

Abstract
Genetically engineered T cells are powerful new medicines, offering hope for curative responses in patients with cancer. Chimeric antigen receptor (CAR) T cells were recently approved by the US Food and Drug Administration and are poised to enter the practice of medicine for leukemia and lymphoma, demonstrating that engineered immune cells can serve as a powerful new class of cancer therapeutics. The emergence of synthetic biology approaches for cellular engineering provides a broadly expanded set of tools for programming immune cells for enhanced function. Advances in T cell engineering, genetic editing, the selection of optimal lymphocytes, and cell manufacturing have the potential to broaden T cell-based therapies and foster new applications beyond oncology, in infectious diseases, organ transplantation, and autoimmunity.
Keywords: chimeric antigen receptor, CAR-T cell, adoptive cell transfer, immune-oncology, leukemia, synthetic biology
INTRODUCTION
Immunotherapy is the latest breakthrough in cancer therapy, thanks to the remarkable clinical results of checkpoint inhibitors (1) and chimeric antigen receptor (CAR) T cells (2). The US Food and Drug Administration (FDA) approval in 2017 of two CAR-T cell therapies for the treatment of B cell malignancies in pediatric and adult patients is a landmark for cancer immunotherapies. In 2018, these therapies were approved in the European Union, the United Kingdom, and Canada.
These novel, effective immunotherapies rely on the ability of T cells to destroy cancer cells. Indeed, T cells, when adequately rewired, are proving to be the most powerful anticancer immune cell. Immunotherapy not only leads to unprecedented clinical responses in patients with otherwise refractory tumors but is also able to ensure long-lasting clinical remission in patients that were historically considered incurable due to their disseminated disease.
Among the different T cell immunotherapies, adoptive cell therapy (ACT) has attracted substantial attention and interest lately. ACT is a personalized therapy in which a patient’s own immune cells are removed from, expanded in vitro to large numbers, and reinfused back into the patient to eradicate tumors. In the first attempts to apply this therapeutic strategy, tumor-infiltrating lymphocytes (TILs) were isolated from melanoma tumors. The infusion of TILs resulted in complete and durable responses in patients with metastatic melanoma (3). Rapid advances in the field of gene therapy, gene editing, T cell biology, and target identification make it now possible to genetically modify T cells to render them tumor specific. By using T cells easily collected from patients’ blood and genetically modifying them with synthetic antigen-recognizing receptors, including T cell receptors (TCRs) or CARs, it is now potentially possible to target any type of cancer. The best clinical results with ACT have been obtained mostly with CAR-T cells for CD19+ leukemia and lymphoma (4–9). CAR-T cells targeting CD22 and B cell maturation antigen (BCMA) have also led to promising responses in B cell acute lymphoblastic leukemia (B-ALL) (10) and multiple myeloma (11), respectively. Of note, T cells engineered to express a TCR against NY-ESO-1 induced tumor regression in patients with synovial cell sarcoma and melanoma (12).
Despite exciting results in certain human malignancies, only a few trials have shown responses in solid tumors. Therefore, both academia and the pharmaceutical industry are intensively investigating how to improve ACT and develop next-generation T cell immunotherapies. Thanks to the novel gene-editing technologies and new combinatorial strategies, ACT has the potential to target virtually every cancer type. Moreover, T cells can be modified to express transgenes that could further enhance their effector functions and persistence and/or reduce toxicity, opening a myriad of possibilities in the emerging field of synthetic biology. Here, we highlight the most recent results and lessons learned in clinical trials with ACT, and we discuss the principles of effective treatment that are guiding the design of next-generation T cell therapies (Figure 1).
Figure 1.
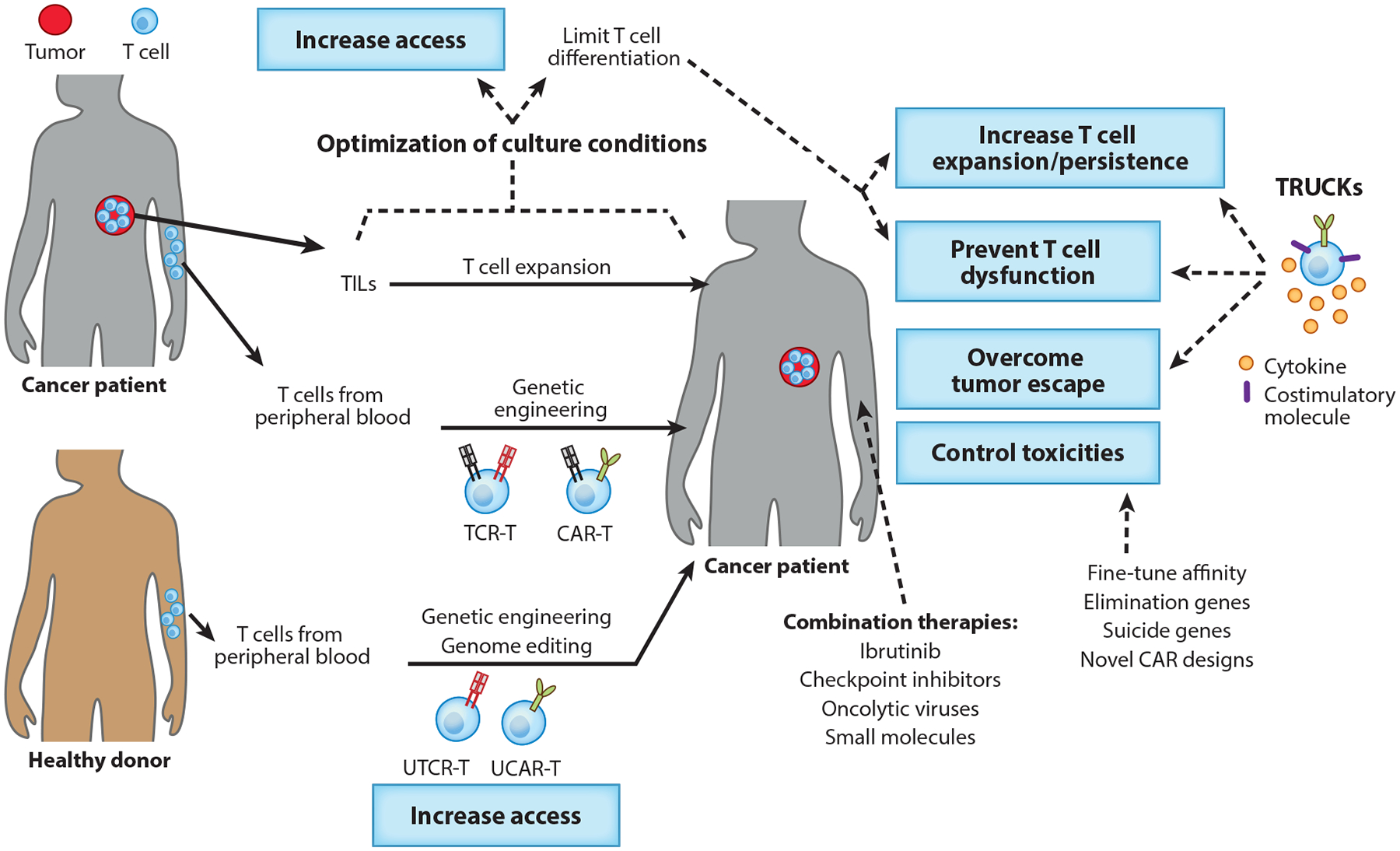
Adoptive T cell transfer therapies. Tumor-specific T cells can be isolated from tumors or generated by genetic engineering of peripheral T cells. To eliminate the tumor, infused therapeutic T cells need to reach cancer cells, proliferate, and survive in a hostile tumor microenvironment. Research approaches to overcome these challenges while preventing toxicity and ensuring access to every patient are exemplified. Abbreviations: CAR, chimeric antigen receptor; TCR, T cell receptor; TIL, tumor-infiltrating lymphocyte; TRUCK, T cell redirected for universal cytokine-mediated killing; UCAR-T, universal CAR-T cell; UTCR-T, universal TCR-T cell.
ADOPTIVE T CELL THERAPY: LESSONS LEARNED
Tumor-Infiltrating Lymphocytes
Adoptive transfer of naturally occurring tumor-specific T cells has been used in melanoma patients for over 20 years. The rationale behind this approach is that melanoma tumors are often infiltrated by tumor-specific T cells, but they appear to be unresponsive and fail to control the tumor. Removing these T cells from the immunosuppressive tumor microenvironment and culturing them in vitro with IL-2 enables their activation and large-scale expansion before they are infused back into the patient. Current efforts are focused on enhancing the quality and characterization of the final T cell product used for the treatments and simplifying the methods to obtain these tumor-specific T cells (Figure 2). Accumulating evidence suggests that tumor regression following TIL transfer is mediated by T cells targeting tumor-specific antigens, also known as neoantigens (13, 14). TILs are a heterogeneous population, and thus enrichment of the final product in neoantigen-reactive T cells may enhance clinical efficacy. In this regard, PD-1 and 4–1BB have been proposed as markers for the tumor-reactive repertoire in the tumor (13, 15, 16). Whether isolation and infusion of these neoantigen-specific T cells will be more effective than bulk TIL transfer requires clinical investigation. A more recent report showed that expression of PD-1 identifies neoantigen-reactive lymphocytes in the peripheral blood of melanoma patients (17), providing a novel noninvasive strategy to develop T cell-based personalized therapies. Alternatively, tumor-specific T cells could potentially be isolated following vaccination or treatment with checkpoint blockade therapy. In this regard, personalized vaccines targeting neoantigens have been shown to promote preexisting, as well as de novo, neoantigen-specific T cell immunity (18).
Figure 2.
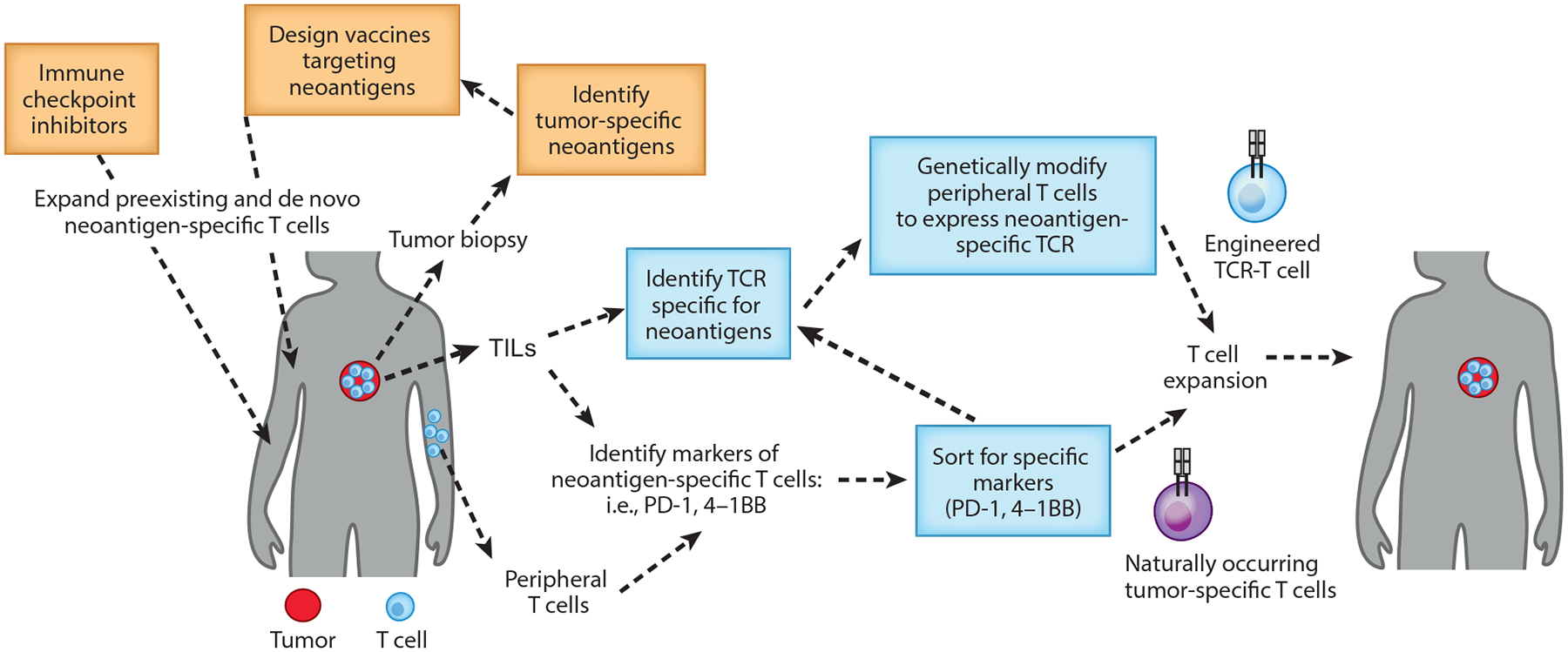
Future strategies with adoptive transfer of neoantigen-reactive T cells. Tumor-reactive T cells are isolated from tumors or peripheral blood after sorting for specific markers (PD-1+ and 4–1BB+ cells), expanded ex vivo, and reinfused into the tumors. Alternatively, neoantigen-specific TCRs can be identified and cloned in viral vectors and used to genetically modify peripheral T cells. These strategies can be combined with immunological-checkpoint inhibitors or neoepitope vaccination. Abbreviations: TCR, T cell receptor; TIL, tumor-infiltrating lymphocyte.
TCR-Engineered Lymphocytes
Despite the exciting results in melanoma patients treated with TILs, and recent progress in the field, the translation of TIL therapy into a setting of routine personalized medicine is still challenging and limited by the logistics of obtaining tumor biopsies. An alternative to the process of isolating TILs is to genetically modify T cells isolated from the blood to express transgenic TCRs able to recognize tumor antigens and induce cancer cell killing. In early approaches, T cells were modified to express TCRs targeting shared antigens, which are expressed at high levels on different cancers but can also be expressed in normal tissues. Treatment with TCR-T cells targeting MART-1, gp100, CEA, and MAGE-A3 resulted in unfavorable safety profiles, including poor antitumor effects and/or severe toxicity (19–24). By contrast, adoptive transfer of high-avidity TCR-T cells targeting NY-ESO-1, a cancer-testis antigen, induced objective clinical responses in patients with synovial cell sarcoma, melanoma, or myeloma, without severe toxicity (12, 25), and this is likely to be the first transgenic TCR to be approved by the FDA.
The observation that T cells recognizing neoantigens have a critical role in tumor regressions after TIL therapy or checkpoint blockade has shifted the focus of TCR-T cell therapy from self-antigens to neoantigens. T cells targeting neoepitopes are not subjected to negative selection and may therefore express high-avidity TCRs with the potential of inducing strong antitumor effects in the absence of toxicity. Recent technological advances in tumor exome sequencing and epitope prediction algorithms permit rapid identification of the immunogenic neoepitopes resulting from somatic gene mutations in tumor cells. Isolation of T cells with reactivity against these targets, and identification of mutation-reactive TCRs that can be genetically introduced into autologous T cells, is being actively explored for the development of personalized therapies.
Although the infusion of neoantigen-specific T cells holds great potential, the current protocols to generate good manufacturing practice (GMP)-grade, neoantigen-specific, personalized autologous T cells are costly, labor intensive, and time consuming. Moreover, limitations of transgenic TCRs (and TIL therapy) are that they are dependent on external costimulation and presentation of the targeted neoepitope via the MHC-I complex, which is often downregulated in cancer cells (26–29). An alternative approach that may address some of these challenges is to genetically modify T cells to express CARs that are MHC independent.
Chimeric Antigen Receptors
CARs are synthetic hybrid receptors created by genetic engineering that combine an extracellular domain, typically derived from an antibody single-chain variable fragment (scFv), with intracellular signaling domains providing activation signals that are derived from endogenous TCRs and costimulatory signals (2). Since antigen recognition by CAR-T cells is based on scFv binding to intact surface antigens, the targeting of tumor cells is neither MHC restricted nor dependent on processing and effective presentation of target epitopes. However, CAR recognition requires surface expression of the tumor antigen.
The extracellular glycoprotein CD19 is the most common B cell target for CAR-T therapies. Within the past few years, clinical trials using CD19-targeting T cells (CD19-CAR-T cells) have shown high rates of sustained complete responses that are unprecedented in relapsed and refractory B-ALL (7, 30, 31, 32). Significant responses have been observed also in chronic lymphocytic leukemia (CLL) (33, 34) and non-Hodgkin lymphoma (NHL) (9, 35, 36). Some key lessons were learned in these trials: (a) An effective proliferative response is the best predictor of clinical efficacy—T cells have to expand and survive (33); (b) CD19-CAR-T cells show differential rates of complete responses among different B cell neoplasms, with ~80% complete-response rate in pediatric and young adult patients with B-ALL and 30–50% complete-response rate in adult patients with diffuse large B cell lymphoma; (c) CAR-T cell dysfunction (37) and loss of CD19 in leukemia cells (38) are the main relapse mechanism in patients treated with CD19-CAR-T cells; (d) CAR-T treatment can cause significant toxicities, such as cytokine-release syndrome (CRS) and neurotoxicity (35); and (e) the use of anti-IL-6R antibody (tocilizumab) and steroids can successfully control these adverse events in most patients (39).
Importantly, CAR-T cells are also being evaluated in other hematologic malignancies that do not express CD19 (40). For acute myeloid leukemia (AML) several targets are under investigation, such as CD33 (41), CD123 (42–47), and CD44v6 (48); however, off-tumor expression of these antigens suggests potential toxicities. Clinical data have shown some efficacy of anti-CD30 CAR-T therapy for Hodgkin lymphoma (49). In multiple myeloma anti-BCMA CAR-T cell therapies (50) are leading to excellent clinical responses in multiple clinical trials (51, 52).
The success shown by CAR-T cells in hematologic cancers supports the translation of this technology to the challenging setting of solid tumors that are responsible for more than three-quarters of cancer-related deaths in humans and therefore represent a large unmet medical need. Several clinical trials testing CAR-T cells for the treatment of solid tumors are ongoing. Early clinical trials reported poor CAR-T cell efficacy with different levels of toxicity (53–55). However, recent reports of patients with glioblastoma, pancreatic cancer, mesotheliomas, and sarcomas have provided evidence supporting the feasibility of CAR-T cells, with transient activity and absence of serious adverse events (56–60). Of note, in a recent report CAR-T cells directed to IL-13Rα2 induced a complete regression of metastatic glioblastoma in one patient (61). Some of the valuable lessons learned by initial clinical trials with solid tumors that will drive the design of future CAR-T cell therapies include the following: (a) Despite CAR-T cells trafficking to the tumor, initially proliferating in situ and exerting some direct antitarget activity, substantial antitumor responses are rarely observed (57, 59); (b) antitumor efficacy is often limited by a lack of significant expansion and/or survival of CAR-T cells in the tumor (55, 58, 62); (c) significant decrease or loss of targeted-antigen expression has been observed following CAR-T cell administration, suggesting some level of transient CAR-T cell activity and pointing at antigen heterogeneity and antigen loss as major barriers to overcome (57, 59); and (d) severe on-target off-tumor toxicity has been observed in some trials, even when antigens are expressed at very low levels. As CAR-T cells can dramatically proliferate in vivo, great caution should be taken when designing and implementing novel CAR-T cell therapies to mitigate toxicities (53, 54, 62).
CURRENT ROADBLOCKS FOR T CELL IMMUNOTHERAPIES
As discussed, the efficacy of ACT needs to be improved in solid tumors, but in CD19+ neoplasms additional optimization of the treatment could improve the long-term success and safety of CD19-CAR-T cells. Finally, when these new exciting products are commercialized, fair access to them should be ensured to all patients. To better understand how to improve ACT, a deep knowledge of the current roadblocks is required, and optimal strategies need to be developed.
T Cell Persistence and Expansion
Significant expansion and persistence of adoptively transferred T cells are considered critical predictors of clinical efficacy. Several clinical trials testing the infusion of TILs or genetically modified T cells have confirmed the correlation between the persistence of the infused T cells in the circulation and the likelihood of tumor regression (37, 63). By contrast, most ACT clinical trials where lack of overall efficacy was observed have reported poor T cell persistence, especially in the context of solid tumors. Likewise, lack of response in some patients and relapse in patients responding to ACT are often associated with loss of tumor-reactive T cells. Several factors can influence the persistence of adoptively transferred T cells, including patient preconditioning, ex vivo culture conditions, development of T cell exhaustion, and lack of costimulation or host immune responses against the cellular infusion product.
Early trials with melanoma patients demonstrated that sustained persistence of TILs is only achieved after preparative lymphodepletion of the patient before TIL transfer (64). Preclinical models suggest that lymphodepletion creates a homeostatic space for T cell expansion, depletion of regulatory T cells, and activation of the innate immune cells that leads to a better engraftment and stimulation of infused antitumor CD8+ T cells (65, 66). Clinical studies indicate that the intensity of lymphodepletion is an important variable governing the expansion and persistence of CAR-T cells (7).
Some of the protocols currently used to culture T cells for ACT result in extensive in vitro proliferation and T cell differentiation. Moreover, TILs may contain exhausted or terminally differentiated T cell populations. T cell differentiation is associated with telomere shortening, phenotypic changes (including loss of expression of CD27, CD28, and CCR7), and loss of replicative capacity. In this regard, studies in melanoma patients using biological correlates indicated that short TIL culture duration, longer telomeres of the infused cells, and the number of CD8+CD27+ cells infused are associated with objective responses (67). Similarly, one study conducted at the University of Pennsylvania evaluating the biomarkers predictive of clinical response to CD19-CAR-T cells in patients with CLL found that the preinfusion product of responding patients was enriched in less-differentiated T cells (CD27+CD45RO−) and Th17 cells (37). In this same study, long-term remissions were associated with infusions containing transcriptomic signatures of early memory CAR-T cells, while T cells from nonresponding patients were enriched in genes related with terminal differentiation and exhaustion. In line with this, preclinical studies in animal models have shown that adoptive transfer of less-differentiated T cell subsets, including naive and memory stem cells, results in superior in vivo expansion, persistence, and antitumor response compared to the more-differentiated T cell subsets. The rationale behind this is that less-differentiated T cells persist longer and are a continuous source of effector cytotoxic cells. Other studies suggest that Th17 cells may have enhanced antitumor effects when used in ACT due to their higher in vivo survival, self-renewal capacity, and resistance to senescence compared to Th1 or nonpolarized T cells (68, 69).
T Cell Dysfunction
Some patients fail ACT despite tumor-specific T cells accumulating within tumors that express the target antigen. A growing body of evidence suggests that this inability of T cells to eliminate tumors may be due to a progressive loss of T cell effector functions that can be caused by both intrinsic (T cell fitness) or extrinsic (tumor microenvironment) factors (70).
T cell dysfunction, including T cell anergy, exhaustion, and senescence, arises during many chronic infections and cancer and is driven in part by chronic persistence of antigen (71). Dysfunctional T cells are characterized by impaired proliferation and effector function and expression in different degrees of various inhibitory receptors, including PD-1, TIM-3, LAG-3, TIGIT, and KLGR-1 (71, 72). In melanoma patients, the majority of tumor-reactive cells express PD-1 when isolated from tumors (16). Clinical results with adoptive transfer of TILs or immune checkpoint blockade demonstrate that T cell dysfunction can be partially reverted and that reinvigorated T cells can mediate complete responses in patients with solid tumors, underscoring the therapeutic potential of counteracting immune inhibition.
T cell exhaustion has been mainly studied in virus-specific or tumor-specific, naturally occurring T cells. Exhausted T cells have distinct genetic and epigenetic reprogramming states that appear to be stable and not reversible (73). CAR-T cells have been modified with chimeric receptors that do not exist in nature, and thus the dynamics and features of T cell exhaustion may be different compared to the signaling through the TCR. The presence of exhaustion markers on CD19-CAR-T cells at the time of infusion predicts poor response to therapy in CLL patients. Whether chronic antigen exposure can induce T cell exhaustion in patients treated with CAR-T cells is currently unknown, but it is likely that preventing T cell exhaustion will be important to improve treatment.
An important mechanism of T cell exhaustion in CAR-T cells that has not been described in naturally occurring T cells is antigen-independent tonic signaling due to spontaneous clustering of CAR molecules. Emerging evidence suggests that the optimal configuration of all the elements within a CAR, including the scFv, hinge, transmembrane (TM) domain, and intracellular domains, is essential for optimal CAR-T cell function and persistence. Depending on the selection and configuration of CAR modules, and the levels of CAR expression on the cell surface, some CAR-T cells can have tonic signaling during ex vivo expansion (74–78). Tonic signaling is associated with augmented T cell apoptosis, early T cell differentiation and exhaustion, and impaired antitumor effects. CAR-T cells with tonic signaling have been used in preclinical and clinical trials, leading to poor antitumor activity and results that have been difficult to interpret.
Tumor Heterogeneity and Antigen Loss
In the presence of fully functional tumor-specific T cells, tumor cells can develop adaptive resistance to avoid being eliminated by the immune system. Tumor evasion due to antigen loss, heterogeneity of antigen expression, or impaired antigen presentation has been widely reported after ACTs in both preclinical and clinical studies, and these are considered main causes of treatment failure.
In the case of engineered T cells targeting a single antigen, to avoid tumor escape, the target antigen should be expressed in all tumor cells; alternatively, its expression should be essential for tumor growth. However, even in this ideal scenario, tumor escape can potentially occur. For example, in the setting of CAR-T cells targeting CD19, a cell surface antigen that is uniformly and highly expressed on B lineage cells, emergence of antigen-negative leukemic cells is observed in a significant subset of treated patients (30). It is unclear if these cells are present in the initial cancer and overgrow thanks to a selective advantage under pressure from the T cell therapy, or if they develop de novo mutations to escape the selective pressure. Lack of surface expression of CD19 may be due to mutations (79, 80), alternative splicing in CD19 (exon 2 skipping) (80), or mutations in the B cell receptor protein CD81 (81, 82). New evidence suggests that loss of antigen expression might contribute to treatment failure when targeting not only CD19 but also other antigens like CD22. Similar to CD19, CD22 is uniformly expressed in B lineage cells and has been proposed as an alternative antigen to target CD19-negative relapsed ALL patients (83, 84). Diminished CD22 surface expression on ALL B cells without detectable CD22 mutations has been recently identified as a mechanism of relapse following CD22-CAR-T therapy (10). Sequential loss of both CD19 and CD22 following sequential CAR-T cell treatment has also been observed in one patient with NHL (85). A novel mechanism of CD19-negative immune escape includes lineage switch from lymphoid to myeloid leukemia with consequent loss of expression of B lymphoid lineage antigens (86, 87).
Solid tumors are characterized by heterogeneous antigen expression with regard to intensity and distribution, which increases the possibility of tumor escape when compared to hematologic malignancies. CAR-T cell therapies preferentially kill tumor cells with high targeted-antigen expression but may fail to eliminate cells with lower antigen densities (88–90). Diminished expression of the targeted antigen after T cell therapy in patients with solid tumors has been reported in several trials with both CAR-T cells and TCR-T cells (12, 57, 59). Targeting multiple tumor antigens, using combination therapies, or implementing strategies to promote epitope spreading could mitigate tumor escape.
TIL therapy or infusion of patient-specific engineered TCR-T cells targeting different neoantigens could provide a solution to the difficulties of finding single tumor antigens to serve as universal targets. However, the mutation or decreased expression of genes involved in antigen-presentation pathways, including specific deletion of HLA class I alleles or loss of β2 microglobulin (β2m), may also lead to tumor escape (26, 27, 29, 91). An interesting example of this was reported by Tran et al. (28) after treatment of one patient with metastatic colorectal cancer with TILs targeting mutant KRAS G12D. Despite an evident clinical benefit, one metastatic lesion relapsed by acquiring uniparental disomy, which maintains the surface expression of irrelevant MHC molecules (preventing tumor cell elimination by natural killer cells) but precludes the presentation of the mutant KRAS peptide.
Tumor Contamination Leading to Resistance
Tumor contamination in the cellular therapy product is known to correlate with relapse in the setting of autologous stem cell transplantation (92). CD19-CAR-T cells are generated from the peripheral blood lymphocytes and, for patients with acute leukemia, leukemic blasts can often be present in the peripheral blood at the time of collection. Usually by the end of the CD19-CAR-T cell expansion, no residual B cell blasts are detected by flow cytometry, as CD19-CAR-T cells effectively purged them ex vivo. However, we recently reported a patient treated with CD19-CAR-T cells (CTL019) who relapsed nine months after infusion with CD19-negative leukemia that aberrantly expressed the anti-CD19 CAR protein (93). The CD19-CAR-T gene was unintentionally introduced into a single leukemic B cell by lentiviral transduction during CD19-CAR-T cell manufacturing. Interestingly, the CAR19+ leukemic cells were able to survive the CD19-CAR-T cell purging during expansion as the CAR protein expressed on the surface of leukemic cells bound in cis to the CD19 epitope, masking it from recognition by CD19-CAR-T cells. Although very rare, this case warrants careful evaluation of the minimal residual disease in the ACT products (93).
Altogether, these results highlight the strong immune selective pressure that targeted T cells can impose, as well as the great capacity of tumor cells to find new mechanisms to evade the immune system. Several lessons from the infectious diseases field are instructive in strategies that will be required to overcome resistance to ACT (94).
Toxicity
Together with the remarkable clinical activity, ACT has demonstrated significant and unique toxicities (95). Potent immune activation and general inflammation are triggered by the infused T cells recognizing tumor cells. In the setting of CD19-CAR-T cell therapy, CRS is observed in the majority of B-ALL patients and in subsets of B-CLL and B-NHL patients (6, 8, 9, 33, 96–98). This syndrome is characterized by increased levels of cytokines (IL-6, TNF-α, and others) and other inflammatory markers (ferritin, C-reactive protein), alongside with fever, hypotension, myalgia, and additional systemic symptoms. In some patients treated with CD19-CAR-T cells, CRS is followed within days by neurotoxicity of unknown etiology. While this syndrome is generally self-limiting and resolves within days, in rare instances it can result in fatal cerebral edema (99, 100). The prompt use of an anti-IL-6R monoclonal antibody, tocilizumab, can control CRS in the majority of patients (101, 102), but it does not prevent or treat neurotoxicity. New murine models that recapitulate key features of CRS and neurotoxicity suggest that blocking IL-1 may abolish both CRS and neurotoxicity, enabling new therapeutic interventions (103–105).
Another important toxicity is on-target, off-tumor toxicity that occurs when healthy tissues bearing the same antigen being targeted by the cellular therapy are also damaged. An example of this adverse event is B cell aplasia, caused by the presence of CD19 on normal B cells and subsequent killing by CAR-T cells (106). This event can be predicted in most cases but can be an unexpected clinical adverse event. Treatment with CAR-T cells targeting Her2/neu antigen, which is expressed at low levels in the lungs, resulted in rapid and fatal toxicity in one early trial (54). Off-target, off-tumor aberrant reactivity, also known as cross-reactive toxicity, describes unexpected targeting of healthy tissues by T cells that had not been anticipated, in particular with TCR-T cells. In this regard, two different trials have reported severe toxicity, including lethal events, after treatment with T cells redirected to a MAGE-A3 peptide (23, 24). In one trial, T cells induced neurotoxicity due to TCR cross-reaction with MAGE-A12 (107), while in the other, T cells recognized an unrelated muscle-specific protein, Titin, leading to cardiac toxicity (108, 109).
Solid Tumors
With rare exceptions, the outcomes of ACT in patients with nonmelanoma, solid tumors remain poor. Clinical trials conducted with CAR-T cells targeting solid tumors suggest that CAR-T cells can engraft in the peripheral blood, traffic to tumors, and respond to antigen but fail to expand, persist, and mediate objective responses (56, 58, 59, 61). Compared to hematologic malignancies, solid tumors pose extra barriers of complexity (110, 111). First, T cells must traffic and infiltrate into the solid mass. On arrival at the tumor, T cells encounter an immunosuppressive environment that includes immunosuppressive cells (regulatory T cells, tumor-associated macrophages, myeloid-derived suppressor cells), hypoxia, necrosis, nutrient shortage, and an array of immunosuppressive molecules (PD-L1, IL-10, TGF-β). Moreover, many enzymes with immune regulatory activity are up-regulated in cancer patients, either in the tumor environment or in draining lymph nodes, such as arginase 1, inducible nitric oxide synthase, and IDO, and production of nitric oxide and reactive oxygen and nitrogen species is increased (112, 113). These conditions can lead to inadequate T cell activation and/or T cell inhibition, restricting the persistence of tumor-specific T cells within the tumor and limiting the benefits of ACTs (114). Furthermore, if the tumor persists, continuous antigen exposure could lead to the T cell-intrinsic program of exhaustion. Finally, in the case that T cells are able to survive in the immunosuppressive tumor environment and maintain their effector functions, loss of antigen or the components in the antigen-presentation pathway may result in overgrowth of antigen-negative populations and tumor escape.
Financial Toxicity
In developed economies, there is an emerging debate about rising prices for cancer drugs (115, 116). In the United States the price tag for all new cancer drugs approved in 2017 is more than $100,000 per year. ACT is a powerful approach that provides durable responses and the potential for cure in patients with previously refractory cancers. Two CD19-CAR-T cell products were approved for clinical use in 2017 in the United States at a price of $373,000 to $475,000. Due to their personalized nature, the bespoke GMPs of TILs, CAR-T cells, and TCR-T cells are associated with high development and manufacturing costs, stringent regulatory requirements pertaining to gene transfer, and reimbursement challenges. The current high cost of CAR-T cell manufacturing might be mitigated by an approach such as annuity payments with risk sharing, also known as pay-for-performance (117). Novartis has implemented this approach in the United States and charges for tisagenlecleucel CAR-T cells only upon clinical benefit in patients with ALL (118). There is an urgent need to control the escalating costs of cancer therapy, and in the case of ACT, improved cell manufacturing is the most direct strategy to lower the cost and improve access to this emerging technology.
STRATEGIES TO IMPROVE ADOPTIVE CELL THERAPY
Enhancing Persistence
Most efforts directed at enhancing T cell persistence have been focused on optimizing CAR constructs to contain intracellular domains that support T cell persistence and/or identifying strategies to enrich the final T cell product in T cell subsets with enhanced regenerative capacity.
The molecular design of CARs can strongly influence T cell expansion and persistence. While first-generation CARs containing only the CD3ζ chain persisted long-term in HIV patients (119), they failed to persist in cancer patients (55). The addition of a costimulatory domain in second-generation CARs greatly enhanced T cell persistence and antitumor effects. CARs containing either CD28 or 4–1BB costimulatory domains have been the most widely used in the clinic, and both CAR designs yielded significant tumor regressions in patients with hematologic malignancies. Several studies indicate that CD28-based CARs have greater initial antitumor activity, while 4–1BB signaling enhances CAR-T cell persistence and mitigates exhaustion. The enhanced persistence of CAR-T cells containing the 4–1BB signaling domain may be explained, at least in part, by the increased mitochondrial biogenesis and oxidative metabolism induced by 4–1BB signaling (120). Other signaling domains, including CD27, OX40, and ICOS, are also being investigated in preclinical trials. CD27, which like 4–1BB is a member of the tumor necrosis factor receptor (TNFR) family, has been shown to promote T cell survival similarly to 4–1BB (121). Signaling through ICOS, a member of the CD28 family, has a key role in enhancing the persistence of CD4+ T cells (78, 122). Third-generation CARs combining two costimulatory domains, usually one from the CD28 family (CD28 or ICOS) and one from the TNFR family (4–1BB or OX40), can activate different downstream signals, combining potency with long-term persistence, and are promising candidates for clinical evaluation (78).
A different strategy to enhance persistence is to enrich the infusion product in T cells with stem cell properties. Current approaches focus on shortening the expansion protocols and optimizing them to include cytokines or agents able to delay T cell differentiation. Rapid-expansion protocols used to expand TILs induce rapid T cell differentiation, while TILs that undergo shorter expansion ex vivo persist longer (123, 124). A recent work proposes to shorten the expansion protocol for CAR-T cells from day 9 to day 3, which would limit T cell differentiation while simplifying production methods and increasing access (125). Exposure to IL-7, IL-15, or IL-21 during expansion protocols has been shown to preserve a less-differentiated state and improve in vivo persistence (126–128). However, whether enriching the final product in certain subsets, rather than using a sorted population, will be sufficient to increase ACT efficacy remains unknown. In this regard, it has been proposed that T cell subsets can synchronize their differentiation state, and therefore the presence of antigen-experienced subsets in the initial product can promote the differentiation of naive cells (129). This process is mediated by T cell-T cell interaction through a nonapoptotic Fas signal, resulting in Akt-driven cellular differentiation. Blocking the interaction between naive T cells and more-differentiated T cells, through sorting of naive T cells or Fas signaling blockade, may be used to prevent differentiation. Alternatively, activation of the wnt signaling (130) or pharmacologic inhibition of Akt can also enhance the expansion of T cells with memory cell characteristics (131, 132). Another strategy to enhance persistence is to enrich the infusion product in Th17 cells by culturing T cells with Th17 polarizing cytokines and optimized costimulation. ICOS signaling plays a key role in the function of Th17 cells (133), and CARs containing the ICOS ICD have been shown to polarize T cells toward a Th1/Th17 phenotype, which results in enhanced persistence of CD4+ T cells and superior antitumor effects (78, 122).
Recent work shows that in exceptional conditions, the progeny of a single CAR-T cell is sufficient to mediate potent antitumor effects in advanced leukemia, suggesting that quality of the infused T cells may be more critical than quantity (134). However, the majority of ongoing current trials with ACT use bulk T cells. The advantage of using selected populations with less-differentiated phenotypes awaits confirmation in clinical trials.
Manipulating Host Hematopoiesis
The absence of cancer-restricted surface markers is a major impediment to antigen-specific immunotherapy for solid tumors and several hematologic malignancies. As an example, anti-CD33 CAR-T cells can effectively kill CD33+ AML cells, but as CD33 is also expressed in the hematopoietic stem cell, they result in severe myelotoxicity. Kim et al. (135) preclinically demonstrated that CD33 can be safely targeted by deleting CD33 from normal hematopoietic stem and progenitor cells, generating a hematopoietic progeny resistant to CART33 (Figure 3). Therefore, infusion of CD33-deleted hematopoietic stem cells could allow efficient elimination of CD33+ leukemia cells by CART33 without myelotoxicity.
Figure 3.
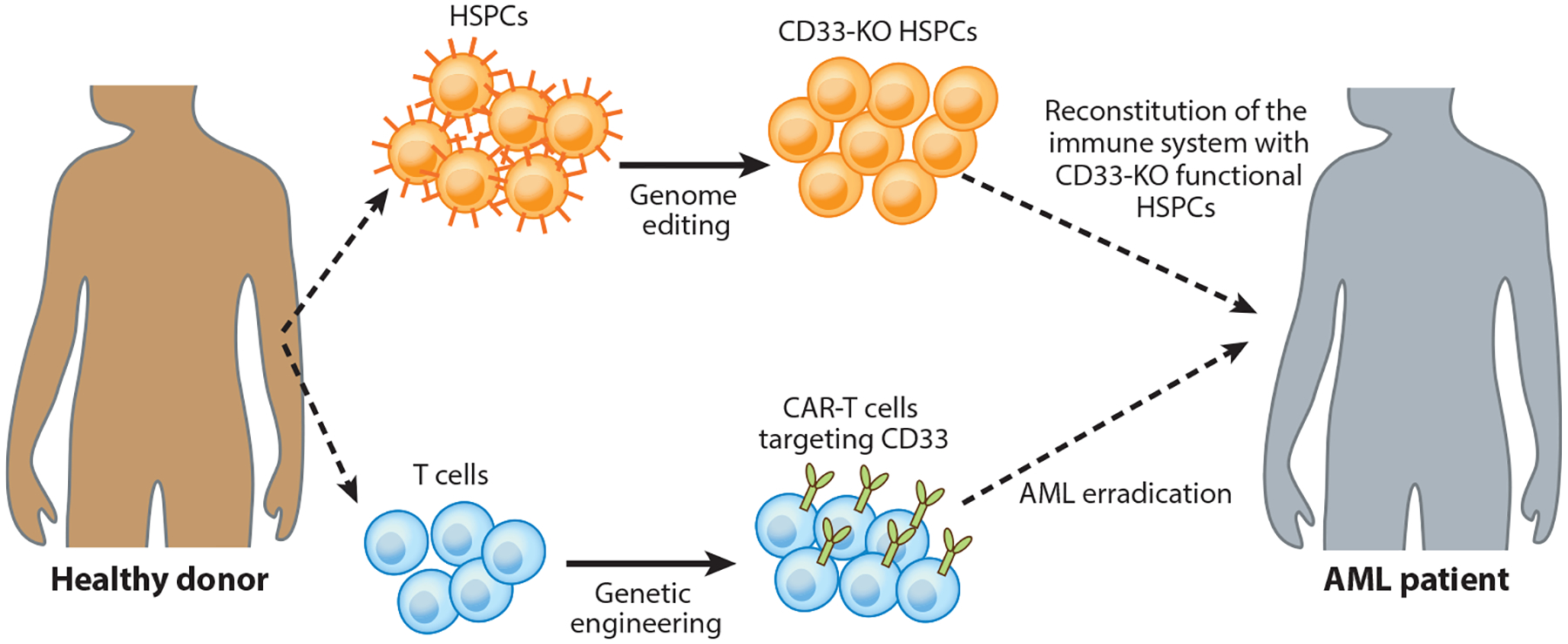
Reconstitution of the patient’s immune system with genome-edited hematopoietic stem cells to prevent on-target, off-tumor toxicity after CAR-T cell treatment. CD33 is expressed in both AML cells and normal myeloid progenitors, and therefore CAR-T cells targeting CD33 could induce myelotoxicity. In this strategy, CD33 is genetically disrupted in hematopoietic stem and progenitor cells to regenerate an antigen-negative myeloid system that is resistant to CAR-T cell therapy. Anti-CD33 CAR-T cells can eradicate AML while sparing CD33-deficient hematopoietic stem and progenitor cells. Abbreviations: AML, acute myeloid leukemia; CAR, chimeric antigen receptor; HSPC, hematopoietic stem and progenitor cell; KO, knockout.
Overcoming T Cell Dysfunction
One possible strategy to overcome T cell dysfunction is to shield adoptively transferred T cells from inhibitory signals. Infused tumor-specific T cells undergo activation-induced upregulation of coinhibitory receptors, including PD-1, TIM-3, LAG-3, and CD200, although the role of these markers is not fully understood yet. On the other hand, tumor cells can augment the expression of coinhibitory ligands, such as PD-L1, upon T cell activation-mediated release of Th1 cytokines. Strategies that combine CAR-T cells with checkpoint blockade, most of them focusing on the PD-1/PD-L1 interaction, are being actively investigated. PD-1/PD-L1 pathway interference through short hairpin RNA blockade, PD-1 dominant negative receptor, or chimeric switch-receptors have been shown to augment the efficacy of CAR-T cells in both solid tumors and hematologic malignancies (136). Using genome editing, it is now possible to delete the PD-1 gene locus on T cells. Preclinical studies indicated that PD-1 editing enhanced anticancer activity of engineered T cells, with promising evidence of efficacy in solid tumors (137, 138). A first-in-human phase 1 CRISPR gene-editing clinical trial is now testing the safety and efficacy of NY-ESO-specific TCR-T cells lacking both the endogenous TCR and PD-1 in patients with melanoma, synovial sarcoma, and multiple myeloma (NCT03399448). However, although expression of inhibitory receptors is a hallmark of exhausted T cells, it is unclear whether signaling through these receptors directly causes T cell exhaustion. Genetic absence of PD-1 has been shown to promote accumulation of terminally exhausted CD8+ T cells, highlighting a novel role of PD-1 in preserving T cells from overstimulation, excessive proliferation, and terminal differentiation (139). A better understanding on how these inhibitory receptors affect exhaustion of engineered T cells, together with the identification of novel pathways implicated in T cell dysfunction, will be required to further enhance the effector functions and persistence of CAR-T cells in the tumor microenvironment. Moreover, whether the abrogation of negative immune regulators could lead to increased challenges of autoimmunity or transformation needs to be addressed in clinical trials. Relevant to the later possibility, a recent study has shown that PD-1 functions as a haploinsufficient tumor suppressor in mouse T cells (140). Whether this is true for human T cells remains unknown.
A distinct class of strategies to prevent or delay dysfunction in the context of CAR-T cells involves optimizing the design and regulatory expression of CAR constructs. The choice of the type and position of the costimulatory domain, the spacer length, and/or the promoter or vector used to express the CAR has been shown to influence the level of tonic signaling. Therefore, optimization of the CAR constructs is essential to avoid early T cell dysfunction. A newer approach to regulate CAR expression involves targeting the insertion of a CAR-coding sequence into the native TCR locus, placing it under the control of endogenous regulatory elements. Targeting the CAR to the TCR established effective internalization and reexpression of the CAR upon antigen recognition, reduced tonic signaling, delayed T cell differentiation and exhaustion, and enhanced potency of CAR-T cells (141).
Another way to curb environmental immunosuppression is to target non-cancer cell components of the tumor. Tumor stromal fibroblasts and tumor-associated macrophages can induce T cell dysfunction by secreting immunosuppressive proteins, expressing ligands for inhibitory receptors, and/or developing a fibrotic extracellular matrix that can impair T cell penetration and function. One approach currently undergoing clinical evaluation is to generate CAR-T cells targeting the fibroblast activation protein, which is overexpressed in nearly all solid tumors by a subset of immunosuppressive tumor fibroblasts (142–144). A more ideal, but also more challenging, strategy is to target an antigen that is overexpressed in both malignant and neighboring immunosuppressive cells. In this regard, CAR-T cells targeting CD123 have been shown to overcome immunosuppression in preclinical models of Hodgkin lymphoma by simultaneous targeting of malignant cells and tumor-associated macrophages (145). A different approach to enhance the activity of CAR-T cells in patients with stroma-rich solid tumors is to further modify CAR-T cells to overexpress enzymes able to degrade the extracellular matrix (146).
Overcoming Loss and/or Heterogeneity of Antigen Expression
In both B-ALL (30) and glioblastoma (59) patients treated with CAR-T cells, antigen loss has been observed as a frequent escape mechanism. In B-ALL the majority of relapses after CD19-CAR-T therapy are characterized by the loss of CD19. While several mechanisms are being described (80, 87), targeting more than one antigen in leukemic cells might reduce antigen-negative escape. Possible targets expressed in both normal B cells and B-ALL blasts are CD22, CD20, and CD123. Several preclinical studies (88, 147–149) have been published using bi- or trispecific CAR-T cells, all showing that multiple-targeting is highly effective in preventing escape. Pools of CAR-T cells, i.e., mixtures of CAR-T cells with different specificities; or dual CAR-T cells where a single cell can engage several antigens with distinct CAR constructs; or a single CAR with two binding-domains in frame can serve this purpose (Figure 4). Ruella et al. (150) demonstrated in a clinically relevant preclinical model of CD19-negative leukemia escape that cotargeting CD19 and CD123 can treat and also prevent antigen loss, and dual CAR-T cells are more potent than pooled CAR-T cells. Other important targets in addition to CD123 that are being evaluated for cotargeting to prevent antigen escape are CD22 (10, 151) and CD20 (152). More recently, switchable CARs, such as biotin-binding immune receptors and UniCARs, have been developed by creating an array of soluble, modular, tagged scFv domains, equally fitting a constant CAR stalk and supplied as required, thus allowing extra flexibility and multispecificity in a timely controlled manner. A main drawback of these approaches is that selecting safe solid tumor targets can be challenging due to on-target, off-tumor side effects.
Figure 4.
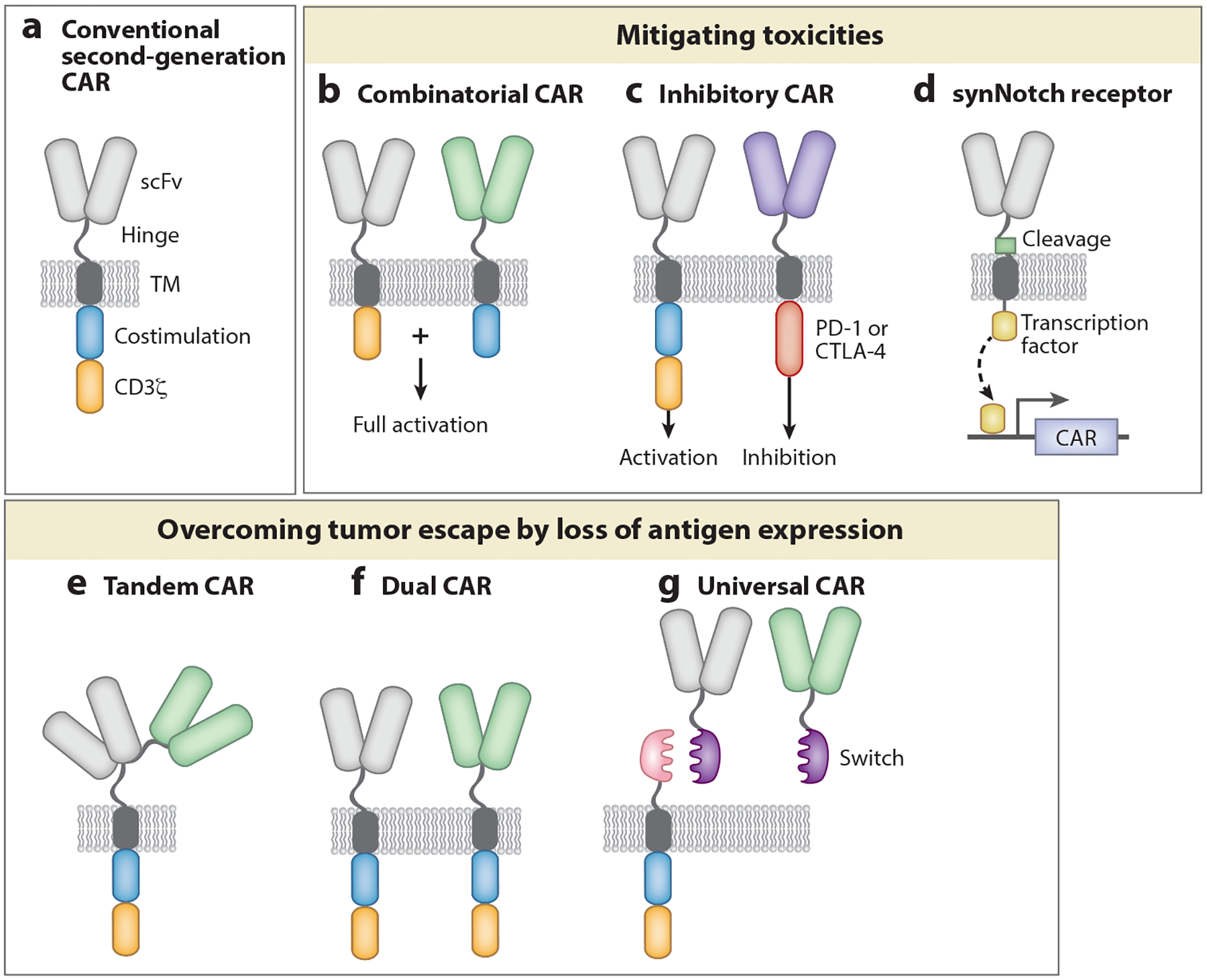
Next-generation CAR-T cells. Novel CAR designs can mitigate toxicities and/or address tumor escape. (a) Second-generation CARs consist of an extracellular domain that recognizes the tumor antigen, a hinge region, a transmembrane domain, and two intracellular signaling domains that transmit the signal. Optimization of each module is required for optimal T cell function and persistence. (b) Combinatorial CARs consist of two separate receptors targeting two different tumor antigens. One receptor bears the CD3ζ intracellular domain, and the other bears the costimulatory domain. Full activation is only achieved after recognition of both antigens. (c) Inhibitory CARs contain the intracellular domain from inhibitory molecules. Antigen recognition in normal cells by inhibitory CARs turns T cells off. (d) Synthetic Notch receptors involve a combination of two receptors. Activation of one receptor induces the expression of a second receptor (a CAR or TCR) that mediates cell killing. Efficient T cell activation occurs only when both antigens are recognized. (e) Tandem CARs contain two different scFvs in a single CAR construct. These receptors can be activated by two different cognate antigens. (f) Dual CARs consist of two separate second-generation CARs targeting two different tumor antigens. (g) Universal CAR constructs include T cells engineered to express the universal CAR, an antibody that will recognize the tumor antigen, and a switch that acts as a bridge between the target cell and the CAR, allowing targets to be switched without reengineering the T cells. Abbreviations: CAR, chimeric antigen receptor; scFv, single-chain variable fragment; synNotch, synthetic Notch; TCR, T cell receptor; TM, transmembrane.
Cytokine Armed T Cells: Armored CARs and TRUCKs
One possible strategy to overcome some of the obstacles mentioned above is to further modify T cells to release cytokines, express costimulatory ligands, or secrete checkpoint-blocking scFvs. These novel engineered T cells are known as TRUCKs (T cells redirected for universal cytokine-mediated killing) or armored T cells. The selected transgene may be constitutively expressed or induced after T cell activation following recognition of the tumor-targeted antigen. Cytokine-secreting tumor-specific T cells would offer the opportunity to harness the adjuvant effect of recombinant cytokines as living pharmacies, with the advantages of dispensing the drug locally at the tumor site and obtaining maximal activity in situ with minimal systemic toxicity. Expression of IL-12 by TILs (153), virus-specific CTLs (154), and CAR-modified T cells (155, 156) has been proposed as a mechanism to avoid tumor escape. Because systemic administration of IL-12 has proven to induce severe side effects (157), it is important to limit its expression in the tumor microenvironment, ideally by using an inducible promoter. IL-12 expression in the tumor microenvironment has been shown to improve T cell cytolytic activity, mitigate regulatory T cell-mediated immunosuppression, and recruit and activate an innate immune cell response toward antigen-negative cancer cells that could otherwise escape T cell therapy. IL-12-armored T cells are superior to their unarmored counterparts in a number of preclinical studies, thus providing the foundation for further clinical investigation (158). However, a first clinical trial infusing TILs modified to express IL-12 under the NFAT promoter reported lack of T cell persistence with severe dose-limiting toxicities (159). Given the potential IL-12 toxicity, expression of other cytokines, such as IL-15 (160) or IL-18 (161–163), has been proposed as a safer strategy to armor T cells. Both IL-15 and IL-18 have been shown to enhance the antitumor effects of infused T cells by prolonging T cell persistence and expansion.
Similarly, engineered T cells can be modified to constitutively express ligands for costimulatory molecules. Expression of CD40L in CAR-T cells has been proposed as a method to recruit an endogenous antitumor immune response (164). In a different strategy, Zhao et al. (165) tested different CAR designs and combinations of second-generation CARs with costimulatory ligands with the aim of clarifying the optimal costimulatory support to engineered CD19-CAR-T cells. Armoring a CD28-based CAR with 4–1BBL preserved the superior killing capacity of CD28-based CARs with the extended T cell persistence provided by the 4–1BB signaling.
Reducing Toxicity
The most obvious strategy to prevent toxicities is to target antigens that are expressed in tumor cells but not in normal tissue. In this regard, neoantigen-specific T cells have been shown to induce tumor regression in the absence of adverse effects (28). An elegant example of this was reported by Tran et al. (28), who showed that T cells modified to express a TCR against mutant KRAS G12D are able to kill such molecule-expressing tumor cells while ignoring HLA molecules that have wild-type KRAS peptides. Another promising strategy is to redirect CAR-T cells against cancer-specific neoepitopes formed by aberrant glycosylation, such as the Tn antigen on MUC-1 (166). However, the reality is that the majority of engineered T cells currently used, especially in the context of solid tumors, target overexpressed shared antigens. One alternative to target shared antigens is to generate affinity-optimized CAR variants that can discriminate between aberrant and healthy cells based on different levels of expression of the target antigen (167, 168). Specifically, low-affinity CARs have been proven to mediate strong cytotoxicity against transformed cells expressing high levels of the relevant antigen with minimal or no reactivity against antigen-low, normal cells (169, 170). Whether using low-affinity CARs will increase the chance of tumor escape should be further studied.
In case of T cell-mediated toxicities, the current clinical approach relies on the administration of tocilizumab and nonspecific pharmacological immunosuppression such as corticosteroid therapy. However, several approaches to actively eliminate toxic engineered T cells without killing beneficial immune cells are currently in clinical trials. Selective termination of CAR-T cell activity has been achieved by introducing suicide and/or elimination switches. Suicide genes include proteins like herpes simplex virus thymidine kinase or inducible caspase-9, which trigger apoptosis upon induction with ganciclovir or specific synthetic dimerizer drug, respectively (171, 172). Elimination switches are surface antigens, e.g., CD20 and EGFR-derived extracellular epitopes, that can mediate in vivo depletion by clinical-grade monoclonal antibodies. Although powerful, the other side of in vivo CAR-T cell ablation would be the concomitant loss of immunosurveillance. Moreover, these strategies are reactionary, focused on treating rather than preventing toxicities. Prophylactic strategies to prevent severe toxicity due to inadequate T cell specificity are also being explored (Figure 4). The introduction of microcircuits and transcriptional loops to modulate CAR expression (173), the use of trans second-generation CARs with split costimulatory configuration or multi-CAR platforms, and combining conventional activating constructs with inhibitory CARs that carry CTLA-4- or PD-1-derived inhibitory domains could potentially add further precision and ensure that the effector cells reach full activation only against targets expressing a defined array of tumor-associated antigens (174, 175).
Finally, the assessment of the potential toxicity of novel CAR-T cells can be conducted in clinical trials by infusing T cells with transient expression of the CAR construct by means of RNA-electroporation (176). In the context of solid tumors, local delivery of genetically modified T cells could also mitigate toxicities while increasing efficacy (57, 177, 178).
Allogeneic Cell Infusions to Increase Access
An interesting alternative to current clinical approaches using autologous T cells is to develop universal CAR-T cells or TCR-engineered T cells that could be used “off the shelf” (179). An off-the-shelf product would allow one to start with T cells from healthy donors to create large quantities of universal tumor-specific T cells that could be used in any patient without the need for HLA matching. This strategy would reduce costs, speed drug administration, and make the T cell products accessible to lymphopenic and critically ill cancer patients that often do not have sufficient numbers of healthy T cells for treatment. To this end, strategies to avoid donor T cell rejection by the recipient immune system and to reduce the graft-versus-host disease (GVHD) caused by donor T cells are needed. Zinc-finger nucleases (180), TAL effector nuclease (TALEN) (181–183), or CRISPR/Cas9 (137, 138, 184) gene-editing techniques have been used to disrupt the cell surface expression of TCRs (to prevent GVHD) and β2m or HLA-I molecules (to prevent rejection). An initial clinical trial testing universal CAR-T cells targeting CD19 showed effective separation of graft-versus-leukemia and GVHD effects in B-ALL pediatric patients (182), and other clinical trials to test universal CAR-T and TCR-T cells are underway. A limitation of this approach is that the absence of HLA molecules may trigger a natural killer cell response against universal T cells, and therefore strategies to avoid this eventual rejection are being explored. Finally, preclinical studies suggest that CAR-TCR interactions are a prerequisite for optimal CAR-driven T cell activation (185, 186). A better knowledge of the interaction between the CAR and the endogenous TCR is required to understand whether absence of TCRs might impair the function of universal CAR-T cells.
Lastly, another strategy that is currently being explored is the use of induced pluripotent stem cells to generate universal CAR-T cells. By inserting the CAR in the TCR locus of a single pluripotent cell, one can obtain and bank a TCR-deficient CAR-T cell product with unlimited capacity to self-renew. While this promising strategy is still in its infancy, it could offer several advantages over current strategies and warrants further evaluation.
Combination Therapies
Despite the promising results of ACT, a single optimized T cell therapy may not be sufficient to induce sustained complete responses in the majority of cancers, and therefore combination therapies may be needed to treat cancer. Combining treatments that have different mechanisms of action has the potential to increase antitumor effects and reduce the chance of resistance to ACT. However, while new therapies emerge, the options to combine therapies are many. To ensure the best possible responses, rational combinations will need to be designed upon an understanding of the mechanisms underlying tumor resistance to ACT. Here, we review three emerging therapies with special potential for synergy with ACTs.
A first logical strategy to increase the potency of T cell therapies is to take advantage of checkpoint inhibitors. Adoptively infused tumor-specific T cells are susceptible to immune-checkpoint inhibitory signals and combinatorial therapy with anti-PD1 monoclonal antibodies has shown the ability to relieve immunosuppression and restore T cell function in preclinical studies (136, 187, 188). A phase 1/2 clinical trial evaluating the effect of a PD-1-blocking antibody in patients with B cell lymphoma who failed treatment with CD19-CAR-T cells showed clinical efficacy in a subset of patients, with reexpansion of the CD19-CAR-T cells (NCT02650999) (189). Multiple clinical trials investigating the combination of ACT and checkpoint blockade in both hematologic malignancies and solid tumors are ongoing.
Another strategy to increase the potency of CAR-T therapy is combination with other novel therapies that have proved efficacious in lymphomas and leukemias, such as ibrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase, a key component of B cell signaling and growth. Preclinical data have shown that concurrent ibrutinib treatment enhances CAR-T cell antitumor activity and engraftment in xenograft models of leukemia and lymphoma (190, 191). A clinical trial evaluating this combination is currently recruiting patients at the University of Pennsylvania (NCT02640209).
Oncolytic viruses (OVs) have received considerable attention recently due to their potential to synergize with cancer immunotherapies. OVs can specifically kill cancer cells by viral lysis without causing damage to normal cells. Similar to engineered T cells, these therapeutic agents are considered living therapies, as OVs can replicate in cancer cells and release their progeny, which results in an exponential increase of the virus dose. Another interesting aspect of OVs is that they can be genetically modified to deliver external transgenes locally in the tumor microenvironment, combining direct tumor debulking with gene delivery. Also, viruses provide a danger signal with the potential to overcome tolerance and revert immunosuppression, which makes them an appealing partner for cancer immunotherapies. In different preclinical studies, administration of an armed OV enhanced the immune functions of CAR-T cells. OVs expressing cytokines have been shown to enhance the survival of tumor-bearing mice by increasing CAR-T cell accumulation in the tumor and modulating the tumor microenvironment (192, 193). In a different strategy, expression of a PD-L1-blocking minibody from an OV could block the PD-1:PD-L1 interaction between CAR-T cells and tumor cells, resulting in enhanced tumor control (194). Finally, release of a bispecific-T cell engager by an OV enhanced CAR-T cell homing and activation in the tumor, addressing tumor escape caused by loss of antigen or heterogeneity of antigen expression and enhancing survival (90, 195).
CONCLUSIONS
The field of ACT is exponentially growing, and new strategies and ideas are being developed at a rapid pace. The scientific community is intensively studying strategies to increase T cell efficacy, to optimize T cell production, to develop off-the-shelf T cells, to reduce clinical toxicity, to reduce T cell exhaustion, to avoid tumor-microenvironment immunosuppression, and finally to test commercialization approaches. With the FDA approval of tisagenlecleucel in 2017, the ACT field has capitalized on the multitude of lessons learned as a result of the commitment, insights, and innovations of a multitude of scientists combined with the profound bravery and generosity of patients. In the last few years, ACT has progressively demonstrated dramatic potency in clinical trials of TILs and redirected TCR-T cells and CAR-T cells, leading to complete and durable responses in patients with late-stage and treatment-refractory disease. However, subsets of patients are still not responding, are relapsing, or do not have an adequate ACT strategy for their disease. There is therefore much to be done to ensure that most cancer patients respond in the long term.
Immunotherapy is now a pillar of cancer treatment and will likely be the backbone of future treatment algorithms. ACT has led to excellent results in some cancers, and ongoing research efforts will hopefully broaden its application to most cancers. For cancer with high mutational burden, checkpoint inhibitors and TILs are viable options (196). When the mutational burden is low, redirected high-affinity TCR-T cells might be a strategy, together with CAR-T cells when an appropriate surface marker is available.
Looking at the broader history of cancer therapy, where it is rare for a single agent to achieve long-term remission—with the exception of tyrosine kinase inhibitors for a subset of patients with chronic myeloid leukemia—it is likely that CAR-T therapy and indeed ACT will ultimately be of the most clinical utility in combination with novel therapies. The combination of CAR-T therapy, in particular with immune-based therapies, is eagerly awaited.
Finally issues of scale-up, automation, commercialization, intellectual property (197), and regulatory hurdles unique to cell therapy (198) need to be addressed, as more ACT products are being approved for clinical use and patients treated at large scale.
DISCLOSURE STATEMENT
S.G., M.R., and C.H.J. are inventors on patents related to CAR-T cell therapy, filed by the University of Pennsylvania and licensed to Novartis. C.H.J. and M.R. report receiving commercial research grants from Tmunity. C.H.J. has ownership interest (including patents) in Tmunity, and is a consultant/advisory board member for Tmunity, Immune Design, Carisma, and Celldex Therapeutics. M.R. reports receiving commercial research grants from Tmunity. C.H.J. has sponsored research grants from Novartis.
LITERATURE CITED
- 1.Ribas A, Wolchok JD. 2018. Cancer immunotherapy using checkpoint blockade. Science 359:1350–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June CH, Sadelain M. 2018. Chimeric antigen receptor T cells. N. Engl. J. Med 379:64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, et al. 2011. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res 17:4550–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, et al. 2011. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med 3:95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, et al. 2013. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med 5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, et al. 2014. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med 371:1507–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, et al. 2016. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig 126:2123–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, et al. 2017. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl. J. Med 377:2531–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, et al. 2017. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med 377:2545–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, et al. 2018. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med 24:20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mikkilineni L, Kochenderfer JN. 2017. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood 130:2594–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, et al. 2015. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med 21:914–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, et al. 2017. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356:200–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, et al. 2014. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344:641–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye Q, Song DG, Poussin M, Yamamoto T, Best A, et al. 2014. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res 20:44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, et al. 2014. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig 124:2246–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, et al. 2016. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med 22:433–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, et al. 2015. Cancer immunotherapy: A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348:803–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, et al. 2006. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bos R, van Duikeren S, Morreau H, Franken K, Schumacher TN, et al. 2008. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res 68:8446–55 [DOI] [PubMed] [Google Scholar]
- 21.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, et al. 2009. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, et al. 2011. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther 19:620–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, et al. 2013. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother 36:133–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, et al. 2013. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122:863–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, et al. 2015. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin. Cancer Res 21:1019–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, et al. 2018. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 8:730–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, et al. 2016. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375:819–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, et al. 2016. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375:2255–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gettinger S, Choi J, Hastings K, Truini A, Datar I, et al. 2017. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 7:1420–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, et al. 2018. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378:439–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, et al. 2015. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385:517–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JH, Riviere I, Gonen M, Wang X, Senechal B, et al. 2018. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med 378:449–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, et al. 2015. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med 7:303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, et al. 2017. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J. Clin. Oncol 35:3010–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, et al. 2018. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat. Rev. Clin. Oncol 15:47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kochenderfer JN, Somerville RPT, Lu T, Yang JC, Sherry RM, et al. 2017. Long-duration complete remissions of diffuse large B cell lymphoma after anti-CD19 chimeric antigen receptor T cell therapy. Mol. Ther 25:2245–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, et al. 2018. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med 24:563–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruella M, Maus MV. 2016. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput. Struct. Biotechnol. J 14:357–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frey NV, Porter DL. 2016. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016:567–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rotolo A, Karadimitris A, Ruella M. 2017. Building upon the success of CART19: chimeric antigen receptor T cells for hematologic malignancies. Leuk. Lymphoma 59:2040–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q-S, Wang Y, Lv H-Y, Han Q-W, Fan H, et al. 2015. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther 23:184–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith BD, Roboz GJ, Walter RB, Altman JK, Ferguson A, et al. 2014. First-in man, phase 1 study of CSL362 (anti-IL3Rα / anti-CD123 monoclonal antibody) in patients with CD123+ acute myeloid leukemia (AML) in CR at high risk for early relapse. Blood 124:120 [Google Scholar]
- 43.Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, et al. 2013. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 122:3138–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, et al. 2013. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br. J. Haematol 161:389–401 [DOI] [PubMed] [Google Scholar]
- 45.Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, et al. 2014. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 28:1596–605 [DOI] [PubMed] [Google Scholar]
- 46.Zhou L, Liu X, Wang X, Sun Z, Song XT. 2016. CD123 redirected multiple virus-specific T cells for acute myeloid leukemia. Leuk. Res 41:76–84 [DOI] [PubMed] [Google Scholar]
- 47.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, et al. 2014. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 123:2343–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casucci M, Falcone L, Camisa B, Gentner B, Naldini L, et al. 2013. CD44v6 is required for in vivo tumorigenesis of human AML and MM cells: role of microenvironmental signals and therapeutic implications. Blood 122:605 [Google Scholar]
- 49.Wang C-M, Wu Z-Q, Wang Y, Guo Y-L, Dai H-R, et al. 2017. Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory Hodgkin lymphoma: an open-label phase I trial. Clin. Cancer Res 23:1156–66 [DOI] [PubMed] [Google Scholar]
- 50.Bu DX, Singh R, Choi EE, Ruella M, Nunez-Cruz S, et al. 2018. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget 9:25764–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Agostino M, Boccadoro M, Smith EL. 2017. Novel immunotherapies for multiple myeloma. Curr. Hematol. Malig. Rep 12:344–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, et al. 2018. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol 36:2267–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, et al. 2006. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol 24:e20–22 [DOI] [PubMed] [Google Scholar]
- 54.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. 2010. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18:843–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, et al. 2006. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res 12:6106–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, et al. 2011. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118:6050–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, et al. 2015. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res 21:4062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, et al. 2015. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol 33:1688–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, et al. 2017. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med 9:eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, et al. 2018. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 155:29–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, et al. 2016. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med 375:2561–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, et al. 2017. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother 66:1425–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, et al. 2004. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol 173:7125–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. 2005. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol 26:111–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, et al. 2005. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med 202:907–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg SA, Restifo NP. 2015. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, et al. 2011. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 35:972–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bowers JS, Nelson MH, Majchrzak K, Bailey SR, Rohrer B, et al. 2017. Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight 2:e90772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, et al. 2016. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45:389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15:486–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crespo J, Sun H, Welling TH, Tian Z, Zou W. 2013. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol 25:214–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, et al. 2016. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354:1160–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, et al. 2015. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 21:581–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, et al. 2015. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res 3:356–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, et al. 2016. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 5:e1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, et al. 2017. Tonic 4–1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep 21:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, et al. 2018. Enhancing CAR T cell persistence through ICOS and 4–1BB costimulation. JCI Insight 3:96976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Zelm MC, Reisli I, van der Burg M, Castano D, van Noesel CJ, et al. 2006. An antibody-deficiency syndrome due to mutations in the CD19 gene. N. Engl. J. Med 354:1901–12 [DOI] [PubMed] [Google Scholar]
- 80.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, et al. 2015. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 5:1282–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, et al. 2010. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J. Clin. Investig 120:1265–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Braig F, Brandt A, Goebeler M, Tony HP, Kurze AK, et al. 2017. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood 129:100–4 [DOI] [PubMed] [Google Scholar]
- 83.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, et al. 2008. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol 180:7028–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, et al. 2013. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121:1165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shalabi H, Kraft IL, Wang HW, Yuan CM, Yates B, et al. 2018. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica 103:e215–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, et al. 2016. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun 7:12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, et al. 2016. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127:2406–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, et al. 2014. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther 22:623–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ Jr. 2016. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J. Hematol. Oncol 9:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, et al. 2018. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol. Res 6:605–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, et al. 2017. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun 8:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tarella C, Corradini P, Astolfi M, Bondesan P, Caracciolo D, et al. 1999. Negative immunomagnetic ex vivo purging combined with high-dose chemotherapy with peripheral blood progenitor cell autograft in follicular lymphoma patients: evidence for long-term clinical and molecular remissions. Leukemia 13:1456–62 [DOI] [PubMed] [Google Scholar]
- 93.Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, et al. 2018. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med 24:1499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Glickman MS, Sawyers CL. 2012. Converting cancer therapies into cures: lessons from infectious diseases. Cell 148:1089–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.June CH, Warshauer JT, Bluestone JA. 2017. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat. Med 23:540–47 [DOI] [PubMed] [Google Scholar]
- 96.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, et al. 2012. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119:2709–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, et al. 2016. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov 6:664–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Davila ML, Riviere I, Wang X, Bartido S, Park J, et al. 2014. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med 6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marshall A, ed. 2017. CAR-T death strikes Kite. Nat. Biotechnol 35:492. [DOI] [PubMed] [Google Scholar]
- 100.Gust J, Hay KA, Hanafi LA, Li D, Myerson D, et al. 2017. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov 7:1404–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, et al. 2013. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med 368:1509–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maude SL, Barrett D, Teachey DT, Grupp SA. 2014. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J 20:119–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ruella M, June CH. 2018. Predicting dangerous rides in CAR T cells: bridging the gap between mice and humans. Mol. Ther 26:1401–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, et al. 2018. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med 24:739–48 [DOI] [PubMed] [Google Scholar]
- 105.Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. 2018. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med 24:731–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ruella M, June CH. 2016. Chimeric antigen receptor T cells for B cell neoplasms: choose the right CAR for you. Curr. Hematol. Malig. Rep 11:368–84 [DOI] [PubMed] [Google Scholar]
- 107.Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, et al. 2011. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J. Immunol 186:685–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, et al. 2013. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med 5:197ra03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raman MC, Rizkallah PJ, Simmons R, Donnellan Z, Dukes J, et al. 2016. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci. Rep 6:18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, et al. 2013. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol 25:268–76 [DOI] [PubMed] [Google Scholar]
- 111.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. 2011. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol 29:235–71 [DOI] [PubMed] [Google Scholar]
- 112.Rabinovich GA, Gabrilovich D, Sotomayor EM. 2007. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol 25:267–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Arina A, Bronte V. 2015. Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Curr. Opin. Immunol 33:120–25 [DOI] [PubMed] [Google Scholar]
- 114.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, et al. 2014. Multifactorial T-cell hypo-function that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res 20:4262–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morrison C 2015. ‘Financial toxicity’ looms as cancer combinations proliferate. Nat. Biotechnol 33:783–84 [DOI] [PubMed] [Google Scholar]
- 116.Chen Q, Jain N, Ayer T, Wierda WG, Flowers CR, et al. 2017. Economic burden of chronic lymphocytic leukemia in the era of oral targeted therapies in the United States. J. Clin. Oncol 35:166–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carr DR, Bradshaw SE. 2016. Gene therapies: the challenge of super-high-cost treatments and how to pay for them. Regen. Med 11:381–93 [DOI] [PubMed] [Google Scholar]
- 118.Hettle R, Corbett M, Hinde S, Hodgson R, Jones-Diette J, et al. 2017. The assessment and appraisal of regenerative medicines and cell therapy products: an exploration of methods for review, economic evaluation and appraisal. Health Technol. Assess 21:1–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, et al. 2012. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med 4:132ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, et al. 2016. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44:380–90 [DOI] [PubMed] [Google Scholar]
- 121.Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ Jr. 2012. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 119:696–706 [DOI] [PubMed] [Google Scholar]
- 122.Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, et al. 2014. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 124:1070–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, et al. 2010. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin. Cancer Res 16:6122–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Donia M, Junker N, Ellebaek E, Andersen MH, Straten PT, Svane IM. 2012. Characterization and comparison of ‘standard’ and ‘young’ tumour-infiltrating lymphocytes for adoptive cell therapy at a Danish translational research institution. Scand. J. Immunol 75:157–67 [DOI] [PubMed] [Google Scholar]
- 125.Ghassemi S, Nunez-Cruz S, O’Connor RS, Fraietta JA, Patel PR, et al. 2018. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res 6:1100–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, et al. 2013. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 121:573–84 [DOI] [PubMed] [Google Scholar]
- 127.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, et al. 2014. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123:3750–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Santegoets SJ, Turksma AW, Suhoski MM, Stam AG, Albelda SM, et al. 2013. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J. Transl. Med 11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, et al. 2016. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig 126:318–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, et al. 2011. A human memory T cell subset with stem cell-like properties. Nat. Med 17:1290–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, et al. 2015. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 75:296–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van der Waart AB, van de Weem NM, Maas F, Kramer CS, Kester MG, et al. 2014. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood 124:3490–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, et al. 2010. The inducible costimulator (ICOS) is critical for the development of human TH17 cells. Sci. Transl. Med 2:55ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, et al. 2018. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558:307–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, et al. 2018. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell 173:1439–53.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, et al. 2016. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig 126:3130–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. 2017. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res 23:2255–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ren J, Zhang X, Liu X, Fang C, Jiang S, et al. 2017. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8:17002–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. 2015. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med 212:1125–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, et al. 2017. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 552:121–25. Erratum. 2017. Nature 553:238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, et al. 2017. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543:113–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, et al. 2013. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med 210:1125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, et al. 2014. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res 2:154–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gulati P, Ruhl J, Kannan A, Pircher M, Schuberth P, et al. 2018. Aberrant Lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin. Cancer Res 24:3981–93 [DOI] [PubMed] [Google Scholar]
- 145.Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, et al. 2017. Overcoming the immunosuppressive tumor microenvironment of Hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov 7:1154–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, et al. 2015. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med 21:524–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, et al. 2013. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther 21:2087–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. 2013. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum. Gene Ther 24:717–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, et al. 2016. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig 126:3036–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, et al. 2016. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig 126:3814–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Qin H, Haso W, Nguyen SM, Fry TJ. 2015. Preclinical development of bispecific chimeric antigen receptor targeting both CD19 and CD22. Blood 126:4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. 2016. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res 4:498–508. Addendum. 2016. Cancer Immunol. Res. 4:639–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, et al. 2011. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther 19:751–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wagner HJ, Bollard CM, Vigouroux S, Huls MH, Anderson R, et al. 2004. A strategy for treatment of Epstein-Barr virus-positive Hodgkin’s disease by targeting interleukin 12 to the tumor environment using tumor antigen-specific T cells. Cancer Gene Ther 11:81–91 [DOI] [PubMed] [Google Scholar]
- 155.Chmielewski M, Kopecky C, Hombach AA, Abken H. 2011. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 71:5697–706 [DOI] [PubMed] [Google Scholar]
- 156.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, et al. 2012. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119:4133–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cohen J 1995. IL-12 deaths: explanation and a puzzle. Science 270:908. [DOI] [PubMed] [Google Scholar]
- 158.Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. 2015. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med 13:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, et al. 2015. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res 21:2278–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, et al. 2016. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. PNAS 113:E7788–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hu B, Ren J, Luo Y, Keith B, Young RM, et al. 2017. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep 20:3025–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chmielewski M, Abken H. 2017. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep 21:3205–19 [DOI] [PubMed] [Google Scholar]
- 163.Kunert A, Chmielewski M, Wijers R, Berrevoets C, Abken H, Debets R. 2017. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 7:e1378842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, et al. 2015. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol. Ther 23:769–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, et al. 2015. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell 28:415–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Posey AD Jr., Schwab RD, Boesteanu AC, Steentoft C, Mandel U, et al. 2016. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 44:1444–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Johnson LA, June CH. 2017. Driving gene-engineered T cell immunotherapy of cancer. Cell Res 27:38–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. 2004. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol 173:7647–53 [DOI] [PubMed] [Google Scholar]
- 169.Liu X, Jiang S, Fang C, Yang S, Olalere D, et al. 2015. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 75:3596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, et al. 2015. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 75:3505–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lupo-Stanghellini MT, Provasi E, Bondanza A, Ciceri F, Bordignon C, Bonini C. 2010. Clinical impact of suicide gene therapy in allogeneic hematopoietic stem cell transplantation. Hum. Gene Ther 21:241–50 [DOI] [PubMed] [Google Scholar]
- 172.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, et al. 2011. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med 365:1673–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lim WA, June CH. 2017. The principles of engineering immune cells to treat cancer. Cell 168:724–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sadelain M, Rivière I, Riddell S. 2017. Therapeutic T cell engineering. Nature 545:423–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fedorov VD, Themeli M, Sadelain M. 2013. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med 5:215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, et al. 2015. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 29:1637–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, et al. 2014. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med 6:261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, et al. 2017. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res 5:1152–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ruella M, Kenderian SS. 2017. Next-generation chimeric antigen receptor T-cell therapy: going off the shelf. BioDrugs 31:473–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, et al. 2012. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119:5697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Berdien B, Mock U, Atanackovic D, Fehse B. 2014. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther 21:539–48 [DOI] [PubMed] [Google Scholar]
- 182.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, et al. 2017. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med 9:eaaj2013 Erratum. 2017. Sci. Transl. Med. 9:aam9292 [DOI] [PubMed] [Google Scholar]
- 183.Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, et al. 2015. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res 75:3853–64 [DOI] [PubMed] [Google Scholar]
- 184.Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, et al. 2014. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15:643–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. 2010. The optimal antigen response of chimeric antigen receptors harboring the CD3ζ transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol 184:6938–49 [DOI] [PubMed] [Google Scholar]
- 186.Bridgeman JS, Ladell K, Sheard VE, Miners K, Hawkins RE, et al. 2014. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: implications for optimization of receptor structure for adoptive cell therapy. Clin. Exp. Immunol 175:258–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.John LB, Kershaw MH, Darcy PK. 2013. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2:e26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, et al. 2016. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin. Cancer Res 22:436–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, et al. 2017. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129:1039–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, et al. 2016. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 127:1117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ruella M, Kenderian SS, Shestova O, Fraietta JA, Qayyum S, et al. 2016. The addition of the BTK inhibitor ibrutinib to anti-CD19 chimeric antigen receptor T cells (CART19) improves responses against mantle cell lymphoma. Clin. Cancer Res 22:2684–96 [DOI] [PubMed] [Google Scholar]
- 192.Watanabe K, Luo Y, Da T, Guedan S, Ruella M, et al. 2018. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 3:99573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, et al. 2014. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res 74:5195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, et al. 2017. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res 77:2040–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, et al. 2017. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res 77:2052–63 [DOI] [PubMed] [Google Scholar]
- 196.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, et al. 2016. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351:1463–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Levine BL, June CH. 2013. Perspective: assembly line immunotherapy. Nature 498:S17. [DOI] [PubMed] [Google Scholar]
- 198.McGarrity GJ, Hoyah G, Winemiller A, Andre K, Stein D, et al. 2013. Patient monitoring and follow-up in lentiviral clinical trials. J. Gene Med 15:78–82 [DOI] [PubMed] [Google Scholar]