

The major transcription factors that regulate how stem cells commit to become T lymphocytes are functionally defined
Abstract
T cell factor 1 (Tcf1) is the first T cell–specific protein induced by Notch signaling in the thymus, leading to the activation of two major target genes, Gata3 and Bcl11b. Tcf1 deficiency results in partial arrests in T cell development, high apoptosis, and increased development of B and myeloid cells. Phenotypically, seemingly fully T cell–committed thymocytes with Tcf1 deficiency have promiscuous gene expression and an altered epigenetic profile and can dedifferentiate into more immature thymocytes and non-T cells. Restoring Bcl11b expression in Tcf1-deficient cells rescues T cell development but does not strongly suppress the development of non-T cells; in contrast, expressing Gata3 suppresses their development but does not rescue T cell development. Thus, T cell development is controlled by a minimal transcription factor network involving Notch signaling, Tcf1, and the subsequent division of labor between Bcl11b and Gata3, thereby ensuring a properly regulated T cell gene expression program.
INTRODUCTION
T cells are disease-fighting leukocytes that, similar to all blood cells, originate from hematopoietic stem cells (HSCs). However, whereas all other blood cell lineages develop in the bone marrow in specific niches, T cells develop in the thymus, a specialized organ located in the chest where progenitor cells migrate from the bone marrow and definitively commit to the T cell lineage, ultimately forming mature T cells (1). The development of T cells within the thymus is a highly complex process involving successive stages in which the expression of CD4 and CD8 co-receptors occurs in distinct microenvironments (2). Via a series of progressive developmental stages, T cell precursors (i.e., thymocytes) differentiate from double-negative (DN; CD4−CD8−) cells into intermediate immature single-positive (ISP; CD8+CD3−CD4−) cells, then into double-positive (DP; CD4+CD8+) cells, and finally into single-positive (SP; CD8+CD4−CD3+ or CD4+CD8−CD3+) cells. In the DN stage, developing thymocytes can be further subdivided into four stages of differentiation based on their expression levels of CD44 and CD25: DN1 (CD44+CD25−), DN2 (CD44+CD25+), DN3 (CD44−CD25+), and DN4 (CD44−CD25−). Early stages are not committed to the T cell lineage (i.e., fate restricted), allowing other lineages to develop (3). B cells, dendritic cells, myeloid cells, and natural killer (NK) cells can all be generated from CD44+CD25−c-kithi early thymic progenitors (ETPs) (4, 5), DN1 cells, and—albeit to a lesser extent—DN2 cells (6). These multipotent cells, which can enter a number of differentiation programs, are directed toward the T cell lineage via a process called specification. The irreversible capacity to develop solely into T cells occurs somewhat later and is referred to as T lineage commitment; this process also involves the active repression of non-T cell lineages (7–9).
The microenvironment of the thymus provides a cellular context that drives T cell development. This process is initially driven by the expression of Notch ligands, particularly delta-like protein 4 (DLL4) (10), and later in the DP stage by providing the signals required to control positive selection [for self-MHC (major histocompatibility complex)] and negative selection (against autoreactive T cell clones). The various stages in T cell development have been investigated in great detail using flow cytometry and genomic analyses; thus, T cell development serves as a paradigm for the molecular regulation of cell fate (11, 12). The fact that T cell development occurs in an anatomically separate niche has allowed researchers to study the detailed successive steps that underlie lineage specification and commitment. All of the events that establish the identity of T cell precursors are driven by Notch signaling (13), involving binding of the transcription factor RBP-J (also known as CBF1) to intracellular Notch ligands, thereby forming an active transcription factor complex in ETPs.
The subsequent stages of T cell development are governed by several key transcription factors that form an intricate gene regulatory network (14). The core set of transcription factors in the early phases of T cell development is T cell factor 1 (Tcf1) (encoded by the gene confusingly termed Tcf7), Gata3, Bcl11b, and two members of the E2A family (E2A and HEB), Ikaros and Runx1 (14–17). The Tcf7 gene is a direct Notch signaling target and the first T cell–specific transcription factor induced by Notch signaling (18); in contrast, Bcl11b drives T cell commitment by limiting the NK cell fate and activating the T cell developmental gene program at the DN2-DN3 stage (19), leading to expression of the fully rearranged T cell receptor β (TCRβ) gene at the DN3 stage. Rothenberg and colleagues (14) showed that four transcription factors—Tcf1, Gata3, Notch/RBP-J, and, to a lesser extent, Runx1—are required for the timed expression of Bcl11b. Of these four transcription factors, Tcf1 is the most complex, as it can act as both a transcriptional repressor (e.g., when bound by a co-repressor such as Groucho) and a transcriptional activator by binding β-catenin to respond to canonical Wnt signals (20). Tcf1 also acts as a tumor suppressor gene (21, 22), and it can be functionally replaced—at least partially—by Lef1, a related transcriptional regulator expressed at approximately 50-fold lower levels than Tcf1 (23). Additional complexity arises from many alterative splice forms and alternative promoter usage, leading to at least six different Tcf1 isoforms that are differentially expressed throughout the T cell lineage.
The precise role that Tcf1 plays in regulating T cell specification and commitment, and its interaction with other core regulatory factors in T cell development, is not fully understood. Therefore, we examined the role of Tcf1 at the earliest stages of T cell development, focusing initially on fully committed DN3 cells. We found that Tcf1 is necessary for driving thymocytes down the T cell developmental path even after the T cell commitment stage, as Tcf1-deficient DN3 thymocytes can dedifferentiate into DN1/2-like cells that can then develop into the myeloid and B cell lineages. In addition, we found that Tcf1 supports this “lineage fidelity” via two direct—and functionally complementary—target genes, Gata3 and Bcl11b. An epistasis analysis using retroviral gene complementation in Tcf1-deficient stem cells revealed that the role of Gata3 in immature T cells is to repress B cell and myeloid fate, whereas Bcl11b establishes the T cell lineage program, and its expression can overcome the defect in T cell development in Tcf1-deficient thymocytes.
RESULTS
Tcf1 deficiency leads to several arrests in T cell development with increased non-T cells
Tcf1 deficiency results in multiple incomplete blocks in T cell development that vary from mouse to mouse. Besides the well-documented block at the ISP stage (24–26), T cell development can be arrested at DN1, DN2, and DN3 stages (fig. S1A). In contrast to these partial arrests in developing mice, transplanting Tcf1-deficient stem cells into adult recipient mice led to a complete block in T cell development at the DN1-DN2 transition (fig. S1B), presumably the result of an insufficient compensatory expression of Lef1 in these cells (27). We also observed increased percentages of non-T cell lineages, most notably B cells and myeloid cells (fig. S1, C and D), consistent with previous reports of ex vivo cultured Tcf1-deficient cells.
Phenotypically, fully committed DN3 Tcf1-deficient thymocytes have promiscuous gene expression and altered chromatin
Given the effects of Tcf1 deficiency on sequential stages of T cell development, we initially focused on those stages where thymocytes should be fully T cell committed. Therefore, we compared gene expression profiles between Tcf1-deficient thymocytes and wild-type (WT) thymocytes. The T cell commitment process starts at the DN2 stage and continues to the DN3a (CD25+CD44−CD27−) stage, in which a rearranged Tcrb gene is expressed in combination with pTA to form the pre-TCR complex in a process known as β selection. After β selection, the cells rapidly proliferate, express CD27, and are fully T cell–committed based on expression of a functional, rearranged Tcrb gene (28). We consider thymocyte αβ T cells committed when they express a fully rearranged TCRβ. We realize that there are definitions where T cell commitment occurs at earlier stages but phenotypically defined DN3(b) cells are here considered as the candidate population for committed T cells. We performed whole-transcriptome RNA sequencing (RNA-seq) on DN3b cells obtained from Tcf1-deficient and WT littermates (Fig. 1A), reasoning that at DN3b thymocytes should be fully T cell lineage–committed (see fig. S2B; CD27 and Ptcra). We found 108 genes with down-regulated expression [>1.5-fold, false discovery rate (FDR) < 0.05] in the Tcf1−/− DN3b thymocytes but also 97 up-regulated genes (table S1). For visualization, the top 100 differentially expressed genes are shown, and the absence of Tcf7 expression was confirmed in Tcf1-deficient DN3b cells. Furthermore, the RNA-seq analysis shows fewer rearranged Tcrb genes than in WT control DN3b thymocytes, as shown for the Trbj expressed gene segments. We used the genes differentially expressed between Tcf1-deficient and WT DN3b cells in a gene set enrichment analysis (GSEA) and used published gene sets of T cell developmental stages to establish DN1 and DN2 signatures (29). The genes highly expressed in Tcf1−/− DN3b clustered strongly with the DN2-specific gene set (DN2a and DN2b, but not DN1), indicating that they share many characteristics of earlier developmental stages that are less T cell–committed (Fig. 1A and fig. S2A). The RNA-seq data also indicated that many of the T cell commitment genes were low or not expressed, while genes involved in non-T cell lineages (Pax5, Pu.1, and Blc11a) were highly expressed in the Tcf1-deficient cells compared to the control DN3b cells. On the basis of these data, we validated the expression of a number of important T cell developmental genes by quantitative polymerase chain reaction (qPCR) on sorted DN1, DN2, DN3, and DN4 thymocytes. These results validated the RNA-seq data and showed lower expression (twofold change) of the T cell–specific transcription factors Gata3 (DN1 to DN4) and Bcl11b (DN2 stage) (with higher expression of its functional counterpart Bcl11a), while the B cell commitment marker CD19 and the myeloid-associated factor Pu.1 were significantly higher expressed in the Tcf1-deficient thymocytes (Fig. 1B and fig. S2B). In addition, genes known to be associated with stem/progenitor cells [sometimes referred to as legacy genes (1)] such as c-kit were also significantly higher expressed (Fig. 1B), while both Wnt and Notch target genes (HES-1 and Axin2) were decreased. Collectively, these data showed that while in some regard Tcf1−/− DN3b thymocytes were T cell–committed (phenotypic markers and expression of some Tcrb genes), they also showed lineage infidelity, with expression of master regulatory genes from non-T cells.
Fig. 1. Tcf1-deficient DN3b cells show promiscuous gene expression compared to WT littermate controls.

(A) Heat map of the top 100 differentially expressed gene as determined by RNA-seq of sorted DN3b cells from WT and Tcf1-deficient thymi. GSEA of the differentially expressed genes (Tcf1−/− KO over Tcf1 WT for DN3b) is enriched for DN2 genes (DN2a and DN2b with NES +1.23 and + 1.53, respectively). (B) qPCR validation of RNA-seq data for selected T cell–specific genes, genes expressed in non-T cells, and legacy genes whose expression is inherited from stem cells/multipotent progenitors. The levels of expression are normalized by ABL-2 expression as housekeeping gene. (Mann-Whitney U test; *P < 0.05, **P < 0.01, and ***P < 0.001. Error bars represent the SD of three pooled mice and from two independent experiments.)
The strongly reduced number of thymocytes due to the lack of Tcf1 is explained not only by the developmental arrests and differentiation into non-T cells but also by high levels of apoptosis. Compared to WT cells, we found increased levels of apoptosis in Tcf1-deficient cells at nearly every stage (fig. S3A), as well as decreased cell proliferation in the DN2 and DN4 stages (fig. S3B).
Gata3 and Bcl11b are direct targets of Tcf1 and down-regulated in Tcf1-deficient thymocytes
The down-regulated mRNA expression levels of the transcription factors Gata3 and Bcl11b in various DN thymocyte stages in Tcf1-deficient mice suggested that these factors may be direct target genes of Tcf1. In accordance, the Bcl11b and Gata3 promoter/enhancer sequences contain conserved Tcf/Lef binding sites (30, 31). To check whether in ex vivo DN thymocytes these promoters are regulated in a Tcf-dependent manner, we performed chromatin immunoprecipitation using a monoclonal antibody specific for Tcf1 (Fig. 2A) followed by qPCR. This revealed binding of Tcf1 to the Gata3 and Bcl11b promoter sequences in WT DN thymocytes, but not in Tcf1-deficient thymocytes, consistent with both genes being direct target genes of Tcf1. This supports previous reports on OP9-DL1 cultures (18) and reporter gene assays.
Fig. 2. Chromatin accessibility analysis in Tcf1-deficient versus WT DN thymocytes.

(A) Chromatin immunoprecipitation with an antibody specific for Tcf1 revealed that the Gata3 promoter and the Bcl11b enhancer are occupied by Tcf1 in vivo, whereas in Tcf1 KO DN thymocytes, no binding can be detected. Negative controls with IgG instead of anti-Tcf1 showed no enrichment. (Multiple t test. Error bars represent the SD of at least three pooled mice and from two independent experiments.) (B) Heat map of DESeq2 normalized read counts of ATAC-seq shows differentially accessible regions between WT and Tcf1−/− in DN3a and DN3b. Motif analysis was performed in the differentially accessible regions using HOMER showing the three highest scores and Tcf1 score. (C) ATAC-seq data mined for the Bcl11b, Gata3, and Trbj (T cell Receptor Beta) genomic regions. Per locus, the relative abundance of transposase accessible regions is indicated. The individual ATAC-seq profile from each genotype is shown. Data are shown as normalized read density.
This finding was further substantiated by ATAC-seq (assay for transposase-accessible chromatin sequencing) data, which indicate chromatin accessibility. In total, 68,883 and 30,357 peaks were found in WT samples, and for Tcf1−/− samples, 40,716 and 68,605 peaks were found (fig. S2C).
To find regions with differentially chromatin accessibility between Tcf1−/− and WT for DN3a and DN3b thymocytes, we looked for peaks statistically different between the conditions. For this analysis, only differential peaks with FDR less than 0.05 were taken into account. In DN3a, 564 accessible sites were lost in Tcf1−/− cells, from which 141 were Tcf1 binding sites. Only eight sites were statistically significantly higher in Tcf1−/− containing three Tcf1 binding sites. In the case of DN3b, extra sites were lost in Tcf1−/− compared to Tcf1 WT (4950 in total), including 756 Tcf1 binding sites. Twenty-one sites were more accessible, but no Tcf1 binding sites were found. These results indicate that global chromatin accessibility was higher in WT thymocytes than in Tcf1-deficient thymocytes (Fig. 2B). Both DN3a and DN3b share the fact that Runx motifs seem to be abundantly lost upon Tcf1 deficiency (Fig. 2B), in accordance with the diminished Runx1 expression shown in the RNA-seq data (fig. S2B).
Focusing on the Bcl11b and Gata3 promoter/enhancer sequences, the chromatin in these promoters was less accessible compared to WT littermate control DN3b cells (Fig. 2C). Similarly, the TCRB loci were much less accessible in accordance with the RNA-seq data. The full genome-wide data analysis is provided in table S2. No major differences in chromatin accessibility were found at genes involved in alternative lineages, indicating that expression of these genes was not regulated at the level of chromatin opening. Collectively, these data show profound differences because of the lack of Tcf1 in chromatin accessibility and expression of genes and promoters associated with T cell commitment.
Phenotypically, fully committed DN3 Tcf1-deficient thymocytes dedifferentiate into DN1 thymocytes, B cells, and myeloid cells
On the basis of the hypothesis derived from these results, that Tcf1-deficient DN3 thymocytes may not be fully T cell–committed, we sought to better investigate the differentiation capacity of Tcf1−/− DN3 thymocytes. Therefore, DN3 cells were sorted and cultured under conditions with strong T cell–inducing capacity (OP9-DL1 system). Most of the WT DN3 thymocytes differentiated further into DN4 cells, with a smaller part remaining DN3 (Fig. 3, A and B). Unexpectedly, most Tcf1−/− DN3 thymocytes dedifferentiated into DN1 and DN2 cells, with extensive B and myeloid development, while only a minority of cells remained DN3 without any further development along the T cell lineage (Fig. 3, A and B). In particular, development into B cells was extensive, with up to 60% of DN3 thymocytes developing into B cells (Fig. 3, A and B). These dedifferentiated DN1 and DN2 cells were not a contaminating fraction in the sorted DN3 cells that expanded, as intracellular staining for Tcrb revealed high Tcrb expression in these DN1/2 cells at similar levels to cells remaining in DN3 stage and WT DN3 and DN4 cells (Fig. 3C). Therefore, these DN1- and DN2-like cells were derived from the sorted “fully” committed DN3 cells. Similarly, non-T cells (B and myeloid cells) developing in the assay expressed intracellular TCR, indicating that they also derived from the seeded DN3 Tcf1−/− promiscuous cells. We conclude that Tcf1 knockout (KO) cells dedifferentiate to less committed cells and exhibit lineage infidelity with significant development into alternative (non-T) lineages. When ETP cells rather than DN3 cells were seeded on OP9-DL1, as expected, Tcf1-deficient cells were arrested in development at DN1 (fig. S4A), with abundant B and myeloid development, whereas WT stem cells differentiated along the T cell lineage with many fewer non-T cells (fig. S4B).
Fig. 3. Tcf1-deficient DN3 cells dedifferentiate into DN1/2-like cells with multipotent lineage capacity.

(A) WT DN3 cells sorted and seeded on OP9 WT/OP9-DL1 (10:1) cells develop largely further into DN4 or remain DN3 after 7 days in culture, while Tcf1-deficient cells develop into DN1 and DN2 cells (pregated Thy1+ Lin− cells) with prominent B cell (B220+ CD19+) and myeloid cell (CD11b+ Gr1+) development. (B) Quantification of the developmental plasticity and dedifferentiation effects of DN3 Tcf1-deficient thymocytes into DN1, DN2, myeloid, and B cells. (C) Intracellular TCRβ staining reveals the dedifferentiated DN1 and DN2 cells to be derived from DN3 cells. (Mann-Whitney U test. Error bars represent the SD of three samples from three independent experiments.)
Dedifferentiation into alternate lineages can be prevented by expressing Gata3 in Tcf1-deficient thymocytes
Epistasis analysis is a powerful genetic tool, often used in model organisms such as Drosophila to investigate hierarchical relationships between genes (32). It can be more complex to perform in mammals such as mice, where not only expression per se but also gene dosage is important. For instance, while complete loss of Gata3 blocks T cell development at the earliest stages, transgenic overexpression of Gata3 can lead to development of mast cells in the thymus (33–36). We therefore expressed Gata3 and Blc11b using recombinant retroviruses as they have a broad range of expression that would allow different phenotypes to be selected under the strong developmental pressure of the thymic microenvironment. We used retroviruses encoding green fluorescent protein (GFP) only, Gata3 together with GFP, or Bcl11b together with GFP to investigate complementation of the Tcf1 phenotype by either Gata3 or Bcl11b (Figs. 4A and 5A). We used retroviruses solely encoding GFP as negative controls.
Fig. 4. Reexpression of Gata3 suppresses B and myeloid development in Tcf1 deficiency.

(A) Layout of retroviral complementation experiments with GFP control and/or Gata3. (B) Gata3 expression partially overcomes the DN1 thymocyte block and (C) suppressed the enhanced non-T cell lineages (B and myeloid cell development) after 7 days in the OP9-DL1 culture system. FACS shows representative plots and graphs quantitative data from replicate measurements. (Multiple t test analysis. Error bars represent the SD from two independent experiments.) (D) In vivo complementation [Ly5.2 Tcf1 (WT or KO)–transduced stem cells transplanted into Ly5.1 recipient mice] reveals suppression of B cell development also in the thymus (right) 8 weeks after transplantation but minimal and partial rescue of T cell development in the thymus (left and middle). (Middle: Multiple t test analysis. Right: Paired t test. Error bars represent the SD from three individual mice per group.)
Fig. 5. Reexpression of Bcl11b rescues T cell development in Tcf1-deficient stem cells.

(A) Layout of retroviral complementation experiments with Bcl11b. (B) Thy1 expression is rescued by Tcf1 deficiency by expression of Bcl11b after 14 days in OP9-DL1 culture (pregated Lin−GFP+ cells). (C) Bcl11b fully rescues T cell development from Tcf1−/− stem cells that otherwise are arrested in DN1 (pregated Thy1+Lin−GFP+). (D) Intracellular TCRβ expression can be restored in Tcf1-deficient cells by expression of Bcl11b (pregated Thy1+Lin−GFP+ DN subset). (E) Bcl11b overexpression does not affect myeloid and B cell development. (Two-way ANOVA. Error bars represent the SD from three independent experiments.)
Reexpression of Gata3 could partially rescue the development of Tcf1−/− thymocytes from a DN1 arrest to an apparent CD25+ DN2 stage but not further (Fig. 4B). However, as Thy1 expression was not increased on the apparent DN2 cells (fig. S5A), they cannot be considered real DN2 cells. Similarly, key T cell lineage–specific (CD3 and PtA) gene expression was not induced upon forced Gata3 expression (fig. S6, A and B). Strikingly, high Gata3 expression strongly suppressed the enhanced development of B and myeloid cells (granulocytes and monocytes) from Tcf1−/− thymocytes. This also occurred to some extent when starting with WT cells (Fig. 4C). Competitive stem cell transplantation (Ly5.2 Tcf1 stem cells/Ly5.1 recipient mice) was used to assess whether reexpression of GATA3 could rescue T cell development in the thymus in vivo. The suppression of B cell development (Ly5.2 B cells) in the thymus was also observed in vivo when Gata3-complemented Tcf1-deficient stem cells were transplanted in irradiated recipient mice (Fig. 4D, right). However, thymic T cell development (Ly5.2 T cells) again was arrested at a DN1/2 transition, barely different than GFP control transduced cells (Fig. 4D, left and middle). Thus, the major role of Gata3 in earliest DN development is the suppression of non-T cell development with only a minor feed forward role into the T cell program.
The T cell lineage–specific defects caused by Tcf1 deficiency can be rescued by expressing Bcl11b
Enforced expression of Bcl11b (Fig. 5A), in contrast, rescued the T cell developmental defect of Tcf1-deficient cells virtually completely. Bcl11b transduced Tcf1-deficient stem cells developed readily into Thy1-positive (Fig. 5B and fig. S5B) cells and could develop into DN2 and DN3 thymocytes to a similar degree as WT thymocytes (Fig. 5C and fig. S5C) [while nontransduced Tcf1-deficient cells are arrested at the DN1/DN2 stage as the control cells (Fig. 5D)]. In addition, expression of TCRβ by intracellular flow cytometry was also restored to WT levels in DN3 and DN4 by expressing Bcl11b in the Tcf1 KO background (Fig. 5D). Accordingly, TCR gene expression was rescued upon Bcl11b overexpression in Tcf1-deficient cells (fig. S6B). In contrast, expression of Bcl11b did not markedly influence B and myeloid development from Tcf1-deficient cells (Fig. 5E and fig. S5C). Overexpression of Blc11b did suppress the development of NK cells (fig. S5E), consistent with its described role in promoting T cell fate over NK cell fate at the DN2 stage (19).
DISCUSSION
T cell development has been used as a classic example of a relatively ordered pathway to study cell fate determination (16), thereby giving the impression that transcriptional regulation during T cell development is a well-understood process. Despite this general belief, however, and compared to other developmental processes (for example, B cell development, which has similar requirements in terms of proliferation, lineage restriction, immune receptor rearrangement, and checkpoints for premature and mature immune receptors), the roles of the major transcription factors in T cell development are rather poorly understood. In B cell development, a clearly defined linear hierarchical relationship exists between E2A, EBF1, and Pax5 (37–44). However, with respect to early T cell development, whether the Notch (RBP-J), Gata3, Bcl11b, Runx1, E2A, Tcf1/Lef1, Ikaros, and/or Hox genes play unique, redundant, or synergistic roles remains unclear and is the subject of intense research that focuses largely on either individual factors or the collective activity of these factors using computational biology. Considering that Notch signaling is required for T cell development and given that the first T cell–specific target gene is Tcf7 (18), which encodes Tcf1, we investigated the process of T cell lineage commitment in Tcf1-deficient mice.
The study of Tcf1-deficient mice is generally complicated by three factors. First, in the absence of Tcf1, the HMG box transcription factor Lef1—which is expressed in the thymus, albeit at much lower levels than Tcf1—plays a compensatory role (fig. S2B) (23, 27). This low-level expression of Lef1 causes incomplete penetrance of the Tcf1-deficient phenotype. However, if adult Tcf1-deficient stem cells are either transplanted into recipient mice or cultured on OP9-DL1 cells to induce T cell differentiation, a complete block occurs at the DN1 stage (see Fig. 1D), as Lef1 expression is believed to result from reaming fetal expression in the thymus (21, 22, 27). Therefore, in our experiments, we used bone marrow–derived cells obtained from Tcf1-deficient mice. Second, Tcf1-deficient mice are prone to developing T cell lymphomas in the thymus (22), which is similar to T cell acute lymphoblastic leukemia in patients. As discussed above, this issue can be overcome by using Tcf1-deficient stem cells instead of thymocytes. The third issue associated with studying Tcf1-deficient mice is that Tcf1 functions as both a transcriptional repressor and a transcriptional activator (for example, when bound to the Wnt mediator β-catenin). When Tcf1-dependent promoters were tested using in vitro reporter systems, transcription occurred only when β-catenin was also expressed (45, 46). Consistent with this notion, Tcf1 binds to the promoter/enhancer regions of the target genes Gata3 and Blcl11b, and it seems likely that Tcf1 binds to β-catenin at these promoter regions. Co-chromatin immunoprecipitation experiments provide initial evidence for this notion, as β-catenin can also be found at active promoters where Tcf1 binds. In addition, DN stages of T cell development show high canonical Wnt signaling, which is driven by β-catenin and Tcf/Lef (47). Of note, expression of the Wnt target gene Axin2 was markedly reduced in thymocytes lacking Tcf1 (Fig. 1B). On the other hand, some of Tcf1’s functions in the earliest stages of T cell development are independent of β-catenin (18), possibly due to the redundant role of Lef1.
A seminal study by Busslinger and colleagues (42, 43, 48) revealed that Pax5 is a major lineage commitment factor in the development of B lymphocytes. Thus, B cells that lack Pax5 can dedifferentiate into multipotent progenitor cells that can replenish all hematopoietic lineages, even in vivo. In this respect, our findings are somewhat analogous, as Tcf1-deficient DN3 cells—which seemingly are fully committed—have promiscuous gene expression and can dedifferentiate into immature cells that can give rise to non-T cell lineages, including B cells and myeloid cells. In the T cell lineage, such dedifferentiation has also been shown to occur in E2A- or HEB-deficient thymocytes (49, 50). Key transcription factors that drive alternate lineages (e.g., the transcription factors Bcl11a, Pax5, and Pu.1) are robustly expressed in Tcf1-deficient DN3 and DN4 cells, but not in WT cells. In contrast with Pax5-deficient cells, however, only a small number of Tcf1-deficient cells survive the dedifferentiation process, which is likely due to the high level of apoptosis in Tcf1-deficient thymocytes (fig. S3). In addition, the assessment of chromatin status by ATAC-seq revealed that in Tcf1-deficient thymocytes, the chromatin is more condensed and several key T cell–specific loci (for instance, the Tcrb locus) are less accessible and therefore likely not as readily transcribed and expressed (Fig. 2, B and C). Therefore, the mechanisms underlying dedifferentiation in Pax5 deficiency and as reported here in Tcf1 deficiency appear to be mechanistically different. It should also be noted that formal proof of dedifferentiation in Tcf1 deficiency would require use of a conditional KO model using a floxed allele with a Cre enzyme under control of a late acting promoter during thymocyte differentiation. As commitment implies loss of plasticity and the capacity to give rise to only one cell type but not to others, Tcf1 deficiency, in contrast, is associated with lineage infidelity and lack of commitment.
Recent work has investigated the epigenetic status of DP thymocytes in Tcf1 deficiency, similar to our experiments using DN3 thymocytes (51). In agreement, Tcf1−/− DN thymocytes also display more condensed chromatin (Fig. 2B). Yet, Tcf1 in the context of T cell commitment and immature thymocyte development seems to act mostly as a transcription factor regulating expression of other key T cell–specific genes than acting as a chromatin-modifying factor per se. An intrinsic histone deacetylase (HDAC) activity has been shown for Tcf1 in CD8+ cells (52). Our analysis in DN3 T cell populations revealed that only a very small number of sites containing a Tcf1 motif (n = 3 in DN3a and n = 0 in DN3b) gained accessibility in Tcf1−/− cells. This supports an activator rather than a suppressor function for Tcf1 in early T cells. Similar observations, i.e., most of the sites (80%) lost accessibility in Tcf1−/− DP cells, were reported by others in total DP thymocytes (51), again consistent with a function of Tcf1 as a transcriptional activator. One explanation could be that the HDAC activity of Tcf1 is differentially required (e.g., cell type–specific, context-dependent manner) and would be different in developing T cells in the thymus versus effector cell maturation in CD8+ peripheral cells. This is consistent with the observation that HDAC-deficient Tcf1 could largely restore differentiation into the CD4+ lineage (52). Nevertheless, further analyses will be required to fully understand the activator/repressor functions of Tcf1 in immune cell development.
Given that both Bcl11b and Gata3 are key target genes for Tcf1, we expressed these transcription factors in Tcf1-deficient cells in an attempt to rescue the thymic phenotype. Similar analyses of epistasis have been used previously in model organisms (e.g., Drosophila) to delineate both hierarchical and functional relationships. The expression of exogenous Gata3 has been shown to suppress B cell development in the WT thymus (35, 53, 54); furthermore, we found that Gata3 also suppresses myeloid fate in DN thymocytes. Gata3 does not suppress myeloid fate in the bone marrow, whereas the effect on B cell development also occurs outside of the thymus.
Our finding that the constitutive expression of Bcl11b in Tcf1-deficient cells fully rescued T cell development suggests a division of labor between Bcl11b and Gata3, with Gata3 suppressing non-T cell lineages and Bcl11b inducing the expression of T cell–specific genes. This is schematically illustrated in Fig. 6. Together, the data from our group and others indicate a gene network in which Notch signaling via RBP-Jκ drives the expression of Tcf1, which, in turn, activates Gata3 and Bcl11b, most likely in collaboration with Notch signals that can also act directly on these genes’ promoters. In addition to its requirement for initiating the T cell commitment process, Tcf1 expression is also required to maintain lineage fidelity. In skin stem cells, lineage infidelity increases the likelihood of malignancy (55). Thus, given that loss of Tcf1 leads to the rapid development of T cell lymphomas (22, 23), lineage infidelity may also serve as a previously unrecognized factor in leukemogenesis.
Fig. 6. Hierarchy of the core transcription factors in immature T cell development.
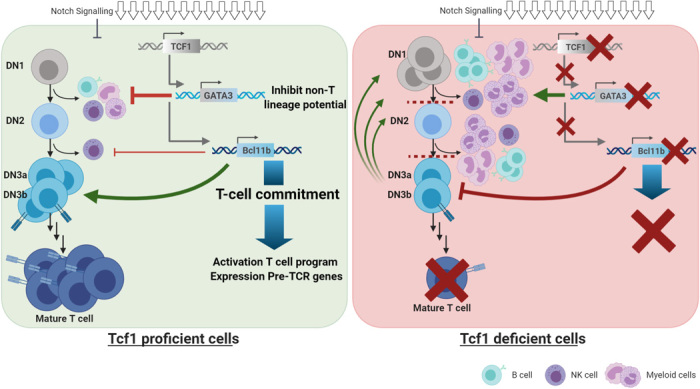
On the basis of the proven functional interactions shown in Figs. 4 and 5. Notch signaling (indicated by the open arrow symbols) induces Tcf1 expression that subsequently has two target genes: Gata3 and Bcl11b. Gata3 has a minor role in supporting development along the T cell linage but mainly acts to suppress the myeloid and B cell fates. In contrast, Bcl11b induces a T cell–specific program but has minor roles in suppressing alternative lineages with exception of NK cell development that is suppressed by Bcl11b. Collectively, there is a clear functional hierarchy of transcription factors. Potential additional roles for Runx1 and E2A are not shown here.
MATERIALS AND METHODS
Mice
C57Bl/6 TCF-1−/− ΔVII/ΔVII mice were originally described by Verbeek et al. (26), and C57Bl/6-Ly5.1 mice were purchased from Charles River Laboratories. Mice were bred and maintained in the animal facility of Leiden University Medical Center. All animal experiments were performed in accordance with legal regulations in The Netherlands and with approved protocols of the Dutch animal ethical committee.
Mice used for transplantation assay were kept in specific pathogen–free section and fed with special food and antibiotic water. Genotyping assay of newborn Tcf1 mice was performed with DNA samples from earpieces using a GoTaq Flexi DNA Polymerase kit (Promega) according to the manufacturer’s instructions.
Flow cytometry and cell sorting
Single-cell suspensions from thymus, spleen, bone marrow, and blood were stained with monoclonal antibodies against CD3e, CD4, Cd8a/Ly-2, CD11b/Mac-1, CD19, CD25, CD27, CD44/Ly-24, CD45.1/Ly-5.1, CD45.2/Ly-5.2, B220/CD45R, CD90.2/Thy1.2, CD117/c-kit, CD135/Flt3, Gr1/Ly-6G-6C, NK1.1, Sca1/Ly-6A, TCRβ, TCRvβ5.1/5.2, TCRβ6, TCRβ8, and Ter-119/Ly-76 (see table S3). All antibodies used were directly conjugated to biotin, fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll-a protein (PerCP), PE-Cy7, allophycocyanin (APC), APC-Cy7, or efluor450. Biotinylated antibodies were revealed with streptavidin-conjugated antibodies (PE, efluor450, APC-Cy7, APC, or Pe-Cy7) (all antibodies were purchased from BD Biosciences, BioLegend, or eBioscience).
Cells were blocked with normal mouse serum (NMS; Invitrogen) for 10 min at room temperature, and subsequently, cell surface staining was performed in two steps. First, cells were incubated for 30 min at 4°C in the dark with the antibody-mix solution including directly conjugated antibodies at the optimal working solution in fluorescence-activated cell sorting (FACS) buffer [phosphate-buffered saline (PBS; pH 7.4), 0.1% azide, and 0.2% bovine serum albumin (BSA)]. After washing with FACS buffer, a second 30-min incubation step at 4°C was performed with the streptavidin-conjugated antibody mix.
Cell apoptosis was assessed by annexin V and 7AAD (7-Aminoactinomycin D) staining, which was performed following the PE Annexin V Apoptosis Detection Kit protocol (BD Pharmingen) after the cell surface staining. Proliferation assay was performed by intracellular Ki67 staining [mouse immunoglobulin G (mIgG) as control] with PE Mouse Anti-Human Set protocol (BD Pharmingen). For that purpose, cells were initially stained for cell surface markers as described previously and subsequently fixated and permeabilized by using fixation/permeabilization buffer (eBioscience) for an hour at 4°C. Cells were then washed with permeabilization (eBioscience) buffer with 2% NMS and stained with Ki67 or IgG1 solution for 30 min at 4°C in the dark. The same procedure was used to assess intracellular TCRβ expression.
DP CD4 and CD8 cells before DN cell sorting and lineage-positive cells before LSK/LK (Lineage–Sca+cKit–/Lineage–cKit–)–sorting were depleted using magnetic-activated cell sorting (autoMACS, Miltenyi Biotec). For DN sorting, thymocytes were first stained with anti-CD4 and CD8-biotin, followed by streptavidin microbead staining according to the manufacturer’s instruction (Miltenyi Biotec). For LSK/LK cell sorting, lineage depletion kit (Miltenyi Biotec) was used according to the manufacturer’s instruction. Subsequently, depleted cells were stained again for DNs or LSKs as described before. Cell sorting was performed on FACSAria II (BD Biosciences), or stained cells were measured with FACSCanto II and LSR Fortessa X-20 (BD Biosciences). Data were analyzed using FlowJo (Tree Star). All different hematopoietic populations were defined as described in table S4 and fig. S7.
Cell culture
Bone marrow–derived stromal cell line OP9 and OP9-DL1 cells that ectopically express the Notch ligand delta-like 1 (DL1) were used as described by J. C. Zuñiga-Pflucker. Sorted DN cells were cultured on OP9 or OP9 WT/OP9-DL1 (10:1) confluent monolayers in αMEM (Minimum Essential Medium Eagle - alpha modification) (Lonza)–10% fetal calf serum (FCS), 1% penicillin/streptomycin (Life Technologies), and GlutaMAX (Life Technologies) medium complemented with recombinant mouse Flt3-ligand (rmFlt3L) (50 ng/ml), recombinant mouse stem cell factor (rmSCF) (50 ng/ml), recombinant mouse interleukin-7 (rmIL-7) (10 to 1 ng/ml), and 50 μM β-mercaptoethanol (β-ME; Sigma-Aldrich) (all cytokines were purchased from R&D Systems). Cells were harvested after 7 to 14 days of coculture and analyzed by flow cytometry.
Transduced LSK and LK with LZRS-ires-eGFP (control), LZRS-Gata3-eGFP, or LZRS-Bcl11b-GFP vector were cultured on OP9-DL1 monolayer for 6 to 14 days in αMEM–10% FCS complemented with rmIL7 (10 ng/ml), rmFlt3L (50 ng/ml), rmSCF (10 ng/ml), and β-ME (50 μM). Harvested cells were analyzed by flow cytometry or sorted.
Retroviral production
LZRS-Gata3 and Bcl11b plasmids were obtained from Addgene and cloned into LZRS-ires-eGFP vector (Addgene, control vector). Control, Gata3, and Bcl11b retroviruses were generated using Phoenix ecotropic and amphotropic packaging cell line (ATCC). Cells were cultured in IMDM (Iscove’s Modified Dulbecco’s Medium) (Lonza)–10% FCS–1% penicillin/streptomycin–1% glutamine and transfected with plasmids using X-treme Gene9 DNA transfection reagent (Roche) protocols. Selection of transfected cells was performed with puromycin (1 mg/ml; Sigma-Aldrich) for a week, and viral supernatant was harvested at 24 and 48 hours.
Retroviral transduction
LSK and LK sorted cells were stimulated overnight in StemSpan serum-free expansion medium (STEMCELL Technologies) supplemented with rmTPO (recombinant mouse thrombopoietin) (10 ng/ml; R&D Systems), rmFlt3L (50 ng/ml; R&D Systems), and rmSCF (100 ng/ml; R&D Systems). Hematopoietic progenitors were transduced using RetroNectin (Takara Bio Inc.)–coated wells according to the manufacturer’s instructions. Nontissue culture plates were coated with RetroNectin overnight at 4°C and then blocked with 2% BSA in PBS for 30 min. Retroviral supernatant (24 or 48 hours) was centrifuged at 1500g for 1 hour at 32°C and incubated an extra hour at 37°C. After coating, viral supernatant was removed and stimulated cells were immediately added on the virus-coated plates. Cells were cultured in StemSpan medium supplemented with rmTPO (10 ng/ml), rmFlt3L (50 ng/ml), and rmSCF (100 ng/ml) and transduced overnight at 37°C. LZRS-ires-eGFP–, LZRS-Gata3-ires-eGFP–, and LZRS-Bcl11b-ires-eGFP–transduced cells were used for in vitro and in vivo approaches.
Quantitative real-time PCR
RNA from sorted cells was purified using a Micro RNeasy kit (Qiagen) and reverse-transcribed into complementary DNA (cDNA) using a SuperScript III kit (Invitrogen). Reverse transcription PCR (RT-PCR) was performed using TaqMan Universal Master Mix II in combination with specific probes for indicated genes from Universal Probe Library (Roche). Specific primers for ABL-2, Bcl11a, Bcl11b, Gata3, Pax5, PU.1/Spfi1, IL-7Ra, CD117/c-kit, ID2, Axin-2, Hes1, CD3e, CD3d, pTa, and ZAP70 were designed and purchased from Sigma-Aldrich (see specific gene sequences in table S5). Samples were analyzed with a StepOnePlus RT-PCR system (Life Technologies). Relative transcript abundance was determined by ΔCt, and expression levels were normalized for the endogenous reference gene ABL-1. All samples were run at least in duplicate.
RNA sequencing
RNA from sorted DN3b cells (Lin−CD25+CD44−CD27+) from Tcf1−/− and WT littermate thymi was isolated using the Mini RNeasy Kit (Qiagen) The integrity (scores >9.0) of the RNA was determined on the Agilent 2100 Bioanalyzer (Agilent). Total RNA enrichment for sequencing poly(A) RNAs was performed with the TruSeq mRNA Sample Preparation Kit (Illumina). One microgram of total RNA for each sample was used for poly(A) RNA selection using magnetic beads coated with poly-dT, followed by thermal fragmentation. The fragmented poly(A) RNA–enriched samples were subjected to cDNA synthesis using an Illumina TruSeq preparation kit. cDNA was synthesized by reverse transcriptase (SuperScript II) using poly-dT and random hexamer primers. The cDNA fragments were then blunt-ended through an end-repair reaction, followed by dA-tailing. Subsequently, specific double-stranded barcoded adapters were ligated, and library amplification for 15 cycles was performed. The pooled cDNA library consisted of equal concentration barcoded samples. The pooled library was sequenced in one-lane, 360–base pair (bp) single read on HiSeq2500 (Illumina). Raw RNA-seq reads are accessible on SRA (Sequence Read Archive) by accession number SRP158670.
RNA-seq data processing
FASTQ files were aligned to the mm10 genome using STAR 2.5.1b (56). Transcript counts were quantified and annotated using HTSeq-0.6.1. WT sample 3 was removed because of a low number of aligned reads.
Differential expression and statistical analysis
Differential expression of DN3b WT versus TCF-1−/− was identified by using DESeq2 (57), after filtering for genes with a low read count (>5 reads per sample), resulting in 205 differential expressed genes (97 up-regulated in KO and 108 down-regulated in KO) at a P value of <0.05 (FDR adjusted) and a log2 fold change of >1.5.
Gene set enrichment
RNA-seq results from mouse T cell precursors in different developmental stages including DN1, DN2a, DN2b, DN3, and DP [Gene Expression Omnibus accession: GSE89198; (58)] were used to create DN1 and DN2 gene sets. Of this RNA-seq dataset, log2-transformed FPKM (Fragments Per Kilobase Million) values of 25 DN2 and 8 DN3 WT mice were used for differential expression analysis with Limma. Genes that were differentially up-regulated [P < 0.05 and log fold change (FC) > 2] between DN1 versus DN3 (365 genes), DN2a versus DN3 (342 genes), DN2b versus DN3 (120 genes), and DN2a/b combined versus DN3 (141 genes) were used as gene sets for GSEA. GSEAPreranked (GSEA 4.0.3, Broad) was run on all expressed WT versus TCF−/− RNA-seq genes, which were ranked by the P value and log FC generated by DESeq2. DN3b TCF−/− was negatively associated with the DN1 gene set [normalized enrichment score (NES) of −1.04] and positively associated with all of the DN2 gene sets (DN2a NES 1.23, DN2b NES 1.53, and DN2a/b combined NES 1.36).
Assay for transposase-accessible chromatin sequencing
Sorted DN3a (15,000) (Lin−CD25+CD44−CD27−) and DN3b (Lin−CD25+CD44−CD27+) cells were washed one time with cold PBS. Pellets were spin down at 500g for 5 min at 4°C, and the supernatant was removed carefully. Twenty microliters of transposase mix [10 μl of 2× TD (Tagment DNA) buffer, 1 μl of TDE (Tagment DNA Enzyme) (Nextera DNA Library Prep Kit; Illumina), 0.2 μl of digitonin (G9441, Promega), and 8.8 μl of nuclease-free water] was added to the cells. Reactions were incubated at 37°C for 30 min. Transposed DNA was purified using the MinElute Reaction Cleanup Kit (28204, Qiagen), amplified, and again purified according to published protocols (59). Size selection was performed using Low Range Ultra agarose (161-3107, Bio-Rad). Fragments between 150 and 600 bp in size were used for further analysis. Quality and quantity of the libraries were assessed by the Bioanalyzer High Sensitivity DNA Analysis Kit (Agilent) before sequencing. Libraries were sequenced 50 bp, paired-end, on HiSeq4000.
The reads were filtered by quality using TrimGalore tool (60) (default values), and the quality control was driven by FastQC (61) and MultiQC (62). The remaining reads were mapped to mm10 using bowtie2 (63) with –very-sensitive parameter. After all, before the peak calling, the read duplicates and multiple mapping reads were removed using Picard tools (http://broadinstitute.github.io/picard). The peaks for two WT and two Tcf1−/− samples were called using MACS2 (64) with the following parameters: -g mm -B –shift -100 -ext 200 –nomodel –q 0.05 and BigWig-tracks with FPKM were generated by deepTools (65). Coverage plots and heat maps were generated with deepTools using the BigWig tracks previously generated with the following parameters: --binSize 100 -m 3000 -b 1000 -a 1000. To find differential open chromatin regions, the differential peaks between WT and Tcf1−/− conditions were calculated by DiffBind R/Bioconductor package (66), and only the statistically significant peaks (FDR < 0.05) were taken into account for downstream analysis. Motif analysis on the differentially accessible regions was performed using Homer (http://homer.ucsd.edu/homer/) using the parameters : size given. MEME-FIMO (67) and Tcf1 position probability matrix (MA07769.1) from JASPAR (http://jaspar.genereg.net/) were used to analyze the distribution of the Tcf1 motif on the differentially accessible regions.
Chromatin immunoprecipitation
DN thymocytes (CD8−CD4−) from Tcf1−/− and WT littermates were sorted and subsequently cross-linked with formaldehyde (Sigma). Cross-linking was quenched with glycine, and after cell lysis, chromatin was sonicated into fragments. Sonicated chromatin was precleared and incubated with TCF-1 antibodies (C46C7; #2206, Cell Signaling Technology). Immunoprecipated chromatin complexes were purified and quantified by real-time PCR using FastStart Universal SYBR Green Master Mix (Roche). (See specific gene sequences in table S5.)
Stem cell transplantation
Competitive transplantation assay is used to determine HSC development and functionality in vivo by measuring multilineage reconstitution of hematopoiesis in irradiated transplanted mice. Competitive transplantation Ly5.2/Ly2.1 was used to assess whether in vivo reexpression of Gata3 could rescue T cell development in the thymus. Total 52,500 Ly5.2 Tcf1 (WT or −/−) transduced cells (mixed LSK and LK progenitors cells) were transplanted into lethally irradiated (8.07 Gy) Ly5.1 recipient mice (8 to 12 weeks), together with 300,000 splenocytes (Ly5.1) as support cells. Chimerism and peripheral T cells were analyzed at week 6 after transplantation in peripheral blood. Mice were sacrificed for analysis 7 weeks after transplantation to evaluate hematopoietic system repopulation. Mice were considered repopulated when ≥1% multilineage Ly5.2 Tcf1 cells could be detected. Single-cell suspension from the thymus, spleen, and bone marrow, as well as lysate blood, was analyzed by flow cytometry as described previously.
Statistical methods
All statistics were calculated, and all graphs were generated using GraphPad Prism 6 (GraphPad Software). Statistical significance was determined by Mann-Whitney U test (*P < 0.05, **P < 0.01, and ***P < 0.001), multiple t test, or two-way analysis of variance (ANOVA) depending on the experimental setting.
Supplementary Material
Acknowledgments
We are indebted to S. Verbeek and K. Cante for critically reading the manuscript and many suggestions and to H. Clevers for continued support. We thank S. Vloemans for expert assistance with the constructs. Funding: This work was supported, in part, by a TOP grant from ZonMW to F.J.T.S.; a H2020 grant SCID-Net to F.J.T.S., K.P.-O., and L.G.-P.; and a ZonMW E-RARE grant to F.J.T.S., M.C., and K.P.-O. Author contributions: Conceptualization: F.F., L.G.-P., and F.J.T.S.; methodology: F.J.T.S., K.P.-O., and L.D.; investigation: F.F., L.G.-P., M.v.E., H.W., M.B., J.C., D.S.L.G., M.C., and M.M.T.; writing (original draft): L.G.-P., K.P.-O., and F.J.T.S.; writing (review and editing): F.F., L.G.-P., M.B., M.C., M.M.T., K.P.-O., L.D., and F.J.T.S.; resources: M.B. and M.C.; supervision: F.J.T.S., K.P.-O., and L.G.-P. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw RNA-seq reads are accessible on SRA by accession number SRP158670. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaaw7313/DC1
REFERENCES AND NOTES
- 1.Rothenberg E. V., Moore J. E., Yui M. A., Launching the T-cell-lineage developmental programme. Nat. Rev. Immunol. 8, 9–21 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weerkamp F., Pike-Overzet K., Staal F. J., T-sing progenitors to commit. Trends Immunol. 27, 125–131 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Jenkinson E. J., Jenkinson W. E., Rossi S. W., Anderson G., The thymus and T-cell commitment: The right niche for Notch? Nat. Rev. Immunol. 6, 551–555 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Holländer G., Gill J., Zuklys S., Iwanami N., Liu C., Takahama Y., Cellular and molecular events during early thymus development. Immunol. Rev. 209, 28–46 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Wu L., Antica M., Johnson G. R., Scollay R., Shortman K., Developmental potential of the earliest precursor cells from the adult mouse thymus. J. Exp. Med. 174, 1617–1627 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luc S., Luis T. C., Boukarabila H., Macaulay I. C., Buza-Vidas N., Bouriez-Jones T., Lutteropp M., Woll P. S., Loughran S. J., Mead A. J., Hultquist A., Brown J., Mizukami T., Matsuoka S., Ferry H., Anderson K., Duarte S., Atkinson D., Soneji S., Domanski A., Farley A., Sanjuan-Pla A., Carella C., Patient R., de Bruijn M., Enver T., Nerlov C., Blackburn C., Godin I., Jacobsen S. E. W., The earliest thymic T cell progenitors sustain B cell and myeloid lineage potential. Nat. Immunol. 13, 412–419 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhandoola A., von Boehmer H., Petrie H. T., Zuniga-Pflucker J. C., Commitment and developmental potential of extrathymic and intrathymic T cell precursors: Plenty to choose from. Immunity 26, 678–689 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Keefe R., Dave V., Allman D., Wiest D., Kappes D. J., Regulation of lineage commitment distinct from positive selection. Science 286, 1149–1153 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Rothenberg E. V., Telfer J. C., Anderson M. K., Transcriptional regulation of lymphocyte lineage commitment. Bioessays 21, 726–742 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Koch U., Fiorini E., Benedito R., Besseyrias V., Schuster-Gossler K., Pierres M., Manley N. R., Duarte A., MacDonald H. R., Radtke F., Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J. Exp. Med. 205, 2515–2523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothenberg E. V., Stepwise specification of lymphocyte developmental lineages. Curr. Opin. Genet. Dev. 10, 370–379 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Rothenberg E. V., Transcriptional control of early T and B cell developmental choices. Annu. Rev. Immunol. 32, 283–321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deftos M. L., Bevan M. J., Notch signaling in T cell development. Curr. Opin. Immunol. 12, 166–172 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Kueh H. Y., Yui M. A., Ng K. K. H., Pease S. S., Zhang J. A., Damle S. S., Freedman G., Siu S., Bernstein I. D., Elowitz M. B., Rothenberg E. V., Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat. Immunol. 17, 956–965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engel I., Johns C., Bain G., Rivera R. R., Murre C., Early thymocyte development is regulated by modulation of E2A protein activity. J. Exp. Med. 194, 733–745 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothenberg E. V., Negotiation of the T lineage fate decision by transcription-factor interplay and microenvironmental signals. Immunity 26, 690–702 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Georgopoulos K., Moore D. D., Derfler B., Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science 258, 808–812 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Weber B. N., Chi A. W. S., Chavez A., Yashiro-Ohtani Y., Yang Q., Shestova O., Bhandoola A., A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476, 63–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L., Leid M., Rothenberg E. V., An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329, 89–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staal F. J., Meeldijk J., Moerer P., Jay P., van de Weerdt B. C., Vainio S., Nolan G. P., Clevers H., Wnt signaling is required for thymocyte development and activates Tcf-1 mediated transcription. Eur. J. Immunol. 31, 285–293 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Staal F. J., Clevers H., Tales of the unexpected: Tcf1 functions as a tumor suppressor for leukemias. Immunity 37, 761–763 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Tiemessen M. M., Baert M. R. M., Schonewille T., Brugman M. H., Famili F., Salvatori D. C. F., Meijerink J. P. P., Ozbek U., Clevers H., van Dongen J. J. M., Staal F. J. T., The nuclear effector of Wnt-signaling, Tcf1, functions as a T-cell–specific tumor suppressor for development of lymphomas. PLOS Biol. 10, e1001430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu S., Zhou X., Steinke F. C., Liu C., Chen S. C., Zagorodna O., Jing X., Yokota Y., Meyerholz D. K., Mullighan C. G., Knudson C. M., Zhao D.-M., Xue H.-H., The TCF-1 and LEF-1 transcription factors have cooperative and opposing roles in T cell development and malignancy. Immunity 37, 813–826 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schilham M. W., Wilson A., Moerer P., Benaissa-Trouw B. J., Cumano A., Clevers H. C., Critical involvement of Tcf-1 in expansion of thymocytes. J. Immunol. 161, 3984–3991 (1998). [PubMed] [Google Scholar]
- 25.Schuijers J., Clevers H., Adult mammalian stem cells: The role of Wnt, Lgr5 and R-spondins. EMBO J. 31, 2685–2696 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verbeek S., Izon D., Hofhuis F., Robanus-Maandag E., te Riele H., van de Watering M., Oosterwegel M., Wilson A., Robson MacDonald H., Clevers H., An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature 374, 70–74 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Okamura R. M., Sigvardsson M., Galceran J., Verbeek S., Clevers H., Grosschedl R., Redundant regulation of T cell differentiation and TCRα gene expression by the transcription factors LEF-1 and TCF-1. Immunity 8, 11–20 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Gravestein L. A., van Ewijk W., Ossendorp F., Borst J., CD27 cooperates with the pre-T cell receptor in the regulation of murine T cell development. J. Exp. Med. 184, 675–685 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yui M. A., Feng N., Rothenberg E. V., Fine-scale staging of T cell lineage commitment in adult mouse thymus. J. Immunol. 185, 284–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu Q., Sharma A., Oh S. Y., Moon H. G., Hossain M. Z., Salay T. M., Leeds K. E., du H., Wu B., Waterman M. L., Zhu Z., Sen J. M., T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-γ. Nat. Immunol. 10, 992–999 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L., Zhang J. A., Dose M., Kueh H. Y., Mosadeghi R., Gounari F., Rothenberg E. V., A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood 122, 902–911 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van de Wetering M., Cavallo R., Dooijes D., van Beest M., van Es J., Loureiro J., Ypma A., Hursh D., Jones T., Bejsovec A., Peifer M., Mortin M., Clevers H., Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell 88, 789–799 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Hendriks R. W., Nawijn M. C., Engel J. D., van Doorninck H., Grosveld F., Karis A., Expression of the transcription factor GATA-3 is required for the development of the earliest T cell progenitors and correlates with stages of cellular proliferation in the thymus. Eur. J. Immunol. 29, 1912–1918 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Hosoya T., Kuroha T., Moriguchi T., Cummings D., Maillard I., Lim K.-C., Engel J. D., GATA-3 is required for early T lineage progenitor development. J. Exp. Med. 206, 2987–3000 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taghon T., Yui M. A., Rothenberg E. V., Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat. Immunol. 8, 845–855 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ting C. N., Olson M. C., Barton K. P., Leiden J. M., Transcription factor GATA-3 is required for development of the T-cell lineage. Nature 384, 474–478 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Allman D., Li J., Hardy R. R., Commitment to the B lymphoid lineage occurs before DH-JH recombination. J. Exp. Med. 189, 735–740 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bain G., Maandag E. C. R., Riele H. P. J., Feeney A. J., Sheehy A., Schlissel M., Shinton S. A., Hardy R. R., Murre C., Both E12 and E47 allow commitment to the B cell lineage. Immunity 6, 145–154 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Enver T., B-cell commitment: Pax5 is the deciding factor. Curr. Biol. 9, R933–R935 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Hagman J., Lukin K., Transcription factors drive B cell development. Curr. Opin. Immunol. 18, 127–134 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Lin H., Grosschedl R., Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature 376, 263–267 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Mikkola I., Heavey B., Horcher M., Busslinger M., Reversion of B cell commitment upon loss of Pax5 expression. Science 297, 110–113 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Nutt S. L., Heavey B., Rolink A. G., Busslinger M., Commitment to the B-lymphoid lineage depends on the transcription factor Pax5 [see comments]. Nature 401, 556–562 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Rolink A. G., Nutt S. L., Melchers F., Busslinger M., Long-term in vivo reconstitution of T-cell development by Pax5- deficient B-cell progenitors. Nature 401, 603–606 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Roose J., Molenaar M., Peterson J., Hurenkamp J., Brantjes H., Moerer P., van de Wetering M., Destrée O., Clevers H., The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature 395, 608–612 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Van de Wetering M., Castrop J., Korinek V., Clevers H., Extensive alternative splicing and dual promoter usage generate Tcf-1 protein isoforms with differential transcription control properties. Mol. Cell. Biol. 16, 745–752 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luis T. C., Naber B. A. E., Roozen P. P. C., Brugman M. H., de Haas E. F. E., Ghazvini M., Fibbe W. E., van Dongen J. J. M., Fodde R., Staal F. J. T., Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 9, 345–356 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Urbánek P., Wang Z.-Q., Fetka I., Wagner E. F., Busslinger M., Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking Pax5/BSAP. Cell 79, 901–912 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Miyazaki T., Muller U., Campbell K. S., Normal development but differentially altered proliferative responses of lymphocytes in mice lacking CD81. EMBO J. 16, 4217–4225 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Braunstein M., Anderson M. K., HEB-deficient T-cell precursors lose T-cell potential and adopt an alternative pathway of differentiation. Mol. Cell. Biol. 31, 971–982 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson J. L., Georgakilas G., Petrovic J., Kurachi M., Cai S., Harly C., Pear W. S., Bhandoola A., Wherry E. J., Vahedi G., Lineage-determining transcription factor TCF-1 initiates the epigenetic identity of T cells. Immunity 48, 243–257.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xing S., Li F., Zeng Z., Zhao Y., Yu S., Shan Q., Li Y., Phillips F. C., Maina P. K., Qi H. H., Liu C., Zhu J., Pope R. M., Musselman C. A., Zeng C., Peng W., Xue H.-H., Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nat. Immunol. 17, 695–703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson M. K., Hernandez-Hoyos G., Dionne C. J., Arias A. M., Chen D., Rothenberg E. V., Definition of regulatory network elements for T cell development by perturbation analysis with PU.1 and GATA-3. Dev. Biol. 246, 103–121 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Garcia-Ojeda M. E., Klein Wolterink R. G. J., Lemaître F., Richard-Le Goff O., Hasan M., Hendriks R. W., Cumano A., Di Santo J. P., GATA-3 promotes T-cell specification by repressing B-cell potential in pro-T cells in mice. Blood 121, 1749–1759 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Ge Y., Gomez N. C., Adam R. C., Nikolova M., Yang H., Verma A., Lu C. P.-J., Polak L., Yuan S., Elemento O., Fuchs E., Stem cell lineage infidelity drives wound repair and cancer. Cell 169, 636–650.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Longabaugh W. J. R., Zeng W., Zhang J. A., Hosokawa H., Jansen C. S., Li L., Romero-Wolf M., Liu P., Kueh H. Y., Mortazavi A., Rothenberg E. V., Bcl11b and combinatorial resolution of cell fate in the T-cell gene regulatory network. Proc. Natl. Acad. Sci. U.S.A. 114, 5800–5807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buenrostro J. D., Wu B., Chang H. Y., Greenleaf W. J., ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.F. Krueger, Trim galore, A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. 516, 517 (2015).
- 61.S. Andrews, FastQC: A Quality Control Tool for High Throughput Sequence Data (2010); http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 62.Ewels P., Magnusson M., Lundin S., Käller M., MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B., Salzberg S., Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramírez F., Dündar F., Diehl S., Grüning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.R. Stark, G. Brown, DiffBind: Differential binding analysis of ChIP-Seq peak data (2011); http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf
- 67.Grant C. E., Bailey T. L., Noble W. S., FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/31/eaaw7313/DC1