

Abstract
N6-methyladenosine (m6A) RNA modification regulates multiple biological functions. Methyltransferase like 3 (METTL3), one of the major N6-methyltransferases, is highly expressed in gastric cancer, but its potential role in disease is unclear. The current study knocked out METTL3 (METTL3-KO) in human gastric cancer AGS cells using CRISPR/Cas9. METTL3-KO AGS cells exhibited decreased m6A methylation levels. A significant inhibition of cell proliferation was observed in METTL3-KO AGS cells. Silencing METTL3 in AGS cells altered the expression profile of many effector molecules that were previously demonstrated to serve key roles in AGS cell proliferation, including the suppressor of cytokine signaling (SOCS) family of proteins. The results further demonstrated that SOCS2 upregulation in METTL3-KO AGS cells was associated with a decreased RNA decay rate. Furthermore, SOCS2 KO or SOCS2 overexpression caused a significant increase and decrease in AGS cell proliferation, respectively. The current data suggested that METTL3-KO in gastric cancer cells resulted in the suppression of cell proliferation by inducing SOCS2, suggesting a potential role of elevated METTL3 expression in gastric cancer progression.
Keywords: m6A modification, methyltransferase like 3, suppressor of cytokine signaling 2, gastric cancer, cell proliferation
Introduction
Increasing evidence supports that RNA methylation is a widespread phenomenon and a critical regulator of transcript expression (1,2). The most prevalent RNA methylation, N6-methyladenosine (m6A), is a reversible RNA post-transcriptional modification and occurs in approximately 25% of transcripts at the genome-wide level (1). RNA m6A modification regulates RNA splicing, translocation, stability, and translation into protein (3–6). Dynamic regulation of the m6A epitranscriptome is involved in diverse cellular functions, including heat shock, DNA damage, cancer, stem cell differentiation, circadian rhythm, spermatogenesis and oogenesis, response to interferon-γ, and viral infections (2,3,7).
m6A dynamics and functions are executed by three groups of proteins: Methyltransferases or ‘writers’, demethylases or ‘erasers’, and m6A-binding proteins or ‘readers’ (2,3,7). In most cell types, m6A modification is catalyzed by the methyltransferase complex consisting of the methyltransferase like 3 (METTL3) and METTL14 and their cofactors (3,7). The erasers include the fat mass and obesity-associated protein (FTO) and alpha-ketoglutarate-dependent dioxygenase alkB homolog 3 (ALKBH3) and ALKBH5 (2,3,7). Increasing evidence supports that m6A modification may play an important role in tumorigenesis (8,9). For example, METTL3 has been found to be upregulated in human glioblastoma tissues, and high levels of METTL3 expression in tumor tissues predict poor patient survival (10). Suppression of METTL3 or METTL14 decreases hepatocellular carcinoma cell proliferation, migration and colony formation in vitro, as well as hepatocellular carcinoma tumorigenicity and lung metastasis in vivo (11). FTO is over-expressed in human cervical squamous cell carcinoma tissues, and high levels of FTO expression correlate with poor patient prognosis (12). ALKBH5 is highly expressed in glioblastoma stem-like cells and demethylates FOXM1 nascent transcripts, leading to FOXM1 over-expression, stem-like cell proliferation and tumorigenesis (13).
Gastric cancer is one of the most prevalent and deadly malignancies that threatens global health (14). Previous studies demonstrated that the expression level of METTL3 is elevated in many gastric cancer cell lines and tumor tissues (15,16). The elevated level of METTL3 expression is clinically correlated with the procession of gastric cancer (15,16). Results from previous studies indicate that gastric cancer cell proliferation is associated with aberrant expression of various effector molecules, such as leucine rich repeat containing G protein coupled receptor 5 (LGR5), RAD17 checkpoint clamp loader component (RAD17), facilitated trehalose transporter Tret1-2 homolog (TRET1-2), ATPase Na+/K+ transporting subunit beta 1 (ATP1B1), matrix metallopeptidase 3 (MMP3), HEPANAS_3, interferon induced transmembrane protein 3 (IFITM3), and S100 calcium binding protein A4 (S100A4) (17–19). In addition, increasing evidence supports that the activity of the suppressor of cytokine signaling (SOCS) family proteins correlates with the progression and poor prognosis in various cancers, including gastric cancer (20). SOCS2 is well defined as a negative feedback regulator in multiple proliferation-related pathways and may act as a tumor suppressor in multiple malignancies (21–26). However, the role of METTL3 in gastric cancer progression and whether METTL3 can modulate SOCS expression to regulate gastric cancer cell proliferation are still not fully understood. In this study, we report that upregulation of METTL3 in gastric cancer may maintain gastric cancer tumorigenicity through suppressing SOCS2 to promote cell proliferation.
Materials and methods
Gastric cancer cell line culture
The AGS cells (the gastric cancer cell line) were purchased from American Type Culture Collection (ATCC). Cells were cultured in the medium with L-15 medium supplemented with 10% fetal bovine serum and 100 U penicillin/streptomycin as recommended.
CRISPR-Cas9 knockout METTL3
We took the CRISPR/Cas9 approach to knock out the METTL3 gene (NCBI Gene ID 56339) in AGS cells and generate stable cell lines. AGS cells were transfected with the METTL3 CRISPR/Cas9 and Homology-Directed Repair (HDR) plasmids (Santa Cruz Biotech, Inc.). Stably transfected cells were selected, collected and further confirmed and validated by real-time PCR and Western blot analysis, as in our previous studies (27).
Cell transfection
The specific siRNA to SOCS2 (si-SOCS2) was obtained from Santa Cruz Biotech, Inc. (sc-40998). A non-specific siRNA of a scrambled sequence from Santa Cruz was used as the control. siRNA was mixed with Lipofectamine RNAiMax (Thermo Fisher Scientific) and transfected into cells, as we previously reported (28). The pCMV6 plasmid with full-length SOCS2 sequence (pCMV6-SOCS2) was purchased from Origene (RC203163). The pCMVv6 empty vector from Origene was used as the control. AGS cells were transfected with the plasmid via Lipofectamine 2000 (Thermo Fisher Scientific) (28). Knockdown or overexpression of SOCS2 in AGS cells was validated by real-time PCR at 48 h after transfection.
Cell proliferation and apoptotic death assays
AGS cells were initially seeded in 96-well plate at a density of 1×103 cells/well. At various culture hours of incubation after seeding, the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent was added cells (Promega) for MTS assay. After 1 h of incubation, the OD490 value was measured with a SpectraMax (Molecular Devices Corporation). AGS proliferation was also measured by direct count of cell numbers. Cells were plated at 1×104 per well in 12-well plate. At various culture hours of incubation after seeding, cells were trypsinized and total numbers were counted in a double-blind manner. The Annexin V-FITC apoptosis staining/detection kit (Abcam) was used to detect apoptotic cell death.
Real-time PCR
RNA expression levels were quantified by using real-time PCR. Total RNA was extracted with the Trizol reagent (Invitrogen). A total of 500 ng RNA was used for reverse transcription (Invitrogen). PCR reactions were performed using the SYBR Green reagent (Bio-Rad) on a Bio-Rad CFX96 platform. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was utilized as internal control. Each sample was run in triplicate. The relative abundance of each RNA was calculated using the 2−ΔΔCt method and normalized to GAPDH as we previously reported (27,28). The sequences of all the primers were listed in Table I.
Table I.
The sequences of real-time PCR primers.
| Primer | Sequence (5′-3′) |
|---|---|
| METTL3-F | CTTTGCCAGTTCGTTAGTCTC |
| METTL3-R | CTGACCTTCTTGCTCTGTTGT T |
| SOCS1-F | TCTGTAGGATGGTAGCACACA |
| SOCS1-R | GGAAGAGGAGGAAGGTTCTG |
| SOCS3-F | GACGATAGCAACCACAAGTG |
| SOCS3-R | AGATTCCCTGGCAGTTCTC |
| SOCS2-si-F | AGGGAATGGCAGAGACACT |
| SOCS2-si-R | TGGCAGAGAGAGAAGGGAT |
| SOCS2-OE-F | CGAATACCAAGACGGAAA |
| SOCS2-OE-R | CAGATGAACCACACTGTCA |
| MMP7-F | ATGAGGATGAACGCTGGA |
| MMP7-R | TGTCCCATACCCAAAGAATG |
| S100A4-F | AGAGGGTGACAAGTTCAAGC |
| S100A4-R | GTCCAAGTTGCTCATCAGC |
| TERT-F | GCTCCCATTTCATCAGCA |
| TERT-R | CTGCGTTCTTGGCTTTCA |
| Rad17-F | TGCCATACCTTGCTCTACTAAC |
| Rad17-R | TTCAATCTTCCAAAGTGTCG |
| Heparanase-F | TGCTATCCGACACCTTTGC |
| Heparanase-R | CTTGCCTCATCACCACTTCTAT |
| MMP7-F | CCAACCTATGGAAATGGAGA |
| MMP7-R | GAATGGATGTTCTGCCTGA |
| ATP1B1-F | TCAAACCTAAGCCTCCCA |
| ATP1B1-R | CTCCACATTTCCAACTTTATCC |
| IFITM3-F | TCGCCTACTCCGTGAAGTCT |
| IFITM3-R | GGATGACGATGAGCAGAATG |
| LGR5-F | CTACATGGTCGCTCTCATCTTG |
| LGR5-R | ATATTCTCCAGGTCTCCCTTGTC |
| GAPDH-F | TGCACCACCAACTGCTTAGC |
| GAPDH-R | GGCATGGACTGTGGTCATGAG |
F, forward, R, reverse.
Western blot
AGS cell cultures were harvested and lysed with the M-PER mammalian protein extraction reagent (Thermo Fisher Scientific). Samples were run in 10% acrylamide gel (30 µg total protein/well) and transferred to the PVDF membranes (Millipore). Membranes were then subjected to the following primary antibody at 4°C overnight: Anti-METTL3 (1:1,000, 15073-1-AP; Proteintech), anti-SOCS1 (Proteintech), anti-SOCS2 (1:1,000, 2779, CST, MA, USA), anti-SOCS3 (Proteintech), anti-tyrosine phosphorylation (Santa Cruz Biotech Inc.), anti-beta actin (Santa Cruz Biotech Inc.) or anti-GAPDH (Santa Cruz Biotech Inc.). Details for Western blot are as described in our previous studies (28,29).
m6A quantification
Total RNA extracted from cells was purified as described above for real-time PCR analysis. The rRNA and noncoding RNAs were removed using the Dynabeads mRNA Purification kit (Thermo Fisher Scientific). A total of 200 ng mRNA of each sample was used for m6A quantification using the EpiQuik m6A Methylation Quantification kit (Colorimetric; Epigentek), according to the manufacturer's protocol.
RNA stability
AGS cells were seeded in 12-well plates at a density of 2×104/well. After overnight adhesion, cells were treated with actinomycin (A9415; Sigma Aldrich) at a final concentration of 10 µg/ml. Cells were collected and total RNA isolated at different time points after actinomycin treatment (0, 30 min or 2 h). Expression levels of specific genes of interest were quantified by using real-time PCR.
Statistical analysis
All experiments were performed as three independent replications. Experiment data were presented as the mean ± SEM. Student's t-test (for differences between two groups) or one-way ANOVA (for multiple group comparisons) was used to analyze the data using SPSS software (v20.0; SPSS, Inc.). Tukey post hoc tests were conducted following one-way ANOVA to determine significant differences between specific groups in datasets containing 3 or more groups. P<0.05 was considered to be statistically significant.
Results
The establishment of stable METTL3-knockout (METTL3-KO) gastric cancer cell lines
We took the CRISPR-Cas9 approach to generate stable knockout gastric cancer cells using the well-documented human AGS cell line. Two generated cell lines of METTL3-KO, METTL3-KO-C20 and METTL3-KO-C43, were generated and knockout of METTL3 in the cells was confirmed by using real-time PCR and Western blot (Fig. 1A and B). No real-time PCR product was detected in the KO cells using the primer sets covering the targeted region of METTL3 (Fig. 1A). Western blot showed a decrease in signaling of METTL3 band in the METTL3-KO-C20 cells and a truncated form of METTL3 protein for the METTL3-KO-C43 cells (Fig. 1B). A significant decrease in the global m6A levels was detected in METTL3-KO-C20 and METTL3-KO-C43 cells, compared with the wild-type AGS cells (Fig. 1C).
Figure 1.
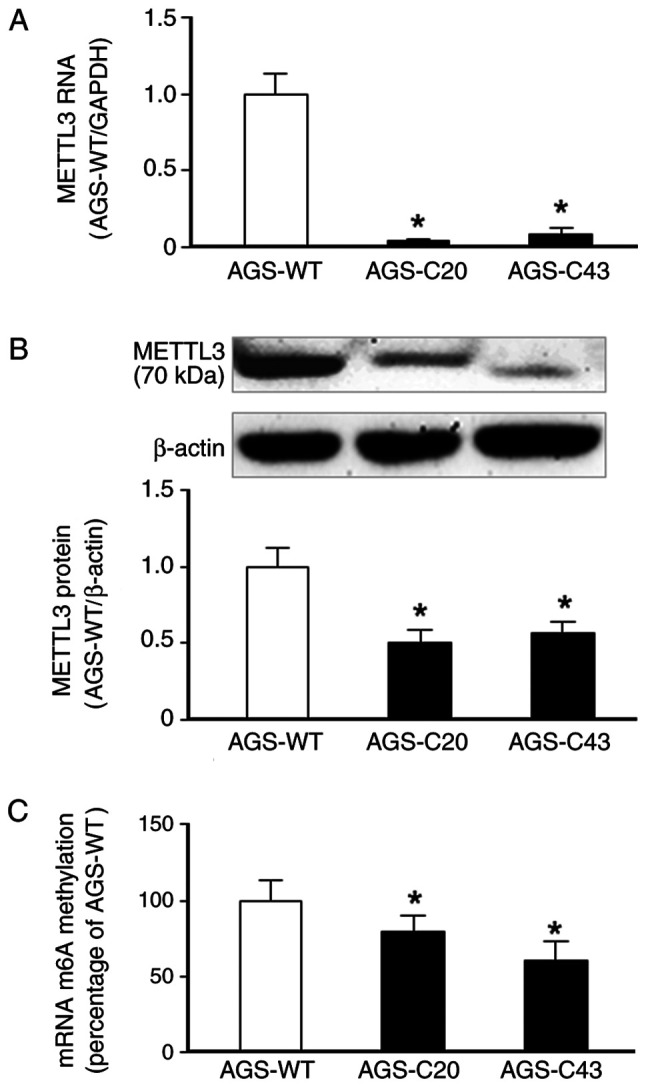
Establishment of METTL3-KO AGS cells. AGS cells were transfected with METTL3 CRISPR/Cas9 and homology-directed repair plasmids. Colonies were selected and confirmed by detecting METTL3 expression and global m6A levels. (A) Stable METTL3-KO AGS cells (AGS-C20 and AGS-C43) were selected and METTL3-KO was confirmed via PCR analysis of METTL3 RNA. (B) Stable METTL3-KO AGS cells (AGS-C20 and AGS-C43) exhibited a significant decrease or a truncated expression of METTL3 protein, as determined via western blotting. Representative images are presented. Densitometric levels of positive bands were quantified and are expressed as the ratio to β-actin. (C) A significant decrease in global m6A levels was detected in METTL3-KO AGS cells. *P<0.05 vs. AGS-WT. METTL3-KO, methyltransferase like 3 knocked down; WT, wild type.
METTL3-KO cells elicit an inhibitory effect on cell proliferation
We then examined the effects of METTL3 KO on AGS cell proliferation. The METTL3-KO-C20 and -C43 cells showed a decrease in cell growth as revealed by cell number counting and by MTS assay, respectively (Fig. 2A and B). Decrease in cell number of METTL3-KO AGS cells is not due to increased cell death as no significant difference in apoptotic cell death was observed in the wild-type and METTL3-KO cells (data not shown). These data suggest that METTL3 may play an important role in regulating AGS cell proliferation.
Figure 2.

Inhibition of METTL3-KO AGS cell proliferation. AGS-WT and AGS METTL3-KO cells were cultured for up to 96 h. (A) METTL3-KO C20 and C43 cells demonstrated a reduced proliferation rate as determined via cell counting. (B) The MTS assay revealed a decreased cell proliferation in METTL3-KO-C20 and -C43 AGS cells. *P<0.01 vs. AGS-WT. METTL3-KO, methyltransferase like 3 knocked down; WT, wild type; OD, optical density.
Altered expression pattern of effector molecules associated in cell proliferation in METTL3 KO AGS cells
To explore the underlying mechanisms of METTL3-mediated cell proliferation suppression in AGS cells, we asked if METTL3 can affect the expression levels of these effector molecules that are associated with gastric cancer cell proliferation demonstrated from previous studies (17–19). We screened the expression levels of these effector molecules in our METTL3-KO AGS cells, compared with that in the wild-type cells. Interestingly, many of them showed an increased expression level in METTL3-KO-C20 cells (Fig. 3A) and METTL3-KO-C43 cells (data not shown). Whereas a decrease in HEPANAS_3 was detected in the METTL3-KO AGS cells, no detectable changes in IFITM3 and S100A4 expression levels was observed between METTL3-KO cells and wild-type AGS cells (Fig. 3A). The above data suggest that METTL3-mediated RNA methylation may impact the expression of genes that can subsequently influence AGS cell proliferation.
Figure 3.

Expression of effector molecules associated with AGS cell proliferation in METTL3-KO AGS cells. (A) Expression of selected genes associated with AGS cell proliferation were measured using reverse transcription-quantitative PCR. Several of these genes, including SOCS1, SOCS2, SOCS3 and LGR5, exhibited decreased expressions in METTL3-KO cells when compared with METTL3-WT cells. (B) Inhibition of RAD17, LGR5 and SOCS2 RNA stability in METTL3-KO AGS cells. AGS-WT and AGS METTL3-KO cells were cultured in the presence of actinomycin for 2 h. Levels of RAD17, LGR5 and SOCS2 were measured in cells following actinomycin treatment and compared with cells tested prior to actinomycin administration. Reduced RNA levels represents degradation rate. A lower degradation rate was observed for RAD17, LGR5 and SOCS2 in METTL3-KO AGS cells compared with AGS-WT. *P<0.01 vs. AGS-WT. METTL3-KO, methyltransferase like 3 knocked down; WT, wild type; SOCS, suppressor of cytokine signaling; LGR5, leucine-rich repeat-containing G-protein coupled receptor 5; RAD17, RAD17 checkpoint clamp loader component.
We then aimed to explore the underlying molecular mechanisms by which METTL3 may control expression of these genes. Previous studies show that RNA m6A methylation exerts its function via modulation of RNA stability (4). To investigate this potential, we carried out the RNA stability assay. After blocking new RNA synthesis with actinomycin, a slower decay rate for RAD17, LGR5 and SOCS2 RNAs was observed in METTL3-KO AGS cells (Fig. 3B), suggesting modulation of their stability through m6A modification.
Aberrant expression of SOCS2 may contribute to METTL3-mediated cell proliferation in AGS cells
Given the fact that several SOCS family members are among these effector molecules that are upregulated in the METTL3-KO AGS cells (Fig. 3), we hereafter focused on the SOCS family members to explore their potential role in METTL3-associated AGS cell proliferation. To further confirm the expression of SOCSs associated with METTL3 in AGS cells, we expanded our measurement of SOCS1, SOCS2, and SOCS3 at both RNA level and protein levels in METTL3-KO cells. The expression of SOCS1, SOCS2, and SOCS3 at the RNA level was significantly higher in the METTL3-KO cells, compared with that in the wild-type AGS cells (Fig. 4A-C). Nevertheless, at the protein level, only SOCS2, but not SOCS1 and SOCS3, showed an increased content in the METTL3-KO cells, compared with that in the wild-type AGS cells (Fig. 4D-F).
Figure 4.

Expression of SOCS1, SOCS2 and SOCS3 at the RNA and protein levels in METTL3-WT and METTL3-KO AGS cells. Elevated RNA expressions of (A) SOCS1, (B) SOCS2 and (C) SOCS3 were detected in METTL3-KO AGS cells, as determined via reverse transcription-quantitative PCR and compared with METTL3-WT AGS cells. METTL3-KO resulted in increased levels of (E) SOCS2, but not (D) SOCS1 and (F) SOCS3 (F) protein in AGS cells, as determined via western blotting and compared with METTL3-WT AGS cells. β-actin was also immunoblotted to ensure equal loading of proteins to each lane. Representative blotting images are presented. Densitometric levels of positive bands for SOCS1, SOCS2 and SOCS3 were quantified and are expressed as the ratio to β-actin. *P<0.01 vs. AGS-WT. SOCS, suppressor of cytokine signaling; METTL3, methyltransferase like 3; WT, wild type; KO, knocked down.
To further exclude the potential involvement of SOCS1 and SOCS3 in METTL3-mediated cell proliferation, we measured the activation of SOCS1 and SOCS3 in METTL3-KO AGS cells. One of its key downstream transcription factors for the SOCS signaling is the STAT family proteins (30). The activation of the STAT signaling pathway involves tyrosine phosphorylation of STAT proteins (31). We then measured by Western blot the expression of STAT1 and STAT3 at the protein level in METTL3-KO AGS cells in response to IL6 stimulation. No detectable changes in the protein content of STAT1 (Fig. 5A) and STAT3 (Fig. 5B) was observed in METTL3-KO AGS cells following IL-6 stimulation, compared with the wild-type cells. Moreover, we also blotted for the tyrosine phosphorylation ratio relative to the total protein levels in METTL3-KO AGS cells. We failed to detect any difference in the tyrosine phosphorylation status of STAT1 and STAT3 in IL-6-treated MELLT3-KO cells, compared with that in the wild-type AGS cells (Fig. 5A and B).
Figure 5.

Tyrosine phosphorylation of STAT1 and STAT3 in METTL3-WT and METTL3-KO AGS cells. METTL3-WT and METTL3-KO AGS cells were collected after IL-6 stimulation for up to 60 min, followed by Western blotting for total and tyrosine phosphorylation of STAT1 and STAT3. No detectable changes in the total and tyrosine phosphorylation of (A) STAT1 and (B) STAT3 were detected in METTL3-KO AGS cells following IL-6 stimulation compared with WT AGS cells. Representative blotting images from three independent experiments are presented. STAT, signal transducer and activator of transcription; METTL3, methyltransferase like 3; WT, wild type; KO, knocked down; IL, interleukin; p, phosphorylated; t, total.
Given the significant increase in SOCS2 protein level in METTL3-KO in AGS cells, we further tested the potential role of SOCS2 in METTL3-mediated cell proliferation by manipulating its expression. We took the RNAi approach to knockdown SOCS2 and used the pCMV6-SOCS2 plasmid to transfect cells to upregulate SOCS2 expression in AGS cells (Fig. 6A). Accordingly, a significant increase or decrease in cell proliferation was detected in the SOCS2 siRNA-treated AGS cells and cells overexpressing SOCS2, respectively (Fig. 6B), further supporting the role of SOCS2 in regulating AGS cell proliferation.
Figure 6.

Manipulation of SOCS2 expression alters the proliferation of AGS cells. Knockdown of SOCS2 in AGS-WT cells promoted cell proliferation in AGS cells. Cells were treated with SOCS2-siRNA to knock down SOCS2. Cells treated with a non-specific control siRNA were used as controls. AGS cell treatment with SOCS2-siRNA significantly knocked down SOCS2 expression in AGS-WT cells at (A) RNA and (B) protein levels. *P<0.05 vs. Ctrl siRNA. (C) A significantly higher cell proliferation rate was detected by using an MTS assay in cells treated with siRNA-SOCS2 when compared with cells treated with Ctrl-siRNA. *P<0.05 vs. Ctrl siRNA. Overexpression of SOCS2 inhibited cell AGS cell proliferation. Cells were transfected with pCMV6-SOCS2 (full-length-SOCS2) to overexpress SOCS2. Cells treated with the pCMV6 empty vector were used as controls. SOCS2 overexpression in AGS-WT cells following transfection of pCMV6-SOCS2 was confirmed via reverse transcription-quantitative PCR and western blotting, respectively, at (D) RNA and (E) protein levels. *P<0.05 vs. pCMV6-Ctrl. A significantly lower cell proliferation rate was detected using an MTS assay in cells transfected with pCMV6-SOCS2 compared with pCMV6 empty vector-treated cells (F) Representative blotting images are presented in B and E. Densitometric levels of positive band for SOCS2 were quantified and are expressed as the ratio to GAPDH. *P<0.05 vs. pCMV6-Ctrl. SOCS, suppressor of cytokine signaling; WT, wild type; siRNA, small interfering RNA; Ctrl, control; OD, optical density.
Discussion
m6A modification is the most prevalent internal modification decorating RNA molecules and may play a critical regulatory role in a series of biological processes, such as RNA decay, splicing, translation and transportation (3–6). Emerging evidence indicates that the cellular status of m6A modification may impact the pathologic processes of various cancers (8,9,32). METTL3, a major writer of m6A modification, is often upregulated in numerous malignancies and can epigenetically silence specific gene expression, eliciting a potential oncogenic function (11,33). Previous studies demonstrated that expression levels of METTL3 can critically change the cell cycle and apoptosis processes in hepatic cancer and acute myeloid leukemia (11,34). METTL3 is usually upregulated in gastric cancer, which should maintain a higher m6A RNA modification level in tumor cells. In this report, we demonstrate that knockdown of METTL3 in gastric cancer cells results in suppression of cell proliferation through induction of SOCS2, suggesting a potential role for elevated METTL3 expression in gastric cancer progression.
Given the elevated expression level of METTL3 in gastric cancer tissues (15,16), we took the CRISPR/Cas9 approach to generate stable cell lines of gastric cancer cells deficient in METTL3 function. The CRISPR/Cas9 approach may provide several advantages over the siRNA approach, as it may be more specific and can generate stable KO cells (35). Of these stable METTL3-KO cells that generated from this approach, we selected two clones, METTL3-KO-C20 and METTL3-KO-C43, which showed an almost complete deletion of METTL3 RNA expression as evident by the PCR analysis using the primer set covering the CRISP/Cas9 target region of METTL3. Interestingly, Western blot indicated a significant decrease in signaling of METTL3 band in the METTL3-KO-C20 cells and a truncated form of METTL3 protein for the METTL3-KO-C43 cells. Nevertheless, both cell lines demonstrated a significant suppression of global m6A methylation, compared with the wild-type cells. Functionally, knockdown of METTL3 significantly reduced AGS cell proliferation.
The process of AGS cell proliferation involves a complex regulatory network of multiple signaling pathways, involving key effector molecules, such as SOCS1, SOCS2, SOCS3, LGR5, RAD17, TRET_2, ATP1B1, MMP7_3, HEPANAS_3, IFITM3, and S100A4 (17–19). Interestingly, knockdown of METTL3 in AGS cells showed an increased expression level for many of these effector molecules, implicating that METTL3-mediated RNA methylation may influence the expression of key components of multiple signaling pathways to modulate AGS cell proliferation. Regulation of RNA stability, particularly for SOCS2, may be involved in METTL3-mediated gene expression associated with AGS cell proliferation.
SOCS2, a member of the SOCS family that regulates multiple cytokine-induced intracellular signal pathways, is involved in the regulation of numerous biological processes, including immune responses (36,37). SOCS2 can be activated through tyrosine phosphorylation and may act as a downstream factor of the JAK/STAT pathway, providing a negative feedback regulation to this signaling pathway (38). Indeed, the JAK/STAT signal pathway is often dysregulated in gastric cancer (39). The JAK/STAT signaling pathway participates in multiple cancerous cellular processes: apoptosis, proliferation, metastasis and inflammation (40–43). Aberrant expression of SOCS2 is of important pathologic significance in development of many diseases (38,44). Overexpression of SOCS2 showed a proliferation-suppression effect in gastric cancer cells (45). In this study using AGS cells in culture, we failed to detect any difference in the tyrosine phosphorylation status of STAT1 and STAT3 in the MELLT3-KO AGS cells, compared with that in the wild-type cells. Instead, we observed the negative association of SOCS2 and cell proliferation in gastric cancer cells. Knockdown of METTL3 resulted in an increased expression of SOCS2, accompanied with a decreased cell proliferation in AGS cells. Experimentally induced suppression or forced expression of SOCS2 caused reciprocal alterations in AGS cell proliferation, indicating that SOCS2 downregulation, at least partially, contributes to METTL3-mediated cell proliferation in cultured AGS cells. Several recent reports indicate that METTL3 may contribute to the proliferation and migration of gastric cancer (16,46–48). Results from our study further support that an elevated expression of METTL3 may increase AGS cell proliferation, promoting cancer progression. Therefore, targeting METTL3-mediated RNA methylation and SOCS signaling may be of therapeutic potential for gastric cancer. Future experiments should investigate the molecular mechanisms of METTL3-mediated gastric cancer progression, including in vivo experiments on the role of SOCS2 in METTL3-mediated cancer cell proliferation.
Acknowledgements
The authors would like to thank Ms. Barbara L. Bittner (Creighton University) for her assistance in English language editing with the manuscript.
Glossary
Abbreviations
- m6A
N6-methyladenosine
- METTL3
methyltransferase like 3
- METTL14
methyltransferase like 14
- FTO
fat mass and obesity-associated protein
- ALKBH5
a-ketoglutarate-dependent dioxygenase alkB homolog 5
- SOCS
suppressor of cytokine signaling
- KO
knockout
- LGR5
leucine rich repeat containing G protein-coupled receptor 5
- ACD
Actinomycin D
Funding
The present study was supported by the Nebraska Cancer and Smoking Disease Research Program (grant no. LB595; to XMC).
Availability of data and materials
The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.
Authors' contributions
LJ, TC, LX, JHX, AYG and XMC conceived the project. LJ, TC, LX, JHX, BD, GW, KZ, EL and AYG conducted the experiments. LJ, JHX, TC, LX, NWM, AYG and XMC analyzed and interpreted the data. LJ, JHX, NWM and XMC wrote the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–1355. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2013;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 3.Batista PJ. The RNA modification N6-methyladenosine and its implications in human disease. Genomics Proteomics Bioinformatics. 2017;15:154–163. doi: 10.1016/j.gpb.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Cgen K, Shi H, He C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi H, Wei J, He C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–650. doi: 10.1016/j.molcel.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W, Li H, Qiu M, Xu Y, Xia W, et al. Roles of RNA methylation by means of N6-methyladenosine (m6A) in human cancers. Cancer Lett. 2017;408:112–120. doi: 10.1016/j.canlet.2017.08.030. [DOI] [PubMed] [Google Scholar]
- 9.Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m6A methylation in cancer. Cancer Res. 2019;79:1285–1292. doi: 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- 10.Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, Somasundaram K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–533. doi: 10.1038/onc.2017.351. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 12.Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, Zhe H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–597. doi: 10.1002/mc.22782. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyons K, Le LC, Pham YT, Borron C, Park JY, Tran CTD, Tran TV, Tran HT, Vu KT, Do CD, et al. Gastric cancer: Epidemiology, biology, and prevention: A mini review. Eur J Cancer Prev. 2019;28:397–412. doi: 10.1097/CEJ.0000000000000480. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Zheng D, Wang F, Xu Y, Yu H, Zhang H. Expression of demethylase genes, FTO and ALKBH1, is associated with prognosis of gastric cancer. Dig Dis Sci. 2019;64:1503–1513. doi: 10.1007/s10620-018-5452-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu T, Yang S, Sui J, Xu SY, Cheng YP, Shen B, Zhang Y, Zhang XM, Yin LH, Pu YP, Liang GY. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J Cell Physiol. 2020;235:548–562. doi: 10.1002/jcp.28994. [DOI] [PubMed] [Google Scholar]
- 17.Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review) Int J Onco. 2017;51:1357–1369. doi: 10.3892/ijo.2017.4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aggarwal A, Leong SH, Lee C, Kon O, Tan P. Wavelet transformations of tumor expression profiles reveals a pervasive genome-wide imprinting of aneuploidy on the cancer transcriptome. Cancer Res. 2005;65:186–194. [PubMed] [Google Scholar]
- 19.Xu QW, Zhao W, Wang Y, Sartor MA, Han DM, Deng J, Ponnala R, Yang JY, Zhang QY, Liao GQ, et al. An integrated genome-wide approach to discover tumor-specific antigens as potential immunologic and clinical targets in cancer. Cancer Res. 2012;72:6351–6361. doi: 10.1158/0008-5472.CAN-12-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Xu J, Wang Z, Yuan Y, Li Y, Cai S, He Y. Low expression of SOCS-1 and SOCS-3 is a poor prognostic indicator for gastric cancer patients. J Cancer Res Clin Oncol. 2015;141:443–452. doi: 10.1007/s00432-014-1838-5. [DOI] [PubMed] [Google Scholar]
- 21.Inagaki-Ohara K, Kondo T, Ito M, Yoshimura A. SOCS, inflammation, and cancer. JAKSTAT. 2013;2:e24053. doi: 10.4161/jkst.24053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, Campbell IG, Visvader JE. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 2004;23:7726–7733. doi: 10.1038/sj.onc.1207787. [DOI] [PubMed] [Google Scholar]
- 23.Slattery ML, Lundgreen A, Hines LM, Torres-Mejia G, Wolff RK, Stern MC, John EM. Genetic variation in the JAK/STAT/SOCS signaling pathway influences breast cancer-specific mortality through interaction with cigarette smoking and use of aspirin/NSAIDs, the breast cancer health disparities study. Breast Cancer Res Treat. 2014;147:145–158. doi: 10.1007/s10549-014-3071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoefer J, Kern J, Ofer P, Eder IE, Schäfer G, Dietrich D, Kristiansen G, Geley S, Rainer J, Gunsilius E, et al. SOCS2 correlates with malignancy and exerts growth-promoting effects in prostate cancer. Endocr Relat Cancer. 2014;21:175–187. doi: 10.1530/ERC-13-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das R, Gregory PA, Fernandes RC, Denis I, Wang Q, Townley SL, Zhao SG, Hanson AR, Pickering MA, Armstrong HK, et al. MicroRNA-194 promotes prostate cancer metastasis by inhibiting SOCS2. Cancer Res. 2017;77:1021–1034. doi: 10.1158/0008-5472.CAN-16-2529. [DOI] [PubMed] [Google Scholar]
- 26.Vitali C, Bassani C, Chiodoni C, Fellini E, Guarnotta C, Miotti S, Sangaletti S, Fuligni F, De Cecco L, Piccaluga PP, et al. SOCS2 controls proliferation and stemness of hematopoietic cells under stress conditions and its deregulation marks unfavorable acute leukemias. Cancer Res. 2015;75:2387–2399. doi: 10.1158/0008-5472.CAN-14-3625. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Gong AY, Ma S, Chen X, Strauss-Soukup JK, Chen XM. Delivery of parasite Cdg7_Flc_0990 RNA transcript into intestinal epithelial cells during cryptosporidium parvum infection suppresses host cell gene transcription through epigenetic mechanisms. Cell Microbiol. 2017;19:e12760. doi: 10.1111/cmi.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ming Z, Gong AY, Wang Y, Zhang XT, Li M, Mathy NW, Strauss-Soukup JK, Chen XM. Involvement of cryptosporidium parvum Cdg7_FLc_1000 RNA in the attenuation of intestinal epithelial cell migration via trans-suppression of host cell SMPD3. J Infect Dis. 2017;217:122–133. doi: 10.1093/infdis/jix392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Gong AY, Ma S, Chen X, Li Y, Su CJ, Norall D, Chen J, Strauss-Soukup JK, Chen XM. Delivery of parasite RNA transcripts into infected epithelial cells during cryptosporidium infection and its potential impact on host gene transcription. J Infect Dis. 2017;215:636–643. doi: 10.1093/infdis/jiw607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci. 2019;40:298–308. doi: 10.1016/j.tips.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N6-methyladenosine modification in cancers: Current status and perspectives. Cell Res. 2018;28:507–517. doi: 10.1038/s41422-018-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369–1376. doi: 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lino CA, Harper JC, Carney JP, Timlin JA. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018;25:1234–1257. doi: 10.1080/10717544.2018.1474964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimitriou ID, Clemenza L, Scotter AJ, Chen G, Guerra FM, Rottapel R. Putting out the fire: Coordinated suppression of the innate and adaptive immune systems by SOCS1 and SOCS3 proteins. Immunol Rev. 2008;224:265–283. doi: 10.1111/j.1600-065X.2008.00659.x. [DOI] [PubMed] [Google Scholar]
- 37.Palmer DC, Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009;30:592–602. doi: 10.1016/j.it.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Letellier E, Haan S. SOCS2: Physiological and pathological functions. Front Biosci (Elite Ed) 2016;8:189–204. doi: 10.2741/E760. [DOI] [PubMed] [Google Scholar]
- 39.Khanna P, Chua PJ, Wong BSE, Yin C, Thike AA, Wan WK, Tan PH, Baeg GH. GRAM domain-containing protein 1B (GRAMD1B), a novel component of the JAK/STAT signaling pathway, functions in gastric carcinogenesis. Oncotarget. 2017;8:115370–115383. doi: 10.18632/oncotarget.23265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong D, Wang Y. Knockdown of lncRNA HULC inhibits proliferation, migration, invasion, and promotes apoptosis by sponging miR-122 in osteosarcoma. J Cell Biochem. 2018;119:1050–1061. doi: 10.1002/jcb.26273. [DOI] [PubMed] [Google Scholar]
- 41.Lui AJ, Geanes ES, Ogony J, Behbod F, Marquess J, Valdez K, Jewell W, Tawfik O, Lewis-Wambi J. IFITM1 suppression blocks proliferation and invasion of aromatase inhibitor-resistant breast cancer in vivo by JAK/STAT-mediated induction of p21. Cancer Lett. 2017;399:29–43. doi: 10.1016/j.canlet.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho KH, Jeong KJ, Shin SC, Kang J, Park CG, Lee HY. STAT3 mediates TGF-β1-induced TWIST1 expression and prostate cancer invasion. Cancer Lett. 2013;336:167–173. doi: 10.1016/j.canlet.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 43.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trengove MC, Ward AC. SOCS proteins in development and disease. Am J Clin Exp Immunol. 2013;2:1–29. [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou X, Xia Y, Li L, Zhang G. MiR-101 inhibits cell growth and tumorigenesis of helicobacter pylori related gastric cancer by repression of SOCS2. Cancer Biol Ther. 2015;16:160–169. doi: 10.4161/15384047.2014.987523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, Gong J, Shen L. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019;8:4766–4781. doi: 10.1002/cam4.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, Chen Y, Wang H. METTL3 promotes the proliferation and mobility of gastric cancer cells. Open Med (Wars) 2019;14:25–31. doi: 10.1515/med-2019-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He H, Wu W, Sun Z, Chai L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m6A-caused stabilization of SEC62. Biochem Biophys Res Commun. 2019;517:581–587. doi: 10.1016/j.bbrc.2019.07.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed in the present study are available from the corresponding author on reasonable request.