3′; PTGS2, F: 5′-CTTCACGCATCAGTTTTTCAAGAGC-3′, R: 5′-TCACCGTAAATATGATTTAAGTCCAC-3′; CSF-2, F: 5′-TCTCAGAAATGTTTGACCTCCA-3′, R: 5′-GCCCTTGAGCTTGGTGAG-3′; GAPDH, F: 5′-AGCCACATCGCTCAGACAC-3′, R: 5′-GCCCAATACGACCAAATCC-3′.
PCR Array
HUVEC were treated with or without 2.0 mmol/L indoxyl sulfate for 20 h, followed by stimulated with TNF-α for 2 h. Total RNA was extracted, and cDNA was synthesized as described above. Expression of 90 genes was quantified using RT2 pro-filer PCR Array Human Signal Transduction Pathway Finder (PARS-014A: SABioscience, Qiagen, Hilden, Germany).
Transfection
siAhR or siCont (10 pmol each) and 3 µL Lipofectin (Life Technologies, Inc., Gaithersburg, MD, USA) were diluted in 100 µL Opti-MEM® I Reduced Serum Medium, incubated for 30 min, and then mixed with each other, followed by incubation for 15 min. The mixture was added to the HUVEC seeded onto a 6-well plate at 0.8 × 105 cells/well the day before the transfection. After 4 h, the reagent was replaced with EGM-2, and the cells were incubated for 20 h.
Leukocyte Adhesion Assay
HL-60 cells pre-labeled with the fluorescent dye 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (1.6 × 106 cells/well) in RPMI + 1% FCS were added to monolayers of siAhR- or siCont-transfected HUVEC in 6-well plates. After incubation under non-static adhesion assay conditions (rotation at 64 rpm, 10 min, 22°C–25°C), non-adherent HL-60 cells were removed by washing 3 times with RPMI + 1% FCS. Monolayer-associated HL-60 cells were then collected in Hank's balanced salt solution (Wako, Osaka, Japan) containing 5 mM ethylenediaminetetraacetic acid and 4 mM ethylene glycol tetraacetic acid. Fluorescence was measured using a fluorescence microplate reader (ARVO X3; Perkin Elmer, Waltham, MA).
Detection of Reactive Oxygen Species (ROS)
The intracellular formation of ROS was detected by the fluorescent probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) as previously described13, 35). The intensity of the fluorescence was measured using an ARVO X3 plate reader. Oxidative stress was also detected using dihydroethidium (DHE)23).
Plasmid Preparation
The XRE-pGL4 and CRE-pGL4 reporter plasmids were prepared by inserting the corresponding sequences into pGL4.24 [luc2P/minP] vector (Pro-mega, Madison, WI, USA) containing the firefly luciferase reporter gene via the SacI and BglII restriction sites. Double-stranded XRE and CRE nucleotide sequences were prepared by annealing the following synthetic oligomers. For the XRE-pGL4 vector: XREluc-1, 5′- TTGCGTGACAAGCTTGCGTGACAAGCTTGCGTGACAAGCTTGCGTGACAAGC
TTGCGTGACAAGCTTGCGTGACAAGCTTGCGTGACAAGCTTGCGTGACAAGC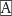 -3′; and XRE-luc-2, 5′-
-3′; and XRE-luc-2, 5′- GCTTGTCACGCAAGCTTGTCACGCAAGCTTGTCACGCAAGCTTGTCACGCAA
GCTTGTCACGCAAGCTTGTCACGCAAGCTTGTCACGCAAGCTTGTCACGCAA -3′. For the CRE-pGL4 vector: CRE-luc1, 5′- TGACGTCAATCCACTGACGTCAATCCACTGACGTCAATCCAC-3′; and CRE-luc-2, 5′-GTGGATTGACGTCAGTGGATTGACGTCAGTGGATTGACGTCA-3′ (the restriction sites are boxed, while XRE and CRE sequences are underlined). The annealed oligonucleotides were phosphorylated by T4 polynucleotide kinase (Takara) and ligated into the vector using ligation high ver.2 (Toyobo, Osaka, Japan).
-3′. For the CRE-pGL4 vector: CRE-luc1, 5′- TGACGTCAATCCACTGACGTCAATCCACTGACGTCAATCCAC-3′; and CRE-luc-2, 5′-GTGGATTGACGTCAGTGGATTGACGTCAGTGGATTGACGTCA-3′ (the restriction sites are boxed, while XRE and CRE sequences are underlined). The annealed oligonucleotides were phosphorylated by T4 polynucleotide kinase (Takara) and ligated into the vector using ligation high ver.2 (Toyobo, Osaka, Japan).
Preparation of the Plasmid Containing the E-Selectin Promoter Region
The E-selectin promoter region in the pKM2L vector (pKM2L-phESEL) was obtained from Riken BioResource Center (Gene Engineering Division, Ibaraki, Japan). The region was restricted with SacI and MluI and transferred to the pGL3-basic vector. The sequence containing base pairs −166 to +107 from the promoter region of E-selectin, including the nuclear factor-endothelial adhesion molecule 1 (NF-ELAM-1) site, was amplified by PCR using the following primers: F, 5′- C GAGACAGAGTTTCTGACATC-3′ (restriction sites are boxed), and common reverse, 5′-CTTTATGTTTTTGGCGTCTTCCA-3′. The amplicon was inserted into the original vector via SacI and MluI sites. The resulting vector was named NF-ELAM-1 vector. The non-functional mutated NF-ELAM-1 vector (mNFELAM-1), vector with the NF-ELAM-1 site carrying an activator protein-1 (AP-1)-binding site (NF-ELAM-AP-1), and vector with the NF-ELAM-1 site carrying a cyclic AMP response element (CRE) (NF-ELAM-CRE) were prepared by replacing the original insert with the PCR product obtained using the above common reverse primer and the following forward primers: mNFELAM-1 F, 5′- CGAGACAGAGTTTCGTCGAGCCTTG-3′, NFELAM-AP1 F, 5′- CGAGACAGAGTTTCTGACTCATTG-3′, and NF-ELAMCRE F, 5′-CGAGACAGAGTTTCTGACGTCATTG-3′, respectively (restriction sites are boxed and mutated nucleotides are underlined).
GAGACAGAGTTTCTGACATC-3′ (restriction sites are boxed), and common reverse, 5′-CTTTATGTTTTTGGCGTCTTCCA-3′. The amplicon was inserted into the original vector via SacI and MluI sites. The resulting vector was named NF-ELAM-1 vector. The non-functional mutated NF-ELAM-1 vector (mNFELAM-1), vector with the NF-ELAM-1 site carrying an activator protein-1 (AP-1)-binding site (NF-ELAM-AP-1), and vector with the NF-ELAM-1 site carrying a cyclic AMP response element (CRE) (NF-ELAM-CRE) were prepared by replacing the original insert with the PCR product obtained using the above common reverse primer and the following forward primers: mNFELAM-1 F, 5′- CGAGACAGAGTTTCGTCGAGCCTTG-3′, NFELAM-AP1 F, 5′- CGAGACAGAGTTTCTGACTCATTG-3′, and NF-ELAMCRE F, 5′-CGAGACAGAGTTTCTGACGTCATTG-3′, respectively (restriction sites are boxed and mutated nucleotides are underlined).
Luciferase Reporter Gene Assay
HUVEC were cultured in 24-well plates and transiently transfected using Lipofectin. Briefly, 120 ng firefly luciferase vector, 50 ng internal control pRL-TK, and 2 pmol siRNA were co-transfected into HUVEC as previously reported23).
Electrophoresis Mobility Shift Assay
HUVEC were treated with 2 mmol/L indoxyl sulfate, followed by treatment with TNF-α, and the nuclear fraction was extracted by NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rock-ford, IL, USA). Biotin-labeled NF-ELAM-1 probe was prepared by annealing the following synthetic 3′-biotinylated oligonucleotides: 5′-GAGTTTCTGACATCATTGTAAT-3′, and 5′-ATTACAATGATGTCAGAAACTC-3′ (NF-ELAM-1 sequence is underlined). The nuclear extracts, probe, and antibodies against ATF-2 or AhR were mixed and incubated, followed by electrophoresis on 6% polyacrylamide gel. After transferring to membranes at 200 mA for 60 min, cross-linking was performed by exposure to ultraviolet light for 15 min. Protein-DNA complexes were detected with chemiluminescence (Panomics, Fremont, CA, USA) according to the manufacturer's protocol.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP was performed as previously described36, 37). In brief, cells were crosslinked with 1% formaldehyde for 10 min at room temperature before glycine was added to a final concentration of 0.125 M. Chromatin was sheared to 200–1000 bp by sonication. The chromatin solution was then incubated with antibody-bound Dynabeads (Dynal Biotech) at 4°C. Immune complexes were eluted from the beads and reverse cross-linked at 65°C. The samples were treated with proteinase K, DNA was purified using the MinElute PCR purification kit (Qiagen) and analyzed by real-time PCR with specific primers for E-selectin promoter region; F, 5′-AGGCATGGACAAAGGTGAAG-3′ and R 5′-GTCCACATCCAGTAAAGAGG-3′; for the E-selectin extreme 3′ region; F, 5′-TGGCAGGCAGAGGAATGG-3′, and R, 5′-CCATTTTCCACACCGCTATGA-3′34). For the CSF-2 promoter region; F, 5′-ATAAGGGCCAGGAGATTCCA-3′, R, 5′-AGCCACATCCTCCAGAGAACT-3′. Negative control RPL30 primers were obtained from cell signaling technology.
Other Methods
Western blotting was performed as per standard protocols using ECL prime reagent (GE Healthcare, Uppsala, Sweden).
Statistical Analysis
Data are expressed as mean values ± standard error of the mean (SEM) or standard deviation (SD). One-way analysis of variance with the Tukey's post-hoc test or two-tailed unpaired t-test was used to estimate statistical significance. A P value of <0.05 was considered to represent statistical significance.
Results
Indoxyl Sulfate-Enhanced Leukocyte–Endothelial Interactions were Abrogated in Endothelial-Specific AhR Null Mice.
To determine the physiological role of AhR, we investigated the effect of endothelial cell-specific AhR deficiency on the indoxyl sulfate-enhanced leukocyte–endothelial interactions using an IVM system23, 29–33). First, we purified endothelial cells from lungs taken from AhRfl/fl and eAhR KO mice to confirm the expression of AhR. Flow cytometric analysis indicated that over 90% of the isolated cells were positive for CD31 (Fig. 1A), an endothelial cell marker. AhR expression was decreased in endothelial cells of eAhR KO mice when compared with those of AhRfl/fl mice. On the other hand, AhR expression in liver was not significantly different (Fig. 1B). Injection of indoxyl sulfate induced mRNA expression of cyp1a1, a gene downstream of AhR, in endothelial cells of control AhRfl/fl mice, but not those of eAhR KO mice (Fig. 1C).
Fig. 1.
Effects of indoxyl sulfate on leukocyte-endothelial interactions in AhRfl/fl and eAhR KO mice. (A) Flow cytometry analysis of pulmonary cells stained with phycoerythrin-conjugated CD31 antibody before (dashed line) and after (line) endothelial cell isolation using CD31 microbeads. Gray filled-in line indicates cells stained with isotype control. (B) Immunoblotting images using anti-AhR antibody of isolated endothelial cells and liver from AhRfl/fl and eAhR KO mice. The mRNA expression of cre recombinase and 18S ribosomal (r)RNA was detected by reverse transcriptase-PCR. (C) AhRfl/fl and eAhR KO mice were subcutaneously injected with (+) 4 mmol/kg of indoxyl sulfate or left untreated (−) for 3 h, followed by isolation of endothelial cells from lung tissue. Cyp1a1 mRNA expression was determined by real-time PCR. (D, E, F, and G) Representative snapshots from intravital video microscopic analysis of leukocyte adhesive interactions in the femoral arteries (margins of vessels are indicated with dashed lines) of AhRfl/fl mice treated (D) with indoxyl sulfate or untreated (E) as well as eAhR KO mice treated (F) with indoxyl sulfate or untreated (G). White spots represent fluorescently labeled leukocytes visualized by intravenous injection of rhodamine 6G. (H) Quantitative analyses of leukocyte adhesion interactions in the femoral arteries. The data are expressed as means ± SEM (n = 6). *P < 0.05 versus (vs.) AhRfl/fl not treated with indoxyl sulfate, †P < 0.05 vs. AhRfl/fl treated with indoxyl sulfate.
eAhR KO and control AhRfl/fl mice were either untreated or administrated with indoxyl sulfate for 2 weeks, followed by injection with TNF-α. We found that treatment with TNF-α increased its serum concentration (data not shown). There were no significant differences between these groups in urea, cholesterol, and triglyceride levels. As expected, the indoxyl sulfate-treated groups exhibited significantly higher serum indoxyl sulfate levels than the buffer control groups (Table 1). We then performed IVM analysis to evaluate leukocyte adhesion to the vascular wall in the femoral artery. We first confirmed that no leukocyte interaction was observed in AhRfl/fl mice (Supplemental video 1: supplemental movie provided journal website) and that TNF-α treatment slightly induced leukocyte adhesion (Supplemental video 2). Administration of indoxyl sulfate dramatically enhanced leukocyte recruitment to the vascular wall in TNF-α-treated AhRfl/fl mice [Fig. 1DE, Supplemental video 2: AhRfl/fl, indoxyl sulfate (−) and Supplemental video 3: AhRfl/fl, indoxyl sulfate (+)]. In contrast, indoxyl sulfate failed to stimulate leukocyte - endothelial interactions in the eAhR KO animals [Fig. 1FG, Supplemental video 4: eAhR KO, indoxyl sulfate (−) and Supplemental video 5: eAhR KO, indoxyl sulfate (+)]. Additionally, the number of adhered leukocytes were quantified, as shown in Fig. 1H.
Table 1. Comparison of characteristics of AhRfl/fl mice, AhRfl/fl mice administered indoxyl sulfate, endothelial cell-specific AhR knockout (eAhR KO) mice, and eAhR KO mice receiving indoxyl sulfate.
| AhRfl/fl (n = 6) | AhRfl/fl + indoxyl sulfate (n = 6) | eAhR KO (n = 6) | eAhR KO + indoxyl sulfate (n = 6) | |
|---|---|---|---|---|
| Urea (mmol/L) | 10.2 ± 0.8 | 10.9 ± 0.8 | 9.7 ± 1.3 | 11.2 ± 0.9 |
| Total cholesterol (mmol/L) | 1.505 ± 0.089 | 1.384 ± 0.139 | 1.719 ± 0.142 | 1.719 ± 0.260 |
| HDL cholesterol (mmol/L) | 1.158 ± 0.064 | 1.058 ± 0.090 | 1.344 ± 0.178 | 1.293 ± 0.200 |
| Triglycerides (mmol/L) | 0.4 ± 0.1 | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.7 ± 0.1 |
| Indoxyl sulfate (mmol/L) | 0.005 ± 0.001 | 0.208 ± 0.041* | 0.005 ± 0.001 | 0.152 ± 0.086† |
Data are means ± SD.
p<0.01 vs. AhRfl/fl
p<0.05 vs. eAhR KO
Abbreviations: AhR, aryl hydrocarbon receptor; KO, knockout; HDL, high-density lipoprotein
Indoxyl Sulfate Induced AhR Transcriptional Activity in the Vascular Endothelium.
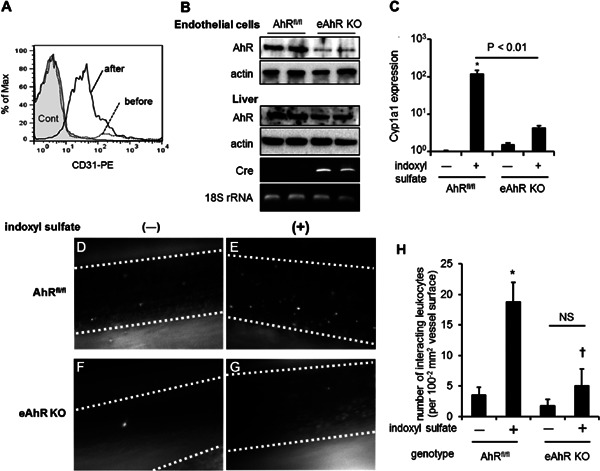
To elucidate the underlying mechanisms, we first examined whether indoxyl sulfate induced AhR transcriptional activation in HUVEC. Treatment with indoxyl sulfate led to the translocation of AhR from the cytosol to the nucleus (Fig. 2A). To confirm the direct involvement of AhR activation by indoxyl sulfate, we used an AhR-responsive luciferase reporter (XRE reporter vector). As shown in Fig. 2B, indoxyl sulfate induced XRE-dependent luciferase activity in transfected HUVEC in the same manner as AhR ligands TCDD and 3-metylchoranthlene (3MC), which were used as positive controls (Fig. 2B). We further confirmed that indoxyl sulfate significantly increased the expression of cyp1a1 and cyp1b1 mRNA, which are AhR targets, whereas a knockdown of AhR in HUVEC by transfection with siAhR inhibited their expression (Fig. 2C–E).
Fig. 2.
Indoxyl sulfate induced AhR transcriptional activity that enhanced TNF-α-induced leukocyte–endothelial interactions and cell adhesion molecule expression. (A) HUVEC were treated with 0.2 or 2.0 mmol/L indoxyl sulfate for 20 h. Cell lysates were fractionated into nuclear and cytosol protein and subjected to western blotting using the anti-AhR, lamin A/C, and tubulin antibodies. (B) HUVEC were transfected with XRE-pGL4.24 and pRL-TK vectors, followed by treatment with 0.2 or 2.0 mmol/L indoxyl sulfate (left graph) or the indicated concentration of an AhR agonist, such as 2,3,7,8-tetrachlorodibenzo-pdioxin (TCDD) or 3-metylchoranthlene (3MC) (right graph). The luciferase reporter assay was carried out as described in Methods. Relative promoter activity was measured based on firefly luciferase activity and then normalized with Renilla luciferase activity derived from pRL-TK experiments. The values are means from 3 experiments. *P < 0.05 vs. indoxyl sulfate (−), **P < 0.01 vs. indoxyl sulfate (−). (C–E) siAhR decreased indoxyl sulfate-induced mRNA expression of AhR target genes, cyp1a1 (D) and cyp1b1 (E). HUVEC were transfected with siAhR or siCont and treated with indoxyl sulfate for 24 h, followed by real-time quantitative PCR. The data are means ± SD (n = 3). The expression level of AhR in HUVEC was confirmed by western blotting (C).
AhR is Involved in Indoxyl Sulfate-Enhanced Leukocyte–Endothelial Interactions and E-Selectin Expression.
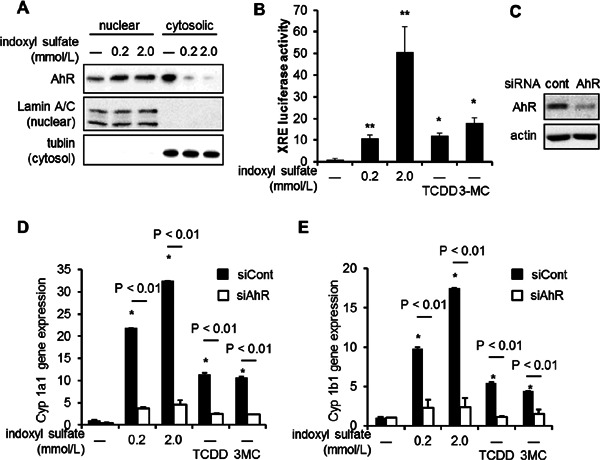
We evaluated the involvement of AhR in indoxyl sulfate-induced leukocyte–endothelial cell interactions under flow conditions by suppressing AhR expression. We found that treatment with indoxyl sulfate enhanced HL-60 adhesion to activated HUVEC. siAhR significantly reduced the indoxyl sulfate-enhanced leukocyte adhesion to HUVEC (Fig. 3A). As shown in Fig. 3BC, indoxyl sulfate-enhanced protein or mRNA expression of E-selectin was reduced by the knockdown of AhR. In contrast, minimal effects were observed for ICAM-1 and VCAM-1 expression. Furthermore, treatment with TCDD or 3MC, an AhR agonist, increased TNF-α-induced E-selectin expression in HUVEC, which was decreased by siAhR (Fig. 3DE).
Fig. 3.
siAhR suppressed indoxyl sulfate-enhanced vascular inflammation. (A-C) The results of the adhesion assay (A), western blotting detection of adhesion molecules (B) and mRNA expression of E-selectin (C) in HUVEC treated with 0.2 or 2.0 mmol/L indoxyl sulfate for 20 h, followed by tumor necrosis factor-α (TNF-α; 5.7 pmol/L). (D and E) HUVEC were transfected with siAhR or siCont and treated with 10 nmol/L 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (D) or 100 nmol/L 3-methylcholanthrene (3MC) (E) for 20 h, and then with TNF-α (5.7 pmol/L), followed by real-time quantitative PCR. (F). HUVEC were transfected with siAhR or siCont and treated with indoxyl sulfate for 20 h and then with TNF-α (5.7 pmol/L). mRNA expression of colony-stimulating factor 2 (CSF-2) by real-time quantitative PCR. Data are means ± SD (n = 3). *P < 0.05 vs. siCont and TNF-α (+) and without indoxyl sulfate; **P < 0.01 vs. siCont and TNF-α (+) and without indoxyl sulfate. Data shown are representative of 3 independent experiments. (G) Chromatin was prepared from HUVECs treated with indicated concentration of indoxyl sulfate, followed by stimulation with TNF-α for 30 min and the ChIP assay using anti-AhR antibody. Real-time PCR analysis of the ChIP samples was performed using primers designed to amplify the CSF-2 promoter region and ribosomal protein L30 exon3 as negative control. Values are expressed as means means ± SD (n = c3) of a representative of 2 independent experiments. *P < 0.05 vs. TNF-α (+) and without indoxyl sulfate.
Next, we examined the role of AhR in other endothelial genes induced by indoxyl sulfate. PCR array revealed that the mRNA expression of 12 out of 90 genes was increased more than 1.3-fold in HUVEC treated with indoxyl sulfate as compared to control (Table 2). Real-time quantitative PCR showed that colony-stimulating factor-2 (CSF-2) expression was also increased by indoxyl sulfate, which was inhibited by siAhR (Fig. 3F). ChIP analysis revealed that the CSF-2 promoter bound to AhR induced by indoxyl sulfate (Fig. 3G).
Table 2. Genes up-regulated by indoxyl sulfate.
| Gene | Relative gene expression |
|---|---|
| PTGS2 | 2.44 |
| EGR1 | 2.2 |
| CSF-2 | 1.76 |
| SELE | 1.56 |
| BIRC3 | 1.53 |
| IL4R | 1.52 |
| CXCL9 | 1.49 |
| VEGFA | 1.49 |
| IL1A | 1.43 |
| TANK | 1.39 |
| TCF7 | 1.31 |
| WNT2 | 1.31 |
Human umbilical vein endothelial cells were treated with or without 2 mmol/L indoxyl sulfate for 20 h, followed by TNF-α (5.7 pmol/L) and finally real-time quantitative PCR. Data are expressed relative to TNF-α-treated control only and genes up-regulated more than 1.3-fold higher by indoxyl sulfate are presented.
Abbreviations: PTGS2, prostaglandin-endoperoxide synthase 2; EGR1, early growth response protein 1; CSF-2, colony stimulating factor 2; SELE, E-selectin; BIRC3, Baculovirus inhibitors of apoptosis-repeat containing 3; IL4R, interleukin-4 receptor; CXCL9, CXC chemokine ligand 9; VEGFA, Vascular endothelial growth factor-A; IL1A, interleukin-1 alpha; TANK, TRAF family member-associated NFκB activator; TCF7, transcription factor 7; WNT2, Wingless-type MMTV integration site family member 2.
Indoxyl Sulfate-Induced ROS Production, JNK Phosphorylation, and NF-κB Activation were not Altered by siAhR.
We then evaluated the role of AhR in indoxyl sulfate-mediated ROS production23, 38, 39) by CM-H2DCFDA, which mainly reacts with free radicals such as hydrogen peroxide, hydroxyl radical, and peroxynitrite, while DHE is a probe for the detection of superoxide. Although the level of intracellular ROS detected by CM-H2DCFDA was increased by indoxyl sulfate in a dose-dependent manner, neither siAhR nor CH-223191, a chemical inhibitor of AhR, had any effect (Fig. 4AB). Similar results were obtained when ROS was detected by DHE (Fig. 4C). As reported previously18, 23), ROS detected by DHE was inhibited by apocynin, an inhibitor of nicotinamide adenine dinucleotide phosphate oxidase39, 40). To validate DHE measurement, we confirmed that apocynin suppressed oxidative stress caused by indoxyl sulfate (Fig. 4D).
Fig. 4.
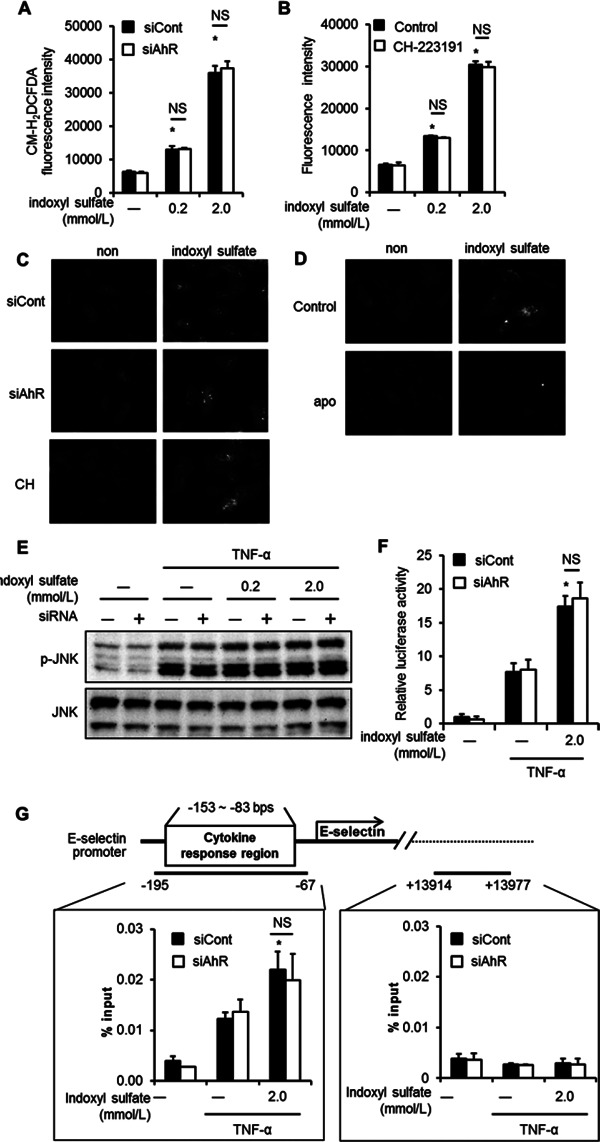
AhR does not mediate reactive oxygen species (ROS) production, JNK phosphorylation, and NF-κB activation. (A and B) Detection of ROS generation by CM-H2DCFDA. HUVEC were transfected with siCont or siAhR (A), or treated with CH-223191 (B); then, they were treated with CM-H2DCFDA for 30 min, followed by stimulation with 0.2 or 2.0 mmol/L indoxyl sulfate for 5 h. (C and D) Detection of ROS generation by dihydroethidium (DHE). HUVEC were treated with siCont, siAhR, CH-223191 (C), or apocynin (D); then, they were stimulated with 2.0 mmol/L indoxyl sulfate, followed by treatment with DHE for 30 min. (E) Detection of phosphorylated (p)-JNK by western blotting. HUVEC were incubated in the presence of the indicated concentrations of indoxyl sulfate for 20 h and then stimulated with TNF-α (5.7 pmol/L) for 15 min before further assays. Quantities of phosphorylated and total JNK were determined by immunoblotting. (F) The NF-κB luciferase reporter assay was carried out as described in Experimental Procedures. The values are means from 3 independent experiments. *P < 0.01 vs. siCont and TNF-α (+) and without indoxyl sulfate. (G) Schema of the E-selectin gene and location of the primers used in the ChIP assay. Chromatin was prepared from HUVEC transfected with siCont or siAhR, followed by treatment with (+) or without (−) 2.0 mmol/L indoxyl sulfate for 20 h, and then stimulation by TNF-α (5.7 pmol/L) for 30 min. ChIP assay was performed using the anti-NF-κB antibody. Real-time PCR analysis of the ChIP samples was performed using primers designed to amplify the E-selectin promoter region (−195 to −67 bps), which contains the cytokine response region and the E-selectin extreme 3′ region (+13914 to +13977 bps). *P < 0.05 vs siCont, TNF (+), indoxyl sulfate (−). Values are expressed as means ± SD (n = 3) of a representative of 2 independent experiments. *P < 0.05 vs. TNF-α (+) and without indoxyl sulfate.
We also examined the contribution of JNK, a member of the mitogen-activated protein kinase family, which is closely associated with E-selectin expression23). Western blotting analyses revealed that indoxyl sulfate enhanced JNK phosphorylation in the presence of TNF-α; however, treatment with siAhR did not change the phosphorylation level (Fig. 4E). The luciferase reporter assay revealed that the activation of NF-κB by indoxyl sulfate was not affected by siAhR (Fig. 4F).
To further elucidate the role of AhR in nuclear factor-κB (NF-κB) activation associated with E-selectin expression, we performed ChIP analysis of the E-selectin promoter region containing cytokine response region34). This region is located within the first 160 bp upstream of the transcriptional start site of the E-selectin gene and contains binding sites for NF-κB and ATF-2/c-jun41–43). ChIP analysis showed that NF-κB bound to the E-selectin promoter after TNF-α stimulation and indoxyl sulfate enhanced the activation; however, siAhR did not affect the enhancement (Fig. 4G, left panel). TNF-α-induced NF-κB activation was not detected at the extreme 3′ region of the E-selectin gene (Fig. 4G, right panel).
AP-1 but not Cyclic AMP Response Element (CRE) is Involved in Indoxyl Sulfate-Mediated AhR Activation of E-Selectin Transcription.
Previous reports have demonstrated that NF-ELAM-1, positioned at base pairs −153 to −146 from start point of the E-selectin transcription was essential for its expression44). Therefore, we examined the effect of indoxyl sulfate on transcriptional activation of NF-ELAM-1. We confirmed that indoxyl sulfate activated the NF-ELAM-1 site (Fig. 5A).
Fig. 5.
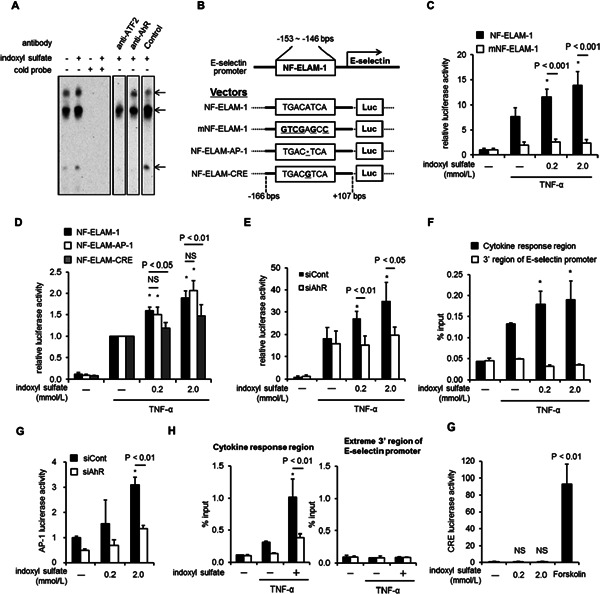
Activator protein-1 (AP-1) and not cyclic AMP response element (CRE) is involved in indoxyl sulfate-mediated E-selectin expression. (A) Electrophoresis mobility shift assay using the nuclear factor-endothelial adhesion molecule 1 (NF-ELAM-1) probe. HUVEC were treated with 2 mmol/L indoxyl sulfate for 20 h, followed by stimulation with tumor necrosis factor-α (5.7 pmol/L) for 2 h. The nuclear extracts, biotin-labeled probe, cold probe, and antibodies were incubated and subjected to electrophoresis, followed by chemiluminescence detection. Arrowheads indicate putative complexes between the transcription factors and the probe. (B) Summary of the E-selectin promoter region and the corresponding luciferase vector. The detailed sequences of the NF-ELAM-1 site, NF-ELAM-1 site inactivated by mutagenesis (mNF-ELAM-1), and NF-ELAM-1 site carrying an AP-1 binding site (NF-ELAM-AP-1) or CRE (NF-ELAM-CRE) are presented in the boxes. (C and D) Luciferase assays were carried out using the NF-ELAM-1 vector and the mNF-ELAM-1 vector (C) or NF-ELAMAP-1 and NF-ELAM-CRE (D). The values are means from 4 experiments. *P < 0.01 vs. NF-ELAM-1 without indoxyl sulfate. (E) The results of the luciferase assay using NF-ELAM-1-AP-1 transfected with siAhR or siCont. The values are means from 3 experiments. *P < 0.01 vs. siCont without indoxyl sulfate. (F) Chromatin was extracted from HUVEC treated with (+) 2.0 mmol/L of indoxyl sulfate or without (−) for 20 h, followed by stimulation with TNF-α (5.7 pmol/L) for 30 min, and the ChIP assay was performed using anti-AhR antibody. Real-time PCR analysis of the ChIP samples was performed using primers designed to amplify the E-selectin promoter region (−195 to −67 bps), which contains the cytokine response region and the E-selectin extreme 3′ region (+13914 to +13977 bps). *P < 0.05 vs TNF-α (+), indoxyl sulfate (−). Values are expressed as means ± SD (n = 3) of a representative of 2 independent experiments. *P < 0.05 vs. TNF-α (+) and without indoxyl sulfate. (G) The AP-1 luciferase reporter assay was performed after transfection with siAhR or siCont. The values are means from 3 experiments. *P < 0.05 vs. siCont without indoxyl sulfate. (H) Chromatin was extracted from HUVEC transfected with siCont or siAhR, followed by treatment with (−) 2.0 mmol/L indoxyl sulfate or without (+) for 20 h, followed by stimulation with TNF-α (5.7 pmol/L) for 30 min. ChIP assay was performed using the anti-ATF-2 antibody. Real-time PCR analysis of the ChIP samples was performed using primers designed to amplify the E-selectin promoter region containing cytokine response region and the E-selectin extreme 3′ region. *P < 0.01 vs. siCont, TNF (+), indoxyl sulfate (−). Values are expressed as means ± SD (n = 3) of a representative of two independent experiments. *P < 0.05 vs. TNF-α (+) and without indoxyl sulfate. (I) The CRE luciferase reporter assay was carried out with indoxyl sulfate or Forskolin, a CRE activator. The values are means from 3 independent experiments.
Fig. 5B illustrates the promoter region of E-selectin and its luciferase vector. The luciferase assay using functional NF-ELAM-1 and mNF-ELAM-1 revealed that the NF-ELAM-1 site was necessary for transcription of E-selectin by both TNF-α and indoxyl sulfate (Fig. 5C). We also produced mutant NF-ELAM-1 constructs carrying AP-1 and CRE sequences. The AP-1 construct and not the CRE construct was responsive to indoxyl sulfate treatment (Fig. 5D). In addition, we found that indoxyl sulfate-activated NF-ELAM-AP1 was suppressed by siAhR (Fig. 5E). ChIP analysis showed that AhR bound to E-selectin promoter after indoxyl sulfate stimulation (Fig. 5F).
Treatment with indoxyl sulfate without TNF-α induced AP-1 transcriptional activity, while siAhR caused a decrease (Fig. 5G). We confirmed that ATF-2, a component of AP-1, directly bound to cytokine response region of E-selectin and AhR modulated their transcriptional activity (Fig. 5H). On the other hand, indoxyl sulfate failed to induce CRE-related transcription (Fig. 5I).
Discussion
In this study, we demonstrated a pivotal role of endothelial AhR in indoxyl sulfate-enhanced vascular endothelial inflammation via AP-1 dependent transcriptional regulation. AP-1 plays an important role in endothelial inflammation by activating the expression of proinflammatory cytokines and other proteins relevant to vascular pathology, such as selectins and monocyte chemoattractant protein-145, 46). Several studies have demonstrated that the NF-ELAM-1 site activated by proinflammatory cytokines is recognized by AP-1 and not by the CRE-binding protein43, 47). We further confirmed that not only ATF-2 but also AhR associated with the NF-ELAM-1 site by electrophoresis mobility shift assay and ChIP assay (Fig. 5A, F, H), suggesting that the NF-ELAM-1 site activated by indoxyl sulfate was required for AP-1 and AhR. Moreover, we found that although indoxyl sulfate induced AP-1 activation via an AhR-dependent pathway, it did not activate mRNA expression of AP-1 components such as ATFa, ATF-2, ATF-3, c-jun, junB, and junD. Furthermore, immunoprecipitation experiments failed to detect physical binding between AhR and ATF-2 or c-jun, major components of AP-1 (data not shown). Thus, additional regulatory element(s) may be present and mediate AhR-dependent activation of AP-1.
Surprisingly, indoxyl sulfate-induced AhR activation did not involve oxidative stress or NF-κB but rather involved AP-1-dependent transcriptional activation to induce E-selectin in the vascular endothelium. Although previous reports have suggested that AhR mediates ROS generation in other tissues such as hepatocytes48), we failed to detect the involvement of AhR in indoxyl sulfate-triggered oxidative stress in the vascular endothelium. We obtained similar findings using 3MC, another AhR ligand, in the endothelial model (data not shown). These observations suggest that AhR-dependent signaling pathway activated by indoxyl sulfate is independent of oxidative stress, at least in the vascular endothelium. This observation is in agreement with previous reports where AhR is shown to not be associated with indoxyl sulfate-induced activation of NF-κB and phosphorylation of JNK because oxidative stress plays an important role in these processes49, 50).
Fig. 6 summarizes the effect of indoxyl sulfate on vascular inflammation based on the current findings. Indoxyl sulfate induces ROS production, which leads to TNF-α-triggered JNK and NF-κB activation. Indoxyl sulfate-induced AhR activation, on the other hand, stimulates AP-1 transcriptional activity. AP-1 activation and signal transduction via the oxidative stress pathway synergistically up-regulate E-selectin expression, leading to the aggravation of leukocyte–endothelial interactions. CSF-2 is also known to be downstream of AP-151), suggesting that the indoxyl sulfate–AhR pathway is operative in broad endothelial activation. Several studies have also demonstrated that AhR mediates indoxyl sulfate-induced expression of inflammatory genes such as IL-6, tissue factor, and monocyte chemoattractant protein-152–54).
Fig. 6.
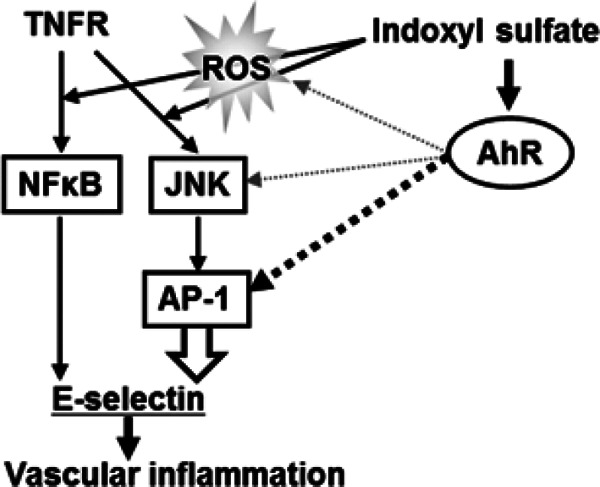
A model of the mechanism through which AhR mediates indoxyl sulfate-enhanced E-selectin expression via an activator protein-1 (AP-1)-dependent pathway. Indoxyl sulfate induces ROS production, which leads to TNF-α-triggered JNK and NF-κB activation. Indoxyl sulfate-induced AhR stimulates AP-1 transcriptional activity, but it is not associated with ROS production and JNK or NF-κB activation. AP-1 transcriptionally up-regulates E-selectin expression, followed by an increase in leukocyte-endothelial interactions.
E-selectin has been shown to play a central role in indoxyl sulfate- and kidney disease-related vascular inflammation. In fact, circulating E-selectin is a strong predictor of death and cardiovascular events in patients with end-stage renal disease55, 56). We also reported previously that blockade of E-selectin by neutralizing antibody dramatically reduced indoxyl sulfate-enhanced leukocyte adhesion to vascular endothelium in vivo and in vitro23). Further, an oral adsorbent, AST-120, reduces circulating and tissue indoxyl sulfate, which suppresses the adhesion of monocytes and inhibits plaque formation in apolipoprotein E-deficient mice, with reduced renal function57, 58). IVM analysis suggested that AhR plays a crucial role in induction of leukocyte recruitment by indoxyl sulfate. TCDD has been shown to enhance the expression of adhesion molecules such as P-selectin as well as the migration of macrophages into the plaque in apolipoprotein E-deficient mice26). These results suggest that indoxyl sulfate-induced leukocyte adhesion via AhR plays a role during the development of atherosclerosis in patients with renal dysfunction. This further suggests that indoxyl sulfate–AhR pathway can be considered as a new therapeutic target for vascular inflammation in patients with renal dysfunction. Indeed, AST-120, which decreases serum level of indoxyl sulfate, has been shown to reduce arterial stiffness and wall thickening in patients with chronic kidney disease59).
To mimic the clinical situation of patients with chronic kidney disease and those requiring dialysis, we administered TNF-α to mice in this study because the serum concentration of proinflammatory cytokines such as TNF-α and IL-1β is increased in patients with renal disease19). The serum concentration of indoxyl sulfate in these mice (approximately 0.2 mmol/L) was equivalent to that in dialysis patients35, 60). Furthermore, stimulation with 0.02 mmol/L indoxyl sulfate, which is comparable to that in patients at Stage 4 to 5 of chronic kidney disease12, 61), enhanced the expression of E-selectin in HUVEC (data not shown). Thus, our findings indicate an important molecular background underlying the inflammatory process in chronic kidney disease including dialysis with high circulating proinflammatory cytokines and uremic toxins. Therefore, our findings are relevant to the mechanisms of cardiovascular disease in patients with renal disease. The potential role of the indoxyl sulfate– AhR axis in atherosclerosis will be investigated in our future studies.
In conclusion, AhR mediates indoxyl sulfate-enhanced leukocyte–endothelial interactions through AP-1 transcriptional activity. These results provide a novel mechanistic insight into the vascular inflammation in patients with kidney disease.
Supplementary Material
Acknowledgments
We thank Kaori Kikuchi for measurement of indoxyl sulfate and Michiyo Deushi for the technical support with the endothelial cells cultures. We also thank Katsuhiko Hamada, Sumie Goto and Satoshi Yamashita for the excellent assistance with DNA work including ChIP assay and Naoki Sawada for providing mice.
Sources of Funding
This study was supported in part by a JSPS Grand-in-Aid for Scientific Research (C) (No. 13210388). This work was also supported in part by a grant from Kureha Corporation.
Conflict of Interest
S.I., T. E., and Y.I. are employees of Kureha Corporation. M.O. and M.Y. have no conflict of interest associated with this manuscript.
Footnotes
The abbreviations used are: 3MC, 3-metylchoranthlene; AhR, aryl hydrocarbon receptor; AP-1, activator protein-1; ATF, activating transcription factor; ChIP, chromatin immunoprecipitation; CM-H2DCFDA, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester; CRE, cyclic AMP response element; CSF-2, colony-stimulating factor-2; Cyp, cytochrome p450; DHE, dihydroethidium; eAhR KO, endothelial cell-specific AhR knockout; F, forward; HUVEC, human umbilical vein endothelial cells; ICAM-1, intercellular cell adhesion molecule 1; IVM, intravital microscopy; NF-ELAM-1, nuclear factorendothelial adhesion molecule-1; NF-ELAM-AP-1, vector with the NF-ELAM-1 site carrying an AP-1binding site; NF-ELAM-CRE, vector with the NF-ELAM-1 site carrying CRE; NF-κB, nuclear factor-γB; mNF-ELAM-1, non-functional mutated NF-ELAM-1 vector; PCR, polymerase chain reaction; R, reverse; ROS, reactive oxygen species; RPL-30, ribosomal protein L-30; siAhR, siRNA specific to AhR; TCDD, 2,3,7,8-Tetrachlorodibenzo-p-dioxin VCAM-1, vascular cell adhesion molecule 1; XRE, xenobiotic response element.
References
- 1). Foley RN, Parfrey PS, Sarnak MJ: Epidemiology of cardiovascular disease in chronic renal disease. J Am Soc Nephrol, 1998; 9: S16-23 [PubMed] [Google Scholar]
- 2). Stack AG, Bloembergen WE: Prevalence and clinical correlates of coronary artery disease among new dialysis patients in the United States: a cross-sectional study. J Am Soc Nephrol, 2001; 12: 1516-1523 [DOI] [PubMed] [Google Scholar]
- 3). Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY: Chronic kidney disease and the risks of death, cardio vascular events, and hospitalization. N Engl J Med, 2004; 351: 1296-1305 [DOI] [PubMed] [Google Scholar]
- 4). Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH: Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med, 2004; 164: 659-663 [DOI] [PubMed] [Google Scholar]
- 5). Clementi A, Virzi GM, Goh CY, Cruz DN, Granata A, Vescovo G, Ronco C: Cardiorenal syndrome type 4: a review. Cardiorenal Med, 2013; 3: 63-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Ronco C, Chionh CY, Haapio M, Anavekar NS, House A, Bellomo R: The cardiorenal syndrome. Blood Purif, 2009; 27: 114-126 [DOI] [PubMed] [Google Scholar]
- 7). Ronco C, Haapio M, House AA, Anavekar N, Bellomo R: Cardiorenal syndrome. J Am Coll Cardiol, 2008; 52: 1527-1539 [DOI] [PubMed] [Google Scholar]
- 8). Ross R: Atherosclerosis--an inflammatory disease. N Engl J Med, 1999; 340: 115-126 [DOI] [PubMed] [Google Scholar]
- 9). Vanholder R, De Smet R: Pathophysiologic effects of uremic retention solutes. J Am Soc Nephrol, 1999; 10: 1815-1823 [DOI] [PubMed] [Google Scholar]
- 10). Niwa T, Nomura T, Sugiyama S, Miyazaki T, Tsukushi S, Tsutsui S: The protein metabolite hypothesis, a model for the progression of renal failure: an oral adsorbent lowers indoxyl sulfate levels in undialyzed uremic patients. Kidney Int Suppl, 1997; 62: S23-28 [PubMed] [Google Scholar]
- 11). Niwa T, Ise M: Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med, 1994; 124: 96-104 [PubMed] [Google Scholar]
- 12). Barreto FC, Barreto DV, Liabeuf S, Meert N, Glorieux G, Temmar M, Choukroun G, Vanholder R, Massy ZA: Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol, 2009; 4: 1551-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Itoh Y, Ezawa A, Kikuchi K, Tsuruta Y, Niwa T: Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal Bioanal Chem, 2012; 403: 1841-1850 [DOI] [PubMed] [Google Scholar]
- 14). Ito S, Yoshida M: Protein-bound uremic toxins: new culprits of cardiovascular events in chronic kidney disease patients. Toxins (Basel), 2014; 6: 665-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Lekawanvijit S, Adrahtas A, Kelly DJ, Kompa AR, Wang BH, Krum H: Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J, 2010; 31: 1771-1779 [DOI] [PubMed] [Google Scholar]
- 16). Lekawanvijit S, Kompa AR, Wang BH, Kelly DJ, Krum H: Cardiorenal syndrome: the emerging role of protein-bound uremic toxins. Circ Res, 2012; 111: 1470-1483 [DOI] [PubMed] [Google Scholar]
- 17). Shimizu H, Hirose Y, Nishijima F, Tsubakihara Y, Miyazaki H: ROS and PDGF-beta [corrected] receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am J Physiol Cell Physiol, 2009; 297: C389-396 [DOI] [PubMed] [Google Scholar]
- 18). Ito S, Higuchi Y, Yagi Y, Nishijima F, Yamato H, Ishii H, Osaka M, Yoshida M: Reduction of indoxyl sulfate by AST-120 attenuates monocyte inflammation related to chronic kidney disease. J Leukoc Biol, 2013; 93: 837-845 [DOI] [PubMed] [Google Scholar]
- 19). Adesso S, Popolo A, Bianco G, Sorrentino R, Pinto A, Autore G, Marzocco S: The Uremic Toxin Indoxyl Sulphate Enhances Macrophage Response to LPS. PLoS One, 2013; 8: e76778-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20). Lekawanvijit S: Role of Gut-Derived Protein-Bound Uremic Toxins in Cardiorenal Syndrome and Potential Treatment Modalities. Circ J, 2015; 79; 2088-2097 [DOI] [PubMed] [Google Scholar]
- 21). Koizumi M, Tatebe J, Watanabe I, Yamazaki J, Ikeda T, Morita T: Aryl hydrocarbon receptor mediates indoxyl sulfate-induced cellular senescence in human umbilical vein endothelial cells. J Atheroscler Thromb, 2014; 21: 904-916 [DOI] [PubMed] [Google Scholar]
- 22). Shimizu H, Bolati D, Adijiang A, Muteliefu G, Enomoto A, Nishijima F, Dateki M, Niwa T: NF-kappaB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am J Physiol Cell Physiol, 2011; 301: C1201-1212 [DOI] [PubMed] [Google Scholar]
- 23). Ito S, Osaka M, Higuchi Y, Nishijima F, Ishii H, Yoshida M: Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J Biol Chem, 2010; 285: 38869-38875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Schroeder JC, Dinatale BC, Murray IA, Flaveny CA, Liu Q, Laurenzana EM, Lin JM, Strom SC, Omiecinski CJ, Amin S, Perdew GH: The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry, 2010; 49: 393-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Haarmann-Stemmann T, Bothe H, Abel J: Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol, 2009; 77: 508-520 [DOI] [PubMed] [Google Scholar]
- 26). Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, Van Winkle L, Matsumura F, Vogel CF: Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arterioscler Thromb Vasc Biol, 2011; 31: 1260-1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Sawada N, Salomone S, Kim HH, Kwiatkowski DJ, Liao JK: Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res, 2008; 103: 360-368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA: Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med, 2001; 193: 741-754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29). Hagita S, Osaka M, Shimokado K, Yoshida M: Oxidative stress in mononuclear cells plays a dominant role in their adhesion to mouse femoral artery after injury. Hypertension, 2008; 51: 797-802 [DOI] [PubMed] [Google Scholar]
- 30). Hagita S, Osaka M, Shimokado K, Yoshida M: Adipose inflammation initiates recruitment of leukocytes to mouse femoral artery: role of adipo-vascular axis in chronic inflammation. PLoS One, 2011; 6: e19871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31). Hagita S, Osaka M, Shimokado K, Yoshida M: Combination of amlodipine and atorvastatin synergistically reduces leukocyte recruitment to mechanically injured mouse femoral artery. Hypertens Res, 2011; 34: 450-455 [DOI] [PubMed] [Google Scholar]
- 32). Mizuko Osaka SH, Masayuki Yoshida: In Vivo Imaging of Leukocyte Recruitment to the Atheroprone Femoral Artery Reveals Anti-Inflammatory Effect of Rosuvastatin. BioMed Research International, 2012; 2013: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Osaka M, Hagita S, Haraguchi M, Kajimura M, Suematsu M, Yoshida M: Real-time imaging of mechanically injured femoral artery in mice reveals a biphasic pattern of leukocyte accumulation. Am J Physiol Heart Circ Physiol, 2007; 292: H1876-1882 [DOI] [PubMed] [Google Scholar]
- 34). Hamada K, Osaka M, Yoshida M: Cell density impacts epigenetic regulation of cytokine-induced E-selectin gene expression in vascular endothelium. PLoS One, 2014; 9: e90502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Kikuchi K, Itoh Y, Tateoka R, Ezawa A, Murakami K, Niwa T: Metabolomic search for uremic toxins as indicators of the effect of an oral sorbent AST-120 by liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 2010; 878: 2997-3002 [DOI] [PubMed] [Google Scholar]
- 36). Yamashita S, Miyaki S, Kato Y, Yokoyama S, Sato T, Barrionuevo F, Akiyama H, Scherer G, Takada S, Asahara H: L-Sox5 and Sox6 proteins enhance chondrogenic miR140 microRNA expression by strengthening dimeric Sox9 activity. J Biol Chem, 2012; 287: 22206-22215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Yamashita S, Andoh M, Ueno-Kudoh H, Sato T, Miyaki S, Asahara H: Sox9 directly promotes Bapx1 gene expression to repress Runx2 in chondrocytes. Exp Cell Res, 2009; 315: 2231-2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38). Adelibieke Y, Shimizu H, Muteliefu G, Bolati D, Niwa T: Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J Ren Nutr, 2012; 22: 86-89 [DOI] [PubMed] [Google Scholar]
- 39). Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F, Brunet P: The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost, 2007; 5: 1302-1308 [DOI] [PubMed] [Google Scholar]
- 40). Shimoishi K, Anraku M, Kitamura K, Tasaki Y, Taguchi K, Hashimoto M, Fukunaga E, Maruyama T, Otagiri M: An oral adsorbent, AST-120 protects against the progression of oxidative stress by reducing the accumulation of indoxyl sulfate in the systemic circulation in renal failure. Pharm Res, 2007; 24: 1283-1289 [DOI] [PubMed] [Google Scholar]
- 41). Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T: Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J, 1995; 9: 899-909 [PubMed] [Google Scholar]
- 42). Whitley MZ, Thanos D, Read MA, Maniatis T, Collins T: A striking similarity in the organization of the E-selectin and beta interferon gene promoters. Mol Cell Biol, 1994; 14: 6464-6475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Kaszubska W, Hooft van Huijsduijnen R, Ghersa P, DeRaemy-Schenk AM, Chen BP, Hai T, DeLamarter JF, Whelan J: Cyclic AMP-independent ATF family members interact with NF-kappa B and function in the activation of the E-selectin promoter in response to cytokines. Mol Cell Biol, 1993; 13: 7180-7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Montgomery KF, Osborn L, Hession C, Tizard R, Goff D, Vassallo C, Tarr PI, Bomsztyk K, Lobb R, Harlan JM, et al. Activation of endothelial-leukocyte adhesion molecule 1 (ELAM-1) gene transcription. Proc Natl Acad Sci U S A, 1991; 88: 6523-6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). Schleicher E, Friess U: Oxidative stress, AGE, and atherosclerosis. Kidney Int Suppl, 2007; S17-26 [DOI] [PubMed] [Google Scholar]
- 46). De Caterina R, Massaro M, Scoditti E, Annunziata Carluccio M: Pharmacological modulation of vascular inflammation in atherothrombosis. Ann N Y Acad Sci, 2010; 1207: 23-31 [DOI] [PubMed] [Google Scholar]
- 47). Brostjan C, Anrather J, Csizmadia V, Natarajan G, Winkler H: Glucocorticoids inhibit E-selectin expression by targeting NF-kappaB and not ATF/c-Jun. J Immunol, 1997; 158: 3836-3844 [PubMed] [Google Scholar]
- 48). Joshi AD, Carter DE, Harper TA, Jr, Elferink CJ: Aryl hydrocarbon receptor-dependent stanniocalcin 2 induction by cinnabarinic acid provides cytoprotection against endoplasmic reticulum and oxidative stress. J Pharmacol Exp Ther, 2015; 353: 201-212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49). Martindale JL, Holbrook NJ: Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol, 2002; 192: 1-15 [DOI] [PubMed] [Google Scholar]
- 50). Wang X, Martindale JL, Liu Y, Holbrook NJ: The cellular response to oxidative stress: influences of mitogenactivated protein kinase signalling pathways on cell survival. Biochem J, 1998; 333 (Pt 2): 291-300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51). Munoz C, Pascual-Salcedo D, Castellanos MC, Alfranca A, Aragones J, Vara A, Redondo JM, de Landazuri MO: Pyrrolidine dithiocarbamate inhibits the production of interleukin-6, interleukin-8, and granulocyte-macrophage colony-stimulating factor by human endothelial cells in response to inflammatory mediators: modulation of NF-kappa B and AP-1 transcription factors activity. Blood, 1996; 88: 3482-3490 [PubMed] [Google Scholar]
- 52). Adelibieke Y, Yisireyili M, Ng HY, Saito S, Nishijima F, Niwa T: Indoxyl sulfate induces IL-6 expression in vascular endothelial and smooth muscle cells through OAT3-mediated uptake and activation of AhR/NF-kappaB pathway. Nephron Exp Nephrol, 2014; 128: 1-8 [DOI] [PubMed] [Google Scholar]
- 53). Gondouin B, Cerini C, Dou L, Sallee M, Duval-Sabatier A, Pletinck A, Calaf R, Lacroix R, Jourde-Chiche N, Poitevin S, Arnaud L, Vanholder R, Brunet P, Dignat-George F, Burtey S: Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int, 2013; 84: 733-744 [DOI] [PubMed] [Google Scholar]
- 54). Watanabe I, Tatebe J, Namba S, Koizumi M, Yamazaki J, Morita T: Activation of aryl hydrocarbon receptor mediates indoxyl sulfate-induced monocyte chemoattractant protein-1 expression in human umbilical vein endo thelial cells. Circ J, 2013; 77: 224-230 [DOI] [PubMed] [Google Scholar]
- 55). Malatino LS, Stancanelli B, Cataliotti A, Bellanuova I, Fatuzzo P, Rapisarda F, Leonardis D, Tripepi G, Mallamaci F, Zoccali C: Circulating E-selectin as a risk marker in patients with end-stage renal disease. J Intern Med, 2007; 262: 479-487 [DOI] [PubMed] [Google Scholar]
- 56). Testa A, Benedetto FA, Spoto B, Pisano A, Tripepi G, Mallamaci F, Malatino LS, Zoccali C: The E-selectin gene polymorphism and carotid atherosclerosis in end-stage renal disease. Nephrol Dial Transplant, 2006; 21: 1921-1926 [DOI] [PubMed] [Google Scholar]
- 57). Inami Y, Hamada C, Seto T, Hotta Y, Aruga S, Inuma J, Azuma K, Io H, Kaneko K, Watada H, Tomino Y: Effect of AST-120 on Endothelial Dysfunction in Adenine-Induced Uremic Rats. Int J Nephrol, 2014; 2014: 164125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58). Yamamoto S, Zuo Y, Ma J, Yancey PG, Hunley TE, Motojima M, Fogo AB, Linton MF, Fazio S, Ichikawa I, Kon V: Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol Dial Transplant, 2011; 26: 2491-2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59). Nakamura T, Kawagoe Y, Matsuda T, Ueda Y, Shimada N, Ebihara I, Koide H: Oral ADSORBENT AST-120 decreases carotid intima-media thickness and arterial stiffness in patients with chronic renal failure. Kidney Blood Press Res, 2004; 27: 121-126 [DOI] [PubMed] [Google Scholar]
- 60). Niwa T, Tsukushi S, Ise M, Miyazaki T, Tsubakihara Y, Owada A, Shiigai T: Indoxyl sulfate and progression of renal failure: effects of a low-protein diet and oral sorbent on indoxyl sulfate production in uremic rats and undialyzed uremic patients. Miner Electrolyte Metab, 1997; 23: 179-184 [PubMed] [Google Scholar]
- 61). Huang ST, Shu KH, Cheng CH, Wu MJ, Yu TM, Chuang YW, Chen CH: Serum total p-cresol and indoxyl sulfate correlated with stage of chronic kidney disease in renal transplant recipients. Transplant Proc, 2012; 44: 621-624 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.