

Abstract
Although there are molecularly distinct subtypes of prostate cancer, no molecular classification system is used clinically. The ribonucleotide reductase small subunit M2 (RRM2) gene plays an oncogenic role in many cancers. Our previous study elucidated comprehensive molecular mechanisms of RRM2 in prostate cancer (PC). Given the potent functions of RRM2, we set out to determine whether the RRM2 signature can be used to identify aggressive subtypes of PC. We applied gene ontology and pathway analysis in RNA‐seq datasets from PC cells overexpressing RRM2. We refined the RRM2 signature by integrating it with two molecular classification systems (PCS and PAM50 subtypes) that define aggressive PC subtypes (PCS1 and luminal B) and correlated signatures with clinical outcomes in six published cohorts comprising 4000 cases of PC. Increased expression of genes in the RRM2 signature was significantly correlated with recurrence, high Gleason score, and lethality of PC. Patients with high RRM2 levels showed higher PCS1 score, suggesting the aggressive PC feature. Consistently, RRM2‐regulated genes were highly enriched in the PCS1 signature from multiple PC cohorts. A simplified RRM2 signature (12 genes) was identified by intersecting the RRM2 signature, PCS1 signature, and the PAM50 classifier. Intriguingly, inhibition of RRM2 specifically targets PCS1 and luminal B genes. Furthermore, 11 genes in the RRM2 signature were correlated with enzalutamide resistance by using a single‐cell RNA‐seq dataset from PC circulating tumor cells. Finally, high expression of RRM2 was associated with an immunosuppressive tumor‐immune microenvironment in both primary prostate cancer and metastatic prostate cancer using CIBERSORT analysis and LM22, a validated leukocyte gene signature matrix. These data demonstrate that RRM2 is a driver of aggressive prostate cancer subtypes and contributes to immune escape, suggesting that RRM2 inhibition may be of clinical benefit for patients with PC.
Keywords: molecular subtyping, PAM50, PCS subtyping, prostate cancer, RRM2
RRM2 is essential for DNA synthesis and repair. We integrated clinical and experimental datasets to generate RRM2 signatures that not only define subtypes with poor outcomes but also predict enzalutamide resistance. RRM2 overexpression is associated with an immunosuppressive tumor‐immune microenvironment. RRM2 signatures may be novel biomarkers for aggressive prostate cancer subtypes, and targeting RRM2 could be a useful therapeutic strategy.

Abbreviations
- AR
androgen receptor
- CIBERSORT
cell‐type identification by estimating relative subsets of RNA transcripts
- CRPC
castration‐resistant prostate cancer
- CTCs
circulating tumor cells
- DFS
disease‐free survival
- dNTPs
deoxyribonucleotide triphosphates
- EMT
epithelial–mesenchymal transition
- ENZ
enzalutamide
- FC
fold change
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- GO
gene ontology
- GRID
Genomic Resource Information Database
- GSEA
gene set enrichment analysis
- GSVA
gene set variation analysis
- PCS
prostate cancer subtype
- PCS1
prostate cancer subtype 1
- PCS2
prostate cancer subtype 2
- PCS3
prostate cancer subtype 3
- PSA
prostate‐specific antigen
- RNA‐seq
RNA sequencing
- RRM2
ribonucleotide reductase subunit M2
- ssGSEA
single‐sample gene set enrichment analysis
- SU2C/PCF
Stand Up To Cancer/Prostate Cancer Foundation
- TCGA
The Cancer Genome Atlas
- TIL
tumor‐infiltrating lymphocyte
- TIME
tumor‐immune microenvironment
1. Introduction
Prostate cancer is a heterogeneous disease and the third leading cause of cancer death among American men. Clinical decision making has been largely driven by clinical and pathologic variables, such as tumor stage, Gleason score, and serum prostate‐specific antigen (PSA) levels (Falzarano and Magi‐Galluzzi, 2011; Gleason and Mellinger, 1974). Inhibition of androgen receptor (AR) signaling is the mainstay of therapy for recurrent or advanced prostate cancer (Assikis and Simons, 2004) but is limited in its utility because of acquired resistance (Attard et al., 2016). There is an unmet clinical need to identify patients with aggressive and drug‐resistant prostate cancer and develop therapies to treat these patients.
Molecular classification has been successfully applied in many cancers and is routinely used to guide treatment decisions (Perou et al., 2000). In contrast, molecular subtyping of prostate cancer is based on the underlying genomic alterations and is less established as a determinant of prognosis and guide to treatment. Multiple studies have attempted to establish individual biomarkers or gene expression signatures to predict aggressive cases of prostate cancer (Bibikova et al., 2007; Cuzick et al., 2011; Glinsky et al., 2005; Penney et al., 2011), but these studies were limited by the small number of samples analyzed. Recently, You et al. reported a novel molecular classification of prostate cancer subtypes (PCS) that was generated from transcriptomic data from more than 4600 prostate cancer specimens. This classification categorizes prostate cancer into three distinct molecular subtypes (PCS1, PCS2, and PCS3) and was validated in ten independent prostate cancer cohorts and several preclinical in vitro and in vivo prostate cancer models (You et al., 2016). The PCS classification system appears useful in distinguishing aggressive disease using both the tumor and blood of patients with prostate cancer. In addition to the PCS signatures, the PAM50 classifier, which was commercially developed as Prosigna to assess breast cancer risk (Nielsen et al., 2014), was recently proven to segregate prostate cancer into three subtypes (luminal A, luminal B, and basal) in retrospective and prospective cohorts totaling 3782 samples (Zhao et al., 2017).
Both the PCS1 and luminal B signatures can be used to effectively identify cases of prostate cancer with poor prognosis, but treating these patients will require an understanding of the molecular drivers of these subtypes. Although the FOXM1 pathway was recently identified as a key driver of PCS1 tumors (Ketola et al., 2017), small molecules targeting transcription factors are difficult to develop, and there are no specific FOXM1 inhibitors for clinical application. Similar to FOXM1, ribonucleotide reductase subunit M2 (RRM2) is a highly expressed gene in the PCS1 and luminal B signatures. RRM2 maintains the deoxyribonucleotide triphosphate (dNTP) pool to support DNA synthesis and repair (Kumar et al., 2011) and is overexpressed in multiple cancers (Grade et al., 2011; Kretschmer et al., 2011). We previously reported the significant prognostic value of RRM2 in prostate cancer by analyzing 11 prostate cancer cohorts (Mazzu et al., 2019). We elucidated the molecular mechanisms underlying its potent oncogenic function by knocking down or overexpressing RRM2 in multiple prostate cancer cell lines. Additionally, we demonstrated that COH29, an RRM2 inhibitor currently in clinical trials for solid tumors, had efficacy against prostate cancer cells in vitro and in vivo.
In this study, we further demonstrated that RRM2 is a druggable driver of PCS1 and luminal B tumors. Bioinformatic analysis revealed that RRM2‐regulated genes are highly enriched in PCS1 genes and are significantly correlated with clinical outcomes. Tumors with high expression of RRM2 have tumor‐infiltrating lymphocyte (TIL) populations consistent with an immunosuppressive microenvironment. Finally, we demonstrated that targeting RRM2 specifically inhibits the expression of genes in the PCS1 and luminal B signatures.
2. Materials and methods
2.1. Clinical cohort summary
All publicly available prostate cancer cohorts used in this study are summarized in Table 1.
Table 1.
Details of the prostate cancer clinical cohorts that were used in the study. aCGH, array comparative genomic hybridization; BCR, biochemical recurrence; dbGaP, database of Genotypes and Phenotypes; NCI GDC, National Cancer Institute Genomic Data Commons; OS, overall survival; PRAD, prostate adenocarcinoma; RPPA, reverse‐phase protein array; WES, whole‐exome sequencing.
| Cohort name | Benign/normal tissue number | Tumor number | Primary number | Metastasis number | Clinical outcome | Data type | Year | Accession number | Reference |
|---|---|---|---|---|---|---|---|---|---|
| TCGA | 0 | 333 | 333 | 0 | BCR | WES, RNA‐seq, RPPA | 2015 | TCGA‐PRAD (NCI GDC Data Portal) | CGA Research Network, TCGA Data Portal |
| Taylor | 29 normal | 216 | 131 | 19 | BCR | aCGH, RNA‐seq | 2010 | GSE21032 (GEO) | Taylor et al. (2010) |
| SU2C/PCF | 0 | 150 | 0 | 150 | BCR | WES, RNA‐seq | 2015 | Phs000915.v1.p1 (dbGaP) | Robinson et al. (2015) |
| Kumar | 176 benign | 176 | 22 | 154 | BCR | aCGH, WES, microarray | 2016 | GSE77930 (GEO) | Kumar et al. (2016) |
| Grasso | 28 benign | 122 | 59 | 35 | OS | aCGH, microarray | 2012 | GSE35988 (GEO) | Grasso et al. (2012) |
| Setlur | 0 | 363 | 363 | 0 | OS | microarray | 2008 | GSE8402 (GEO) | Setlur et al. (2008) |
2.2. Cell culture
LNCaP (RRID: CVCL_0395) and PC‐3 (RRID: CVCL_0035) cells were purchased from ATCC (Manassas, VA, USA). C4‐2 (LNCaP C4‐2, RRID: CVCL_4782) cells were obtained from VitroMed (Burlington, NC, USA). As previously described (Mazzu et al., 2019), lentiviral vectors encoding RRM2 were infected in LNCaP and PC‐3 cells, and stable cell lines were generated and maintained using puromycin selection. Efficiency of overexpression was verified by qPCR and western blot. All cells were maintained in media with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 2 mm of l‐glutamine (Thermo Fisher Scientific) and 100 U·mL−1 penicillin/streptomycin (Thermo Fisher Scientific) at 37 °C in 5% CO2. Cell line authentication was performed by human short‐tandem repeat profiling at the Memorial Sloan Kettering Cancer Center Integrated Genomics Operation within the last 3 years. Experiments were performed in mycoplasma‐free cell lines.
2.3. Gene silencing and overexpression
SMARTpool siRNAs (Dharmacon, Lafayette, CO, USA) were used for transfection with RNAiMAX (Thermo Fisher Scientific) to knock down target gene expression. For overexpression, cells were transduced with lentiviral vectors encoding RRM2 and selected by treatment with puromycin as described previously (Mazzu et al., 2019). Efficiency of knockdown and overexpression was verified after 2 or 3 days by qPCR and western blot.
2.4. RNA sequencing
Total RNA was extracted from cells and analyzed as previously described (Zhang et al., 2011). RNA sequencing (RNA‐seq) was performed by 50 million 2 × 50 bp reads at the Memorial Sloan Kettering Cancer Center Integrated Genomics Operation, and data were analyzed in partek flow software (St. Louis, MO, USA). The data are available from GEO (GSE117921–GSE117924).
2.5. Bioinformatic analysis of clinical cohorts
Bioinformatic analysis of the clinical cohorts was performed using data obtained from cBioPortal for Cancer Genomics (Gao et al., 2013) and Oncomine (Rhodes et al., 2004). Heat maps and volcano plots were generated using r version 3.4.3 (https://www.R‐project.org). Pathway analysis from RNA‐seq data was performed using gene set enrichment analysis (GSEA) and ToppGene (Chen et al., 2009; Subramanian et al., 2005).
The enrichments function in cBioPortal was used to identify genes with expression that was significantly correlated with RRM2 overexpression (RRM2: EXP > 1.5, z‐score) in prostate cancer clinical cohorts. Only genes with expression that positively correlated with RRM2 levels [R > 0.5, false discovery rate (FDR) < 0.05] from published prostate cancer cohorts [The Cancer Genome Atlas (TCGA), Kumar, and Stand Up To Cancer/Prostate Cancer Foundation (SU2C/PCF)] were selected (Kumar et al., 2016; Network, 2015; Robinson et al., 2015). These genes (n = 626) were intersected with gene expression data from the PC‐3 and LNCaP cell lines with stable RRM2 overexpression to develop the RRM2 signature.
Prostate cancer subtype scores were calculated with gene set variation analysis (GSVA) using single‐sample GSEA (ssGSEA) (Barbie et al., 2009). Briefly, PCS signature scores were defined by the quantification of the composite expression of each gene in the signature in each sample. We computed a z‐score for the expression of each gene in each sample by subtracting the pooled mean from the RNA‐seq expression values and dividing by the pooled standard deviation. The overall survival analysis with the 12‐gene signature was performed using KM plotter (www.kmplot.com/mirpower) (Lanczky et al., 2016).
2.6. TIL maps and cell‐type identification by estimating relative subsets of RNA transcripts analysis
In each cohort, samples were categorized as RRM2 high (upper quantile) or low (lower quantile) based on mRNA expression. The fraction of TILs in TCGA cases was determined with a machine‐learning algorithm that uses digital hematoxylin and eosin (H&E) slides (Saltz et al., 2018). The abundance of immune cell fractions in each sample was determined using cell‐type identification by estimating relative subsets of RNA transcripts (CIBERSORT) and LM22, a validated leukocyte gene signature matrix (Newman et al., 2015).
2.7. Statistical analysis
Results are reported as mean ± standard deviation. Comparisons between groups were performed using an unpaired two‐sided Student's t‐test or Wilcoxon rank‐sum test (P < 0.05 was considered significant). Disease‐free survival (DFS) was examined using the Kaplan–Meier method. Patients were divided into two groups (upper and lower quartile based on RRM2 expression or RRM2 signature score), and Kaplan–Meier curves were generated for each group. The log‐rank test was used to determine significance. Cox proportional hazard regression was performed, adjusting for clinical and demographic factors. The significance of the correlation between gene expression and enzalutamide resistance was analyzed by Fisher's exact test. The significance of the differences in the abundance of immune cell types between groups was determined using Wilcoxon's rank‐sum test with Benjamini–Hochberg correction. Statistical analysis was completed using r version 3.4.3 (https://www.R‐project.org).
2.8. Data accessibility
RNA‐seq data are available from the Gene Expression Omnibus (GEO: GSE117921, GEO: GSE117922, GEO: GSE117923, and GEO: GSE117924).
3. Results
3.1. Defining the RRM2 signature and its clinical relevance in prostate cancer
Our prior study reported the potent oncogenic activity and clinical significance of RRM2 in prostate cancer (Mazzu et al., 2019). Although we demonstrated that there was a significant correlation between increased RRM2 levels and poor clinical outcomes, we believed that the prognostic value of RRM2 had been underestimated because RRM2 expression is strictly regulated during the cell cycle, with levels peaking during S‐phase, followed by rapid degradation (Chabes and Thelander, 2000). However, its potent oncogenic activity is maintained to support tumor survival and progression (Fujita et al., 2010; Lee et al., 2014; Su et al., 2014). We hypothesized that an RRM2 signature would further elucidate the function of RRM2. To modulate RRM2 activity in prostate cancer cells, we developed two prostate cancer cell lines with stable overexpression of RRM2 (PC‐3‐RRM2 and LNCaP‐RRM2) and used siRNA and COH29, a small molecule inhibitor of RRM2 (Mazzu et al., 2019). Using these cellular models, we were able to explore the transcriptomic changes induced by RRM2, define the downstream mechanisms through which RRM2 functions, and identify an RRM2 signature.
To uncover downstream pathways, genes deregulated with manipulation of RRM2 [FDR < 0.05, −1.5 > fold change (FC) > 1.5] were subjected to gene ontology (GO) analysis (Fig. 1A). To identify an RRM2 signature, these genes were also compared to the genes with expression that correlated with RRM2 levels in prostate cancer clinical cohorts. The clinical significance of the RRM2 signature was evaluated in multiple prostate cancer cohorts. To determine whether RRM2 is a driver of PCSs with poor prognosis, the signatures of two well‐established prostate cancer classifications (PCS and PAM50) were compared to the RRM2 signature.
Fig. 1.
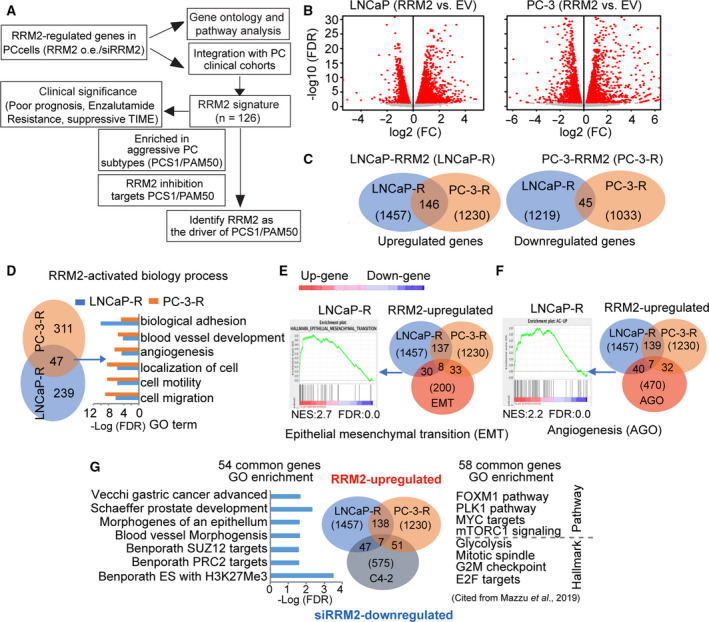
RRM2 function is disease‐state‐specific. (A) Schematic of the experimental design. As previously reported (Mazzu et al., 2019), transcriptomic changes induced by RRM2 overexpression or inhibition (FDR < 0.05, −1.5 > FC > 1.5) from cellular models were integrated with prostate cancer clinical cohorts to generate an RRM2 signature. Here, we applied the PCS and PAM50 gene sets to further characterize the signature. (B) Volcano plots show transcriptomic changes induced by RRM2 overexpression in LNCaP (left) and PC‐3 (right) cells. (C) Venn diagrams show the overlap of genes upregulated (left) and downregulated (right) with RRM2 overexpression in LNCaP and PC‐3 cells (FDR < 0.05, −1.5 > FC > 1.5). (D) Venn diagram (left) depicts the overlap of GO analysis of genes upregulated by RRM2 overexpression. Bar graphs (right) show common biological processes activated in 2 cell lines with RRM2 overexpression. (E) Enrichment of RRM2‐upregulated genes from LNCaP in EMT and (F) angiogenesis. GSEA results are from LNCaP cells, and Venn diagrams show the overlap between pathway genes and genes upregulated in LNCaP and PC‐3 cells that overexpress RRM2. (G) GO enrichment of common genes deregulated in LNCaP‐RRM2/C4‐2‐siRRM2 (left) and PC‐3‐RRM2/C4‐2‐siRRM2 (right).
3.2. RRM2 function is disease‐state‐specific in prostate cancer
We have previously analyzed the transcriptomic changes in PC‐3 cells, an AR‐negative cell line, that overexpress RRM2 (PC‐3‐RRM2); we used this as a castration‐resistant cellular model (Mazzu et al., 2019). To compare RRM2 function in different disease states, we performed RNA‐seq of LNCaP cells, an AR‐positive cell line, that overexpress RRM2 (LNCaP‐RRM2). We confirmed overexpression of RRM2 in both PC‐3 and LNCaP cells in our previous study (Mazzu et al., 2019). In both stable cell lines, a similar number of genes were deregulated by RRM2 overexpression (Fig. 1B). Among the 1230 PC‐3 and 1457 LNCaP upregulated genes, there were 146 genes that were upregulated in both cell lines. Only 45 downregulated genes were shared among the 1033 PC‐3 and 1219 LNCaP downregulated genes in either cell line (Fig. 1C). Overall, less than 10% of genes were regulated by RRM2 in both LNCaP and PC‐3 cells, indicating the underlying function of RRM2 may be disease‐state‐specific, as these two prostate cancer cell lines may represent different disease states because of their AR status.
Given the previously reported strong oncogenic role of RRM2 in prostate cancer (Mazzu et al., 2019), we performed GO analysis on the RRM2‐upregulated genes in LNCaP and PC‐3 cells and found that 47 biological processes were activated in both cell lines (Fig. 1D). The top six were related to tumor metastasis, which is consistent with our prior report of RRM2‐induced epithelial–mesenchymal transition (EMT) phenotypes in both cell lines (Mazzu et al., 2019). Unlike GO analysis, GSEA provides enrichment scores that signify the enrichment of the specific gene set. GSEA demonstrated that RRM2‐upregulated genes in LNCaP cells were significantly enriched in EMT and angiogenesis gene sets (Fig. 1E,F), which is similar to the phenotype we previously reported in PC‐3‐RRM2 cells (Mazzu et al., 2019). Surprisingly, only eight of the 38 enriched genes in the EMT gene set and only seven of the 47 enriched genes in the angiogenesis gene set are shared by the two cell lines, suggesting that RRM2 regulates both pathways in LNCaP and PC‐3 cells through distinct gene sets.
To further understand the molecular mechanisms regulated by RRM2, we integrated transcriptomic datasets from siRRM2‐treated C4‐2 cells (C4‐2‐siRRM2), LNCaP‐RRM2, and PC‐3‐RRM2. Changes in RRM2 expression levels in these cell lines were shown in our prior study (Mazzu et al., 2019). Previously, our ToppGene analysis revealed that the 58 common genes that were upregulated in PC‐3‐RRM2 and downregulated in C4‐2‐siRRM2 were significantly enriched in oncogenic pathways and cancer hallmarks (Fig. 1G) (Mazzu et al., 2019). The 54 shared genes between LNCaP‐RRM2 and C4‐2‐siRRM2 cells were enriched in gene sets related to prostate development, gastric cancer progression, angiogenesis, and H3K27me3 (Fig. 1G). Only seven genes were shared between the 58 upregulated genes in PC‐3‐RRM2 and the 54 upregulated genes in LNCaP‐RRM2. These results support the hypothesis that RRM2 may play a similar oncogenic role in PC‐3 and LNCaP cells by regulating distinct gene sets in different biological contexts.
3.3. Clinical relevance of the RRM2 signature
To validate RRM2‐regulated genes in prostate cancer clinical samples, we compared genes upregulated in cells that overexpress RRM2 to those with expression that positively correlated with RRM2 levels in the TCGA (localized prostate cancer), Kumar [metastatic castration‐resistant prostate cancer (CRPC)], and SU2C/PCF (metastatic CRPC) cohorts (Kumar et al., 2016; Network, 2015; Robinson et al., 2015) (Fig. 2A). There were approximately 2000–3000 genes with expression that positively correlated with RRM2 levels in each of the three cohorts. Among these genes, more were upregulated in PC‐3‐RRM2 (< 250) than in LNCaP‐RRM2 (< 116, Fig. 2A). When we compared genes from the three cohorts with the genes identified in the cell lines that overexpress RRM2, there were 126 genes in PC‐3‐RRM2 and only seven genes in LNCaP‐RRM2 that were shared with the clinical cohorts (Fig. 2B and Table S1). Using ssGSEA, we previously reported (Mazzu et al., 2019) that the expression of 126 genes was highly correlated with poor DFS in the Taylor cohort (Taylor et al., 2010). Here, we confirmed this result in the TCGA cohort (Fig. 2C) and found that increased expression of the 126‐gene signature was significantly correlated with higher Gleason score and lethal disease in the Setlur cohort (Fig. 2D), which has long‐term outcome data (Setlur et al., 2008).
Fig. 2.
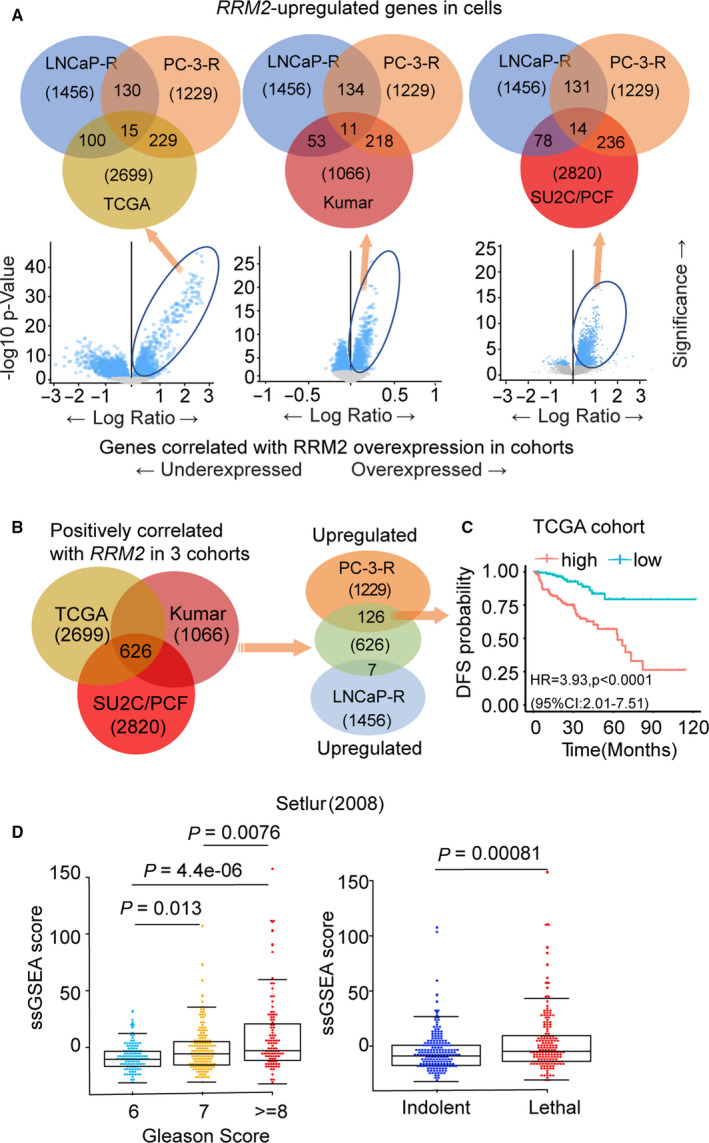
Integration of prostate cancer cell line transcriptomic data with clinical outcomes. (A) Venn diagrams (top) depicting the overlap of genes with expression that positively correlated with RRM2 levels in TCGA (left), Kumar (middle), and SU2C/PCF (right) cohorts with upregulated genes in LNCaP‐RRM2 or PC‐3‐RRM2 cells. Below, plots show the genes with expression that correlates with RRM2 expression level in each prostate cancer cohort. (B) RRM2 signature: The 626 genes with expression that correlated with RRM2 levels in the three clinical cohorts (left) were compared with genes upregulated in PC‐3‐RRM2 or LNCaP‐RRM2 (right) to identify RRM2 signature (126 genes). (C) Clinical significance of expression of the 126‐gene RRM2 signature in the TCGA cohort. Samples were ranked based on expression of the 126‐gene RRM2 signature, and Kaplan–Meier curves were used to estimate survival differences between patients in the top and bottom 25th percentiles of expression. The log‐rank test was calculated to determine significance. Cox proportional hazard regression was performed, adjusting for clinical and demographic factors. (D) Association between RRM2 signature (126 genes) level with Gleason score (left) and lethality (right) in the Setlur cohort. Comparisons between groups were performed using Wilcoxon's rank‐sum test.
3.4. High RRM2 expression is correlated with the poor prognosis prostate cancer subtype PCS1
Among the three prostate cancer subtypes (PCS1–PCS3), PCS1 is the most aggressive and lethal, and PCS1 tumors progress more rapidly to metastatic disease than PCS2 or PCS3 tumors (You et al., 2016). The FOXM1 pathway was recently reported as the master regulator of the PCS1 subtype (Ketola et al., 2017). We previously reported that RRM2 is not only a target of FOXM1 but also regulates the FOXM1 pathway (Mazzu et al., 2019). Furthermore, RRM2 is one of the most highly expressed genes in the PCS1 signature. To test our hypothesis that overexpression of RRM2 could contribute to the development of PCS1 tumors, ssGSEA was performed to determine the correlation between RRM2 expression level and PCS score in multiple prostate cancer cohorts. In each patient sample, scores of PCS1, PCS2, and PCS3 gene expression were calculated using ssGSEA. In these analyses, RRM2 was removed from the PCS1 signature to avoid a false‐positive correlation. There was a significant association between high RRM2 expression and high PCS1 score and low PCS3 score in patient samples in the TCGA and Taylor cohorts (Fig. 3). Intriguingly, the strong correlation between RRM2 expression level and PCS1 score was also seen in the SU2C/PCF cohort in which all samples are of metastatic CRPC (and RRM2 expression levels are already high), suggesting that RRM2 is not only associated with an aggressive PCS but may also regulate multiple key PCS1 genes.
Fig. 3.
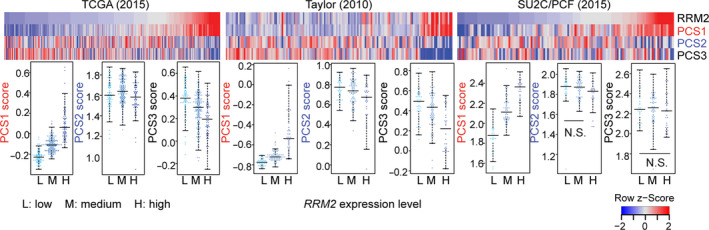
RRM2 levels are highly correlated with PCS1 gene expression. Correlation of RRM2 level with PCS gene expression in TCGA (left), Taylor (middle), and SU2C/PCF (right) cohorts. Each individual patient sample is indicated by a single column (top plot) and a single dot (bottom plot). PCS scores were calculated with GSVA using the ssGSEA method, and the values were compared to RRM2 mRNA expression levels divided by quantiles. The differences between pairs are statistically significant except for those labeled as N.S. (not significant). Significance was determined using Wilcoxon's rank‐sum test.
3.5. RRM2 may be a driver of PCS1 tumors
Because tumors with high RRM2 expression have high PCS1 scores, we assessed whether RRM2‐regulated genes correlated with PCS signatures. GSEA demonstrated that the 126 RRM2‐regulated genes (Fig. 2B) were highly enriched in PCS1 genes (Fig. 4A). Fifty (40%) of the 126 genes overlapped with the 86 PCS1 genes (Fig. 4A and Table S1). Additionally, the PAM50 classifier, which is used in determining breast cancer prognosis, has also been reported to consistently segregate prostate cancer into luminal and basal subtypes that correlate with clinical outcome (Zhao et al., 2017). Interestingly, all the overlapping genes in the PCS1 signature and PAM50 classifier are luminal B genes (Table S1). Fourteen genes in the 126‐gene signature overlap with the 50 genes of the PAM50 signature. Among them, 12 genes were shared with both PCS1 and PAM50 genes (Fig. 4A and Table S1). The 126‐gene signature did not share any genes with the PCS2 and PCS3 signatures (Fig. 4A), demonstrating that the signature is predictive of the aggressive subtype of prostate cancer.
Fig. 4.
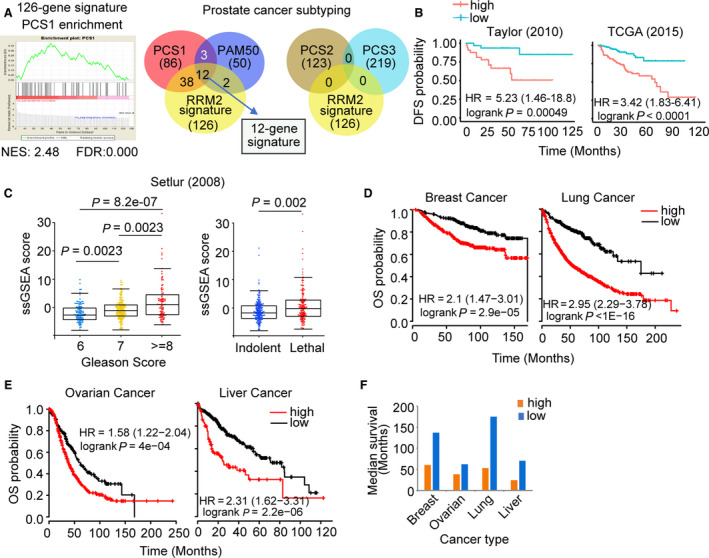
Clinical significance of the 12‐gene RRM2 subsignature. (A) GSEA plot (left) shows high enrichment of PCS1 genes in the RRM2 signature. Venn diagrams (right) depict the overlap between genes in the RRM2 signature with PCS1 and PAM50 genes (left) and PCS2 and PCS3 genes (right). The 12 genes shared by PCS1, PAM50, and RRM2 signature comprise the 12‐gene subsignature. (B) Correlation of expression of the 12‐gene signature with DFS in the Taylor (left) and TCGA (right) cohorts. (C) Correlation of the 12‐gene signature ssGSEA score with Gleason score (left) and lethality (right) in the Setlur cohort. (D) Correlation between 12‐gene signature expression and probability of overall survival (OS) was analyzed in breast and lung cancer and (E) ovarian and liver cancer. Samples were ranked based on expression of the 12‐gene subsignature, and Kaplan–Meier curves were used to estimate survival differences between patients in the top and bottom 25th percentiles of expression. The log‐rank test was calculated to determine significance. Cox proportional hazard regression was performed, adjusting for clinical and demographic factors. Significance was determined using Wilcoxon's rank‐sum test. (F) Median survival time was compared between cases with low (blue) or high (orange) expression of the 12‐gene panel.
We have shown that high expression of the 126‐gene signature is associated with higher Gleason score and shorter patient survival (Fig. 2C,D) (Mazzu et al., 2019). Similarly, we found that high levels of the 12‐gene RRM2/PCS1 subsignature were associated with a significant decrease in DFS in the TCGA and Taylor cohorts (Fig. 4B). High expression of the 12 genes was also associated with increased Gleason score and lethality in the Setlur cohort (Fig. 4C). The oncogenic function of RRM2 has been confirmed in breast, ovarian, lung, and liver cancers, and we assessed whether the 12‐gene signature was associated with poor outcomes in these tumors (Aird et al., 2014; Shah et al., 2014; Xu et al., 2008). High expression of the signature was significantly correlated with worse overall survival in all four cancer types (Fig. 4D,E), with 1.4‐ to 3.3‐fold shorter median survival (Fig. 4F). Altogether, these data suggest that the 12‐gene panel is the core set of genes downstream of RRM2 that control tumor progression and affect clinical outcomes in prostate cancer and tumors of other cellular origins.
3.6. Inhibition of RRM2 activity specifically targets aggressive prostate cancer subtypes
To further evaluate how the regulation of RRM2 affects PCS signatures, we integrated our RRM2‐regulated transcriptome profiling from cell lines with gene expression data from prostate cancer clinical cohorts. We validated that the distinct gene profiling patterns of PCS genes correlated with tumor type in both the Taylor and Grasso (Grasso et al., 2012) cohorts (Fig. 5A). PCS1 genes were highly upregulated in metastatic tumors compared with normal prostate and primary tumors, PCS2 genes had high expression in primary tumors, and PCS3 genes were downregulated in prostate cancer compared with normal prostate. PCS genes also showed different profiling patterns in the Kumar cohort, which is mostly composed of metastatic cases (154/176). This suggests that PCS signatures not only distinguish normal, primary, and metastatic samples, but they may also define a subset of metastatic samples (Fig. 5A).
Fig. 5.
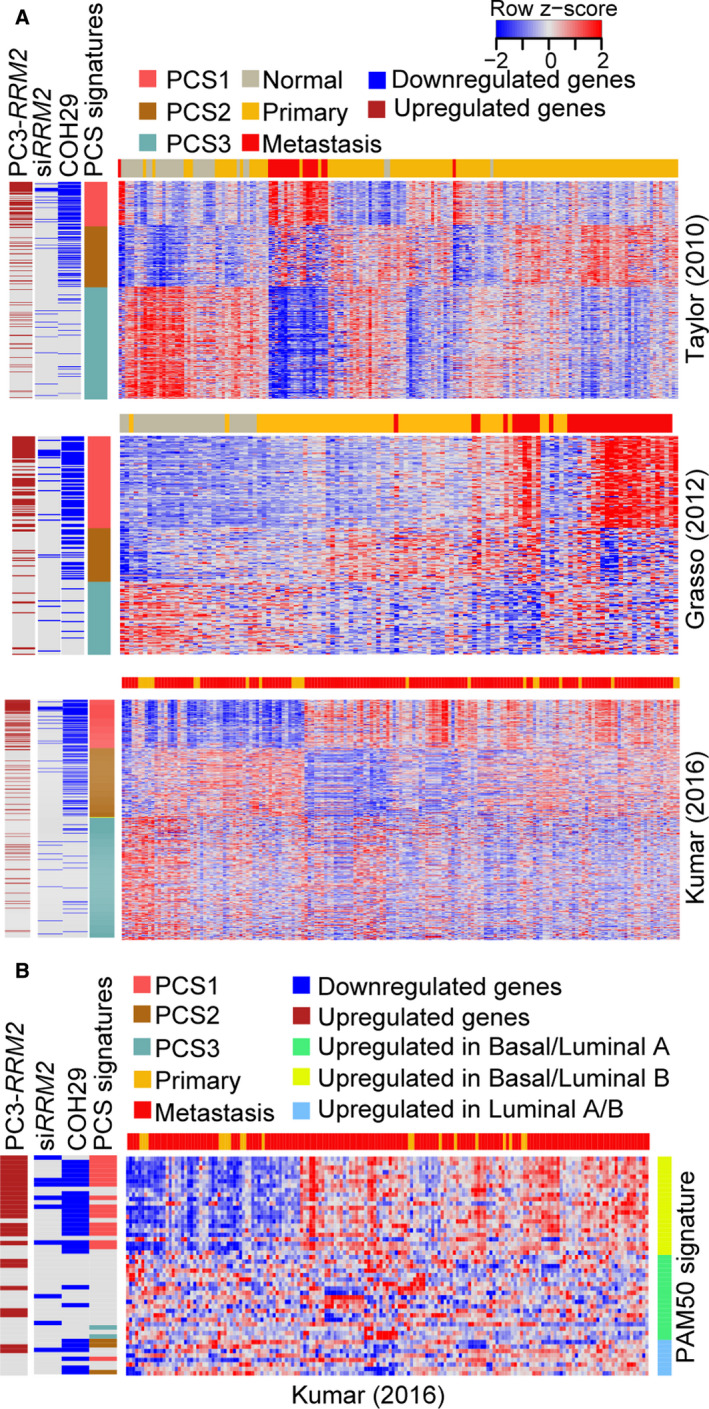
Inhibition of RRM2 specifically targets genes that define poor prognostic subtypes of prostate cancer. (A) Supervised hierarchical clustering of prostate cancer cases in the Taylor (top), Grasso (middle), and Kumar (bottom) cohorts, based on expression of PCS genes. Genes deregulated with RRM2 overexpression (PC‐3‐RRM2) and inhibition of RRM2 (by COH29) are shown. (B) Supervised hierarchical clustering of prostate cancer cases from the Kumar cohort, based on expression of PAM50 classifier genes. Genes deregulated with RRM2 overexpression (PC‐3‐RRM2) and inhibition of RRM2 (by COH29) are shown.
Prostate cancer subtype genes were compared to genes downregulated with RRM2 inhibition and genes upregulated with RRM2 overexpression. Strikingly, COH29 treatment specifically inhibited the expression of most PCS1 genes and also targeted PCS2 genes (Fig. 5A). In addition to the PCS signatures, we also applied the PAM50 classifier in our analysis. In the Kumar cohort, we observed some separation of basal and luminal subtypes (Fig. 5B). The majority of PCS1 genes overlapped with genes upregulated in luminal B tumors; these cases have the poorest clinical prognoses (Zhao et al., 2017). Genes targeted by RRM2 inhibition or genes upregulated by RRM2 overexpression in PC‐3 cells were highly enriched in luminal B genes (Fig. 5B). Together, these data suggest that RRM2 is a driver of the aggressive PCSs and that inhibition of RRM2 could specifically target the subtypes of prostate cancer with the worst prognosis.
3.7. The RRM2 signature may predict enzalutamide resistance in prostate cancer circulating tumor cells
Circulating tumor cells (CTCs) detach from the primary or secondary tumor sites and invade the bloodstream, and they have been reported to be useful prognostic biomarkers to aid prostate cancer diagnosis, treatment decision making, and patient follow‐up (Chung et al., 2019; De Laere et al., 2019; Nimir et al., 2019). The prognostic value of CTCs collected by the epithelial marker‐dependent method CellSearch has been established in the context of metastatic PC (Hegemann et al., 2016). Given the prognostic significance of the RRM2 signature in prostate cancer, we further investigated whether the RRM2 signature had clinical significance in prostate cancer CTCs.
Based on single‐cell RNA‐seq data of CTCs from patients with CRPC, a 37‐gene panel was reported to identify patients with resistance to the AR antagonist enzalutamide (Miyamoto et al., 2015; You et al., 2016). As previously described (Miyamoto et al., 2015), patients who did not receive enzalutamide treatment before CTC collection were denoted as enzalutamide naïve, and patients whose cancer showed radiographic and/or PSA progression during enzalutamide therapy were denoted as enzalutamide‐resistant. We assessed whether our 126‐gene RRM2 signature could also predict enzalutamide resistance in CTCs. Of the 126‐gene signature, 21 genes were detectable in the CTC dataset (FDR < 0.05). Unsupervised hierarchical clustering based on expression of the 21 genes revealed two groups of CTCs (Fig. 6A). In Group I, 21 (36%) of the 59 CTCs were from enzalutamide‐resistant patients. In Group II, 15 (83%) of the 18 CTCs were from enzalutamide‐resistant patients. Increased expression of 11 of the 21 genes significantly correlated with enzalutamide resistance (Fig. 6A and Table S1); eight of the 11 genes were upregulated in the enzalutamide‐resistant CTCs of Group II (Fig. 6A). Surprisingly, only three genes of the 11‐gene panel from the RRM2 signature overlapped with the reported 37‐gene PCS panel (You et al., 2016). These results suggest that the 11‐gene panel derived from the RRM2 signature could be useful in predicting enzalutamide resistance in the CTCs of patients with CRPC. Furthermore, high expression of the 11‐gene signature is significantly associated with poor clinical outcomes (e.g., Gleason score and lethality in the Setlur cohort and DFS in the TCGA cohort; Fig. S1).
Fig. 6.
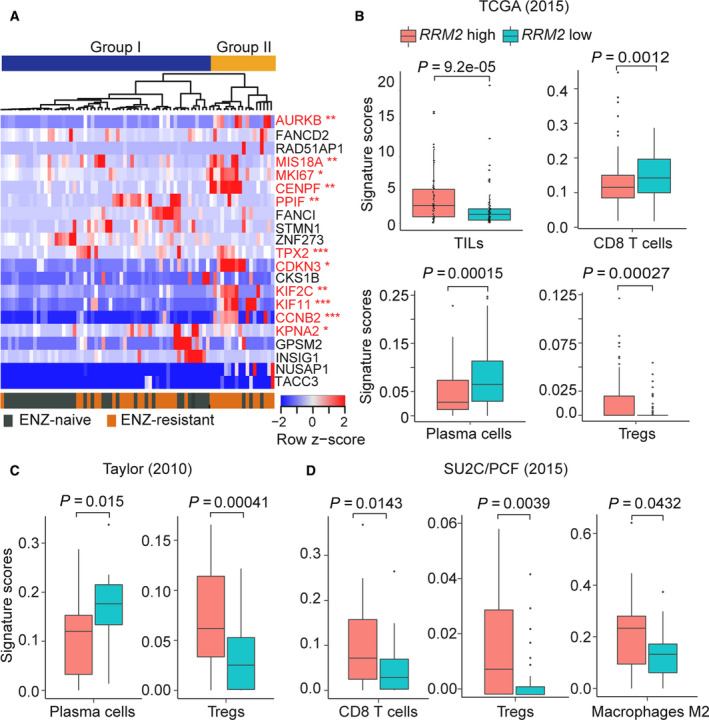
RRM2 overexpression contributes to enzalutamide (ENZ) resistance and an immunosuppressive TIME. (A) Unsupervised hierarchical clustering of single‐cell RNA‐seq data from 77 CTCs from 13 patients with CRPC treated with enzalutamide (from GSE67980) based on expression of 21 genes from the RRM2 signature. Genes with expression that was significantly upregulated in the ENZ‐resistant CTCs of Group II are shown in red (11‐gene panel). *P < 0.05, **P < 0.01, ***P < 0.001. (B) Profiling of immune cells by CIBERSORT in the TCGA, (C) Taylor, and (D) SU2C/PCF cohorts. Significance was determined using Wilcoxon's rank‐sum test with Benjamini–Hochberg correction.
3.8. RRM2 overexpression creates an immunosuppressive tumor‐immune microenvironment in prostate cancer
The tumor‐immune microenvironment (TIME), which can alter tumor progression and clearance, is affected by the genomic alterations of the tumor (Thorsson et al., 2018). We previously reported that overexpression of RRM2 is highly correlated with copy number alteration (Mazzu et al., 2019); we therefore analyzed the correlation between RRM2 overexpression and infiltration of immune cells in patients with prostate cancer from multiple cohorts. TIL scores were calculated in RRM2‐high and RRM2‐low groups from the TCGA cohort using deep‐learning models that integrate H&E staining of tissues (Saltz et al., 2018). Intriguingly, TIL enrichment was significantly greater in the RRM2‐high group than the RRM2‐low group (P = 9.2e‐05, Fig. 6B). To further examine the correlation between RRM2 level and immune cell infiltration, we applied CIBERSORT analysis, a method of estimating the composition and abundance of immune cells from tumor biopsies (Newman et al., 2015). In the TCGA cohort, the infiltration of antitumor immune cells was significantly lower in the RRM2‐high group than in the RRM2‐low group (CD8+ T cells, P = 0.0012; plasma cells, P = 0.00015), whereas immunosuppressive regulatory T cells (Tregs) were more abundant in RRM2‐high tumors (P = 0.00027, Fig. 6B). Similarly, RRM2‐high tumors in the Taylor cohort, which is mostly composed of primary prostate cancer, had significantly fewer plasma cells (P = 0.015) and Tregs (P = 0.00041) than RRM2‐low tumors (Fig. 6C). In the SU2C/PCF cohort, which includes only metastatic CRPC, RRM2‐high tumors had significantly more immunosuppressive M2 macrophages (P = 0.0431) and Tregs (P = 0.0039) than RRM2‐low tumors (Fig. 6D). There was also greater infiltration of antitumor CD8+ T cells (P = 0.031) in RRM2‐high tumors. The signature scores of the 22 types of immune cells in the LM22 signature in the three prostate cancer cohorts are shown (Figs [Link], [Link], [Link]). Altogether, the high infiltration of immunosuppressive immune cells is suggestive of dysfunctional or exhausted cytotoxic T cells in RRM2‐high tumors.
4. Discussion
Molecular subtyping based on genomic alterations or oncogenic signatures has been successfully applied in multiple cancers. However, the heterogeneous nature of prostate cancer is a major impediment to developing a classification system with clinical relevance. Compared to individual biomarkers or other oncogenic signatures, the prostate cancer classification systems PCS and PAM50 are significantly better at identifying aggressive and resistant cases of prostate cancer.
The PCS classification system was developed and validated in 4600 samples from patients with prostate cancer. TCGA genomic subtypes (e.g., ERG, ETV1/4, SPOP, FOXA1, and others) were present across all the PCS categories (You et al., 2016). PCS1 is highly enriched with the SPOP subtype, whereas PCS2 tumors were overrepresented in ERG cancers (You et al., 2016). In the Genomic Resource Information Database (GRID) cohorts, PCS1 was enriched for Tomlins/ETS+ and Tomlins/SPINK1+ subtypes (You et al., 2016). Importantly, in the GRID cohorts, patients with PCS1 tumors had significantly shorter metastasis‐free survival than patients with PCS2 and PCS3 tumors, but no difference in metastatic progression was seen among the Tomlins categories (You et al., 2016). The PAM50 classification, which was developed using 3782 samples from patients with prostate cancer, was also recently shown to predict associations with clinical outcomes and response to treatment (Zhao et al., 2017).
This study expands upon our prior work on RRM2 in prostate cancer and demonstrates that RRM2 is a master driver of poor prognosis prostate cancer identified by both the PCS1 and PAM50 classification systems. RRM2 is essential for DNA synthesis and repair by producing dNTPs. Its level is rigorously regulated during the cell cycle, and delayed degradation may lead to genomic instability (D'Angiolella et al., 2012). RRM2 is expressed at low levels in normal prostate tissue, but increased expression of RRM2 is highly correlated with poor clinical outcomes in prostate cancer (Huang et al., 2014; Mazzu et al., 2019). We have previously demonstrated that RRM2 is an oncogene in prostate cancer cells, regulates multiple oncogenic signaling pathways, and promotes EMT and angiogenesis (Mazzu et al., 2019). Although common pathways were activated by RRM2 overexpression in LNCaP and PC‐3 cells, the majority of the upregulated genes were different (Fig. 2D–F), suggesting that RRM2‐regulated genes may be disease‐state‐specific.
Genomic alterations that occur in primary prostate cancer may not be enough to predict clinical behavior. The additional and distinct genomic alterations that develop over time add to the molecular heterogeneity of the primary disease and promote metastatic CRPC phenotypes. Therefore, it is not surprising that RRM2 regulates distinct gene sets in two cell lines that may represent different disease states. LNCaP‐RRM2 cells share a greater number of upregulated genes with the TCGA cohort, which only includes localized prostate cancer, than with the Kumar and SU2C/PCF cohorts, which mainly include metastatic CRPC (Fig. 2A). PC‐3 cells are more aggressive than LNCaP cells and may be more representative of advanced CRPC. This is supported by our data on the TCGA, Kumar, and SU2C/PCF cohorts, which demonstrates that a greater number of upregulated genes in PC‐3‐RRM2 cells overlap with genes with expression that correlates with RRM2 than LNCaP‐RRM2 cells (126 genes in PC‐3; seven genes in LNCaP; Fig. 2B). These results further support the idea that RRM2 may function by regulating distinct gene sets in different disease stages of prostate cancer.
Although FOXM1 was identified as a key regulator of the most aggressive subtype of prostate cancer (PCS1), it is difficult to target pharmacologically. Given that RRM2, a gene in the PCS1 and PAM50 signatures, has significant prognostic value in prostate cancer, we evaluated whether it could be another key regulator of aggressive subtypes. Intriguingly, expression of PCS1 genes is highly correlated with RRM2 levels in prostate cancer cohorts; genes upregulated by RRM2 overexpression in prostate cancer cells are also significantly enriched in the PCS1 signature. Furthermore, the 12 genes of the RRM2 signature that are also in the PCS1 and PAM50 signatures are luminal B genes. These results indicate that RRM2 may be a master driver of the aggressive subtypes PCS1 and luminal B by directly or indirectly regulating the expression of critical genes. Ribonucleotide reductase inhibitors have been developed for cancer treatment (Knighton et al., 2018), and we previously reported the potency of the novel RRM2 inhibitor COH29 in prostate cancer (Mazzu et al., 2019). In this study, we confirmed that inhibiting RRM2 activity by siRNA or small molecule (COH29) specifically targets PCS1 and luminal B genes (Fig. 5).
Interestingly, the PAM50 classifier was recently reported as a pan‐carcinoma luminal/basal subtyping across epithelial tumors, and luminal B tumors were more sensitive to the ribonucleotide reductase inhibitor gemcitabine than the other subtypes (Zhao et al., 2019). Because gemcitabine‐induced amplification of RRM2 is a mechanism of gemcitabine resistance (Duxbury et al., 2004; Zhou et al., 2001), we propose that RRM2‐specific inhibitors (e.g., COH29) may be more effective than gemcitabine for multiple epithelial cancers with similar luminal and basal subtypes. Additionally, we demonstrated that RRM2 overexpression may contribute to AR antagonist resistance, suggesting that inhibition of RRM2 may delay the development of resistance.
Because the response rates to immunotherapy in prostate cancer are low, biomarkers are needed to determine which patients will respond. As a driver of aggressive prostate cancer, RRM2 may have a major impact on the TIME. Here, we demonstrated that tumors with high expression of RRM2 have more TILs, but the concomitant enrichment of immunosuppressive immune cells suggests that these TILs may be dysfunctional. Metastatic cases of prostate cancer with high RRM2 levels have increased infiltration of immunosuppressive M2 macrophages, which may contribute to immune escape. It will be critical to validate the association between RRM2 overexpression and changes in the TIME by histologic staining in prostate cancer tissue. Patients with RRM2‐high tumors may be good candidates to receive immunotherapy because of increased TIL infiltration. Combination treatment of RRM2 inhibitors with immunomodulators to stimulate cytotoxic T cells and inhibit immunosuppressive cells may sensitize these tumors to immunotherapies.
5. Conclusions
In summary, we have shown that the genes shared by the PCS1 and luminal B signatures are regulated by RRM2. This suggests that RRM2 is a master driver of aggressive PCSs. Targeting RRM2 may be an effective therapeutic option to reprogram the TIME and treat the subtypes of prostate cancer with poor prognosis.
Conflict of interest
YZM has filed a patent application relevant to the work that is the subject of this paper: U.S. Provisional Patent Application No. 62/834914, RRM2 Signature as a Prognostic Marker for Prostate Cancer Survival, filed April 16, 2019; MSK Ref.: SK2019‐043‐01. As of January 29, 2020, PWK reports the following disclosures for the last 24‐month period: He has investment interest in Context Therapeutics LLC, DRGT, Placon, Seer Biosciences; is a company board member for Context Therapeutics LLC; and is a consultant/scientific advisory board member for Bavarian Nordic Immunotherapeutics, DRGT, GE Healthcare, Janssen, OncoCellMDX, Progenity, Seer Biosciences, and Tarveda Therapeutics; and serves on data safety monitoring boards for Genentech/Roche and Merck.
Author contributions
YZM and PWK conceived and designed the study. YZM developed the methodology. YZM, JA, GC, LEJ, YY, MA, NK, NS, G‐SML, and PWK acquired the data, acquired and managed patient cohort data, and provided facilities. YZM, JA, SN, and PWK analyzed and interpreted the data such as statistical analysis, biostatistics, and computational analysis. YZM, JA, SN, and PWK wrote, reviewed, and/or revised the manuscript. YZM, JA, GC, YY, and LEJ involved in administrative, technical, or material support such as reporting or organizing the data. PWK and YZM supervised the data.
Supporting information
Fig. S1. Clinical significance of 11‐gene signature in patient tissues.
Fig. S2. Profiling of immune cells in RRM2‐high and RRM2‐low prostate cancer samples from the TCGA cohort.
Fig. S3. Profiling of immune cells in RRM2‐high and RRM2‐low prostate cancer samples from the Taylor cohort.
Fig. S4. Profiling of immune cells in RRM2‐high and RRM2‐low prostate cancer samples from the SU2C/PCF cohort.
Table S1. Genes in the RRM2 signature and derived sub‐signatures.
Supplementary Material
Acknowledgements
We thank Sara E. DiNapoli and Margaret McPartland for editing and members of the Kantoff laboratory for help and discussion. This research was funded in part through an NIH/NCI Cancer Center Support Grant to Memorial Sloan Kettering Cancer Center (P30 CA008748).
Contributor Information
Ying Z. Mazzu, Email: mazzuy@mskcc.org.
Philip W. Kantoff, Email: kantoff@mskcc.org.
References
- Aird KM, Li H, Xin F, Konstantinopoulos PA and Zhang R (2014) Identification of ribonucleotide reductase M2 as a potential target for pro‐senescence therapy in epithelial ovarian cancer. Cell Cycle 13, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assikis VJ and Simons JW (2004) Novel therapeutic strategies for androgen‐independent prostate cancer: an update. Semin Oncol 31, 26–32. [DOI] [PubMed] [Google Scholar]
- Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, Drake CG and de Bono JS (2016) Prostate cancer. Lancet 387, 70–82. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C et al (2009) Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Chudin E, Arsanjani A, Zhou L, Garcia EW, Modder J, Kostelec M, Barker D, Downs T, Fan JB et al (2007) Expression signatures that correlated with Gleason score and relapse in prostate cancer. Genomics 89, 666–672. [DOI] [PubMed] [Google Scholar]
- Chabes A and Thelander L (2000) Controlled protein degradation regulates ribonucleotide reductase activity in proliferating mammalian cells during the normal cell cycle and in response to DNA damage and replication blocks. J Biol Chem 275, 17747–17753. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ and Jegga AG (2009) ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JS, Wang Y, Henderson J, Singhal U, Qiao Y, Zaslavsky AB, Hovelson DH, Spratt DE, Reichert Z, Palapattu GS et al (2019) Circulating tumor cell‐based molecular classifier for predicting resistance to abiraterone and enzalutamide in metastatic castration‐resistant prostate cancer. Neoplasia 21, 802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, Mesher D, Speights VO, Stankiewicz E, Foster CS et al (2011) Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol 12, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP and Pagano M (2012) Cyclin F‐mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Laere B, Rajan P, Gronberg H, Dirix L and Lindberg J (2019) Androgen receptor burden and poor response to abiraterone or enzalutamide in TP53 wild‐type metastatic castration‐resistant prostate cancer. JAMA Oncol 5, 1060–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW and Whang EE (2004) RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene 23, 1539–1548. [DOI] [PubMed] [Google Scholar]
- Falzarano SM and Magi‐Galluzzi C (2011) Prostate cancer staging and grading at radical prostatectomy over time. Adv Anat Pathol 18, 159–164. [DOI] [PubMed] [Google Scholar]
- Fujita H, Ohuchida K, Mizumoto K, Itaba S, Ito T, Nakata K, Yu J, Kayashima T, Souzaki R, Tajiri T et al (2010) Gene expression levels as predictive markers of outcome in pancreatic cancer after gemcitabine‐based adjuvant chemotherapy. Neoplasia 12, 807–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason DF and Mellinger GT (1974) Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol 111, 58–64. [DOI] [PubMed] [Google Scholar]
- Glinsky GV, Berezovska O and Glinskii AB (2005) Microarray analysis identifies a death‐from‐cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 115, 1503–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grade M, Hummon AB, Camps J, Emons G, Spitzner M, Gaedcke J, Hoermann P, Ebner R, Becker H, Difilippantonio MJ et al (2011) A genomic strategy for the functional validation of colorectal cancer genes identifies potential therapeutic targets. Int J Cancer 128, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC et al (2012) The mutational landscape of lethal castration‐resistant prostate cancer. Nature 487, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann M, Stenzl A, Bedke J, Chi KN, Black PC and Todenhofer T (2016) Liquid biopsy: ready to guide therapy in advanced prostate cancer? BJU Int 118, 855–863. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu X, Wang YH, Yeh SD, Chen CL, Nelson RA, Chu P, Wilson T and Yen Y (2014) The prognostic value of ribonucleotide reductase small subunit M2 in predicting recurrence for prostate cancers. Urol Oncol 32, 51.e9–19. [DOI] [PubMed] [Google Scholar]
- Ketola K, Munuganti RSN, Davies A, Nip KM, Bishop JL and Zoubeidi A (2017) Targeting prostate cancer subtype 1 by Forkhead box M1 pathway inhibition. Clin Cancer Res 23, 6923–6933. [DOI] [PubMed] [Google Scholar]
- Knighton LE, Delgado LE and Truman AW (2018) Novel insights into molecular chaperone regulation of ribonucleotide reductase. Curr Genet 65, 477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer C, Sterner‐Kock A, Siedentopf F, Schoenegg W, Schlag PM and Kemmner W (2011) Identification of early molecular markers for breast cancer. Mol Cancer 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T et al (2016) Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 22, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA and Chabes A (2011) Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res 39, 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanczky A, Nagy A, Bottai G, Munkacsy G, Szabo A, Santarpia L and Gyorffy B (2016) miRpower: a web‐tool to validate survival‐associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat 160, 439–446. [DOI] [PubMed] [Google Scholar]
- Lee B, Ha SY, Song DH, Lee HW, Cho SY and Park CK (2014) High expression of ribonucleotide reductase subunit M2 correlates with poor prognosis of hepatocellular carcinoma. Gut Liv 8, 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzu YZ, Armenia J, Chakraborty G, Yoshikawa Y, Coggins S, Nandakumar S, Gerke T, Pomerantz M, Qiu X, Zhao H et al (2019) A novel mechanism driving poor‐prognosis prostate cancer: overexpression of the DNA repair gene, ribonucleotide reductase small subunit M2 (RRM2). Clin Cancer Res 25, 4480–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, Desai R, Fox DB, Brannigan BW, Trautwein J et al (2015) RNA‐Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 349, 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network CGAR (2015) The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M and Alizadeh AA (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen T, Wallden B, Schaper C, Ferree S, Liu S, Gao D, Barry G, Dowidar N, Maysuria M and Storhoff J (2014) Analytical validation of the PAM50‐based prosigna breast cancer prognostic gene signature assay and nCounter analysis system using formalin‐fixed paraffin‐embedded breast tumor specimens. BMC Cancer 14, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimir M, Ma Y, Jeffreys SA, Opperman T, Young F, Khan T, Ding P, Chua W, Balakrishnar B, Cooper A et al (2019) Detection of AR‐V7 in liquid biopsies of castrate resistant prostate cancer patients: a comparison of AR‐V7 analysis in circulating tumor cells, circulating tumor RNA and exosomes. Cells 8(7), 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penney KL, Sinnott JA, Fall K, Pawitan Y, Hoshida Y, Kraft P, Stark JR, Fiorentino M, Perner S, Finn S et al (2011) mRNA expression signature of Gleason grade predicts lethal prostate cancer. J Clin Oncol 29, 2391–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA et al (2000) Molecular portraits of human breast tumours. Nature 406, 747–752. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A and Chinnaiyan AM (2004) ONCOMINE: a cancer microarray database and integrated data‐mining platform. Neoplasia 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G et al (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltz J, Gupta R, Hou L, Kurc T, Singh P, Nguyen V, Samaras D, Shroyer KR, Zhao T, Batiste R et al (2018) Spatial organization and molecular correlation of tumor‐infiltrating lymphocytes using deep learning on pathology images. Cell Rep 23, 181–193.e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, Perner S, Sboner A, Pawitan Y, Andren O, Johnson LA et al (2008) Estrogen‐dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst 100, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KN, Mehta KR, Peterson D, Evangelista M, Livesey JC and Faridi JS (2014) AKT‐induced tamoxifen resistance is overturned by RRM2 inhibition. Mol Cancer Res 12, 394–407. [DOI] [PubMed] [Google Scholar]
- Su YF, Wu TF, Ko JL, Tsai HT, Tee YT, Chien MH, Chou CH, Lin WL, Low HY, Chou MY et al (2014) The expression of ribonucleotide reductase M2 in the carcinogenesis of uterine cervix and its relationship with clinicopathological characteristics and prognosis of cancer patients. PLoS ONE 9, e91644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B et al (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA Data Portal . https://tcga‐data.nci.nih.gov/docs/publications/tcga. Accessed February 6, 2020.
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta‐Pardo E, Gao GF, Plaisier CL, Eddy JA et al (2018) The immune landscape of cancer. Immunity 48, 812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Page JL, Surtees JA, Liu H, Lagedrost S, Lu Y, Bronson R, Alani E, Nikitin AY and Weiss RS (2008) Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res 68, 2652–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You S, Knudsen BS, Erho N, Alshalalfa M, Takhar M, Al‐Deen Ashab H, Davicioni E, Karnes RJ, Klein EA, Den RB et al (2016) Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res 76, 4948–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ and Stein GS (2011) A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci USA 108, 9863–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SG, Chang SL, Erho N, Yu M, Lehrer J, Alshalalfa M, Speers C, Cooperberg MR, Kim W, Ryan CJ et al (2017) Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol 3, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao SG, Chen WS, Das R, Chang SL, Tomlins SA, Chou J, Quigley DA, Dang HX, Barnard TJ, Mahal BA et al (2019) Clinical and genomic implications of luminal and basal subtypes across carcinomas. Clin Cancer Res 25, 2450–2457. [DOI] [PubMed] [Google Scholar]
- Zhou B, Mo X, Liu X, Qiu W and Yen Y (2001) Human ribonucleotide reductase M2 subunit gene amplification and transcriptional regulation in a homogeneous staining chromosome region responsible for the mechanism of drug resistance. Cytogenet Cell Genet 95, 34–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Clinical significance of 11‐gene signature in patient tissues.
Fig. S2. Profiling of immune cells in RRM2‐high and RRM2‐low prostate cancer samples from the TCGA cohort.
Fig. S3. Profiling of immune cells in RRM2‐high and RRM2‐low prostate cancer samples from the Taylor cohort.
Fig. S4. Profiling of immune cells in RRM2‐high and RRM2‐low prostate cancer samples from the SU2C/PCF cohort.
Table S1. Genes in the RRM2 signature and derived sub‐signatures.
Supplementary Material
Data Availability Statement
RNA‐seq data are available from the Gene Expression Omnibus (GEO: GSE117921, GEO: GSE117922, GEO: GSE117923, and GEO: GSE117924).