

Applying functional principal component analyses to time series genome-wide association studies increases the power and accuracy of gene identification in a sorghum association panel.
Abstract
The phenotypes of plants develop over time and change in response to the environment. New engineering and computer vision technologies track these phenotypic changes. Identifying the genetic loci regulating differences in the pattern of phenotypic change remains challenging. This study used functional principal component analysis (FPCA) to achieve this aim. Time series phenotype data were collected from a sorghum (Sorghum bicolor) diversity panel using a number of technologies including conventional color photography and hyperspectral imaging. This imaging lasted for 37 d and centered on reproductive transition. A new higher density marker set was generated for the same population. Several genes known to control trait variation in sorghum have been previously cloned and characterized. These genes were not confidently identified in genome-wide association analyses at single time points. However, FPCA successfully identified the same known and characterized genes. FPCA analyses partitioned the role these genes play in controlling phenotypes. Partitioning was consistent with the known molecular function of the individual cloned genes. These data demonstrate that FPCA-based genome-wide association studies can enable robust time series mapping analyses in a wide range of contexts. Moreover, time series analysis can increase the accuracy and power of quantitative genetic analyses.
Quantitative genetic approaches are widely used across all domains of biology to identify genetic loci controlling variation in target traits. Many genes where naturally occurring functionally variable alleles control variation in agronomically relevant traits have been identified. Both quantitative trait locus (QTL) mapping (structured populations) or genome-wide association studies (GWAS; association panels) have been used to identify these loci (Huang et al., 2010; Chen et al., 2016; Navarro et al., 2017). Collecting phenotypic data from the hundreds to thousands of accessions required for QTL mapping or GWAS has historically been a time- and resource-intensive undertaking. Hence, most attempts to identify genes controlling variation in a target trait employ data from a single time point, usually either at maturity or after a fixed number of days from planting. However, recent engineering advances, such as wearable devices, automated phenotyping greenhouses, field phenotyping robots, unmanned aerial vehicles, and computer vision advances, are lowering the barriers and activation energy required to score traits from multiple points throughout development (Furbank and Tester, 2011; Moore et al., 2013; Honsdorf et al., 2014; Holman et al., 2016; Fernandez et al., 2017; Oren et al., 2017; Feldman et al., 2017, 2018). Plant growth and development is a dynamic process, responding to environmental perturbations. It is regulated by different suites of genes at different times and in different environments (Yan et al., 1998; Wu and Lin, 2006; Moore et al., 2013; Würschum et al., 2014; Feldman et al., 2017, 2018; Muraya et al., 2017). The availability and use of time series trait data from mapping and association populations has the potential to increase the accuracy and power of gene mapping studies (Moore et al., 2013; Sang et al., 2019). It may also provide greater insight into the biologically distinct roles different loci play in determining final phenotypes. However, integrating time series data into statistical frameworks originally envisioned for single-trait measurements across large populations is not straightforward. A range of approaches are currently being explored by the research community.
There are several approaches to the use of time-series phenotypic data in mapping studies. The most straightforward is to conduct QTL mapping or GWAS separately at each time point and then summarize the mapping results (Yan et al., 1998; Wu et al., 2010; Osman et al., 2013). However, this approach is not robust when faced with missing data or when subsets of the population are scored on alternating time points. This procedure also requires complex approaches to multiple testing correction given the partially correlated nature of both linked genetic markers and measurements of the same trait in the same individuals at multiple time points. A second widely employed method is to summarize patterns of change over time using predefined functions with discrete numbers of variables (Ma et al., 2002; Wu et al., 2004; Paine et al., 2012). QTL mapping or GWAS can then be conducted for the values of the different variables within the equation, as that function is fit to data from different individuals. This approach can be powerful and is able to impute missing data points in individuals. However, this approach can fail when the pattern of phenotypic change over time follows an unknown function, is too complex to fit, or does not conform to the expected function (Feldman et al., 2018). Functional principal component analysis (FPCA), as a more general method, provides some of the strengths of fitting parametric functions sharing data across time points. This includes the ability to impute missing values without requiring that patterns of phenotypic change over time fit any particular function (Kwak et al., 2016; Xu et al., 2018a, 2018b). A variation of nonparametric functional principal component-based mapping has been successfully employed to identify loci controlling the gravitropism response in Arabidopsis (Arabidopsis thaliana) seedlings (Moore et al., 2013; Kwak et al., 2016). Kwak et al. (2016) concluded that dimensional reduction via FPCA may increase the power to detect QTL in recombinant inbred populations relative to prior approaches that include trait data from each time point. Muraya et al. (2017) employed an FPCA method adapted from Kwak et al. (2016) to identify several trait-associated single nucleotide polymorphisms (SNP) for maize (Zea mays) biomass accumulation in vegetative development. This represented an advancement over parametric regression, which had not been able to identify any trait-associated SNPs using the same trait and marker dataset. However, none of the identified loci coincided either with markers identified in single time point analyses or with known loci controlling the trait of interest.
Three-cloned sorghum (Sorghum bicolor) genes controlling height (dwarf1 [Dw1], dwarf2 [Dw2], and dwarf3 [Dw3]) are segregating in the sorghum association panel (SAP). Dw1 (Sobic.009G229800) was cloned by two independent fine-mapping studies (Hilley et al., 2016; Yamaguchi et al., 2016). The function of the Dw1 gene appears to act in the brassinosteroid signaling pathway to influence sorghum plant height by reducing cell proliferation activity in the internodes (Hirano et al., 2017). Dw2 (Sobic.006G067700) on chromosome 6 is in tight linkage with maturity1 (Ma1; Sobic.006G057866), a gene with a large effect on sorghum flowering time. In many cases, both loci are introgressed together during the backcrossing and conversion process of adapting tropical sorghum germplasm to grow in temperate latitudes (Quinby, 1974; Higgins et al., 2014). The Dw2 gene encodes a protein kinase that is homologous to KIPK in Arabidopsis and influences sorghum plant height by reducing the length of internodes (Hilley et al., 2017). Ma1 gene has the largest impact on flowering time through encoding the pseudoresponse regulator protein 37 (PRR37) to modulate flowering time in sorghum (Murphy et al., 2011). Dw3 (Sobic.007G163800) encodes a MDR transporter orthologous to brachytic2 in maize and appears to influence cell elongation through a role in polar auxin transport (Multani et al., 2003). Previous sorghum height mapping studies identified epistatic interactions between Dw1 and Dw3, but no significant epistatic interactions have been reported between Dw2 and either Dw1 or Dw3 (Brown et al., 2008; Hilley et al., 2016; Yamaguchi et al., 2016).
In this study, we employed a new approach to FPCA of nonparametric regression data (Xu et al., 2018a). The procedure’s effectiveness at both controlling false positives and identifying known true positives in sorghum was evaluated. A phenotypically constrained subset of the SAP was utilized (Casa et al., 2008). This population was grown and imaged through vegetative and reproductive development in a high-throughput phenotyping facility. Organ-level semantic segmentation from hyperspectral images was employed to extract phenotypic values (Miao et al., 2020). Genome-wide association using the functional principal component scores derived from the FPCA approach described by Xu et al. (2018a) could be used to successfully identify all three known dwarf genes. Three novel signals were also detected. The signal on chromosome 3 was independently validated in data from a previous study of a different sorghum population. These results favorably contrasted with terminal phenotyping or time point by time point analyses in the same population and provide additional insight into the distinct biological roles of three known dwarf genes in determining sorghum height.
RESULTS
Loss of Association Power in Phenotypically Constrained Populations
The classical sorghum dwarfing genes have large effects on plant height and are segregating within the SAP (Morris et al., 2013; Li et al., 2015). Field-collected plant height data for 357 lines from the SAP correctly identified both Dw1 and Dw2 (Fig. 1A) as well as a third previously reported locus known to influence variation in height in this population (Li et al., 2015). Thirty-eight lines from the field study exceeded the physical limit on maximum height for the imaging facility employed in this study (Fig. 1B). After the exclusion of these 38 lines as well as an additional 27 lines that failed to germinate or thrive in the greenhouse, field-measured height data for the remaining 292 lines was still sufficient to identify Dw1, albeit with substantially reduced statistical significance. However, after the removal of these phenotypically extreme lines, signals from Dw2 and the other previously reported height-controlling loci were no longer statistically significant (Fig. 1C). Dw3, located on the long arm of chromosome 7, was not identified in association analyses using either field data on terminal plant heights from the complete set of 357 SAP lines or in analyses of data only from the phenotypically constrained subset of SAP lines employed for greenhouse experiments. Excluding lines in the mapping population not only decreased the total population size, but also reduced the minor allele frequency of SNPs near genes known to control variation in sorghum height (Supplemental Fig. S1).
Figure 1.

Reduced power to identify causal loci in phenotypically constrained populations. A, A genome-wide association analyses for plant height, defined as the distance between the soil surface and the top of the panicle at maturity, using field collected data for 357 lines from SAP and the set of genotype call data used in this study. The location of Dw1, Dw2, and Ma1 are indicated with dashed lines, as is an additional known height locus (KHL) identified in multiple prior GWAS conducted on height in this population using different genetic marker data. B, Distribution of observed heights for the 357 lines employed for association analysis in A. The set of 38 lines above 2 m in height are marked in red. C, A genome-wide association analysis identical to that shown in A but with the exclusion of lines with the field heights >2 m (38 lines) and those which we were not able to successfully germinate and phenotype in this study (27 lines).
Genetic Associations with Sorghum Height at Different Time Points
The phenotypically constrained subset of 292 SAP lines was imaged at the University of Nebraska’s Greenhouse Innovation Center. Imaging lasted ∼1 month and centered on reproductive development. Each plant was imaged using a set of five cameras, including top views and side views from multiple angles and hyperspectral imaging from a single side view perspective (Ge et al., 2016; Liang et al., 2018; Miao et al., 2020). Two methods were employed to extract plant height at individual time points. The first was whole-plant segmentation, an approach widely used in plant image analysis (Gehan et al., 2017): segmentation of an image into plant pixels and not plant pixels and measuring height by the difference between the minimum and maximum y axis values for plant pixels (Fig. 2A). The second approach was a recently described method (Miao et al., 2020) to semantically segment plant pixels of sorghum plants into separate stalk, leaf, and panicle classes (Fig. 2B; Supplemental Fig. S2). There are many different ways to measure the plant height (Brown et al., 2008; Miao et al., 2020). Generally, researchers in the field have preferred to benchmark on the height of stalk, height to the uppermost leaf collar, or the height to the tip of the inflorescence rather than the height to the highest leaf tip. An advantage of the latter method, semantic segmentation, is that it can be used to measure a version of plant height, which more closely approximates how height is measured in the field (Miao et al., 2020). Sorghum plant height measured by whole-plant segmentation tended to oscillate over time as new leaves emerged, whereas sorghum plant height measured via the semantic segmentation method tended to be monotonically increasing (Fig. 2C). Generally, researchers in the field have preferred to measure the height of stalk or the height of inflorescence rather than the height to the highest point on the plant, which is often a leaf tip. The semantic segmentation approach makes it possible to measure height using a definition for plant height that corresponds to the definition of plant height used by sorghum geneticists in the field (Miao et al., 2020).
Figure 2.

Different methods to define and measure plant height produce different outcomes. A, Conventional RGB images of a single sorghum plant (PI 576401) taken on eight different days spanning the transition from vegetative to reproductive development. Pixels identified as “plant” through whole plant segmentation are outlined in red. Measured plant height, defined as the distance between the plant pixels with the smallest and greatest y axis value, is indicated by the horizontal blue bar in each image. B, Semantically segmented images of same plant taken on the same day from a moderately different viewing angle using a hyperspectral camera. Pixels classified as “leaf” are indicated in green, pixels classified as “stem” are indicated in orange, and pixels classified as “panicle” are indicated in purple. Measured plant height, defined as the distance between stem or panicle pixels with the smallest and greatest y axis value, is indicated by the horizontal red bar in each image. C, Observed and imputed plant heights for the same sorghum plant on each day within the range of phenotypic data collection. Blue and red circles indicate measured height values from whole plant segmentation of RGB images and semantic segmentation of hyperspectral images, respectively. Solid blue and solid red lines indicate height values imputed for unobserved time points using nonparametric regression for whole plant and semantic height datasets, respectively.
A second challenge faced by many time series imaging studies is the maximum daily throughput provided by fixed imaging infrastructure. In this particular study, individual plants were divided into two groups imaged on alternating days. Therefore, there was no single time point at which image or height data were available for all individual plants (Supplemental Fig. S3A). In order to estimate the height of each individual plant at each individual time point, nonparametric regression was employed to impute all missing data points (Supplemental Fig. S3B). To assess the accuracy of this imputation, the percent of variance in height explained by genetic factors was assessed at individual time points for subsets of the population using either height measured directly from images or height imputed from nonparametric curves. In most cases, the proportion of variance in imputed height that could be explained by genetic factors matched or exceeded the equivalent value for directly measured height (Supplemental Fig. S4). This is consistent with previous work, which found that missing values in time-series plant phenomics data can be imputed accurately from nonparametric curves (Xu et al., 2018b).
Independent genome-wide association analyses were conducted for imputed sorghum height as measured via semantic segmentation on each day from 41 to 76 d after planting (DAP). Consistent with observations from field-collected plant height data, almost no significant trait/marker associations were observed in the analyses for 65 to 76 DAP (Supplemental Fig. S5; Supplemental File S1). In the early days of the experiment, statistically significant signals were identified all over the genome, likely as a result of extreme height values for a small number of lines. Those lines experienced reproductive transition and panicle emergence prior to the start of imaging (Supplemental Fig. S6). Excluding extreme lines led to the identification of signals near Dw1, Dw3, and Ma3 in data from some early time points mixed in among approximately a dozen other repeatedly identified loci across the genome. A loss of detectable genetic associations between 54 and 66 DAP corresponded roughly to booting and panicle exertion in many sorghum accessions. The peduncle length is under the control of a distinct set of genetic factors from plant height below the flag leaf (Li et al., 2015), and the timing of panicle exertion is under the control of a third distinct set of factors, namely maturity genes (Quinby and Karper, 1945; Quinby, 1967). We speculate that, once a large proportion of lines advanced to the booting stage or beyond, the role these three sets of genetic factors played in determining plant height diluted the total variance attributable to any one single genetic locus, reducing power to identify statistically significant associations.
A second set of analyses were conducted where nonparametric curves calculated for individual plants were aligned based on time relative to panicle emergence (e.g. days after panicle emergence [DAPE]) rather than time relative to planting (DAP; Fig. 3; Supplemental File S1). Given variation in the date of panicle emergence and the dangers of extrapolating nonparametric curves beyond the range of observed data points, it must be noted that this approach meant that data were available only for distinct subsets of the original 292 phenotyped sorghum lines at any given time point. Day-by-day GWASs were conducted from 14 d prior to panicle emergence to 10 d after panicle emergence, as described above (Fig. 4). No known sorghum flowering-time loci were identified using this approach, with the exception of Ma1. However, due to the strong linkage between Ma1 and Dw2 genes based on previous studies, it is not possible to exclude the possibility that the significantly associated markers near Ma1 reflect the effect of Dw2 on plant height (Quinby, 1974; Higgins et al., 2014). The signal corresponding to Dw2 was identified in the DAPE GWAS analysis for 6 d between −14 and −5 DAPE with the signal 323 kb from the known locus. The signal corresponding to Dw1 was identified from −14 to +1 DAPE and was located only 15 kb away from the known location of Dw1 on sorghum chromosome 9. This was closer than the 114-kb distance between the gene and the closest significant SNP identified in DAP GWAS results (Supplemental Fig. S6). Dw3 did not show any statistically significant signal in the DAPE-based analysis but was identified in some time points when plant data were compared using DAP. Hence, whereas it was possible to identify all three known true-positive genes controlling sorghum plant height through genome-wide association analyses at individual time points, in no case were all three identified in a single analysis across the ∼60 total GWASs either conducted at different time points or using different developmental landmarks. Many other confounding associations were also identified. In many cases, both the single most significantly associated SNP and the “hot zone” of SNPs all showing strong significant association with trait variation were quite distant from the known causal locus. However, they were still within the range expected given observed linkage disequilibrium decay rates in sorghum (Morris et al., 2013; Miao et al., 2019).
Figure 3.

Comparison of change in plant height over time for members of the SAP population when anchoring either on planting date (DAP) or panicle emergence date (DAPE). A, Growth curves imputed using nonparametric regression for 20 representative sorghum genotypes, anchored for comparison based on sharing the same date of planting. B, Growth curves imputed using nonparametric regression for the same 20 sorghum genotypes shown in A, anchored for comparison based on sharing the same date of panicle emergence. Lines with identical colors in A and B indicate data taken from the same plants. Regression lines are not extended beyond the range of observed data points. As panicle emergence occurred less than 18 d after the start of imaging for some lines and less than 17 d before the last day of imaging for others, curves in B are incomplete.
Figure 4.

Several known causal loci show statistically significant associations with plant height when sequential genome-wide association studies are conducted using data anchored to the date of panicle emergence. A summary of where statistically significant trait-associated SNPs were identified in separate genome-wide association studies conducted using height data for each day between −14 to +10 DAPE. Each vertical column summarizes the results from one of the 25 independently conducted genome-wide association studies. Each sorghum chromosome is divided into 16 bins containing equal numbers of SNP markers. Each cell in each vertical column is color coded based on the single most significant P-value observed for any marker within that bin on that day. Light pink cells indicate bins that contain no markers that exceed the multiple testing-corrected threshold for statistical significance. The locations of the two cloned dwarf genes and the one cloned maturity gene which were successfully identified in analysis of data from at least one time point are indicated with horizontal dashed lines.
Mapping Genes Controlling Variation in Growth Curves
All three known true positive height genes were identified by sequential time point-based GWAS. However, there was no single time point, nor any single treatment of the data (DAP or DAPE), where all three of said genes were identified. Many other loci not previously reported to be linked to height were also identified with equal or greater statistical support to known true positive genes across many time points. In order to conduct the individual time point analyses, it was necessary to fit nonparametric curves to each individual plant to impute unobserved values. Functional principal component analyses (Fig. 5A) was employed to decompose variation among the curves into four functional principal components that combined to explain >99% of the total variance in curve shape among the plants in the population. The first two functional principal components were able to explain >97% of total variation (Fig. 5, B and C).
Figure 5.

Two functional principal components explain >97% of variance in the sorghum growth curves observed in this study. Functional principal component analysis seeks to describe the pattern of change in height over time of each observed plant using a mean function combined with variable weightings of a set of eigenfunctions. A, Comparison of the empirical mean function (red) for all growth curves observed in this study (gray) and the mean function estimated using functional principal component analysis (blue). B, Illustration of how changing the score for functional principal component one alters the resulting growth curve. C, Illustration of how changing the score for functional principal component two alters the resulting growth curve.
As the first two functional principal components described the vast majority of the variation in patterns of change in plant growth over time, genome-wide association analyses were conducted to identify genes controlling variation in these two phenotypic descriptors (Fig. 6; Supplemental File S1). Sorghum lines with negative scores of the first functional principal component tended to be taller overall and exhibited greater increases in height during panicle emergence than lines with positive scores of the second functional principal component (Fig. 6, A and B). Both Dw1 and Dw2 showed statistically significant associations with variation in the first functional principal component, as did a single locus on the short arm of chromosome 3 (Fig. 6E). Sorghum lines with negative functional principal component two scores tended to be taller prior to panicle emergence but exhibited limited additional increases in height during panicle emergence. Moreover, lines with positive functional principal component two scores started out shorter but exhibited big increases in height during panicle emergence. This second set of lines was taller at the end of the experiment (Fig. 6, C and D). Dw3 showed a statistically significant association with variation in the second functional principal component, as did two other regions of the genome close to the centromeres of sorghum chromosomes 5 and 9 (Fig. 6F). As was seen in Figure 3, none of the hits identified here overlapped with known sorghum maturity genes with the exception of Ma1. However, another potential explanation was that previously uncharacterized loci were controlling flowering time in the environmental conditions used to conduct this study. A genome-wide association analysis for days to panicle emergence identified three well-supported loci: Ma1 on chromosome 6 and two loci on sorghum chromosomes 1 and 10. These loci do not correspond to known sorghum maturity genes (Supplemental Fig. S7). However, with the exception of Ma1, these loci did not overlap with regions of the genome associated with height or patterns of change in height when using panicle emergence as a developmental landmark (Figs. 3 and 6).
Figure 6.

Mapping genes associated with variation in functional principal component scores among sorghum genotypes. A, Distribution of functional principal component one scores among the 292 genotypes phenotyped as part of this study. Genotypes with the most negative values for functional principal component one are indicated in red, and genotypes with the most positive values for functional principal component one are indicated in blue. B, Growth curves for a subset of genotypes with the most negative values for functional principal component one are indicated in red. Growth curves for a subset of genotypes with the most positive values for functional principal component one are indicated in blue. C, Distribution of functional principal component two scores among the 292 genotypes phenotyped as part of this study. Genotypes with the most negative values for functional principal component two are indicated in red, and genotypes with the most positive values for functional principal component two are indicated in blue. D, Growth curves for a subset of genotypes with the most negative values for functional principal component two are indicated in red. Growth curves for a subset of genotypes with the most positive values for functional principal component two are indicated in blue. E, Results of conducting a genome-wide association analysis for functional principal component one scores. F, Results of conducting a genome wide association analysis for functional principal component two scores. In E and F, the positions of three cloned dwarf genes Dw1, Dw2, and Dw3 as well as the cloned maturity gene Ma1 are indicated using vertical dash lines. Horizontal dash lines indicate multiple testing corrected cutoff of a statistically significant association.
The distance between the nearest SNP significantly associated with functional principal component one and Dw1 (Sobic.009G229800) is approximately 35 kb. The hot region of SNPs on chromosome 6, which is significantly associated with functional principal component one, spans both Dw2 (Sobic.006G067700) and Ma1 (Sobic.006G057866). The novel hit for functional principal component one on chromosome 3 was also identified in sequential DAPE GWAS analysis (Fig. 4). The same small interval on chromosome 3 where this novel hit was identified was previously linked to variation in sorghum height in an independent QTL mapping population (Brown et al., 2006). This hit is associated with a large (17 complete genes or gene fragments) tandem array of wall-associated kinase (WAK) genes. WAKs play a role in cell elongation and expansion, and previous antisense experiments targeting genes in this family in Arabidopsis produced dwarf phenotypes (Lally et al., 2001; Wagner and Kohorn, 2001). The distance between the nearest SNP significantly associated with functional principal component two and Dw3 (Sobic.007G163800) is approximately 39 kb. The other two significant signals for functional principal component two on chromosome 5 and 9 were not associated with any immediately obvious candidate genes. This may reflect the location of these hits in low-recombination/high linkage-disequilibrium regions of the genome, expanding the potential distance between a trait-associated genetic marker and the causal locus.
DISCUSSION
Engineering and computational advances are making it increasingly practical to dynamically monitor the trait values for organisms across time and development. Time series data provide opportunities to understand the role individual genetic loci play in shaping phenotype. At the same time, extracting the greatest possible insight from time series data requires modifications to quantitative genetic approaches traditionally employed for linking genotypic variation to phenotypic variation across an entire population at a single point in time. Time series data from a phenotypically constrained diversity population in sorghum was used to evaluate a number of approaches for leveraging time series data to identify and characterize how different loci play different roles in determining phenotype at different stages of development. A key feature that enabled the analyses was that three known large-effect loci controlling height were segregating in the population of plants analyzed. However, these loci were not consistently identified in conventional single-point GWAS given the phenotypically constrained set of the plants employed in the study (Fig. 1). In field studies, it is often necessary to restrict the range of phenotypic diversity exhibited by an association population key trait in order to obtain meaningful and comparable trait data in a single environment (Hansey et al., 2011). Artificial selection also tends to reduce the range of phenotypic variation present within elite populations used for crop improvement (Yamasaki et al., 2007).
Different plants proceed through different stages of their life cycle at different rates. Comparisons across varieties must, explicitly or implicitly, employ life-cycle landmarks to enable comparisons across individuals. In many field studies, traits are collected from terminal-stage plants, using physiological maturity as the life-cycle landmark for comparisons across individuals. In several other studies, including many greenhouse- or growth chamber-based ones, comparisons are performed at a fixed number of days after planting or days after germination, i.e. using planting or germination as the life cycle landmark. The SAP employed in this study is sampled primarily from sorghum conversion lines collected from around the world (Casa et al., 2008). Despite the introgression of photoperiod insensitivity loci as part of the conversion process, lines vary significantly in total days spent in vegetative development before transitioning to reproductive development. We experimented with using either time of planting or time of panicle emergence as the life cycle landmark for phenotypic comparisons. For these particular analyses, using panicle emergence as a landmark provided significantly cleaner results with identification of known height-related loci (Fig. 4). In general, time series trait data enable researchers to experiment with using different landmarks for comparisons across individuals in a population. The correct choice in any given case will depend on the specific goal of the analyses.
Plant heights at different time points were extracted from hyperspectral images using a semantic segmentation approach (Miao et al., 2020). Compared to widely used thresholding-based whole-plant segmentation approaches in RGB (red-green-blue) images, semantic segmentation is able to classify each plant pixel to the corresponding organ, which provides more plant height estimations at different stages of development. These estimations are more comparable to measurements made by hand by biologists. The approach shows generalizability within sorghum between mature plants and plants at the late vegetative stage, prior to visible reproductive development but after clear leaf and stalk separations (Miao et al., 2020). However, one source of error with this classification approach is a tendency to misclassify leaf midribs as stem pixels, particularly early in development. In early vegetative development of sorghum, the shoot apical meristem remains below the soil surface, and the apparent stem is simply a whorl of multiple leaf sheaths. As the dataset evaluated here consists of plants from 40 DAP onwards, it is not possible to draw strong conclusions about how this approach would perform for sorghum seedlings.
Simulation studies based on fitting parametric models have demonstrated that integrating longitudinal measures of phenotypes in a single population can provide increased resolution and power to identify QTLs (Sang et al., 2019). Here, we employed nonparametric models that required fewer initial assumptions about the pattern of how phenotypes change over time (Yang et al., 2009). Like parametric models, nonparametric models can be used to accurately impute trait values at unobserved time points (Xu et al., 2018b). This enables the combined analyses of time data from larger populations given a fixed capacity in terms of number of individuals phenotyped per day. Relative to previously published functional principal component analyses, the statistical approach employed here is robust to small with uneven numbers of time point observations per sample.
GWASs based on functional principal component weightings assigned to the time series data from individual plants, collected on separate days, successfully identified all three known true positive genes controlling height in sorghum. Both Dw1 and Dw2 were associated with variation in the first functional principal component of sorghum height, which exhibited a consistent effect on height both before and after panicle emergence and exertion (Fig. 6, A, B, and E). Dw3 was instead associated with the second functional principal component of sorghum height, which exhibited opposite directions of effect on height before and after panicle emergence and exertion (Fig. 6, C, D, and F). Previous studies suggest that epistasis exists between Dw1 and Dw3, but they are thought to act through different mechanisms or pathways to influence sorghum plant height (Yamaguchi et al., 2016; Hirano et al., 2017). Dw1 influences plant height through the brassinosteroid signaling pathway (Hirano et al., 2017). It reduces the length of all internodes by reducing the number of cells in each internode (Hilley et al., 2016; Yamaguchi et al., 2016). However, Dw3 and its maize ortholog brachytic2 appear to influence plant height through auxin transport. Dw3 primarily reduces the lengths of lower internodes rather than all the internodes and acts by decreasing the length of cells rather than decreasing the total cell number (Multani et al., 2003; Yamaguchi et al., 2016). The molecular function of Dw2 (Sobic.006G067700) is still not clear, but the Sobic.006G067700 loss of function phenotype includes reductions in the length of all internodes, which is similar to the loss-of-function phenotype of Dw1(Sobic.009G229800) and dissimilar to the loss-of-function phenotype of Dw3 (Sobic.007G163800; Hilley et al., 2017). These known phenotypes and molecular functions of the three sorghum dwarfing genes are consistent with the pattern observed in the FPCA-based GWAS analyses where Dw1 and Dw2 are associated with a consistent effect before and after panicle emergence. At the same time, Dw3 is associated with a functional principle component that switches the direction of effect before and after panicle emergence.
In addition to successfully identifying all three known true positive height genes in sorghum, functional principal component-based mapping for plant height exhibited much greater enrichment for these genes. Three statistically significant loci were identified outside of the predefined known true positive set: the signal on the short arm of chromosome 3 for the first functional principal component and two signals in the pericentromeric regions of chromosomes 5 and 9 for the second functional principal component. Given the broader mapping intervals and slower linkage disequilibrium decay in pericentromeric regions, it was not possible to confidently conclude whether the signals on chromosomes 5 and 9 correspond to specific previous reports from QTL mapping or genome-wide association in sorghum. However, the chromosome three signal associated with WAK genes was also identified in the DAPE-based sequential GWAS results around panicle emergence (Fig. 4). This indicates it may play an important role in a shift in the speed that booting stage occurs. This signal was also validated as corresponding to a tightly mapped sorghum plant height QTL identified in a separate recombinant inbred line population (Brown et al., 2006).
Compared to the GWAS results on terminal height collected from the field in the phenotypically constrained population (Fig. 1C), FPCA-based GWAS analyses recovered Dw1 and Dw2 identified using the whole population and also identified Dw3 as well as several additional loci (Figs. 1A and 6, E and F). However, the known height locus on chromosome 6, which was identified in the field-based data presented in this paper as well as in previous GWAS for sorghum height, was not recovered (Li et al., 2015). Mapping genes controlling height or most other highly polygenic traits in different field seasons or locations will tend to identify only partially overlapping sets of loci. This can be a result of genotype by environment interactions, where certain loci influence variation in a given trait in some environments but not others. It can also be a result of stochastic noise in phenotypic measurements. The statistical approaches used for GWAS are designed to minimize false positives and accept a very high false-negative rate. As a result, modest-effect loci will rise above the noise in some experimental replications but not others. Consistent with either of these explanations, the known height locus on chromosome 6 was not statistically significant in a GWAS analysis for genes controlling sorghum height conducted by Morris et al. (2013) but was statistically significant in a separate analysis conducted for the same trait by Li et al. (2015).
Time series trait data are rapidly becoming more widely available and collected in a broad range of contexts: model organisms, crops, livestock, humans, etc. Integrating functional principal component analyses into GWASs allows the identification of known true positive genes controlling phenotypic variation in populations where these genes cannot be confidently identified from data at any single time point. In addition, this study demonstrates that the associations of different known true positive genes with different functional principal components describing variation in the target trait are consistent with the known biological roles and previous quantitative genetic associations of those genes. This in turn suggests that functional principal component-based GWASs of time series data can provide greater insight into the distinct roles different trait-associated loci play in determining variation in a single phenotype. A statistical approach to decomposing patterns of phenotypic variation over time into functional principal components was employed that is robust to incomplete and used uneven observations of different subsets of the population at different time points. This robust approach should aid the broader adoption of functional principal component-based genome-wide association in a wider range of quantitative genetics contexts.
MATERIALS AND METHODS
Plant Materials and Growth Conditions
Three-hunderd and fifty seven sorghum (Sorghum bicolor) lines from the SAP were planted, grown, and phenotyped at (latitude 41.162°, longitude −96.407°) part of the University of Nebraska-Lincoln’s Eastern Nebraska Research and Extension Center in 2016. Plant height, defined as the distance from emergence from the soil to the top of the uppermost panicle, was manually scored at maturity. Due to the height limitation of the imaging chamber equipped in the high-throughput phenotyping facility in the University of Nebraska-Lincoln’s Greenhouse Innovation Center (UNL-GIC; latitude 40.83°, longitude −96.69°), 38 lines with heights in the field >2 m were excluded from subsequent greenhouse experiments. Seeds from the remaining 319 sorghum SAP lines were sown in the greenhouse of UNL-GIC on June 15, 2016. Sorghum seeds were sown in 9.46-L pots with Fafard germination mix supplemented with 8.8 kg 3- to 4-month Osmocote and 8.8 kg 5- to 6-month Osmocote, 1 tablespoon (15 mL) of Micromax micronutrients, and 1800 g lime per 764.5 L (1 cubic yard) of soil. Twenty-seven sorghum lines failed to germinate or failed to grow healthily under greenhouse conditions. These sorghum lines were omitted from downstream analyses, leaving a total of 292 lines for phenotyping. Phenotyped plants were grown under a target photoperiod of 14:10 day:night with supplementary light provided by light-emitting diode growth lamps from 7 am to 7 pm each day. The target temperature of the growth facility was between 20°C to 28.3°C. After growing in the greenhouse for 40 d, all the plants were moved on to the conveyor belt, which transferred each pot to the imaging chamber every 2 d and to the watering station each day to keep all the plants growing under a good condition. At the watering station, plants were weighed once per day and watered back to a target weight, including pot, soil, carrier, and plant of 6,300 g from July 25 to August 9, 7000 g from August 9 until the termination of the experiment August 31 to 76 DAP.
Image Data Acquisition
The imaging of all phenotyped sorghum lines commenced on July 26 and continued until August 31, 41 to 76 DAP. Image data were collected using the high-throughput phenotyping facility in UNL-GIC previously described (Ge et al., 2016). Each plant was imaged every other day by a visible camera (piA2400-17gm, BASLER, with PENTAX lens) and a hyperspectral camera (Headwall Photonics). The visible camera was used to capture RGB images with the resolution 2354 × 2056 including two different zoom levels. The first zoom level was applied at 41 to 53 DAP. Each pixel represented an area of approximately 0.45 mm2 for objects in the range between the camera and the pot containing the plant. The second zoom level with each pixel representing an area of approximately 1.8 mm2 was applied after 54 DAP until the termination of the experiment (76 DAP). The hyperspectral camera has a spectral range of 546 to 1700 nm and 243 image bands. Plants were arranged so that most leaves perpendicularly face to the hyperspectral camera. A hyperspectral cube for each plant was captured at a resolution of 320 × 560 pixels. For each hyperspectral cube, a total of 243 separate intensity values were captured for each pixel spanning the range of light wavelengths between 546 to 1700 nm. A constant zoom level was applied for all the hyperspectral images throughout the whole experiment, with each pixel representing an area of ∼8.8 mm2.
Height Extraction from RGB and Hyperspectral Images
The estimate of plant height in RGB images uses the green index threshold method based on the Equation 2 ×green/(red + blue; Ge et al., 2016). In this study, the green index threshold 1.12 was applied to separate plant pixels from background pixels. After the whole-plant segmentation was done, the number of pixels from the lowermost to the uppermost of the plant in the vertical direction were counted to represent the plant height in the pixel unit. The stalk plus panicle height from hyperspectral images were estimated using a semantic segmentation method based on the hyperspectral signatures of different sorghum organs (Miao et al., 2020). A linear discriminant analysis model was adopted from the previous sorghum semantic segmentation project to classify each pixel to background, leaf, stalk, or panicle (Miao et al., 2020). After the semantic segmentation, the stalk plus panicle plant height in the pixel unit was estimated by counting the number of stalk and panicle pixels on the vertical direction. Manual proofing of growth curves was used to identify and correct a modest number of cases in early time points where leaf midrib pixels were misclassified as stalk pixels, resulting in incorrect estimation of plant heights. These cases could also be identified as poor fits of nonparametric regression curves to the problematic data point relative to its neighbors. The real plant heights from RGB and hyperspectral images were obtained by using the pixel height multiplied by the ratio of the real size and pixel size based on the corresponding zoom level.
Nonparametric Fitting of Growth Curves and Missing Heights Imputation
The growth curve of each sorghum line was obtained by fitting the heights at different time points using the nonparametric regression. The missing heights were also imputed. Let Yij be the jth observed phenotype of the ith plant, made at day tij, i = 1, 2, ..., n, j = 1, 2, ..., mi, where mi is the total number of days observed for the ith plant. To model the plant growth, the following nonparametric model was employed:
| (1) |
where µ(·) is a mean function of the phenotype development and e(tij) is a zero-mean process associated with ith plant observed at tij. Let B(t) = (B1, ..., BK)T (t) be a vector of B-spline basis functions, where K is the number of basis functions. The estimated mean function can be expressed as 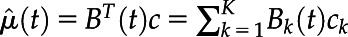 , where c is a vector of coefficients of length K obtained using penalized least-squares approach (Ramsay and Silverman, 2005; Xu et al., 2018b).
, where c is a vector of coefficients of length K obtained using penalized least-squares approach (Ramsay and Silverman, 2005; Xu et al., 2018b).
FPCA
The plant growth curves over time were summarized using functional principle component scores. In FPCA, the process e(tij) in Equation 1 is decomposed into two parts:
| (2) |
where ξi,l are zero-mean principal components scores with variance λl, 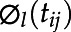 are eigenfunctions corresponding to principal components scores, and εij are zero-mean measurement errors with constant variance. In FPCA, eigenfunctions are orthonormal, namely ∫
are eigenfunctions corresponding to principal components scores, and εij are zero-mean measurement errors with constant variance. In FPCA, eigenfunctions are orthonormal, namely ∫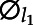 (t)
(t)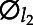 (t)dt = 0, for all
(t)dt = 0, for all 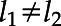 and
and 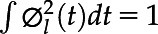 , so the characteristics of phenotype development for the ith genotype can be represented by its principal component scores ξi,l,
, so the characteristics of phenotype development for the ith genotype can be represented by its principal component scores ξi,l,
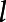 = 1, 2, ..., L. The variance of the principal component scores, λl,
= 1, 2, ..., L. The variance of the principal component scores, λl,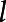 = 1, 2, ..., L, are sorted in decreasing order, so the first few principal component scores usually capture the majority of variation in the phenotype data. B-spline bases were employed for the approximation of eigenfunctions. The variance λl and eigenfunctions
= 1, 2, ..., L, are sorted in decreasing order, so the first few principal component scores usually capture the majority of variation in the phenotype data. B-spline bases were employed for the approximation of eigenfunctions. The variance λl and eigenfunctions 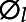 (·) are estimated by eigenvalue decomposition, and the principal components scores ξi,l,
(·) are estimated by eigenvalue decomposition, and the principal components scores ξi,l,
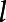 = 1, 2, ..., L are estimated using best linear unbiased prediction (Ramsay and Silverman, 2005; Xu et al., 2018a).
= 1, 2, ..., L are estimated using best linear unbiased prediction (Ramsay and Silverman, 2005; Xu et al., 2018a).
Genotyping the SAP
DNA was extracted from seedling-stage plants grown in the Beadle Center Greenhouse complex at University of Nebraska-Lincoln using the same seed employed for phenotyping. Sequencing libraries generated using a modified tGBS (tuneable-Genotyping-By-Sequencing) protocol (Ott et al., 2017) were constructed and paired-end (151-bp read length) sequenced in eight lanes of an Illumina HiSeqX instrument. This produced an average of 4 million paired-end reads per sample. The quality of raw reads was controlled using the Trimmomatic v0.33 software with the parameters ‘TRAILING:20,’ ‘SLIDINGWINDOW:4:20,’ and ‘MINLEN:40’ (Bolger et al., 2014). High-quality reads were mapped to the sorghum reference genome (V4; McCormick et al., 2018) using the BWA v0.7.17 MEM algorithm with default parameters (Li and Durbin, 2009). After the alignment, the GATK v4.1 HaplotypeCaller function was used to perform SNP calling with default parameters (McKenna et al., 2010) and the raw VCF file including 358 samples was generated. Quality controls of the raw VCF including missing data rate (<0.7), minor allele frequency (>0.01), and heterozygous rate (< 0.05) were applied using customized python scripts (https://github.com/freemao/schnablelab). Missing data were then imputed using Beagle v4.1 with parameters ‘window = 6600, overlap = 1320’ (Browning and Browning, 2016). The sizes of sliding windows and the overlap windows were set to capture 10% of all SNPs on a given chromosome and 2% of all SNPs on a given chromosome, respectively. A final set of 569,305 high-confidence SNPs was generated and employed in all downstream analyses.
GWAS Analyses
Three kinds of GWAS were conducted in this study: (1) sequential GWAS of plant height based on DAP (DAP sequential GWAS); (2) sequential GWAS of plant height based on DAPE (DAPE sequential GWAS); and (3) GWAS of the dynamic trait of the plant growth curve (FPCA GWAS).
For DAP sequential GWAS, the plant heights of 292 lines at each time point were used as the phenotypes. As different subsets of the population were phenotyped on alternating dates, height values for all plants on individual data were drawn from the values for the nonparametrically fit curves for that genotype, as described above. Genotype data for the set of 292 lines imaged at each time point was taken from the complete genotype dataset described above which included all of these 292 lines in addition to some further lines from the SAP which were not phenotyped in this study.. After subsetting to the data from the 292 phenotyped SAP lines, any SNPs that now fell below the previous criteria used to filter SNPs for the full set of 358 lines—minor allele frequency (>0.01) and the heterozygous rate (<0.05)—were excluded. A total of 36 GWAS were conducted from 41 DAP to 75 DAP using the same genotype data. For the DAPE sequential GWAS, x axis (time) values for individual plant growth curves were recentered based on the date of panicle emergence. The panicle emergence was defined when the tip of the panicle is visible from the sorghum flag leaf sheath in the image. In all but 13 cases, panicle emergence occurred within the window in which image data were collected. In one case, panicle emergence occurred prior to the first day of imaging, and in 12 cases, panicle emergence occurred subsequent to the last day of imaging. As a result, the number of lines with observed data was inconsistent across individual time points. Analyses were conducted between −14 and 10 DAPE. Within this interval, observed height values were present for at least 235 lines at each time point. For each time point, genotype information was subset and refiltered based on the set of lines with available trait data, as described above. A total of 25 GWAS were conducted from −14 to 10 DAPE using the corresponding customized marker datasets generated for phenotyped lines at each individual time point. For the FPCA GWAS, the first and second principal component scores of each sorghum line were used as trait values. The genotype data used was identical to that employed for the DAP sequential GWAS analyses.
All the GWASs were conducted using mixed linear models (MLMs) implemented by GEMMA v0.95 (Zhou and Stephens, 2012). The first five principal components derived from the genotype data using Tassel v5.0 (Bradbury et al., 2007) were fitted to MLMs as the fixed effect. Meanwhile, the kinship matrix calculated using the “gemma -gk 1” command was fitted to MLM as the random effect. All 63 GWAS jobs were run on the Holland Computing Center’s Crane cluster at the University of Nebraska-Lincoln. The number of independent SNPs in each genotype data were determined using the GEC v0.2 software (Li et al., 2012). A Bonferroni corrected P-value of 0.05 calculated based on the number of independent SNPs in each specific analysis was applied as the cutoff for statistical significance in each separate GWAS analysis (Yang et al., 2014).
Data and Code Availability
Both imputed and unimputed genotype datasets used in this study have been deposited on Figshare (Miao and Schnable, 2019). Raw image data, including RGB and hyperspectral collected from each plant at each time point are being deposited and disseminated through CyVerse (https://doi.org/10.25739/p39b-dz61). The R code implementing the nonparametric regression and FPCA used in this study has been deposited on GitHub (https://github.com/freemao/FPCA_GWAS).
Accession Numbers
Sequences for the four major genes/proteins mentioned in this paper, Dwarf1 (Sobic.009G229800), Dwarf2 (Sobic.006G067700), maturity1 (Sobic.006G057866), and Dwarf3 (Sobic.007G163800), were retrieved from v3.1.1 of the sorghum genome as provided in Phytozome v12.1.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. The distributions of minor allele frequency of SNPs around dwarf genes in unconstrained (blue) and constrained (yellow) populations.
Supplemental Figure S2. Semantic segmentation of an example sorghum plant using a hyperspectral image.
Supplemental Figure S3. Imputing unobserved height values for sorghum genotypes through nonparametric regression.
Supplemental Figure S4. Proportion of variance explained for observed and imputed plant heights.
Supplemental Figure S5. Summary of significant SNPs identified in independent GWAS for each time point anchoring on days after planting rather than DAPE.
Supplemental Figure S6. Manhattan plots for GWAS on height measured at 41 DAP and 72 DAP.
Supplemental Figure S7. Manhattan plot for GWAS on the date of panicle emergence.
Supplemental Table S1. Significant SNPs identified by DAP Sequential GWAS.
Acknowledgments
J.C.S., P.S.S., and S.L. have equity interests in Data2Bio, a company that provides genotyping services using the same protocol employed for genotyping in this study. The authors affirm that they have no other conflicts of interest.
Footnotes
This work was supported by a University of Nebraska Agricultural Research Division seed grant to J.C.S. and a National Science Foundation award (grant no. OIA–1557417 to J.C.S.). This project was completed utilizing the Holland Computing Center of the University of Nebraska, which receives support from the Nebraska Research Initiative.
Articles can be viewed without a subscription.
References
- Bolger AM, Lohse M, Usadel B(2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES(2007) TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23: 2633–2635 [DOI] [PubMed] [Google Scholar]
- Brown PJ, Klein PE, Bortiri E, Acharya CB, Rooney WL, Kresovich S(2006) Inheritance of inflorescence architecture in sorghum. Theor Appl Genet 113: 931–942 [DOI] [PubMed] [Google Scholar]
- Brown PJ, Rooney WL, Franks C, Kresovich S(2008) Efficient mapping of plant height quantitative trait loci in a sorghum association population with introgressed dwarfing genes. Genetics 180: 629–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR(2016) Genotype imputation with millions of reference samples. Am J Hum Genet 98: 116–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casa AM, Pressoir G, Brown PJ, Mitchell SE, Rooney WL, Tuinstra MR, Franks CD, Kresovich S(2008) Community resources and strategies for association mapping in Sorghum. Crop Sci 48: 30 [Google Scholar]
- Chen W, Wang W, Peng M, Gong L, Gao Y, Wan J, Wang S, Shi L, Zhou B, Li Z, et al. (2016) Comparative and parallel genome-wide association studies for metabolic and agronomic traits in cereals. Nat Commun 7: 12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman MJ, Ellsworth PZ, Fahlgren N, Gehan MA, Cousins AB, Baxter I(2018) Components of water use efficiency have unique genetic signatures in the model c4 grass Setaria. Plant Physiol 178: 699–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman MJ, Paul RE, Banan D, Barrett JF, Sebastian J, Yee MC, Jiang H, Lipka AE, Brutnell TP, Dinneny JR, et al. (2017) Time dependent genetic analysis links field and controlled environment phenotypes in the model C4 grass Setaria. PLoS Genet 13: e1006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez MGS, Bao Y, Tang L, Schnable PS(2017) A high-throughput, field-based phenotyping technology for tall biomass crops. Plant Physiol 174: 2008–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furbank RT, Tester M(2011) Phenomics—technologies to relieve the phenotyping bottleneck. Trends Plant Sci 16: 635–644 [DOI] [PubMed] [Google Scholar]
- Ge Y, Bai G, Stoerger V, Schnable JC(2016) Temporal dynamics of maize plant growth, water use, and leaf water content using automated high throughput RGB and hyperspectral imaging. Comput Electron Agric 127: 625–632 [Google Scholar]
- Gehan MA, Fahlgren N, Abbasi A, Berry JC, Callen ST, Chavez L, Doust AN, Feldman MJ, Gilbert KB, Hodge JG, et al. (2017) PlantCV v2: Image analysis software for high-throughput plant phenotyping. PeerJ 5: e4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansey CN, Johnson JM, Sekhon RS, Kaeppler SM, de Leon N(2011) Genetic diversity of a maize association population with restricted phenology. Crop Sci 51: 704–715 [Google Scholar]
- Higgins RH, Thurber CS, Assaranurak I, Brown PJ(2014) Multiparental mapping of plant height and flowering time QTL in partially isogenic sorghum families. G3 (Bethesda) 4: 1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilley J, Truong S, Olson S, Morishige D, Mullet J(2016) Identification of dw1, a regulator of sorghum stem internode length. PLoS One 11: e0151271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilley JL, Weers BD, Truong SK, McCormick RF, Mattison AJ, McKinley BA, Morishige DT, Mullet JE(2017) Sorghum dw2 encodes a protein kinase regulator of stem internode length. Sci Rep 7: 4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano K, Kawamura M, Araki-Nakamura S, Fujimoto H, Ohmae-Shinohara K, Yamaguchi M, Fujii A, Sasaki H, Kasuga S, Sazuka T(2017) Sorghum DW1 positively regulates brassinosteroid signaling by inhibiting the nuclear localization of BRASSINOSTEROID INSENSITIVE 2. Sci Rep 7: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman F, Riche A, Michalski A, Castle M, Wooster M, Hawkesford M(2016) High throughput field phenotyping of wheat plant height and growth rate in field plot trials using uav based remote sensing. Remote Sens 8: 1031 [Google Scholar]
- Honsdorf N, March TJ, Berger B, Tester M, Pillen K(2014) High-throughput phenotyping to detect drought tolerance QTL in wild barley introgression lines. PLoS One 9: e97047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, et al. (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42: 961–967 [DOI] [PubMed] [Google Scholar]
- Kwak I-Y, Moore CR, Spalding EP, Broman KW(2016) Mapping quantitative trait loci underlying function-valued traits using functional principal component analysis and multi-trait mapping. G3 (Bethesda 6: 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally D, Ingmire P, Tong H-Y, He Z-H(2001) Antisense expression of a cell wall-associated protein kinase, WAK4, inhibits cell elongation and alters morphology. Plant Cell 13: 1317–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R(2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Yeung JM, Cherny SS, Sham PC(2012) Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet 131: 747–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li X, Fridman E, Tesso TT, Yu J(2015) Dissecting repulsion linkage in the dwarfing gene Dw3 region for sorghum plant height provides insights into heterosis. Proc Natl Acad Sci USA 112: 11823–11828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Pandey P, Stoerger V, Xu Y, Qiu Y, Ge Y, Schnable JC(2018) Conventional and hyperspectral time-series imaging of maize lines widely used in field trials. Gigascience 7: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C-X, Casella G, Wu R(2002) Functional mapping of quantitative trait loci underlying the character process: A theoretical framework. Genetics 161: 1751–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick RF, Truong SK, Sreedasyam A, Jenkins J, Shu S, Sims D, Kennedy M, Amirebrahimi M, Weers BD, McKinley B, et al. (2018) The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J 93: 338–354 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao C, Schnable J (2019) Genotype dataset for sorghum association panel (SAP). figshare. Dataset.
- Miao C, Pages A, Xu Z, Rodene E, Yang J, Schnable JC(2020) Semantic segmentation of sorghum using hyperspectral data identifies genetic associations. Plant Phenomics 2020: 4216373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao C, Yang J, Schnable JC(2019) Optimising the identification of causal variants across varying genetic architectures in crops. Plant Biotechnol J 17: 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CR, Johnson LS, Kwak IY, Livny M, Broman KW, Spalding EP(2013) High-throughput computer vision introduces the time axis to a quantitative trait map of a plant growth response. Genetics 195: 1077–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GP, Ramu P, Deshpande SP, Hash CT, Shah T, Upadhyaya HD, Riera-Lizarazu O, Brown PJ, Acharya CB, Mitchell SE, et al. (2013) Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci USA 110: 453–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multani DS, Briggs SP, Chamberlin MA, Blakeslee JJ, Murphy AS, Johal GS(2003) Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302: 81–84 [DOI] [PubMed] [Google Scholar]
- Muraya MM, Chu J, Zhao Y, Junker A, Klukas C, Reif JC, Altmann T(2017) Genetic variation of growth dynamics in maize (Zea mays L.) revealed through automated non-invasive phenotyping. Plant J 89: 366–380 [DOI] [PubMed] [Google Scholar]
- Murphy RL, Klein RR, Morishige DT, Brady JA, Rooney WL, Miller FR, Dugas DV, Klein PE, Mullet JE(2011) Coincident light and clock regulation of pseudoresponse regulator protein 37 (PRR37) controls photoperiodic flowering in sorghum. Proc Natl Acad Sci USA 108: 16469–16474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro JAR, Willcox M, Burgueño J, Romay C, Swarts K, Trachsel S, Preciado E, Terron A, Vallejo Delgado H, et al. (2017) A study of allelic diversity underlying flowering-time adaptation in maize landraces. Nat Genet 49: 476–480 [DOI] [PubMed] [Google Scholar]
- Oren S, Ceylan H, Schnable PS, Dong L(2017) High-resolution patterning and transferring of graphene-based nanomaterials onto tape toward roll-to-roll production of tape-based wearable sensors. Adv Mater Technol 2: 1700223 [Google Scholar]
- Osman KA, Tang B, Wang Y, Chen J, Yu F, Li L, Han X, Zhang Z, Yan J, Zheng Y, et al. (2013) Dynamic QTL analysis and candidate gene mapping for waterlogging tolerance at maize seedling stage. PLoS One 8: e79305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Liu S, Schnable JC, Yeh C-T, Wang C, Schnable PS (2017) Tunable genotyping-by-sequencing (tGBS) enables reliable genotyping of heterozygous loci. bioRxiv 100461, doi: 10.1101/100461 [DOI] [PMC free article] [PubMed]
- Paine CET, Marthews TR, Vogt DR, Purves D, Rees M, Hector A, Turnbull LA(2012) How to fit nonlinear plant growth models and calculate growth rates: An update for ecologists. Methods Ecol Evol 3: 245–256 [Google Scholar]
- Quinby JR.(1967) The maturity genes of sorghum. Adv Agron 19: 267–305 [Google Scholar]
- Quinby JR.(1974) Sorghum Improvement and the Genetics of Growth. Texas Agricultural Experiment Station, College Station, TX [Google Scholar]
- Quinby JR, Karper RE(1945) The inheritance of three genes that influence time of floral initiation and maturity date in milo. J Am Soc Agron 37: 916–936 [Google Scholar]
- Ramsay JO, Silverman BW(2005) Functional Data Analysis, Ed 2 Springer-Verlag, New York [Google Scholar]
- Sang M, Shi H, Wei K, Ye M, Jiang L, Sun L, Wu R(2019) A dissection model for mapping complex traits. Plant J 97: 1168–1182 [DOI] [PubMed] [Google Scholar]
- Wagner TA, Kohorn BD(2001) Wall-associated kinases are expressed throughout plant development and are required for cell expansion. Plant Cell 13: 303–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Lin M(2006) Functional mapping—how to map and study the genetic architecture of dynamic complex traits. Nat Rev Genet 7: 229–237 [DOI] [PubMed] [Google Scholar]
- Wu R, Ma C-X, Lin M, Wang Z, Casella G(2004) Functional mapping of quantitative trait loci underlying growth trajectories using a transform-both-sides logistic model. Biometrics 60: 729–738 [DOI] [PubMed] [Google Scholar]
- Wu X, Wang Z, Chang X, Jing R(2010) Genetic dissection of the developmental behaviours of plant height in wheat under diverse water regimes. J Exp Bot 61: 2923–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würschum T, Liu W, Busemeyer L, Tucker MR, Reif JC, Weissmann EA, Hahn V, Ruckelshausen A, Maurer HP(2014) Mapping dynamic QTL for plant height in triticale. BMC Genet 15: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Li Y, Nettleton D(2018a) Nested hierarchical functional data modeling and inference for the analysis of functional plant phenotypes. J Am Stat Assoc 113: 593–606 [Google Scholar]
- Xu Y, Qiu Y, Schnable JC(2018b) Functional modeling of plant growth dynamics. Plant Phenome J 1: 1–10 [Google Scholar]
- Yamaguchi M, Fujimoto H, Hirano K, Araki-Nakamura S, Ohmae-Shinohara K, Fujii A, Tsunashima M, Song XJ, Ito Y, Nagae R, et al. (2016) Sorghum Dw1, an agronomically important gene for lodging resistance, encodes a novel protein involved in cell proliferation. Sci Rep 6: 28366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Wright SI, McMullen MD(2007) Genomic screening for artificial selection during domestication and improvement in maize. Ann Bot 100: 967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Zhu J, He C, Benmoussa M, Wu P(1998) Molecular dissection of developmental behavior of plant height in rice (Oryza sativa L.). Genetics 150: 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu R, Casella G(2009) Nonparametric functional mapping of quantitative trait loci. Biometrics 65: 30–39 [DOI] [PubMed] [Google Scholar]
- Yang W, Guo Z, Huang C, Duan L, Chen G, Jiang N, Fang W, Feng H, Xie W, Lian X, et al. (2014) Combining high-throughput phenotyping and genome-wide association studies to reveal natural genetic variation in rice. Nat Commun 5: 5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M(2012) Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44: 821–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Both imputed and unimputed genotype datasets used in this study have been deposited on Figshare (Miao and Schnable, 2019). Raw image data, including RGB and hyperspectral collected from each plant at each time point are being deposited and disseminated through CyVerse (https://doi.org/10.25739/p39b-dz61). The R code implementing the nonparametric regression and FPCA used in this study has been deposited on GitHub (https://github.com/freemao/FPCA_GWAS).