

Abstract
Autism spectrum disorder (ASD) is a highly heritable neurodevelopmental disorder. Large genetically informative cohorts of individuals with ASD have led to the identification of a limited number of common genome-wide significant (GWS) risk loci to date. However, many more common genetic variants are expected to contribute to ASD risk given the high heritability. Here, we performed a genome-wide association study (GWAS) on 6222 case-pseudocontrol pairs from the Simons Foundation Powering Autism Research for Knowledge (SPARK) dataset to identify additional common genetic risk factors and molecular mechanisms underlying risk for ASD. We identified one novel GWS locus from the SPARK GWAS and four significant loci, including an additional novel locus from meta-analysis with a previous GWAS. We replicated the previous observation of significant enrichment of ASD heritability within regulatory regions of the developing cortex, indicating that disruption of gene regulation during neurodevelopment is critical for ASD risk. We further employed a massively parallel reporter assay (MPRA) and identified a putative causal variant at the novel locus from SPARK GWAS with strong impacts on gene regulation (rs7001340). Expression quantitative trait loci data demonstrated an association between the risk allele and decreased expression of DDHD2 (DDHD domain containing 2) in both adult and prenatal brains. In conclusion, by integrating genetic association data with multi-omic gene regulatory annotations and experimental validation, we fine-mapped a causal risk variant and demonstrated that DDHD2 is a novel gene associated with ASD risk.
Subject terms: Genetics, Autism spectrum disorders
Introduction
Autism spectrum disorder (ASD) is a common neurodevelopmental disorder characterized by characteristic social deficits, as well as ritualistic behaviors1. Because ASD is highly heritable (~50–80%)2–6, a number of studies have been conducted to identify both rare and common genetic variants contributing to risk for ASD. While previous studies have successfully identified rare de novo and rare inherited presumed loss of function mutations leading to risk for ASD7–14, these de novo variants do not explain the large heritability and therefore are missing an important component of ASD risk.
To identify common inherited genetic risk factors, genome-wide association studies (GWAS) have accumulated over 18,000 individuals with ASD and have begun discovering genome-wide significant (GWS) loci that explain some of the inherited risks for ASD15. The previously discovered three GWS ASD susceptibility loci from the discovery sample of the iPSYCH-PGC study together explain only 0.13% of the liability for autism risk, whereas all common variants are estimated to explain 11.8% of liability15. Therefore, there are more common risk variants to be discovered, which requires larger sample sizes to provide sufficient power to detect risk variants of small effect16–18. The newly established genetic cohort, SPARK (Simons Foundation Powering Autism Research for Knowledge) (https://sparkforautism.org/), is planning to collect and analyze data from 50,000 individuals with ASD19. SPARK has recently released genotype data for over 8000 families or singletons with ASD, which we utilize here to increase the power of ASD GWAS.
Once we identify GWS loci, the critical next step is to understand their biological impact. This is especially challenging because most GWAS identified loci for neurodevelopmental disorders and other traits are located in poorly annotated non-coding regions with presumed gene regulatory function20. In addition, most loci are comprised of multiple single nucleotide polymorphisms (SNPs) that are often inherited together, which makes it difficult to identify the true causal variant(s) and their regulatory effects21,22. To overcome these problems, various experimental validation tools have been developed23–25. One of these tools, a massively parallel reporter assay (MPRA), simultaneously evaluates allelic effects on enhancer activity for many variants. In this assay, exogenous DNA constructs, harboring risk and protective alleles at an associated variant, drive the expression of a barcoded transcript. Differences in barcode counts between the risk and protective alleles indicate the regulatory function of that variant24,25. This assay thus demonstrates the regulatory potential of individual SNPs and provides evidence of causal variants within an associated locus.
Though fine-mapping approaches can suggest causal variants at a locus, they cannot identify target genes affected by those variants. Several approaches are designed to link variants to genes they regulate including expression quantitative trait loci (eQTL)26–28, as well as chromatin interaction (via Hi–C) assays29–31. Recently, we developed Hi–C coupled MAGMA (H-MAGMA) which predicts genes associated with the target phenotype by integrating long-range chromatin interaction with GWAS summary statistics32. Together with existing eQTL resources in the adult and fetal cortex33,34, it is possible to link variants associated with risk for ASD to target genes and functional pathways.
In this study, we increase the sample size of existing ASD GWAS by adding 6222 cases-pseudocontrol pairs from the genetically diverse SPARK project. Our analysis identified five loci associated with risk for ASD, including two novel loci. For one novel locus identified, we used an MPRA to identify the causal variant within the locus. Further, we integrated multi-level functional genomic data obtained from the developing brain, including eQTLs, chromatin interactions, and regulatory elements, to identify DDHD2 as a candidate gene involved in ASD etiology at the MPRA-validated locus.
Methods and materials
This study (analysis of this publicly available dataset) was reviewed by the Office of Human Research Ethics at UNC, which has determined that this study does not constitute human subjects research as defined under federal regulations [45 CFR 46.102 (d or f) and 21 CFR 56.102(c)(e)(l)] and does not require IRB approval.
SPARK dataset
SPARK participants who received any of the following diagnoses: autism spectrum disorder [ASD], Asperger syndrome, autism/autistic disorder and pervasive developmental disorder-not otherwise specified (PDD-NOS) were recruited. The samples were enriched for affected individuals whose parents were also available to participate. Participants registered for SPARK online at www.SPARKforAutism.org or at 25 clinical sites across the country by completing questionnaires on medical history and social communication as described here: https://www.sfari.org/spark-phenotypic-measures/. Thus, case status is based on patient/parent-report.
In this study, participants were drawn from the SPARK 27K release (20190501 ver.) through SFARIBase (https://www.sfari.org/resource/sfari-base/), which included 27,290 individuals (who were genotyped with a SNP array and/or whole-exome sequencing [WES]) with phenotype information such as sex, diagnosis, and cognitive impairment. The data included probands and their family members if applicable (e.g., 3192 quads (2798 families with unaffected siblings, 394 with multiple affected siblings), 2486 trios, and 2448 duos) (Supplementary Fig. 1). Individuals overlapping with either Autism Sequencing Consortium (ASC) cohorts or the Simons Simplex Collection (SSC) were excluded by SPARK. Twenty families in this release overlapped with the Simon’s Variations in Individuals Project (SVIP) cohort and were subsequently removed for the genome-wide association analysis (Supplementary Fig. 2) since the SVIP cohort has targeted probands with 16p11.2 deletions. We also obtained whole-exome sequencing (WES) data to estimate the imputation accuracy. Details on genotyping and whole-exome sequencing data, and pre-imputation quality control are provided in Supplementary Methods.
Genotype phasing and imputation
Phasing was performed using EAGLE v2.4.135 (https://data.broadinstitute.org/alkesgroup/Eagle/) within SPARK samples. Before making pseudocontrols, we removed two individuals, one each from two pairs of monozygotic twins with Identity-By-Descent (PI_HAT) > 0.9, by selecting the individual with lower call rates. We then defined pseudocontrols by PLINK 1.936 (www.cog-genomics.org/plink/1.9/) for trios by selecting the alleles not inherited from the parents to the case37. We re-phased all SPARK samples that passed our QC measures with pseudocontrols. Imputation was performed on the Michigan imputation server38 (https://imputationserver.sph.umich.edu/index.html). Since SPARK participants are genetically diverse, we imputed genotypes using the Trans-Omics for Precision Medicine (TOPMed) Freeze 5b (https://www.nhlbiwgs.org/) reference panel which consists of 125,568 haplotypes from multiple ancestries39,40. Imputation accuracy relative to WES was assessed using a similar approach to previous work41 (Supplementary Fig. 3) as described in Supplementary Methods.
Genome-wide association analysis and meta-analysis with iPSYCH-PGC study
We tested association within the SPARK all case-pseudocontrol pairs (full dataset; Supplementary Table 1) using PLINK2 generalized linear model (--glm) for SNPs with MAF ≥ 0.01 and imputation quality score from minimac4 (R2) > 0.5 (Supplementary Fig. 3). In this model, we did not include any covariates since cases and pseudocontrols are matched on environmental variables and genetic ancestry. We performed secondary GWAS analyses by subsetting to only specific ancestry groups. We called ancestry using multidimensional scaling (MDS) analysis with 988 HapMap3 individuals and one random case from each trio (Supplementary Fig. 4, Supplementary Table 2). Ancestry of individuals from SPARK was called as European, African or East Asian ancestries if they were within 5 standard deviations of defined HapMap3 population (CEU/TSI; YRI/LWK; or CHB/CHD/JPT, respectively) centroids in MDS dimensions 1 and 2. Population-specific GWASs were carried out using the same association model as described above for the SPARK all ancestries dataset. Meta-analyses with iPSYCH-PGC study15 were performed by METAL (release 2018–08–28)42. Additional information for iPSYCH-PGC summary statistics is provided in Supplementary Methods.
Investigation of pleiotropic effects for ASD loci
The pleiotropic effects of identified loci were investigated for phenotypes available in the NHGRI/EBI GWAS catalog (downloaded October 22, 2019)43 (Supplementary Methods).
Linkage disequilibrium score regression analysis
LD SCore regression (LDSC) (v1.0.0)44,45 was used to estimate genome-wide SNP based heritability, heritability enrichment of tissue/cell-type specific epigenetic states, and genetic correlation across phenotypes for GWAS meta-analysis results (Supplementary Methods). Prior to the analyses, we filtered SNPs to those found in HapMap3 and converted to LDSC input files (.sumstats.gz) using munge_sumstats.py. The pre-computed LD scores for Europeans were obtained from https://data.broadinstitute.org/alkesgroup/LDSCORE/eur_w_ld_chr.tar.bz2. For all LDSC analyses, we used individuals from European ancestry as described in the “Genome-wide association analysis (GWAS)” section above.
Estimating polygenic risk score
Polygenic risk scores (PRSs) were calculated based on the iPSYCH-PGC study15 using PRSice-246 (https://www.prsice.info/). Details on generation of PRS, sex-stratified and family-type PRS, and parental origin PRS analyses are provided in Supplementary Methods.
H-MAGMA
SNP to Ensembl gene annotation was carried out by Hi–C coupled MAGMA (H-MAGMA) (https://github.com/thewonlab/H-MAGMA) by leveraging chromatin-interaction generated from fetal and adult brain Hi–C33,47 as previously described32. Details on H-MAGMA and functional analyses of H-MAGMA genes are provided in Supplementary Methods.
Construction of a massively parallel reporter assay (MPRA) library
Because the novel SPARK associated locus (chr8:38.19M–chr8:38.45M) was also detected in a previous schizophrenia GWAS which is better powered, we obtained a set of credible SNPs for the locus based on schizophrenia GWAS results48 (see Supplementary Methods). Ninety-eight credible SNPs were detected in this locus. We obtained 150 bp sequences that flank each credible SNP with the SNP at the center (74 bp + 75 bp). Because each SNP has risk and protective alleles, this resulted in 196 total alleles to be tested. We seeded HEK293 cells (ATCC® CRL-11268™) in 6 wells (total 6 replicates) to be 70–90% confluent at transfection. We used lipofectamine 2000 (Invitrogen cat#11668) with our final MPRA library following the manufacturer’s instructions. Additional information for construction of MPRA library is available in Supplementary Methods. MPRA data was analyzed by the mpra package in R49,50 (https://github.com/hansenlab/mpra) with more details in Supplementary Methods.
Functional annotation of rs7001340 locus with multi-omic datasets
To investigate the target gene(s) affected by allelic variation at rs7001340, we used two expression quantitative trait loci (eQTL) datasets derived from fetal cortical brain tissue34 and adult dorsolateral prefrontal cortex33. We also used chromatin accessibility profiles from primary human neural progenitor cells and their differentiated neuronal progeny51, as well as HEK293T cells (GSM1008573)52. Further information is provided in Supplementary Methods.
Results
GWAS in SPARK dataset identified a new locus associated with ASD risk
We obtained genotype and clinical diagnosis of ASD via self-report or parent-report from 27,290 individuals who participated in the SPARK project19. The majority of data comprised families, including those where both parents and multiple children were genotyped (quads; N = 3192 families), where both parents and one child were genotyped (trios; N = 2486 families), or where one parent and one child were genotyped (duos; N = 2448 families) (Supplementary Fig. 1). Only 68 individuals were ascertained without family members (singletons). After genotyping quality control (Supplementary Methods), 375,918 variants from 26,883 individuals were retained. Because the SPARK dataset did not genotype unrelated controls, we created pseudocontrols from the alleles not transmitted from parents to probands37. Case-pseudocontrol design requires genotyping of both parents, so singletons and duos were excluded from the analysis. Due to the diverse ancestry in the cohort (Supplementary Fig. 4, Supplementary Table 1), genotypes of all individuals including pseudocontrols were imputed to a diverse reference panel (TOPMed Freeze 5b reference panel consisting of 125,568 haplotypes). After imputation quality control (Methods; Supplementary Fig. 2, 3), 8,992,756 autosomal SNPs were tested for association in 6222 case-pseudocontrol pairs (SPARK full dataset) consisting of 4956 males and 1266 females from multiple ancestries including European (N = 4535), African (N = 37), East Asian (N = 83) and other ancestries/admixed individuals (N = 1567) (Supplementary Fig. 2, Supplementary Table 2). We observed no inflation of test statistics (λGC = 1.00) (Supplementary Fig. 5), indicating population stratification was well-controlled when using this case-pseudocontrol design. We identified two SNPs at one locus (index SNP: rs60527016-C; OR = 0.84, P = 4.70 × 10–8) at genome-wide significance (P < 5.0 × 10–8) (Fig. 1a, Table 1, Supplementary Table 3), which were supported by the previous largest ASD GWAS15 (OR = 0.95, P = 0.0047) derived from the PGC and iPSYCH cohorts (Supplementary Fig. 6).
Fig. 1. Genome-wide association of ASD in the SPARK dataset.

a GWAS result from SPARK full dataset (Ncase+pseudocontrol = 12,444). b Genetic correlations across ASD GWAS. From left to right, iPSYCH versus PGC53, SPARK EUR versus iPSYCH, SPARK EUR versus PGC and SPARK EUR versus iPSYCH-PGC study15. c GWAS results from the meta-analysis (SPARK European population and iPSYCH-PGC, Nmax_case+control = 55,420). For Manhattan plots (a, c), the x-axes indicate the chromosomal position and y-axes indicate the significance of associations. The blue and red lines denote the significance threshold at suggestive (P < 1 × 10–6) and significant (P < 5 × 10–8) levels. SNPs with P < 1 × 10–6 are shown as a filled circle. Rs number indicates index SNPs from independent loci (1 MB apart from each other) at P < 1 × 10–8). Index SNPs at P < 5 × 10–8 are shown as diamonds. d Common variant risk burden is higher in cases compared to pseudocontrols. e Comparison of PRS across family types (from left to right, families with multiple affected children with affected parent(s), multiple affected children with unaffected parents, one affected child with affected parent(s), and one affected child with unaffected parents) shows no evidence for a higher common variant burden in multiplex families. f Comparison of PRS between male and female probands shows no evidence of enrichment of common variants impacting risk for ASD in females. g There is no evidence for a difference in the transmission of common variant risk burden from mother versus father.
Table 1.
Genome-wide significant loci associated with ASD risk.
| SNPa | Position(hg38) | EA | OA | EAF | SPARK | iPSYCH + PGC | Meta(EUR)b | |||
|---|---|---|---|---|---|---|---|---|---|---|
| OR(95%CI) | P | OR(95%CI) | P | OR(95%CI) | P | |||||
| Genome-wide significant loci (P < 5 × 10–8) | ||||||||||
| rs716219 | 1:96104001 | T | C | 0.34 | 1.08 (1.03–1.14) | 0.003 | 1.08 (1.05–1.11) | 3.99 × 10–7 | 1.08 (1.05–1.11) | 6.42×10–9 |
| rs10099100 | 8:10719265 | C | G | 0.31 | 1.08 (1.02–1.14) | 0.008 | 1.09 (1.06–1.12) | 1.07×10–8 | 1.08 (1.05–1.11) | 7.65×10–9 |
| rs60527016 | 8:38442106 | C | T | 0.21 | 0.84 (0.79–0.90) | 4.70×10−8 | 0.95 (0.92−0.99) | 0.00466 | 0.93 (0.91–0.96) | 3.05 × 10−6 |
| rs112436750 | 17:45887763 | A | AT | 0.21 | 1.07 (1.01−1.14) | 0.027 | 1.09 (1.05−1.12) | 1.23 × 10−6 | 1.09 (1.06–1.12) | 2.62×10−8 |
| rs1000177 | 20:21252560 | T | C | 0.24 | 1.08 (1.02−1.15) | 0.014 | 1.10 (1.07−1.14) | 3.32×10−9 | 1.09 (1.06–1.13) | 1.34×10−9 |
| Suggestive loci (5 × 10−8 ≤ P < 1 × 10−6) | ||||||||||
| rs6701243 | 1:98627228 | A | C | 0.38 | 0.99 (0.94−1.00) | 0.610 | 0.93 (0.90−0.96) | 3.07 × 10−7 | 0.94 (0.91–0.96) | 5.90 × 10--7 |
| rs6743102 | 2:158521946 | G | A | 0.34 | 0.94 (0.89−0.99) | 0.021 | 0.94 (0.91–0.97) | 8.99 × 10−6 | 0.94 (0.91–0.96) | 4.07 × 10−7 |
| rs33966416 | 4:170285452 | CA | C | 0.50 | 0.95 (0.90−1.00) | 0.038 | 0.94 (0.91–0.96) | 2.73 × 10−6 | 0.94 (0.92−0.96) | 6.99 × 10−7 |
| rs4916723 | 5:88558577 | A | C | 0.40 | 1.10 (1.00–1.10) | 0.062 | 1.07 (1.04–1.10) | 1.92 × 10−6 | 1.07 (1.04–1.09) | 6.90 × 10−7 |
| rs416223 | 5:104655775 | C | A | 0.40 | 1.00 (0.96–1.10) | 0.730 | 1.07 (1.04–1.10) | 3.84 × 10−7 | 1.07 (1.04–1.09) | 3.56 × 10−7 |
| rs67248478 | 6:134711094 | C | T | 0.34 | 0.94 (0.90–1.10) | 0.032 | 0.94 (0.91–0.96) | 3.22 × 10−6 | 0.94 (0.91–0.96) | 3.22 × 10−7 |
| rs76569799 | 9:73565191 | C | T | 0.15 | 1.10 (0.99–1.10) | 0.076 | 1.09 (1.05–1.13) | 3.90 × 10−6 | 1.08 (1.05–1.12) | 9.99 × 10−7 |
| rs4750990 | 10:128689762 | T | C | 0.36 | 1.00 (0.98–1.10) | 0.250 | 1.07 (1.04–1.10) | 1.37 × 10−6 | 1.07 (1.04–1.09) | 4.89 × 10−7 |
| rs2224274 | 20:14780101 | C | T | 0.43 | 1.00 (0.97–1.10) | 0.310 | 1.07 (1.04–1.10) | 2.86 × 10−7 | 1.07 (1.05–1.10) | 5.56 × 10−8 |
Genome-wide significant and suggestive loci in any of the GWAS analyses and meta-analysis of SPARK European ancestries and iPSYCH+PGC participants are shown.
EA effect allele, OA other allele, EAF effect allele frequency in SPARK full dataset.
aIndex SNPs from loci that survived genome-wide significance in any of the GWASs including meta-analysis.
bMeta-analysis of SPARK European ancestries and iPSYCH+PGC.
P-values < 5 × 10−8 are shown in bold.
Replication of genetic risk factors for ASD
Given the phenotypic heterogeneity of ASD and potential technical differences such as genotyping platforms or data processing, we assessed the replication of genetic risk factors across cohorts by comparing previous major ASD studies including PGC and iPSYCH cohort15,53 with the SPARK dataset subset to individuals of European descent (EUR) (Fig. 1b). Although each study included multiple ASD subtypes including ASD from DSM5, Asperger’s, autism/autistic disorder, and Pervasive Developmental Disorder–Not otherwise specified (PDD-NOS) from DSM IV, and approaches differed across these samples from requiring community diagnosis to best-estimate diagnosis based on standardized assessment, we obtained high genetic correlations between the SPARK EUR dataset and the largest iPSYCH-PGC GWAS (rg = 0.82; P = 5.27 × 10–14), suggesting the genetic risk factors for autism are largely shared among different ASD GWAS and are generalizable despite differences in diagnostic criteria and batch effects.
We next performed meta-analysis with SPARK EUR samples and iPSYCH-PGC samples (Ncase = 18,381 and Ncontrol = 27,969) to maximize power. The meta-analysis identified four additional loci associated with risk for ASD (Supplementary Figs. 7–10). These included three previously reported loci15 and one novel locus on chromosome 17, where a gene-based test from the iPSYCH-PGC study has previously shown association with risk for ASD15 (Fig. 1c, Table 1, Supplementary Fig. 9). This novel locus was also reported to be associated with more than 60 phenotypes including neuroticism54–58, educational attainment59 and intracranial volume60 (index SNPs r2 > 0.8 in 1 KG EUR) (Supplementary Table 4), indicating highly pleiotropic effects. The SNP based heritability in SPARK EUR samples was estimated (h2G) to be 0.117 (s.e. = 0.0082) for population prevalence of 0.01215,61 which was comparable with the previous report (h2G = 0.118; s.e. = 0.010)15.
The generalization of effects across ancestries for the five index SNPs identified (Table 1) was examined (Supplementary Fig. 6, and Supplementary Table 5). The association results from the cross-ancestry dataset were mainly driven by the European population, as expected given the larger sampling from this population. We found that some regions showed differences in allele frequencies based on population. For example, rs10099100 was more common in European and African populations (MAF = 0.33, 0.39 from tested samples, respectively) than in East Asians (MAF = 0.02 from tested samples), necessitating a further investigation of genetic risk factors for ASD in populations of diverse ancestry62,63.
The generalization of genetic effects on risk for ASD was also confirmed by polygenic risk scores (PRSs) derived from the iPSYCH-PGC GWAS that showed higher scores in SPARK EUR cases (N = 4097) compared to pseudocontrols (N = 4097) (P = 1.61 × 10–19; p value threshold = 0.01; Nagelkerke’s R2 = 1.4%) (Fig. 1d, Supplementary Fig. 11).
Investigation of common variant burden impacting risk for ASD
We next used PRSs to compare common variant risk burden among family types, sex, and parent of origin (Fig. 1e-g). Because ASD families with multiple affected siblings were shown to have different segregation patterns compared with simplex families that have a higher burden of de novo mutations11,64,65, we compared the distribution of PRSs across four family types (Fig. 1e, Supplementary Table 1). Our results showed no evidence for a difference in common variant burden impacting risk for ASD in multiplex families as compared to simplex families. We note that multiplex/simplex status was indicated by either enrollment or self-report in a questionnaire and may be underestimated due to missing survey data.
As the prevalence of ASD is higher in males than in females (OR = 4.20)66, and previous studies have reported that females with ASD have a higher burden of de novo variants9,67–69, we also investigated the potential contribution of common variants to the female protective effect by comparing PRS between sexes. We did not find evidence that ASD common variant risk burden differs between females and males (Fig. 1f).
A previous study hypothesized that a new mutation in a mother, who is less susceptible to developing autism because of the female protective effect, may be more likely to transmit risk factors to their children with ASD70. We, therefore, examined the over-transmission of common variant risk for ASD from mother to offspring. We found no evidence of the over-transmission of common variant risk burden from either mothers or fathers to their affected children (Fig. 1g).
Contribution of cortical development to risk for ASD
Previous studies suggest an important role of brain development in ASD15,71. To characterize tissue types relevant to risk for ASD, we next evaluated heritability enrichment within active enhancer or promoter regions in different tissues72 (Supplementary Fig. 12A, Table 5). Significant enrichment of heritability was observed in regulatory elements of brain germinal matrix, as well as primary cultured neurospheres from the fetal cortex (FDR = 0.004 and 0.015, respectively, Supplementary Table 6), suggesting that disruption of gene regulation in these tissues increases the risk for ASD. We further examined SNP heritability in the developing cortex using differentially accessible peaks between the neuron-enriched cortical plate and the progenitor-enriched germinal zone73 (Supplementary Fig. 12B). We found significant enrichment in peaks more accessible in the germinal zone (FDR = 0.008), but not in the cortical plate, replicating previous reports that genetically mediated alterations in cortical development play a crucial role in ASD etiology15.
H-MAGMA identified genes and pathways impacting risk for ASD
To identify genes associated with risk for ASD from meta-analysis (EUR only), we applied Hi–C coupled MAGMA (H-MAGMA)32, which aggregates SNP-level P-values into a gene-level association statistic with an additional assignment of non-coding SNPs to their chromatin-interacting target genes generated from fetal brain Hi–C47 (Fig. 2a). We identified 567 genes associated with ASD (FDR < 0.1), including 263 protein coding genes (Fig. 2b, Supplementary Table 7). Five genes implicated from common variant evidence (KMT2E, RAI1, BCL11A, FOXP1, and FOXP2) also harbored an excess of rare variants associated with ASD74. This overlap between rare and common ASD risk variants was more than expected by chance (hypergeometric P = 0.01; Fig. 2c), corroborating previous findings that common and rare variation converge on the same genes and pathways32,75,76. We also found that 14 H-MAGMA genes were also differentially expressed in the post-mortem cortex between individuals with ASD and neurotypical controls (upregulated in ASD: NFKB2, BTG1, RASGEF1B, TXNL4B, IFI16, WDR73, and C2CD4A; downregulated in ASD: PAFAH1B1, SEMA3G, DDHD2, GTDC1 ASH2L, USP19, and ARIH2; FDR < 0.05)77 (Fig. 2d). Rank-based gene ontology enrichment analysis78 suggested that ASD risk genes were enriched in 188 terms including telencephalon development and regulation of synapse organization (Fig. 2E, Supplementary Tables 8, 9).
Fig. 2. H-MAGMA identified 263 protein-coding genes linked to ASD.

a Schematic diagram of H-MAGMA. SNP based association P-values were aggregated to gene-based P-values using positional information, as well as chromatin interaction in the fetal brain. b Gene-based association results from H-MAGMA. The x-axis indicates the start position of genes (hg19). c Overlap of ASD risk genes harboring common and rare variants74. d Overlapped genes with differential expression from post-mortem brains in individuals with ASD patients and neurotypical controls77. e Gene ontologies enriched for ASD linked genes (top 20). f Developmental expression pattern of ASD risk genes118.
Since heritability enrichment analyses suggested genetically mediated impacts on cortical development contribute to ASD risk (Supplementary Fig. 12), we explored whether the expression level of ASD risk genes from H-MAGMA is different between prenatal and postnatal cortex. In this analysis, we combined H-MAGMA genes from either adult or fetal brain Hi–C (Supplementary Fig. 13) to ensure that the enrichment is not driven by the use of Hi–C from only one developmental time period, such as observing higher prenatal expression levels of H-MAGMA identified ASD risk genes exclusively due to the use of fetal brain Hi–C (Supplementary Table 7,10). As shown previously15,32, we found ASD risk genes exhibited higher expression in the prenatal cortex as compared to the postnatal cortex (P = 2.77 × 10–62) (Fig. 3f). In particular, the expression level of ASD risk genes was highest between 20 and 30 post-conception weeks (Supplementary Fig. 14, Supplementary Table 11). Taken together, our results demonstrate common risk variants for ASD play an important role in the developing cortex.
Fig. 3. Genetic correlation of ASD against twelve brain and behavioral phenotypes.
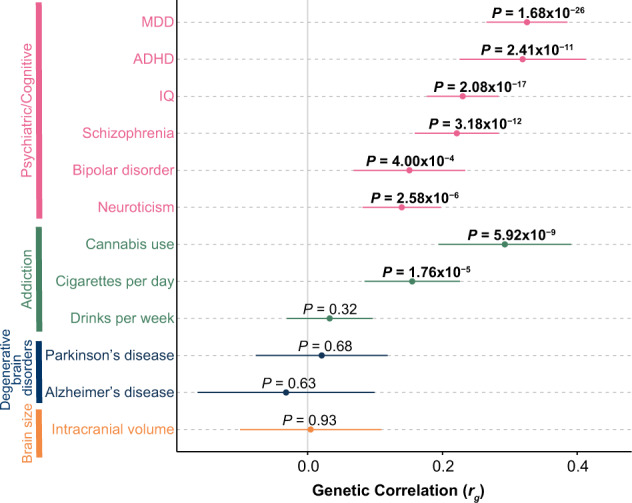
The x-axis represents an estimate of the genetic correlation (rg). Error bars represent the 95% confidence interval. P-values at FDR < 0.05 are shown in bold. MDD major depressive disorder, ADHD attention-deficit/hyperactivity disorder.
Genetic correlation between ASD and 12 brain and behavioral phenotypes
Both epidemiological studies and genetic studies suggested the phenotypic comorbidity79–82 or genetic correlation15,83 of ASD with various brain and behavioral phenotypes. Thus, we evaluated the pleiotropic effect of ASD risk SNPs with twelve other brain and behavioral phenotypes48,57,60,84–91 (Fig. 3, Supplementary Table 12). We observed a novel genetic correlation between ASD and cigarettes per day (rg = 0.16, P = 8.80 × 10–5), indicating a partially shared genetic basis between risk for ASD and addictive smoking behavior. We also replicated positive genetic correlations previously detected for seven phenotypes (FDR < 0.05)15, providing further support for a shared genetic basis of multiple neuropsychiatric disorders83,92.
Functional validation to fine-map causal variants and prioritize genes
Interestingly, the novel locus identified by the SPARK full dataset (rs60527016 at chr8:38.19M–chr8:38.45M, Figs. 1a, 4a) was previously identified as a pleiotropic locus in a recent cross-disorder meta-analysis on eight psychiatric disorders83, as well as a schizophrenia GWAS48 (Supplementary Fig. 15). This locus was not only associated with ASD but also with schizophrenia, bipolar disorder and obsessive-compulsive disorder (OCD), suggesting that understanding the regulatory mechanism at this locus may reveal the basis for pleiotropic effects across psychiatric disorders. The index SNP (rs60527016) was located within a 300 kb LD block (r2 > 0.5 in SPARK full dataset) that contains seven genes (Fig. 4a). To prioritize causal variants within this locus, we performed a massively parallel reporter assay (MPRA)24,25 on 98 credible SNPs in this region in HEK 293 cells (Supplementary Fig. 15, 16, 17). MPRA measures barcoded transcriptional activity driven by each allele in a high-throughput fashion (Supplementary Fig. 16). Surprisingly, SNP rs7001340 exhibited the strongest allelic difference in barcoded expression (P = 1.51 × 10–24) even though it is 37 kb away from the GWAS index SNP (r2 = 0.85 with the index SNP in SPARK full dataset) (Fig. 4a, b, Supplementary Table 13), demonstrating the regulatory potential of this SNP and suggesting its causal role in psychiatric disorders, including ASD. While MPRA was performed in HEK cells, the SNP was located in a regulatory element present in both HEK cells and neural progenitors, with higher chromatin accessibility in human neural progenitors compared with postmitotic neurons51 (Fig. 4a, Supplementary Fig. 17), indicating its regulatory potential in the developing brain. The risk allele (T) at this SNP was associated with downregulation of barcoded expression in MPRA (Fig. 4b, Supplementary Fig. 16), and was predicted to disrupt two transcription factor binding motifs93 (TBX1 and SMARCC1) (Supplementary Fig. 18), providing a possible mechanism of action of this variant. We next investigated potential target genes impacted by regulatory changes at this SNP by using eQTL data from fetal34 and adult brain tissues33. Expression levels of three eGenes were significantly associated with this SNP (DDHD2 from the fetal brain and DDHD2, LSM1, LETM2 from the adult brain) (Fig. 4a, Supplementary Fig. 19). Of these three genes, two genes (DDHD2, LETM2) showed the direction of the effect expected from the MPRA result (risk allele downregulates the eGene). It is of note that DDHD2 was identified in both fetal and adult brain eQTL datasets (beta = −0.080, P = 2.212 × 10–13; beta = −0.177, P = 1.38 × 10–20, respectively; Fig. 4c, d). We further validated the association between DDHD2 and ASD by additional transcriptome wide association study (TWAS) in the brain (PrediXcan94–96 and FUSION97) (Supplementary Fig. 20)28,98–101. Notably, DDHD2 was also significantly downregulated in the post-mortem cortex of individuals with autism (logFC = −0.28, FDR = 0.013), providing an added layer of evidence supporting its role in ASD risk77. DDHD2 was also identified by H-MAGMA (Fig. 2f), and a copy number variation (CNV) containing DDHD2 (deletions) was found in proband-sibling pairs with discordant social-behavior phenotypes102. Collectively, by integrating existing multi-level functional genomic resources and an experimental fine-mapping approach using MPRA, we suggest DDHD2 as a strong candidate gene impacting risk for ASD.
Fig. 4. Identification of putative causal variant and gene impacting risk for ASD.

a Annotated locus plot near rs60527016 ASD risk index variant, from top panel to bottom, ASD associations within SPARK full dataset (n = 6222 case-pseudocontrol pairs), eQTL for DDHD2 in fetal brains (n = 235) and adult brain (n = 1387), MPRA expression (n = 6), ATAC-seq averaged depth in neuron (n = 61) and progenitor (n = 73). Differential open chromatin accessibility peaks from ATAC-seq, and gene model (NCBI Refseq). LD was calculated to rs7001340 within SPARK parents of cases, fetal brain donors, or 1 KG EUR and colored accordingly. b The barcoded expression level of mRNA based from each allele at rs7001340 from the MPRA experiment. c The expression level of DDHD2 by rs7001340 genotypes in the fetal brain. d The expression level of DDHD2 by rs7001340 genotypes in adult brain. Individuals with allele dosage (0–0.1 as C/C, 0.9–1.1 as C/T, 1.9–2.0 as T/T) are shown. For b to d, ASD risk allele for rs7001340 is T and protective allele is C.
Discussion
In this study, we increased sample sizes for ASD GWAS to Ncase(max) = 24,063, Ncontrol(max) = 34,191 and identified five loci associated with risk for ASD (four from European only meta-analysis, one locus from SPARK project alone), including two new loci (marked by index SNPs rs60527016 and rs112436750). These loci have pleiotropic effects on multiple psychiatric disorders including schizophrenia (for rs60527016 and rs112436750), bipolar disorder, and OCD (for rs60527016).
Using a PRS derived from a previous study15, we found enrichment of risk variants in SPARK cases, indicating the contribution of common genetic risk factors to ASD is consistent across cohorts. However, despite several hypotheses that rare variants associated with risk for ASD are enriched in certain subgroups of individuals with ASD, such as in females compared to males (female protective effect)9,67–69,103, multiplex families compared to simplex families11,64,65, or maternal alleles compared to paternal alleles10,70, we do not find evidence to support the increased burden of common risk variants within those subgroups. This result indicates potential rare and common variant differences in contribution to subgroup risk for ASD. However, it is notable that similar to GWAS in other neuropsychiatric disorders104,105, PRS explained only a small percent of variance in risk (1.4%). Moreover, given the small sample size of specific subgroups (N = 835 in female whereas N = 3262 in male, N = 14 for families with multiple affected children versus N = 3618 with one affected children), our study may have limited power to identify the differences among subgroups. Thus, a larger sample size would be warranted to compare the difference in the role of common variants within these categories.
Identifying locations in the genome associated with risk for ASD does not in itself lead to insights into what tissues or developmental time points are crucial for the etiology of ASD. Here, by integrating SNP association statistics with existing annotations of regulatory elements active during specific developmental time periods or within specific brain regions, we found an excess of common genetic risk for ASD in the fetal brain regulatory elements (brain germinal matrix and primary cultured neurospheres from the fetal cortex), and progenitor enriched germinal zone of the developing cortex, confirming previous findings that alterations of gene regulation in the prenatal cortex play a key role in ASD etiology15.
To further understand genes leading to risk for ASD, we applied a recently developed platform, H-MAGMA32 and identified 263 putative candidate protein-coding risk genes. H-MAGMA genes are highly expressed in the prenatal brain, similar to the enrichment of ASD risk genes with rare variations during neurodevelopment106. This result suggests potential molecular convergence regardless of classes of mutation, which is supported by five genes (previously identified KMT2E and newly identified RAI1, BCL11A, FOXP1, and FOXP2) that are affected by both rare and common variation.
Since identification of a GWS locus does not elucidate the causal variant(s), we performed MPRA and identified a potential causal SNP (rs7001340) at a novel ASD locus discovered in the SPARK sample. Interestingly, the individual variant with the strongest regulatory effect (rs7001340; r2 = 0.85 with the index SNP in SPARK full dataset) was different from the SNP with the strongest association with ASD (rs60527016), highlighting the importance of experimental validation in identifying causal variants. It should be noted that the regulatory effects of these variants were assessed in non-neural (HEK) cells. Although this regulatory element was found in both HEK cells and neural progenitors (Supplementary Fig. 17), further validation of these effects in ASD-relevant cell types would provide increased confidence in declaring this SNP as causal. The experimentally validated regulatory SNP (rs7001340) is in the intron of LETM2, and is also an eQTL for LETM2, LSM1 (247 kb away) and DDHD2 (173 kb away), indicating that the SNP functions as a distal regulatory element. The risk allele (T) was associated with decreased expression of barcoded transcripts in the MPRA and downregulation of DDHD2 from eQTL in both fetal and adult brains, implying a consistent direction of the allelic effects on gene regulation. The risk allele showed the same direction of effect for LETM2 in adult brain tissue, but was not significantly associated in fetal brain tissue (P-value = 0.33). Intriguingly, DDHD2 was also downregulated in the cortex from individuals with ASD compared to neurotypical controls77, providing an additional level of support for this gene as a risk factor for ASD. DDHD2 (DDHD domain-containing protein 2), also known as KIAA0725p, encodes a phospholipase and is localized in the Golgi107. DDHD2 plays a role in the efficient regulation of membrane trafficking between the Golgi and cytosol107 and is highly expressed in the brain108–110. Mutations in DDHD2 have been previously found in individuals with spastic paraplegia type 54 (SPG)110–112. Ddhd2 null mice exhibited motor and cognitive impairments113, which are frequent comorbidities of ASD114. We, therefore, conclude DDHD2 is a strong candidate risk gene for ASD through multiple lines of evidence.
There is still a large amount of common variant heritability not explained in this study indicating that further increases in sample size will be necessary to explain the common inherited component of ASD risk. While the combination of TOPMed imputation and the case-pseudocontrol study model enabled us to include individuals from multiple ancestries, the case-pseudocontrol model is lower powered compared to case-unscreened control models because a pseudocontrol might have greater liability for ASD than the average individual in the population115. In addition, the case-pseudocontrol model cannot incorporate duos or singletons due to the lack of parental genotype information, which resulted in over 2000 individuals with ASD with genotyping information in the SPARK project not being included in our analysis. Moreover, this model has the disadvantage that X-chromosome cannot be analyzed due to lack of untransmitted genotype information from the father. Future studies could potentially solve this problem and also increase power by including all cases available in SPARK and using unscreened population matched controls116. Secondly, although we performed population stratified GWAS, the limited number of individuals for some populations (e.g., 37 from AFR and 83 from EAS) may lead to a large standard error in the estimate of the effect size. Also, subsequent analyses including PRS, LDSC regression, and H-MAGMA were limited to individuals from European ancestries only, because most resources and software are designed to be used only within one population, generally European ancestry117. Including other ancestries for these analyses will be able to uncover risk factors shared or specific to existing human populations.
In summary, ASD GWAS in the SPARK dataset and meta-analysis with previous GWAS identified two new susceptibility loci. By integrating existing multi-level functional genomic resources and experimental tools such as MPRA and eQTL, we highlight DDHD2 as a novel high confidence ASD risk gene impacted by a distal common variant within a regulatory element present in neural progenitors of the developing cortex. This strategy can be broadly applied to common variant risk loci of multiple neuropsychiatric disorders to identify causal variant(s), regulatory regions, cell-types, and genes whose misregulation leads to risk for neuropsychiatric disorders.
Supplementary information
Acknowledgements
This work was supported by a grant from the Simon Foundation (SFARI[Award #: 605259], H.W. and J.L.S.), NIMH (R00MH113823 and DP2MH122403 to H.W. and R01MH118349, R01MH120125, R01MH121433 to J.L.S.), NIGMS (DP2GM114829 to S.K.) and the NARSAD Young Investigator Award (H.W.). L.M.R. was supported by T32 HL129982. We are grateful to all of the families in SPARK, the SPARK clinical sites and SPARK staff. We appreciate obtaining access to genetic and phenotypic data on SFARI Base. Approved researchers can obtain the SPARK population dataset described in this study (more details available at https://www.sfari.org/resource/spark/) by applying at https://base.sfari.org. Trans-Omics in Precision Medicine (TOPMed) program imputation panel (version Freeze5) was supported by the National Heart, Lung and Blood Institute (NHLBI); see www.nhlbiwgs.org. TOPMed study investigators contributed data to the reference panel, which can be accessed through the Michigan Imputation Server; see https://imputationserver.sph.umich.edu. The panel was constructed and implemented by the TOPMed Informatics Research Center at the University of Michigan (3R01HL-117626–02S1; contract HHSN268201800002I). The TOPMed Data Coordinating Center (3R01HL-120393–02S1; contract HHSN268201800001I) provided additional data management, sample identity checks, and overall program coordination and support. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
Data availability
Summary statistics are available at https://bitbucket.org/steinlabunc/spark_asd_sumstats.
Code availability
Code is available at https://github.com/thewonlab/GWAS_ASD_SPARK.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Hyejung Won, Jason L. Stein
Contributor Information
Hyejung Won, Email: hyejung_won@med.unc.edu.
Jason L. Stein, Email: jason_stein@med.unc.edu
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41398-020-00953-9).
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). (American Psychiatric Publication, 2013).
- 2.Lee SH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaugler T, et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016;22:345–361. doi: 10.1038/nm.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandin S, et al. The heritability of autism spectrum disorder. JAMA. 2017;318:1182–1184. doi: 10.1001/jama.2017.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai, D. et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry10.1001/jamapsychiatry.2019.1411 (2019). [DOI] [PMC free article] [PubMed]
- 7.Michaelson JJ, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders SJ, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leppa VM, et al. Rare inherited and de novo CNVs reveal complex contributions to ASD risk in multiplex families. Am. J. Hum. Genet. 2016;99:540–554. doi: 10.1016/j.ajhg.2016.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruzzo EK, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–866.e26. doi: 10.1016/j.cell.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satterstrom FK, et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 2019;22:1961–1965. doi: 10.1038/s41593-019-0527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Satterstrom FK, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–584.e23. doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grove J, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019;51:431–444. doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park J-H, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 2010;42:570–575. doi: 10.1038/ng.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson MR, Wray NR, Visscher PM. Explaining additional genetic variation in complex traits. Trends Genet. 2014;30:124–132. doi: 10.1016/j.tig.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.SPARK Consortium. SPARK: A US Cohort of 50,000 families to accelerate autism research. Neuron. 2018;97:488–493. doi: 10.1016/j.neuron.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenet. Chromatin. 2015;8:57. doi: 10.1186/s13072-015-0050-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018;19:491–504. doi: 10.1038/s41576-018-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muerdter F, Boryń M, Arnold CD. STARR-seq—principles and applications. Genomics. 2015;106:145–150. doi: 10.1016/j.ygeno.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Inoue F, Ahituv N. Decoding enhancers using massively parallel reporter assays. Genomics. 2015;106:159–164. doi: 10.1016/j.ygeno.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis, J. E., Insigne, K. D., Jones, E. M., Hastings, Q. B. & Kosuri, S. Multiplexed dissection of a model human transcription factor binding site architecture. bioRxiv 625434 10.1101/625434 (2019).
- 26.Rockman MV, Kruglyak L. Genetics of global gene expression. Nat. Rev. Genet. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- 27.Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013;368:20120362. doi: 10.1098/rstb.2012.0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.GTEx Consortium. et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin F, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sey, N. Y. A. et al. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci.10.1038/s41593-020-0603-0 (2020). [DOI] [PMC free article] [PubMed]
- 33.Wang, D. et al. Comprehensive functional genomic resource and integrative model for the human brain. Science362, 6420 (2018). [DOI] [PMC free article] [PubMed]
- 34.Walker RL, et al. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell. 2019;179:750–771.e22. doi: 10.1016/j.cell.2019.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loh P-R, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cordell HJ, Barratt BJ, Clayton DG. Case/pseudocontrol analysis in genetic association studies: a unified framework for detection of genotype and haplotype associations, gene-gene and gene-environment interactions, and parent-of-origin effects. Genet. Epidemiol. 2004;26:167–185. doi: 10.1002/gepi.10307. [DOI] [PubMed] [Google Scholar]
- 38.Das S, et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv 563866 10.1101/563866 (2019). [DOI] [PMC free article] [PubMed]
- 40.Kowalski, M. H. et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. bioRxiv 683201 10.1101/683201 (2019). [DOI] [PMC free article] [PubMed]
- 41.Sariya S, et al. Rare variants imputation in admixed populations: comparison across reference panels and bioinformatics tools. Front. Genet. 2019;10:239. doi: 10.3389/fgene.2019.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buniello A, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bulik-Sullivan BK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi, S. W. & O’Reilly, P. F. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience8, giz082 (2019). [DOI] [PMC free article] [PubMed]
- 47.Won H, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–527. doi: 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pardiñas AF, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018;50:381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Law CW, Chen Y, Shi W, Smyth G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myint L, Avramopoulos DG, Goff LA, Hansen KD. Linear models enable powerful differential activity analysis in massively parallel reporter assays. BMC Genom. 2019;20:209. doi: 10.1186/s12864-019-5556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang, D. et al. Cell-type specific effects of genetic variation on chromatin accessibility during human neuronal differentiation. bioRxiv 2020.01.13.904862 10.1101/2020.01.13.904862 (2020). [DOI] [PMC free article] [PubMed]
- 52.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism. 2017;8:21. doi: 10.1186/s13229-017-0137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith DJ, et al. Genome-wide analysis of over 106 000 individuals identifies 9 neuroticism-associated loci. Mol. Psychiatry. 2016;21:749–757. doi: 10.1038/mp.2016.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luciano M, et al. Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat. Genet. 2018;50:6–11. doi: 10.1038/s41588-017-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagel M, Watanabe K, Stringer S, Posthuma D, van der Sluis S. Item-level analyses reveal genetic heterogeneity in neuroticism. Nat. Commun. 2018;9:905. doi: 10.1038/s41467-018-03242-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagel M, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018;50:920–927. doi: 10.1038/s41588-018-0151-7. [DOI] [PubMed] [Google Scholar]
- 58.Kichaev G, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JJ, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018;50:1112–1121. doi: 10.1038/s41588-018-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adams HHH, et al. Novel genetic loci underlying human intracranial volume identified through genome-wide association. Nat. Neurosci. 2016;19:1569–1582. doi: 10.1038/nn.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen SN, Overgaard M, Andersen PK, Parner ET. Estimating a population cumulative incidence under calendar time trends. BMC Med. Res. Methodol. 2017;17:7. doi: 10.1186/s12874-016-0280-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peterson RE, et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179:589–603. doi: 10.1016/j.cell.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Duncan L, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019;10:3328. doi: 10.1038/s41467-019-11112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 2014;15:133–141. doi: 10.1038/nrg3585. [DOI] [PubMed] [Google Scholar]
- 66.Loomes R, Hull L, Mandy WPL. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry. 2017;56:466–474. doi: 10.1016/j.jaac.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 67.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 68.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong S, et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep. 2014;9:16–23. doi: 10.1016/j.celrep.2014.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao X, et al. A unified genetic theory for sporadic and inherited autism. Proc. Natl Acad. Sci. USA. 2007;104:12831–12836. doi: 10.1073/pnas.0705803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hazlett HC, et al. Early brain development in infants at high risk for autism spectrum disorder. Nature. 2017;542:348–351. doi: 10.1038/nature21369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roadmap Epigenomics Consortium, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de la Torre-Ubieta L, et al. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell. 2018;172:289–304.e18. doi: 10.1016/j.cell.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kyle Satterstrom, F. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. bioRxiv 484113 10.1101/484113 (2019). [DOI] [PMC free article] [PubMed]
- 75.Zhou J, et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019;51:973–980. doi: 10.1038/s41588-019-0420-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mah, W. & Won, H. The three-dimensional landscape of the genome in human brain tissue unveils regulatory mechanisms leading to schizophrenia risk. Schizophr. Res. 10.1016/j.schres.2019.03.007 (2019). [DOI] [PMC free article] [PubMed]
- 77.Parikshak NN, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423–427. doi: 10.1038/nature20612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Simonoff E, et al. Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry. 2008;47:921–929. doi: 10.1097/CHI.0b013e318179964f. [DOI] [PubMed] [Google Scholar]
- 80.van Steensel FJA, Bögels SM, Perrin S. Anxiety disorders in children and adolescents with autistic spectrum disorders: a meta-analysis. Clin. Child Fam. Psychol. Rev. 2011;14:302–317. doi: 10.1007/s10567-011-0097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Antshel KM, Zhang-James Y, Wagner KE, Ledesma A, Faraone SV. An update on the comorbidity of ADHD and ASD: a focus on clinical management. Expert Rev. Neurother. 2016;16:279–293. doi: 10.1586/14737175.2016.1146591. [DOI] [PubMed] [Google Scholar]
- 82.Romero M, et al. Psychiatric comorbidities in autism spectrum disorder: a comparative study between DSM-IV-TR and DSM-5 diagnosis. Int. J. Clin. Health Psychol. 2016;16:266–275. doi: 10.1016/j.ijchp.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cross-Disorder, Group Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179:1469–1482.e11. doi: 10.1016/j.cell.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Demontis D, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019;51:63–75. doi: 10.1038/s41588-018-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Savage JE, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018;50:912–919. doi: 10.1038/s41588-018-0152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stahl EA, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019;51:793–803. doi: 10.1038/s41588-019-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Howard DM, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019;22:343–352. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pasman JA, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat. Neurosci. 2018;21:1161–1170. doi: 10.1038/s41593-018-0206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu M, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 2019;51:237–244. doi: 10.1038/s41588-018-0307-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jansen IE, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019;51:404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nalls, M. A. et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. bioRxiv 388165 (2019).
- 92.Brainstorm Consortium et al. Analysis of shared heritability in common disorders of the brain. Science360, eaap8757 (2018). [DOI] [PMC free article] [PubMed]
- 93.Coetzee SG, Coetzee GA, Hazelett DJ. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015;31:3847–3849. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gamazon ER, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barbeira A, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wheeler HE, et al. Survey of the heritability and sparse architecture of gene expression traits across human tissues. PLoS Genet. 2016;12:e1006423. doi: 10.1371/journal.pgen.1006423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gusev A, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lonsdale J, et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fromer M, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016;19:1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huckins LM, et al. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat. Genet. 2019;51:659–674. doi: 10.1038/s41588-019-0364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krumm N, et al. Transmission disequilibrium of small CNVs in simplex autism. Am. J. Hum. Genet. 2013;93:595–606. doi: 10.1016/j.ajhg.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jacquemont S, et al. A higher mutational burden in females supports a ‘female protective model’ in neurodevelopmental disorders. Am. J. Hum. Genet. 2014;94:415–425. doi: 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ripke S, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Conde LC, et al. A direct test of the diathesis-stress model for depression. Eur. Neuropsychopharmacol. 2019;29:S805–S806. [Google Scholar]
- 106.Parikshak NN, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sato S-I, Inoue H, Kogure T, Tagaya M, Tani K. Golgi-localized KIAA0725p regulates membrane trafficking from the Golgi apparatus to the plasma membrane in mammalian cells. FEBS Lett. 2010;584:4389–4395. doi: 10.1016/j.febslet.2010.09.047. [DOI] [PubMed] [Google Scholar]
- 108.Nagase T, et al. Prediction of the coding sequences of unidentified human genes. XII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 1998;5:355–364. doi: 10.1093/dnares/5.6.355. [DOI] [PubMed] [Google Scholar]
- 109.Nakajima K-I, et al. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J. Biol. Chem. 2002;277:11329–11335. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 110.Schuurs-Hoeijmakers JHM, et al. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gonzalez M, et al. Mutations in phospholipase DDHD2 cause autosomal recessive hereditary spastic paraplegia (SPG54) Eur. J. Hum. Genet. 2013;21:1214–1218. doi: 10.1038/ejhg.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Novarino G, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Inloes JM, et al. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc. Natl Acad. Sci. USA. 2014;111:14924–14929. doi: 10.1073/pnas.1413706111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sokhadze EM, Tasman A, Sokhadze GE, El-Baz AS, Casanova MF. Behavioral, cognitive, and motor preparation deficits in a visual cued spatial attention task in autism spectrum disorder. Appl. Psychophysiol. Biofeedback. 2016;41:81–92. doi: 10.1007/s10484-015-9313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peyrot WJ, Boomsma DI, Penninx BWJH, Wray NR. Disease and polygenic architecture: avoid trio design and appropriately account for unscreened control subjects for common disease. Am. J. Hum. Genet. 2016;98:382–391. doi: 10.1016/j.ajhg.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bodea CA, et al. A method to exploit the structure of genetic ancestry space to enhance case-control studies. Am. J. Hum. Genet. 2016;98:857–868. doi: 10.1016/j.ajhg.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177:1080. doi: 10.1016/j.cell.2019.04.032. [DOI] [PubMed] [Google Scholar]
- 118.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics are available at https://bitbucket.org/steinlabunc/spark_asd_sumstats.
Code is available at https://github.com/thewonlab/GWAS_ASD_SPARK.