

Cancer cells with high mTORC1 activity rely on the RUVBL1/2-TTT pathway for survival, thereby providing a therapeutic window.
Abstract
Despite considerable efforts, mTOR inhibitors have produced limited success in the clinic. To define the vulnerabilities of mTORC1-addicted cancer cells and to find previously unknown therapeutic targets, we investigated the mechanism of piperlongumine, a small molecule identified in a chemical library screen to specifically target cancer cells with a hyperactive mTORC1 phenotype. Sensitivity to piperlongumine was dependent on its ability to suppress RUVBL1/2-TTT, a complex involved in chromatin remodeling and DNA repair. Cancer cells with high mTORC1 activity are subjected to higher levels of DNA damage stress via c-Myc and displayed an increased dependency on RUVBL1/2 for survival and counteracting genotoxic stress. Examination of clinical cancer tissues also demonstrated that high mTORC1 activity was accompanied by high RUVBL2 expression. Our findings reveal a previously unknown role for RUVBL1/2 in cell survival, where it acts as a functional chaperone to mitigate stress levels induced in the mTORC1-Myc-DNA damage axis.
INTRODUCTION
The mechanistic target of rapamycin (mTOR) is a serine/threonine kinase that belongs to the phosphatidylinositol 3-kinase (PI3K)–related protein kinase (PIKK) family. mTOR forms two distinct multiprotein complexes referred to as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The mTORC1 signaling network functions as a sensor of various environmental cues and responds to growth factors, nutrients, energy status, cellular stress, and oxygen levels. mTORC1 integrates this cellular and environmental information and coordinates appropriate cellular responses, which include regulation of protein translation, metabolism, and proliferation (1). Meanwhile, mTORC2 is a major upstream regulator of Akt, protein kinase C (PKC), and serum/glucocorticoid-regulated kinase (SGK) kinases, which contribute to proliferation, migration, metabolism, and survival via separate and overlapping effector pathways (2). Because of its central role in these processes, aberrant mTOR signaling is frequently implicated in cancer pathogenesis, with oncogenes including PI3K and Ras demonstrated to up-regulate mTOR activation in proliferating cells (3, 4). The frequent loss of tumor suppressors including p53, PTEN, TSC1/2, and LKB1 in malignant cells further promotes mTOR pathway activation (3, 5, 6). The understanding of these pivotal roles of mTOR in cancer has provided rationale for the development of small molecules that can selectively suppress cancer cells with high mTOR activity (7–9).
The mTOR inhibitor, rapamycin, and its analogs (rapalogs) have demonstrated anticancer efficacy in preclinical models. However, rapalogs have generated only modest clinical outcomes in various tumor types (8, 10–13). Some clinical responses to rapalogs have been observed in renal cell carcinoma and mantle-cell lymphoma studies; however, these are typically short-lasting partial responses (14–16). Also, despite the initial positive response in some tumors that have high mTOR activity, the tumors regrow after discontinuation of therapy, as rapalogs cause only cytostatic responses (8, 13, 17, 18). An unmet need therefore exists for the development of combination therapies or new approaches that can elicit cytotoxic responses in mTOR-hyperactive cancers (8, 9, 13).
In the current study, we screened for small molecules targeting cells with a hyperactive mTORC1 phenotype and identified piperlongumine (PL). Using PL as a chemical probe, we sought to explore the underlying mechanism of PL to identify potential therapeutic targets and to understand the vulnerabilities of mTORC1-addicted cancer cells.
RESULTS
Identification of compounds with synthetic lethality against cancer cells with mTORC1 hyperactivation
To identify candidate small molecules with strong potency toward a hyperactivated mTORC1 phenotype, we conducted a chemical library screen of U.S. Food and Drug Administration (FDA)–approved drugs and bioactive small molecular compounds against cells with high mTORC1 or low mTORC1 activities. On the basis of the differential levels of mTORC1 activity, the SW480 cancer cell line was chosen for the cell with low mTORC1 activity from a set of human cancer cells (fig. S1A). Since the Tuberous sclerosis complex (TSC) complex functions as the key negative regulator of mTORC1 signaling (19, 20), we generated stable Tsc2 knockdown SW480 cells (TSC2 KD) in which mTORC1 is hyperactive and compared the effect with shControl SW480 cells (TSC2 WT) (fig. S1, B and C). We designed and focused on screening compounds displaying selective cytotoxicity in mTORC1-high cells by measuring the capability to induce cell death rather than to reduce cell proliferation, for reduction in cell number can also arise from cytostatic compounds (Fig. 1A). Through comparing the different levels of cytotoxicity induced in KD (mTORC1-high) and WT (mTORC1-low) cells, we were able to identify compounds that specifically caused cell death in mTORC1-high cells (Fig. 1B and table S1). Among the top hits, PL particularly exhibited a strong and selective cytotoxic phenotype toward SW480 KD (high mTORC1 activity) cells (Fig. 1B). Manual validation using trypan blue exclusion assay further confirmed the finding. When TSC2 was knocked down using short hairpin RNA to elevate mTORC1 activity in SW480 cells, a marked increase in PL-induced cell death was observed (Fig. 1C and fig. S1D), and this increase in cell death matched well with the increase in mTORC1 activity (Fig. 1D). Assessment with a broader panel of various human normal and cancer cells showed that the cytotoxic effect of PL was selective against cancer cells with high levels of mTORC1 activity (Fig. 1E). Although PL has been reported to be effective against a broad range of cancer cell types (21, 22), our data illustrate that at least some cancer cells harboring low mTORC1 activity may exhibit low susceptibility toward PL due to minimal induction of cell death (Fig. 1E). We next asked whether reduction of mTORC1 activity could decrease PL sensitivity in mTORC1-hyperactive T24 cells. Pretreatment with rapamycin, an mTOR inhibitor, suppressed PL-mediated cell death in T24 cells (Fig. 1F and fig. S1E). In addition, overexpressing TSC1/2 in mTORC1-hyperactive T24 cells reduced PL-mediated cell death (fig. S1, F and G). Collectively, these results suggest that the sensitivity of PL highly depends on mTORC1 activity (Fig. 1G).
Fig. 1. High-throughput screening for compounds with synthetic lethality against mTORC1-high cancer cells.
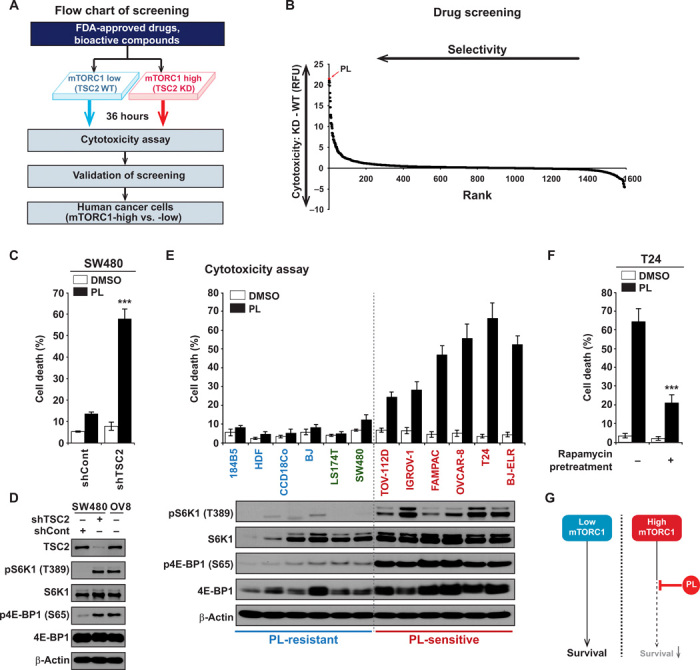
(A) Scheme of screening. Chemical library containing 1576 FDA-approved drugs and bioactive compounds were treated to SW480 shControl (WT) and SW480 shTSC2 (KD) cells. shControl or shTSC2 viral vectors were infected to SW480 cells and cells were selected with puromycin. Capability to induce cell death was measured to rule out cytostatic compounds. Hits were validated and further confirmed in the cells. (B) Cytotoxicity in WT and KD cells were measured after compounds were treated for 36 hours in 96-well plates in duplicate. Fluorescence-based cytotoxicity assay (CellTox Green) was used and to determine the selectivity, and the level of cell death induced in WT cells was subtracted from the level of cell death induced in KD cells. Higher values indicate higher selective cytotoxicity against mTORC1-high cells. RFU, relative fluorescence unit. (C) Increase in mTORC1 activity enhances PL-mediated cell death. SW480 shControl and SW480 shTSC2 cells were treated with PL (10 μM) for 48 hours, and cell viability was measured. (D) Analysis of mTORC1 signaling in SW480 shControl and SW480 shTSC2 cells. OVCAR-8 (OV8) cells were used as a control for comparison. (E) Correlation between PL-induced cell death and mTORC1 levels. Cell death was measured in various normal and cancer cells after 48 hours of PL (10 μM) treatment. Normal or nonmalignant immortalized cells, 184B5, human dermal fibroblast (HDF), CCD-18Co, and BJ-hTERT (BJ); low mTORC1 cancer cells, LS174T and SW480; high mTORC1 cancer cells, TOV-112D, IGROV-1, FAMPAC, OVCAR-8, T24, and BJ-ELR. Immunoblot results show increased mTORC1 activity in PL-sensitive cells. (F) Inhibition of mTORC1 activity reduces PL-mediated cell death. Dimethyl sulfoxide (DMSO) or 100 nM rapamycin was pretreated for 16 hours, and then PL 10 μM was treated to T24 cells. (G) PL selectively kills cancer cells with high mTORC1 activity. All data are presented as means ± SD. Significant differences were calculated by one-way analysis of variance (ANOVA) compared with shControl group or rapamycin-untreated group (***P < 0.001).
PL preferentially suppresses the growth of mTORC1-high tumors in vivo
To determine whether the relationship between PL efficacy and the level of mTORC1 activity is also observed in vivo, we performed tumorigenesis assays in nude mice. PL markedly suppressed tumors derived from mTORC1-high OVCAR-8 cells (Fig. 2A), while only marginally inhibiting mTORC1-low SW480 derived tumors (Fig. 2B). The antitumor effect of PL was significantly enhanced when mTORC1 activity was elevated via TSC2 knockdown in SW480 xenografts (Fig. 2B). In addition, since cancer patient-derived xenografts (PDXs) more closely recapitulates characteristics of actual human cancers (23, 24), we sought to test the selective inhibition of PL against mTORC1-high tumors using different types of PDX models. A panel of cancer cells isolated form patients was sorted on the basis of their mTORC1 activity (fig. S2). PL exerted significant inhibitory effects against the growth of mTORC1-high tumors (HJG152 and HJG172), whereas there was no noticeable effect toward mTORC1-low tumors (HJG78) in colon cancer PDX models (Fig. 2C and fig. S2). Furthermore, we examined the selectivity of PL in lung cancer and esophageal cancer PDXs to demonstrate that our findings are tissue agnostic (25). PL inhibited the growth of mTORC1-high tumors (lung: LG70; esophagus: LEG139) but not that of mTORC1-low tumors (lung: LG43 and LG55; esophagus: LEG8 and LEG110) (Fig. 2, D and E, and fig. S2). Therefore, these results indicate that PL selectively targets cancer cells with high mTORC1 activity in vivo.
Fig. 2. PL inhibits mTORC1-high tumor growth in vivo.

(A) The effect of PL on OVCAR-8 xenograft. PL (7 mg/kg) was administered intraperitoneally everyday to nude mice injected with OVCAR-8 cells. Eight mice per group were used. (B) The effect of PL on SW480 xenograft. PL (7 mg/kg) was administered intraperitoneally everyday to nude mice injected with SW480 shControl and SW480 shTSC2 cells. Eight mice per group were used. (C) The effect of PL on colon cancer PDX. PL (7 mg/kg) was administered intraperitoneally everyday to female SCID mice implanted with HJG152, HJG172, or HJG78 tumors. Seven mice per group for HJG152 and HJG78 and six mice per group for HJG172 were used. (D) The effect of PL on lung cancer PDX. PL (7 mg/kg) was administered intraperitoneally everyday to female severe combined immunodeficient (SCID) mice implanted with LG70, LG43, or LG55 tumors. Six mice per group for LG70 and seven mice per group for LG43 and LG55 were used. (E) The effect of PL on esophageal cancer PDX. PL (7 mg/kg) was administered intraperitoneally everyday to female SCID mice implanted with LEG139, LEG8, or LEG110 tumors. Seven mice per group for LEG139 and LEG8 and five mice per group for LEG110 were used. The volume and weight were measured as described in Materials and Methods. All data are presented as means ± SD (one-way ANOVA; *P < 0.05, **P < 0.01, significant difference compared to the vehicle group.)
PL targets RUVBL1/2 and prevents formation of the functional RUVBL1/2-TTT complex
Although some previous reports have attributed the anticancer effect of PL to reactive oxygen species (ROS), none of the ROS inducers in our screening was selected as a hit (table S1), and recent studies have demonstrated the possibility of ROS-independent mechanisms of PL-mediated cell death (26, 27). In an effort to understand the molecular mechanism responsible for the selective effects of PL in mTORC1-hyperactive cancer cells, we constructed biotin-labeled PL to identify target proteins by analyzing proteins pulled-down from the cell lysate (Fig. 3A and fig. S3). Among the binding candidates, RuvB Like AAA ATPase 1 (RUVBL1) and RuvB Like AAA ATPase 2 (RUVBL2) attracted our attention, because of their recent reports in connection with the mTOR pathway (28, 29). Pull-down assays using biotin-labeled PL confirmed that PL binds to RUVBL1 and RUVBL2 and competes with nonlabeled PL in solution (Fig. 3B), and overexpression of RUVBL1/2 led to a reduction in PL-induced cell death (fig. S5A). The TTT (Tel2, TTI1, and TTI2) complex forms a multiprotein complex in association with RUVBL1/2 and regulates the stability and the assembly of the PIKK-containing complexes (30, 31). In addition, previous reports have shown that the chaperonin function of the RUVBL1/2 is essential for the formation of RUVBL1/2-TTT complex (29). Therefore, we then examined the possibility that PL might interfere with the stability of the RUVBL1/2-TTT complex. As shown in Fig. 3C, the addition of PL to cells followed by immunoprecipitation with Flag-Tel2 demonstrated a decreased interaction of Tel2 with TTI1 and RUVBL1, the components of the RUVBL1/2-TTT complex, indicating that PL caused disassembly of the RUVBL1/2-TTT complex. Concomitant with dissociation of the complex, we also observed that PL treatment reduced protein expression of all the TTT components in a time-dependent manner with no apparent sign of decrease in RUVBL1/2 proteins (Fig. 3D). The activity and stability of PIKK members are regulated by the RUVBL1/2-TTT complex, and treatment with PL suppressed ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related protein (ATR), and mTOR activities, suggesting a subsequent blockage of the PIKK pathways as a consequence of RUVBL1/2-TTT inhibition (Fig. 3, D and E). ATM and ATR are the master transducer of DNA damage response including checkpoint signaling and downstream repair (32, 33). We found that PL can inhibit phosphorylations of checkpoint proteins Chk1 and Chk2 induced by doxorubicin (DOX) or camptothecin (CPT; Fig. 3E). ATM, ATR, and DNA-dependent protein kinase (DNAPK) are redundantly responsible for the phosphorylation of γH2A histone family member X (γH2AX) in response to DNA damage (34–36). We found that PL causes time-dependent increase of γH2AX in the cells without DNA damage reagent treatment (fig. S4A), but partially decreases γH2AX at the early time point after DOX or CPT treatment (Fig. 3E and fig. S4C), and mildly increases γH2AX at the late time point after DNA damage treatment (fig. S4, E to G). We think that the complicated effect of PL on γH2AX may be caused by the combinational effect of PL-induced ROS and the DNA signaling defect caused by decrease of ATM and ATR. Without exogenous damage reagents, PL-induced ROS could generate more DNA damages and increase γH2AX. At the early time point after DNA damage, the initial phosphorylation of γH2AX could be delayed due to PL-induced decrease of ATM and ATR. However, at the late time point, increased ROS and residual or redundant kinase activity finally increase the phosphorylation of γH2AX.
Fig. 3. PL targets the RUVBL1/2-TTT pathway.

(A) Target identification using mass spectrometry. Cell lysates were mixed with or without PL, and biotin-labeled PL was used to detect PL-binding proteins. Avidin beads were used to pull down PL-biotin adduct and was subjected to mass spectrometry analysis. (B) PL-biotin binds to RUVBL1 and RUVBL2 and free PL competes with this binding. Glyceraldehyde-3-phosphate dehydrogenase was used as a negative control. (C) The effect of PL on RUVBL1/2-TTT complex formation. T24 cells and T24 cells overexpressing Flag-Tel2 were used for immunoprecipitation with anti-Flag antibody and affinity agarose beads. PL (10 μM) was treated for 2 and 4 hours. (D) The effect of PL on RUVBL1/2-TTT proteins. T24 and OVCAR-8 cells were treated with PL (10 μM) for 1, 3, 6, and 12 hours and examined for RUVBL1/2-TTT proteins and its downstream PIKK pathway by immunoblotting. (E) PL suppresses doxorubicin (DOX)– and camptothecin (CPT)–induced DNA damage response signaling in T24 cells. PL was pretreated for 40 min before the addition of DOX (2 μM) and CPT (1 μM) for 1 hour. pChk1, pChk2, and pH2AX were examined by immunoblotting. (F) PL inhibits ionizing radiation (IR)–induced 53BP1, BRCA1, and Rad51 foci formation. PL was pretreated for 1 hour before IR at 4 gray for 3 hours.
We further investigate whether PL affects the downstream repair process. PL inhibits DOX- or CPT-induced foci of 53BP1 and Rad51 (fig. S4, B to E). It suggests that PL may block the downstream repair signaling by abolishing the accumulation of DNA damage response factors. Consistently, PL also blocks ionizing radiation (IR)–induced foci of 53BP1, breast cancer type 1 susceptibility protein (BRCA1), and Rad51 (Fig. 3F and fig. S4, F to H). The levels of 53BP1, BRCA1, and Rad51 foci in PL-treated cells were lower than those in dimethyl sulfoxide (DMSO)–treated control cells, suggesting that PL-treated cells failed to respond to intrinsic DNA damage (Fig. 3F and fig. S4, F to H). Collectively, these results demonstrate that PL treatment reduced the functions of the DNA damage response–initiating ATM/ATR kinases, leading to compromised downstream events in DNA damage signaling and DNA repair. In addition, as the adenosine triphosphatase (ATPase) activities of RUVBL1/2 are required for the proper functioning and assembly of the RUVBL1/2-TTT complex (29), we tested whether PL had an effect on RUVBL1/2 ATPase activity. The addition of PL to the RUVBL1/2 complexes in vitro inhibited their ATPase activity (fig. S5B). Notably, despite generation of cellular ROS by various ROS inducers at the levels similar to or greater than that caused by PL (fig. S6A), none of these ROS inducers recapitulated the inhibitory effects of PL on TTT proteins, suggesting little involvement of ROS in PL-induced inhibition of the RUVBL1/2-TTT pathway (fig. S6B). In addition, since N-acetylcysteine causes Michael addition of a thiol (SH) to a double bond in the six-membered ring of PL leading to direct binding and sequestration of PL (fig. S6, C and D) (26, 37), we have used other well-known antioxidants, catalase and Trolox, to determine the involvement of ROS in PL-induced cell death. Pretreatment with catalase and Trolox showed some protective effects against PL-induced cell death in T24 and OVCAR-8 cells, but a large portion of cells were not protected, further demonstrating ROS-independent mechanisms in PL-mediated cytotoxicity (fig. S6, E and F).
RUVBL1/2 knockdown selectively kills cancer cells with high mTORC1 activity
Since PL targets the RUVBL1/2-TTT complex and exerts selective cytotoxicity toward mTORC1-high cancer cells, we further investigated the relationship between RUVBL1/2 and mTOR signaling with regard to cell survival. While knockdown of RUVBL1 or RUVBL2 showed minimal cell death in cells with low mTORC1 activity [CCD-18co, human dermal fibroblast (HDF), and SW480], knockdown of either RUVBL1 or RUVBL2, in IGROV-1, OVCAR-8, and T24 cancer cells with high levels of mTORC1 activity, significantly induced cell death (Fig. 4A and fig. S7, A to C). Increasing mTORC1 activity in SW480 cells by knocking down TSC2 made these cells highly sensitive toward RUVBL1/2 knockdown (7, 11, 14, and 10% increase of cell death in shControl cells compare to 24, 33, 29, and 32% increase of cell death in SW480 shTSC2 cells) (Fig. 4B), further supporting the conclusion that inhibition of RUVBL1/2 selectively targets cancer cells with hyperactivated mTORC1 activity. As knocking down RUVBL1/2 promotes selective cell death in cancer cells with high mTORC1, we examined whether artificially lowering mTORC1 level in cancer cells naturally harboring hyperactive mTORC1 would protect from RUVBL1/2 inhibition–induced cytotoxicity. Notably, lowering mTOR activity with pretreatment of the mTORC1-high T24 or OVCAR-8 cells with mTOR inhibitors, Torin1 or rapamycin, prevented RUVBL1 and RUVBL2 knockdown–induced cell death (Fig. 4C). Knockdown of either RUVBL1 or RUVBL2 led to the down-regulation of Tel2, TTI1, and TTI2 and its dependent PIKK members including, DNA-PKcs, ATM, ATR, and mTOR, demonstrating the interdependency between RUVBL1 and RUVBL2 and their similar impact on cell signaling (Fig. 4D). In addition, as the TTT complex proteins were down-regulated after RUVBL1/2 knockdown, we examined the effect of silencing TTT proteins. Similar to RUVBL1/2 inhibition, knockdown of Tel2 also induced cell death in OVCAR-8 and T24 cells (fig. S7, D and E). We next analyzed RUVBL2 expression in human cancer biopsies and found that RUVBL2 was overexpressed in various individual tumors in comparison to corresponding normal tissues, which was in line with previous findings (Fig. 4E and figs. S8 and S9) (38–40). There was a significant correlation (Pearson correlation coefficient, 0.70) between levels of mTORC1 activity (assessed by phospho-S6 expression) and RUVBL2 expression in human cancer tissues (Fig. 4F). This finding further supports the hypothesis that when cancer cells acquire high mTORC1 activity, they require the support of RUVBL1/2 to survive and manifest their oncogenic phenotype (Fig. 4G). Analysis on the expression levels for the components of the RUVBL1/2-TTT complex in cell lines and primary patient-derived tumors was also performed. Results demonstrate that the expression pattern of RUVBL1/2 and TTT proteins was generally elevated in cells/tissues with higher mTORC1 activity (fig. S8, D and E).
Fig. 4. Cancer cells with high mTORC1 activity have increased dependency on RUVBL1/2 for survival.

(A) The effect of RUVBL1/2 depletion on cell viability. Cell death was measured 4 days (T24) or 6 days (CCD-18co, HDF, SW480, IGROV-1, and OVCAR-8) after siRUVBL1 (si#1, si#2) or siRUVBL2 (si#1, si#2) transfection. (B) Increase in mTORC1 activity enhances sensitivity to RUVBL1/2 silencing–induced cell death. Cell death was measured 6 days after siRUVBL2 transfection to SW480 shControl and SW480 shTSC2 cells. (C) Reduction in mTOR activity prevents cell death induced by RUVBL1 or RUVBL2 knockdown. To reduce mTOR activity in T24 cells, rapamycin or Torin1 (100 nM) was pretreated to T24 cells 16 hours before siRUVBL1/2 transfection. Cell death was measured 4 days (T24) or 6 days (OVCAR8) after transfection. (D) The effect of RUVBL1/2 knockdown on TTT and PIKK family proteins. Protein levels of RUVBL1/2, TTT, and PIKK family members were examined 48 hours after knockdown of RUVBL1-#2 and RUVBL2-#1. (E) RUVBL2 is overexpressed in human cancer tissues. Various human cancer and corresponding normal tissues were stained with RUVBL2 (green) and phospho-S6 (red), and RUVBL2 expression levels were quantified using ImageJ. Representative picture of RUVBL2 (green) and phospho-S6 (red) staining are shown from esophagus cancer and corresponding normal tissue. (**P < 0.01, significant difference of expression between normal and cancer tissues). (F) mTORC1 activity (p-S6) and RUVBL2 expression show positive correlation in human cancer tissues. (Pearson correlation coefficient = 0.70, P < 0.001). (G) Cancer cells with high mTORC1 activity require RUVBL1/2 for survival. All data are presented as means ± SD. Significant differences were calculated by one-way ANOVA compared with DMSO-treated group for each siRNA treatment (***P < 0.001).
High mTORC1 intensifies DNA damage stress via c-Myc, increasing dependency on RUVBL1/2 for survival
Activation of the mTORC1 pathway leads to induction of transcription, translation, ribosome biogenesis, and anabolic metabolism, subsequently causing an increase in cell mass and size through macromolecule biosynthesis (2, 3). These high-energy–consuming processes can generate high cellular stress conditions and render cells to vastly depend on various stress support pathways (41). We hypothesized that RUVBL1/2 plays a crucial role in maintaining cell integrity by mitigating stress levels arising from pathways downstream of mTORC1. As RUVBL1/2 has been reported to regulate chromatin remodeling, transcription, and the DNA damage response (38, 39), we sought to examine whether its inhibition influences DNA damage status of the cells. RUVBL2 knockdown in CCD-18co and HDF normal cells did not cause any significant increase in DNA damage (Fig. 5A). Notably, RUVBL2 silencing in T24 and SW480 shTSC2 cells (with high mTORC1 activity) induced a marked increase in DNA damage (Fig. 5A). SW480 shControl cells (with low mTORC1 levels) had higher levels of basal DNA damage compared to normal cells (CCD-18co and HDF); however, its increase in DNA damage compared to SW480 shTSC2 cells was small, suggesting that the increase in mTORC1 activity could render cells more sensitive to DNA damage stress control pathways (Fig. 5A). In addition, while etoposide was capable of inducing DNA damage in HDF cells, treatment of PL showed minimal induction of DNA damage in HDF and SW480 shControl cells (Fig. 5B and fig. S10). In contrast, SW480 shTSC2 and T24 cells were subjected to significant increases in DNA damage after PL treatment (Fig. 5B). To further understand the connection between DNA damage stress and mTORC1, we evaluated the relationship of mTORC1 activity to the level of DNA damage. We observed that elevating mTORC1 activity by TSC2 knockdown in SW480 cells increased levels of DNA damage represented by phospho-H2AX (S139) (Fig. 5C). Conversely, reducing mTORC1 levels via treating a direct mTOR inhibitor, Torin1, led to a reduction in DNA damage (Fig. 5D). These results suggest a common link between heightened mTORC1 activity, increases in DNA damage, and an increased dependency on stress control pathways that mitigate DNA damage stress.
Fig. 5. Hyperactive mTORC1 causes overreliance on RUVBL1/2 for maintenance of DNA damage stress.
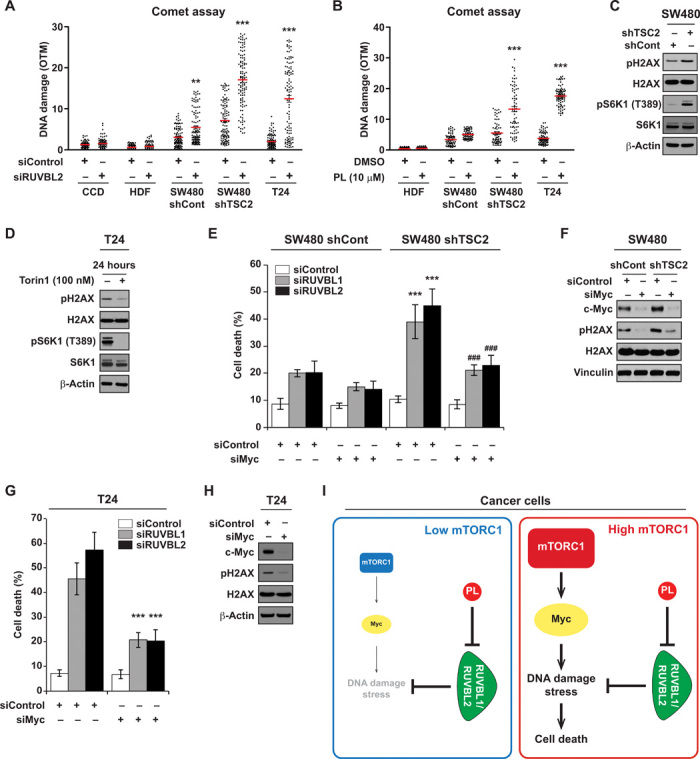
(A) Differential DNA damage induction by RUVBL2 knockdown (siRUVBL2-#1). DNA damage was measured using comet assay. DNA damage was quantified on the basis of the olive tail movement (OTM) value, automatically calculated using a computer program, CometScore. OTM is computed as the summation of each tail intensity integral value, multiplied by its relative distance from the center of the head, and divided by the total comet intensity. A minimum of 80 cells or more were analyzed per group. (B) Differential DNA damage induction by PL. Cells were treated with PL for 14 hours before the occurrence of evident cell death and collected for DNA damage measurement using comet assay. A minimum of 70 cells or more were analyzed per group. DNA damage was quantified on the basis of the OTM value. (C) mTORC1 activity affects DNA damage levels. SW480 cells stably expressing shControl or shTSC2 were analyzed for phospho-histone H2AX (Ser139). Histone H3 and β-actin were used as loading control. (D) T24 cells were harvested 24 hours after Torin1 treatment (100 nM). (E) Depletion of c-Myc reduces RUVBL1/2 silencing–mediated cell death in SW480 cells. Cells were transfected with either siControl or siMyc (si pool) at seeding and were subsequently transfected with siRUVBL1-#2 or siRUVBL2-#1 24 hours after seeding. Cell viability was measured 6 days after transfection. Significant differences between shControl and shTSC2 cells transfected with siRUVBL1/2 (***P < 0.001) and significant differences between shTSC2 cells transfected with siRUVBL1/2-only and siRUVBL1/2 and siMyc (###P < 0.001). (F) SW480 cells infected with shControl or shTSC2 were transfected with siMyc. Cells were lysed 48 hours after transfection, and proteins were analyzed by immunoblotting. (G) Depletion of c-Myc reduces RUVBL1/2 silencing–mediated cell death in T24 cells. Cells were transfected with either siControl or siMyc (si pool) at seeding and were subsequently transfected with siRUVBL1-#2 or siRUVBL2-#1 24 hours after seeding. Cell viability was measured 4 days after transfection. (H) T24 cells were analyzed for immunoblot after siMyc transfection (48 hours). (I) Model for synthetic lethality of RUVBL1/2 inhibition in cancer cells with mTORC1 hyperactivation. Cancer cells with high mTORC1 activity have increased DNA damage stress, which is partially through c-Myc. Proper functioning of RUVBL1/2 is critical in mitigating the stress. Blockage of RUVBL1/2 selectively kills cancer cells with high mTORC1 activity. All data are presented as means ± SD. Significant differences were calculated by one-way ANOVA compared with DMSO-treated group for each siRNA treatment (***P < 0.001).
This intriguing link between mTORC1 activity and DNA damage stress led us to question the responsible factors involved. Translation efficiency of the proto-oncogene c-Myc is specifically up-regulated under mTORC1 activation (42, 43), and c-Myc overexpression has been implicated to enhance DNA double-strand breaks and genomic instability (44, 45). Silencing c-Myc reduced RUVBL1 or RUVBL2 knockdown induced cell death in SW480 shTSC2 cells (Fig. 5E). We also found that increasing mTORC1 activity in SW480 cells resulted in up-regulation of c-Myc and subsequent DNA damage levels, whereas silencing c-Myc led to reduction in DNA damage levels in both SW480 shControl and shTSC2 cells (Fig. 5F). Also, knockdown of c-Myc protected T24 cells from RUVBL1/2 knockdown–induced cytotoxicity and reduced DNA damage levels (Fig. 5, G and H). These results demonstrate that mTORC1-induced c-Myc levels at least partially contribute to the DNA damage stress arising in cancer cells with hyperactive mTORC1. Collectively, our results show that cancer cells with high mTORC1 activity have up-regulated levels of c-Myc and DNA damage stress, generating overreliance on RUVBL1/2 to maintain cellular integrity, and this provides a therapeutic window to selectively kill cancer cells addicted to the mTOR pathway by RUVBL1/2 inhibitors (Fig. 5I).
DISCUSSION
The continued activation of mTOR is a costly process that consumes substantial amount of energy for the induction of transcription, translation, and ribosome biogenesis, promoting cells to generate macromolecules and progress through the cell cycle (2, 3). We found that high mTORC1 activity also correlates with elevated levels of DNA damage and that mTORC1-hyperactive cancer cells are highly dependent on RUVBL1/2 to survive and counteract this stress. Elevation of mTORC1 activity markedly enhanced the sensitivity to RUVBL1/2 inhibition, while reduced mTORC1 activity diminished cytotoxic effects driven by RUVBL1/2 depletion. Cancer cells with hyperactive mTORC1 generate DNA damage/replicative stress partially via c-Myc and require support pathways to cope with high stress driven by mTORC1 activity, thereby causing an addictive phenotype toward RUVBL1/2.
The RUVBL1/2 complex participates in diverse cellular processes, including a major role in the regulation of DNA damage responses. RUVBL1/2 and the TTT complex together regulate the activity and stability of PIKK family members including ATM, ATR, and DNA-PKcs, key factors in DNA damage sensing and the amplification of DNA damage response signals (30, 31). ATM and ATR also participate in checkpoint activation that can block cell cycle progression in the presence of DNA damage (46). Therefore, any impairment of RUVBL1/2 function would conceivably hinder the DNA damage response and cause checkpoint malfunctions leading to deleterious effects. Knockdown of RUVBL1/2 also appears to destabilize Tel2, TTI1, and TTI2, while the knockdown of either Tel2 or TTI1 had no effect on RUVBL1/2 levels, suggesting that RUVBL1/2 regulates TTT complex stability and not vice versa. This suggests that the down-regulation of TTT proteins after RUVBL1/2 inhibition could have additional impact independently and additionally from suppressed ATM, ATR, and DNA-PKcs. Previous reports have shown that in addition to RUVBL1/2, the TTT proteins themselves are involved in maintenance of the telomere and genome integrity (31, 47). RUVBL1/2 also have been identified in TIP60, Ino80, and Swr1 complexes, which localize to sites of DNA damage to modify the chromatin to participate in the DNA repair procedures. Studies have shown that yeast and mammalian cells lacking these functional complexes are hypersensitive to DNA damage due to dysfunctional repair systems (38, 48, 49). High mTOR activity could perceivably increase chromatin remodeling activities, adding more DNA damage/mitotic stress (50, 51) and thus pushing cancer cells to increase their dependency on support pathways such as RUVBL1/2-TTT. As inhibiting the RUVBL1/2-TTT pathway prevents proper sensing and repairing of DNA damage, cells with hyperactive mTORC1 appears to suffer from maintaining DNA damage stress to a sublethal level.
There exist further mechanisms that could be potentially responsible for cell death in cancer cells as a result of RUVBL1/2 inhibition. RUVBL1/2 function is connected to mTOR activity in other ways in addition to supporting DNA damage stress. mTOR signaling induces gene expression involved in nucleotide, protein and lipid synthesis, mitochondrial oxidation, and glycolysis (52). Hence, the activation of mTOR leads to activation of multiple RNA polymerases and various transcription regulatory machineries. RUVBL1/2 are present in a number of chromatin remodeling complexes that play a major role in regulating transcription. It has been shown that RUVBL1/2 are essential for Ino80- and NuA4/TIP60-dependent chromatin remodeling activity (53, 54). In addition, RUVBL1/2 have been reported to be associated with RNA polymerase I and II assembly and subsequent activity (39, 55). In addition, one of the major outcomes of mTOR activation is ribosome biogenesis via the generation of ribosomal proteins and the synthesis of ribosomal RNA (rRNA) (56). Small nucleolar ribonucleolar proteins (snoRNPs) cleave and modify small nuclear RNA and rRNA (57), and RUVBL1/2 is required for localization and assembly of snoRNPs (58, 59), further emphasizing the link between RUVBL1/2’s function and mTOR activity. Together, the downstream processes elicited by mTOR activation generate cellular stresses and cells must develop mechanisms to counteract these imbalances. Clinical cancer tissue samples also revealed that high mTORC1 activity correlates with high RUVBL2 expression, suggesting that cancer cells with high mTOR activity are under selective pressure to increase RUVBL1/2 expression for their oncogenic manifestation. We propose that in mTOR-hyperactive cancer cells an overreliance on RUVBL1/2 develops, and this represents a weakness that can be exploited.
In the current study, we have found that cancer cells with high mTORC1 activity exhibit an increased dependency on RUVBL1/2 function for survival. mTOR hyperactivation up-regulates multiple metabolic pathways that promote cancer cell growth and proliferation but, as a result, also increases levels of cellular stresses. This provides a selective therapeutic window for targeting cancer cells with high mTOR activity. Our results demonstrate that adequate functioning of RUVBL1/2 is not only necessary for the assembly of mTOR complexes but also required to guarantee the integrity of the cell after mTOR activation. Therefore, RUVBL1/2 seems to function as a chaperone in the initiation and in the healthy execution step of the mTOR pathway to secure the full activation of mTOR-driven processes. Our findings highlight the potential advantage of targeting RUVBL1/2 for selective killing of cancer cells addicted to the mTOR pathway and suggest a therapeutic strategy for a biomarker-based personalized treatment.
MATERIALS AND METHODS
Materials
Chemicals were purchased from the following companies: PL (Indofine Chemical Company), Torin1 (Cayman), DOX (Sigma-Aldrich), l-buthionine-sulfoximine (Sigma-Aldrich), tert-butyl hydroperoxide (Santa Cruz Biotechnology), hydrogen peroxide (Thermo Fisher Scientific Inc.), and erastin (EMD Millipore). Antibodies against phospho-S6K1 (#9205), S6K1 (#2708), phospho-4E-BP1 (#9451), 4E-BP1 (#9644), TSC2 (#4308), phospho-Akt (#9271), mTOR (#2972), glyceraldehyde-3-phosphate dehydrogenase (#2118), phospho-H2AX (#2577), H2AX (#7631), Histone H3 (#9715), phospho-Chk1 (#2348), phospho-Chk2 (#2661), 53BP1 (#4937), cleaved Caspase-3 (Asp175, #9661), cleaved poly(ADP-ribose) polymerase 1 (Asp214, #5625), ATM (#2873), ATR (#2790), DNA-PKcs (#4602), and c-Myc (#5605) were purchased from Cell Signaling Technology. Antibodies for RUVBL1 (10210-2-AP), RUVBL2 (10195-1-AP), TSC1 (20988-1-AP), and Tel2 (15975-1-AP) were obtained from ProteinTech. Antibodies against Rad51 (sc-8349), α-tubulin (sc-69969), Vinculin (sc-73614), p38 (sc-535), and TTI1 (sc-85605) were purchased from Santa Cruz Biotechnology. Antibody to detect TTI2 (A303-476A) was purchased from Bethyl Laboratories. Antibody against hemagglutinin–horseradish peroxidase (12013819001) was purchased from Roche. Antibodies for BRCA1 (OP92), Flag (F3165), and β-actin (A5441) were purchased from Sigma-Aldrich. TSC1, TSC2, and RUVBL1/Pontin plasmids were obtained from Addgene, and RUVBL2/Reptin plasmid was provided by J. Rosenbaum (Université de Bordeaux).
Cell culture
All cells were cultured in media containing penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C in an incubator with an atmosphere of 5% CO2. Primary cultured HDFs, BJ-hTERT, BJ-ELR, LS174T, SW480, TOV-112D, FAMPAC, and T24 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Cellgro) with 10% fetal bovine serum (FBS) (Gibco). CCD-18co cells were cultured in MEM (Cellgro) with 10% FBS. RPMI 1640 (Cellgro) supplemented with 10% FBS was used for OVCAR-8 and IGROV-1. 184B5 was cultured in DMEM/F12 (Cellgro) supplemented with insulin, epidermal growth factor, bovine pituitary extract, FBS, ascorbic acid, transferrin, hydrocortisone, and cholera toxin. The following cell lines were provided by: BJ-hTERT and BJ-ELR from W. C. Hahn (Dana Farber Cancer Institute); IGROV-1 from M. J. Birrer (Massachusetts General Hospital Cancer Center); FAMPAC from C. Goldring (University of Liverpool); OVCAR-8 from National Cancer Institute (Molecular Radiation Therapeutics); HDFs from J.-H. Jung (Seoul National University, College of Medicine). All other cell lines were obtained from American Type Culture Collection. Negative mycoplasma contamination status was verified using the MycoAlert Mycoplasma Detection Kit (Lonza). Short-tandem repeat (STR) profiling was performed for cell line authentication by Genetica DNA Laboratories.
Chemical screening
SW480 shControl or SW480 shTSC2 cells were seeded to 96-well plates in DMEM with 10% FBS and penicillin/streptomycin. After overnight culturing, transfer of small-molecule library was performed. At 36-hour after compound addition, 50 μl of CellTox Green (Promega) was added to each well. The plates were shaken and allowed to sit for 10 min before being read on the Varioskan multimode microplate reader (Thermo Fisher Scientific).
Cell viability assay
Cells were seeded in six-well plates or 6-cm dishes at a concentration that becomes confluent after 3 days and incubated overnight before treatment. Cell viability was measured after indicated time points using trypan blue. Briefly, cells were collected and mixed with trypan blue solution and loaded into Countess chamber slides. Live and dead cell concentration was measured using the Countess II FL Automated Cell Counter (Life Technologies).
Immunoblot
Harvested cells were lysed with 0.5% NP-40 lysis buffer [50 mM tris-HCl (pH 8.0), 100 mM NaCl, 5 mM EDTA, and 0.5 mM dithiothreitol (DTT)] or with 1% NP-40 lysis buffer [50 mM tris-HCl (pH 8.0), 0.5 M NaCl, 2 mM EDTA, 0.2% SDS, and 1% sodium deoxycholate] with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche). Supernatant fractions were harvested after centrifugation (13,000 rpm, 10 min). The protein concentration was determined using a dye-binding protein assay reagent (Bio-Rad). Equal amount of protein lysates was subjected to SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% nonfat dry milk in 1× tris-buffered saline containing 0.05% Tween 20 and incubated with a specific primary antibody at 4°C overnight. Protein bands were visualized by Western Lightning Plus ECL (PerkinElmer) after incubation with a secondary antibody.
RNA interference
Cells were grown in six-well plates or 60-mm dishes and transfected with RNA interference oligonucleotides using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s protocol. The small interfering RNA (siRNA) sequences for RUVBL1, RUVBL2, and Tel2 were the following: RUVBL1 #1, 5′-GUUUACUCAACUGAGAUCA-3′; RUVBL1 #2, 5′-GGUGAAGUCACAGAGCUAA-3′; RUVBL2 #1, 5′-GAUGAUUGAGUCCCUGACCAA-3′; RUVBL2 #2, 5′-GAAACGCAAGGGUACAGAA-3′; Tel2 #1, 5′-AGAACUGUGAGGUCAGAUAGUCGGC-3′; Tel2 #2, 5′-AGGUGCUUCUGCAUCUGGAGGAGAA-3′. SMARTpool siRNA (mixture of four siRNAs; ACGGAACUCUUGUGCGUAAUU, GAACACACAACGUCUUGGAUU, AACGUUAGCUUCACCAACAUU, and CGAUGUUGUUUCUGUGGAAUU) for human c-Myc was purchased from Dharmacon. As a negative control, scrambled oligonucleotides (si-scrambled; cat no: #SN-1003 Bioneer) were used.
Lentiviral infections
Lentiviruses were generated by transfecting pLKO.1-puro-shControl or pLKO.1-puro-shTSC2 vectors with lentivirus packaging vectors into 293 T cells. The pLKO.1-puro-shTSC2 vector was a gift from W. Du (The University of Chicago). Viruses were collected and infected to SW480 cells in the presence of polybrene. Noninfected cells were eliminated later in the presence of puromycin (4 μg/ml).
Xenograft models
Animal experiments were reviewed and approved by the Incheon National University Subcommittee on Research Animal Care (INU-ANIM-2018-20) and approved by the Subcommittee on Research Animal Care and Zhengzhou University Institutional Animal Care and Use Committee. For cell-based xenograft tumor models, OVCAR-8 (7 × 106) and SW480 (4 × 106) cells in 100 μl of media were mixed with 100 μl of Matrigel (BD Biosciences). Cells were injected subcutaneously in the hind flank of each nude mouse (NCr nude, 5 to 6 weeks old). Animals were randomly assigned to each group. Mice were treated when their tumor volume reached approximately 50 mm3 as measured using calipers, and volumes were estimated using the equation V = l × w × h × 0.5 (l, length; w, width; h, height). For PDXs, colon, lung, and esophageal cancer tissue fragments were collected from the First Affiliated Hospital of Zhengzhou University (Zhengzhou, Henan, China) and implanted into female severe combined immunodeficient (SCID) mice for passage. Once the tumor volume reached approximately 150 mm3, mice were divided into different groups. PL at 7 mg/kg was administered intraperitoneally everyday. Five to eight mice were used per experimental group. Tumor volume was measured every other day, and tumor weight was measured after excision on the final day of the experiment.
ROS measurement
Cells were treated with compounds for indicated time and incubated with H2DCF-DA (Life Technologies) for 30 min at 37°C in an incubator. Cells were washed with phosphate-buffered saline (PBS) and immediately analyzed by flow cytometry.
Immunofluorescence microscopy and quantitative fluorescence analysis
For immunostaining, cells were washed with PBS and then incubated with pre-extraction buffer [10 mM Pipes (pH 6.8), 100 mM NaCl2, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, and 0.2% Triton X-100] for 5 min. After wash with PBS, the cells were fixed with 3% paraformaldehyde in PBS for 15 min, washed with PBS, and fixed with cold methanol (−20°C) for 10 min. Subsequently, cells were washed with PBS, permeabilized with 0.5% Triton X-100 for 5 min, washed a second time with PBS, and blocked in PBST (1× PBS containing 0.05% Tween 20) containing 3% bovine serum albumin (BSA) or 10% milk and 2% BSA for 1 hour at room temperature. Cells were further incubated with the primary antibody diluted in PBST containing 3% BSA or 10% milk and 2% BSA at 4°C for overnight. Coverslips were washed three times with PBST before the incubation with the fluorescent secondary antibody for 1 hour. Coverslips were washed three times with PBST and stained with 4′,6-diamidino-2-phenylindole (DAPI) at the second time. The images were acquired using a Nikon 90i microscope with a Nikon Plan Apo Lambda 20× or 60× objective. Signals of pH2AX of each nucleus in DAPI-stained area were quantified using the Fiji software.
Acetylcysteine and PL adduct formation
N-acetylcysteine (10.2 mg, 62 μmol; Fluka) and PL (20 mg, 63 μmol; Indofine) were dissolved in DMSO (2 ml) and stirred at 40°C in a 1-dram glass vial with a screw cap on, without any special precautions to maintain an inert atmosphere over the reaction mixture. The reaction was monitored by periodically sampling 10 to 50 μl of the mixture, diluting with deionized water (1 ml) and injecting into Waters liquid chromatography–mass spectrometry (LC-MS) (Alliance 2795, Waters, Milford, MA). Ratio of reactant and adduct was measured by ultraviolet absorbance at 210 nm. Identity was determined on an single quadrupole (SQ) mass spectrometer by positive electrospray ionization. Mobile phase A consisted of 0.01% formic acid in water, while mobile phase B consisted of 0.01% formic acid in acetonitrile. The gradient ran from 5 to 95% mobile phase B over 7.5 min at 1.75 ml/min. An Agilent Poroshell 120 EC-C18 column (2.7 μm, 3.0 mm by 30 mm) was used with column temperature maintained at 40°C. A total of 2.1 μl of sample solution was injected. The peaks corresponding to PL at mass of 318.095 and to adduct at 481.089 in the positive ionization mode were identified. The diode array curves in the region corresponding to these masses were integrated. The percent adduct formation was calculated from the ratio of integrated area corresponding to adduct and the sum of integrated areas of adduct and the starting material.
Synthesis of PL-biotin adduct
Demethylation
AlCl3 [2.65 g, 19.85 mmol, 7 equivalent (equiv)] was added in small portions to a solution of PL (900 mg, 2.84 mmol, 1 equiv) in dichloromethane (25 ml, 0.113 M) (fig. S12). The reaction was stirred at room temperature until complete consumption of the starting material. The reaction mixture was partitioned between water and dichloromethane, organic layer was dried and evaporated, and the product was purified by silica gel flash column chromatography using ethylacetate in dichloromethane (0 to 20% gradient; product eluting at 10% Ethyl acetate (EtOAc); main by-product being bis-phenol). The product was obtained as yellowish solid (578 mg, 67% yield).
Mitsunobu coupling
Diethylazadicaroboxylate (41.1 mg, 39.9 μl, 0.203 mmol, 1.1 equiv) was slowly added to a solution of (E)-1-(3-(4-hydroxy-3,5-dimethoxyphenyl)acryloyl)-5,6-dihydropyridin-2(1H)-one (56 mg, 0.185 mmol, 1 equiv), tert-butyl 3-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)propanoate (51.4 mg, 48.8 μl, 0.185 mmol, 1 equiv), and triphenylphosphine (53.3 mg, 0.203 mmol, 1.1 equiv) in THF (1.85 ml, 0.1 M) at 0°C, and the reaction was stirred at 0°C and slowly warmed up to room temperature as the ice bath warmed up. Volatiles were evaporated in vacuo, and the product was purified by silica gel flash column chromatography using EtOAc in hexanes (0 to 100% gradient; product eluting around 70% EtOAc). The product was obtained as a colorless oil (111 mg, 107% yield).
Deprotection of tert-butyl group
The product from the previous reaction (105 mg, 0.186 mmol, 1 equiv) was dissolved in dichloromethane (1.86 ml, 0.1 M). Trifluoroacetic acid (435 mg, 287 μl, 3.73 mmol, 20 equiv) was added to this solution. After several hours, the volatiles were evaporated in vacuo, and the product was purified by silica gel flash column chromatography using EtOAc in hexanes (0 to 100% gradient; product eluting around 90% EtOAc) yielding 51 mg, 54% of theoretical mass.
Acid activation as NHS ester and coupling with amine-PEG2-biotin
The product from the previous reaction (9.8 mg, 0.019 mmol, 1 equiv), and N-hydroxysuccinimide (2.67 mg, 0.023 mmol, 1.2 equiv) were dissolved in DMSO (0.2 ml, 0.097 M). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) (4.50 mg, 0.029 mmol, 1.5 equiv) was added to this solution. The reaction was monitored by LC-MS until the starting material was consumed.
In a separate vial, (+)-biotinyl-3,6-dioxaoctanediamine (6.81 mg, 0.018 mmol; Thermo Fisher Scientific #21346) was dissolved in DMSO (100 μl), and triethylamine (9.21 mg, 12.68 μl, 5 equiv) was added to this solution. The solution was transferred to the solution of activated acid prepared above. The final product was purified on preparative high-performance liquid chromatography with mass detector yielding 6.4 mg, 40.7% of the theoretical mass.
1H NMR (400 MHz, CHLOROFORM-d) δ ppm 1.45 (br. s., 2 H), 1.68 (br. s., 3 H), 2.25 (br. s., 2 H), 2.51 (d, J = 5.62 Hz, 4 H), 2.76 (d, J = 12.72 Hz, 1 H), 2.92 (d, J = 8.80 Hz, 1 H), 3.17 (br. s., 1 H), 3.33 to 3.47 (m, 4 H), 3.49 to 3.62 (m, 9 H), 3.63 to 3.81 (m, 13 H), 3.88 (s, 6 H), 4.05 (t, J = 6.11 Hz, 2 H), 4.17 (br. s., 2 H), 4.37 (br. s., 1 H), 4.55 (br. s., 1 H), 6.05 (d, J = 9.54 Hz, 1 H), 6.80 (s, 2 H), 6.90 to 7.02 (m, 1 H), 7.41 (d, J = 15.41 Hz, 1 H), 7.66 (d, J = 15.41 Hz, 1 H).
Measurement of apoptosis using flow cytometry
Apoptosis was evaluated using the Annexin V-FITC Apoptosis Detection Kit from MBL International Corporation, Watertown, MA. Cells were harvested with 0.025% trypsin + 5 mM EDTA in PBS, and 2.5% FBS in PBS + 5 mM EDTA was added as soon as the cells were released from the dish. Then, the cells were transferred to a centrifuge tube, washed with PBS, and incubated for 5 min at room temperature with Annexin V-FITC and propidium iodide following the protocol included in the kit. Cells were analyzed on a Becton Dickinson FACS Calibur flow cytometer (BD Biosciences, San Jose, CA), placing the FITC signal in FL1 and the PI signal in FL2. Intact cells were gated in the FSC/SSC plot to exclude small debris.
PL-biotin pull-down assay
T24 cells were lysed with 0.3% CHAPS buffer [40 mM Hepes (pH 7.4), 120 mM NaCl, and 4 mM MgCl2] with protease inhibitor cocktail (Thermo Fisher Scientific) and phosphatase inhibitor cocktail (Sigma-Aldrich) and subsequently incubated with or without PL (100 μM). PL-biotin (10 μM) was subsequently added to the lysate and incubated at 4°C for 1 hour. Biotin was purified using the Pierce Monomeric Avidin Kit (Thermo Fisher Scientific) following the manufacturer’s instruction. Purified proteins were concentrated using Amicon Ultra centrifugal filter (EMD Millipore). Proteins were mixed with Bolt LDS Sample Buffer (Thermo Fisher Scientific) and boiled to be further analyzed by immunoblotting. For mass spectrometry analysis, biotin was purified using NeutrAvidin (Thermo Fisher Scientific) and loaded on a gel and sent for analysis.
Immunoprecipitation
Cells were washed with cold PBS twice and lysed using 0.3% CHAPS buffer [40 mM Hepes (pH7.4), 120 mM NaCl, 0.5 mM DTT, and 1 mM EDTA] supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche). The cell lysates were spun at 13,000 rpm in a microcentrifuge, supernatants were collected, and protein concentration was quantified using a dye-binding protein assay kit (Bio-Rad). Cell lysates were precleared with protein G agarose beads (GenDepot) by rocking for 30 min at 4°C and coimmunoprecipitated with appropriate antibodies at 4°C overnight. After incubation with protein G agarose beads for 2 hours at 4°C and wash with the lysis buffer, immunoprecipitated proteins were recovered from the beads by boiling in NuPAGE LDS Sample Buffer (Life Technologies). Proteins were analyzed by immunoblotting.
ATPase assay
His-Flag–tagged RUVBL1 and RUVBL2 were expressed in Rosetta (DE3) pLysS competent cells and purified using Ni-NTA agarose (Life Technologies). Expression vectors were provided by M. Grigoriev (Washington University School of Medicine). ATPase assay was performed using an ADP-Glo Max Assay kit (Promega). Proteins were incubated with the compound for 10 min and added to the ATPase buffer to a final concentration of 50 mM tris-HCl (pH 7.5), 10 mM MgCl2, 20 mM NaCl, 1 mM ATP, and BSA (0.05 mg/ml). Reagents included in the kit were subsequently applied as shown in the manufacturer’s instruction to detect the adenosine 5′-diphosphate formed in the assay. The luminescence was measured using a Varioskan multimode microplate reader (Thermo Fisher Scientific).
Comet assay
DNA damage was measured using the OxiSelect Comet Assay Kit (Cell Biolabs Inc.) based on the manufacturer’s instructions. Briefly, cells were transfected with siRNA or treated with compounds and collected at 1.5 × 105 cells/ml in cold PBS (without MG2+ and Ca2+). Cells were mixed with comet agarose and placed on comet slides. Slides were transferred to 4°C in dark for 15 min and then were incubated in lysis buffer and subsequently in alkaline solution. The slides were electrophoresed in a horizontal chamber and then dried and stained with a DNA dye. All buffers, dye, agarose, and slides were provided by the kit. The comet was observed under a confocal microscope (Carl Zeiss). Quantification of the comet tail was performed using an automated computer program, CometScore (TriTek Corp.)
Immunostaining of human tissue array
Human tissue slides containing various cancer tissues with corresponding normal tissues (AccuMax Array, ISU ABXIS CO) were used to analyze RUVBL2 and phospho-S6 levels. Detailed information of the tissues used in the array is in the Supplementary Materials. Tissue slide was deparaffinized with xylene and dehydrated through a graded alcohol bath. The deparaffinized section was boiled in citric acid buffer and tris-EDTA buffer for antigen retrieval. After blocking with donkey serum, the slide was incubated with anti-RUVBL2 (BD Biosciences) and anti–phospho-S6 (Alexa Fluor 647 Conjugate) (Cell Signaling, #4851) primary antibodies overnight at 4°C. After washing, Alexa Fluor 488 (Life Technologies) and Hoechst (Life Technologies) were subsequently applied. Images were taken with a spinning disk confocal microscopy (Nikon), and ImageJ was used for quantification.
Statistical analyses
Graphs are presented as means of technical replicates with error range indicated. Software used was GraphPad Prism v.6 or MATLAB. Two-tailed unpaired Student t test or analysis of variance (ANOVA) was used to compare between groups. As indicated in each figure legend, data are presented as mean values ± SD. P values are indicated in each case. The number of animal chosen for xenograft experiments was based on preliminary data using other tumor cells with the same compound. No blinding was used for animal study. Experiments using cells shown are representative and have been repeated a minimal of three times.
Gene expression analysis
Gene expressions of ruvbl1 and ruvbl2 in tumor and normal human tissue samples from three datasets [Gene Expression Omnibus (GEO), ArrayExpress, and Expression Project for Oncology] were analyzed using GENT (60). A total of 883 tumor and normal human tissue samples of bladder, liver, ovary, and pancreas were filtered and compared with one-way ANOVA. The accession numbers of bladder, liver, and pancreas tissue arrays are as follows: E-TAMB-118, E-TAMB-176, E-TAMB-276, E-TAMB-282, E-TAMB-302, GSE10138, GSE10191, GSE10245, GSE10258, GSE10281, GSE10282, GSE10300, GSE10334, GSE10358, GSE10445, GSE10609, GSE10616, GSE10780, GSE10792, GSE10799, GSE10810, GSE10820, GSE10846, GSE10927, GSE10971, GSE11001, GSE11024, GSE11045, GSE11083, GSE11135, GSE11151, GSE11166, GSE11190, GSE11375, GSE1145, GSE11499, GSE11504, GSE11783, GSE11831, GSE11877, GSE11882, GSE12090, GSE12172, GSE12187, GSE12195, GSE12276, GSE12326, GSE12345, GSE12368, GSE12408, GSE12417, GSE12606, GSE12667, GSE12734, GSE12763, GSE12814, GSE12902, GSE12992, GSE13027, GSE13041, GSE13067, GSE13070, GSE13136, GSE13159, GSE13205, GSE13294, GSE13353, GSE13355, GSE13367, GSE13471, GSE13501, GSE13506, GSE13564, GSE13671, GSE13732, GSE13785, GSE13787, GSE13911, GSE13933, GSE13985, GSE14001, GSE14017, GSE14038, GSE14062, GSE14245, GSE14333, GSE14378, GSE14407, GSE14479, GSE14580, GSE14642, GSE14762, GSE14827, GSE14879, GSE14880, GSE14905, GSE14924, GSE14973, GSE15061, GSE15083, GSE15090, GSE15238, GSE15258, GSE15431, GSE15459, GSE15471, GSE15490, GSE15578, GSE15645, GSE15695, GSE15773, GSE15824, GSE15913, GSE15960, GSE16015, GSE16020, GSE16024, GSE16032, GSE16059, GSE16134, GSE16155, GSE16161, GSE16191, GSE16236, GSE16237, GSE16363, GSE16382, GSE16391, GSE1643, GSE16455, GSE16461, GSE16515, GSE16524, GSE16538, GSE16593, GSE16615, GSE16759, GSE16879, GSE17061, GSE17170, GSE17183, GSE17187, GSE17306, GSE17351, GSE17368, GSE17372, GSE17459, GSE17536, GSE17537, GSE17612, GSE17743, GSE17855, GSE17861, GSE17891, GSE17907, GSE17920, GSE17951, GSE18015, GSE18018, GSE18088, GSE18239, GSE18462, GSE18497, GSE18521, GSE18583, GSE18681, GSE18696, GSE18728, GSE18736, GSE18781, GSE18842, GSE18864, GSE18897, GSE18997, GSE19067, GSE19188, GSE19234, GSE19246, GSE19293, GSE19332, GSE19352, GSE19407, GSE19420, GSE19429, GSE19475, GSE19577, GSE19578, GSE19615, GSE19650, GSE19665, GSE19667, GSE19681, GSE19697, GSE19728, GSE19784, GSE19804, GSE19826, GSE19959, GSE19963, GSE19982, GSE20086, GSE20141, GSE20146, GSE20565, GSE20667, GSE20685, GSE20916, GSE20950, GSE21029, GSE21050, GSE2109, GSE21138, GSE21261, GSE21293, GSE21349, GSE21354, GSE21369, GSE21422, GSE21452, GSE21497, GSE21510, GSE21653, GSE21779, GSE22035, GSE22056, GSE22138, GSE22377, GSE22459, GSE22501, GSE22513, GSE22541, GSE22544, GSE22779, GSE22927, GSE22968, GSE23117, GSE23177, GSE2328, GSE23343, GSE23618, GSE23720, GSE24006, GSE24223, GSE24235, GSE24244, GSE25407, GSE25414, GSE25462, GSE25550, GSE26339, GSE26378, GSE26440, GSE26495, GSE26511, GSE26526, GSE26673, GSE26682, GSE26713, GSE26760, GSE26787, GSE26906, GSE26910, GSE26966, GSE26969, GSE27651, GSE27838, GSE28044, GSE2817, GSE2842, GSE3292, GSE3325, GSE3467, GSE3629, GSE3678, GSE3744, GSE4036, GSE4039, GSE4107, GSE4172, GSE4183, GSE4237, GSE4290, GSE4488, GSE4554, GSE4587, GSE4588, GSE4757, GSE4780, GSE4845, GSE4888, GSE5058, GSE5059, GSE5081, GSE5109, GSE5281, GSE5460, GSE5563, GSE5675, GSE5764, GSE5787, GSE5900, GSE6004, GSE6222, GSE6269, GSE6338, GSE6364, GSE6369, GSE6532, GSE6551, GSE6764, GSE6791, GSE6798, GSE6891, GSE7023, GSE7307, GSE7392, GSE7440, GSE7476, GSE7515, GSE7553, GSE7621, GSE7696, GSE7741, GSE7757, GSE7788, GSE7846, GSE7869, GSE7904, GSE8023, GSE8052, GSE8157, GSE8167, GSE8271, GSE8514, GSE8545, GSE8581, GSE8671, GSE8977, GSE9103, GSE9195, GSE9250, GSE9254, GSE9419, GSE9438, GSE9452, GSE9511, GSE9599, GSE9686, GSE9843, GSE9899, GSE9960. The accession numbers of ovary tissue arrays are as follows: E-AFMX-5, E-CBIL-30, E-MEXP-114, E-MEXP-120, E-MEXP-1327, E-MEXP-1690, E-MEXP-231, E-MEXP-313, E-MEXP-383, E-MEXP-833, E-MEXP-97, E-MTAB-57, E-TABM-117, E-TABM-125, E-TABM-15, E-TABM-26, E-TABM-292, E-TABM-36, E-TABM-53, GSE10072, GSE1010, GSE10172, GSE10255, GSE10320, GSE10325, GSE10615, GSE10631, GSE10760, GSE10783, GSE10935, GSE11038, GSE11121, GSE1124, GSE1133, GSE1140, GSE1147, GSE1152, GSE11582, GSE1159, GSE11681, GSE11691, GSE11904, GSE11965, GSE11971, GSE12021, GSE12093, GSE12288, GSE12417, GSE12627, GSE12630, GSE12648, GSE12649, GSE12685, GSE12845, GSE12907, GSE1296, GSE1297, GSE12995, GSE13041, GSE13083, GSE13280, GSE13411, GSE13471, GSE13591, GSE13996, GSE1420, GSE1427, GSE1428, GSE14286, GSE14317, GSE14323, GSE1456, GSE14577, GSE14618, GSE1462, GSE1466, GSE14764, GSE1542, GSE15456, GSE1551, GSE1561, GSE15641, GSE1577, GSE15777, GSE15852, GSE1650, GSE1729, GSE1751, GSE1786, GSE1789, GSE1849, GSE1869, GSE1993, GSE2004, GSE2006, GSE2034, GSE2113, GSE2138, GSE2152, GSE2240, GSE2280, GSE2326, GSE2351, GSE2361, GSE2395, GSE2443, GSE2503, GSE2510, GSE2549, GSE2603, GSE2665, GSE2712, GSE2719, GSE2779, GSE2841, GSE3112, GSE3149, GSE3167, GSE3185, GSE3189, GSE3218, GSE3297, GSE3307, GSE3365, GSE3494, GSE3524, GSE3585, GSE3593, GSE362, GSE3726, GSE3790, GSE3910, GSE3912, GSE4045, GSE4119, GSE4271, GSE4412, GSE4475, GSE4573, GSE4667, GSE4698, GSE473, GSE474, GSE4780, GSE4845, GSE4922, GSE5060, GSE5090, GSE5122, GSE5222, GSE5287, GSE5327, GSE5364, GSE5390, GSE5392, GSE5462, GSE5580, GSE5667, GSE5788, GSE5808, GSE5820, GSE5847, GSE5967, GSE6008, GSE6011, GSE6012, GSE6095, GSE6236, GSE6253, GSE6269, GSE6272, GSE6306, GSE6344, GSE635, GSE6365, GSE6401, GSE6477, GSE6481, GSE6532, GSE6613, GSE6691, GSE6710, GSE674, GSE6740, GSE6772, GSE6883, GSE7123, GSE7148, GSE7208, GSE7390, GSE7429, GSE7529, GSE7638, GSE7670, GSE7803, GSE781, GSE7893, GSE7895, GSE8218, GSE8397, GSE8401, GSE8440, GSE8441, GSE8650, GSE8692, GSE8835, GSE8970, GSE8987, GSE9006, GSE9335, GSE9476, GSE9536, GSE9574, GSE9662, GSE9676, GSE974, GSE9750, GSE9782, GSE9874, GSE994, GSE9963.
Data availability
Bioinformatics data that support the findings of this study were derived from GENT database (60) that uses GEO datasets. Accession number of every GEO dataset is provided in the “Gene expression analysis” section.
Supplementary Material
Acknowledgments
We thank Fanxiang Yin [China-US (Henan) Hormel Cancer Institute] for helping with the PDX experiments. Funding: This research was funded by NIH grant 1RO1CA142805 to S.W.L. and National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2017R1C1B1006072) to S.B. Author contributions: S.H.S., J.S.L., and S.B. conducted most of the experimental work. J.-M.Z., S.C., Z.V.B., J.H.K, M.J., J.H.L., M.P., R.Z., M.S., R.W., J.T., and S.G.K. made experimental contributions. M.-H.L., S.G.K., L.Z., S.W.L., and S.B. designed the experimental plans and analyzed and interpreted the data. S.W.L. and S.B. designed and directed the project. S.B., J.-M.Z., and S.G.K. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaay9131/DC1
REFERENCES AND NOTES
- 1.Guertin D. A., Sabatini D. M., Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Shimobayashi M., Hall M. N., Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 15, 155–162 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Laplante M., Sabatini D. M., mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plichta K. A., Mathers J. L., Gestl S. A., Glick A. B., Gunther E. J., Basal but not luminal mammary epithelial cells require PI3K/mTOR signaling for Ras-driven overgrowth. Cancer Res. 72, 5856–5866 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budanov A. V., Karin M., p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw R. J., Bardeesy N., Manning B. D., Lopez L., Kosmatka M., De Pinho R. A., Cantley L. C., The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., Wang S., Ren P., Martin M., Jessen K., Feldman M. E., Weissman J. S., Shokat K. M., Rommel C., Ruggero D., The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markman B., Dienstmann R., Tabernero J., Targeting the PI3K/Akt/mTOR pathway--beyond rapalogs. Oncotarget 1, 530–543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meric-Bernstam F., Gonzalez-Angulo A. M., Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 27, 2278–2287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan S., Scheulen M. E., Johnston S., Mross K., Cardoso F., Dittrich C., Eiermann W., Hess D., Morant R., Semiglazov V., Borner M., Salzberg M., Ostapenko V., Illiger H.-J., Behringer D., Bardy-Bouxin N., Boni J., Kong S., Cincotta M., Moore L., Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. 23, 5314–5322 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Galanis E., Buckner J. C., Maurer M. J., Kreisberg J. I., Ballman K., Boni J., Peralba J. M., Jenkins R. B., Dakhil S. R., Morton R. F., Jaeckle K. A., Scheithauer B. W., Dancey J., Hidalgo M., Walsh D. J.; North Central Cancer Treatment Group , Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A north central cancer treatment group study. J. Clin. Oncol. 23, 5294–5304 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Margolin K., Longmate J., Baratta T., Synold T., Christensen S., Weber J., Gajewski T., Quirt I., Doroshow J. H., CCI-779 in metastatic melanoma: A phase II trial of the california cancer consortium. Cancer 104, 1045–1048 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Wander S. A., Hennessy B. T., Slingerland J. M., Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Invest. 121, 1231–1241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hess G., Herbrecht R., Romaguera J., Verhoef G., Crump M., Gisselbrecht C., Laurell A., Offner F., Strahs A., Berkenblit A., Hanushevsky O., Clancy J., Hewes B., Moore L., Coiffier B., Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 27, 3822–3829 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Kwitkowski V. E., Prowell T. M., Ibrahim A., Farrell A. T., Justice R., Mitchell S. S., Sridhara R., Pazdur R., FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 15, 428–435 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motzer R. J., Escudier B., Oudard S., Hutson T. E., Porta C., Bracarda S., Grünwald V., Thompson J. A., Figlin R. A., Hollaender N., Urbanowitz G., Berg W. J., Kay A., Lebwohl D., Ravaud A.; RECORD- Study Group , Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 372, 449–456 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Bissler J. J., McCormack F. X., Young L. R., Elwing J. M., Chuck G., Leonard J. M., Schmithorst V. J., Laor T., Brody A. S., Bean J., Salisbury S., Franz D. N., Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 358, 140–151 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormack F. X., Inoue Y., Moss J., Singer L. G., Strange C., Nakata K., Barker A. F., Chapman J. T., Brantly M. L., Stocks J. M., Brown K. K., Lynch J. P. III, Goldberg H. J., Young L. R., Kinder B. W., Downey G. P., Sullivan E. J., Colby T. V., McKay R. T., Cohen M. M., Korbee L., Taveira-Da Silva A. M., Lee H.-S., Krischer J. P., Trapnell B. C.; National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group , Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N. Engl. J. Med. 364, 1595–1606 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dibble C. C., Manning B. D., Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 15, 555–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoki K., Li Y., Zhu T., Wu J., Guan K.-L., TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Han S.-S., Son D.-J., Yun H., Kamberos N. L., Janz S., Piperlongumine inhibits proliferation and survival of Burkitt lymphoma in vitro. Leuk. Res. 37, 146–154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J. M., Pan F., Li L., Liu Q. R., Chen Y., Xiong X. X., Cheng K., Yu S. B., Shi Z., Yu A. C.-H., Chen X. Q., Piperlongumine selectively kills glioblastoma multiforme cells via reactive oxygen species accumulation dependent JNK and p38 activation. Biochem. Biophys. Res. Commun. 437, 87–93 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Byrne A. T., Alférez D. G., Amant F., Annibali D., Arribas J., Biankin A. V., Bruna A., Budinská E., Caldas C., Chang D. K., Clarke R. B., Clevers H., Coukos G., Dangles-Marie V., Eckhardt S. G., Gonzalez-Suarez E., Hermans E., Hidalgo M., Jarzabek M. A., de Jong S., Jonkers J., Kemper K., Lanfrancone L., Mælandsmo G. M., Marangoni E., Marine J.-C., Medico E., Norum J. H., Palmer H. G., Peeper D. S., Pelicci P. G., Piris-Gimenez A., Roman-Roman S., Rueda O. M., Seoane J., Serra V., Soucek L., Vanhecke D., Villanueva A., Vinolo E., Bertotti A., Trusolino L., Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 17, 254–268 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Li S., Shen D., Shao J., Crowder R., Liu W., Prat A., He X., Liu S., Hoog J., Lu C., Ding L., Griffith O. L., Miller C., Larson D., Fulton R. S., Harrison M., Mooney T., McMichael J. F., Luo J., Tao Y., Goncalves R., Schlosberg C., Hiken J. F., Saied L., Sanchez C., Giuntoli T., Bumb C., Cooper C., Kitchens R. T., Lin A., Phommaly C., Davies S. R., Zhang J., Kavuri M. S., Eachern D. M., Dong Y. Y., Ma C., Pluard T., Naughton M., Bose R., Suresh R., Dowell R. M., Michel L., Aft R., Gillanders W., De Schryver K., Wilson R. K., Wang S., Mills G. B., Gonzalez-Angulo A., Edwards J. R., Maher C., Perou C. M., Mardis E. R., Ellis M. J., Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemery S., Keegan P., Pazdur R., First FDA approval agnostic of cancer site - when a biomarker defines the indication. N. Engl. J. Med. 377, 1409–1412 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Adams D. J., Dai M., Pellegrino G., Wagner B. K., Stern A. M., Shamji A. F., Schreiber S. L., Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc. Natl. Acad. Sci. U.S.A. 109, 15115–15120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boskovic Z. V., Hussain M. M., Adams D. J., Dai M., Schreiber S. L., Synthesis of piperlogs and analysis of their effects on cells. Tetrahedron 69, 7559–7567 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izumi N., Yamashita A., Iwamatsu A., Kurata R., Nakamura H., Saari B., Hirano H., Anderson P., Ohno S., AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci. Signal. 3, ra27 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Kim S. G., Hoffman G. R., Poulogiannis G., Buel G. R., Jang Y. J., Lee K. W., Kim B.-Y., Erikson R. L., Cantley L. C., Choo A. Y., Blenis J., Metabolic stress controls mTORC1 lysosomal localization and dimerization by regulating the TTT-RUVBL1/2 complex. Mol. Cell 49, 172–185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurov K. E., Cotta-Ramusino C., Elledge S. J., A genetic screen identifies the Triple T complex required for DNA damage signaling and ATM and ATR stability. Genes Dev. 24, 1939–1950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Izumi N., Yamashita A., Ohno S., Integrated regulation of PIKK-mediated stress responses by AAA+ proteins RUVBL1 and RUVBL2. Nucleus 3, 29–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maréchal A., Zou L., DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 5, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blackford A. N., Jackson S. P., ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Furuta T., Takemura H., Liao Z.-Y., Aune G. J., Redon C., Sedelnikova O. A., Pilch D. R., Rogakou E. P., Celeste A., Chen H. T., Nussenzweig A., Aladjem M. I., Bonner W. M., Pommier Y., Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 278, 20303–20312 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Stiff T., O'Driscoll M., Rief N., Iwabuchi K., Löbrich M., Jeggo P. A., ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 64, 2390–2396 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Wang H., Wang M., Wang H., Böcker W., Iliakis G., Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell. Physiol. 202, 492–502 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Halasi M., Wang M., Chavan T. S., Gaponenko V., Hay N., Gartel A. L., ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 454, 201–208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jha S., Dutta A., RVB1/RVB2: Running rings around molecular biology. Mol. Cell 34, 521–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenbaum J., Baek S. H., Dutta A., Houry W. A., Huber O., Hupp T. R., Matias P. M., The emergence of the conserved AAA+ ATPases Pontin and Reptin on the signaling landscape. Sci. Signal. 6, mr1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rousseau B., Ménard L., Haurie V., Taras D., Blanc J.-F., Moreau-Gaudry F., Metzler P., Hugues M., Boyault S., Lemière S., Canron X., Costet P., Cole M., Balabaud C., Bioulac-Sage P., Zucman-Rossi J., Rosenbaum J., Overexpression and role of the ATPase and putative DNA helicase RuvB-like 2 in human hepatocellular carcinoma. Hepatology 46, 1108–1118 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Luo J., Solimini N. L., Elledge S. J., Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 136, 823–837 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Csibi A., Lee G., Yoon S.-O., Tong H., Ilter D., Elia I., Fendt S.-M., Roberts T. M., Blenis J., The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. 24, 2274–2280 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.West M. J., Stoneley M., Willis A. E., Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene 17, 769–780 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Dominguez-Sola D., Ying C. Y., Grandori C., Ruggiero L., Chen B., Li M., Galloway D. A., Gu W., Gautier J., Dalla-Favera R., Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Karlsson A., Deb-Basu D., Cherry A., Turner S., Ford J., Felsher D. W., Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. U.S.A. 100, 9974–9979 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S., Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Stirling P. C., Bloom M. S., Solanki-Patil T., Smith S., Sipahimalani P., Li Z., Kofoed M., Ben-Aroya S., Myung K., Hieter P., The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLOS Genet. 7, e1002057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerhold C.-B., Hauer M. H., Gasser S. M., INO80-C and SWR-C: Guardians of the genome. J. Mol. Biol. 427, 637–651 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Ikura T., Ogryzko V. V., Grigoriev M., Groisman R., Wang J., Horikoshi M., Scully R., Qin J., Nakatani Y., Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102, 463–473 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Foster D. A., Yellen P., Xu L., Saqcena M., Regulation of G1 cell cycle progression: Distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 1, 1124–1131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hills S. A., Diffley J. F. X., DNA replication and oncogene-induced replicative stress. Curr. Biol. 24, R435–R444 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Magnuson B., Ekim B., Fingar D. C., Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 441, 1–21 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Jónsson Z. O., Jha S., Wohlschlegel J. A., Dutta A., Rvb1p/Rvb2p recruit Arp5p and assemble a functional Ino80 chromatin remodeling complex. Mol. Cell 16, 465–477 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Taubert S., Gorrini C., Frank S. R., Parisi T., Fuchs M., Chan H.-M., Livingston D. M., Amati B., E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol. Cell. Biol. 24, 4546–4556 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boulon S., Pradet-Balade B., Verheggen C., Molle D., Boireau S., Georgieva M., Azzag K., Robert M.-C., Ahmad Y., Neel H., Lamond A. I., Bertrand E., HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol. Cell 39, 912–924 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mayer C., Grummt I., Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 25, 6384–6391 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Holley C. L., Topkara V. K., An introduction to small non-coding RNAs: miRNA and snoRNA. Cardiovasc Drugs Ther. 25, 151–159 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Boulon S., Marmier-Gourrier N., Pradet-Balade B., Wurth L., Verheggen C., Jády B. E., Rothé B., Pescia C., Robert M.-C., Kiss T., Bardoni B., Krol A., Branlant C., Allmang C., Bertrand E., Charpentier B., The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J. Cell Biol. 180, 579–595 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Machado-Pinilla R., Liger D., Leulliot N., Meier U. T., Mechanism of the AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 18, 1833–1845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shin G., Kang T.-W., Yang S., Baek S.-J., Jeong Y.-S., Kim S.-Y., GENT: Gene expression database of normal and tumor tissues. Cancer Inform. 10, 149–157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaay9131/DC1
Data Availability Statement
Bioinformatics data that support the findings of this study were derived from GENT database (60) that uses GEO datasets. Accession number of every GEO dataset is provided in the “Gene expression analysis” section.