

Abstract
Lysinuric protein intolerance (LPI) is an inborn error of cationic amino acid (arginine, lysine, ornithine) transport caused by biallelic pathogenic variants in SLC7A7, which encodes the light subunit of the y+LAT1 transporter. Treatments for the complications of LPI, including growth failure, renal disease, pulmonary alveolar proteinosis, autoimmune disorders and osteoporosis, are limited. Given the early lethality of the only published global Slc7a7 knockout mouse model, a viable animal model to investigate global SLC7A7 deficiency is needed. Hence, we generated two mouse models with global Slc7a7 deficiency (Slc7a7em1Lbu/em1Lbu; Slc7a7Lbu/Lbu and Slc7a7em1(IMPC)Bay/em1(IMPC)Bay; Slc7a7Bay/Bay) using CRISPR/Cas9 technology by introducing a deletion of exons 3 and 4. Perinatal lethality was observed in Slc7a7Lbu/Lbu and Slc7a7Bay/Bay mice on the C57BL/6 and C57BL/6NJ inbred genetic backgrounds, respectively. We noted improved survival of Slc7a7Lbu/Lbu mice on the 129 Sv/Ev × C57BL/6 F2 background, but postnatal growth failure occurred. Consistent with human LPI, these Slc7a7Lbu/Lbu mice exhibited reduced plasma and increased urinary concentrations of the cationic amino acids. Histopathological assessment revealed loss of brush border and lipid vacuolation in the renal cortex of Slc7a7Lbu/Lbu mice, which combined with aminoaciduria suggests proximal tubular dysfunction. Micro-computed tomography of L4 vertebrae and skeletal radiographs showed delayed skeletal development and suggested decreased mineralization in Slc7a7Lbu/Lbu mice, respectively. In addition to delayed skeletal development and delayed development in the kidneys, the lungs and liver were observed based on histopathological assessment. Overall, our Slc7a7Lbu/Lbu mouse model on the F2 mixed background recapitulates multiple human LPI phenotypes and may be useful for future studies of LPI pathology.
Introduction
Lysinuric protein intolerance (LPI, MIM #222700) is an inborn error of amino acid metabolism that causes impaired function of the y+LAT1 transporter, which is required for the cellular efflux of the cationic amino acids, arginine (Arg), lysine (Lys) and ornithine (Orn) in some cell types (1,2). LPI is caused by biallelic pathogenic variants in SLC7A7, which encodes the light subunit of the y+LAT1 transporter (3). The y+LAT1 transporter is primarily located on the basolateral membrane of intestinal and renal epithelial cells (3). In addition, SLC7A7 is highly expressed in certain non-epithelial cells, such as monocytes and macrophages (3,4). Impaired intestinal absorption and renal reabsorption of Arg, Orn and Lys lead to reduced plasma concentrations and increased urinary excretion of the cationic amino acids, and these laboratory findings are the biochemical hallmarks of LPI (3,4).
Reduced plasma levels of Arg and Orn in LPI are hypothesized to be the cause of secondary urea cycle dysfunction (4). As a result of this urea cycle dysfunction, individuals with LPI are at risk for hyperammonemia and often exhibit dietary protein aversion. Unlike other urea cycle disorders, individuals with LPI may present with a wide variety of other phenotypes, including failure to thrive (4), short stature (4), early onset osteoporosis (5–9), renal disease (10, 11), pulmonary alveolar proteinosis (12), hepatosplenomegaly and immunological disorders, such as systemic lupus erythematosus (SLE) (13–15) and hemophagocytic lymphohistiocytosis (HLH) (16). The frequency, age of onset and severity of these clinical manifestations vary widely among individuals with LPI (4).
The primary therapeutic approach for LPI is to treat the urea cycle dysfunction with dietary protein restriction, L-citrulline supplementation and nitrogen scavenging agents (4). L-Lys supplementation to treat growth failure in LPI has been described (17,18). Although improved Lys concentrations with L-Lys supplementation has been reported (18,19), growth did not improve and ~50% of the subjects with LPI discontinued L-Lys supplementation due to gastrointestinal symptoms (17,18). Consistent with growth failure, growth hormone (GH) deficiency or reduced concentrations of insulin-like growth factor-1 (IGF-1) in some individuals with LPI have been reported (19–23). rGH has been used in children with LPI, aged 6–16 years, but with inconsistent results (21–23). No specific therapies targeting other phenotypes of LPI have been reported.
To date, the majority of published studies investigating the disease mechanisms of LPI has utilized in vitro models and primarily focused on the mechanistic contributions of SLC7A7-deficient macrophages to the complications of LPI (24–28). Specifically, studies in monocytes and monocyte-derived macrophages from individuals with LPI showed marked reductions in efflux of Arg and reduced phagocytosis compared to controls (26,28). Increased RNA expression and secretion of pro-inflammatory factors, such as TNFα and IL1β, have also been reported in SLC7A7-deficient THP-1 cells (stable cell line of human monocytes), which are consistent with inflammatory cascades observed in immunological disorders that have been associated with LPI, such as SLE and HLH (27).
The first published mouse model of LPI was characterized by significant intrauterine growth restriction and perinatal lethality (29). The intrauterine growth restriction in the Slc7a7−/− global knockout mouse model (C57BL/6 background) was associated with downregulation of Igf1 in the fetal liver, but the expression of Igf1 in adult livers from two Slc7a7−/− mice was similar to wild-type (WT) controls (29). Importantly, reduced viability of the Slc7a7−/− mouse model was noted, and only two of 18 Slc7a7−/− mice survived to adulthood of the 606 mice generated from >200 Slc7a7+/− intercrosses (29). Similar findings of reduced survival and poor weight gain were noted in an adult-inducible global Slc7a7 knockout mouse model, and these phenotypes were attenuated with L-citrulline supplementation (30).
Our objective was to develop a viable mouse model of LPI that recapitulates human LPI phenotypes for use in future mechanistic and therapeutic studies. To this end, we generated a global Slc7a7-deficient mouse model (Slc7a7Lbu/Lbu) on a mixed genetic background (129/SvEv × C57BL/6 F2) that exhibits improved survival. Importantly, our Slc7a7Lbu/Lbu mouse model recapitulates the biochemical phenotype of LPI and exhibits many of the characteristic features, such as growth failure, renal disease and delayed skeletal development, observed in individuals with LPI.
Results
Reduced survival and growth failure in Slc7a7 knockout mouse models
Two global Slc7a7 knockout mouse models with homozygous deletion of exons 3 and 4 were generated using CRISPR/Cas9 technology (Fig. 1). The homozygous deletion of exons 3 and 4 in Slc7a7 is predicted to introduce a frameshift and early termination of translation. Our laboratory and the Baylor College of Medicine Knockout Mouse Production and Phenotyping Program (KOMP2) independently initiated the generation of Slc7a7 conditional knockout alleles on the C57BL/6 and C57BL/6NJ backgrounds, respectively. Although no founder mice with loxP sites in trans were produced, both groups produced multi-exon deletion, null allele founder mice from which independent lines were established (Supplemental Material, Fig. S1). Critically, perinatal lethality was observed in Slc7a7Bay/Bay mice (C57BL/6NJ genetic background) and Slc7a7Lbu/Lbu mice (C57BL/6 genetic background) compared to heterozygous and WT littermates on their respective inbred genetic backgrounds (Table 1).
Figure 1.
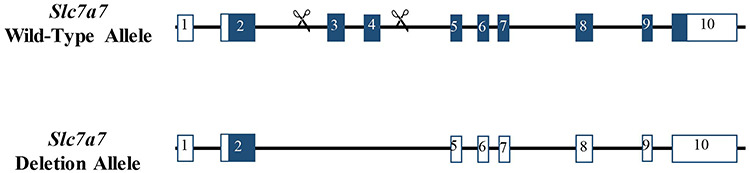
Schematic representation of the Slc7a7 WT and deletion alleles.
Table 1.
Early lethality observed in Slc7a7-deficient mice on four genetic backgrounds
| Allele | Mouse background | Age | WT, n (%) | Slc7a7 +/−, n (%) | Slc7a7 −/−, n (%) |
|---|---|---|---|---|---|
| Slc7a7em1(IMPC)Bay | C57BL/6NJ | E18.5 | 8 (19) | 27 (62) | 8 (19) |
| Slc7a7em1(IMPC)Bay | C57BL/6NJ | P13 | 42 (36) | 76 (64) | 0 (0) |
| Slc7a7em1Lbu | C57BL/6 | E18.5 | 19 (28) | 37 (54) | 12 (18) |
| Slc7a7em1Lbu | C57BL/6 | P21 | 21 (30) | 50 (70) | 0 (0) |
| Slc7a7em1Lbu | FVB/N × C57BL/6 F2 | P21 | 28 (47) | 31 (53) | 0 (0) |
| Slc7a7em1Lbu | 129 Sv/Ev × C57BL/6 F2 | P10 | 242 (33) | 436 (59) | 57 (8) |
IMPC, International Mouse Phenotyping Consortium.
Slc7a7Bay/Bay mice demonstrated perinatal lethality; however, it was determined that 8 of 43 (19%) of murine embryos had the homozygous Slc7a7 mutation at E18.5. In addition to the reduced body length and volume of Slc7a7Bay/Bay embryos at E18.5 (Fig. 2A–C), several phenotypes were identified, including hydrocephalus (n = 8/8), cleft palate and/or short snout (n = 2/8), ventricular hypertrophy (n = 1/8) and ventricular septal defect (n = 1/8). Cleft palate, short snout, ventricular hypertrophy and ventricular septal defects were not observed in the four WT embryos at E18.5; however, hydrocephalus was noted in one of four WT embryos. Anophthalmia was observed in one of eight Slc7a7Bay/Bay embryos and none of the four WT embryos, but this finding may not be unusual given the C57BL/6NJ genetic background (31–33).
Figure 2.
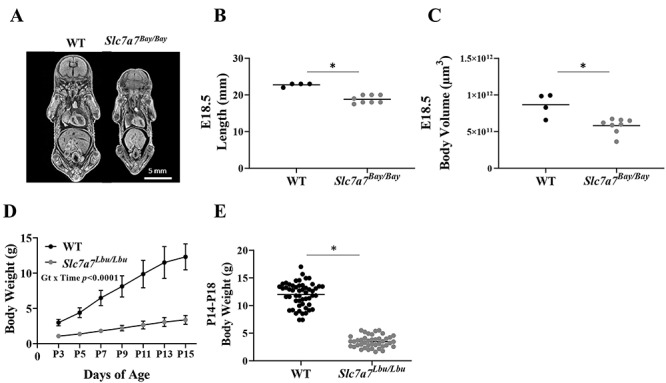
Intrauterine growth restriction and postnatal growth failure with Slc7a7 deficiency. (A) Representative micro-CT images of WT and Slc7a7Bay/Bay embryos (C57BL/6NJ background) at E.18.5. (B) Slc7a7Bay/Bay embryos were shorter in length (anterior to posterior) than WT mice at E18.5. Sample sizes included four WT and eight Slc7a7Bay/Bay embryos (Kruskal–Wallis test, P = 0.006). (C) Slc7a7Bay/Bay embryos demonstrated lower total body volume than WT embryos at E18.5 (unpaired t-test, P = 0.004). (D) Growth curve from P3 through P15 demonstrated postnatal growth failure of Slc7a7Lbu/Lbu versus WT mice (129/SvEv × C57BL/6 F2 background, n = 4–7/genotype), such that WT mice had significantly greater body weights at every time point from P3 through P15 (genotype, P < 0.0001; time, P < 0.0001; genotype × time, P < 0.0001). Statistical analysis included repeated measures ANOVA with main effects for genotype and time and the genotype by time interaction. Values are means ± SD. (E) Male and female Slc7a7Lbu/Lbu mice (129/SvEv × C57BL/6 F2 background) had significantly lower body weights compared to male and female WT mice (genotype, P < 0.0001; sex, P = 0.49; genotype by sex, P = 0.30). Body weight (g), means ± SD; WT/males, 12.0 ± 2.3, n = 31; WT/females, 12.0 ± 2.1, n = 22; Slc7a7Lbu/Lbu/males, 3.2 ± 0.9, n = 25; Slc7a7Lbu/Lbu/females, 3.8 ± 1.2, n = 17. Bars indicate means. Statistical analysis included a two-way ANOVA with main effects for genotype (WT or Slc7a7Lbu/Lbu) and sex and the genotype by sex interaction. Bars indicate means. Asterisks indicate groups that are significantly different (P < 0.05). Gt, genotype.
Similar to the Slc7a7Bay/Bay mice on the C57BL/6NJ background, Slc7a7Lbu/Lbu mice on the C57BL/6 background also demonstrated perinatal lethality (Table 1). Because perinatal lethality was observed in Slc7a7Bay/Bay and Slc7a7Lbu/Lbu mice on the C57BL/6NJ and C57BL/6 genetic backgrounds, respectively, we aimed to generate Slc7a7Lbu/Lbu mice on a mixed genetic background to test whether a mixed genetic background might introduce genetic modifiers that promote survival in a subset of animals (34–36). To this end, we attempted to generate Slc7a7Lbu/Lbu mice on two mixed genetic backgrounds (FVB/N × C57BL/6 F2 and 129/SvEv × C57BL/6 F2). Although survival rates of Slc7a7Lbu/Lbu mice did not improve when generated on the FVB/N × C57BL/6 F2 background, improved survival of Slc7a7Lbu/Lbu mice at P14–18 was observed on the 129/SvEv × C57BL/6 F2 background, but postnatal growth failure occurred (Table 1, Fig. 2D and E).
Biochemical features of LPI observed in Slc7a7Lbu/Lbu mice
We confirmed reduced RNA expression of Slc7a7 in spleen, liver, lungs, kidney, the proximal small intestines and calvaria in Slc7a7Lbu/Lbu versus WT mice (Fig. 3). Because the homozygous deletion of exons 3 and 4 of Slc7a7 is predicted to introduce a frameshift that leads to nonsense-mediated decay, we predict that the residual RNA expression of Slc7a7 in the tissues obtained from Slc7a7Lbu/Lbu mice likely reflects mutant transcripts that did not go through nonsense-mediated decay. Consistent with the characteristic biochemical phenotype of human LPI, Slc7a7Lbu/Lbu mice demonstrated lower plasma concentrations and increased urinary excretion of Arg, Lys and Orn (Table 2). Plasma concentrations of the full amino acid panel are provided in Supplementary Material, Table S1.
Figure 3.
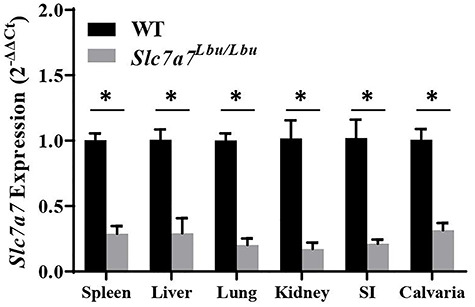
Reduced RNA expression of Slc7a7 in the spleen, liver, lung, kidney, small intestines and calvaria in Slc7a7Lbu/Lbu versus WT mice. Sample sizes included n = 3–4 per genotype per tissue. Values are means ± SE. Gene expression was assessed using the 2−ΔΔCt method. Statistical analyses included unpaired t-tests. Asterisks indicate groups that are significantly different (P < 0.05). SI, small intestines.
Table 2.
Plasma and urine cationic amino acid concentrations in WT and Slc7a7Lbu/Lbu mice
| Laboratory measure | WT | Slc7a7Lbu/Lbu | P | ||
|---|---|---|---|---|---|
| Mean ± SD | Rangea | Mean ± SD | Rangea | ||
| Plasma (μmol/l) | |||||
| Arginine | 183 ± 20 | (125–243) | 33 ± 11 | (22–46) | 0.001 |
| Lysine | 534 ± 111 | (377–678) | 299 ± 113 | (22–46) | 0.01 |
| Ornithine | 95 ± 26 | (64–117) | 24 ± 6 | (16–30) | 0.003 |
| Urine (μmol/g creatinine) | |||||
| Arginine | 37 ± 43 | (5–113) | 5583 ± 4014 | (1352–9410) | 0.0001 |
| Lysine | 199 ± 111 | (77–372) | 15 278 ± 9486 | (6425–25 343) | <0.0001 |
| Ornithine | 23 ± 18 | (9–52) | 1907 ± 777 | (942–2841) | 0.003 |
aRange indicates minimum to maximum. Sample size included n = 4–5 per genotype. Statistical analysis included unpaired t-tests for plasma concentrations and ANOVA for urine excretion of the cationic amino acids.
Proximal tubular dysfunction and delayed renal development in Slc7a7Lbu/Lbu mice
To investigate renal, hepatic and pulmonary microarchitecture, hematoxylin and eosin (H&E) and periodic acid–Schiff (PAS) staining and electron microscopy were performed in Slc7a7Lbu/Lbu and WT mice. Histological alterations in the kidneys of Slc7a7Lbu/Lbu versus WT mice included overall loss of the renal cortex, cortical tubular atrophy, tubular dilation in the renal cortex and medulla, in addition to loss of brush border and vacuolation in the proximal tubules (Fig. 4A and B). Electron microscopy revealed that the vacuoles in the proximal tubules contained lipid (Supplementary Material, Fig. S2A). More secondary lysosomes were also observed in the Slc7a7Lbu/Lbu mouse versus one WT mouse (n = 1/genotype) based on electron microscopy, which may suggest renal injury (Supplementary Material, Fig. S2B) (37,38). However, evaluation of additional kidney samples using electron microscopy would be needed to confirm this finding. Furthermore, Slc7a7Lbu/Lbu mice demonstrated generalized aminoaciduria (Table 3), which in combination with histological alterations in the proximal tubules suggests proximal tubular dysfunction. Despite these alterations in the renal microarchitecture of Slc7a7Lbu/Lbu mice, kidney weights as a percentage of body weight (g kidney weight/100 g body weight) were similar in WT and Slc7a7Lbu/Lbu mice (Supplementary Material, Table S2). Although renal tubular atrophy was observed in Slc7a7Lbu/Lbu mice, no evidence of inflammation or fibrosis was noted in the H&E-stained renal sections.
Figure 4.
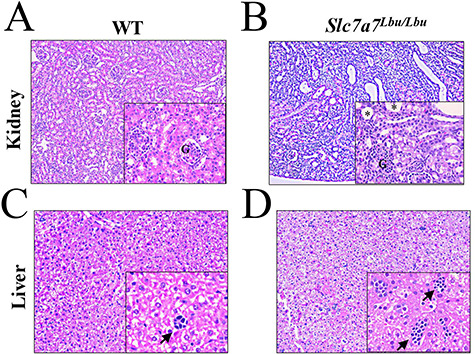
Altered renal and hepatic histological architecture of Slc7a7Lbu/Lbu versus WT mice. Sample sizes included n = 3 per genotype and tissue. The main panels were photographed at the 10× objective and the lower panels were photographed at the 40× objective. (A) Representative H&E-stained renal section of a WT mouse. (B) H&E-stained renal sections of Slc7a7Lbu/Lbu mice demonstrated loss of brush border and vacuolation in the proximal tubules (main panel), in addition to immature glomeruli (lower panel). Asterisks indicate vacuoles. (C) Representative H&E-stained liver section of a WT mouse. (D) H&E-stained liver sections from Slc7a7Lbu/Lbu mice showed more hematopoietic aggregates that appeared larger in size. Arrows indicate hemopoietic aggregates. G, glomerulus.
Table 3.
Generalized aminoaciduria was observed in Slc7a7Lbu/Lbu mice
| Laboratory measure | WT | Slc7a7Lbu/Lbu | ||
|---|---|---|---|---|
| Mean ± SD | Rangea | Mean ± SD | Rangea | |
| Urine (μmol/g creatinine) | ||||
| 1-Methylhistidine | 32 ± 16 | (12–55) | 232 ± 91 | (124–308) |
| 3-Methylhistidine | 94 ± 21 | (71–115) | 157 ± 90 | (56–276) |
| Alanine | 117 ± 72 | (27–227) | 3161 ± 1698 | (56–276) |
| Alpha-aminobutyric acid | 26 ± 39 | (0–95) | 181 ± 131 | (37–337) |
| Asparagine | 28 ± 26 | (1–69) | 543 ± 618 | (108–1456) |
| Aspartic acid | 52 ± 63 | (5–152) | 9739 ± 12 479 | (71–27 845) |
| Citrulline | 57 ± 54 | (2–124) | 5186 ± 3179 | (2236–9489) |
| Cystine | 205 ± 80 | (132–333) | 1953 ± 1012 | (804–3144) |
| Glutamate | 32 ± 15 | (18–58) | 14 731 ± 15 822 | (58–37 098) |
| Glutamine | 70 ± 32 | (37–121) | 9626 ± 6079 | (4357–18 333) |
| Glycine | 280 ± 141 | (173–527) | 6768 ± 6015 | (1943–14 874) |
| Histidine | 29 ± 9 | (20–41) | 3664 ± 2184 | (1482–6666) |
| Isoleucine | 18 ± 9 | (8–28) | 425 ± 430 | (97–1043) |
| Leucine | 114 ± 135 | (12–335) | 1065 ± 871 | (372–2333) |
| Methionine | 56 ± 65 | (8–169) | 1118 ± 749 | (478–2139) |
| Phenylalanine | 29 ± 18 | (14–59) | 388 ± 343 | (129–891) |
| Phosphoethanolamine | 158 ± 109 | (84–346) | 325 ± 184 | (102–505) |
| Phosphoserine | 286 ± 151 | (136–539) | 979 ± 296 | (700–1367) |
| Proline | 51 ± 18 | (33–80) | 6401 ± 5891 | (2236–14 967) |
| Serine | 181 ± 155 | (55–447) | 2278 ± 1730 | (770–4660) |
| Taurine | 15 641 ± 5950 | (9026–24 679) | 30 011 ± 11 251 | (14 922–42 163) |
| Threonine | 152 ± 92 | (95–315) | 3222 ± 2387 | (1104–6610) |
| Tyrosine | 78 ± 53 | (15–161) | 2353 ± 1023 | (1500–3770) |
| Valine | 88 ± 44 | (55–161) | 745 ± 748 | (234–1824) |
aRange indicates minimum to maximum. Sample size included n = 4–5 per genotype.
Slc7a7Lbu/Lbu mice demonstrated delayed renal development as evidenced by immature glomeruli containing podocytes with cuboidal morphology and greater Bowman’s space based on H&E staining (Fig. 4B) (39–41). Consistent with the H&E-stained renal sections, electron microscopy showed greater Bowman’s space in all of the glomeruli assessed in the renal cortex of one Slc7a7Lbu/Lbu mouse (n = 5 glomeruli), whereas the glomeruli assessed in the WT mouse were within normal limits (n = 7 glomeruli) (Supplementary Material, Fig. S2C). In addition, more mesenchymal cells were present in the H&E-stained renal sections of Slc7a7Lbu/Lbu mice (data not shown), which also support the observation of delayed renal development with Slc7a7 deficiency (42,43).
Delayed liver and lung development in Slc7a7Lbu/Lbu mice
In addition to delayed renal development, Slc7a7Lbu/Lbu mice also demonstrated delayed development in the liver and lungs (Figs 4C and D and 5). H&E-stained liver sections revealed increased large hematopoietic aggregates in Slc7a7Lbu/Lbu versus WT mice, which suggests delayed liver development (44,45). Although there was variability in the pulmonary microarchitecture of H&E-stained lung sections (Supplementary Material, Fig. S3), Slc7a7Lbu/Lbu mice demonstrated reduced alveolargenesis (Fig. 5A and B) such that the number of alveoli in Slc7a7Lbu/Lbu mice appeared fewer compared to WT littermates. Quantitative morphometry of the right and left pulmonary lobes demonstrated that the mean linear intercept (MLI) was greater in Slc7a7Lbu/Lbu versus WT mice, which indicates increased alveolar spacing consistent with reduced alveolargenesis and delayed pulmonary development (Fig. 5C) (46,47). No histological findings consistent with pulmonary alveolar proteinosis were observed in the Slc7a7Lbu/Lbu or WT mice at P14–18.
Figure 5.
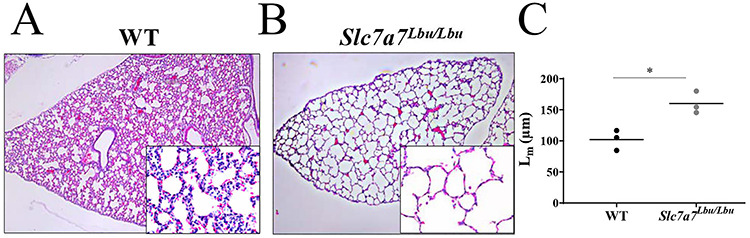
Altered histological architecture of the lungs of Slc7a7Lbu/Lbu versus WT mice at P14–18. Sample size included n = 3 per genotype. The main panels were photographed at the 4× objective and the lower panels were photographed at the 40× objective. (A) H&E-stained sections of the right pulmonary lobe of a WT mouse. (B) The right pulmonary lobe of Slc7a7Lbu/Lbu mice showed reduced alveolargenesis, such that alveoli appear fewer and distended. (C) Slc7a7Lbu/Lbu mice demonstrated a greater MLI (Lm) compared to WT mice (P = 0.01, unpaired t-test), indicating that there were fewer alveoli and reduced alveolargenesis.
Delayed skeletal development in Slc7a7Lbu/Lbu mice
To assess trabecular bone mass and mineralization in WT and Slc7a7Lbu/Lbu mice at P14–P18, we performed micro-computed tomography (micro-CT) analyses of the L4 vertebrae and obtained skeletal radiographs of the spine and lower extremity. Two-dimensional micro-CT images of the L4 vertebrae revealed that the architecture of the L4 vertebrae of Slc7a7Lbu/Lbu mice more closely resembled the L4 vertebrae of WT mice at P5 and P10 versus P14–P18, suggestive of delayed skeletal development (Fig. 6A). Consistent with delayed development in the spine, skeletal radiographs also revealed delayed development in the lower extremity in Slc7a7Lbu/Lbu mice (Supplementary Material, Fig. S4). Furthermore, skeletal radiographs of the spines and lower extremities in Slc7a7Lbu/Lbu mice suggested reduced mineralization compared to WT mice at P14–18 (Fig. 6B, Supplementary Material, Fig. S4). Due to delayed skeletal development and possible reduced mineralization at P14–P18, trabecular bone mass in the spines and lower extremities could not be accurately quantified with micro-CT analyses.
Figure 6.
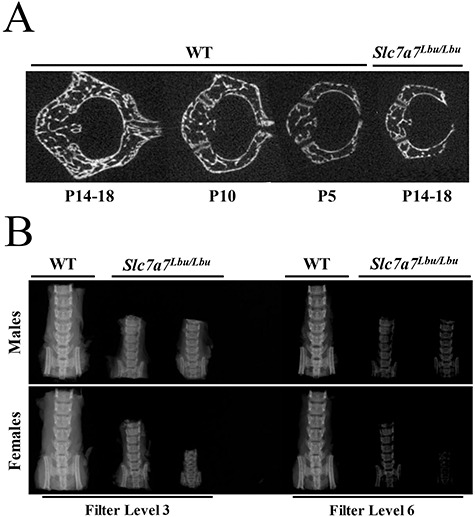
Delayed skeletal development and reduced mineralization of spines in WT and Slc7a7Lbu/Lbu mice at P14–18. (A) Representative two-dimensional micro-CT images of L4 vertebrae show that the architecture of the L4 vertebrae of Slc7a7Lbu/Lbu mice at P14–18 is more similar to WT mice at P5 and P10 than P14–P18. (B) X-ray images, obtained using two X-ray imaging filters, of spines harvested from male and female WT and Slc7a7Lbu/Lbu littermates at P14–18 suggest reduced mineralization with Slc7a7 deficiency.
Altered splenic architecture and hematological studies in Slc7a7Lbu/Lbu mice
Because immune dysfunction is a characteristic phenotype of LPI (4,48), H&E-stained spleen sections and complete blood counts (CBC) were assessed in WT and Slc7a7Lbu/Lbu mice. Slc7a7Lbu/Lbu mice demonstrated the expansion of the red pulp and diminished formation of the germinal center (Fig. 7A and B). Although the CBC panels were generally similar between WT and Slc7a7Lbu/Lbu mice (Supplementary Material, Table S3), Slc7a7Lbu/Lbu mice demonstrated a 94% increase in the percentage of neutrophils (mean ± SD; percent, WT, 16 ± 7; Slc7a7Lbu/Lbu, 31 ± 10; P = 0.03) and a 23% decrease in the percentage of lymphocytes (mean ± SD; percent, WT, 70 ± 10; Slc7a7Lbu/Lbu, 54 ± 8; P = 0.04). Furthermore, average spleen weight (g spleen/100 g body weight) in Slc7a7Lbu/Lbu mice was significantly lower compared to WT mice (Fig. 7C). These findings of altered splenic structures, increased percentage of neutrophils, decreased percentage of lymphocytes and decreased spleen weights may possibly suggest immune dysfunction in Slc7a7Lbu/Lbu mice. Further immunophenotyping and functional studies are needed to confirm immune dysfunction in this Slc7a7Lbu/Lbu mouse model.
Figure 7.
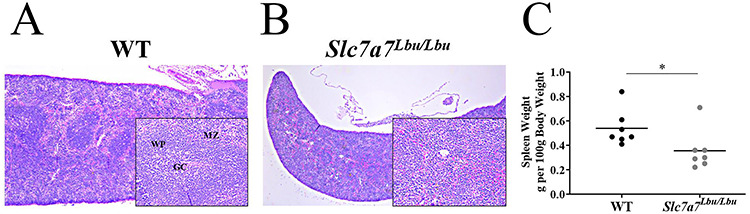
Altered histological architecture of the spleens of Slc7a7Lbu/Lbu versus WT mice at P14–18. Sample size included n = 3 per genotype. The main panels were photographed at the 4× objective and the lower panels were photographed at the 20× objective. (A) Representative H&E-stained spleen section of a WT mouse. (B) Representative H&E-stained spleen section of a Slc7a7Lbu/Lbu mouse. (C) Slc7a7Lbu/Lbu mice had significantly lower spleen weights (g/100 g body weight) compared to WT mice (P = 0.047, unpaired t-test). Asterisks indicate groups that are significantly different. GC, germinal center; MZ, marginal zone; WP, white pulp.
Relationship between IGF-1 and growth delay in Slc7a7Lbu/Lbu mice
In the first published Slc7a7Lbu/Lbu mouse model, reduced RNA expression of Igf1 in Slc7a7Lbu/Lbu fetal livers was reported, and thus, it was suggested that reduced IGF-1 signaling may contribute to intrauterine growth restriction (29). To test if reduced IGF-1 signaling is associated with the postnatal growth failure observed in Slc7a7Lbu/Lbu mice at P14–P18, RNA and protein expression of IGF-1 in the liver were assessed. RNA expression of Igf1 in the liver was significantly lower in Slc7a7Lbu/Lbu versus WT mice (Fig. 8). Protein expression of IGF-1 was assessed in the liver obtained from WT and Slc7a7Lbu/Lbu mice with immunoblots, but no consistent trends were observed (data not shown).
Figure 8.
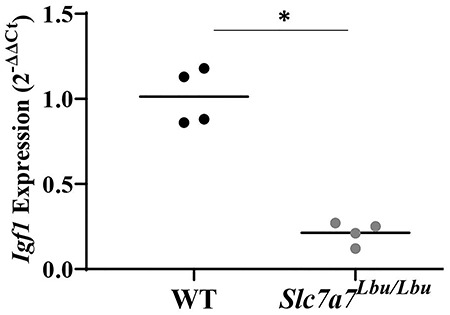
Reduced RNA expression of Igf1 in the livers of Slc7a7Lbu/Lbu versus WT mice at P14–P18 (P = 0.0001). Sample size included n = 4 per genotype. Gene expression was assessed using the 2−ΔΔCt method. Statistical analysis included the unpaired t-test. Asterisks indicate groups that are significantly different (P < 0.05).
Discussion
The pathophysiology of LPI is complex due to its multiorgan involvement, and therefore, tissue-specific mechanisms of this disorder remain poorly understood (4,49). Several biochemical features of LPI, such as reduced plasma concentrations of cationic amino acids and hyperammonemia, might be explained by reduced intestinal absorption of cationic amino acids or reduced hepatic availability of urea cycle intermediates causing urea cycle dysfunction, respectively (4,49). However, neither the characteristic biochemical features of LPI nor the urea cycle dysfunction can explain the diverse phenotypes of LPI, including growth failure, renal disease, pulmonary alveolar proteinosis, osteoporosis and immune dysfunction (autoimmune diseases and immune deficiency) (4). Development of viable animal models of Slc7a7 deficiency is crucial for investigations of the underlying mechanisms of LPI pathology. To this end, we generated a mouse model of LPI that recapitulates key features of human LPI, including growth failure, proximal tubular dysfunction, possible immune dysfunction and delayed skeletal development. Our Slc7a7Bay/Bay and Slc7a7Lbu/Lbu mouse models were generated by introducing a homozygous deletion of exons 3 and 4 leading to a premature frameshift, early termination of translation and predicted nonsense-mediated decay. Although the deletion in our Slc7a7Bay/Bay and Slc7a7Lbu/Lbu mouse models do not precisely recapitulate the variants that have been reported in individuals with LPI, homozygous multi-exon deletions and homozygous loss-of-function variants in SLC7A7 have been reported (1,2,50–53).
Early lethality was a major obstacle in the previously reported global Slc7a7−/− mouse model, with only 18 (3%) live Slc7a7−/− pups of the 606 pups generated from Slc7a7+/− intercrosses (29). Of these 18 live Slc7a7−/− pups, 89% of pups died within 24 hours of birth and only 2 (11%) mice survived into adulthood (29). Despite the reduced viability that we also observed in Slc7a7Bay/Bay mice on the C57BL/NJ genetic background and Slc7a7Lbu/Lbu mice on the C57BL/6 and FVB/N × C57BL/6 F2 genetic backgrounds, we ultimately generated 57 Slc7a7Lbu/Lbu mice on the 129/SvEv × C57BL/6 F2 background for this study.
Consistent with the previously reported global Slc7a7−/− mouse model (29), we demonstrated intrauterine growth restriction in our Slc7a7Bay/Bay and Slc7a7Lbu/Lbu mouse models (Fig. 2). Although intrauterine growth restriction has not been reported as a common feature in individuals with LPI (9), SLC7A7 is known to play a major role in fetal growth, placental function and organ development albeit the precise mechanisms remain unclear (29,54,55). Intrauterine growth restriction, including low birth weight (<2500 g), was reported in 7 of 19 (37%) infants born to 9 individuals with LPI, despite normal placenta size and circulating IGF-1 concentrations (54). Interestingly, downregulation of SLC7A7 expression in the chorionic tissue of placentas obtained from unaffected individuals has been implicated in the development of intrauterine growth restriction with or without preeclampsia (55). Thus, future studies are needed to elucidate the role of SLC7A7 in growth and metabolism during embryonic and fetal development.
In addition to intrauterine growth restriction, we observed postnatal growth failure in our Slc7a7Lbu/Lbu mice on the 129/SvEv × C57BL/6 F2 background (Slc7a7em1Lbu). Failure to thrive and short stature are common phenotypes observed in individuals with LPI (4,9,17,19,48,56). Moreover, GH deficiency of unclear etiology has been observed in a subset of patients (21–23). Improved height velocity with GH replacement (rGH) therapy was demonstrated in a case report describing a 12-year-old girl with LPI and three of four children with LPI in a small clinical study (22,23), but this observation was not consistent (21). Consistent with the alterations in the GH/IGF-1 axis in some individuals with LPI, it has been hypothesized that intrauterine growth restriction and growth delay observed with Slc7a7 deficient mice may be due, in part, to downregulation of Igf1 in the liver (21–23,29). In the previously published global Slc7a7−/− mouse model (29), reduced RNA and protein expression of IGF-1 were observed in fetal, but not adult, livers of Slc7a7−/− mice. In our study, we demonstrated reduced RNA expression of Igf1 in the livers of Slc7a7Lbu/Lbu mice at P14–P18, but we found no consistent trends when protein expression of IGF-1 was assessed. Interestingly, Arg, a cationic amino acid with low circulating concentrations in LPI, is required for the expression and secretion of GH and IGF-1 (57,58). Pituitary GH and hepatic IGF-1 form a tightly regulated negative feedback mechanism that is crucial for cellular growth and development via the activation of the PI3k/Akt/mTOR signaling axis (57,58). We hypothesize that these disturbances in the IGF-1/GH axis may contribute to reduced mTOR signaling thereby hindering growth and development in SLC7A7 deficiency. Consistent with this, Slc7a7-deficient medaka embryos demonstrate impaired left–right symmetry of the heart and liver during organogenesis with depressed mTOR signaling (59). Thus, SLC7A7 may operate as an upstream regulator of the mTOR signaling pathway for organogenesis (59).
The poor prenatal and postnatal growth observed in our Slc7a7Lbu/Lbu mouse model was associated with delayed skeletal development and reduced mineralization at P14–18. Specifically, the architecture of the L4 vertebrae of Slc7a7Lbu/Lbu mice at P14–18 more closely resembled the L4 vertebrae of WT mice at P5 and P10 versus P14–18, which suggests delayed skeletal development. The delayed skeletal development in the spines of Slc7a7Lbu/Lbu mice hindered quantification of trabecular bone mass in the L4 vertebrae of Slc7a7Lbu/Lbu mice and reliable comparison with WT littermates at P14–18. Nonetheless, this delay in skeletal development of Slc7a7Lbu/Lbu mice is interesting, considering that delayed bone age is a common feature in individuals with LPI (5,9,19–22). It is unclear if the apparent reduced mineralization that was observed in male and female Slc7a7Lbu/Lbu mice is driven by delayed skeletal development or a metabolic defect in the bone.
Although early onset osteoporosis is a well-recognized complication of LPI, the pathogenesis of osteoporosis in LPI is poorly understood (4–8,56,60,61). Malnutrition has been proposed as a potential explanation for growth failure and osteoporosis in LPI (4,62). Despite a previous report that average dietary protein intakes of adults and children with LPI meet the American Dietary Reference Intake recommendations (62), cationic amino acid deficiency likely persists due, in part, to impaired intestinal absorption and renal reabsorption of the cationic amino acids with SLC7A7 deficiency (4). Additionally, human clinical studies and case reports have reported improvements in urea cycle dysfunction with L-citrulline supplementation but have not consistently demonstrated improvements in bone mineral density with L-Arg, L-citrulline or L-Lys supplementation (17,18).
Aberrant function of osteoblasts (bone-forming cells), osteoclasts (bone-resorbing cells) and macrophages may also contribute to osteoporosis in LPI (4,63–65). Macrophage activation is a known complication of LPI, and osteoclasts are of myeloid hematopoietic lineage (4,66,67). Therefore, a potential consequence of SLC7A7 deficiency could be increased bone resorption due to increased osteoclastogenesis or osteoclast activation and impaired osteoblast differentiation in response to greater expression and secretion of inflammatory factors by activated macrophages (27,63,65–67). Future in vivo and in vitro studies that dissect the relative contribution of cell autonomous Slc7a7 deficiency versus circulating metabolites to osteoporosis and other characteristic phenotypes of LPI are essential to advance our understanding of disease pathology in order to develop targeted therapeutic strategies.
In addition to delayed development in multiple organs, such as the liver, lungs and skeleton, Slc7a7Lbu/Lbu mice demonstrated delayed renal development and renal dysfunction similar to the renal disease reported in individuals with LPI (10,15,48,68,69). Renal involvement affects as many as 75% of individuals with LPI (48). The most common features of renal involvement in LPI include proximal tubular dysfunction and nephrocalcinosis, but glomerular lesions have also been described (10,15,48,68,69). Consistent with these human studies, we observed proximal tubular dysfunction in our Slc7a7Lbu/Lbu mouse model at P14–18 with marked elevations of several amino acids in the urine. As growth failure is a common feature in children with proximal tubular dysfunction, it is unclear whether this proximal tubular dysfunction is contributing to postnatal growth failure in our mouse model (70–73).
The exact cause for the renal disease in LPI remains unknown. Although reduced renal absorption of the cationic amino acids likely contributes to renal disease in LPI, it has been hypothesized that cytotoxicity of Lys and overproduction of nitric oxide (NO), secondary to intracellular trapping of Lys and Arg, may also contribute to renal disease in LPI (4,48,74–76). An in vitro study demonstrated that L-Lys, in a dose-dependent manner, increased apoptosis and production of reactive oxygen species in HK-2 cells, a stable human cell line of proximal tubular cells, which was attenuated by caspase-3 and -9 inhibitors; however, response to L-Lys in HK-2 cells with SLC7A7 deficiency was not assessed (74). Overproduction of NO, due to intracellular trapping of Arg, may also contribute to renal disease in LPI, given that NO is known to induce apoptosis of renal tubular cells and increase secretion of inflammatory factors (74, 77–80).
Recently, an adult-inducible global knockout mouse model of LPI (Slc7a7fl/fl; UBC-Cre-ERT2+) was generated (30), which, similar to our Slc7a7Bay/Bay and Slc7a7Lbu/Lbu mouse models, results in homozygous deletion of exons 3 and 4 of Slc7a7. Reduced survival (<50%) and body weight were reported in adult Slc7a7fl/fl; UBC-Cre-ERT2+ mice fed an 8% protein diet, which was attenuated with L-citrulline supplementation in the drinking water (1 g L-citrulline/l) (30). Although L-citrulline supplementation also improved hippocampal microarchitecture in adult Slc7a7fl/fl; UBC-Cre-ERT2+ mice, histological findings consistent with pulmonary alveolar proteinosis persisted (30). Given the reduced survival and growth failure that we also observed, we attempted to rescue the postnatal growth failure in our Slc7a7Lbu/Lbu mice by delivering L-citrulline (200 mg/kg) via daily intraperitoneal injections to WT and Slc7a7Lbu/Lbu mice starting at P1. This pilot intervention study was discontinued due to poor survival of the Slc7a7Lbu/Lbu mice (data not shown). Our Slc7a7Lbu/Lbu mouse model (129 Sv/Ev × C57BL/6 F2 genetic background) and this adult-inducible global knockout mouse model of LPI (Slc7a7fl/fl; UBC-Cre-ERT2+) may be useful, complementary tools for future studies of LPI (30).
One limitation of our studies was the small body size and associated poor survival of the Slc7a7Lbu/Lbu mice. Thus, our investigations of Slc7a7Lbu/Lbu mice were limited to P14–18, given the poor survival beyond these time points. The small size and poor survival limited treatment studies and prevented assessment of long-term complications, such as pulmonary alveolar proteinosis. Moreover, a mixed F2 genetic background was required to promote survival in Slc7a7Lbu/Lbu mice. One advantage of the F2 genetic background is that any spontaneous variants or variants that result from off-target effects of the CRISPR/Cas9 engineering approach used to develop the Slc7a7Lbu/Lbu mouse model will segregate in the genetic background of both the F2 WT control mice and F2 Slc7a7Lbu/Lbu littermates, except in the rare case that the variant is closely linked to the Slc7a7 locus. Although the resulting variation in mixed F2 genetic background is more similar to human LPI, the use of a mixed F2 genetic background may have also increased phenotypic variability in our assessment of LPI phenotypes in F2 WT and Slc7a7Lbu/Lbu littermates.
Overall, we demonstrated early lethality in new global Slc7a7 knockout mouse models (Slc7a7Bay/Bay and Slc7a7Lbu/Lbu) on two inbred genetic backgrounds and two mixed F2 genetic backgrounds. We achieved improved survival in our Slc7a7Lbu/Lbu mouse model when generated on a mixed 129 Sv/Ev × C57BL/6 F2 background. We demonstrated that our Slc7a7Lbu/Lbu mouse model recapitulates many characteristic phenotypes of LPI, including postnatal growth failure, renal disease, delayed skeletal development and possible immune dysfunction. This global Slc7a7 knockout mouse model may provide a useful tool to investigate the pathology of LPI phenotypes during growth and development.
Materials and Methods
Generation of Slc7a7-deficient mouse models
C57BL/6 mice harboring an exon 3 and 4 knockout allele of Slc7a7 (Slc7a7em1Lbu/em1Lbu; Slc7a7Lbu/Lbu) were generated using CRISPR/Cas9 technology (Fig. 1). To this end, sgRNA sequences were designed to target the genomic sequences of introns 2 and 4 using upstream sgRNA, chr14: 54379316-54379338, 5′-AGTTTAAGGGGCTAGGGCTTGGG-3′ and downstream sgRNA, chr14: 54377446-54377468, 5′-AGAGGTTCGCTTTTACCGACAGG-3′ (81). As the original objective of the design was a conditional allele, 5′ and 3′ short single-stranded oligodeoxynucleotide donors (ssODNs) harboring loxP sites were also designed and generated (Integrated DNA Technologies, Inc., Coralville, Iowa, USA) (82). The microinjection mix of Cas9 mRNA, sgRNA and ssODN was prepared, and the C57BL/6 zygote microinjections were performed as previously described (82). Founder mice harboring an exon 3 and 4 interval deletion were identified using polymerase chain reaction (PCR; 55°C = annealing temperature) followed by agarose gel electrophoresis (forward primer 5′-CCAGGAATTTTGCCATCTTG-3′; reverse primer 5′-TGTATGGGGTAAGGGTGAGTG-3′). An interval deletion founder mouse was identified and backcrossed to C57BL/6 to generate N1 mice. N1 mice that inherited the deleted allele were identified using PCR with the primers listed above. PCR cloning (p-GEM®-T vector, Promega Corp., Madison, Wisconsin, USA) followed by Sanger sequencing was used to confirm the sequence at the site of the deletion in N1 mice. Although no mice with loxP sites in trans were generated despite repeated injection attempts, a small segment of donor DNA, including a loxP site, was integrated at the site of repair in the founder mouse for our line (Supplementary Material, Fig. S1B). N1 mice were backcrossed to C57BL/6 mice to generate N2 mice. N2 mice were intercrossed to maintain the mouse line. A similar approach was used to generate an independent exon 3 and 4 knockout allele (Slc7a7em1(IMPC)Bay/em1(IMPC)Bay; Slc7a7Bay/Bay) on the C57BL/6NJ inbred background (Supplementary Material, Fig. S1A). Although similar approaches were used to generate the knockout alleles on the C57BL/6NJ and C57BL/6 genetic backgrounds, the precise sequence at the DNA repair junctions differs.
Mouse colony management
All mouse studies were performed in the Transgenic Mouse Facilities at Baylor College of Medicine. The mice were housed in microisolator cages in ventilated racks with a 12:12 hour, light:dark cycle. Although the mice were dissected prior to weaning and presumed to primarily consume maternal breast milk, the WT and Slc7a7Lbu/Lbu mice had ad libitum access to food (Lab Diet 5V5R) and autoclaved water. The mouse studies were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC).
Primary viability studies
We assessed the viability of Slc7a7-deficient mice by examining the distribution of offspring genotypes arising from Slc7a7 heterozygous intercrosses at P13 (Slc7a7Bay/Bay, C57BL/6NJ background) or between P10 and P21 (Slc7a7Lbu/Lbu, C57BL/6 background) (Table 1). Because no Slc7a7Bay/Bay or Slc7a7Lbu/Lbu mice were obtained on either inbred genetic background, we extended these studies to late-stage embryos (E18.5). Additionally, we attempted to generate Slc7a7Lbu/Lbu mice on two mixed genetic backgrounds (FVB/N × C57BL/6 F2 and 129/SvEv × C57BL/6 F2) and examined the distribution of offspring genotypes from Slc7a7Lbu/+ intercrosses between P10 and P21.
Murine embryo preparation for micro-CT imaging and phenotyping
Standard timed mating approaches were used to harvest WT and Slc7a7Bay/Bay murine embryos at E18.5 prior to micro-CT imaging (83,84). Preparation of murine embryos harvested at E18.5 for micro-CT imaging and phenotyping has been previously described (83,85). Briefly, pregnant dams were euthanized according to approved protocols from local IACUC. Embryos were dissected and collected in cold 1× PBS, while the yolk sac from each embryo was collected for genotyping. Each embryo was immersed in individual sample tubes with ice-cold 4% paraformaldehyde and fixed at 4°C for 3 days. After fixation, a hydrogel-based tissue scaffolding protocol (STABILITY) was used to prevent sample shrinkage and deformation (83,85). To scaffold the embryo with hydrogel, each sample was transferred to 50 ml conical tubes, immersed in 20 ml of STABILITY buffer and incubated at 4°C for 3 days to allow the polymer to diffuse through. The samples were then placed in a desiccator to remove the air in the sample tubes with bench top vacuum for 10 minutes, followed by purging with nitrogen gas at 10 psi for 5 minutes. The crosslinking reaction was initialized by incubating the samples at 37°C for 3 hours. After crosslinking, the external hydrogels were removed from the specimens. Samples were stored in 1× PBS with 0.1% w/v sodium azide at 4°C until ready for imaging. Iodine solution (0.1 N) was used to contrast the soft tissue of the embryo for micro-CT imaging. Samples were immersed in iodine solution for staining on a nutating mixer at room temperature. Embryos were stained for at least 3 days and then mounted in 56 mm capped sample tubes in 1% w/v agarose immediately before micro-CT imaging. The raw data for 3D imaging of the samples were acquired by the Bruker SKYSCAN 1272 micro-CT scanner (Bruker Corp., Billerica, MA, USA) along the anterior–posterior (AP) axis and acquired projection images were then reconstructed by NRecon Reconstruction (Version: 1.6.9.8; Bruker Corp.) software. Image processing software CTVox (Bruker Corp.) and Slicer (www.slicer.org) were used for rendering the reconstructed 3D volume data and data analysis.
Animal studies, Slc7a7Lbu/Lbu mouse model on the 129/SvEv × C57BL/6 F2 background
Due to the perinatal lethality observed in Slc7a7Lbu/Lbu mice on the C57BL/6 genetic background, female Slc7a7Lbu/+ mice (C57BL/6 background) were crossed with male 129/SvEv mice, and the Slc7a7Lbu/+ F1 offspring were intercrossed to generate F2 offspring for subsequent studies. Although survival of Slc7a7Lbu/Lbu mice improved on the 129/SvEv × C57BL/6 F2 background, the majority of Slc7a7Lbu/Lbu mice did not survive to the time of weaning (P21). Thus, tissues were collected from male and female F2 WT and Slc7a7Lbu/Lbu littermates at 14–18 days of age. Body weights and spot urine collections were obtained prior to isoflurane anesthesia. Mice were anesthetized with isoflurane and blood was collected via retro-orbital bleeding in lithium heparin-coated microvettes (Sarstedt Ag. & Co. KG, Nümbrecht, Germany). After the blood collection, mice were euthanized by isoflurane overdose and cervical dislocation. The blood was centrifuged at 1500g for 5 minutes after which plasma was collected into 1.5 ml microcentrifuge tubes. The plasma was snap frozen in liquid nitrogen and stored at −80°C.
Liver, kidneys, spleen, lungs, proximal small intestines, femurs, spine and calvaria were collected. Liver, kidneys and spleens were fixed in 10% buffered formalin for histology or snap frozen in liquid nitrogen and stored at −80°C. Lungs that were used for histology were inflated with 4% paraformaldehyde using 25 cm of pressure. The proximal small intestines were flushed with cold 1× phosphate buffered saline prior to fixation. Small intestines and lungs were fixed in liquid nitrogen and stored −80°C. Femurs and spines were fixed in 10% buffered formalin for 48 hours and stored in 70% ethanol at 4°C. After the calvaria was removed, the calvaria was placed in 1 ml of cold 1× phosphate buffered saline and cut with surgical scissors. The supernatant was removed. The calvaria was fixed in 1 ml of TRIzol (Invitrogen Corp, Carlsbad, California, USA) and then snap frozen in liquid nitrogen. Fixed calvaria samples were stored −80°C.
Biochemical and histological assessment
Plasma and urine samples were stored at −80°C until the time of analysis. Amino acid concentrations in plasma and urine were measured with the Biochrom 30 Amino Acid Analyzer (Biochrom, UK) at Baylor Genetics using standard methods (86).
Following fixation in 10% buffered formalin (liver, spleen and kidney) or 4% paraformaldehyde (lungs), tissue samples were stored in 70% ethanol at 4°C until the time of processing. Tissues were bisected longitudinally prior to standard tissue processing, embedding in paraffin, sectioning at 3–7 μm and staining with PAS and/or H&E. H&E-stained sections of the kidney, liver and spleen or lungs were reviewed by a pathologist or pulmonologist, respectively. The pathologist and pulmonologist were blinded to the genotypes of the mice during the histological assessment. To assess MLI in H&E-stained lung sections, two fields from the right pulmonary lobe and two fields from the left pulmonary lobe per mouse were randomly selected on the EVOS M5000 Microscope Imaging System (Thermo Fisher Scientific, Waltham, Massachusetts, USA) (87). Five parallel lines that were 200 μm apart were generated for each field. MLI was calculated by multiplying the length of the image (1068 μm) by the number of lines per field (5 lines per field) and divided by the number of intercepts (87). Thus, four MLI values per mouse were generated and then averaged prior to the statistical analysis.
For transmission electron microscopy, one kidney from one WT mouse and one kidney from one Slc7a7Lbu/Lbu mouse were fixed in Trump’s phosphate buffered glutaraldehyde (Thermo Fisher Scientific, Catalog #11750), postfixed in buffered osmium tetroxide, dehydrated in 100% ethanol and embedded in Araldite resin. The embedded kidneys were sectioned at 60 nm and stained with aqueous uranyl acetate and Reynold’s lead citrate. Stained sections of the renal cortex were studied with a FEI Tecnai Spirit Transmission Electron Microscope (Field Electron and Ion Company, Hillsboro, OR, USA) equipped with a FEI Eagle Image Capture System.
Quantitative real-time PCR
All tissues were fixed in liquid nitrogen. Total RNA was extracted (Quiagen, Hilden, Germany, RNeasy Mini Kit, Catalog #74104, and RNase-Free DNase Set, Catalog #79254), except for calvaria. Because calvaria was fixed in TriZOL, alternative methods were used to extract RNA (Sigma Aldrich, St. Louis, Missouri, USA, GenElute Mammalian Total RNA Miniprep Kit, Catalog #RTN70-1KT). RNA was reversed transcribed using the Superscript III Synthesis System (Invitrogen Corp., Catalog #18080051). Real-time PCR was performed using the Light Cycler 96 (Roche Holding AG, Basel, Switzerland) in triplicate using FastStart Essential DNA Master reagent (Roche Holding AG, Catalog #06402712001). RNA expression of Slc7a7 in tissues was assessed with primers flanking exon 8 (5′TGTGCAAGTTCCCAGTGGTT′3) and exon 9 (5′CCTGCCAAGGTCAGTTTCCT′3). β-2 microglobulin was used as the reference gene. Primers used to assess the expression of β-2 microglobulin included the following: forward, 5′GGTCTTTCTGGTGCTTGTC′3; reverse, 5′CGTATGTATCAGTCTCAGT′3. Fold change in gene expression was calculated using the 2−ΔΔCt method (88).
Radiography and micro-CT imaging of the skeleton
Spine and femurs were fixed in buffered formalin for 48 hours and later stored in 70% ethanol at 4°C. Radiography of the spines and femurs was performed with the Kubtec XPERT 80 system (Kubtec Medical Imaging, Milford, CT, USA). The micro-CT imaging was performed with Scanco μCT 40 System with 50-peak kilovoltage and 145-μA X-ray source (Scanco Medical AG, Bruttisellen, Switzerland). The spines were scanned at 12 μm resolution for quantification of the bone parameters. To determine bone parameters of the L4 vertebrae, the maximum number of slices for each vertebral body was analyzed with the automated thresholding algorithm of the Scanco analysis software using a threshold of 190.
Statistical analysis
All statistical analyses were performed using SAS 9.4 (SAS Institute Inc., 2007, Cary, NC, USA). Repeated measures analysis of variance (ANOVA) using PROC MIXED was performed to assess the growth curves with main effects for genotype (WT or Slc7a7Lbu/Lbu) and time (P3, P5, P7, P9, P11, P13, P15) and the genotype by time interaction. If the data did not meet the statistical assumptions of normality and equal variance, a log transformation was performed. Unpaired t-tests using PROC TTEST (SAS Institute Inc.) were performed to investigate differences due to genotype for plasma concentrations of the cationic amino acids, RNA expression of genes, CBC parameters and protein expression. When data were skewed, the Kruskal–Wallis test was performed. Penalties were not applied to the P-values generated from the pairwise comparisons to assess the CBC parameters in WT and Slc7a7Lbu/Lbu mice due to concern for Type II error with a small sample size (89). One-way ANOVA using PROC GLM was performed to assess urinary excretion of the cationic amino acids with a main effect for genotype (WT or Slc7a7Lbu/Lbu).
Two-way ANOVA using PROC GLM was performed to assess body weights prior to dissection and trabecular bone parameters with main effects for genotype and sex and the genotype by sex interaction. If the data did not meet the statistical assumptions of normality and equal variance, a log transformation was performed. If the data remained skewed after the log transformation, rank variables were calculated using PROC RANK (SAS Institute, Inc.) and the ANOVA was performed within PROC GLM. P-values <0.05 were considered significant.
Supplementary Material
Acknowledgements
We would like to acknowledge M. ‘Sayeed’ Sayeeduddin (Baylor College of Medicine Pathology and Histology Core) for assistance with the histotechnology. We would like to acknowledge the Baylor College of Medicine Optical Imaging and Vital Microscopy Core for assistance with the micro-CT imaging of the WT and Slc7a7Bay/Bay murine embryos. L.C.B. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund.
Conflict of Interest Statement: All authors have no conflicts of interest to declare.
ENSEMBL Accession No. for Slc7a7 is ENSMUST00000000984.8.
Funding
National Institutes of Health (5T32DK007664-28 to B.M.S.); U.S. Public Health Service (grant P30DK56338 to B.M.S.), which funds the Texas Medical Center for Digestive Disease Center; National Institutes of Health (T32GM007526-11 to R.M.); Rolanette and Berdon Lawrence Award of the Bone Disease Program of Texas (to R.M.); BCM Comprehensive Cancer Training Program (CPRIT RP160283 to C.Y.C.); National Institutes of Health (R03DE026233 to I.G.); National Institutes of Health (DK102641, AR071741 to B.L.); National Institutes of Health (K08DK106453 to L.C.B.); Caroline Wiess Law Fund for Research in Molecular Medicine at Baylor College of Medicine and Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute of Child Health & Human Development; Dan L. Duncan Comprehensive Cancer Center P30 (CA125123 to BCM Pathology and Histology Core, Mouse Embryonic Stem Cell Core, and Gastrointestinal Experimental Model Systems (GEMS) Core). National Institutes of Health (UM1 HG006348 to BCM Knockout Mouse Production and Phenotyping Program (KOMP2)). The BCM Integrated Microscopy Core is supported by National Institutes of Health (DK56338, CA125123), CPRIT (RP150578, RP170719), the Dan L. Duncan Comprehensive Cancer Center and the John S. Dunn Gulf Coast Consortium for Chemical Genomics.
References
- 1. Borsani G., Bassi M.T., Sperandeo M.P., De Grandi A., Buoninconti A., Riboni M., Manzoni M., Incerti B., Pepe A., Andria G. et al. (1999) SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat. Genet., 21, 297–301. [DOI] [PubMed] [Google Scholar]
- 2. Torrents D., Mykkanen J., Pineda M., Feliubadalo L., Estevez R., Cid R., Sanjurjo P., Zorzano A., Nunes V., Huoponen K. et al. (1999) Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat. Genet., 21, 293–296. [DOI] [PubMed] [Google Scholar]
- 3. Broer S. (2008) Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev., 88, 249–286. [DOI] [PubMed] [Google Scholar]
- 4. Ogier de Baulny H., Schiff M. and Dionisi-Vici C. (2012) Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol. Genet. Metab., 106, 12–17. [DOI] [PubMed] [Google Scholar]
- 5. Svedstrom E., Parto K., Marttinen M., Virtama P. and Simell O. (1993) Skeletal manifestations of lysinuric protein intolerance. A follow-up study of 29 patients. Skelet. Radiol., 22, 11–16. [DOI] [PubMed] [Google Scholar]
- 6. Carpenter T.O., Levy H.L., Holtrop M.E., Shih V.E. and Anast C.S. (1985) Lysinuric protein intolerance presenting as childhood osteoporosis. Clinical and skeletal response to citrulline therapy. N. Engl. J. Med., 312, 290–294. [DOI] [PubMed] [Google Scholar]
- 7. Posey J.E., Burrage L.C., Miller M.J., Liu P., Hardison M.T., Elsea S.H., Sun Q., Yang Y., Willis A.S., Schlesinger A.E. et al. (2014) Lysinuric protein intolerance presenting with multiple fractures. Mol. Genet. Metab. Rep., 1, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parto K., Penttinen R., Paronen I., Pelliniemi L. and Simell O. (1993) Osteoporosis in lysinuric protein intolerance. J. Inherit. Metab. Dis., 16, 441–450. [DOI] [PubMed] [Google Scholar]
- 9. Simell O., Perheentupa J., Rapola J., Visakorpi J.K. and Eskelin L.E. (1975) Lysinuric protein intolerance. Am. J. Med., 59, 229–240. [DOI] [PubMed] [Google Scholar]
- 10. Esteve E., Krug P., Hummel A., Arnoux J.B., Boyer O., Brassier A., Lonlay P., Vuiblet V., Gobin S., Salomon R. et al. (2017) Renal involvement in lysinuric protein intolerance: contribution of pathology to assessment of heterogeneity of renal lesions. Hum. Pathol., 62, 160–169. [DOI] [PubMed] [Google Scholar]
- 11. Pitkanen H.H., Karki M., Niinikoski H., Tanner L., Nanto-Salonen K., Pikta M., Kopatz W.F., Zuurveld M., Meijers J.C.M., Brinkman H.J.M. et al. (2018) Abnormal coagulation and enhanced fibrinolysis due to lysinuric protein intolerance associates with bleeds and renal impairment. Haemophilia, 24, e312–e321. [DOI] [PubMed] [Google Scholar]
- 12. Kerem E., Elpelg O.N., Shalev R.S., Rosenman E., Bar Ziv Y. and Branski D. (1993) Lysinuric protein intolerance with chronic interstitial lung disease and pulmonary cholesterol granulomas at onset. J. Pediatr., 123, 275–278. [DOI] [PubMed] [Google Scholar]
- 13. Kamoda T., Nagai Y., Shigeta M., Kobayashi C., Sekijima T., Shibasaki M. and Nakamura N. (1998) Lysinuric protein intolerance and systemic lupus erythematosus. Eur. J. Pediatr., 157, 130–131. [DOI] [PubMed] [Google Scholar]
- 14. Aoki M., Fukao T., Fujita Y., Watanabe M., Teramoto T., Kato Y., Suzuki Y. and Kondo N. (2001) Lysinuric protein intolerance in siblings: complication of systemic lupus erythematosus in the elder sister. Eur. J. Pediatr., 160, 522–523. [DOI] [PubMed] [Google Scholar]
- 15. Parsons H., Snyder F., Bowen T., Klassen J. and Pinto A. (1996) Immune complex disease consistent with systemic lupus erythematosus in a patient with lysinuric protein intolerance. J. Inherit. Metab. Dis., 19, 627–634. [DOI] [PubMed] [Google Scholar]
- 16. Parto K., Svedstrom E., Majurin M.L., Harkonen R. and Simell O. (1993) Pulmonary manifestations in lysinuric protein intolerance. Chest, 104, 1176–1182. [DOI] [PubMed] [Google Scholar]
- 17. Rajantie J., Simell O., Rapola J. and Perheentupa J. (1980) Lysinuric protein intolerance: a two-year trial of dietary supplementation therapy with citrulline and lysine. J. Pediatr., 97, 927–932. [DOI] [PubMed] [Google Scholar]
- 18. Tanner L.M., Nanto-Salonen K., Niinikoski H., Huoponen K. and Simell O. (2007) Long-term oral lysine supplementation in lysinuric protein intolerance. Metabolism, 56, 185–189. [DOI] [PubMed] [Google Scholar]
- 19. Awrich A.E., Stackhouse W.J., Cantrell J.E., Patterson J.H. and Rudman D. (1975) Hyperdibasicaminoaciduria, hyperammonemia, and growth retardation: treatment with arginine, lysine, and citrulline. J. Pediatr., 87, 731–738. [DOI] [PubMed] [Google Scholar]
- 20. Goto I., Yoshimura T. and Kuroiwa Y. (1984) Growth hormone studies in lysinuric protein intolerance. Eur. J. Pediatr., 141, 240–242. [DOI] [PubMed] [Google Scholar]
- 21. Evelina M., Grazia M., Francesca O., Marta C., Paolo C., Rossella G., Franco A. and Andrea B. (2015) Growth hormone deficiency and Lysinuric protein intolerance: case report and review of the literature. JIMD Rep., 19, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esposito V., Lettiero T., Fecarotta S., Sebastio G., Parenti G. and Salerno M. (2006) Growth hormone deficiency in a patient with lysinuric protein intolerance. Eur. J. Pediatr., 165, 763–766. [DOI] [PubMed] [Google Scholar]
- 23. Niinikoski H., Lapatto R., Nuutinen M., Tanner L., Simell O. and Nanto-Salonen K. (2011) Growth hormone therapy is safe and effective in patients with lysinuric protein intolerance. JIMD Rep., 1, 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barilli A., Rotoli B.M., Visigalli R., Bussolati O., Gazzola G.C. and Dall'Asta V. (2011) Arginine transport in human monocytic leukemia THP-1 cells during macrophage differentiation. J. Leukoc. Biol., 90, 293–303. [DOI] [PubMed] [Google Scholar]
- 25. Barilli A., Rotoli B.M., Visigalli R., Bussolati O., Gazzola G.C., Gatti R., Dionisi-Vici C., Martinelli D., Goffredo B.M., Font-Llitjos M. et al. (2012) Impaired phagocytosis in macrophages from patients affected by lysinuric protein intolerance. Mol. Genet. Metab., 105, 585–589. [DOI] [PubMed] [Google Scholar]
- 26. Barilli A., Rotoli B.M., Visigalli R., Bussolati O., Gazzola G.C., Kadija Z., Rodi G., Mariani F., Ruzza M.L., Luisetti M. et al. (2010) In lysinuric protein intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J. Rare Dis., 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rotoli B.M., Barilli A., Visigalli R., Ingoglia F., Milioli M., Di Lascia M., Riccardi B., Puccini P. and Dall'Asta V. (2018) Downregulation of SLC7A7 triggers an inflammatory phenotype in human macrophages and airway epithelial cells. Front. Immunol., 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rotoli B.M., Barilli A., Ingoglia F., Visigalli R., Bianchi M.G., Ferrari F., Martinelli D., Dionisi-Vici C. and Dall'Asta V. (2019) Analysis of LPI-causing mutations on y+LAT1 function and localization. Orphanet J. Rare Dis., 14, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sperandeo M.P., Annunziata P., Bozzato A., Piccolo P., Maiuri L., D'Armiento M., Ballabio A., Corso G., Andria G., Borsani G. et al. (2007) Slc7a7 disruption causes fetal growth retardation by downregulating Igf1 in the mouse model of lysinuric protein intolerance. Am. J. Phys. Cell Physiol., 293, C191–C198. [DOI] [PubMed] [Google Scholar]
- 30. Bodoy S., Sotillo F., Espino-Guarch M., Sperandeo M.P., Ormazabal A., Zorzano A., Sebastio G., Artuch R. and Palacin M. (2019) Inducible Slc7a7 knockout mouse model recapitulates Lysinuric protein intolerance disease. Int. J. Mol. Sci., 20, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moore B.A., Roux M.J., Sebbag L., Cooper A., Edwards S.G., Leonard B.C., Imai D.M., Griffey S., Bower L., Clary D. et al. (2018) A population study of common ocular abnormalities in C57BL/6N rd8 mice. Invest. Ophthalmol. Vis. Sci., 59, 2252–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon M.M., Greenaway S., White J.K., Fuchs H., Gailus-Durner V., Wells S., Sorg T., Wong K., Bedu E., Cartwright E.J. et al. (2013) A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol., 14, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith R.S., Roderick T.H. and Sundberg J.P. (1994) Microphthalmia and associated abnormalities in inbred black mice. Lab. Anim. Sci., 44, 551–560. [PubMed] [Google Scholar]
- 34. Simpson E.M., Linder C.C., Sargent E.E., Davisson M.T., Mobraaten L.E. and Sharp J.J. (1997) Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet., 16, 19–27. [DOI] [PubMed] [Google Scholar]
- 35. Tanabe L.M., Martin C. and Dauer W.T. (2012) Genetic background modulates the phenotype of a mouse model of DYT1 dystonia. PLoS One, 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Davie S.A., Maglione J.E., Manner C.K., Young D., Cardiff R.D., MacLeod C.L. and Ellies L.G. (2007) Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res., 16, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Surendran K., Vitiello S.P. and Pearce D.A. (2014) Lysosome dysfunction in the pathogenesis of kidney diseases. Pediatr. Nephrol., 29, 2253–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lynch M.R., Tran M.T., Ralto K.M., Zsengeller Z.K., Raman V., Bhasin S.S., Sun N., Chen X., Brown D., Rovira I.I. et al. (2019) TFEB-driven lysosomal biogenesis is pivotal for PGC1alpha-dependent renal stress resistance. JCI Insight, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ao Y., Sun Z., Hu S., Zuo N., Li B., Yang S., Xia L., Wu Y., Wang L., He Z. et al. (2015) Low functional programming of renal AT2R mediates the developmental origin of glomerulosclerosis in adult offspring induced by prenatal caffeine exposure. Toxicol. Appl. Pharmacol., 287, 128–138. [DOI] [PubMed] [Google Scholar]
- 40. Rodriguez M.M., Gomez A.H., Abitbol C.L., Chandar J.J., Duara S. and Zilleruelo G.E. (2004) Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr. Dev. Pathol., 7, 17–25. [DOI] [PubMed] [Google Scholar]
- 41. Chong E. and Yosypiv I.V. (2012) Developmental programming of hypertension and kidney disease. Int. J. Nephrol., 2012, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dressler G.R. (2009) Advances in early kidney specification, development and patterning. Development, 136, 3863–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brenner-Anantharam A., Cebrian C., Guillaume R., Hurtado R., Sun T.T. and Herzlinger D. (2007) Tailbud-derived mesenchyme promotes urinary tract segmentation via BMP4 signaling. Development, 134, 1967–1975. [DOI] [PubMed] [Google Scholar]
- 44. Crawford L.W., Foley J.F. and Elmore S.A. (2010) Histology atlas of the developing mouse hepatobiliary system with emphasis on embryonic days 9.5-18.5. Toxicol. Pathol., 38, 872–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamiya A., Inoue Y. and Gonzalez F.J. (2003) Role of the hepatocyte nuclear factor 4alpha in control of the pregnane X receptor during fetal liver development. Hepatology, 37, 1375–1384. [DOI] [PubMed] [Google Scholar]
- 46. Dumpa V., Nielsen L., Wang H. and Kumar V.H.S. (2019) Caffeine is associated with improved alveolarization and angiogenesis in male mice following hyperoxia induced lung injury. BMC Pulm. Med., 19, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rozance P.J., Seedorf G.J., Brown A., Roe G., O'Meara M.C., Gien J., Tang J.R. and Abman S.H. (2011) Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am. J. Physiol. Lung Cell Mol. Physiol., 301, L860–L871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Noguchi A. and Takahashi T. (2019) Overview of symptoms and treatment for lysinuric protein intolerance. J. Hum. Genet., 64, 849–858. [DOI] [PubMed] [Google Scholar]
- 49. Sebastio G., Sperandeo M.P. and Andria G. (2011) Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am. J. Med. Genet. C Semin. Med. Genet., 157C, 54–62. [DOI] [PubMed] [Google Scholar]
- 50. Mykkanen J., Torrents D., Pineda M., Camps M., Yoldi M.E., Horelli-Kuitunen N., Huoponen K., Heinonen M., Oksanen J., Simell O. et al. (2000) Functional analysis of novel mutations in y(+)LAT-1 amino acid transporter gene causing lysinuric protein intolerance (LPI). Hum. Mol. Genet., 9, 431–438. [DOI] [PubMed] [Google Scholar]
- 51. Sperandeo M.P., Bassi M.T., Riboni M., Parenti G., Buoninconti A., Manzoni M., Incerti B., Larocca M.R., Di Rocco M., Strisciuglio P. et al. (2000) Structure of the SLC7A7 gene and mutational analysis of patients affected by lysinuric protein intolerance. Am. J. Hum. Genet., 66, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shoji Y., Noguchi A., Shoji Y., Matsumori M., Takasago Y., Takayanagi M., Yoshida Y., Ihara K., Hara T., Yamaguchi S. et al. (2002) Five novel SLC7A7 variants and y+L gene-expression pattern in cultured lymphoblasts from Japanese patients with lysinuric protein intolerance. Hum. Mutat., 20, 375–381. [DOI] [PubMed] [Google Scholar]
- 53. Font-Llitjos M., Rodriguez-Santiago B., Espino M., Sillue R., Manas S., Gomez L., Perez-Jurado L.A., Palacin M. and Nunes V. (2009) Novel SLC7A7 large rearrangements in lysinuric protein intolerance patients involving the same AluY repeat. Eur. J. Hum. Genet., 17, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tanner L., Nanto-Salonen K., Niinikoski H., Erkkola R., Huoponen K. and Simell O. (2006) Hazards associated with pregnancies and deliveries in lysinuric protein intolerance. Metabolism, 55, 224–231. [DOI] [PubMed] [Google Scholar]
- 55. Huang X., Anderle P., Hostettler L., Baumann M.U., Surbek D.V., Ontsouka E.C. and Albrecht C. (2018) Identification of placental nutrient transporters associated with intrauterine growth restriction and pre-eclampsia. BMC Genomics, 19, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mauhin W., Habarou F., Gobin S., Servais A., Brassier A., Grisel C., Roda C., Pinto G., Moshous D., Ghalim F. et al. (2017) Update on Lysinuric Protein Intolerance, a multi-faceted disease retrospective cohort analysis from birth to adulthood. Orphanet J. Rare Dis., 12, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Oh H.S., Oh S.K., Lee J.S., Wu C. and Lee S.J. (2017) Effects of l-arginine on growth hormone and insulin-like growth factor 1. Food Sci. Biotechnol., 26, 1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garza R.G., Morales-Garza L.A., Martin-Estal I. and Castilla-Cortazar I. (2017) Insulin-like growth factor-1 deficiency and cirrhosis establishment. J. Clin. Med. Res., 9, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Asaoka Y., Nagai Y., Namae M., Furutani-Seiki M. and Nishina H. (2016) SLC7 family transporters control the establishment of left-right asymmetry during organogenesis in medaka by activating mTOR signaling. Biochem. Biophys. Res. Commun., 474, 146–153. [DOI] [PubMed] [Google Scholar]
- 60. Gomez L., Garcia-Cazorla A., Gutierrez A., Artuch R., Varea V., Martin J., Pinillos S. and Vilaseca M.A. (2006) Treatment of severe osteoporosis with alendronate in a patient with lysinuric protein intolerance. J. Inherit. Metab. Dis., 29, 1. [DOI] [PubMed] [Google Scholar]
- 61. Parto K., Maki J., Pelliniemi L.J. and Simell O. (1994) Abnormal pulmonary macrophages in lysinuric protein intolerance. Ultrastructural, morphometric, and X-ray microanalytic study. Arch. Pathol. Lab. Med., 118, 536–541. [PubMed] [Google Scholar]
- 62. Tanner L.M., Nanto-Salonen K., Venetoklis J., Kotilainen S., Niinikoski H., Huoponen K. and Simell O. (2007) Nutrient intake in lysinuric protein intolerance. J. Inherit. Metab. Dis., 30, 716–721. [DOI] [PubMed] [Google Scholar]
- 63. Briot K., Geusens P., Em Bultink I., Lems W.F. and Roux C. (2017) Inflammatory diseases and bone fragility. Osteoporos. Int., 28, 3301–3314. [DOI] [PubMed] [Google Scholar]
- 64. Tanaka Y., Nakayamada S. and Okada Y. (2005) Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr. Drug Targets Inflamm. Allergy, 4, 325–328. [DOI] [PubMed] [Google Scholar]
- 65. Yang D.H. and Yang M.Y. (2019) The role of macrophage in the pathogenesis of osteoporosis. Int. J. Mol. Sci., 20, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Raggatt L.J. and Partridge N.C. (2010) Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem., 285, 25103–25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Furuya M., Kikuta J., Fujimori S., Seno S., Maeda H., Shirazaki M., Uenaka M., Mizuno H., Iwamoto Y., Morimoto A. et al. (2018) Direct cell-cell contact between mature osteoblasts and osteoclasts dynamically controls their functions in vivo. Nat. Commun., 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Riccio E. and Pisani A. (2014) Fanconi syndrome with lysinuric protein intolerance. Clin. Kidney J., 7, 599–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Parto K., Kallajoki M., Aho H. and Simell O. (1994) Pulmonary alveolar proteinosis and glomerulonephritis in lysinuric protein intolerance: case reports and autopsy findings of four pediatric patients. Hum. Pathol., 25, 400–407. [DOI] [PubMed] [Google Scholar]
- 70. Silverstein D.M. (2018) Growth and nutrition in pediatric chronic kidney disease. Front. Pediatr., 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mahesh S. and Kaskel F. (2008) Growth hormone axis in chronic kidney disease. Pediatr. Nephrol., 23, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Paschou S.A., Kanaka-Gantenbein C., Chrousos G.P. and Vryonidou A. (2019) Growth hormone axis in patients with chronic kidney disease. Hormones (Athens), 18, 71–73. [DOI] [PubMed] [Google Scholar]
- 73. Haque S.K., Ariceta G. and Batlle D. (2012) Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol. Dial. Transplant., 27, 4273–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Verzola D., Fama A., Villaggio B., Di Rocco M., Simonato A., D'Amato E., Gianiorio F. and Garibotto G. (2012) Lysine triggers apoptosis through a NADPH oxidase-dependent mechanism in human renal tubular cells. J. Inherit. Metab. Dis., 35, 1011–1019. [DOI] [PubMed] [Google Scholar]
- 75. Kurko J., Vaha-Makila M., Tringham M., Tanner L., Paavanen-Huhtala S., Saarinen M., Nanto-Salonen K., Simell O., Niinikoski H. and Mykkanen J. (2015) Dysfunction in macrophage toll-like receptor signaling caused by an inborn error of cationic amino acid transport. Mol. Immunol., 67, 416–425. [DOI] [PubMed] [Google Scholar]
- 76. Mannucci L., Emma F., Markert M., Bachmann C., Boulat O., Carrozzo R., Rizzoni G. and Dionisi-Vici C. (2005) Increased NO production in lysinuric protein intolerance. J. Inherit. Metab. Dis., 28, 123–129. [DOI] [PubMed] [Google Scholar]
- 77. Erez A., Nagamani S.C., Shchelochkov O.A., Premkumar M.H., Campeau P.M., Chen Y., Garg H.K., Li L., Mian A., Bertin T.K. et al. (2011) Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med., 17, 1619–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brune B. (2002) Nitric oxide and apoptosis in mesangial cells. Kidney Int., 61, 786–789. [DOI] [PubMed] [Google Scholar]
- 79. Tiwari M.M., Messer K.J. and Mayeux P.R. (2006) Inducible nitric oxide synthase and apoptosis in murine proximal tubule epithelial cells. Toxicol. Sci., 91, 493–500. [DOI] [PubMed] [Google Scholar]
- 80. Du C., Guan Q., Diao H., Yin Z. and Jevnikar A.M. (2006) Nitric oxide induces apoptosis in renal tubular epithelial cells through activation of caspase-8. Am. J. Physiol. Ren. Physiol., 290, F1044–F1054. [DOI] [PubMed] [Google Scholar]
- 81. Hodgkins A., Farne A., Perera S., Grego T., Parry-Smith D.J., Skarnes W.C. and Iyer V. (2015) WGE: a CRISPR database for genome engineering. Bioinformatics, 31, 3078–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lanza D.G., Gaspero A., Lorenzo I., Liao L., Zheng P., Wang Y., Deng Y., Cheng C., Zhang C., Seavitt J.R. et al. (2018) Comparative analysis of single-stranded DNA donors to generate conditional null mouse alleles. BMC Biol., 16, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hsu C.W., Wong L., Rasmussen T.L., Kalaga S., McElwee M.L., Keith L.C., Bohat R., Seavitt J.R., Beaudet A.L. and Dickinson M.E. (2016) Three-dimensional microCT imaging of mouse development from early post-implantation to early postnatal stages. Dev. Biol., 419, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hsu C.W., Kalaga S., Akoma U., Rasmussen T.L., Christiansen A.E. and Dickinson M.E. (2019) High resolution imaging of mouse embryos and neonates with X-ray micro-computed tomography. Curr. Protoc. Mouse Biol., 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wong M.D., Spring S. and Henkelman R.M. (2013) Structural stabilization of tissue for embryo phenotyping using micro-CT with iodine staining. PLoS One, 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Walker V. and Mills G.A. (1995) Quantitative methods for amino acid analysis in biological fluids. Ann. Clin. Biochem., 32, 28–57. [DOI] [PubMed] [Google Scholar]
- 87. Yuan X., Chang C.Y., You R., Shan M., Gu B.H., Madison M.C., Diehl G., Perusich S., Song L.Z., Cornwell L. et al. (2019) Cigarette smoke-induced reduction of C1q promotes emphysema. JCI Insight, 4, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Livak K.J. and Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- 89. Gelman A., Hill J. and Yajima M. (2012) Why we (usually) don't have to worry about multiple comparisons. J. Res. Edu. Eff., 5, 189–211. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.