

A dual drug–loaded, pH-responsive polymersome can induce endoplasmic reticulum (ER) stress and autophagy blockade in cancer cells.
Abstract
Autophagy is involved in the occurrence and development of tumors. Here, a pH-responsive polymersome codelivering hydroxychloroquine (HCQ) and tunicamycin (Tuni) drugs is developed to simultaneously induce endoplasmic reticulum (ER) stress and autophagic flux blockade for achieving an antitumor effect and inhibiting tumor metastasis. The pH response of poly(β-amino ester) and HCQ synergistically deacidifies the lysosomes, thereby blocking the fusion of autophagosomes and lysosomes and lastly blocking autophagic flux. The function mechanism of regulating autophagy was systematically investigated on orthotopic luciferase gene–transfected, 4T1 tumor–bearing BALB/c mice through Western blot and immunohistochemistry analyses. The Tuni triggers ER stress to regulate the PERK/Akt signaling pathway to increase the autophagic level. The “autophagic stress” generated by triggering ER stress–induced autophagy and blocking autophagic flux is effective against tumors. The reduced expression of matrix metalloproteinase-2 due to ER stress and reduced focal adhesions turnover due to the blockade of autophagic flux synergistically inhibit tumor metastasis.
INTRODUCTION
The important role of autophagy in health and disease has received unprecedented attention (1). As an essential and conservative physiological catabolic process, autophagy is responsible for the removal of protein aggregates, damaged organelles, and foreign bodies that invade cells (2). The autophagy contents are sequestered by double-membraned compartments (autophagosomes). Subsequently, the autophagosomes are fused with lysosomes to form autolysosomes, which degrade and circulate to produce nutrients (amino acids, fatty acids, and nucleotides) to be supplied to cells, and this dynamic process is called autophagic flux (3, 4). The unhindered autophagic flux is of great notable for maintaining homeostasis and protecting cells from attacks (5). The autophagic level is characterized by the amount of autophagy markers (e.g., autophagosomes and autolysosomes) and autophagy protein markers [e.g., LC3 (microtubule-associated protein-1 light chain-3)]. The upstream initiation or downstream blocking of autophagic flux will lead to the increase in autophagy markers (6). Malignant tumors are at a relatively high autophagic level compared with normal tissues to satisfy their metabolic demands, evasion, and resistance and allow tumor growth, survival, and malignancy (7). The involvement of autophagy in the occurrence and development of tumors suggests the reliable prospect of autophagy manipulation as an interventional means for tumor therapy (8). Autophagic flux blockade can disrupt the metabolism cycle of cancer cells, thereby reducing their fitness (9). Hydroxychloroquine (HCQ) and chloroquine (CQ) are the only clinically available autophagic blocking agents and they have been proven to be effective adjuvants for chemotherapeutics to increase their antitumor effects (10). HCQ/CQ, as a lysosomal alkalizing agent, can diffuse into lysosomes, causing the lysosomal pH to rise and dysfunction so that the lysosomes no longer fuse with the autophagosome, thereby blocking autophagic flux (11). However, the use of HCQ/CQ as a monotherapy strategy to block autophagic flux displays limited antitumor activity in clinical treatment (12). However, when HCQ is combined with autophagic stimulus, it can significantly increase its antitumor effect and reduce its dosage (13).
Aberrant endoplasmic reticulum (ER) status triggers autophagy stimulation (14). The ER acts as a reservoir of calcium ions in cells and is responsible for the correct folding and secretion of proteins (15). The accumulation and aggregation of misfolded proteins cause ER stress, triggering unfolded protein response (UPR) to significantly increase the autophagic level to restore homeostasis (16). As an ER stress initiator, tunicamycin (Tuni) blocks N-glycosylation and causes ER stress–induced autophagy to increase the autophagic level through PERK [protein kinase RNA–like ER kinase]/Akt (protein kinase B)/mTOR (mammalian target of rapamycin) signaling pathways, which are also closely related to matrix metalloproteinase-2 (MMP-2) expression (17).
Therefore, the combined application of the autophagic flux blocker HCQ and the ER stress initiator Tuni could cause cancer cells to have a special “autophagic stress,” which can severely disrupt cell homeostasis and cause cell death, resulting in a better therapeutic effect for tumor treatment. However, for a systemic administration of HCQ and Tuni, challenges such as poor hydrophilicity, poor biodistribution profile, low tumor accumulation, and tumor acid microenvironment prevent the drugs from penetrating the cell membranes, further affecting the application of free drugs in vivo (18). Moreover, high-dose HCQ in clinical application can only produce a moderate blockage of autophagic flux due to its poor efficacy, thus producing capricious therapeutic effects (19). Thus, the drug nanocarrier is considered to solve the above problems (20), as it has the following features: co-encapsulating hydrophilic and hydrophobic drugs, accumulating in tumor tissues through an enhanced permeability and retention (EPR) effect (21), entering the cells through the lysosomal pathway, and stimulating drug release with environment-responsive signals (22).
In addition, metastatic tumors undergo local migration and invasion in the early stage of tumor metastasis, and the migration speed depends on the focal adhesions (FAs) turnover (23). FAs are transmembrane multiprotein complexes containing integrins, paxillin, talin, zyxin, etc. Both cell-cell and cell–extracellular matrix (ECM) adhesions form a stable connection via FAs (24). The decomposition of FAs at the cell rear by autophagy is critical for the forward movement and successful migration/invasion of tumor cells (25). In addition, MMPs are also an important factor in the selective regulation of tumor microenvironment to promote tumor metastasis, and are considered to be an inducer of epithelial-mesenchymal transition (26). The ER stress induced by Tuni regulates the PERK/Akt signaling pathway and down-regulates the expression of MMP-2 (27). Both the inhibition of the FAs turnover and the down-regulation of the expression of MMP-2 will reduce tumor metastasis.
Accordingly, in this study, we develop a pH-responsive polymersome for codelivering HCQ and Tuni drugs to simultaneously induce ER stress and block autophagic flux for achieving the antitumor effect and inhibiting tumor metastasis. A dual drug–loaded, pH-responsive polymersome, Tuni/HCQ@CS-PAE, was designed to achieve this objective. The amphiphilic polymer chondroitin sulfate (CS)–poly(β-amino ester) was used to fabricate this polymersome, and hydrophilic HCQ and hydrophobic Tuni were loaded into the inner cavity and outer shell, respectively. Poly(β-amino esters) with acid-stimulated responses are a class of highly biocompatible polymers and are believed to satisfy the performance of drug delivery (28). Repeated tertiary amine groups on the poly(β-amino esters) are protonated by acid stimulation, thereby converting the hydrophobicity of the segment, resulting in the dissociation of the nanostructure and drug release (29). Simultaneously, the protonation process produces a similar HCQ effect to deacidify the lysosomes, swelling and rupturing the lysosomes, which can help the drugs to escape from the lysosomes and block the autophagic flux together with HCQ (30). Polymersomes with both a hydrophilic inner cavity and a hydrophobic shell are considered promising drug delivery platforms (31). In this work, the hydrophilic CS component can mask the surface positive charges of poly(β-amino ester), prolonging the blood circulation time of the polymersomes. These polymersomes reached tumor tissues through the EPR effect, overcoming the nonselective distribution of the Tuni and HCQ drugs in vivo, increasing intratumoral accumulation, entering cells by endocytosis, and remaining in the lysosomes. Because of the pH response of poly(β-amino ester), the polymersomes dissociated and produced a similar alkalization effect in lysosomes with HCQ, destroying and rapidly escaping the lysosomes, releasing the drugs Tuni and HCQ. Under the dual action of poly(β-amino ester) and HCQ, the lysosomes in the tumor cells were destroyed, resulting in the blockade of autophagic flux. Moreover, the released Tuni triggered ER stress, further regulating the PERK/Akt signaling pathway to enhance the autophagic level and down-regulate the MMP-2 expression. The tumor cells were simultaneously attacked by both the inducement of autophagy due to the ER stress and the blockade of autophagic flux due to lysosomal destruction, resulting in a special autophagic stress, which seriously damaged the cell homeostasis and caused cell death.
RESULTS AND DISCUSSION
Synthesis and characterization of the polymersomes
The synthetic routes of CS-poly(β-amino ester) are shown in fig. S1. The chemical structures of the block copolymers were confirmed using Fourier transform infrared spectra and nuclear magnetic resonance (NMR) spectra (figs. S2 to S4). The number-average molecular weight of CS-poly(β-amino ester) was obtained as 8044 g/mol through gel permeation chromatography. The dual drug–loaded Tuni/HCQ@CS-PAE polymersome was prepared by dialysis. The encapsulation efficiency (EE) and encapsulation content (EC) of Tuni were 38.5 and 10.5%, while those of HCQ were 56.1 and 11.2%, respectively. The morphologies of the Tuni/HCQ@CS-PAE polymersomes were characterized using transmission electron microscopy (TEM). Apparent vesicle structures with a size of ~180 nm can be observed in Fig. 1A, and the shell thickness of the vesicle in the enlarged image is ~30 nm. The average hydrodynamic size of the Tuni/HCQ@CS-PAE polymersomes was 230.0 ± 9.3 nm, as measured using dynamic light scattering (DLS), with a polydispersity index of 0.156. The ζ potential was −17.2 mV (Fig. 1B), and the negative charge indicated that it is suitable for drug carriers because it cannot be prematurely cleared in the blood circulation (32). The investigation of stability suggested that the polymersomes showed no significant changes (P > 0.05) in particle size when they were placed in phosphate-buffered saline (PBS) buffer and 10% fetal bovine serum (FBS) for 48 hours at 37°C (fig. S5), indicating their potential for application in vivo. The pH response of the Tuni/HCQ@CS-PAE polymersomes was evaluated in a lysosomal acidic environment, and the CS-poly(β-amino ester) was assayed to determine its pKa (where Ka is the acid dissociation constant) via acid-base titration. The results showed that CS-poly(β-amino ester) had a pKa value of 5.4 (Fig. 1C), which was close to the lysosomal acidity (pH = ~5.0) (33). The TEM image obtained after the Tuni/HCQ@CS-PAE polymersomes were stored at pH 5.0 for 4 hours showed that the polymersome structure disappeared (Fig. 1D), indicating that the pH response of CS-poly(β-amino ester) successfully caused the dissociation of the polymersomes. The 1H-NMR spectrum of CS-poly(β-amino ester) in deuterium chloride at pH 5.0 (fig. S6) showed that the peak of poly(β-amino ester) could be observed, indicating that it was hydrophilic at pH 5.0. DLS was further used to detect the pH response of the Tuni/HCQ@CS-PAE polymersomes. As shown in Fig. 1E, after the polymersomes were stored under the three acidic conditions of pH 7.4, pH 6.8, and pH 5.0 for 4 hours, the particle size distribution demonstrated that the polymersomes at both pH 7.4 and pH 6.8 (tumor ECM acidity) were stable; whereas at pH 5.0, the polymersome structure was destroyed, which is consistent with the TEM results. At pH 5.0, the ζ potential of the Tuni/HCQ@CS-PAE polymersomes significantly shifted from −17.2 to −2.08 mV (Fig. 1F), indicating that the protonation of CS-poly(β-amino ester) resulted in the capture of strong positive charges. The pH response of the polymersomes imparts them the ability to release drugs on-demand. As shown in Fig. 1 (G and H), the release of HCQ and Tuni at pH 6.8 was not significantly different (P > 0.05) from that at pH 7.4, suggesting that the polymersomes were stable in the tumor ECM and would not be released in advance. However, the 24-hour releases of HCQ and Tuni at pH 5.0 (lysosomal acidity) were 86.5 and 76.6%, respectively, which were 7.52 and 6.66 times the releases at pH 7.4, respectively. This result indicates that the Tuni/HCQ@CS-PAE polymersomes can rapidly release drugs in acidic lysosomes.
Fig. 1. Characterizations of Tuni/HCQ@CS-PAE polymersomes.
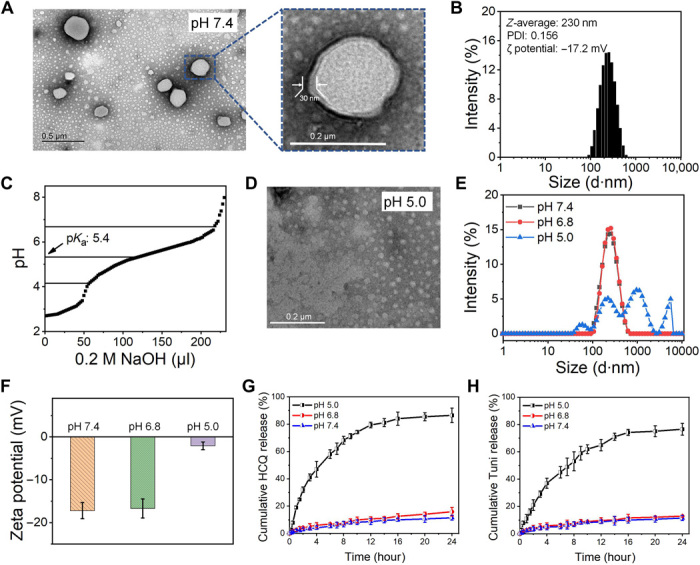
(A) TEM images of the Tuni/HCQ@CS-PAE polymersomes at pH 7.4. (B) Measurement results of the Tuni/HCQ@CS-PAE polymersomes by the Malvern laser particle size analyzer at pH 7.4. (C) Acid-base titration curve of CS-poly(β-amino ester). (D) TEM images of Tuni/HCQ@CS-PAE at pH 5.0. (E) Hydrodynamic particle size distribution of the Tuni/HCQ@CS-PAE polymersomes at pH 7.4, pH 6.8, and pH 5.0. (F) ζ potential of the Tuni/HCQ@CS-PAE polymersomes at pH 7.4, pH 6.8, and pH 5.0. (G) Release profiles of HCQ from the Tuni/HCQ@CS-PAE polymersomes. (H) Release profiles of Tuni from the Tuni/HCQ@CS-PAE polymersomes.
In vitro endocytic pathway
Before applying the polymersomes to cells and animals, both mouse breast cancer cells (4T1) and human umbilical vein endothelial cells (HUVECs) were used to evaluate the cytocompatibility of the polymersome delivery system. The blank material CS-PAE polymersomes exhibited good cytocompatibility at a concentration of 20 to 400 μg/ml (cell viability over 85%, Alamar Blue assay; fig. S7, A and B). Only a small amount of red spots (representing dead cells) was observed in the fluorescence image of cells, with a polymersome concentration of up to 400 μg/ml (live-dead cell staining; fig. S7C), also confirming the low cytotoxicity of the polymersomes.
The endocytic pathway of polymersomes was further examined in vitro. Fluorescein isothiocyanate (FITC)–labeled (green) polymersomes were cocultured with adherent 4T1 cells, and the locations of the polymersomes in the cells and lysosomes labeled by LysoTracker Red DND-99 (red) were observed using fluorescence microscopy at 1 hour (fig. S8A) and 4 hours (fig. S8C), respectively. A large amount of yellow fluorescence in the cells was observed at 1 hour, which was the result of the overlap between green fluorescence and red fluorescence, suggesting that the polymersomes were in the lysosomes. At 4 hours, the yellow fluorescent signal decreased and the separated green and red fluorescent signals increased, indicating that the polymersomes were separated from the lysosomes. The figures (fig. S8, B and D) show the corresponding fluorescence intensity profiles of the white arrow regions in fig. S8 (A and C) obtained using ImagePro Plus, respectively. It can be observed that there was a large overlap between the two fluorescent signals at 1 hour, and their Pearson’s correlation coefficient was calculated to be 0.88, indicating that the polymersomes and the lysosomes were strongly colocalized at 1 hour. At 4 hours, the Pearson’s correlation coefficient was reduced to 0.04 according to fig. S8D, indicating that the polymersomes successfully escaped from the lysosomes. This result indicates that the polymersomes were endocytosed into the cells by the lysosomal pathway and could successfully escape the lysosomes at 4 hours in vitro.
Deacidification of lysosomes
The damage to the lysosomes by poly(β-amino ester) and HCQ was marked by an increase in the lysosomal pH value. The LysoSensor Green-189 can monitor the acidity of the lysosomes, and its fluorescence reaches the highest value in normal lysosomes and decreases with increasing pH value. As shown in fig. S8E, it was observed via fluorescence microscopy that the green fluorescence in the HCQ, blank material CS-PAE polymersomes, and Tuni/HCQ@CS-PAE treatment groups was weakened to varying degrees. The results of the fluorescence-activated cell sorter (FACS) can be more intuitively observed (fig. S8F), and Tuni did not cause a change in the acidity of the lysosomes compared with that of untreated cells (control). Both the HCQ drug and CS-PAE polymersome had an alkalization ability for lysosomes, but HCQ performed better. This can be ascribed to the fact that CS-PAE polymersomes can only destroy lysosomes involved in endocytosis. The Tuni/HCQ@CS-PAE treatment group performed the best, and the median fluorescence intensity was only 14.3% of the control group, suggesting that the double action of HCQ and the polymersomes caused an increase in the intracellular lysosomal pH.
Autophagic level analysis
The relationship between various treatments and autophagy was further examined in vitro. Acridine orange (AO) is an acid-sensitive dye that stains the acidic organelles, including autophagosomes and autolysosomes in the cells red, whereas the DNA and cytoplasm in cells are green. Accordingly, the ratio of red to green signals can be used to evaluate the autophagic level (34). As shown in Fig. 2A, the number of red spots (observed via fluorescence microscopy) is positively correlated with the autophagic level. The red/green ratio calculated using FACS determines the autophagic level in each treatment group (Fig. 2B). The red/green ratios of the treatment groups increased compared with those of the untreated cells (control). The red/green ratio of the Tuni/HCQ@CS-PAE treatment group was approximately 68.0% higher than that of the Tuni/HCQ treatment group and was 1.91 and 2.21 times those of Tuni@CS-PAE and HCQ@CS-PAE, respectively. The red/green ratios of Tuni@CS-PAE and HCQ@CS-PAE also increased by 83.3 and 58.3%, respectively, compared with that of the blank material CS-PAE polymersomes. The red/green ratio of the CS-PAE polymersomes treatment group was also significantly increased by 71.4% compared with that of the control group. The results suggest that both Tuni and HCQ can cause an accumulation of acidic organelles, in addition to CS-PAE polymersomes. Unfortunately, the AO cannot distinguish whether the acidic organelles are autophagosomes or autolysosomes.
Fig. 2. Intracellular autophagic levels and autophagic flux analysis.
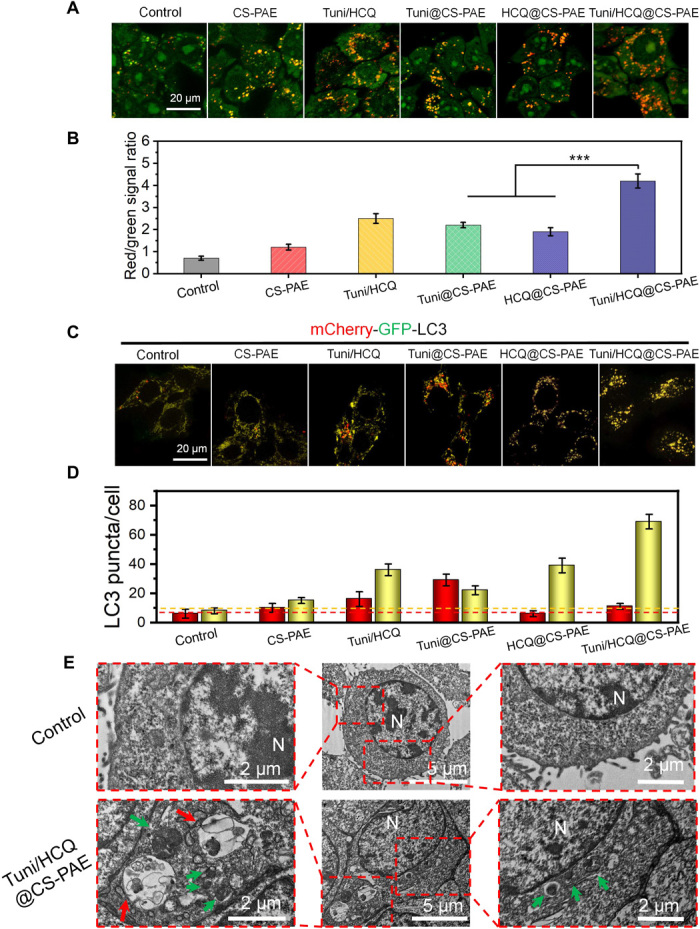
(A) Fluorescence images and (B) FACS analysis of AO-stained 4T1 cells after incubation with different treatments for 24 hours; ***P < 0.001. (C) Fluorescence images of mCherry-GFP-LC3 4T1 cells after incubation with different treatments for 48 hours. (D) Quantification of the number of LC3 puncta per cell (autophagosomes, yellow puncta; autolysosomes, red puncta). (E) TEM images of cells treated with saline or Tuni/HCQ@CS-PAE polymersomes (N, nucleus; green arrow, autophagosomes; red arrow, autolysosomes).
To track the autophagic flux, mCherry–green fluorescent protein (GFP)–LC3 adenovirus–transfected cells were used. When autophagy occurs, a recognized autophagy marker, LC3, aggregates in both the inner and outer membranes of the autophagosomes. The LC3 in the transfected cells simultaneously expresses red fluorescence (mCherry) and green fluorescence (GFP). Thus, it will be observed in the form of yellow puncta in the autophagosomes. When the autophagosomes and lysosomes eventually fuse to form the autolysosomes, GFP is quenched by lysosomal acidity and only exhibits red puncta. Thus, the yellow and red puncta represent the autophagosomes and autolysosomes in the autophagic flux, respectively (Fig. 2C). The LC3 puncta statistic is shown in Fig. 2D. Comparing the LC3 puncta distribution of the Tuni@CS-PAE treatment group with that of the HCQ@CS-PAE treatment group, the former has more red puncta, indicating that the autophagic flux had entered the final stage, and the autolysosome had been formed. The large yellow puncta of the latter indicate that the autophagic flux mainly remained in the autophagosome stage, suggesting that HCQ@CS-PAE destroyed the lysosomes and prevented the autophagosomes from merging with the lysosomes. In contrast to CS-PAE polymersomes, Tuni@CS-PAE increased the autophagosome puncta and autolysosome puncta, indicating that it effectively increased the autophagic level in the cells. Compared with CS-PAE polymersomes, HCQ@CS-PAE showed a significant increase in the autophagosomes, but there was a decrease in the autolysosomes, which proved that HCQ@CS-PAE has a stronger ability to destroy the lysosomes and block the autophagic flux at the autophagosome stage. The autophagic level of the Tuni/HCQ@CS-PAE treatment group was greatly improved compared with that of Tuni/HCQ, whereas the number of autolysosomes was reduced. This result indicated that the dual drug–loaded, pH-responsive polymersomes could increase the autophagic level and block the autophagic flux more evidently than free drugs.
From the TEM images (Fig. 2E), it can be observed that abundant autophagosomes (green arrows) and a few autolysosomes (red arrows) accumulated in the Tuni/HCQ@CS-PAE treatment group, in contrast to the control group (saline treatment). The increase in the autophagosomes and autolysosomes showed that the autophagic level of the Tuni/HCQ@CS-PAE treatment group was significantly enhanced. Furthermore, the amount of autophagosomes was significantly more than the amount of autolysosomes, indicating that the autophagic flux was blocked during the fusion process of the autophagosomes and the lysosomes.
Inhibiting tumor metastasis in vitro
The ability of the polymersomes to resist tumor metastasis was examined. A wound-healing assay (Fig. 3A) and the transwell invasion assay (Fig. 3B) were used to assess the cell migration and invasion in each group of treatments in vitro. The migration area (calculated by ImageJ) in the wound-healing assay is shown in Fig. 3C. The migration area of the Tuni/HCQ@CS-PAE treatment group is only 21.1% (P < 0.001) and 38.4% (P < 0.001) of those of Tuni@CS-PAE and HCQ@CS-PAE, respectively. Matrigel matrix (simulated ECM) was coated in the transwell upper chamber as an in vitro test tool for cell invasion. As shown in Fig. 3D, the number of cells (crystal violet staining) that arrived at the back of the polycarbonate membrane was counted as a quantitative index of invasive ability. The number of invasive cells in the Tuni/HCQ@CS-PAE treatment group was only 6.7% of that in the untreated group, 8.7% of that in the Tuni@CS-PAE treatment group, and 19.3% of that in the HCQ@CS-PAE treatment group. The above results show that the drug delivery system can effectively inhibit cell migration and invasion in vitro. Furthermore, Tuni@CS-PAE has a certain effect in inhibiting cell migration and invasion, but this effect is weaker than that of HCQ@CS-PAE.
Fig. 3. In vitro antimetastasis evaluation.
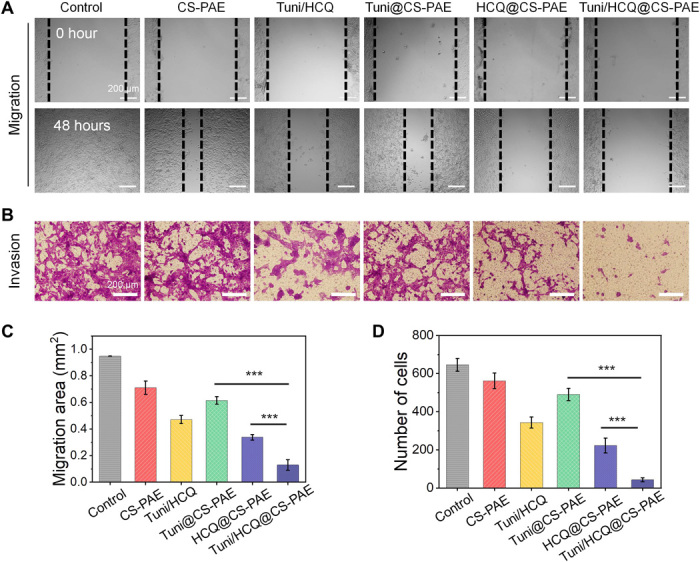
(A) Typical images of wound-healing assay. (B) Cell invasion with the transwell assay (bottom). (C) Migration area of the wound-healing assay. (D) Number of invaded cells by the transwell assay. All data are represented as the means ± SD from three independent experiments; ***P < 0.001.
Antitumor effect
The in vitro cell viability of different treatment groups was firstly investigated. As shown in fig. S9A, the decrease in cell viability was positively correlated with increased autophagic level and autophagic flux blockade. On the basis of the concentration of fig. S9B, the median effect plots (fig. S9, C and D) of Tuni/HCQ and Tuni/HCQ@CS-PAE can be calculated to obtain their half maximal inhibitory concentration (IC50) values. According to the calculation method in the Supplementary Materials, the IC50 value of the Tuni/HCQ@CS-PAE treatment group is 8.2 μM, which is 27.7% lower than that of the Tuni/HCQ treatment group.
The antitumor effect of the pH-responsive polymersome codelivering HCQ and Tuni drugs was further evaluated using the orthotopic luciferase gene–transfected 4T1 (4T1-Luc) tumor–bearing BALB/c mice. The treatment schedule is shown in Fig. 4A. After 7 days of orthotopic 4T1-Luc tumor implantation, the mice were randomly divided into six groups, and the drugs or drug-loaded polymersomes were administered intravenously at 0, 3, 6, and 9 days. The IVIS imaging system was used to monitor the bioluminescence signals of the tumors at days 0 and 30 (Fig. 4B). Ex vivo tumors were photographed on day 30 (Fig. 4C), and tumor volume was measured once in 3 days (Fig. 4D) and ex vivo tumors weighed on day 30 (Fig. 4E). Tumor growth inhibition (TGI; Fig. 4F) was calculated by tumor weight. The results indicated that three of the 4T1 tumors in the Tuni/HCQ@CS-PAE treatment group had successfully ablated, and the tumor weight was only 9.6% (P < 0.001) and 4.6% (P < 0.001) of those treated with Tuni@CS-PAE and HCQ@CS-PAE, respectively, whereas the TGI was as high as 97.5%. This proves that Tuni/HCQ@CS-PAE has an excellent antitumor effect. The body weight of the mice was monitored every 3 days (Fig. 4G), and it showed no difference in each treatment group on day 30 (P > 0.05), indicating the safety of the polymersome delivery system and the potential for application in vivo. Hematoxylin and eosin (H&E) staining of the main organs (heart, liver, spleen, and kidney) (fig. S10) also revealed no significant morphological changes in all the treatment groups.
Fig. 4. In vivo antitumor effect.

(A) Treatment schedule for 4T1 breast tumor in BALB/c mice. (B) Bioluminescence images of 4T1-Luc tumor–bearing BALB/c mice were taken on days 0 and 30 after various treatments. (C) Photographs of the tumors removed from the mice in the different treatment groups at the end of the experiment. (D) Tumor volume growth curves of the different treatment groups. (E) Weight of isolated tumors in the different treatment groups. (F) Tumor growth inhibition (TGI) after the different treatments. (G) Body weight changes of mice in the different treatment groups. (H) Hematoxylin and eosin (H&E) staining and immunohistochemical (IHC) analysis of tumor sections after the different treatments. All statistical data are presented as means ± SD. (n = 5; #P > 0.05; ***P < 0.001). [Photo credit for (B), (C), and (H): Funeng Xu, Southwest Jiaotong University].
H&E staining, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL), and immunohistochemical (IHC) analyses (Ki67) were used to characterize the antitumor effects further (Fig. 4H). The H&E staining sections of the Tuni/HCQ@CS-PAE treatment group showed nuclear shrinkage and fragmentation, and the cell contour disappeared. Its TUNEL-positive signal (characterized apoptosis, brown) was the largest, and its Ki67-positive signal (characterized proliferation, brown) was the smallest. These results suggest that the lysosomal pH-responsive polymersomes entrapped with Tuni and HCQ can achieve excellent antitumor effects in tumor-bearing mouse models, demonstrating the success of autophagy regulation in antitumor applications.
Inhibiting tumor metastasis in vivo
The mouse 4T1 tumor is a metastatic tumor, corresponding mainly to lung and bone metastasis (35). The lung of the mouse was excised on day 30, and the lung metastasis of the tumor was observed using the bioluminescence images (Fig. 5A). There was no bioluminescence signal in the Tuni/HCQ@CS-PAE treatment group, indicating that there was no lung metastasis. Moreover, the pulmonary nodules are visualized using a Bouin’s fixative in Fig. 5B, which also supports this conclusion. The table summarizes the number of metastasis nodes (NOMN) (Fig. 5C), and the lung nodules are categorized by diameter: less than 0.5 mm, 0.5 to 1 mm, 1 to 2 mm, and greater than 2 mm, weighted 1 to 4 in turn. Because of the effective treatment of orthotopic tumors and the effective regulation of autophagy, Tuni/HCQ@CS-PAE has an excellent antimetastatic ability. The results of NOMN are shown in Fig. 5D. The average NOMN of the HCQ@CS-PAE treatment group was 52.89% that of the Tuni@CS-PAE treatment group and 22.49% that of the CS-PAE polymersomes treatment group. This indicates that both HCQ@CS-PAE and Tuni@CS-PAE can inhibit tumor metastasis to a certain extent, but the former performed better than the latter, which is also consistent with the results of in vitro antimetastasis evaluation. The H&E staining of the lungs can also demonstrate the antimetastatic effect (Fig. 5E). The red circle framed the foreign tissues of the lungs, and the foreign tissues were observed to be tumor tissues by comparison with the H&E staining of the tumors. No tumor tissue was observed in the H&E sections of the Tuni/HCQ@CS-PAE treatment group, and the area of the tumor tissue was significantly reduced in the Tuni@CS-PAE and HCQ@CS-PAE treatment groups compared with that in the saline group, which is consistent with the lung metastasis results of Fig. 5 (A and B).
Fig. 5. In vivo antitumor metastasis.
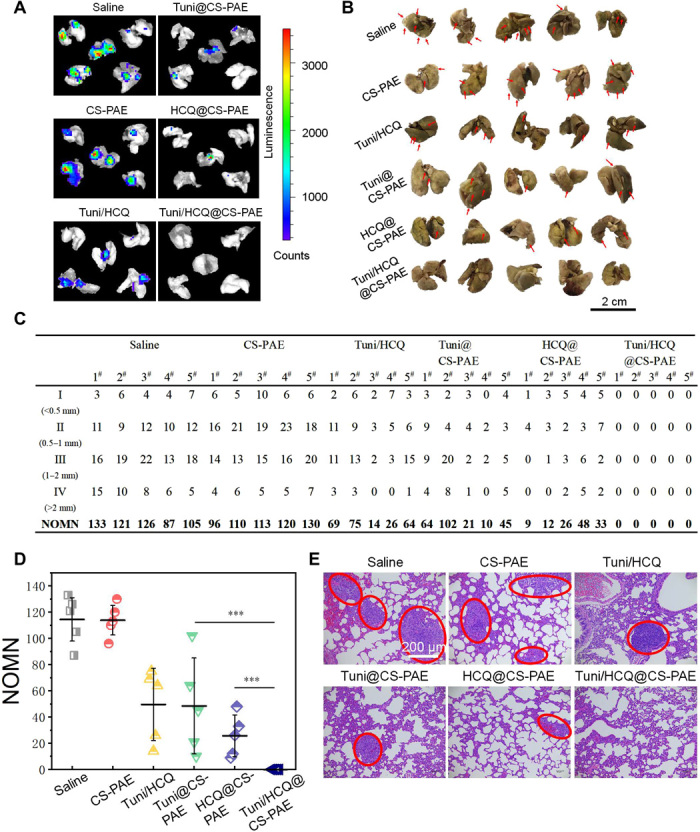
(A) Bioluminescence images of tumor lung metastases in each treatment group in vitro. (B) Photographs of lung tissues; tumor metastasis was visualized by Bouin’s fixative, and metastatic nodules were white (represented by red arrows). (C and D) Counting the number of lung metastasis nodules, measurement of the diameter of metastatic tumors, and performing classification and counting. Number of metastasis nodes (NOMN) = I × 1 + II × 2 + III × 3 + IV × 4 (according to the diameter of the lung nodules for class 4: I < 0.5 mm, 0.5 mm ≤ II < 1 mm, 1 mm ≤ III ≤ 2 mm, and IV > 2 mm). (E) H&E staining of lung tissue after the various treatments. The red circle marks the metastatic tumor tissue. [Photo credit for (A), (B), and (E): Funeng Xu, Southwest Jiaotong University].
Intrinsic signal pathways analysis
The dual drug–loaded, pH-responsive polymersomes (Tuni/HCQ@CS-PAE) have made breakthroughs in the antitumor effect and metastasis inhibition effects in tumor-bearing mice. Western blot (WB) and IHC were further used to explore the mechanism of action of Tuni/HCQ@CS-PAE. ER stress and autophagy are closely related (Fig. 6A). It has been reported that the ER stress enhances the autophagic level by negatively regulating the Akt/mTOR pathway (36). In addition, the expression of MMP-2 is down-regulated by the down-regulation of Akt expression. Therefore, it can be concluded that the Tuni/HCQ@CS-PAE polymersome has two important functions in vivo: increasing autophagic levels and decreasing the MMP-2 expression and blocking the autophagic flow at the autophagosome stage by preventing the fusion of the autophagosomes and the lysosomes.
Fig. 6. Intrinsic signal pathways analysis.
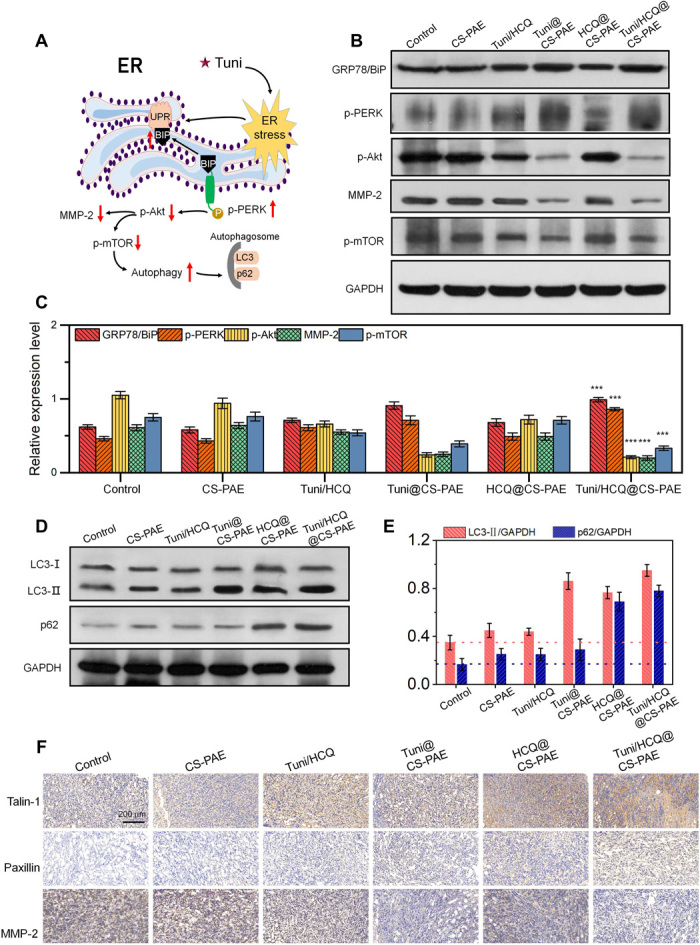
(A) Schematic diagram of Tuni causing ER stress, promoting autophagy, and reducing MMP-2 expression via signaling pathways in vivo. (B) WB analysis of key proteins of ER stress and downstream pathway protein of 4T1 tumor in BALB/c mice. (C) Relative expression level of key proteins in (B); ***P < 0.001 compared with control. (D) Expression of LC3 and p62 lanes of 4T1 tumor in BALB/c mice via WB. (E) Quantification of the ratio of LC3-II to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and p62 to GAPDH expression using ImageJ software. (F) IHC pictures of talin-1, paxillin, and MMP-2 in 4T1 tumor–bearing BALB/c mice.
Under normal physiological conditions, GRP78/BiP (78-kDa glucose-regulated protein/immunoglobulin heavy chain–binding protein) acts as an ER chaperone and binds to ER receptors, which is in an inactive state. However, under ER stress, GRP78/BiP dissociates from ER receptors to activate and trigger UPR (37). The dissociation of GRP78/BiP from PERK (ER transmembrane receptor) triggers kinase dimerization and autophosphorylation to generate activated PERK (p-PERK) (38). Therefore, the expression of GRP78/BiP and p-PERK can be used as an indicator of ER stress. The expression of GRP78/BiP and p-PERK of the Tuni/HCQ@CS-PAE treatment group was significantly increased by 59.7% (P < 0.001) and 87.0% (P < 0.001), respectively, compared with that of the control group (Fig. 6, B and C). It indicated that Tuni/HCQ@CS-PAE can strongly trigger ER stress. The expressions of GRP78/BiP and p-PERK of the Tuni/HCQ treatment group were only 71.7% (P < 0.001) and 70.9% (P < 0.001), respectively, of that of the Tuni/HCQ@CS-PAE treatment group, suggesting that the polymersome system is more efficient than the free drugs at the same dose. The occurrence of ER stress triggered a series of downstream signaling pathways. The expressions of p-Akt and p-mTOR proteins in the Tuni/HCQ@CS-PAE treatment group were reduced by 80.0% (P < 0.001) and 67.2% (P < 0.001), respectively, compared to the control group, suggesting that the increase in the upstream event down-regulates Akt and mTOR activity. The expression of MMP-2 was significantly decreased in the Tuni/HCQ@CS-PAE and Tuni@CS-PAE treatment groups, which were only 32.8 and 41.0% of that of the control group, respectively, which was consistent with the trend of p-Akt expression.
When the autophagy occurs, LC3-I, which is in the cytoplasm of cells, is modified and processed to form LC3-II and expressed on the autophagosome membrane. The expression of p62 (sequestosome-1) as an autophagy substrate can characterize the smoothness of the autophagic flux. When the autophagic level is increased and the autophagic flux is smooth, the expression of LC3-II is increased and the expression of p62 is decreased; however, when the autophagy level is increased and the autophagic flux is blocked, the expression of both LC3-II and p62 is increased (39, 40). As shown in Fig. 6 (D and E), the expression of LC3-II and p62 was up-regulated in all the treatment groups compared with that in the control group, suggesting that the autophagic level of each treatment group was increased, and the autophagic flux was blocked to some extent. The highest expression of LC3-II was observed for Tuni/HCQ@CS-PAE and Tuni@CS-PAE, which was consistent with the results of down-regulation of p-mTOR. The expression of LC3-II of HCQ@CS-PAE was also significantly improved, indicating that, when autophagic flux was blocked by lysosomal destruction, the overall autophagic level of the cells also increased, and the autophagic flux mainly remained in the autophagosome stage. The expression of p62 was the highest in the Tuni/HCQ@CS-PAE and HCQ@CS-PAE treatment groups, i.e., 4.67 (P < 0.001) and 4.13 (P < 0.001) times that of the control group, respectively, suggesting that substrate degradation in the autophagosomes was largely blocked.
Talin-1 and paxillin are the constituent proteins of the FAs. The turnover of FAs is the basis of cell movement. The blockade of the autophagic flux leads to the failure of FAs degradation, which reduces the ability of cell movement. As shown in Fig. 6F, the IHC sections of Tuni/HCQ@CS-PAE and HCQ@CS-PAE showed the largest amounts of talin-1 and paxillin, indicating that the blockade of autophagic flux can significantly prevent FAs turnover. In the IHC section analysis of MMP-2, the expression of MMP-2 of the Tuni/HCQ@CS-PAE and Tuni@CS-PAE treatment groups was significantly inhibited, and the results were also consistent with the WB test in Fig. 6B, indicating that the MMP-2 expression was closely related to the ER stress induced by Tuni. It is suggested that based on the results of in vitro and in vivo antimetastasis experiments, it is known that HCQ@CS-PAE has stronger antimetastatic ability than Tuni@CS-PAE, also suggesting that the reduction in FAs turnover by autophagy affects tumor metastasis more than the decrease in the MMP-2 expression. Thus, Tuni/HCQ@CS-PAE achieved a considerable tumor-metastasis inhibition effect under the combined effect of down-regulation of MMP-2 expression and inhibition of FAs turnover.
CONCLUSIONS
In summary, we presented a proof of concept for a pH-responsive polymersome codelivering HCQ and Tuni drugs, which had the capability of simultaneously inducing ER stress and blocking autophagy for combating malignant tumors and inhibiting tumor metastasis. The pH-responsive function of poly(β-amino ester) of the polymersomes resulted in a dissociation of the polymersomes, leading to the rapid release of the loaded HCQ and Tuni. The combination of the pH response of the polymersomes and the released HCQ action alkalized and damaged the lysosomes to block the autophagic flux. Simultaneously, the released Tuni triggered ER stress and further regulated the PERK/Akt signaling pathway, resulting in an increased autophagic level. The in vivo antitumor effect on orthotopic 4T1-Luc tumor–bearing BALB/c mice demonstrated that ~60.0% of the tumors were completely cured, and an extremely high TGI of 97.5% was achieved. Furthermore, the tumor metastasis was successfully inhibited with the polymersome and the NOMN is 0. The function mechanism investigation through both WB and IHC analyses indicated that the excellent antitumor effect was ascribed to a special autophagic stress, which resulted from the fact that the tumor cells were simultaneously attacked by the autophagy induction due to ER stress and the blockade of autophagic flux; the inhibition of tumor metastasis was mainly due to both the reduced expression of MMP-2 due to ER stress and the reduced FAs turnover due to the blockade of autophagic flux. Therefore, we believe that this dual drug–loaded, pH-responsive polymersome has considerable potential to be developed as nanomedicine for treating tumors and inhibiting tumor metastasis.
MATERIALS AND METHODS
Synthesis of acetylated CS
CS was acetylated to increase its solubility in dimethyl sulfoxide (DMSO). Briefly, CS (1.0 g) was added to a round-bottom flask containing formamide (50.0 ml). The system was heated to 80°C to promote dissolution and then cooled to room temperature. Pyridine (557.0 μl) and acetic anhydride (500.0 μl) were added to the round-bottom flask and magnetically stirred at room temperature for 12 hours. The reaction solution was dialyzed and lyophilized to obtain acetylated CS (Ac-CS; 0.91 g), which was stored for later use.
Synthesis of Had-Ac-CS
Ac-CS (0.41 g), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.16 g), and N-hydroxysuccinimide (0.04 g) were dissolved in reverse osmosis (RO) water. Triethylamine (0.08 g) was added to this mixture, and magnetic stirring was performed to activate the carboxyl group in an ice bath. Subsequently, 1,6-hexanediamine (0.63 g) was added, and the mixture was stirred at room temperature for 24 hours. The product Had-Ac-CS (0.49 g) was obtained via the dialysis and lyophilization of the reaction solution.
Synthesis of CS-poly(β-amino ester)
CS-poly(β-amino ester) was prepared via the Michael addition reaction. Had-Ac-CS (0.37 g), Hexane-1,6-dioldiacrylate (HDDA) (0.5 g), and 3-dibutylamino-1-propylamine (DBPA) (0.37 g) were dissolved in 15 ml of DMSO, and the round-bottom flask was purged with N2. The mixture was stirred at 50°C for 5 days. CS-poly(β-amino ester) (1.1 g) was obtained via the dialysis and lyophilization of the reaction solution.
Polymersome formation
Dialysis was used to prepare the dual drug–loaded polymersomes. First, 20.0 mg of CS-poly(β-amino ester) and 7.0 mg of Tuni were codissolved in a beaker containing 5.0 ml of DMSO. Then, 5.0 ml of RO water containing 5.1 mg of HCQ was added dropwise under high-speed stirring, and the system was continuously stirred for 20 min. The system was transferred to a dialysis bag (molecular weight cutoff, 3500) for 48 hours and then lyophilized for the next experiment. EE and EC were calculated by Eqs. 1 and 2, respectively
| (1) |
| (2) |
Acid-base titration
CS-poly(β-amino ester) was placed in deionized water, and HCl solution was added until it was completely dissolved. Then, 1 to 5 μl of 0.1 M NaOH solution was added dropwise, and the pH was measured after each addition. The pKa of the polymer is the pH at which it is half ionized.
Polymersomes pH sensitivity determination
The pH response of polymersomes was characterized by DLS, TEM, and 1H-NMR, respectively. The polymersomes were placed at pH 5.0, pH 6.8, and pH 7.4, respectively. After 4 hours, DLS was used to measure the particle size distribution and ζ potential. Samples at pH 5.0 were also used for TEM and 1H-NMR detection.
In vitro release profile
The release of Tuni and HCQ in Tuni/HCQ@CS-PAE was investigated under three acidity conditions of pH 7.4, pH 6.8, and pH 5.0 at 37°C. The release solution was taken 1 ml each time at the planned time point, and the same volume of fresh medium was added. The release solution was treated and analyzed by high-performance liquid chromatography.
Stability of Tuni/HCQ@CS-PAE polymersomes
Size changes of Tuni/HCQ@CS-PAE polymersomes after incubation in PBS or in cell culture medium [containing 10% FBS (v/v)] were monitored by DLS. The polymersome concentration was 1 mg ml−1, and the experimental conditions were 37°C.
Cytocompatibility assay
Alamar Blue assay and live-dead staining were used to determine the cytocompatibility of blank polymersomes. In Alamar Blue assay detection, 1 × 104 cells per well of 4T1 and HUVECs were seeded in 48-well plates. After 24 hours of culture, polymersomes with 20 to 400 μg/ml were added to each well. After 48 hours, the culture medium was removed, and 300 μl of Alamar Blue solution [10% Alamar Blue, 80% media 199 (Gibco), and 10% FBS, (v/v)] was added for a further 3-hour incubation. They were then transferred to 96-well plates and detected by automated microplate spectrophotometer. For live-dead staining, 2 × 104 cells per well of 4T1 and HUVECs were seeded in 24-well plates. The cells were stained by 2 mM calcein acetoxymethylester for 10 min and propidium iodide for 5 min after 48 hours incubation, with different concentrations of polymersomes. Live cells were stained green, and dead cells were stained red when visualized by fluorescence microscopy.
In vitro cellular uptake
4T1 cells were cultured in confocal dishes for 12 hours in Dulbecco’s modified Eagle’s medium with 10% FBS. FITC-labeled polymersomes were added to two sets of confocal dishes, and then the medium was discarded at 1 and 4 hours, respectively. Cells were washed three times with PBS before staining, and then lysosomes were labeled with the LysoTracker Red DND-99. The fluorescence signal was observed by fluorescence microscopy.
Lysosome acidity detection
Adherent 4T1 cells were treated with saline, Tuni, HCQ, CS-PAE, and Tuni/HCQ@CS-PAE for 12 hours, and stained with the acid-sensitive dye LysoSensor Green-189. The fluorescence intensity of each group was measured by fluorescence microscope and FACS.
AO staining
The adherent 4T1 cells were treated with saline, CS-PAE, Tuni/HCQ, Tuni@CS-PAE, HCQ@CS-PAE, and Tuni/HCQ@CS-PAE, respectively. After 24 hours, the cells were stained with AO (1 μl) for 15 min and detected by fluorescence microscopy and FACS, respectively.
mCherry-GFP-LC3 adenovirus transfection
4T1 cells were inoculated with 5 × 105 per well in confocal dishes before infection. The density of cells before virus transfection reached 50%, and the amount of virus mother liquor added to the plate was plaque-forming units = cell number × multiplicity of infection (MOI). MOI was 20, 24 hours after infection; 2 ml of fresh medium was added to each well to replace the virus-containing medium.
Bio-TEM
4T1 cells were seeded in culture flasks at a density of 1 × 106 cells/ml for 18 hours. Then, saline and Tuni/HCQ@CS-PAE were added for 48 hours, respectively. Cells were digested, collected by centrifugation, and then fixed overnight in 2.5% glutaraldehyde. Samples were prepared according to TEM standard procedures and photographed.
In vitro cytotoxicity
Alamar Blue assay was used to assess the in vitro cytotoxicity. 4T1 cells were seeded in 48-well plates at a density of 2 × 104 cells per well. After the cells were cultured for 24 hours, various preparations were added to the well plates for 48 hours. After the incubation, 300 μl of Alamar Blue solution was added for further 3 hours, and then the Alamar Blue solution was transferred to a 96-well plate, and the absorbance was measured with an automated microplate spectrophotometer. The median effect plot was a straight line fit with X = log(D) versus Y = log[fa/(1 − fa)] (41). The theoretical IC50 value is the drug concentration corresponding to the x axis intercept of the median effect plot.
Wound-healing assay
4T1 cells were seeded in six-well plates. When the cell confluence reached 100%, scratches were made with a 200-μl pipette tip, and cells were washed three times with PBS to remove the delineated cells. The treatments were added to each group. Various therapeutic agents were added to the treated six-well plates and cultured for 36 hours in serum-free medium. The entire process was monitored with a microscope, and the healing area was calculated by ImageJ.
Transwell invasion assay
One hundred microliters of the diluted Matrigel was added vertically in the center of the transwell upper chamber and incubated at 37°C for 4 hours to form a gel. Six hundred microliters of 10% serum medium was added to the lower chamber, and 100 μl of the cell suspension was added to the upper chamber, and incubation was continued for 24 hours. The transwell chamber was removed, fixed in methanol for 30 min, stained with 0.1% crystal violet for 20 min, and the uninjured cells in the upper layer were gently wiped off with a cotton swab, and the count was observed with a microscope.
In vivo antitumor activity
4T1-Luc cells were injected into the mouse mammary fat pad to establish an orthotopic breast cancer model. The animal experiments were approved by the Institutional Animal Care and Use Committee of Sichuan University and carried out in compliance with its guidelines. When the tumor volume of the mouse reached approximately 35 mm3, it was defined as 0 day of treatment, and the mice were randomly divided into six groups of five mice each. Each group was administered through the tail vein at 0, 3, 6, and 9 days of treatment [Tuni (7.5 mg/kg)]. Tumor volumes were calculated using the following equation: V = 0.5 × A × B2 (A refers to tumor length, and B refers to the tumor width). TGI was calculated using the following equation: TGI (%) = 100 × (mean tumor weight of saline group − mean tumor weight of experimental group)/mean tumor weight of saline group.
Histological and IHC analyses
4T1 tumor–bearing BALB/c mice were sacrificed, and the heart, liver, spleen, lung, kidney, and tumor tissues were excised, fixed with 10% formalin, dehydrated with gradient ethanol, and embedded in paraffin block. After denitrification with xylene, 4-μm-thick tissue sections were stained with H&E, or for TUNEL detection, or for IHC staining with rabbit anti–Ki-67 polyclonal antibody and lastly observed with an optical microscope.
Signal pathway investigation
The potential association of ER stress with autophagy and antimetastatic mechanisms were analyzed by WB and immunohistochemistry. BIP/GRP78, PERK, Akt, mTOR, LC3, and p62 were used for WB analysis to explore the ER stress–autophagy signaling pathway based on relative expression levels. MMP-2, talin, and paxillin were used as indicators of antimetastasis to analyze changes in their expression through WB and immunohistochemistry.
Statistical analysis
SPSS software was used for the statistical data analysis. Data were presented as means ± SD. One-way analysis of variance (ANOVA) was performed to determine statistical significance of the data. The differences were considered significant for #P > 0.05, *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank the Analytical and Testing Center of the Southwest Jiaotong University. Funding: This work was partially supported by the China National Funds for Distinguished Young Scientists (51725303), the National Natural Science Foundation of China (21574105), and the Sichuan Province Youth Science and Technology Innovation Team (2016TD0026). Author contributions: F.X., Y.W., and S.Z. designed research; F.X., X.L., and X.H. performed research; F.X. and J.P. analyzed data; and F.X. and S.Z. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eabb8725/DC1
REFERENCES AND NOTES
- 1.Xu R., Ji Z., Xu C., Zhu J., The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine 97, e12912 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu L., Chen Y., Tooze S. A., Autophagy pathway: Cellular and molecular mechanisms. Autophagy 14, 207–215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doherty J., Baehrecke E. H., Life, death and autophagy. Nat. Cell Biol. 20, 1110–1117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang M., Chen P., Liu J., Zhou S., Kroemer G., Klionsky D. J., Lotze M. T., Zeh H. J., Kang R., Tang D., Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Onorati A. V., Dyczynski M., Ojha R., Amaravadi R. K., Targeting autophagy in cancer. Cancer 124, 3307–3318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J., Qu Y., Zheng T., Tian Y., A dual-mode nanoprobe for evaluation of the autophagy level affected by photothermal therapy. Chem. Commun. 55, 9673–9676 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Deng S., Shanmugam M. K., Kumar A. P., Yap C. T., Sethi G., Bishayee A., Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 125, 1228–1246 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Rabinowitz J. D., White E., Autophagy and metabolism. Science 330, 1344–1348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maiuri M. C., Kroemer G., Therapeutic modulation of autophagy: Which disease comes first? Cell Death Differ. 26, 680–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy J. M. M., Towers C. G., Thorburn A., Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan M. J., Fitzwalter B. E., Owens C. R., Powers R. K., Sottnik J. L., Gamez G., Costello J. C., Theodorescu D., Thorburn A., Metastatic cells are preferentially vulnerable to lysosomal inhibition. Proc. Natl. Acad. Sci. U.S.A. 115, E8479–E8488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thakur P. C., Miller-Ocuin J. L., Nguyen K., Matsuda R., Singhi A. D., Zeh H. J., Bahary N., Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 16, 190 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang J., Li H., Qaed E., Zhang J., Song Y., Wu R., Bu X., Wang Q., Tang Z., Salinomycin, as an autophagy modulator–A new avenue to anticancer: A review. J. Exp. Clin. Cancer Res. 37, 26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song S., Tan J., Miao Y., Zhang Q., Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement of UPR and the core autophagy machinery. J. Cell. Physiol. 233, 3867–3874 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Cheng X., Feng H., Wu H., Jin Z., Shen X., Kuang J., Huo Z., Chen X., Gao H., Ye F., Ji X., Jing X., Zhang Y., Zhang T., Qiu W., Zhao R., Targeting autophagy enhances apatinib-induced apoptosis via endoplasmic reticulum stress for human colorectal cancer. Cancer Lett. 431, 105–114 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Wang H., Zhang G., Endoplasmic reticulum stress-mediated autophagy protects against β,β-dimethylacrylshikonin-induced apoptosis in lung adenocarcinoma cells. Cancer Sci. 109, 1889–1901 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernández A., Ordóñez R., Reiter R. J., González-Gallego J., Mauriz J. L., Melatonin and endoplasmic reticulum stress: Relation to autophagy and apoptosis. J. Pineal Res. 59, 292–307 (2015). [DOI] [PubMed] [Google Scholar]
- 18.McAfee Q., Zhang Z., Samanta A., Levi S. M., Ma X.-H., Piao S., Lynch J. P., Uehara T., Sepulveda A. R., Davis L. E., Winkler J. D., Amaravadi R. K., Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl. Acad. Sci. U.S.A. 109, 8253–8258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Limpert A. S., Lambert L. J., Bakas N. A., Bata N., Brun S. N., Shaw R. J., Cosford N. D. P., Autophagy in cancer: Regulation by small molecules. Trends Pharmacol. Sci. 39, 1021–1032 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Q., Zhou Z., Qiu N., Shen Y., Rational design of cancer nanomedicine: Nanoproperty integration and synchronization. Adv. Mater. 29, 1606628 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Guo X., Wei X., Chen Z., Zhang X., Yang G., Zhou S., Multifunctional nanoplatforms for subcellular delivery of drugs in cancer therapy. Prog. Mater. Sci. 107, 100599 (2020). [Google Scholar]
- 22.Wang Y., Wei G., Zhang X., Xu F., Xiong X., Zhou S., A step-by-step multiple stimuli-responsive nanoplatform for enhancing combined chemo-photodynamic therapy. Adv. Mater. 29, 1605357 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Sharifi M. N., Mowers E. E., Drake L. E., Collier C., Chen H., Zamora M., Mui S., Macleod K. F., Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 15, 1660–1672 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenific C. M., Wittmann T., Debnath J., Autophagy in adhesion and migration. J. Cell Sci. 129, 3685–3693 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mowers E. E., Sharifi M. N., Macleod K. F., Novel insights into how autophagy regulates tumor cell motility. Autophagy 12, 1679–1680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y., Huang L., Wang S., Li J.-L., Li M., Wu Y., Liu T., WFDC2 contributes to epithelial-mesenchymal transition (EMT) by activating AKT signaling pathway and regulating MMP-2 expression. Cancer Manag. Res. 11, 2415–2424 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Chen Z., Wei H., Zhao X., Xin X., Peng L., Ning Y., Wang Y., Lan Y., Zhang Q., Metformin treatment alleviates polycystic ovary syndrome by decreasing the expression of MMP-2 and MMP-9 via H19/miR-29b-3p and AKT/mTOR/autophagy signaling pathways. J. Cell. Physiol. 234, 19964–19976 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Anderson D. G., Peng W., Akinc A., Hossain N., Kohn A., Padera R., Langer R., Sawicki J. A., A polymer library approach to suicide gene therapy for cancer. Proc. Natl. Acad. Sci. U.S.A. 101, 16028–16033 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C., An T., Wang D., Wan G., Zhang M., Wang H., Zhang S., Li R., Yang X., Wang Y., Stepwise pH-responsive nanoparticles containing charge-reversible pullulan-based shells and poly(β-amino ester)/poly(lactic-co-glycolic acid) cores as carriers of anticancer drugs for combination therapy on hepatocellular carcinoma. J. Control. Release 226, 193–204 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Lin Y.-X., Wang Y., Qiao S.-L., An H.-W., Zhang R.-X., Qiao Z.-Y., Rajapaksha R. P. Y. J., Wang L., Wang H., pH-sensitive polymeric nanoparticles modulate autophagic effect via lysosome impairment. Small 12, 2921–2931 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Wang F., Xiao J., Chen S., Sun H., Yang B., Jiang J., Zhou X., Du J., Polymer vesicles: Modular platforms for cancer theranostics. Adv. Mater. 30, e1705674 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Dai Y., Xu C., Sun X., Chen X., Nanoparticle design strategies for enhanced anticancer therapy by exploiting the tumour microenvironment. Chem. Soc. Rev. 46, 3830–3852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Zhang L., Zhang X., Wei X., Tang Z., Zhou S., Precise polymerization of a highly tumor microenvironment-responsive nanoplatform for strongly enhanced intracellular drug release. ACS Appl. Mater. Interfaces 8, 5833–5846 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Hu C., Xu X., Zhang X., Li Y., Li Y., Gu Z., Bioinspired design of stereospecific D-protein nanomimics for high-efficiency autophagy induction. Chem. Mat. 29, 7658–7662 (2017). [Google Scholar]
- 35.Wang W., Belosay A., Yang X., Hartman J. A., Song H., Iwaniec U. T., Turner R. T., Churchwell M. I., Doerge D. R., Helferich W. G., Effects of letrozole on breast cancer micro-metastatic tumor growth in bone and lung in mice inoculated with murine 4T1 cells. Clin. Exp. Metastasis 33, 475–485 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Qin L., Wang Z., Tao L., Wang Y., ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 6, 239–247 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Szegezdi E., Logue S. E., Gorman A. M., Samali A., Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Logue S. E., Cleary P., Saveljeva S., Samali A., New directions in ER stress-induced cell death. Apoptosis 18, 537–546 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Cui Z., Zhang Y., Xia K., Yan Q., Kong H., Zhang J., Zuo X., Shi J., Wang L., Zhu Y., Fan C., Nanodiamond autophagy inhibitor allosterically improves the arsenical-based therapy of solid tumors. Nat. Commun. 9, 4347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martens S., A division of labor in mTORC1 signaling and autophagy. Sci. Signal. 11, eaav3530 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Choudhury D., Ganguli A., Dastidar D. G., Acharya B. R., Das A., Chakrabarti G., Apigenin shows synergistic anticancer activity with curcumin by binding at different sites of tubulin. Biochimie 95, 1297–1309 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eabb8725/DC1