

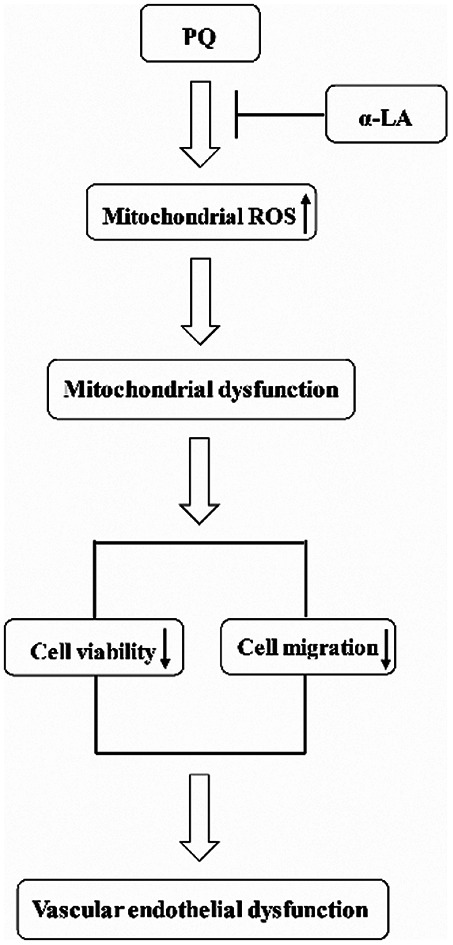
 α-Lipoic acid antagonizes paraquat toxicity.
α-Lipoic acid antagonizes paraquat toxicity.
Abstract
Paraquat (PQ) is a widely used herbicide in the agricultural field. The lack of an effective antidote is the significant cause of high mortality in PQ poisoning. Here, we investigate the antagonistic effects of alpha lipoic acid (α-LA), a naturally existing antioxidant, on PQ toxicity in human microvascular endothelial cells (HMEC-1). All the doses of 250, 500 and 1000 μM α-LA significantly inhibited 1000 μM PQ-induced cytotoxicity in HMEC-1 cells. α-LA pretreatment remarkably diminished the damage to cell migration ability, recovered the declined levels of the vasodilator factor nitric oxide (NO), elevated the expression level of endothelial nitric oxide synthases (eNOS), and inhibited the upregulated expression of vasoconstrictor factor endothelin-1 (ET-1). Moreover, α-LA pretreatment inhibited reactive oxygen species (ROS) generation, suppressed the damage to the mitochondrial membrane potential (ΔΨm) and mitigated the inhibition of adenosine triphosphate (ATP) production in HMEC-1 cells. These results suggested that α-LA could alleviate PQ-induced endothelial dysfunction by suppressing oxidative stress. In summary, our present study provides novel insight into the protective effects and pharmacological potential of α-LA against PQ toxicity in microvascular endothelial cells.
Introduction
Paraquat (PQ; 1,1′-dimethyl 4,4′-bipyridylium dichloride), which belongs to the group of bipyridylium herbicides, is widely used in agricultural fields in various countries due to its herbicidal function and automatic degradation in soil.1 Human poisoning resulting from suicidal or accidental exposure to PQ, with a reported fatality rate of >50%, may cause multiple organ failure.2,3 In the case of severe PQ intoxication, the lung, liver, kidney, brain and heart are damaged functionally and morphologically because these vital organs accumulate PQ via the polyamine transporter system.1 PQ arrives at these vital organs, produces a mass of oxygen free radicals and various inflammatory factors, resulting in oxidative stress and lipid peroxidation. These pathophysiological events occurring in vital organs cause acute multiple organ failure.1,2,4 The lack of effective therapy and medication results in the high mortality of PQ poisoning. The complex mechanisms of PQ toxicity are not fully understood. Thus, developing effective antidotes and exploring the molecular mechanisms for PQ intoxication are urgently required.
Alpha-lipoic acid (1,2-dithiolane-3-pentanoic acid; α-LA), as an essential cofactor for mitochondrial alpha-ketoacid dehydrogenases, plays a pivotal role in mitochondrial energy metabolism.5 α-LA is a naturally occurring disulphide compound that exists in animal-derived foods (red meat and the liver, heart, and kidney) and plant sources (dark green leafy vegetables) and has both hydrophilic and hydrophobic properties.6,7 Previous studies have shown that α-LA, as a potent antioxidant and redox-active metal chelator, can scavenge reactive oxygen species (ROS), promote the generation of endogenous and exogenous antioxidants such as glutathione (GSH), vitamins C and E, and antagonize cytokine-induced inflammation.8–11 Additionally, α-LA has been reported to exert beneficial effects on various diseases such as hypertension, diabetes and its complications, Alzheimer's disease etc.12,13 Notably, several studies have described that α-LA benefits the vascular system through elevating endothelium-dependent NO-mediated vasodilation in diabetes mellitus.14,15 However, a paucity of studies exists on the protective effects of α-LA against PQ toxicity. The underlying mechanisms of action of α-LA remain unclear.16,17
Endothelial cells are the innermost layer of blood vessels that play important roles in homeostasis and vascular function, and they provide a barrier to retain plasma components in the circulation while regulating the exchange of molecules and cells between the lumen and tissues. Endothelial cells are located in the lumen of vessels, have direct contact with cells circulating in the blood, and regulate blood pressure as well as leukocyte trafficking. In many diseases, the physiological function of endothelial cells is disrupted because of oxidative stress, inflammatory reactions, and abnormal expressions of some molecules on the surface of endothelial cells. Therefore, endothelial cells are the direct targets of PQ toxicity when PQ is absorbed into the blood. A few studies indicated that PQ exposure induced dysfunction of the glutathione redox cycle and endothelial hyperpermeability to albumin in microvascular endothelial cells.18,19 Nevertheless, the mechanism of paraquat-induced damage to vascular endothelial cells has not been fully elucidated. Previous studies have demonstrated that endothelial nitric oxide synthase (eNOS) is a critical trigger in the onset of endothelial dysfunction in atherosclerotic cardiovascular and metabolic diseases.20,21 Excessive ROS cause eNOS uncoupling, which generates superoxide instead of nitric oxide (NO), and the superoxide anion reacts with NO, leading to the formation of oxidant peroxynitrite (ONOO–), and aggravates NO bioavailability reduction and aberrant endothelial function.21–23
In the present study, we aimed to investigate the protective effects and underlying mechanisms of α-LA antagonizing PQ toxicity. Our results suggested that α-LA could protect against PQ toxicity in vascular endothelial cells, as indicated by maintaining cell viability, preserving endothelial function, restoring cell migration capacity and suppressing oxidative stress.
Materials and methods
Reagents and chemicals
PQ was purchased from Sigma-Aldrich (USA). α-LA and phosphate-buffered saline (PBS) were purchased from Beyotime (China). Foetal bovine serum (FBS) was purchased from PAN Biotech (Germany). Dulbecco's modified Eagle's medium (DMEM), Hank's balanced salt solution (HBSS), and penicillin–streptomycin were obtained from Gibco (USA). All other reagents were of the highest grade of purity available.
Cell culture and PQ treatment
HMEC-1 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). HMEC-1 cells were cultured in DMEM supplemented with 10% FBS and 1% (v/v) penicillin and streptomycin at 37 °C in the presence of 95% air and 5% CO2. A 1-M PQ stock solution was prepared in sterile ultrapure water, and a 1-M α-LA stock solution was prepared in sterile DMSO (Sigma, USA). Stock solutions were stored at –20 °C and under dark conditions. All the working concentrations were freshly prepared before use and the solvent control also contained 0.1% (v/v) DMSO in the culture media for cell treatment.
Cell viability assay
To assess PQ cytotoxicity, HMEC-1 cells were seeded in 96-well flat-bottomed plates at a density of 7 × 103 cells per well and were cultured overnight, and then PQ was added to the culture medium at the indicated concentrations of 250, 500, 750 and 1000 μM for 0 h, 6 h, 12 h, 24 h, and 48 h, respectively. For the α-LA protective effect study, cells were pretreated with 250, 500 and 1000 μM α-LA for 3 h, followed by treatment with or without PQ for 24 h. Cell viability was detected using CCK-8 (Dojindo Laboratories, Japan) according to the manufacturer's instructions. Briefly, the CCK-8 reagent was mixed in fresh medium to make a 1/10 (v/v) working solution, and 100 μL of the CCK-8 working solution was added to each well, followed by incubation in a cell culture incubator at 37 °C for 1.5 h. The absorbance was read at 450 nm by using a microplate reader (Tecan, Switzerland).
Determination of the intracellular and mitochondrial ROS levels
The levels of intracellular ROS were determined using a well-characterized probe, CM-H2DCFDA (Invitrogen, USA), as previously described.24 A 1 mM CM-H2DCFDA stock solution was prepared in DMSO before the experiments. To measure ROS, cells (7 × 103 cells per well) were plated in a 96-well black plate and treated with α-LA for 3 h, followed by treatment with PQ for 3 h. The treated cells were incubated with CM-H2DCFDA (25 μM) at 37 °C for 30 min in HBSS and were protected from light. The cells were then washed three times with HBSS, and the fluorescence was measured using a microplate reader (Tecan, Switzerland).
The mitochondrial reactive oxygen species (mtROS) levels were determined using a MitoSOX™ Red reagent (Invitrogen, USA), which was dissolved in DMSO to a stock concentration of 5 mM and diluted in HBSS solution.25 The cells were incubated with 10 μM MitoSOX™ Red at 37 °C for 30 min and protected from light. The cells were then washed three times with HBSS, and the fluorescence was measured using a microplate reader (Tecan, Switzerland).
Measurement of the ATP levels
The adenosine triphosphate (ATP) levels were determined using a CellTiter-Lumi™ Plus system (Beyotime, China) according to the manufacturer's instructions. Briefly, 7 × 103 cells per well were seeded in a 96-well plate and were pretreated with α-LA for 3 h, followed by exposure to PQ for 24 h. After treatment, 100 μL of CellTiter-Lumi™ Plus reagent was added to each well, and mixed on an orbital shaker for 2 min. The plate was incubated for 10 min at room temperature in the dark and luminescence was measured using a microplate reader (Molecular Devices, USA).
Mitochondrial membrane potential (ΔΨm) assay
The mitochondrial membrane potential was measured using JC-1 (Beyotime, China) according to the manufacturer's instructions. Briefly, the cells (7 × 103 cells per well) were seeded in a 96-well black plate and were treated with α-LA for 3 h, followed by exposure to PQ for a further 24 h. After treatment, the cells were incubated with JC-1 (1 μg mL–1) in the medium for 20 min in the dark at 37 °C. Next, the cells were washed three times with HBSS, and the fluorescence was measured using a microplate reader (Tecan, Switzerland) using filters of 560 nm excitation and 590 nm emission (red fluorescence), 488 nm excitation and 525 nm emission (green fluorescence). ΔΨm was calculated based on the ratio of red fluorescence divided by green fluorescence.
Determination of the intracellular NO levels
The levels of intracellular NO were measured according to a fluorescent indicator, DAF-FM DA (Beyotime, China), with modifications.26 Cells were cultured in a 96-well black plate and were treated with α-LA for 3 h, followed by exposure to PQ for 24 h. After treatment, the medium was discarded, and the cells were loaded with 100 μL of DAF-FM DA (5 μM) at 37 °C for 30 min in HBSS. Next, the cells were gently washed three times with PBS and maintained in PBS throughout the experiments. The fluorescence intensity was measured using a microplate reader (Tecan, Switzerland) with excitation at 488 nm and emission at 525 nm. The measured fluorescence values were expressed as a percentage of the fluorescence of control cells.
Quantitative real-time PCR analysis
Cells were cultured in 6-well (1.8 × 105 cells per well) plates and were pretreated with α-LA for 3 h, followed by treatment with PQ for 24 h. At the end of the exposure time, total RNA was extracted using an RNA sample Total RNA Kit (TIANGEN, China), according to the manufacturer's protocol, and concentrations were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Next, 1 μg of total RNA was reversed transcribed using the Prime Script RT reagent Kit (Takara, Japan) to synthesize cDNA. The qRT-PCR reactions were performed using the CFX96™ Real-time System (Bio-Rad, USA) with iTaq™ Universal SYBR® Green (Bio-Rad, USA). One microliter of template cDNA was added to the total volume of 20 μL of the reaction mixture. The real-time PCR cycle parameters were 10 min at 95 °C, followed by 40 amplification cycles of 95 °C for 10 s, and 60 °C for 30 s. eNOS, ET-1, and β-actin gene primers were used for qRT-PCR and are shown in Table 1. The expression data were calculated using the 2–ΔΔCt method and were normalized to that of β-actin, which served as an internal housekeeping control.
Table 1. Primers used in quantitative real-time PCR analyses.
| Gene name | Forward primer | Reverse primer |
| ET-1 | 5′-AGAGTGTGTCTACTTCTGCCA-3′ | 5′-CTTCCAAGTCCATACGGAACAA-3′ |
| eNOS | 5′-TGATGGCGAAGCGAGTGAAG-3′ | 5′-ACTCATCCATACACAGGACCC-3′ |
| β-actin | 5′-GCCGACAGGATGCAGAAGG-3′ | 5′-TGGAAGGTGGACAGCGAGG-3′ |
Western blotting analysis
Cells were seeded in 6-well (1.8 × 105 cells per well) plates and were primed with α-LA for 3 h with or without PQ exposure for further 24 h. After treatment, HMEC-1 cells were lysed using RIPA Lysis Buffer (Beyotime, China) with a cocktail of protease inhibitors (Roche, USA), and the concentrations of the proteins were determined using a BCA protein assay kit (Beyotime, China). Equal amounts of protein were separated by 10% (wt/vol) polyacrylamide gel electrophoresis and were electrotransferred onto ImmunoBlot PVDF (Bio-Rad). The membranes were blocked in 5% nonfat dry milk for 2 h at room temperature and were then incubated overnight at 4 °C with the following primary antibodies: rabbit anti-eNOS (1 : 500; Wanleibio, China), rabbit anti-ET-1 (1 : 500, Wanleibio, China), and mouse anti-β-actin (1 : 5000, Beyotime, China). The membranes were then incubated with an appropriate HRP-conjugated secondary antibody for 1.5 h at room temperature. All bands were detected by enhanced chemiluminescence (Millipore, Billerica, MA, USA). The bands were imaged and analysed using the ChemiDoc XRS Plus System with Image Lab Software (Bio-Rad, USA).
Cell migration assay
To determine cell migration, HMEC-1 cells were seeded at a density of 7 × 104 cells per well in 96-well ImageLock plates (Essen BioScience, USA). The cells were pretreated with α-LA for 3 h, and then the WoundMaker system (Essen BioScience, USA) was used to create scratches in all the wells. After creating the scratches, the medium was discarded, and the wells were washed three times with PBS to remove cells from the scratched area. Following the washes, 200 μL of fresh medium with or without α-LA and PQ were added to each well, the plate was placed into the IncuCyte ZOOM apparatus and images of the collective cell spreading were recorded every 6 hours for 48 hours using the IncuCyte ZOOM Live Cell Analysis System (Essen BioScience, USA). The wound width was analysed using IncuCyte Zoom software, and four identically prepared experimental replicates were performed.
Statistical analysis
The data are presented as the means ± SEM of at least three independent experiments. Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad Software, USA). The data were compared using one-way ANOVA followed by Tukey's multiple comparison tests. Differences were considered statistically significant when p < 0.05.
Results
PQ induces cytotoxicity in HMEC-1 cells
To investigate PQ cytotoxicity, cell viability was determined using the CCK-8 assay. Cells were treated with PQ at four concentrations of 250, 500, 750 and 1000 μM, and cell viability was assessed at 6, 12, 24, and 48 h post PQ treatment. As shown in Fig. 1A, PQ significantly inhibited the cell viability dose- and time-dependently. At 24 h post PQ treatment, cell viability began to decline markedly at 250 μM PQ treatment but decreased to 69.3% of the control at 1000 μM PQ treatment. Furthermore, we observed that 1000 μM PQ exposure for 24 h induced morphological damage (Fig. 1B). The cell appearance changed from spindle to round shaped, and cell shrinkage was observed under a microscope. These results indicated that PQ induced significant cytotoxicity in HMEC-1 cells in a dose- and time-dependent manner.
Fig. 1. PQ induces dose- and time-dependent cytotoxicity in HMEC-1 cells. (A) Dose- and time-dependent changes in cell viability after PQ treatment. Cells were treated with 250, 500, 750 and 1000 μM, and cell viability was assessed at 0, 6, 12, 24 and 48 h post PQ treatment. The data are presented as means ± SEM from four independent experiments. *P < 0.05 and **P < 0.01 vs. the control group (0 μM PQ). (B) Morphological changes of HMEC-1 cells exposed to 250, 500, 750 and 1000 μM PQ observed by light phase contrast microscopy with original magnification ×10. Scale bar: 80 μm. Dead cells are pointed out with black arrows.

α-LA alleviates PQ-induced cytotoxicity in HMEC-1 cells
To assess the protective effects of α-LA against PQ-induced cytotoxicity, cells were pretreated with different concentrations of α-LA for 3 h, followed by PQ exposure for 24 h and evaluation of the cell viabilities. As shown in Fig. 2A, α-LA treatment alone at the concentrations of 250, 500, and 1000 μM did not affect cell viability significantly. When the cells were pretreated with these three concentrations of α-LA and exposed to 1000 μM PQ for 24 h, we found that pretreatment with α-LA markedly elevated PQ-induced decline in cell viability compared with PQ treatment. Moreover, 250 μM α-LA pretreatment was potent enough to increase the cell viability and considerably mitigate PQ-induced morphological changes (Fig. 2B). These results demonstrated that α-LA has no effects on cell viability and can antagonize PQ-induced cytotoxicity in HMEC-1 cells.
Fig. 2. α-LA protects against PQ-induced cytotoxicity. (A) Cell viability was determined in cells pretreated with 250, 500, or 1000 μM α-LA for 3 h followed by 1000 μM PQ exposure for 24 h. The data are presented as means ± SEM from four independent experiments. **P < 0.01 vs. the control group; ##P < 0.01 vs. the PQ group. (B) Morphological changes in different treatment groups observed by phase contrast microscopy with original magnification ×10. Scale bar: 80 μm. Dead cells are pointed out with black arrows. Control: solvent, α-LA: 250 μM, PQ: 1000 μM, α-LA + PQ: 250 μM α-LA plus 1000 μM PQ.

α-LA reverses PQ-induced inhibition of cell migration in HMEC-1 cells
The integrity of the endothelial barrier is crucial for the physiological function of the endothelium.27 To maintain the integrity of endothelial function, endothelial cells show dynamic planar migration behaviour of individual cells and cell groups.28 To explore the effect of PQ exposure on migration and evaluate the protective effect of α-LA, we examined the changes in cell migration with 1000 μM PQ exposure in the absence or presence of 250 μM α-LA using the IncuCyte ZOOM Live Cell Analysis System. As shown in Fig. 3, cell migration activity was significantly diminished by PQ treatment. Pretreatment of 250 μM α-LA significantly inhibited the PQ-induced decrease in the cell migration capacity, as indicated by the relative wound density (Fig. 3A). In the wound-healing assay, we observed that α-LA pretreatment could accelerate cell migration and promote wound healing in the presence of PQ exposure (Fig. 3B).
Fig. 3. α-LA antagonizes the PQ-induced damage to the cell migration capacity in HMEC-1 cells. (A) Relative wound density determined in different treatment groups. The cells were seeded at 7 × 104 cells in each well, were pretreated with 250 μM α-LA for 3 h, were mechanically wounded using the WoundMaker system and then were incubated in the medium with 1000 μM PQ. Cell migration was monitored for 48 h using the IncuCyte ZOOM Live Cell Analysis System (Essen BioScience; ×10). The relative wound confluence was analysed. The data are presented as means ± SEM from four independent experiments. **P < 0.01 vs. the control group; ##P < 0.01 vs. the PQ group. (B) Representative images of the wound-healing assay. Scale bar: 300 μm.

α-LA improves PQ-induced endothelial dysfunction in HMEC-1 cells
The vascular endothelium plays an essential physiological role in vascular homeostasis, and endothelial dysfunction is an early pathophysiological characteristic.29 Endothelial function is regulated by complex mechanisms involving many factors. Nitric oxide (NO) is a key vasodilator that regulates vasodilation, and endothelial nitric oxide synthase (eNOS) is the enzyme responsible for controlling NO generation. Endothelin-1 (ET-1) is a major vasoconstrictor that regulates endothelial function. To investigate the damage to the endothelial function by PQ exposure and the antagonistic effects of α-LA in HMEC-1 cells, we determined the changes in the intracellular NO levels. eNOS and ET-1 expression levels at the mRNA and protein levels were also assessed by qRT-PCR and western blotting, respectively. We found that PQ treatment obviously increased ET-1 mRNA and protein expression and significantly suppressed eNOS mRNA and protein expression. Pretreatment with 250 μM α-LA markedly reduced PQ-induced elevation of the ET-1 expression (Fig. 4A and B) and significantly increased the eNOS expression (Fig. 4C and D). Furthermore, pretreatment with 250 μM α-LA could restore the PQ-induced decrease in the NO level to that of the control (Fig. 4E). Taken together, these results suggested that α-LA can remarkably improve PQ-induced endothelial dysfunction.
Fig. 4. α-LA mitigates PQ-induced endothelial dysfunction in HMEC-1 cells. (A) Relative expression levels of ET-1 mRNA determined by qRT-PCR. (B) Expression of ET-1 protein determined by western blotting. (C) Relative expression levels of eNOS mRNA determined by qRT-PCR. (D) Expression of eNOS protein determined by western blotting. (E) Measurement of NO levels in different treatment groups. The data are presented as means ± SEM from four independent experiments. *P < 0.05, **P < 0.01 vs. the control group; #P < 0.05, ##P < 0.01 vs. the PQ group.

α-LA suppresses oxidative stress and protects mitochondrial function in HMEC cells
Previous studies have shown that the major toxic mechanism underlying PQ intoxication is mainly due to the production of excessive superoxide anions which are primarily generated by the mitochondria.30,31 Oxidative stress can cause mitochondrial dysfunction. To elucidate the possible mechanisms of action of α-LA against PQ toxicity in HMEC cells, we determined the influence of α-LA pretreatment on the intracellular ROS and mtROS levels. We used the JC-1 probe to measure the mitochondrial membrane potential (ΔΨm) and detected the intracellular ATP level to indicate mitochondrial function changes. The results showed that PQ exposure significantly elevated the levels of intracellular ROS and mtROS (Fig. 5B and D). Pretreatment at the concentrations of 250, 500, and 1000 μM α-LA reversed the PQ-induced excessive production of ROS (Fig. 5B and D), while α-LA treatment alone at the concentrations of 250, 500, and 1000 μM did not affect the ROS and mtROS levels (Fig. 5A and C). As shown in Fig. 5E and F, cells exposed to 1000 μM PQ for 24 h exhibited reduced mitochondrial membrane potential compared with the control while the ATP levels markedly decreased to 38% of the control after PQ exposure. α-LA pretreatment at the concentrations of 250, 500, and 1000 μM could significantly restore the mitochondrial membrane potential and remarkably elevate the ATP levels Overall, these data suggested that PQ exposure causes mitochondrial oxidative stress and impairs mitochondrial function. α-LA exerts protective effects on PQ toxicity by suppressing oxidative stress and maintaining mitochondrial function.
Fig. 5. α-LA suppresses oxidative stress and preserves mitochondrial function in HMEC cells. (A and B) Total ROS levels determined at 3 h post PQ treatment with or without α-LA pretreatment. (C and D) Mitochondrial ROS levels determined at 3 h post PQ treatment with or without α-LA pretreatment. (E) Mitochondrial membrane potential measured by the JC-1 probe at 3 h post PQ treatment with or without α-LA pretreatment. (F) Total intracellular ATP levels measured at 24 h post PQ treatment with or without α-LA pretreatment. The data are presented as means ± SEM from four independent experiments. **P < 0.01 vs. the control group; #P < 0.05, ##P < 0.01 vs. the PQ group.

Discussion
Severe PQ intoxication by ingestion results in very high mortality primarily because of the damage to the lung and other vital organs.1 Previous studies have demonstrated that the microvascular endothelial barrier might be a critical cellular target of PQ toxicity. However, the underlying mechanisms of PQ-induced damage to vascular endothelial cells remain unclear. There is still a lack of antidotes and treatment regimens for PQ intoxication in clinical practice. Here, we demonstrated that: (i) PQ toxicity in microvascular endothelial cells is characterized by inhibiting the cell viability, suppressing the cell migration capacity, and inducing oxidative stress, resulting in endothelial dysfunction. (ii) α-LA is effective at antagonizing PQ toxicity through suppressing oxidative stress, thus preserving mitochondrial function. (iii) α-LA itself shows no toxic effects on endothelial cells. Taken together, our present study indicated that α-LA can be used as an antidote for PQ toxicity. This finding could be of great importance in the clinical treatment of PQ intoxication.
The endothelium comprises endothelial cells and has an intimal monolayer structure in the vessel lumen that plays a critical role as a barrier between the vessel wall and blood components.32 In the present study, we found that PQ exposure damaged the cell viability in a dose- and time-dependent manner, a finding that is consistent with that in a previous study.19 Our study further revealed that PQ intoxication not only resulted in damage to the cell migration ability but also induced endothelial dysfunction. The migration ability of endothelial cells is crucial to maintain the integrity of the endothelium barrier physiologically. Our study is the first to dynamically record and analyse how PQ exposure inhibits endothelial cell migration. In the experiments to elucidate how PQ exposure affects endothelial function, we found that PQ exposure significantly elevates the level of vasoconstrictor factor ET-1 expression and decreases the level of vasodilator factor NO generation. These toxic effects cause imbalance between vasodilation and vasoconstriction, accelerate endothelial dysfunction, destroy vascular homeostasis, and lead to increased vascular permeability. Increased vascular permeability in the lung may result in pulmonary oedema, which contributes to the development of acute lung injury and acute respiratory distress syndrome in PQ intoxicated patients.33,34 Some previous studies have indicated that PQ exposure increases the protein expression of inducible nitric oxide synthase (iNOS) but does not alter eNOS expression in lung aortas.35,36 However, some studies have shown that PQ induces the upregulation of NO or nitrite concentrations. The reasons for the discrepancies among different studies may be due to the different types of cells with different basal states of iNOS expression. Therefore, the precise mechanisms of regulating endothelial function in the presence of PQ exposure need to be further explored.
In exploring the efficacy of α-LA to antagonize PQ toxicity, we used a significant toxic dose of 1000 μM PQ to induce cell damage after the dose-effect and time-course studies on cell viability. Growing evidence has indicated that α-LA, as a natural antioxidant, has great potential in the prevention and therapy of various diseases, especially chronic illnesses dependent on the oxidative stress. α-LA is also generally considered as a non-toxic endogenous compound. It was proven that α-LA is effective at curing various diseases such as diabetes mellitus and its complications, vascular diseases, hypertension, and neurodegeneration diseases.12,37 Considering the potent antioxidative efficacy of α-LA and well-defined ability of PQ to induce cellular oxidative stress, we speculated that α-LA can provide protection against PQ toxicity via suppressing oxidation. Some reports have studied the protective effects of α-LA against PQ toxicity in human bronchial epithelial cells or Drosophila melanogaster with doses of 200 μM or 430 μM, respectively.16,17 The dose of α-LA used in human bronchial epithelial cells was at the same level of our present study. Our results also suggested that α-LA, at high concentrations, shows no toxic effects on HMEC-1 cells. The protective effects of α-LA against PQ toxicity are characterized by increasing cell viability, preserving cell migration ability, maintaining mitochondrial function and reducing cellular oxidative stress. These results revealed the pharmacological potential of α-LA to restore the microvascular endothelial barrier, maintain endothelial homeostasis, and ameliorate acute PQ toxicity. Regarding the mechanisms of action of α-LA against oxidative stress, some previous investigations have indicated that α-LA has direct and indirect antioxidant properties. The direct antioxidant effects are attributed to scavenging reactive oxygen species (ROS) and reactive nitrogen species (RNS). The indirect actions are due to promoting endogenous antioxidant regeneration and repairing oxidized proteins.38,39 Our present study showed that α-LA increases eNOS expression and elevates NO production, a finding that was consistent with previous studies.40,41 Based on these data, we speculated that α-LA not only significantly increases the NO content through enhancing eNOS expression but also scavenges superoxide anions, decreasing peroxynitrite anion (ONOO–) production to defend the PQ toxicity.
PQ exposure produces oxidative stress by redox cycling, which is the generally accepted mechanism of toxicity.42 Mitochondria are the primary site for superoxide anion production and are susceptible to the oxidative attack. The injured mitochondria could further exacerbate ROS production and oxidative stress. Therefore, the protection of mitochondrial function could serve as an important target to antagonize PQ toxicity. In our present study, we found that PQ exposure not only significantly elevates oxidative stress but also induces mitochondrial dysfunction. α-LA significantly attenuates PQ-induced intracellular and mitochondrial oxidative stress and restores the levels of mitochondrial membrane potential and ATP production. Preservation of mitochondrial function is an effective strategy to antagonize PQ-induced vascular endothelial dysfunction.
Conclusions
Our present study provided new evidence for understanding the characteristics of PQ cytotoxicity in microvascular endothelial cells. Our results highlighted the critical role of oxidative stress in PQ-induced endothelial dysfunction. α-LA could effectively protect against PQ-induced toxicity via suppressing oxidative stress and maintaining mitochondrial function. Our study suggested that α-LA, as a potential therapeutic antioxidant, deserves to be further investigated for clinical use against acute PQ poisoning.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81872596) and Open Grants from the Key Laboratory of Electromagnetic Radiation Protection, Ministry of Education, China (No. 2017DCKF005). We thank Dr H–F Pi for his constructive advice on experimental design, Dr C–H Chen for his instruction on statistical analysis and Dr Lei Zhang for her help in biochemical analysis.
References
- Dinis-Oliveira R. J., Duarte J. A., Sanchez-Navarro A., Remiao F., Bastos M. L., Carvalho F. Crit. Rev. Toxicol. 2008;38:13–71. doi: 10.1080/10408440701669959. [DOI] [PubMed] [Google Scholar]
- Novaes R. D., Goncalves R. V., Cupertino M. C., Santos E. C., Bigonha S. M., Fernandes G. J., Maldonado I. R., Natali A. J. Int. J. Exp. Pathol. 2016;97:114–124. doi: 10.1111/iep.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunnapuk K., Mohammed F., Gawarammana I., Liu X., Verbeeck R. K., Buckley N. A., Roberts M. S., Musuamba F. T. Br. J. Clin. Pharmacol. 2014;78:855–866. doi: 10.1111/bcp.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B., Chen F., Xu L., Xing J., Wang X. Sci. Rep. 2017;7:597. doi: 10.1038/s41598-017-00721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. C., Tsai C. P., Lee C. L., Chen S. Y., Lin G. J., Yen M. H., Sytwu H. K., Chen S. J. Clin. Sci. 2013;125:329–340. doi: 10.1042/CS20120560. [DOI] [PubMed] [Google Scholar]
- Pashaj A., Xia M., Moreau R. Can. J. Physiol. Pharmacol. 2015;93:1029–1041. doi: 10.1139/cjpp-2014-0480. [DOI] [PubMed] [Google Scholar]
- Xia D., Zhai X., Wang H., Chen Z., Fu C., Zhu M. J. Cell. Mol. Med. 2019;23:4088–4096. doi: 10.1111/jcmm.14296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez O., Gonzalez-Castaneda R. E. Nutr. Res. 2006;26:1–5. doi: 10.1016/j.nutres.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atukeren P., Aydin S., Uslu E., Gumustas M. K., Cakatay U. Oxid. Med. Cell. Longevity. 2010;3:206–213. doi: 10.4161/oxim.3.3.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Yang J., Chen J., Zhao S., Wang T., Zou H., Wang Y., Gu J., Liu X., Bian J., Liu Z. Toxicology. 2019;414:1–13. doi: 10.1016/j.tox.2018.12.005. [DOI] [PubMed] [Google Scholar]
- Rochette L., Ghibu S., Richard C., Zeller M., Cottin Y., Vergely C. Mol. Nutr. Food Res. 2013;57:114–125. doi: 10.1002/mnfr.201200608. [DOI] [PubMed] [Google Scholar]
- Gomes M. B., Negrato C. A. Diabetol. Metab. Syndr. 2014;6:80. doi: 10.1186/1758-5996-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay K. P., Moreau R. F., Smith E. J., Smith A. R., Hagen T. M. Biochim. Biophys. Acta. 2009;1790:1149–1160. doi: 10.1016/j.bbagen.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer T., Finckh B., Albers S., Krohn K., Kohlschutter A., Meinertz T. Free Radical Biol. Med. 2001;31:53–61. doi: 10.1016/s0891-5849(01)00551-2. [DOI] [PubMed] [Google Scholar]
- Sena C. M., Nunes E., Louro T., Proenca T., Fernandes R., Boarder M. R., Seica R. M. Br. J. Pharmacol. 2008;153:894–906. doi: 10.1038/sj.bjp.0707474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla E., Medina-Leendertz S., Villalobos V., Molero L., Bohórquez A. Neurochem. Res. 2006;31:1425–1432. doi: 10.1007/s11064-006-9194-8. [DOI] [PubMed] [Google Scholar]
- Kim Y. S., Podder B., Song H. Y. Biol. Pharm. Bull. 2013;36:802–811. doi: 10.1248/bpb.b12-00977. [DOI] [PubMed] [Google Scholar]
- Tsukamoto M., Tampo Y., Sawada M., Yonaha M. Toxicol. Appl. Pharmacol. 2002;178:82–92. doi: 10.1006/taap.2001.9325. [DOI] [PubMed] [Google Scholar]
- Huang Y., He Q. Environ. Toxicol. Pharmacol. 2017;52:62–68. doi: 10.1016/j.etap.2017.01.023. [DOI] [PubMed] [Google Scholar]
- Fukai T., Ushio-Fukai M. Antioxid. Redox Signaling. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incalza M. A., D'Oria R., Natalicchio A., Perrini S., Laviola L., Giorgino F. Vasc. Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Godo S., Shimokawa H. Free Radical Biol. Med. 2017;109:4–10. doi: 10.1016/j.freeradbiomed.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Li H., Forstermann U. Curr. Opin. Pharmacol. 2013;13:161–167. doi: 10.1016/j.coph.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Oparka M., Walczak J., Malinska D., van Oppen L., Szczepanowska J., Koopman W. J. H., Wieckowski M. R. Methods. 2016;109:3–11. doi: 10.1016/j.ymeth.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Chen H., Lu Y., Cao Z., Ma Q., Pi H., Fang Y., Yu Z., Hu H., Zhou Z. Toxicol. Lett. 2016;246:7–16. doi: 10.1016/j.toxlet.2016.01.014. [DOI] [PubMed] [Google Scholar]
- Kojima H., Urano Y., Kikuchi K., Higuchi T., Hirata Y., Nagano T. Angew. Chem., Int. Ed. 1999;38:3209–3212. doi: 10.1002/(sici)1521-3773(19991102)38:21<3209::aid-anie3209>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Michaelis U. R. Cell. Mol. Life Sci. 2014;71:4131–4148. doi: 10.1007/s00018-014-1678-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitorino P., Meyer T. Genes Dev. 2008;22:3268–3281. doi: 10.1101/gad.1725808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Vizcaino F., Duarte J., Andriantsitohaina R. Free Radical Res. 2006;40:1054–1065. doi: 10.1080/10715760600823128. [DOI] [PubMed] [Google Scholar]
- Castello P. R., Drechsel D. A., Patel M. J. Biol. Chem. 2007;282:14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman R. J., Elstner E. F. FEBS Lett. 1981;129:265–268. doi: 10.1016/0014-5793(81)80180-9. [DOI] [PubMed] [Google Scholar]
- Williamson M. R., Shuttleworth A., Canfield A. E., Black R. A., Kielty C. M. Biomaterials. 2007;28:5307–5318. doi: 10.1016/j.biomaterials.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Maniatis N. A., Kotanidou A., Catravas J. D., Orfanos S. E. Vasc. Pharmacol. 2008;49:119–133. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orfanos S. E., Mavrommati I., Korovesi I., Roussos C. Intensive Care Med. 2004;30:1702–1714. doi: 10.1007/s00134-004-2370-x. [DOI] [PubMed] [Google Scholar]
- Zocrato L. B., Capettini L. S., Rezende B. A., Silva J. F., Rodrigues-Machado Mda G., Cortes S. F., Lemos V. S. Toxicol. in Vitro. 2010;24:1019–1025. doi: 10.1016/j.tiv.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Aires R. D., Capettini L. S., Silva J. F., Rodrigues-Machado Mda G., Pinho V., Teixeira M. M., Cortes S. F., Lemos V. S. PLoS One. 2013;8:e73562. doi: 10.1371/journal.pone.0073562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H., Young Park H., Lee H. S., Han C. Y., Lee S. Eur. Rev. Med. Pharmacol. Sci. 2019;23:2587–2595. doi: 10.26355/eurrev_201903_17408. [DOI] [PubMed] [Google Scholar]
- Smith A. R., Shenvi S. V., Widlansky M., Suh J. H., Hagen T. M. Curr. Med. Chem. 2004;11:1135–1146. doi: 10.2174/0929867043365387. [DOI] [PubMed] [Google Scholar]
- Petersen Shay K., Moreau R. F., Smith E. J., Hagen T. M. IUBMB Life. 2008;60:362–367. doi: 10.1002/iub.40. [DOI] [PubMed] [Google Scholar]
- Ying Z., Xie X., Chen M., Yi K., Rajagopalan S. Vasc. Pharmacol. 2015;64:28–35. doi: 10.1016/j.vph.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badran M., Abuyassin B., Golbidi S., Ayas N., Laher I. Oxid. Med. Cell. Longevity. 2019;2019:4093018. doi: 10.1155/2019/4093018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Ayala T., Andérica-Romero A. C., Pedraza-Chaverri J. Free Radic. Res. 2014;48:623–640. doi: 10.3109/10715762.2014.899694. [DOI] [PubMed] [Google Scholar]