

Dorsoventral polarity directs confined cell migration mode and efficiency by tuning nuclear stiffness and RhoA activity.
Abstract
How migrating cells differentially adapt and respond to extracellular track geometries remains unknown. Using intravital imaging, we demonstrate that invading cells exhibit dorsoventral (top-to-bottom) polarity in vivo. To investigate the impact of dorsoventral polarity on cell locomotion through different confining geometries, we fabricated microchannels of fixed cross-sectional area, albeit with distinct aspect ratios. Vertical confinement, exerted along the dorsoventral polarity axis, induces myosin II–dependent nuclear stiffening, which results in RhoA hyperactivation at the cell poles and slow bleb-based migration. In lateral confinement, directed perpendicularly to the dorsoventral polarity axis, the absence of perinuclear myosin II fails to increase nuclear stiffness. Hence, cells maintain basal RhoA activity and display faster mesenchymal migration. In summary, by integrating microfabrication, imaging techniques, and intravital microscopy, we demonstrate that dorsoventral polarity, observed in vivo and in vitro, directs cell responses in confinement by spatially tuning RhoA activity, which controls bleb-based versus mesenchymal migration.
INTRODUCTION
Cell migration represents a key step in the metastatic cascade of events, as it enables tumor cells dissociating from a primary tumor to navigate through interstitial tissues and ultimately colonize distant organs. Cells in vivo migrate either by remodeling their surrounding three-dimensional (3D) extracellular matrix (ECM) to open up migratory paths, by following leader cells, such as cancer-associated fibroblasts, that generate such paths, or by migrating through preexisting, 3D longitudinal channel-like tracks created by various anatomical structures (1). Hence, cells travel through confining pores, which can vary from 1 to 20 μm in diameter, or fiber- and channel-like tracks, which range from 3 to 30 μm in width (2). Longitudinal tracks not only provide the “paths of least resistance” for cell migration (3) but also exert physical cues on cells that initiate intracellular cascades of signaling events that regulate the modes and mechanisms of cell motility (4, 5).
Migration mode plasticity is a crucial element in cancer metastasis, as tumor cells need to adapt to diverse tissue microenvironments presenting different length scales, topographies, stiffness, and composition of ECM to ultimately establish metastatic colonies (1). Tumor cells may migrate with a mesenchymal or elongated migration phenotype, which is characterized by high adhesion and actin-rich protrusions, whereas elevated contractility promotes an amoeboid or bleb-based migration mechanism (1). For instance, vertical compression of cells on a low-adhesion substrate induces a migration mode transition from mesenchymal to amoeboid (5). In 3D collagen gels, cells typically display the classical flat protrusions. However, inside a crosslinked, linearly elastic 3D matrix, cells, such as fibroblasts and protease-inhibited tumor cells, use a pressure-based lobopodial migration mode, characterized by a blunt, cylindrical leading protrusion, that requires high RhoA–ROCK–myosin II–dependent contractility and adhesions containing paxillin and vinculin (6). Inhibition of cell contractility switches the migration mode from lobopodial to lamellipodial (6). In addition, blocking integrins impairs lobopodial migration. Thus, the physical features and the ECM composition of the local microenvironment coupled with the intrinsic properties of the cells regulate the mechanisms and modes of cell motility.
The nucleus has a rate-limiting role in cell migration through confined spaces. Tumor cell motility is halted at pore sizes smaller than ~7 μm2 due to lack of nuclear translocation and can only resume following matrix degradation (7). Nuclear stiffness is considered a key determinant of confined migration, as its reduction via lamin-A knockdown enhances migration through narrow pores (8, 9). However, it is currently unknown how varying nuclear confinement regulates the organization of the cell cytoskeleton and the mode of single-cell migration. Confined migration is commonly studied using microfluidic devices that enable real-time, high-throughput monitoring of cell motility in channel-like tracks of prescribed physical properties (1, 10). Confinement exerts mechanical stress on the nucleus, which can ultimately lead to nuclear envelope rupture and DNA damage with important consequences for genomic integrity (11–13). For cells migrating through openings of the exact same cross-sectional area [height (H) × width (W) = constant], the extent of nuclear rupture is more pronounced when the migration tracks have a low ceiling (small height) rather than a narrow base (small width) (11). Cells are compressed vertically (top to bottom), as opposed to laterally (sidewise) in the former and latter cases, respectively. These observations prompted us to address a fundamental and yet unanswered question: Are migrating cells endowed with the ability to respond to different geometries of migration tracks by changing the speed, the mode, and/or the mechanisms of cell locomotion? We hypothesized that if cells could sense and respond to distinct geometries, this would be due to the intrinsic asymmetry of their molecular machinery and/or cytoskeletal organization along the top-to-bottom cell axis, termed dorsoventral cell polarity. We here show that migrating cells in vivo exhibit dorsoventral polarity. To test the effects of polarity on confined migration, we induced cells with pre-established dorsoventral polarization to migrate inside collagen type I–coated microchannels of a fixed cross-sectional area (30 μm2), which impose either vertical (H × W = 3 × 10 μm2; small height) or lateral (H × W = 10 × 3 μm2; small width) compression on cells. We demonstrate that preexisting dorsoventral polarization directs differential cell responses to distinct geometries by altering key determinants of confined cell locomotion, such as nuclear stiffening, regulation of contractile machinery, and dynamic interconversion of blebbing versus mesenchymal modes of migration.
RESULTS
Cells migrate with different efficiencies through laterally versus vertically confined migration tracks
Previous studies have shown that anterior/posterior polarity of key molecules such as Rho guanosine triphosphatases (GTPases), focal adhesion kinase (FAK), and the microtubule-organizing center (MTOC) is critical for persistent cell migration (14). However, it is unknown whether dorsoventral polarity is present in vivo and how it may regulate confined cell migration. To address this question, HT-1080 fibrosarcoma cells expressing LifeAct-GFP (actin) and H2B-mCherry (histone 2B) were monitored by intravital multiphoton microscopy during invasion into the mouse dermis. In this model, moving cells were predominantly guided by longitudinal tissue interfaces between myofibers and collagen-rich ECM, as described in (2), and developed basolateral polarity with F-actin preferentially distributing toward the myofiber plane (Fig. 1, A to C). Thus, dorsoventrally polarized cells are observed in physiological tissues.
Fig. 1. Cells migrate with different efficiencies through vertically and laterally confined microchannels.

(A) Polarization of actin in HT-1080/LifeAct-GFP/H2B-mCherry fibrosarcoma cell invading the deep dermis along myofiber and fibrillar collagen-rich tissue structures. Images represent overview and detail obtained by multicolor multiphoton time-lapse microscopy 5 days after tumor implantation. Arrowheads, second harmonic generation (SHG)–positive collagen fibers. Asterisks, SHG-positive myofibers. Scale bar, 25 μm. (B) Normalized fluorescence intensities of LifeAct-GFP and SHG along the indicated line scan arrow from a representative cell. (C) Comparison of LifeAct-GFP peak intensities in individual cells relative to the position of myofiber (Myo) and collagen (Col) SHG signals (n = 10 cells; three mice). (D) Schematic representation of a cross-sectional view of vertical and lateral microchannels. (E) Dimensions of vertical and lateral channels, as measured by a profilometer (n = 40 channels). (F) Migration speeds of HT-1080 fibrosarcoma cells in lateral, vertical, and unconfined microchannels (n ≥ 241 cells; four independent experiments). (G) Phase-contrast image of contiguous microchannels. Cells first experience lateral confinement before transitioning to vertical confinement. Scale bar, 40 μm. (H) Migration speeds of HT-1080 cells inside contiguous channels experiencing first lateral and then vertical confinement (left) or vice versa (right) (n = 150 cells; three independent experiments). (I) Migration speeds of HT-1080 cells in lateral/vertical channels when the basal glass slide of the channel is coated with a thin layer of PDMS (n ≥ 101 cells; two independent experiments). Data represent the mean ± SD (E, F, H, and I) or median (C). **P < 0.01 relative to lateral/unconfined control; §§P < 0.05 relative to myofiber.
These results prompted us to hypothesize that cells, due to their intrinsic dorsoventral polarity, would migrate with distinct modes and efficiencies through different confined migration geometries. To test this, we fabricated a microfluidic device consisting of an array of parallel microchannels (4, 15, 16) with a fixed channel length of 200 μm and a constant cross-sectional area of 30 μm2, albeit with distinct aspect ratios. In this device, cells first adhered and spread on a 2D collagen type I–coated seeding area, which induced cells to establish dorsoventral polarity (17) before entering and migrating inside constricted channels. Vertical confinement was applied by inducing cells to migrate through a short and wide(r) channel (H = 3 μm and W = 10 μm), whereas in lateral confinement cells migrated inside a tall and narrow channel (H = 10 μm and W = 3 μm) (Fig. 1D). The microchannels were aligned in a ladder-like configuration and connected orthogonally to two large channels, which served as a cell seeding source and a chemoattractant reservoir. The dimensions of the vertical and lateral channels were verified using a profilometer to confirm that there was no difference in the cross-sectional area of the two channels (Fig. 1E). Using HT-1080 fibrosarcoma cells as a model system, we observed that cells migrated slower in vertical relative to lateral confinement (Fig. 1F and movie S1). Of note, laterally confined cells migrated with the same speed as cells in unconfined (W, H = 10 μm) channels (Fig. 1F), suggesting that vertical confinement induces a less efficient mechanism of cell migration. This observation also held true for other cancer-derived [e.g., human osteosarcoma (HOS) cells] and normal-like cells (e.g., human dermal fibroblasts and aortic smooth muscle) (fig. S1A). In addition to comparing the responses of cell populations in vertical versus lateral confinement, we tracked the motility of individual cells experiencing both types of confinement sequentially. To this end, we fabricated contiguous microchannels in which migrating cells first experienced lateral confinement for 200 μm before passing through a transition region where cells first migrate through a narrow opening (3 μm W × 3 μm H) before being exposed to vertical confinement (Fig. 1G and movie S2). Microchannels were also fabricated with the reverse orientation (movie S2). In both configurations, HT-1080 cells moved slower in vertical confinement (Fig. 1H and movie S2). The differential cell speeds in vertical versus lateral confinement were also maintained when the glass basal surface of the device was coated with a thin (~100 μm) layer of polydimethylsiloxane (PDMS) to generate four-walled PDMS-based channels (Fig. 1I). All four PDMS-based channel walls were coated with collagen type I (20 μg/ml). These data ruled out the possibility that the observed differences in migration speeds were due to surface chemistry differences of the channel walls. Collectively, these data demonstrate that cell migration efficiency is dictated by migration track geometry.
Pre-established dorsoventral cell polarity determines the differential responses to vertical versus lateral compression in confining migration tracks
We next sought to discern the role that dorsoventral cell polarity played in regulating cellular responses to migration track geometries. In our microfluidic assays, cells were plated on a 2D seeding region before entering confining channels. We hypothesized that cells established dorsoventral polarity on this seeding region, which provided a spatial reference for intracellular signaling mechanisms responsible for sensing and responding to the migration track geometries. To evaluate dorsoventral cell polarity, we examined the spatial distribution of actin and focal adhesions (FAs), which are known to display asymmetric organization in cells seeded on unconfined 2D substrates (17). We found that on the 2D seeding area, cells had significantly reduced actin on their dorsal as compared to ventral surfaces (Fig. 2, A and B). In addition, we found that cells maintained their polarization inside microchannels, as evidenced by stronger actin intensity on the ventral than the dorsal cell surface (Fig. 2, C and D). Similarly, cells on 2D only displayed FAs on their ventral surface (fig. S2, A and B), and in vertical channels, more FAs were present on the ventral than dorsal cell surface (fig. S2C). Individual cells maintained this FA polarity during migration through contiguous microchannels. FAs were predominantly observed on the ventral surface of laterally confined cells, and once cells transitioned to the vertical segment of the channel, significantly more FAs were still observed on the ventral relative to dorsal surface (fig. S2D). This suggests that even after changing confinement geometry, cells retain “memory” of their initial dorsoventral polarity. It is noteworthy that the presence or absence of a thin layer of PDMS on the glass basal surface had no effect on the number of FAs in cells inside collagen I–coated confining microchannels (fig. S2E), demonstrating that dorsoventral polarity was not induced by differences in surface chemistry of the channel walls.
Fig. 2. Dorsoventral polarity determines the efficiency of cell migration in confinement.
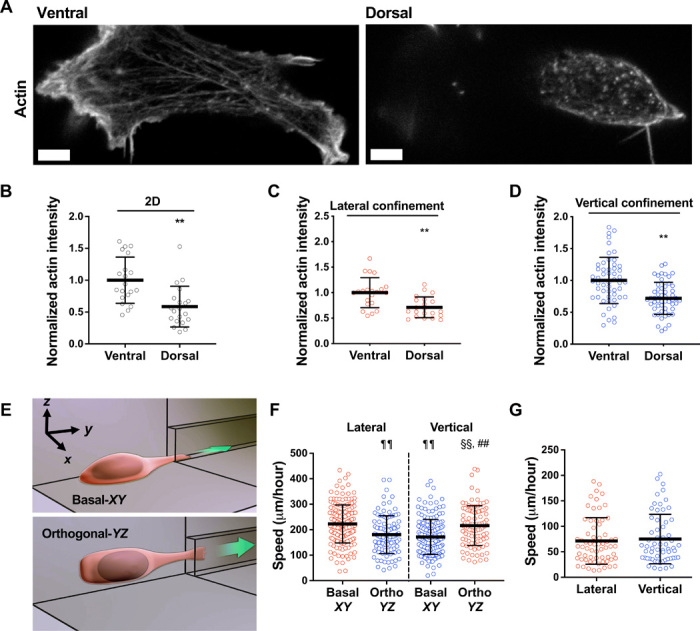
(A) Representative images of HT-1080 cell stained for actin on its ventral and dorsal surfaces on 2D. Scale bars, 5 μm. (B to D) Actin intensity on the ventral and dorsal surfaces of cells fixed and stained with actin phalloidin on 2D (n = 20 cells; two independent experiments) (B) or in lateral (n = 21 cells; two independent experiments) (C) and vertical (n = 53 cells; three or more independent experiments) (D) microchannels. Actin intensity was normalized to the ventral layer. (E) Schematic representation of a microfluidic device in which the 2D cell seeding area is orthogonal (YZ plane) to the typically used basal (XY plane) seeding region. (F) Migration speeds of HT-1080 cells in vertical and lateral microchannels, as assessed after seeding cells on a basal (XY) or orthogonal (YZ) seeding region (n ≥ 96 cells; four independent experiments). (G) Migration speeds of HT-1080 cells in lateral and vertical microchannels treated with poly-l-lysine (PLL) and methoxy poly(ethylene glycol) (mPEG) succinimidyl valerate (n ≥ 61 cells; three independent experiments). Data represent the mean ± SD; **P < 0.01 relative to ventral; ##P < 0.01 relative to vertical basal-XY; ¶¶P < 0.01 relative to lateral basal-XY; §§P < 0.01 relative to lateral orthogonal-YZ.
To validate the role of dorsoventral polarity in confined migration, we fabricated a microfluidic device in which the 2D cell seeding area was orthogonal (YZ plane) to the typically used basal (XY plane) seeding region (Fig. 2E). In this “orthogonal” configuration, the dorsoventral polarity axis pointed in the X direction instead of Z direction (Fig. 2E), and thus, cells sensed lateral confinement as vertical and vice versa, as shown quantitatively for cell speeds (Fig. 2F). This experiment also revealed that (i) cells sensed the four collagen I–coated channel walls identically and (ii) gravity does not play a role in the cell’s distinct responses to different geometries. Elimination of dorsoventral polarity via coating the 2D cell seeding region and the interior of microchannels with poly(ethylene glycol) (PEG) abolished the differential cell speeds in vertical versus lateral confinement (Fig. 2G). Together, our data demonstrate that the ability of cells to sense and respond to changes in the geometry of the local microenvironment depends on their pre-established dorsoventral polarization.
Channel geometry determines migration phenotypes by spatially regulating RhoA activity
Given the differences in cell migration efficiency between vertical and lateral channels, we speculated that established dorsoventral polarity might induce variation in migration phenotype in response to different confinement geometries. To delineate the mode of migration of dorsoventrally polarized cells inside confined microchannels, we either visualized HT-1080 cells fixed and stained with actin phalloidin (Fig. 3, A and B) or performed live cell imaging to track the migration phenotype of HT-1080 cells labeled with LifeAct-GFP and H2B-mCherry (Fig. 3C). In vertical confinement, about 60 to 70% of cells displayed membrane blebs, which were identified as discrete, spherical-like bulges localized at the cell poles (Fig. 3, A to C, and fig. S3A). In contrast, most cells (60 to 70%) maintained a mesenchymal migration phenotype, which was identified by elongated or finger-like protrusions in lateral confinement (Fig. 3, A to C, and fig. S3A). Similar patterns were also observed with HOS cells (fig. S3B). In addition, individual cells migrating through contiguous lateral/vertical channels also transitioned from a mesenchymal phenotype in lateral channels to a blebbing phenotype in vertical channels (Fig. 3C and fig. S3A).
Fig. 3. Channel geometry mediates phenotypic switching of polarized cells by spatially regulating RhoA activity.
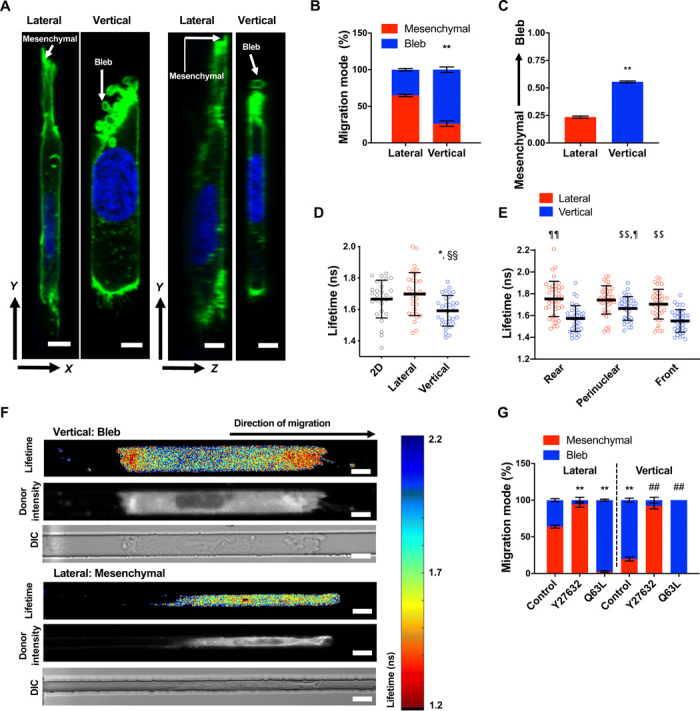
(A) Representative XY/YZ images of a mesenchymal and blebbing cell fixed and stained with actin phalloidin (green) and Hoechst (blue) in lateral and vertical confinement, respectively. Scale bar, 5 μm. (B) Percentage of HT-1080 cells migrating with mesenchymal versus blebbing phenotypes in lateral and vertical confinement (n = 3 independent experiments; ≥20 cells per experiment). (C) Average phenotype score (0, mesenchymal; 1, blebbing) of live LifeAct-GFP–labeled HT-1080 cells during migration through contiguous channels (n = 50 cells; three independent experiments). (D) Donor fluorescence lifetime of RhoA activity biosensor inside vertical and lateral microchannels and on 2D, as measured by FLIM-FRET (n ≥ 27 cells; four independent experiments). (E) Spatial distribution of RhoA activity inside vertical microchannels as measured by FLIM-FRET (n ≥ 35 cells; five independent experiments). (F) Heat map of RhoA activity biosensor of representative cells inside vertical or lateral microchannels, as imaged by FLIM-FRET. ns, nanoseconds; DIC, differential interference contrast. Scale bars, 10 μm. (G) Percentage of control, Y27632-treated (10 μM), or constitutively active RhoA (Q63L)–expressing HT-1080 cells, migrating with mesenchymal versus blebbing phenotypes in lateral and vertical confinement (three or more independent experiments; ≥20 cells per condition). Values represent the mean ± SD (D and E) or the mean ± SEM (B, C, and G); *P < 0.05 and **P < 0.01 relative to lateral control; ##P < 0.01 relative to vertical control; §§P < 0.01 relative to 2D; $$P < 0.01 relative to vertical front; ¶P < 0.05 and ¶¶P < 0.01 relative to vertical rear.
We and others have previously demonstrated that confinement activates RhoA (18) and bleb formation requires the activation of RhoA-dependent contractility (19). We thus hypothesized that dorsoventrally polarized cells might regulate RhoA differently in response to different confinement geometries, resulting in different migration phenotypes. To test this hypothesis, we quantified RhoA activity in cells migrating in vertical and lateral confinement using the Förster resonance energy transfer (FRET)–based RhoA activity biosensor combined with confocal fluorescence lifetime imaging (FLIM), which measured increased RhoA activity through decreased donor fluorescence lifetime and was previously validated using appropriate controls (fig. S3C) (18). Cells in vertical confinement exhibited overall higher levels of RhoA activity than 2D unconfined or laterally constricted cells (Fig. 3D). Vertically confined cells displayed spatial regulation of RhoA, with maximal activity observed at the cell poles where membrane blebs form (Fig. 3, E and F). In contrast, laterally confined cells had relatively uniform RhoA activity (Fig. 3, E and F). In concert with the FLIM/FRET data suggesting that elevated RhoA activity is responsible for membrane blebbing, cells expressing constitutively active RhoA mutant (Q63L) protein displayed nearly exclusively a bleb-based mode of migration irrespective of the type of confinement (Fig. 3G). Conversely, inhibition of Rho-associated protein kinase (ROCK) via cell treatment with Y27632 (10 μM) (Fig. 3G) promoted a mesenchymal phenotype in both vertical and lateral confinement. Despite their mesenchymal mode of migration, Y27632-treated cells moved slower than control cells (fig. S3D), presumably because of their inability to efficiently retract their rear end due to inhibition of contractility. Nevertheless, Y27632-treated cells migrated faster than cells expressing Q63L (fig. S3D), suggesting that cells with a mesenchymal phenotype move faster than cells with a bleb-based phenotype in confined spaces.
As myosin II is downstream of RhoA, we sought to identify its role in promoting a blebbing migration phenotype. Inhibition of actomyosin contractility via cell treatment with blebbistatin (50 μM) (Fig. 4A) promoted a mesenchymal phenotype in both vertical and lateral confinement similar to Y27632-treated cells. Blebbistatin-treated cells moved slower than vehicle control cells (Fig. 4B) because of their inability to efficiently retract their rear end (movie S3). In contrast, partial inhibition of actomyosin contractility via a low concentration of blebbistatin (2 μM) promoted mesenchymal migration and increased cell speed in vertical confinement (Fig. 4, A and B) by enabling efficient retraction of the cell’s rear end (movie S4). These findings suggest that optimal contractility levels at the cell trailing edge are required for efficient migration in confinement. While stable short hairpin RNA (shRNA) knockdown of myosin IIB (MIIB or MYH10) (18) had no effect on the migration phenotype, myosin IIA (MIIA or MYH9) knockdown (18) promoted mesenchymal migration in both types of confinement (Fig. 4C) similar to blebbistatin- or Y27632-treated cells. MIIA strongly accumulated at the poles of cells exhibiting membrane blebs, as we have shown previously (18), further supporting the involvement of MIIA in membrane bleb formation under vertical confinement. We also observed organized myosin fibers around the cell nucleus in vertically confined cells (Fig. 4, D and E, and fig. S4A). MIIA fibers were detected on the dorsal surface of the nucleus and localized to areas of nuclear deformation, as has also been shown previously (18). These fibers were less prominent upon low blebbistatin treatment (Fig. 4, D and E, and fig. S4A). Such perinuclear myosin fibers were not observed around the nuclei of 2D or laterally confined cells (Fig. 4, D and E, and fig. S4A). Myosin activity is well known to promote both cytoskeletal and nuclear tension (20–22). Thus, the localization of myosin both at the cortex and around the nucleus of vertically confined cells suggested that these cells might be under elevated tension. If this were true, then FAs, which are important mechanosensors of myosin II–mediated cytoskeletal tension (21), should mature in vertical confinement. Cells in vertical confinement had more and larger FAs as compared to cells in lateral confinement and larger FAs compared to cells on 2D (Fig. 4, F and G). However, treatment with low doses of blebbistatin significantly reduced the size, but not number, of FAs in vertically confined cells (fig. S4, B to D), suggesting that perinuclear myosin-dependent nuclear and cytoskeletal tension might regulate FA maturation. Of note, more and larger FAs were found at the front of vertically confined cells, where cytoplasmic blebs form, as compared to the perinuclear and rear regions (fig. S4, E and F). FAK is a signaling molecule that becomes activated by tension on FA complexes (23). Cell treatment with a low dose (0.25 μM) of the selective FAK inhibitor 14 decreased the fraction of cells displaying membrane blebbing in vertical confinement (Fig. 4H) and abrogated the differences in cell speeds between vertical and lateral confinement (Fig. 4I). Cumulatively, these results point to a role of intracellular tension in regulating migration mode and efficiency in confinement.
Fig. 4. Optimal levels of contractility promote efficient cell migration in confinement.
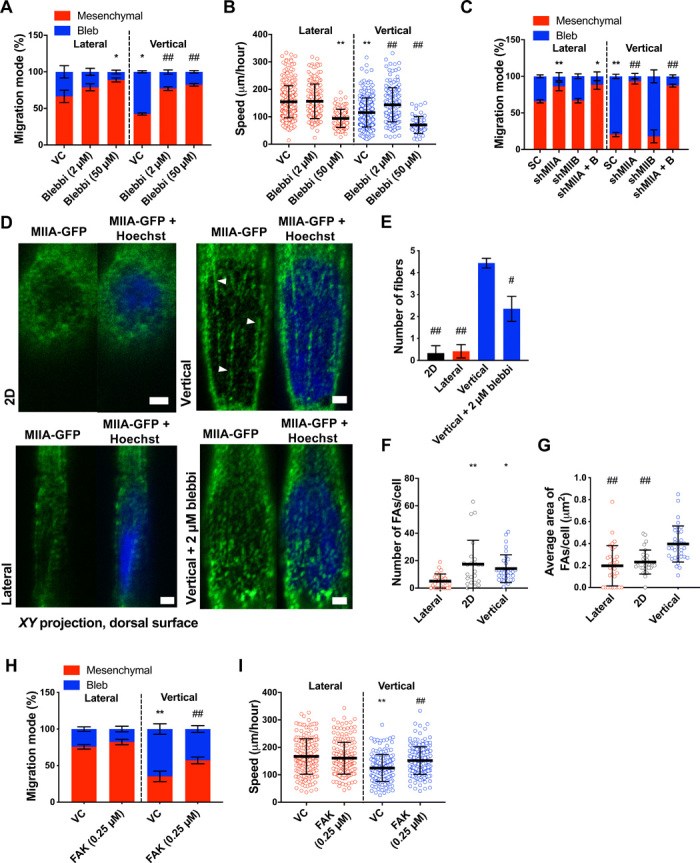
(A) Migration phenotypes of HT-1080 vehicle control and blebbistatin-treated cells (n = 3 independent experiments; ≥20 cells per condition). (B) Migration speeds of vehicle control and blebbistatin-treated (2 or 50 μM) HT-1080 cells (n ≥ 61; three independent experiments). (C) Migration phenotypes of scramble control (SC) and MIIA- and/or MIIB-knockdown HT-1080 cells (n ≥ 3 independent experiments; ≥20 cells per condition). (D) Images and (E) quantification of perinuclear myosin (XY plane, dorsal surface) from representative HT-1080 cells expressing MIIA-GFP and stained with Hoechst on 2D or in vertical (±2 μm of blebbistatin) or lateral confinement. White arrowheads indicate representative myosin fibers (n = 3 independent experiments). Scale bars, 2 μm. (F and G) Average number (F) and area (G) of paxillin-GFP–labeled FAs on 2D and inside lateral and vertical microchannels (n ≥ 24 cells; four independent experiments). (H) Migration phenotypes of vehicle control and low-dose (0.25 μM) FAK-treated HT-1080 cells in vertical and lateral confinement (n = 4 independent experiments; ≥20 cells per experiment). (I) Migration speeds of vehicle control and low-dose FAK-treated HT-1080 cells (n ≥ 170 cells; three independent experiments) in vertical and lateral confinement. Values represent the mean ± SD (B, F, G, and I) or the mean ± SEM (A, C, E, and H). *P < 0.05 and **P < 0.01 relative to lateral control; #P < 0.05 and ##P < 0.01 relative to vertical control.
The nucleus is a mechanical barrier that triggers RhoA activation in vertical confinement
In light of the accumulation of myosin around the nucleus in vertical confinement and a reported link between perinuclear myosin, nuclear tension, and stiffness (22, 24, 25), we hypothesized that the nucleus, which has a rate-limiting role in confined cell migration, might be less efficiently deformed and become stiffer, thus acting as a mechanical barrier in vertically confined cells. Along these lines, live imaging of histone H2B-mCherry–transduced HT-1080 cells showed that longer times were required for both nuclear and cell entry in vertical than lateral confinement (Fig. 5A and fig. S5A). In vertical channels, the nucleus is non-uniformly deformed by actomyosin bundles on its dorsal surface and creates a plug that compartmentalizes the cell posterior and anterior, thereby reducing flow of cytoplasmic constituents from the rear to the front of the cell (18). However, upon photoactivation of green fluorescent protein (GFP) at the cell trailing edge, we found that flow to the cell leading edge was significantly faster in laterally versus vertically confined cells (fig. S5B). This suggests that in the lateral geometry, the perinuclear gaps between the nucleus and channel walls are larger than those in vertical channels, presumably due to enhanced nuclear deformation on the lateral sides of the nucleus as compared to dorsoventral deformation of the nucleus in vertical channels. Along these lines, nuclear volume was significantly reduced in laterally confined cells (fig. S5C). Collectively, these data reveal that the nucleus may be more efficiently deformed in lateral than vertical confinement, suggesting that distinct migration track geometries might differentially regulate nuclear stiffness.
Fig. 5. The nucleus becomes stiffer and acts as a mechanical barrier in vertical confinement.
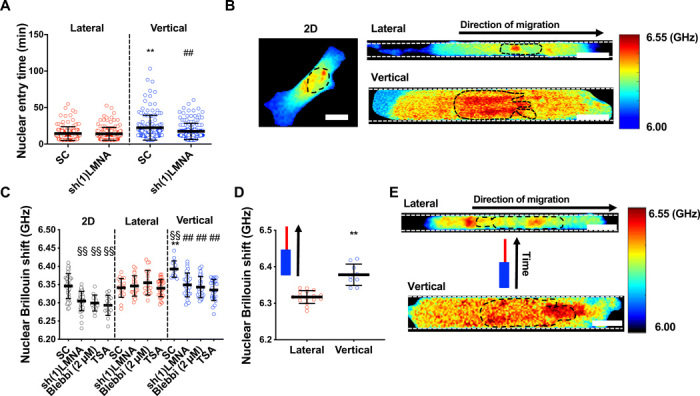
(A) Nuclear entry time of H2B-mCherry–labeled LMNA-KD (knockdown) or scramble control HT-1080 cells (n ≥ 143; three independent experiments) in lateral and vertical channels. (B) Representative heat map of Brillouin shift for 2D, vertically, and laterally confined cells. Scale bars, 10 μm. (C) Nuclear Brillouin shift for scramble control, LMNA-KD–treated, blebbistatin (2 μM)–treated, and TSA (100 ng/μl)–treated HT-1080 cells in 2D, lateral, and vertical confinement (n ≥ 13 cells; two or more independent experiments). (D) Nuclear Brillouin shift of cells in the vertical and lateral segments of contiguous microchannels, where cells experienced vertical and subsequently lateral confinement (n ≥ 9 cells; two independent experiments). (E) Heat map of Brillouin shift for an individual cell migrating, first, through the vertical and, subsequently, the lateral segment of a contiguous microchannel. Scale bars, 10 μm. Values represent the mean ± SD; **P < 0.01 relative to lateral control; ##P < 0.01 relative to vertical control; §§P < 0.01 relative to 2D control.
To investigate how confinement affects nuclear mechanical properties, we used an all-optical technique called confocal Brillouin microscopy to directly sense the nuclear mechanics of confined cells in a noninvasive and noncontact manner. The Brillouin technique is based on the principle of spontaneous Brillouin scattering, which is a physical process that introduces an optical frequency shift (Brillouin shift) to incident light after interaction with matter. Hence, by measuring the Brillouin shift of outgoing scattered light at the local position of a sample in a confocal manner, Brillouin microscopy can directly sense the 3D mechanical properties (longitudinal modulus) of a sample with high spatial resolution (26). We compared the Brillouin shift of control cells on 2D versus in vertical or lateral channels. While there was no difference in nuclear Brillouin shift between 2D and lateral confinement, vertically confined nuclei displayed an increased Brillouin shift (Fig. 5, B and C), suggesting that the nucleus is stiffer in these channels. Of note, cells on 2D and in both vertically and laterally confined channels displayed higher Brillouin shift values in the nucleus compared to the cytoplasm (fig. S5, D to I), and the Brillouin shift decreased gradually within the cytoplasm as distance from the nucleus increased (fig. S5, D to I). We also examined nuclear Brillouin shift of cells migrating through contiguous microchannels, where cells first migrated through vertical confinement before transitioning to lateral confinement. The nuclear Brillouin shift was significantly reduced for cells that had transitioned to the lateral microchannels as compared to the cells in the vertical segment of the channel (Fig. 5D), suggesting that individual cells may alter their nuclear mechanical properties when transitioning between confinement geometries. Tracking the nuclear Brillouin shift of an individual cell revealed that nuclear Brillouin shift significantly decreased after the cell traveled from the vertical to lateral region of the channel (Fig. 5E).
Since nuclear stiffness scales with lamin-A expression (27), we evaluated the effect of lamin-A silencing (fig. S6A) on nuclear Brillouin shift and confined migration. For cells on 2D, the nuclear Brillouin shift was decreased upon lamin-A knockdown (Fig. 5C). A decrease in the elastic modulus of lamin-A knockdown cells was also confirmed using atomic force microscopy (fig. S6B), validating the Brillouin shift as a good metric of nuclear stiffness. Moreover, upon lamin-A knockdown, the nuclear Brillouin shift of vertically confined cells decreased to match levels measured in lateral confinement (Fig. 5C). Lamin-A knockdown had no effect on the cytoplasmic Brillouin shift of 2D or laterally confined cells relative to scramble control cells (fig. S5, E and G), whereas this intervention moderately decreased Brillouin shift in the cytoplasm as compared to the pronounced decrease measured in the nucleus of vertically confined cells (fig. S5I). Given that RhoA is elevated in vertical channels and that lamin-A depletion has been shown to regulate RhoA activity (28), lamin-A may alter cytoplasmic stiffness through RhoA-dependent stress fiber formation, as it has previously been shown that elevated RhoA activity and actin and myosin fiber assembly increase cells’ Young’s modulus (28, 29). Lamin-A knockdown abolished the differences not only in nuclear and cell entry times (Fig. 5A and fig. S5A) but also in the speeds of migration in vertical versus lateral confinement (Fig. 6A). In contrast, this intervention had no effect on nuclear Brillouin shift of laterally confined cells (Fig. 5C). This may suggest that confinement of the nucleus triggers a compensatory mechanism to maintain nuclear stiffness above a certain threshold. Such a mechanism would likely involve the interplay of factors that have been implicated in nuclear stiffening, such as emerin phosphorylation, increases in the ratios of heterochromatin to euchromatin, or increase in perinuclear actomyosin fibers (22, 24, 25, 30). Consequently, nuclear entry times and cell migration speeds were unchanged by lamin-A knockdown in lateral channels (Figs. 5A and 6A). In light of these findings, we next examined whether nuclear stiffness might also be responsible for the elevated levels of RhoA activity exhibited by vertically confined cells. Using confocal FLIM, we demonstrated that lamin-A depletion globally reduced RhoA activity in vertical confinement (Fig. 6B). Consistent with decreased RhoA activity, this molecular intervention increased the percentage of cells displaying a mesenchymal phenotype in vertical confinement (Fig. 6C). While we and others have found that lamin-A/C becomes apically polarized in fibroblasts (31, 32) (fig. S6C) by compressive forces applied to the nucleus by the actin cap (31, 32), lamin-A/C polarity was not detected in HT-1080 fibrosarcoma cells (fig. S6C), suggesting that lamin-A/C polarity is not predominantly responsible for these cells’ geometric sense. To verify that the effects observed with lamin-A knockdown were due to changes in nuclear stiffness and not related to other lamin-A–dependent signaling pathways, we treated cells with trichostatin A (TSA), a histone deacetylase inhibitor that has been shown to increase euchromatin and consequently reduce nuclear stiffness (33). Similar to lamin-A–depleted cells, TSA-treated cells had reduced nuclear Brillouin shift in vertical confinement (Fig. 5C) and displayed a predominantly mesenchymal phenotype in vertical channels (Fig. 6D). It is worth noting that TSA treatment had a more pronounced effect on cell migration phenotype than lamin-A knockdown, suggesting that this pharmacological intervention might promote additional pathways that suppress cytoplasmic blebbing aside from nuclear stiffness. Conversely, treatment of cells with the histone demethylase inhibitor methylstat, which has been shown to increase heterochromatin and nuclear rigidity (30), increased nuclear Brillouin shift of cells on 2D and in lateral and vertical channels relative to 2D control values (fig. S6D). Furthermore, this intervention increased the percentage of cells migrating with a blebbing phenotype in lateral confinement (fig. S6E). Previous work has shown that tension applied to the nucleus promotes nuclear stiffening (25). We thus hypothesized that cells treated with a low dose of blebbistatin, which abolishes speed and phenotype differences in vertical versus lateral confinement (Fig. 4, A and B), would display reduced nuclear stiffness in vertical channels, presumably due to lower myosin II–dependent nuclear tension. This intervention decreased the Brillouin shift of vertically confined cells to levels measured in laterally confined cells (Fig. 5C), pointing to nuclear tension as a regulator of nuclear stiffness. Notably, nuclear Brillouin shift values measured after low blebbistatin treatment were comparable to nuclear Brillouin shift values upon lamin-A knockdown (Fig. 5C). Previous studies have shown that inhibition of myosin contractility can promote lamin-A turnover and nuclear softening via phosphorylation of lamin-A at Ser22 (24). We thus hypothesized that myosin might regulate nuclear stiffness in confinement through a cross-talk with lamin-A. Treatment with low doses of blebbistatin significantly increased phosphorylated lamin-A levels per cell (Fig. 6E). Overall, our data indicate that the nucleus acts as a barrier in vertical channels because of confinement-induced nuclear stiffening, which is regulated via a cross-talk between myosin II and lamin-A.
Fig. 6. Nuclear stiffness regulates RhoA activity and cell migration phenotype in confinement.
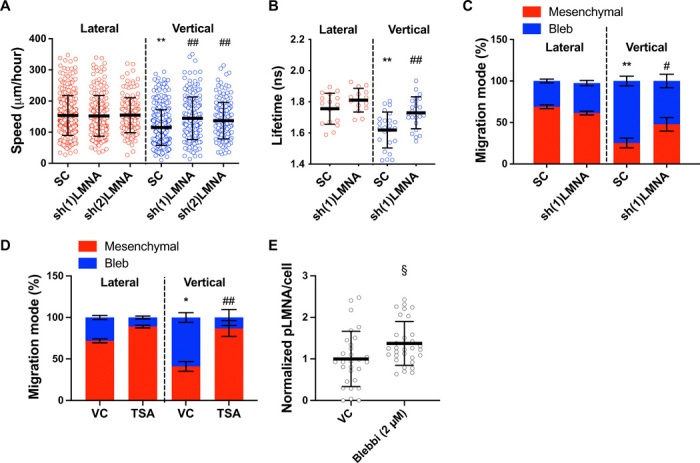
(A) Migration speeds of HT-1080 scramble control and LMNA-KD cells (n ≥ 149; three independent experiments) using two different shRNA sequences. (B) RhoA activity of scramble control and LMNA-KD HT-1080 cells, as measured by FLIM-FRET (n ≥ 14 cells; two or more independent experiments). (C) Migration phenotypes of scramble control and LMNA-KD HT-1080 cells, as assessed after fixing and staining cells with actin phalloidin (n ≥ 3 independent experiments; ≥20 cells per condition). (D) Migration phenotypes of vehicle control and TSA-treated (100 ng/μl) HT-1080 cells, as assessed after fixing and staining cells with actin phalloidin (n ≥ 3 independent experiments; ≥10 cells per condition). (E) Quantification of phosphorylated LMNA (pLMNA) per cell for vehicle control versus blebbistatin-treated (2 μm) cells, as measured from the average intensity projection of cells fixed and stained for pLMNA (n ≥ 29 cells; two independent experiments). Values represent the mean ± SD (A, B, and E) or means ± SEM (C and D). *P < 0.05 and **P < 0.01 relative to lateral control; #P < 0.05 and ##P < 0.01 relative to vertical control; §P < 0.05 relative to 2D control.
DISCUSSION
We here demonstrate that intrinsic dorsoventral polarity directs cell responses to different migration track geometries. Thus, pre-established cell polarization regulates the efficiency of cell motility in confined spaces. In vivo, this intrinsic polarity may be the result of the heterogeneity of the local microenvironment. For instance, HT-1080 fibrosarcoma cells preferentially polarize their perceived ventral and dorsal surfaces toward myofibers versus collagen fibers, respectively. Using in vivo–inspired in vitro channels mimicking the width of invasion-guiding dermal structures, we show that the nucleus acts as a key sensor of confinement in dorsoventrally polarized cells by regulating its stiffness in response to different confinement geometries. This, in turn, modulates the mechanisms of cell migration in vertical and lateral channels. While dorsoventral polarity may not be present in all in vivo environments, polarity cues are likely provided by linear-structured tissue regions, including perimyofiber and perivascular niches, which guide cell migration through in vivo migration tracks.
While previous studies have elucidated the importance of anterior/posterior cell polarity of molecules such as Rho GTPases for the formation of actin-based migratory protrusions and maintenance of persistent migration (14), this study is the first to explore how dorsoventral polarity cues affect cell migration in response to different track geometries. Dorsoventrally polarized cells migrated through either vertically or laterally confining channels, which had the same cross-sectional area but distinct aspect ratios. The dimensions of these channels were chosen to mimic tracks cells might navigate through in vivo, where moving cells encounter asymmetric topographies and ligand conditions. Myofibers are surrounded by a basement membrane, forming an important tissue structure guiding cancer invasion (34), and, as shown here, support ventral actin polarization. Cell polarity, as measured by actin and FA localization, was maintained during migration through vertical and lateral channels and dictated the distinct cell responses to these geometries. Dorsoventrally polarized cells modulate their migration behavior in vertical and lateral channels via nuclear mechanosensing, suggesting a role for nuclear polarity in sensing migration track geometries. While we and others have found that in fibroblasts, lamin-A/C becomes apically polarized via contractile forces generated by a perinuclear actin cap via LINC complexes (31, 32), lamin-A/C polarity was not detected in HT-1080 fibrosarcoma cells. While lamin-A/C polarity specifically may not be responsible for how cells sense vertical versus lateral channels, asymmetry of other aspects of the nuclear or perinuclear region may play a role in cells’ geometric sensing. In addition to lamin-A/C, chromosomes are differentially organized both radially and dorsoventrally inside the nucleus, with stiff heterochromatin located more toward the nuclear periphery and hyperacetylated chromatin located toward the apical nuclear surface (32, 35). In agreement with this, our results using the histone deacetylase inhibitor TSA and the histone demethylase inhibitor methylstat suggest that altering the ratios of heterochromatin to euchromatin in the nucleus can alter cell migration phenotype.
In this work, we examine how vertical and lateral confinement of dorsoventrally polarized cells affect the mechanical properties of the cell nucleus using Brillouin microscopy. Our Brillouin microscopy measurements reveal that vertical, but not lateral, confinement increases nuclear Brillouin shift relative to 2D cells, suggesting increased nuclear stiffness in these channels. Brillouin microscopy has recently emerged as a promising technique for investigating cellular and nuclear mechanical properties in a contact-free manner (26, 36). Although the Brillouin-derived longitudinal modulus is not directly related to traditionally measured elastic modulus, a strong correlation between the two moduli appears in cells (26), indicating that these two moduli change in the same direction in many physiological and pathologic processes. Recent work has benchmarked Brillouin results of the intact cellular nucleus by taking measurements for known effects (i.e., lamin-A/C knockdown and chromatin decondensation) (36), which have been well characterized by conventional methods, such as micropipette aspiration and atomic force microscopy. In these studies, the nuclear mechanical changes measured by the Brillouin technique are consistent with those measured using conventional methods. Here, our data showing reduction in nuclear Brillouin shift upon lamin-A knockdown or TSA treatment and increase in nuclear Brillouin shift upon methylstat treatment further align with these benchmarks. These validations together support Brillouin microscopy as a feasible technique for probing the mechanical properties of cell nucleus in lateral and vertical channels.
Dorsoventrally polarized cells respond to the physical cues of vertical and lateral channels by regulating nuclear tension and stiffness. Differences in nuclear tension of vertically and laterally confined cells arise from the organization of the perinuclear myosin networks in these channels. In vertical channels, MIIA fibers were detected on the dorsal surface of the nucleus and localized to areas of nuclear deformation. In contrast, such perinuclear myosin networks were not detected in cells on 2D or in lateral channels. Recent work using a Nesprin-2G tension sensor revealed that force applied to the nucleus by perinuclear actomyosin fibers, such as those detected in vertical channels, elevates nuclear tension and, in turn, promotes nuclear stiffening (20, 22, 25). Along these lines, our Brillouin microscopy measurements reveal that disruption of perinuclear myosin via low blebbistatin treatment abolishes the elevated nuclear Brillouin shift detected in vertical confinement. Nuclear stiffening in response to tension may be regulated by proteins of the nucleoskeleton, as has been demonstrated by others (25). Specifically, force application to the nucleus recruits lamin-A to the LINC complex and reinforces their physical connection, contributing to nuclear stiffening (25). This recruitment is controlled by Src family kinase phosphorylation of the LEM-domain protein emerin (25). Another mechanism by which the nucleus modulates its stiffness is through lamin-A/C phosphorylation at Ser22, which regulates lamin-A/C turnover and physical properties (24). Studies have shown that mesenchymal stem cells (MSCs) on stiff gels, whose nuclei display a flat morphology resembling the nuclei in vertical channels, have a lower phosphorylated LMNA (pLMNA)/LMNA ratios than MSCs on soft gels, suggesting that high nuclear tension favors lamin-A/C reinforcement, while low nuclear stress promotes lamin-A/C degradation (24). Knockdown of lamin-A or treatment with blebbistatin, which increases phosphorylated lamin-A, abolished migration differences between vertical and lateral channels. These results suggest that variation in the ratios of pLMNA/LMNA in these channels may be one key contributor to differences in nuclear stiffness observed in our system.
Nuclear stiffening tunes both the activity and the spatial distribution of RhoA-dependent actomyosin contractility, thereby regulating cell migration phenotype and efficiency. It has previously been reported that the nucleus can regulate RhoA (23). Both knockdown of lamin-A and disruption of the LINC complex reduce cells’ RhoA activity (23, 28). Studies performed using enucleated cells revealed that the nucleus and the associated perinuclear cytoskeleton regulate whole cell tension (37). Along these lines, previous work has found that compared to wild-type fibroblasts, LMNA−/− fibroblasts have smaller FAs (28), which are important mechanosensors of cytoskeletal tension (21). Therefore, stiffer nuclei, such as those in vertical confinement, should induce higher intracellular tension than softer nuclei, such as those in lateral confinement. In agreement with this, FAs matured to larger sizes in vertical than lateral channels, and inhibition of FAK, a signaling molecule that becomes activated by tension on FA complexes (23), promoted the mesenchymal phenotype and abolished migration differences between vertical and lateral channels. Rho guanine nucleotide exchange factors (GEFs), such as GEFH1, LARG, and p115RhoGeF, can become activated in response to tension on integrins (23, 38). These GEFs are all downstream of FAK signaling and induce RhoA activation (23, 38). Elevated RhoA in vertical confinement is consistent with activation of such GEFs caused by elevated nuclear and cytoskeletal tension. Overall, our data suggest a conceptual model in which dorsoventrally polarized cells differentially regulate their nuclear and consequently cytoplasmic tension in response to confined migration geometry. This leads to changes in the activity of the RhoA pathway, which regulates cells’ modes and mechanisms of migration.
Regulation of RhoA and actomyosin contractility plays a central role in mediating trailing edge retraction and dictating cell migration mode (39). We observed that treating cells with high doses of blebbistatin completely abrogates contraction of the cell rear, while treatment with low doses of blebbistatin, which only partially reduces myosin II activity, allows for essentially normal rear-end contraction. Our data suggest that optimal levels of RhoA-dependent contractility are required for efficient migration in confinement. Because contractility levels in confinement are partially regulated by nuclear stiffness, this suggests that optimal nuclear stiffness facilitates confined migration. If a cell’s nucleus is extremely soft or absent, low intracellular tension may reduce cell migration. In line with this, a recent study revealed that enucleation significantly reduces 3D cell migration (37), presumably because the nucleus plays a key role in an integrated molecular clutch model and is important for promoting contractile energy and traction stress in the cell, which controls cell sensitivity to mechanical cues (37). On the other hand, the nucleus has been shown to be a major obstacle during 3D cell migration (7). Elevated nuclear stiffness in confinement may exacerbate this problem and hyperactivate cell contractility, resulting in a slow bleb-based migration. Partial reduction of nuclear stiffness in vertical channels via treatment with low blebbistatin or lamin-A knockdown was sufficient to promote a mesenchymal phenotype and increase migration speeds.
In summary, we propose a model in which preexisting dorsoventral polarity, observed both in vitro and in vivo, directs cell responses to distinct migration track geometries by spatially regulating RhoA activity, which, in turn, controls cell plasticity. Vertical confinement, directed along the axis of dorsoventral polarity, induces myosin II–dependent nuclear tension and nuclear stiffening, leading to polarized RhoA activation at the cell poles. The activation of the RhoA-dependent pathways mediates a slow bleb-based migration. In lateral confinement, reduced nuclear stiffness leads to lower and nonpolarized RhoA activity and promotes a fast mesenchymal migration. While dorsoventral polarity is required for sensing distinct physical features of the local microenvironment, migration track geometries can also regulate anterior/posterior cell polarity via intracellular signaling pathways that integrate biochemical and mechanical cues. Our work enhances our understanding of the complex process of confined cell migration and provides a perspective on how cells sense and respond to different geometries of migration tracks.
MATERIALS AND METHODS
Experimental methods
Cell culture and pharmacological inhibitors
Human HT-1080 fibrosarcoma cells, dermal fibroblasts, and HOS cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing glucose, l-glutamine, and sodium pyruvate (Gibco) and supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin (10,000 U/ml; Gibco). Human aortic smooth muscle cells (provided by S. Gerecht at Johns Hopkins) were cultured in Smooth Muscle Cell Growth Medium (PromoCell). Cells were grown in an incubator maintained at 37°C and 5% CO2 and passaged every 2 to 4 days.
In select experiments, cells were treated with the following pharmacological agents and corresponding vehicle controls. The following reagents were obtained from Sigma-Aldrich: Y27632 (10 μM), blebbistatin (2 or 50 μM), TSA (100 ng/μl), methylstat (2 μM), and latrunculin A (2 μM). For experiments with TSA and methylstat, cells were pretreated overnight or for two nights. All pharmacological agents were added to medium in inlet wells of microfluidic device.
Cloning, lentivirus preparation, transduction, and transfection
To generate shRNA lentiviral vectors, the target sequences were subcloned into pLVTHM (Addgene, Cambridge, MA; plasmid #12247) using Mlu I and Cla I as restriction sites or pLKO.1 (Addgene, Cambridge, MA; plasmid #8453) using Age I and Eco RI as restriction sites. The target sequences are as follows: scramble control: sh1 (GCACTACCAGAGCTAACTCAGATAGTACT), human MYH9 (ACGGAGATGGAGGACCTTATG), human MYH10 (GGATCGCTACTATTCAGGA), human LMNA sh1(CTCATCTATCTCAATCCTAAT), and human LMNA sh2 (GATGATCCCTTGCTGACTTAC).
pLenti.PGK.LifeAct-GFP.W (plasmid #51010), pLenti.PGK.H2B-mCherry (plasmid #51007), MYH9-GFP (plasmid #11347), tetO-FUW-eGFP-RHOA-Q63L (plasmid #73081), tetO-FUW-eGFP (plasmid #73083), FUdeltaGW-rtTA (plasmid #19780), RhoA2G FRET biosensor (plasmid #40176 and #40179), psPAX2 (plasmid #12260), and pMD2.G (plasmid #12259) were purchased from Addgene. Lentivirus production and infection and transient transfections were performed as described previously (18).
In vivo experiments
Dorsal skin-fold chambers were surgically transplanted onto 10- to 14-week-old athymic nu/nu mice (MD Anderson Experimental Radiation Oncology Breeding Core). Identity of the HT-1080 cells was verified by SNP_ID assay (Sequenom, MassARRAY System; Characterized Cell Line Core Facility, MD Anderson Cancer Center, Houston, TX, USA), and lack of contamination with mycoplasma was routinely verified using the MycoAlert Mycoplasma Detection Kit (Lonza). One day after surgery, 2.5 × 105 to 5.0 × 105 HT-1080 (dual color) human fibrosarcoma cells expressing H2B-mCherry (nuclear reporter) and LifeAct-GFP (F-actin reporter) were injected into the dermis adjacent to the deep dermal vascular plexus with a 30-gauge needle, as described previously (3). Tumor growth was then monitored by epifluorescence and multiphoton microscopy for up to 14 days. For intravital multiphoton microscopy, mice were anesthetized with isoflurane (1 to 3% in oxygen) and the skin-fold chamber was stably mounted on a temperature-controlled stage (37°C). Animal studies were approved by the Institutional Animal Care and Use Committee of the University of Texas, MD Anderson Cancer Center (protocol 00001002) and performed according to the institutional guidelines for animal care and handling.
Imaging was performed on a customized multiphoton microscope (TriM Scope II, LaVision BioTec), equipped with three tunable Ti:Sa (Coherent Ultra II Titanium:Sapphire) lasers and two optical parametric oscillators (Coherent APE). A long-working distance, 25× NA (numerical aperture) 1.05 water immersion objective (Olympus) was used for image acquisition. Multispectral detection was performed using four photomultipliers in the backward configuration using single excitation wavelengths in consecutive scans to separate the following excitation and emission channels: LifeAct-GFP (920 nm; 525/50 nm), H2B-mCherry (1090 or 1180 nm; 620/60 nm), second harmonic generation (SHG) (1090 nm; 525/50 nm), and Alexa Fluor 750 (1280 nm; 810/90 nm). 3D time-lapse acquisitions were performed by acquiring multichannel z stacks with step size of 4 μm every 8.5 min. All time-lapse images were drift-corrected when necessary using Fiji/ImageJ2 plugin Correct 3D Drift (40) using the SHG signal as the reference channel. Single–time point, 3D multichannel z-stacks were acquired with z step sizes of 4 or 5 μm. LifeAct-GFP and SHG line scan profiles were obtained on single z slice images and normalized accordingly, (intensityi − intensitymin)/intensitymax. For comparison of normalized LifeAct-GFP fluorescence of cell regions proximal to SHG signals from myofibers and collagen, the average of three data points from a line scan centered on the LifeAct peak was calculated for both myofiber and collagen proximal positions.
Photolithography and device fabrication
PDMS-based microfluidic devices, which consist of an array of parallel vertical or lateral channels with a fixed channel length of 200 μm and a constant cross-sectional area of 30 μm2, were fabricated as described previously (41–43). Vertical confinement was applied by inducing cells to migrate through a channel with a height, H, of 3 μm (and a width, W, of 10 μm), whereas in lateral confinement the width was set to 3 μm (and H = 10 μm). To generate basal and orthogonal surfaces for cell entry, an entry area (W = 50 μm, H = 50 μm) was added in front of the vertical and lateral channels. These devices were fabricated following the same procedure. In select experiments, contiguous PDMS-based microchannels were fabricated, in which cells encountered sequentially lateral and vertical confinement or vice versa. In this case, the triple-layer silicon master was fabricated in an additive fashion beginning with the smallest feature (vertical confinement, height: 3 μm), followed by the intermediate feature (lateral confinement, 10 μm), and finishing with the tallest feature (seeding/exit regions, 50 μm). Both vertical and lateral features are each 200 μm in length with a 50-μm overlapping transition region that intersects and separates the two channels. Hence, the final channel length is 450 μm. The dimensions of all channels were verified using a profilometer. For cell migration experiments, channels were coated with collagen I (20 μg/ml) (Collagen I Rat Protein, Tail; Thermo Fisher Scientific).
Microfluidic device seeding and live cell imaging
Cells were prepared as described previously (18). Briefly, cells were detached [0.05% trypsin-EDTA (Gibco)], centrifuged (300g for 5 min), and resuspended in DMEM (1% penicillin/streptomycin, no FBS) to a concentration of 5 × 106 cells/ml. Cell suspension (10 to 20 μl) was seeded into the microfluidic device via pressure-driven flow. For independent lateral and vertical channel devices (200 μm in length), the bottom three wells were filled with DMEM (no FBS, 1% penicillin/streptomycin), while the top well was filled with DMEM (10% FBS, 1% penicillin/streptomycin) to create a chemotactic gradient within the device. For contiguous lateral/vertical channel devices, all wells were filled with DMEM (10% FBS, 1% penicillin/streptomycin). Devices were incubated at 37°C, 5% CO2 before imaging.
To PEGylate our microchannel devices, we first treated the surface with poly-l-lysine (100 μg/ml) (Sigma-Aldrich) at room temperature for 30 min. Next, the devices were rinsed with Hepes buffer [100 mM (pH 8 to 8.5)] and coated with methoxy PEG (mPEG)–succinimidyl valerate [Laysan Bio; 50 mg/ml in Hepes buffer (pH 8 to 8.5)] at room temperature for 1 hour. After washing with phosphate-buffered saline (PBS) (Gibco), cells were seeded in the device as described above.
Cells were imaged every 10 to 20 min for 4 to 12 hours on an inverted Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan) with automated controls (NIS-Elements, Nikon) and a ×10/0.45 NA Ph1 objective using time-lapse microscopy. Cells were maintained on a temperature- and CO2-controlled stage top incubator (Okolab, Pozzuoli, Italy; Tokai Hit, Shizuoka-hen, Japan) during these experiments. For select experiments, fluorescein isothiocyanate (FITC) and tetramethyl rhodamine isothiocyanate (TRITC) filters were used to excite cell fluorescence.
Cell migration tracking and analysis
The Manual Tracking (Cordelières F, Institut Curie, Orsay, France) and MTrackJ (44) plugins of ImageJ (National Institutes of Health, Bethesda, MD) were used for cell migration tracking. Cell migration was recorded from the time of complete entry into the microchannel until contact was made with the end of the microchannel. Cell migration speed was analyzed using a custom-made MATLAB (MathWorks, Natick, MA). Cell and nuclear entry time were calculated manually. Dividing or apoptotic cells were excluded from analysis. For each condition, approximately 45 to 60 cells were analyzed for each of three or more independent trials unless otherwise noted.
For migration phenotype classification, cells were observed with an inverted Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan) using a 40× air objective. Migration phenotype was assessed from cells fixed 4 to 8 hours after channel entry and stained with actin phalloidin, unless otherwise noted. Cell migration phenotype was manually tabulated.
For the real-time phenotype analysis, the contiguous microchannel device was used. The 450-μm-long channel was segmented into nine 50-μm-long segments. The middle 50-μm-long segment corresponding to the transition/overlap region was neglected. Cells in each segment were assigned a “0” value for each time point if the leading edge of that cell exhibited a mesenchymal phenotype in the respective 50-μm segment. In addition, cells received a “1” value if they exhibited a blebbing phenotype at the leading edge of the cell in the corresponding segment at a given time point. Segments were then averaged together for each confining feature (4 segments × 50 μm for vertical and 4 × 50 μm for lateral) for all time points throughout the duration of the experiment, giving average phenotype values for cells as they traverse the length of each confining feature.
Actin staining and immunofluorescence
For actin staining, cells were fixed with 4% paraformaldehyde (Affymetrix Inc.), permeabilized in 0.1% Triton X-100 (Sigma-Aldrich), and blocked in 1% bovine serum albumin (Sigma-Aldrich). Cells were stained with rhodamine or Alexa Fluor 488 phalloidin (1:100; Invitrogen) and Hoechst (1:2500; Invitrogen). For lamin-A/lamin-B staining, cells were fixed with 4% paraformaldehyde (Affymetrix Inc.), permeabilized in 0.2% Triton X-100 (Sigma-Aldrich), and blocked in 5% bovine serum albumin (Sigma-Aldrich)/0.2% Triton X-100 (Sigma-Aldrich). Cells were stained with anti–lamin-A + lamin-C antibody (131C3) (1:200; Abcam; mouse) and anti–lamin-B1 antibody (Abcam; rabbit, 1 μg/ml). Goat anti-rabbit immunoglobulin G (IgG) H&L, Alexa Fluor 488 (1:200; Thermo Fisher Scientific, A11034); goat anti-rabbit IgG H&L, Alexa Fluor 568 (1:200; Thermo Fisher Scientific, A11011); goat anti-mouse IgG H&L, Alexa Fluor 488 (1:200; Thermo Fisher Scientific, A11001); and goat anti-mouse IgG H&L, Alexa Fluor 568 (1:200; Thermo Fisher Scientific, A21043) were used as secondary antibodies. All antibodies were prepared in blocking buffer. Nuclei were also stained with Hoechst (1:2500; Invitrogen). For phosphorylated lamin-A staining, cells were fixed with 4% paraformaldehyde (Affymetrix Inc.) and blocked in 5% normal goat serum (Cell Signaling)/0.3% Triton X-100 (Sigma-Aldrich). Cells were stained with phospho–lamin-A/C (Ser22) antibody (1:800; Cell Signaling, rabbit), and goat anti-rabbit IgG H&L, Alexa Fluor 488 (1:200; Thermo Fisher Scientific, A11034) was used as a secondary antibody. Nuclei were also stained with Hoechst (1:2500; Invitrogen). Antibodies were prepared in 1% bovine serum albumin (Sigma-Aldrich)/0.3% Triton X-100 (Sigma-Aldrich).
Confocal imaging
Cells were imaged using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan) using a 63× oil objective with a 1.4 NA or a 40× water objective with a 1.15 NA and a resolution of 1024 pixels × 1024 pixels. The 567-, 488-, and 405-nm lasers were used for imaging.
Actin intensity, myosin fiber, and FA quantification
Each cell was imaged using confocal image slices spaced 0.5 or 1 μm apart. Using ImageJ (National Institutes of Health, Bethesda, MD), images were resliced from the left to create yz projection slices along the x axis of the cell. A region of dorsal or ventral actin was selected, and mean and integrated density were measured within the region. A corrected total fluorescence measurement was calculated by subtracting background using the following formula: Corrected total fluorescence = Integrated density − (Area of selected cell × Mean fluorescence of background readings). This procedure was repeated for 10 Z positions in each region of interest and was averaged to generate an overall intensity value. Normalized actin intensity was calculated relative to the average intensity of ventral surface.
FAs and perinuclear myosin fibers were quantified using custom macros within the General Analysis command of NIS-Elements (Nikon). Adhesions with a max ferret value less than 0.25 μm and greater than 10 μm in area were excluded. Myosin fibers with a derived length of the medial axis less than 5 μm and greater than 100 μm or elongation less than 3 μm were excluded.
FLIM of RhoA FRET sensors
Confocal FLIM of live cells that were stably expressing the RhoA2G sensor was performed as described previously (18) using a Zeiss LSM 780 microscope and a PicoQuant system consisting of the PicoHarp 300 time-correlated single photon counting (TCSPC) module, two-hybrid PMA-04 detectors, and Sepia II laser control module.
FLIM reconvolution, image segmentation, and segmentation quantification
The FLIM data were processed as described previously (18) using SymPhoTime 64 (PicoQuant) software.
PA-GFP imaging
Photoactivatable (PA)–GFP imaging and analysis were performed as described previously (18) using a Nikon A1 confocal microscope (Nikon, Tokyo, Japan) with a 60× oil objective and the curve fitting tools in GraphPad Prism 6 and 7 software.
Nuclear volume measurements
Nuclear volume was measured as described in (18). Briefly, confocal Z-stacks with a step of 1 μm were taken of H2B-mCherry–tagged nuclei using an LSM 800, 63× oil immersion, 1.2/1.4 NA objective, 567-nm laser. Nuclear volume was then measured from Z-stacks using a custom MATLAB (MathWorks, Natick, MA) script.
Confocal Brillouin microscopy
Cells were imaged with an integrated fluorescent and confocal Brillouin microscope based on a commercial microscope stand (IX81; Olympus) (45). A ~15-mW continuous-wave laser (Torus, Laser Quantum; 660 nm) light was used to excite the Brillouin signal. The objective lens with NA of 0.95 (Olympus) was used to focus the laser beam into a spot of 0.42 μm by 0.42 μm by 0.73 μm in the lateral and axial directions. The scattered Brillouin signal was collected with the same objectives and sent into a two-stage VIPA (virtually imaged phased array)–based Brillouin spectrometer via a single mode fiber (Thorlabs). The Brillouin spectrometer can determine the Brillouin shift with a precision of 10 MHz. To obtain the co-registered fluorescence/Brillouin image, cells were first imaged by fluorescent channel and then mapped by Brillouin channel with a pixel dwell time of 40 ms. The switch between channels was achieved by automatically changing dichroic mirrors within the microscope turret. The Brillouin shift of the nucleus was extracted by averaging the nuclear region of the Brillouin map based on the co-registered fluorescent image. The Brillouin shift of the cytoplasm was obtained by subtracting the nuclear region from the whole-cell Brillouin image, and the extracted cytoplasmic region was then averaged to represent the cytoplasmic Brillouin shift of the cell. The relationship between Brillouin shift ΩB and mechanical properties (i.e., longitudinal modulus) of the sample is , where n is the refractive index of the sample, θ is the scattering angle, λ is the wavelength of the incoming light, M′ is the longitudinal modulus, and ρ is the mass density. For various cell activities, the refractive index and mass density have been reported to change in the same direction, and the ratio can be approximated to a constant change (26). Therefore, we used Brillouin shift to represent the behavior of the longitudinal modulus.
Western blotting
Western blots were performed as previously described (18, 42) using NuPage 3 to 8% or 4 to 12% gels and the following antibodies: primary antibodies: anti–lamin-A/C (4C11) mouse (1:2000; Cell Signaling, 4777). β-Actin was used as a loading control (1:10,000; purified mouse anti-actin Ab-5; BD Biosciences, 612656). The secondary antibody used is anti-mouse IgG, horseradish peroxidase (HRP)–linked antibody (1:2000; Cell Signaling, 7076S).
Statistical analysis
Data represent the mean ± SEM or SD from three or more independent experiments unless otherwise noted. The following statistical tests were used to determine statistical significance (P < 0.05) as appropriate: Student’s t test or a one-way analysis of variance (ANOVA) test followed by a Tukey’s test for multiple comparisons. In select experiments, we performed a ROUT test to remove statistical outliers. Analysis was performed using GraphPad Prism 6, 7, or 8 software.
Supplementary Material
Acknowledgments
We thank J. Yang and D. Robinson for insightful discussions and M. Rietveld for help with the schematic depictions of the vertical and lateral microchannels. Funding: This work was supported, in part, by an NSF Graduate Research Fellowship (DGE-1746891 to E.O.W.), an American Heart Association Postdoctoral Fellowship (18POST34080345 to P.M.), and grants from the National Cancer Institute (R01CA183804, R33CA204582, K25HD097288, U01CA202177, and U54CA210173) and NSF (CMMI 1929412). P.F. was supported by the European Research Council (617430-DEEPINSIGHT) and the Cancer Genomics Center (CGC.nl). Author contributions: E.O.W. designed the study, performed most experiments, analyzed data, and wrote the manuscript. P.M. designed the study, supervised the study, performed most experiments, analyzed data, and wrote the manuscript. K.B., R.A.L., S.T., A.A., and R.Z. performed select experiments and analyzed data. J.Z., M.N., and G.S. performed and analyzed Brillouin microscopy experiments, provided critical input, and edited the manuscript. M.W., M.P., and P.F. performed and analyzed in vivo experiments, provided critical input, and edited the manuscript. D.W. provided critical input, supplied reagents, and edited the manuscript. P.K. provided critical input, supplied reagents, assisted with FLIM experiments, and edited the manuscript. K.K. designed and supervised the study and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba6505/DC1
REFERENCES AND NOTES
- 1.Paul C. D., Mistriotis P., Konstantopoulos K., Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 17, 131–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weigelin B., Bakker G. J., Friedl P., Intravital third harmonic generation microscopy of collective melanoma cell invasion: Principles of interface guidance and microvesicle dynamics. Dermatol. Int. 1, 32–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alexander S., Koehl G. E., Hirschberg M., Geissler E. K., Friedl P., Dynamic imaging of cancer growth and invasion: A modified skin-fold chamber model. Histochem. Cell Biol. 130, 1147–1154 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Hung W. C., Chen S. H., Paul C. D., Stroka K. M., Lo Y. C., Yang J. T., Konstantopoulos K., Distinct signaling mechanisms regulate migration in unconfined versus confined spaces. J. Cell Biol. 202, 807–824 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y. J., Le Berre M., Lautenschlaeger F., Maiuri P., Callan-Jones A., Heuze M., Takaki T., Voituriez R., Piel M., Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Petrie R. J., Gavara N., Chadwick R. S., Yamada K. M., Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol. 197, 439–455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf K., Te Lindert M., Krause M., Alexander S., Te Riet J., Willis A. L., Hoffman R. M., Figdor C. G., Weiss S. J., Friedl P., Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 201, 1069–1084 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harada T., Swift J., Irianto J., Shin J. W., Spinler K. R., Athirasala A., Diegmiller R., Dingal P. C., Ivanovska I. L., Discher D. E., Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 204, 669–682 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khatau S. B., Bloom R. J., Bajpai S., Razafsky D., Zang S., Giri A., Wu P. H., Marchand J., Celedon A., Hale C. M., Sun S. X., Hodzic D., Wirtz D., The distinct roles of the nucleus and nucleus-cytoskeleton connections in three-dimensional cell migration. Sci. Rep. 2, 488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paul C. D. H., Hung W.-C., Wirtz D., Konstantopouos K., Engineered models of confined cell migration. Annu. Rev. Biomed. Eng. 18, 159–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denais C. M., Gilbert R. M., Isermann P., McGregor A. L., te Lindert M., Weigelin B., Davidson P. M., Friedl P., Wolf K., Lammerding J., Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raab M., Gentili M., de Belly H., Thiam H. R., Vargas P., Jimenez A. J., Lautenschlaeger F., Voituriez R., Lennon-Dumenil A. M., Manel N., Piel M., ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362 (2016). [DOI] [PubMed] [Google Scholar]
- 13.J. Irianto, C. R. Pfeifer, Y. Xia, A. Athirasala, I. L. Ivanovska, R. E. Greenberg, D. E. Discher, Constricted cell migration causes nuclear lamina damage, DNA breaks, and squeeze-out of repair factors. bioRxiv 035626 [Preprint]. 30 December 2015.
- 14.Raftopoulou M., Hall A., Cell migration: Rho GTPases lead the way. Dev. Biol. 265, 23–32 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Balzer E. M., Tong Z., Paul C. D., Hung W. C., Stroka K. M., Boggs A. E., Martin S. S., Konstantopoulos K., Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 26, 4045–4056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stroka K. M., Jiang H., Chen S. H., Tong Z., Wirtz D., Sun S. X., Konstantopoulos K., Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doyle A. D., Petrie R. J., Kutys M. L., Yamada K. M., Dimensions in cell migration. Curr. Opin. Cell Biol. 25, 642–649 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mistriotis P., Wisniewski E. O., Bera K., Keys J., Li Y., Tuntithavornwat S., Law R. A., Perez-Gonzalez N. A., Erdogmus E., Zhang Y., Zhao R., Sun S. X., Kalab P., Lammerding J., Konstantopoulos K., Confinement hinders motility by inducing RhoA-mediated nuclear influx, volume expansion, and blebbing. J. Cell Biol. 218, 4093–4111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahai E., Marshall C. J., Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 5, 711–719 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Arsenovic P. T., Ramachandran I., Bathula K., Zhu R., Narang J. D., Noll N. A., Lemmon C. A., Gundersen G. G., Conway D. E., Nesprin-2G, a component of the nuclear LINC complex, is subject to myosin-dependent tension. Biophys. J. 110, 34–43 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasapera A. M., Schneider I. C., Rericha E., Schlaepfer D. D., Waterman C. M., Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 188, 877–890 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woroniuk A., Porter A., White G., Newman D. T., Diamantopoulou Z., Waring T., Rooney C., Strathdee D., Marston D. J., Hahn K. M., Sansom O. J., Zech T., Malliri A., STEF/TIAM2-mediated Rac1 activity at the nuclear envelope regulates the perinuclear actin cap. Nat. Commun. 9, 2124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burridge K., Monaghan-Benson E., Graham D. M., Mechanotransduction: From the cell surface to the nucleus via RhoA. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 374, 20180229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buxboim A., Swift J., Irianto J., Spinler K. R., Dingal P. C., Athirasala A., Kao Y. R., Cho S., Harada T., Shin J. W., Discher D. E., Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 24, 1909–1917 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guilluy C., Osborne L. D., Van Landeghem L., Sharek L., Superfine R., Garcia-Mata R., Burridge K., Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 16, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scarcelli G., Polacheck W. J., Nia H. T., Patel K., Grodzinsky A. J., Kamm R. D., Yun S. H., Noncontact three-dimensional mapping of intracellular hydromechanical properties by Brillouin microscopy. Nat. Methods 12, 1132–1134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swift J., Ivanovska I. L., Buxboim A., Harada T., Dingal P. C., Pinter J., Pajerowski J. D., Spinler K. R., Shin J. W., Tewari M., Rehfeldt F., Speicher D. W., Discher D. E., Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hale C. M., Shrestha A. L., Khatau S. B., Stewart-Hutchinson P. J., Hernandez L., Stewart C. L., Hodzic D., Wirtz D., Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys. J. 95, 5462–5475 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gavara N., Chadwick R. S., Relationship between cell stiffness and stress fiber amount, assessed by simultaneous atomic force microscopy and live-cell fluorescence imaging. Biomech. Model. Mechanobiol. 15, 511–523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stephens A. D., Liu P. Z., Banigan E. J., Almassalha L. M., Backman V., Adam S. A., Goldman R. D., Marko J. F., Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 29, 220–233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ihalainen T. O., Aires L., Herzog F. A., Schwartlander R., Moeller J., Vogel V., Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat. Mater. 14, 1252–1261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D. H., Wirtz D., Cytoskeletal tension induces the polarized architecture of the nucleus. Biomaterials 48, 161–172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toth K. F., Knoch T. A., Wachsmuth M., Frank-Stohr M., Stohr M., Bacher C. P., Muller G., Rippe K., Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci. 117, 4277–4287 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Beunk L., Brown K., Nagtegaal I., Friedl P., Wolf K., Cancer invasion into musculature: Mechanics, molecules and implications. Semin. Cell Dev. Biol. 93, 36–45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Croft J. A., Bridger J. M., Boyle S., Perry P., Teague P., Bickmore W. A., Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119–1131 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J., Alisafaei F., Nikolic M., Nou X. A., Kim H., Shenoy V. B., Scarcelli G., Nuclear mechanics within intact cells is regulated by cytoskeletal network and internal nanostructures. Small 16, 1907688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham D. M., Andersen T., Sharek L., Uzer G., Rothenberg K., Hoffman B. D., Rubin J., Balland M., Bear J. E., Burridge K., Enucleated cells reveal differential roles of the nucleus in cell migration, polarity, and mechanotransduction. J. Cell Biol. 217, 895–914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guilluy C., Swaminathan V., Garcia-Mata R., O'Brien E. T., Superfine R., Burridge K., The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 13, 722–727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petrie R. J., Yamada K. M., At the leading edge of three-dimensional cell migration. J. Cell Sci. 125, 5917–5926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rueden C. T., Schindelin J., Hiner M. C., DeZonia B. E., Walter A. E., Arena E. T., Eliceiri K. W., ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong B. S., Shea D. J., Mistriotis P., Tuntithavornwat S., Law R. A., Bieber J. M., Zheng L., Konstantopoulos K., A direct podocalyxin-dynamin-2 interaction regulates cytoskeletal dynamics to promote migration and metastasis in pancreatic cancer cells. Cancer Res. 79, 2878–2891 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yankaskas C. L., Thompson K. N., Paul C. D., Vitolo M. I., Mistriotis P., Mahendra A., Bajpai V. K., Shea D. J., Manto K. M., Chai A. C., Varadarajan N., Kontrogianni-Konstantopoulos A., Martin S. S., Konstantopoulos K., A microfluidic assay for the quantification of the metastatic propensity of breast cancer specimens. Nat. Biomed. Eng. 3, 452–465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao R., Afthinos A., Zhu T., Mistriotis P., Li Y., Serra S. A., Zhang Y., Yankaskas C. L., He S., Valverde M. A., Sun S. X., Konstantopoulos K., Cell sensing and decision-making in confinement: The role of TRPM7 in a tug of war between hydraulic pressure and cross-sectional area. Sci. Adv. 5, eaaw7243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meijering E., Dzyubachyk O., Smal I., Methods for cell and particle tracking. Methods Enzymol. 504, 183–200 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Nikolic M., Scarcelli G., Long-term Brillouin imaging of live cells with reduced absorption-mediated damage at 660 nm wavelength. Biomed. Opt. Express 10, 1567–1580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaba6505/DC1