

Abstract
Secondary lymphedema is a common complication of cancer treatment resulting in progressive fibroadipose tissue deposition, increased risk of infections, and, in rare cases, secondary malignancies. Until recently, the pathophysiology of secondary lymphedema was thought to be related to impaired collateral lymphatic formation after surgical injury. However, more recent studies have shown that chronic inflammation-induced fibrosis plays a key role in the pathophysiology of this disease. In this review, we will discuss the evidence supporting this hypothesis and summarize recent publications demonstrating that lymphatic injury activates chronic immune responses that promote fibrosis and lymphatic leakiness, decrease collecting lymphatic pumping, and impair collateral lymphatic formation. We will review how chronic mixed T-helper cell inflammatory reactions regulate this process and how this response may be used to design novel therapies for lymphedema.
Introduction
Lymphedema (LE) is a chronic disease characterized by progressive soft tissue swelling and fibroadipose tissue deposition (Figure 1). The disorder may be caused by genetic abnormalities of the lymphatic system, a condition known as primary LE, or may develop secondary to traumatic, infectious, or other external insults. Secondary LE is by far the most common form of LE in developed countries, usually occurring after lymph node dissection for cancer treatment. Breast cancer, due to its high incidence, is the most common cause of secondary LE, with the disease developing in 20–30% of patients who undergo axillary lymph node dissection. In these procedures, lymph nodes in the axilla draining the breast (and the ipsilateral upper extremity) are removed for local tumor control and cancer staging. LE also occurs in other cancer survivors, afflicting 1 in 6 patients treated for solid tumors.1,2 Because LE is a lifelong disease and cancer treatment options have improved survival, the total number of patients who suffer from secondary LE is increasing every year. Moreover, although improvements in surgical management such as lymphatic mapping and sentinel lymph node biopsy have decreased the need for lymph node dissection, these advances are offset by changes in patient and disease factors that increase the risk of LE development, including increasing rates of obesity, an aging population, and increased use of adjuvant radiation.3, 4 It is estimated that 5–6 million Americans suffer from LE; however, this estimate is likely conservative.5 Nevertheless, this number is significant, since it exceeds the total number of Americans who suffer from systemic lupus erythematosus, rheumatoid arthritis, Parkinson’s disease, and amyotrophic lateral sclerosis combined.6–9 The number of patients with LE is roughly equivalent to the number of Americans with Alzheimer’s disease10 or glaucoma11. (Figure 2). Thus, LE is a major biomedical burden, and development of novel therapies for LE is an important goal.
Figure 1: Lymphedema chronic inflammatory disease results in swollen limbs with fibroadipose tissue deposition.
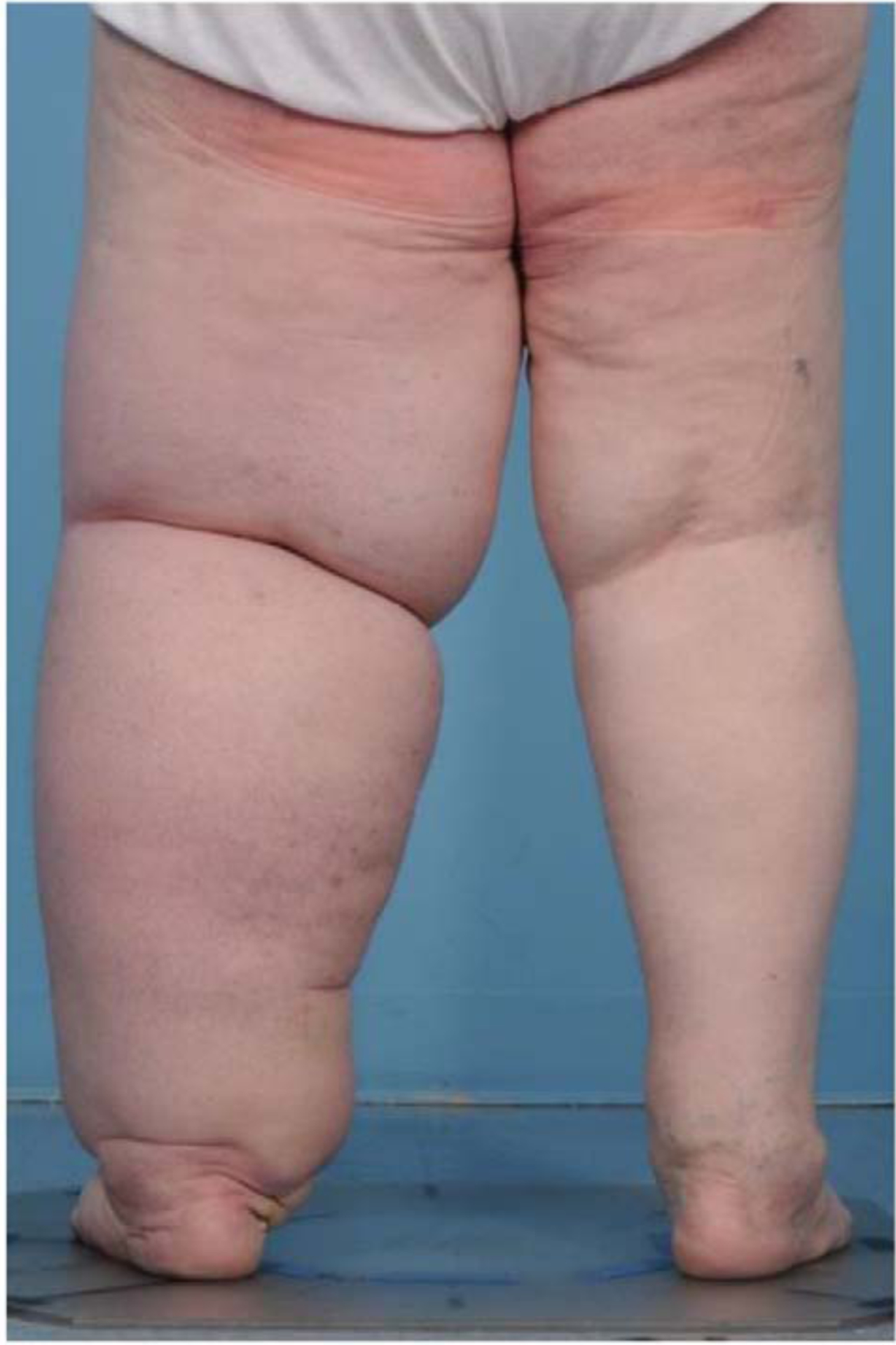
This patient underwent surgical resection of a cervical cancer with pelvic lymph node dissection. Her course was complicated by a rapidly progressing and severely debilitating lymphedema of the lower extremity.
Figure 2: Number of patients in the United States who suffer from lymphedema versus other common chronic disorders.
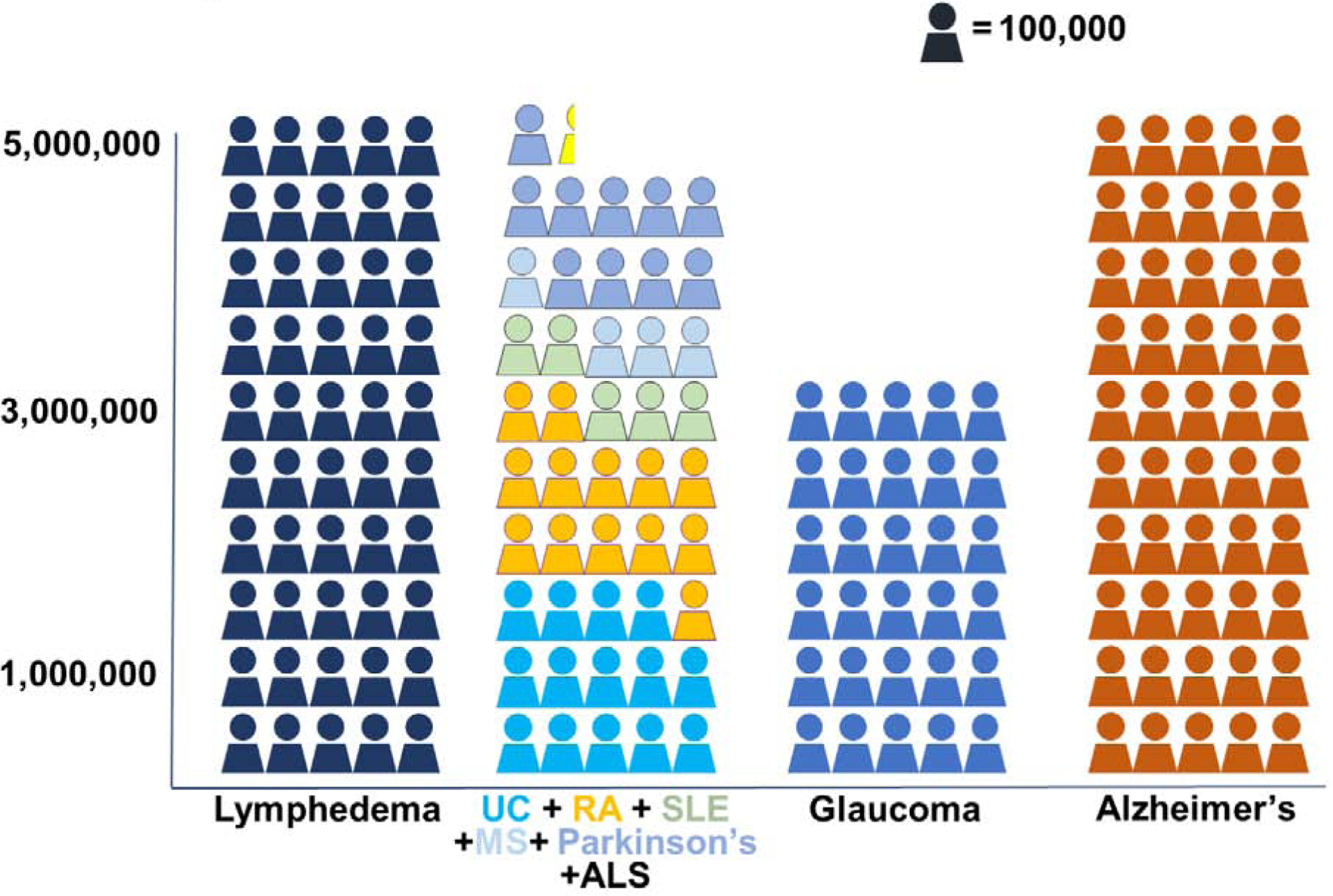
UC, ulcerative colitis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; MS, multiple sclerosis; ALS, amyotrophic lateral sclerosis.
Patients with LE complain of pain, decreased function in the affected extremity, recurrent infections that often require hospitalization and intravenous antibiotics, and, rarely, deadly secondary malignancies.12 Many describe LE as worse than their initial cancer diagnosis because while cancer treatment is finite and predictable, LE is a lifelong disease and unpredictable. The fear of LE progression is a major source of anxiety and decreased quality of life among cancer survivors who have LE.13 Traditionally, LE treatment has been palliative, aiming to prevent swelling by using compressive garments and physical therapy. Importantly, compression and physical therapy do not reverse the underlying abnormalities in the lymphatic system but rather control the symptoms of the disease. These treatments are expensive, increasing direct patient care costs by more than $10,000 per year/patient.14 Often, these costs are not covered by insurance, and this fact, together with the time commitment necessary for LE care, results in high rates of non-compliance and disease progression. More recently, surgical treatments for LE have been developed. These treatments are effective in some patients but can be invasive and require long recovery periods.
Lymphedema is not just lymphatic injury
It has long been assumed that lymphatic injury and failure of lymphatics to regenerate is the defining feature of the pathophysiology of LE. Indeed, the discovery of lymphangiogenic growth factors such as vascular endothelial growth factor-C (VEGF-C) and its homologue VEGF-D led to numerous attempts in preclinical studies to improve LE outcomes in a variety of animal models by delivering exogenous VEGF-C (or other lymphangiogenic growth factors).15, 16 These efforts were strengthened by the finding that Milroy’s disease, a form of primary LE, results from heterozygous inactivating mutations of the receptor for VEGF-C (VEGFR3).17, 18 However, more recent studies have paradoxically shown that VEGF-C expression in lymphedematous tissues and in the serum of patients with LE is increased (suggesting that a deficiency of VEGF-C is not causal) and that this response may contribute to the pathologic changes of the disease by increasing inflammation and blood vessel leakiness.19–21 These findings suggest that lymphatic injury or a deficiency of lymphangiogenic growth factors alone is insufficient to cause the development of LE, and that other pathologic cells and events that either inhibit regeneration of functional lymphatic vessels or soft tissue changes that impair lymphatic transport function (or both) are necessary for the development of the disease.22
The hypothesis that lymphatic injury alone is insufficient for the development of LE is supported by the fact that LE only develops in a subset of patients who undergo lymph node dissection. If lymphatic injury was the defining feature, one would expect LE to develop in all patients, albeit with variable severity, not just a subset. The reason for LE development only in certain percentage of people after lymph node dissection or lymphatic injury is largely unknown. But we can speculate that factors like radiation therapy after surgery, external antigenic stimuli, patient’s inflammatory and immune tolerogenic status could play a critical role in determining the fate of LE progression. Age and BMI also can determine the lymphedema propensity after surgery. Similarly, if lymphatic injury is the primary event, then why do some patients develop LE after seemingly minor lymphatic injury, as occurs following sentinel lymph node biopsy where only a few lymph nodes are removed? Finally, and most importantly, lymphatic injury as the sole pathophysiologic determinant of secondary LE development fails to explain the time course of LE development, since most patients who develop the disease do so in a delayed fashion, often months and sometimes years after surgery.23,12, 24 These epidemiologic characteristics suggest that lymphatic injury is simply an initiating event and that a series of other pathologic changes are required for manifestation of the disease. This concept is supported by the fact that animal models of LE have been notoriously hard to produce. Although many researchers attempted to recreate LE by excising lymph nodes in a variety of animal species (dogs, rats, mice, rabbits, sheep, pigs), these studies were reported as failures since swelling was, in most cases, transient and modest (at best).25 It is probably more accurate to conclude that these animal study findings mimicked clinical changes (i.e., transient edema that resolves only to recur in a subset of individuals months to years later). Thus, animal models of LE rely on other interventions (e.g., extensive skin excision, external beam radiation therapy, etc.) to cause more significant lymphatic damage and accelerate LE development. There is a dearth of perfect animal models to understand LE pathophysiology that recapitulates the human LE pathology completely. Probably, a combination of lymphatic injury with radiation therapy might be a good model to study and understand LE in a detailed fashion.
Consistent with the fact that lymphatic injury at the site of lymph node removal is the initiating event in the development of LE is the finding that the lymphatic vasculature of the entire limb (not just the lymphatics in the axilla) is destroyed in late-stage disease. Improved imaging technologies such as magnetic resonance imaging and indocyanine green (ICG) lymphangiography have documented that as LE progresses, lymphatic vessels (both capillary and collecting lymphatics) become increasingly leaky.26 There is also evidence of lymphatic hyperplasia with development of aberrant lymphatic channels and obstruction at multiple levels. Eventually, in late-stage disease, there is a near complete loss of functional lymphatics, resulting in widespread interstitial fluid accumulation in the skin/dermis27, 28 (Figure 3).
Figure 3: Indocyanine green (ICG) lymphangiography of a normal (upper) and lymphedematous (lower) upper extremity.
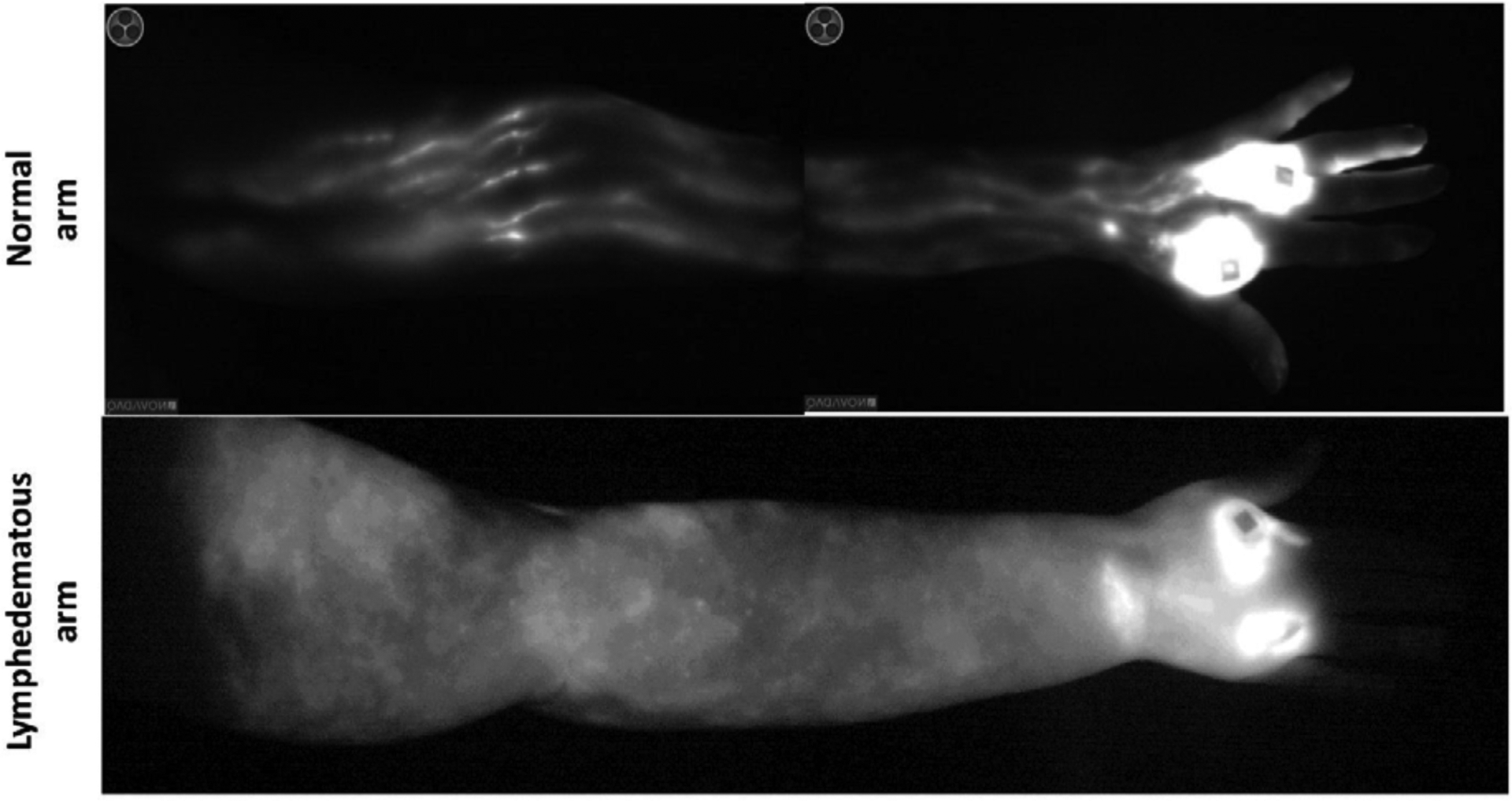
Note linear lymphatic collectors draining from the hand (bright spots are the injection sites) up towards the axilla in the normal limb. In contrast, note complete absence of lymphatic channels and accumulation of ICG dye in the skin of the lymphedematous limb.
Is lymphedema fibrotic organ failure of the lymphatic system?
Because the lymphatic vessels transport interstitial fluid and the initial presentation of LE is fluid accumulation, most lay individuals consider LE to be a “fluid” problem. However, long-term chronic LE is a disease characterized by excessive fibroadipose tissue deposition. In fact, this is the physiologic event that makes compression garments and physiotherapy less effective in patients with long-standing or advanced disease: fat is not compressible.
Fibroadipose tissue deposition and skin fibrosis in LE is progressive, and we and others have shown that the initial lymphatics become encased by thick collagen bundles in this process (Figure 4). Other clinical studies have shown that collecting lymphatics in late-stage LE become increasingly sclerosed, with proliferation of smooth muscle cells and obliteration of the lumen.29, 30 Based on these observations, we have hypothesized that LE is end-stage fibrotic organ failure of the lymphatic system in which functional parenchyma (i.e., lymphatic vessels) is gradually replaced by scar. Thus, patients who develop LE following lymph node dissection do so in a delayed manner (i.e., months after the initial surgery), since the fibrosis necessary to cause clinical symptoms takes time to develop. Our hypothesis is supported by the fact that fibrosis is a common cause of end-organ failure in most organ systems. The clinical features of fibroproliferative diseases is also like LE in that disease development is unpredictable, such that some patients exposed to a particular insult (even sometimes in seemingly trivial doses) develop organ fibrosis, while others with similar exposure do not. The progression of fibroproliferative disorders is also like LE in that the disease process is usually progressive once initiated. Fibrosis also provides a rationale for the clinical risk factors of LE such as obesity or radiation, since these stimuli independently increase tissue fibrosis.31 Indeed, we have previously reported that inhibition of radiation-induced fibrosis is more effective in preserving lymphatic function than strategies that directly protect lymphatic endothelial cells from radiation injury. In addition, we have found that treatments that inhibit profibrotic growth factors such as transforming growth factor beta-1 (TGF-B1) are also effective in preventing the development of LE in preclinical models.32 Based on these findings, we hypothesize that lymphatic injury initiates downstream changes that promote fibrosis and, once a critical threshold is reached, then LE develops. If this threshold is not reached for whatever reason, then the patient remains asymptomatic. This may be the reason why some patients develop LE years after the initial injury, and a minor inciting event, such as an infection or injury, precedes the development at that time.
Figure 4: Lymphedema results in collagen deposition and fibrosis of capillary (upper panel) and collecting (lower panel) lymphatics in a mouse model of tail lymphedema.
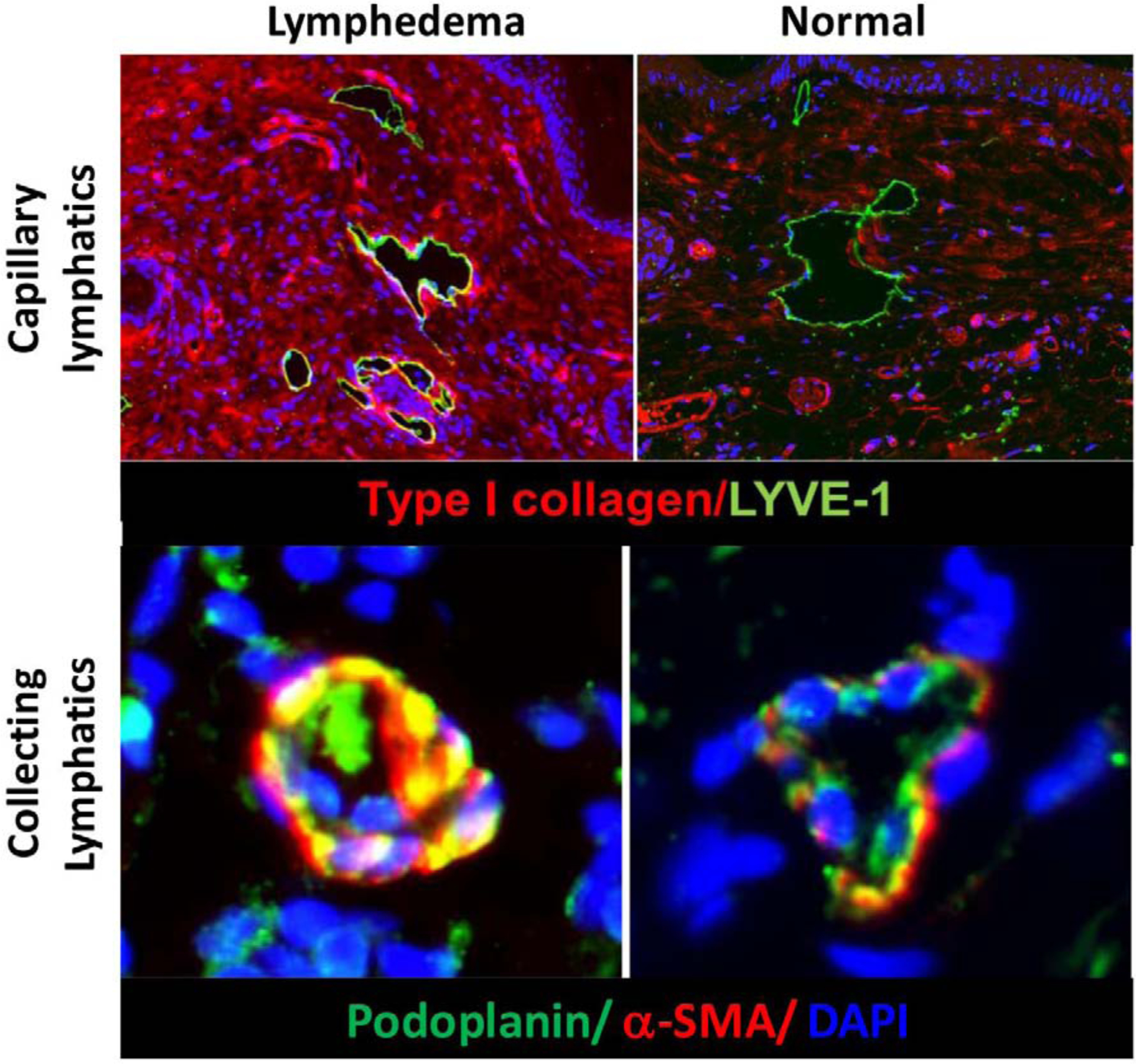
Note accumulation of type I collagen fibers encasing capillary lymphatics in the skin (upper left). Also note proliferation of a-sma positive smooth muscle cells and obliteration of the lumen of collecting lymphatics in lymphedematous tissues (lower left).
How is fibrosis in lymphedema regulated?
A large body of literature supports an important role for T helper cells in the regulation of fibrotic organ failure. Naïve T helper cells, in response to different antigenic stimuli, interaction with antigen-presenting cells, and through exposure to cytokines, can differentiate along multiple pathways, including T helper type 1 (Th1), Th2, Th17, and regulatory T cells, among others. These cells have different cytokine profiles and mediate a variety of immune responses. Th2 cells ordinarily play a key role in parasitic responses; however, chronic Th2 responses can also cause immunosuppression, inhibit lymphatic growth, and promote tissue fibrosis by increasing collagen production, decreasing matrix breakdown, and increasing expression of other profibrotic growth factors such as interleukin-4 (IL-4), IL-13, and TGF-B1.33–35 In contrast, Th1 cells are thought to play an anti-fibrotic role, in some scenarios, balancing the Th2 response. This concept has been referred to as the Th1/Th2 paradigm and was first put forth by Thomas Wynn who, in a series of elegant publications, showed that Th2-biased chronic inflammatory reactions regulate development of fibrosis in a variety of clinical settings, including liver and kidney fibrosis. Thus, despite the wide variability in the clinical presentation and etiology of various fibroproliferative disorders, the mechanisms that regulate fibrosis are remarkably conserved.36–38
Early studies by Stanley Rockson from Stanford University demonstrated that LE, like other fibrotic disorders, is also characterized by chronic inflammatory reactions.39,40 Inhibition of inflammation in preclinical models using ketoprofen, a non-steroidal anti-inflammatory medication, was effective in decreasing the severity of LE.41 These findings led to a clinical trial with ketoprofen, initially as an open-label study with 21 patients and subsequently with 34 patients randomized to a double-blind placebo-controlled trial. Although the study failed to show improvements in arm volumes (the primary end point), the authors did report improvements in skin histopathology (primarily fibrotic changes) and skin thickness.42 Subsequent studies by this group went on to show that the primary benefit of ketoprofen was due to inhibition of leukotriene B4, enabling to use of more targeted anti-inflammatory treatments.43 A new clinical trial with leukotriene B4 inhibitors is underway.
Our lab has focused on characterizing the inflammatory cell infiltrate in clinical LE biopsy samples and mouse models of LE using a variety of techniques, including immunofluorescence, flow cytometry, and polymerase chain reaction (PCR). These studies have shown that most inflammatory cells present in lymphedematous tissues are CD4+. Moreover, we found that the severity of breast cancer-related LE positively correlated with the number of infiltrating CD4+ cells. Thus, patients with more severe LE (e.g., stage 3) had a statistically increased number of CD4+ cells, as compared with women with milder forms of the disease (stage 1 or 2). Even minor lymphatic injury, in the form of lymph node dissection in which there is very limited tissue swelling, resulted in accumulation of CD4+ cells.44,45 These studies have since been confirmed by at least two other labs.46,47 We then showed that mice lacking T cells in general or mice with targeted deletion of CD4, in contrast to control mice, did not develop tissue fibrosis or subsequent LE, suggesting that CD4+ cells play a causal role in the development of the disease. Adoptive transfer of CD4+ cells derived from bone marrow of wild-type mice to lethally irradiated CD4 knockout mice restored the phenotype such that the fibrosis and LE that developed following tail surgery in these mice was indistinguishable from wild-type controls.48
Because knockout mice can have changes in their immune responses that may be independent of the target gene, we performed additional experiments on wild-type mice in which we depleted inflammatory cells using neutralizing antibodies. These mice underwent tail skin and lymphatic excision, and control mice were treated with isotype control antibodies. Experimental mice were treated with neutralizing antibodies that depleted CD4+ cells, CD8+ cells, or B cells. In other experiments, we used clodronate liposomes to deplete macrophages. As expected, these treatments were highly effective in depleting target cells, as confirmed by flow cytometry. Interestingly, however, we found that only depletion of CD4+ cells prevented development of LE. Depletion of other inflammatory cells had either no effect on overall fibrosis or adipose tissue deposition (anti-CD8, anti-B cell) or made the phenotype more severe (macrophage depletion).44,49
More recently, we have shown that topical application of tacrolimus, a calcineurin inhibitor that decreases IL-2 activity, as well as T-cell activation and proliferation, also markedly decreases fibrosis in the mouse tail model of LE. When tacrolimus was applied shortly after lymphatic injury, this treatment completely prevented development of tail LE. In contrast, control mice treated with vehicle alone developed the predictable pattern of tail swelling and fibrosis. Application of tacrolimus 6 weeks following tail LE onset was also beneficial, decreasing the severity of LE significantly; however, as would be expected with any chronic disease, this treatment was not as effective as preventing the development of LE. Importantly, we found that application of topical tacrolimus resulted in marked decreases in CD4+ cell infiltration, even though systemic absorption was below levels known to cause immune defects.50 These findings have translational relevance, since they suggest that treatments for LE can be directly applied to the affected area, thereby decreasing systemic toxicity.
Additional characterization studies showed that the CD4+ response, similar to fibrosis in other organ systems, was a mixed cell population consisting of Th1, Th2, and regulatory T cells.51 T helper cells in lymphedematous tissues tended to cluster around lymphatic vessels (both capillary and collecting lymphatics) and produced abundant amounts of profibrotic cytokines. including IL-4, IL-13, and TGF-B. Inhibition of Th2 differentiation with neutralizing antibodies against IL-4 or IL-13 effectively prevented development of LE in the mouse tail model of LE when antibodies were administered shortly after surgery.45 This treatment was also highly effective in treating established LE after it’s onset. In our recent study, we have found that mice with genetic defects that impair Th2 differentiation (signal transducer and activator of transcription 6 [STAT-6] knockout), in contrast to mice with impaired Th1 differentiation (T-bet knockouts), are protected from LE development, displaying decreased tissue fibrosis, adipose deposition, and decreased overall inflammatory responses.52 The translational relevance of these studies is derived from the fact that immunotherapy has revolutionized the treatment of many chronic inflammatory diseases, including psoriatic arthritis, rheumatoid arthritis, and Crohn’s disease, among others. Targeting Th17 differentiation or Th17 cytokines in rheumatoid arthritis and inhibiting tumor necrosis factor-α in Crohn’s disease showed significantly better clinical outcomes. Also, injecting certain peptide-modified autologous dendritic cells (DCs) reduced disease activity in rheumatoid arthritis patients.53–55These therapies blocking specific immune response are targeted, well tolerated, and highly effective. A similar immunotherapy approach may be feasible in patients with LE to prevent development of the disease or as an adjunct to other treatments in patients with established LE.
How are T cells activated?
T cells are part of the adaptive immune system and respond and proliferate after exposure to antigenic stimuli by antigen-presenting cells. In a series of experiments utilizing adoptively transferred tagged cells to mice treated with popliteal lymph node dissection, we have found that lymphatic injury rapidly causes activation of DCs in the skin of the limb distal to the site of lymphatic injury.48 Adoptively transferred DCs injected into the systemic circulation home to the skin in this area, become activated, increasing expression of CCL7 and CD86, and migrate to regional lymph nodes (usually one lymph node basin away from the site of injury). However, what are the potential factors that activate DCs during lymphatic injury are yet to be studied. We speculate that, DCs could be activated by bacterial pathogens that take tissue entry during surgery and by inflammatory mediators that are released due to surgery induced tissue damage. Pathogen Associated Molecular Patterns (PAMPs) and Damage Associated Molecular Patterns (DAMPs) can indirectly activate DCs. In addition, other first responder cells like neutrophils, dead, necrotic cells and their cellular products can activate DCs during lymphatic injury. DC activation in LE is a critical aspect that requires a more detailed study in future research. Activated DCs in the lymph node interact with naïve CD4+ cells and induce a mixed Th1/Th2 differentiation. This interaction is necessary for T cell activation, since adoptive transfer of DCs from CD28 knockout mice (a co-receptor necessary for T cell activation by DCs) did not result in Th1/Th2 differentiation. Activated T cells are released into the blood circulation from the lymph node. These activated T cells then home to the skin distal to the zone of injury by expressing specific cell surface receptors (cutaneous lymphocyte antigen, chemokine receptor [CCR] 4, CCR10; unpublished observations) that bind chemokines expressed in blood vessels (vascular cell adhesion molecule [V-CAM], E-Selectin) and keratinocytes (CCL17, CCL27). Importantly, we found that inhibition of T cell release from the lymph node by S1P1 antagonist prevents the development of LE, further supporting a causal role for this inflammatory cell type in the pathogenesis of the disease (Figure 5 summary diagram).48
Figure 5: Summary diagram denoting immune cell activation in lymphedema.
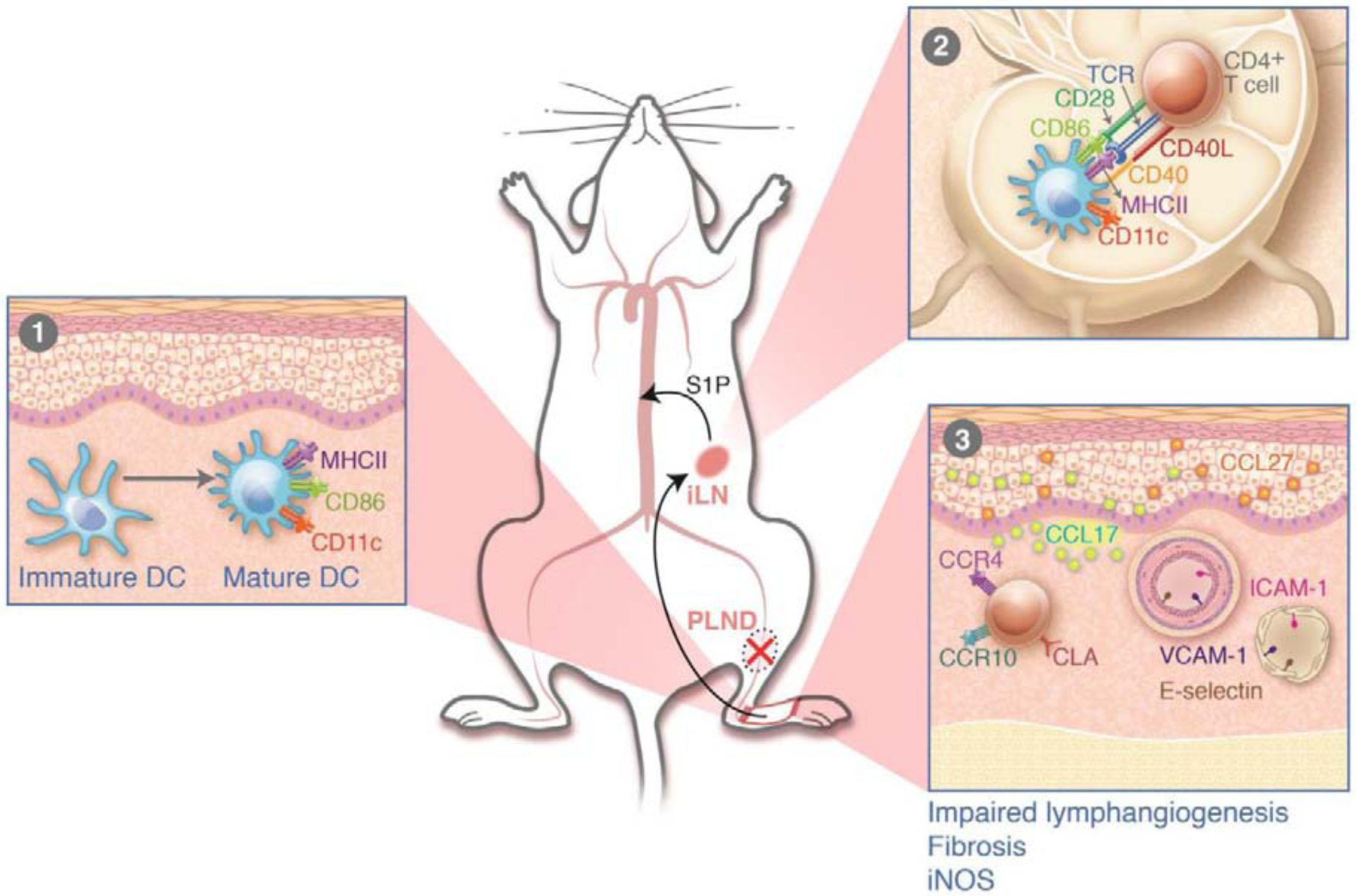
Panel 1 demonstrates activation of dendritic cells (DCs) in the skin; panel 2 shows migration of activated DCs to regional lymph nodes and activation of naïve T cells with T cell receptor and co-stimulatory molecule interaction. Activated T cells home to the skin in the lymphedematous limb in response to chemokines expressed by keratinocytes and leukocyte adhesion molecules by blood endothelial cells. Reprinted with permission from Garcia Nores GD et al; Nature Communications. 2018;9(1):1970
Unresolved questions and where do we go from here?
There are many unresolved questions. For example, how are DCs activated by lymphatic injury? What are the antigens that activated DCs (and T cells) respond to? In addition to fibrosis, what are the mechanistic events that regulate lymphatic function? How do chronic inflammatory reactions regulate collecting vessel phenotype, leakage, and valve structure? Why is the pattern of LE in the extremity so highly variable, with some patients displaying disease in the upper arm, others around the elbow, and yet others in the hands and fingers? Why do some patients with LE suffer from chronic infections, and what is the effect of these infections on the lymphatic system? Just like most other things in life, sometimes it seems as if the more we know, the less we know! Nevertheless, studying the pathophysiology of a disease process is the only way to develop rational and targeted therapies. Determining how the sequence of events is regulated, as well as the key cellular and molecular events that regulate the ultimate expression of the disease, is the best way to translate our knowledge clinically and design novel therapies.
Conclusions
LE is a complex disease, with variable clinical presentation and unpredictable course. Previously, it was thought that LE occurred as a consequence of abnormal or aberrant lymphatic regeneration following injury. However, more recent studies suggest that lymphatic injury is simply an initiating event, setting off a series of downstream changes that in some patients results in the development of LE. These changes include chronic T cell inflammatory reactions, infiltration of a mixed Th1/Th2 cell population, production of proinflammatory and profibrotic cytokines, and progressive fibroadipose tissue deposition. Developing therapies targeting these pathways may provide novel treatment options for a disease in which there has been little progress over the past 40 years.
Funding:
This review was supported by NIH R01 HL111130 grant and P30 CA008748 cancer center support grant.
Abbreviations:
- LE
lymphedema
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor 3
- ICG
indocyanine green
- TGF-β
transforming growth factor beta-1
- Th
helper type T lymphocyte
- IL
interleukin
- PCR
polymerase chain reaction
- STAT
signal transducer and activator of transcription
- T-bet
transcription factor expression in B-cell precursor cell line
- DCs
dendritic cells
- CCR
chemokine receptor
- ICAM
intercellular adhesion molecule
- VCAM
vascular cell adhesion molecule
- S1P
spingosine-1-phosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: All authors have read the journal’s policy on disclosure of potential conflicts of interest. The authors have no conflicts of interest to declare.
References
- 1.Petrek JA, Heelan MC. Incidence of breast carcinoma-related lymphedema. Cancer 1998;83:2776–81. [DOI] [PubMed] [Google Scholar]
- 2.Petrek JA, Senie RT, Peters M, Rosen PP. Lymphedema in a cohort of breast carcinoma survivors 20 years after diagnosis. Cancer 2001;92:1368–77. [DOI] [PubMed] [Google Scholar]
- 3.Tsai RJ, Dennis LK, Lynch CF, Snetselaar LG, Zamba GK, Scott-Conner C. The risk of developing arm lymphedema among breast cancer survivors: a meta-analysis of treatment factors. Ann Surg Oncol 2009;16:1959–72. [DOI] [PubMed] [Google Scholar]
- 4.Cormier JN, Askew RL, Mungovan KS, Xing Y, Ross MI, Armer JM. Lymphedema beyond breast cancer: a systematic review and meta-analysis of cancer-related secondary lymphedema. Cancer 2010;116:5138–49. [DOI] [PubMed] [Google Scholar]
- 5.Rockson SG, Rivera KK. Estimating the population burden of lymphedema. Ann N Y Acad Sci 2008;1131:147–54. [DOI] [PubMed] [Google Scholar]
- 6.Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum 2008;58:15–25. [DOI] [PubMed] [Google Scholar]
- 7.Somers EC, Marder W, Cagnoli P, Lewis EE, DeGuire P, Gordon C, et al. Population-based incidence and prevalence of systemic lupus erythematosus: the Michigan Lupus Epidemiology and Surveillance program. Arthritis Rheumatol 2014;66:369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marras C, Beck JC, Bower JH, Roberts E, Ritz B, Ross GW, et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 2018;4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun 2016;7:12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.2016 Alzheimer’s disease facts and figures. Alzheimers Dement 2016;12:459–509. [DOI] [PubMed] [Google Scholar]
- 11.McDermott RJ, Sarvela PD, Hoalt PN, Bajracharya SM, Marty PJ, Emery EM. Multiple correlates of cigarette use among high school students. J Sch Health 1992;62:146–50. [DOI] [PubMed] [Google Scholar]
- 12.Dayan JH, Ly CL, Kataru RP, Mehrara BJ. Lymphedema: pathogenesis and novel therapies. Annu Rev Med 2018;69:263–76. [DOI] [PubMed] [Google Scholar]
- 13.Gjorup CA, Groenvold M, Hendel HW, Dahlstroem K, Drzewiecki KT, Klausen TW, et al. Health-related quality of life in melanoma patients: Impact of melanoma-related limb lymphoedema. Eur J Cancer 2017;85:122–32. [DOI] [PubMed] [Google Scholar]
- 14.Shih YC, Xu Y, Cormier JN, Giordano S, Ridner SH, Buchholz TA, et al. Incidence, treatment costs, and complications of lymphedema after breast cancer among women of working age: a 2-year follow-up study. J Clin Oncol 2009;27:2007–14. [DOI] [PubMed] [Google Scholar]
- 15.Yoon YS, Murayama T, Gravereaux E, Tkebuchava T, Silver M, Curry C, et al. VEGF-C gene therapy augments postnatal lymphangiogenesis and ameliorates secondary lymphedema. J Clin Invest 2003;111:717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lahteenvuo M, Honkonen K, Tervala T, Tammela T, Suominen E, Lahteenvuo J, et al. Growth factor therapy and autologous lymph node transfer in lymphedema. Circulation 2011;123:613–20. [DOI] [PubMed] [Google Scholar]
- 17.Irrthum A, Karkkainen MJ, Devriendt K, Alitalo K, Vikkula M. Congenital hereditary lymphedema caused by a mutation that inactivates VEGFR3 tyrosine kinase. Am J Hum Genet 2000;67:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brice G, Child AH, Evans A, Bell R, Mansour S, Burnand K, et al. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet 2005;42:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fink AM, Kaltenegger I, Schneider B, Fruhauf J, Jurecka W, Steiner A. Serum level of VEGF-D in patients with primary lymphedema. Lymphology 2004;37:185–9. [PubMed] [Google Scholar]
- 20.Jensen MR, Simonsen L, Karlsmark T, Lanng C, Bulow J. Higher vascular endothelial growth factor-C concentration in plasma is associated with increased forearm capillary filtration capacity in breast cancer-related lymphedema. Physiol Rep 2015;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gousopoulos E, Proulx ST, Bachmann SB, Dieterich LC, Scholl J, Karaman S, et al. An important role of VEGF-C in promoting lymphedema development. J Invest Dermatol 2017;137:1995–2004. [DOI] [PubMed] [Google Scholar]
- 22.Kataru RP, Kim H, Jang C, Choi DK, Koh BI, Kim M, et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity 2011;34:96–107. [DOI] [PubMed] [Google Scholar]
- 23.Planinsek Rucigaj T, Tlaker Zunter V. Lymphedema after breast and gynecological cancer - a frequent, chronic, disabling condition in cancer survivors. Acta Dermatovenerol Croat 2015;23:101–7. [PubMed] [Google Scholar]
- 24.Rockson SG. Lymphedema after breast cancer treatment. N Engl J Med 2018;379:1937–44. [DOI] [PubMed] [Google Scholar]
- 25.Hadamitzky C, Pabst R. Acquired lymphedema: an urgent need for adequate animal models. Cancer Res 2008;68:343–5. [DOI] [PubMed] [Google Scholar]
- 26.Mortimer PS, Rockson SG. New developments in clinical aspects of lymphatic disease. J Clin Invest 2014;124:915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutkowski JM, Moya M, Johannes J, Goldman J, Swartz MA. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, VEGF-C upregulation, and the protective role of MMP-9. Microvasc Res 2006;72:161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaleska MT, Olszewski WL. Imaging lymphatics in human normal and lymphedema limbs-Usefulness of various modalities for evaluation of lymph and edema fluid flow pathways and dynamics. J Biophotonics 2018;11:e201700132. [DOI] [PubMed] [Google Scholar]
- 29.Mihara M, Hara H, Hayashi Y, Narushima M, Yamamoto T, Todokoro T, et al. Pathological steps of cancer-related lymphedema: histological changes in the collecting lymphatic vessels after lymphadenectomy. PLoS One 2012;7:e41126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardenier JC, Hespe GE, Kataru RP, Savetsky IL, Torrisi JS, Nores GD, et al. Diphtheria toxin-mediated ablation of lymphatic endothelial cells results in progressive lymphedema. JCI Insight 2016;1:e84095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwan ML, Darbinian J, Schmitz KH, Citron R, Partee P, Kutner SE, et al. Risk factors for lymphedema in a prospective breast cancer survivorship study: the Pathways Study. Arch Surg 2010;145:1055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Avraham T, Yan A, Zampell JC, Daluvoy SV, Haimovitz-Friedman A, Cordeiro AP, et al. Radiation therapy causes loss of dermal lymphatic vessels and interferes with lymphatic function by TGF-beta1-mediated tissue fibrosis. Am J Physiol Cell Physiol 2010;299:C589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin K, Kataru RP, Park HJ, Kwon BI, Kim TW, Hong YK, et al. TH2 cells and their cytokines regulate formation and function of lymphatic vessels. Nat Commun 2015;6:6196. [DOI] [PubMed] [Google Scholar]
- 34.Savetsky IL, Ghanta S, Gardenier JC, Torrisi JS, Garcia Nores GD, Hespe GE, et al. Th2 cytokines inhibit lymphangiogenesis. PLoS One 2015;10:e0126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheever AW, Williams ME, Wynn TA, Finkelman FD, Seder RA, Cox TM, et al. Anti-IL-4 treatment of Schistosoma mansoni-infected mice inhibits development of T cells and non-B, non-T cells expressing Th2 cytokines while decreasing egg-induced hepatic fibrosis. J Immunol 1994;153:753–9. [PubMed] [Google Scholar]
- 37.Chiaramonte MG, Cheever AW, Malley JD, Donaldson DD, Wynn TA. Studies of murine schistosomiasis reveal interleukin-13 blockade as a treatment for established and progressive liver fibrosis. Hepatology 2001;34:273–82. [DOI] [PubMed] [Google Scholar]
- 38.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 2007;117:524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabibiazar R, Cheung L, Han J, Swanson J, Beilhack A, An A, et al. Inflammatory manifestations of experimental lymphatic insufficiency. PLoS Med 2006;3:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ly CL, Kataru RP, Mehrara BJ. Inflammatory manifestations of lymphedema. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakamura K, Radhakrishnan K, Wong YM, Rockson SG. Anti-inflammatory pharmacotherapy with ketoprofen ameliorates experimental lymphatic vascular insufficiency in mice. PLoS One 2009;4:e8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rockson SG, Tian W, Jiang X, Kuznetsova T, Haddad F, Zampell J, et al. Pilot studies demonstrate the potential benefits of antiinflammatory therapy in human lymphedema. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian W, Rockson SG, Jiang X, Kim J, Begaye A, Shuffle EM, et al. Leukotriene B4 antagonism ameliorates experimental lymphedema. Sci Transl Med 2017;9. [DOI] [PubMed] [Google Scholar]
- 44.Zampell JC, Yan A, Elhadad S, Avraham T, Weitman E, Mehrara BJ. CD4(+) cells regulate fibrosis and lymphangiogenesis in response to lymphatic fluid stasis. PLoS One 2012;7:e49940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Avraham T, Zampell JC, Yan A, Elhadad S, Weitman ES, Rockson SG, et al. Th2 differentiation is necessary for soft tissue fibrosis and lymphatic dysfunction resulting from lymphedema. FASEB J 2013;27:1114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gousopoulos E, Proulx ST, Scholl J, Uecker M, Detmar M. Prominent lymphatic vessel hyperplasia with progressive dysfunction and distinct immune cell infiltration in lymphedema. Am J Pathol 2016;186:2193–203. [DOI] [PubMed] [Google Scholar]
- 47.Gousopoulos E, Proulx ST, Bachmann SB, Scholl J, Dionyssiou D, Demiri E, et al. Regulatory T cell transfer ameliorates lymphedema and promotes lymphatic vessel function. JCI Insight 2016;1:e89081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia Nores GD, Ly CL, Cuzzone DA, Kataru RP, Hespe GE, Torrisi JS, et al. CD4(+) T cells are activated in regional lymph nodes and migrate to skin to initiate lymphedema. Nat Commun 2018;9:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghanta S, Cuzzone DA, Torrisi JS, Albano NJ, Joseph WJ, Savetsky IL, et al. Regulation of inflammation and fibrosis by macrophages in lymphedema. Am J Physiol Heart Circ Physiol 2015;308:H1065–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gardenier JC, Kataru RP, Hespe GE, Savetsky IL, Torrisi JS, Nores GD, et al. Topical tacrolimus for the treatment of secondary lymphedema. Nat Commun 2017;8:14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia Nores GD, Ly CL, Savetsky IL, Kataru RP, Ghanta S, Hespe GE, et al. Regulatory T cells mediate local immunosuppression in lymphedema. J Invest Dermatol 2018;138:325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ly CL, Nores GDG, Kataru RP, Mehrara BJ. T helper 2 differentiation is necessary for development of lymphedema. Transl Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walters TD, Kim MO, Denson LA, Griffiths AM, Dubinsky M, Markowitz J, et al. Increased effectiveness of early therapy with anti-tumor necrosis factor-alpha vs an immunomodulator in children with Crohn’s disease. Gastroenterology 2014;146:383–91. [DOI] [PubMed] [Google Scholar]
- 54.Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Transl Med 2015;7:290ra87. [DOI] [PubMed] [Google Scholar]
- 55.Roeleveld DM, Koenders MI. The role of the Th17 cytokines IL-17 and IL-22 in Rheumatoid Arthritis pathogenesis and developments in cytokine immunotherapy. Cytokine 2015;74:101–7. [DOI] [PubMed] [Google Scholar]