

With well-designed protocols and guide RNAs, off-target changes induced by site-directed nucleases are negligible with fewer genetic differences than from standing variation or induced mutagenesis.
Abstract
Site-directed nucleases (SDNs) used for targeted genome editing are powerful new tools to introduce precise genetic changes into plants. Like traditional approaches, such as conventional crossing and induced mutagenesis, genome editing aims to improve crop yield and nutrition. Next-generation sequencing studies demonstrate that across their genomes, populations of crop species typically carry millions of single nucleotide polymorphisms and many copy number and structural variants. Spontaneous mutations occur at rates of ∼10−8 to 10−9 per site per generation, while variation induced by chemical treatment or ionizing radiation results in higher mutation rates. In the context of SDNs, an off-target change or edit is an unintended, nonspecific mutation occurring at a site with sequence similarity to the targeted edit region. SDN-mediated off-target changes can contribute to a small number of additional genetic variants compared to those that occur naturally in breeding populations or are introduced by induced-mutagenesis methods. Recent studies show that using computational algorithms to design genome editing reagents can mitigate off-target edits in plants. Finally, crops are subject to strong selection to eliminate off-type plants through well-established multigenerational breeding, selection, and commercial variety development practices. Within this context, off-target edits in crops present no new safety concerns compared to other breeding practices. The current generation of genome editing technologies is already proving useful to develop new plant varieties with consumer and farmer benefits. Genome editing will likely undergo improved editing specificity along with new developments in SDN delivery and increasing genomic characterization, further improving reagent design and application.
PLANT GENETIC VARIABILITY
Genetic differences between individuals are the basis of adaptation and evolution. Plant breeding, as a form of directed evolution, has a long history of using genetic diversity for crop improvement. During the process of crop domestication, humans selected individual plants with favorable traits that resulted from novel mutations or standing variation in the ancestral species. The process of selecting plant varieties with favorable characteristics for cultivation and consumption continues to the present day. Modern plant breeding is a more directed process than the crop improvement that occurred through the history and prehistory of most cultivated species, but it continues to involve lengthy development cycles. Progress in our understanding of the genetic basis of traits and refined approaches to select superior progeny have contributed to increased yield for most major crops. Globally, crop yields have increased by >1% each year for much of the past century (Xu et al., 2017). Technological innovations, including those that increased our understanding of nucleic acid sequence and function, have contributed to this progress. Advances have primarily been incremental, with few step changes (Bernardo, 2016). The relatively recent introduction of genome editing, which permits targeted genetic changes, is part of a continuum of development of plant breeding techniques (Lusser et al., 2012). Its primary appeal is that in contrast to established mutagenesis techniques, changes can be targeted to desired genes.

Over the past several decades, there has been rapid development of genome editing techniques (also referred to as gene editing or genome engineering; Gaj et al., 2013; Jaganathan et al., 2018; Zhang et al., 2018). These technologies make use of site-directed nucleases (SDNs) and have broad applicability, including applications in many crop plants (Podevin et al., 2013; Voytas, 2013; Weeks et al., 2016). As described in greater detail below, SDNs are enzymes that create DNA double-strand breaks (DSBs) at specific, predetermined nucleotide sequence sites, permitting targeted genetic modifications (Voytas, 2013; Weeks et al., 2016). The most robust and best-characterized SDNs are the Zinc-Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and the type II Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated proteins (e.g. Cas9 or Cas12a) systems, hereafter referred to as “CRISPR” (Gaj et al., 2013; Jaganathan et al., 2018; Zhang et al., 2018). In all cases, the targeted DSBs generated by these precision nucleases are repaired by the cell’s native DNA break repair mechanisms: the nonhomologous end-joining (NHEJ) or the homologous recombination (HR) pathways (Danner et al., 2017).
One area of consideration for genome editing of plants and animals, and in human (Homo sapiens) therapeutic applications, is the potential for “off-target” edits that could lead to unintended mutations at untargeted loci in the genome. An off-target edit is an unintended, nonspecific mutation that occurs at a site with sequence similarity to the targeted edit region. Off-target edits are distinct from untargeted mutations that occur at sites without similarity to the target region and that are usually associated with spontaneous or other types of background variation. The fidelity of SDNs to only the targeted sequence has been questioned, particularly in mammalian genome editing applications (Carroll, 2011, 2013; Pattanayak et al., 2011; Wu et al., 2014; Zhang et al., 2015a; Tsai and Joung, 2016; Kosicki et al., 2018). The potential to introduce DNA sequence changes at nontargeted loci in patients undergoing genome editing procedures has become an essential consideration for clinical and disease treatment applications (Araki and Ishii, 2016). Furthermore, unintended mutations in treated human somatic cells could pose health risks such as unregulated cell proliferation (cancer; Cox et al., 2015). Off-target edits were detected in the early stages of CRISPR-Cas9 research involving human cancer cell lines (Fu et al., 2013). The off-target edit frequency in human cells ranged from 0 to 150 mutations per genome, and this frequency was closely linked to the single-guide RNA (sgRNA) specificity (Frock et al., 2015). The potential for off-target edits and rate at which they could occur in plants are discussed below.
Plants differ from animals in substantive ways that alter the impact of induced changes. First, somatic cell changes in plants differ in consequences from those in mammals. Somatic changes in a portion of the plant are less likely to affect critical (irreplaceable) tissues. Unlike in many animals, genetic changes in juvenile plants can be transmitted to reproductive tissues (Schmid-Siegert et al., 2017). However, plants frequently develop multiple independent reproductive structures, only a portion of which may be affected by any new mutations. Also, breeding to develop new lines for commercial release involves an intensive process of selection of individual plants with useful phenotypes while eliminating individuals with undesirable mutations or phenotypes (commonly known as “off-types”; ASTA, 2016; Glenn et al., 2017; Kaiser et al., 2020). For these reasons, off-target edits in crops present fewer safety concerns than those that could arise with therapeutic applications of genome editing.
The goal of this review is a quantitative comparison of genetic variation that could arise through genome editing with variation that exists in crop species or is generated by current breeding practices. The review examines our current understanding of the nature and abundance of standing and induced genetic variation in crops and how they compare to the potential for off-target changes during genome editing.
SOURCES AND TYPES OF INHERENT (STANDING) GENOMIC VARIATION IN PLANTS
Before DNA was identified as the molecular basis for transmission of heritable genetic variation, crop domestication and plant breeding involved visible inspection and selection of desired phenotypes. There was limited knowledge of the types and extent of underlying genomic variation in crop plants and their wild relatives (Zohary, 1999). The development of genomic sequencing methodologies has provided an unprecedented understanding of the molecular basis of diversity in plant species, ranging from single nucleotide variants to macro-scale events such as genomic rearrangements or whole-genome duplication leading to polyploidy and transposon-directed DNA transposition (Wendel et al., 2016; Gabur et al., 2019).
Our understanding of the primary sources of genetic diversity, namely mutations and recombination, and the cellular processes involved in generating genetic variants have increased as both DNA sequencing technologies and genome assemblies have improved (Bevan et al., 2017). Mutation creates new variants while recombination arranges variants into new combinations (Morrell et al., 2006); both processes rely upon endogenous DNA repair mechanisms (see Box 1 on DSB repair of DNA). Genetic drift and both positive and purifying selection remove variants from populations, so that an equilibrium is maintained that permits variation to persist at relatively constant levels in stable populations. Much of the process of plant breeding involves the creation of varieties that combine favorable variants from their parental lines.
Box 1.

The importance of double-strand break repairs. Pacher and Puchta, 2017; Puchta, 2005; Schuermann et al., 2005; Waterworth et al., 2010; Waterworth et al., 2011; Waterworth et al., 2016; Friedberg et al., 2006; Voytas, 2013; Brunet and Jasin, 2018; Čermák et al., 2017; Knoll et al., 2014; Gaut et al., 2007; Hajjar and Hodgkin, 2007.
Reference Genomes and Assessment of Genomic Variation
Standing variation in a species can be estimated at the whole-genome level by comparing a sample of individuals using next-generation sequencing (NGS). High-throughput DNA sequencing, whole-genome reference sequences and assembly, and improved bioinformatic tools have provided an unprecedented understanding of the variation in populations of the world’s major field and vegetable crop species (Morrell et al., 2011; Huang and Han, 2014; Bevan et al., 2017). NGS and reference-based read mapping have also been used to identify de novo sequence variation in lines accumulating induced and spontaneous mutations over several generations (Ossowski et al., 2010; Belfield et al., 2012; Henry et al., 2014). Many of the field crop and vegetable species that constitute a major part of the global diet now have a high-quality reference genome sequence (Michael and VanBuren, 2015). Reference genomes have several limitations, the most apparent being that all genes or gene variants are not present in any single accession (Hirsch et al., 2016; Jiao et al., 2017). Means of accounting for structural variation and gene content are rapidly evolving, but they generally involve representing multiple genomes of a single species in a graph-like structure (Church et al., 2015; Paten et al., 2017).
A list of the classes of variation detected in cultivated plants is presented in Table 1. Single-nucleotide variations (SNVs; referred to as single-nucleotide polymorphisms [SNPs] when segregating in populations), are the most abundant class (see Innan et al., 1996). Because of their abundance, SNPs are the most commonly genotyped polymorphism for breeding applications and comparative studies in crops (Morrell et al., 2011). Insertion and deletion changes (indels) are also common forms of variation, but because indel events are superimposed over one another, over evolutionary time it becomes increasingly difficult to distinguish the boundaries of individual indel events (Clegg and Morrell, 2004). These naturally occurring changes are similar to those induced by SDNs, with SNVs being equivalent to base edits (see below) and indels resembling the gene knockouts induced by CRISPR edits.
Table 1. Common types of genomic variation in the breeding populations of field crops and vegetables.
| Variant Type | Variation | Description/Comment |
|---|---|---|
| Nucleotide polymorphisms | SNPs | A variant at a single position where one base replaces another base. SNPs arise from errors during DNA replication or repair. Also referred to as SNV or single-base substitution. |
| Multinucleotide polymorphisms (MNP) | Changes at two or more adjacent nucleotides, which are relatively rare. | |
| Structural variants | Indels | A site where DNA is lost (deletion) or gained (insertion), typically describing relatively small (1–20 bp but up to 1,000 bp) changes. Usually classified as sequence differences smaller than PAVs. |
| Simple sequence repeats (SSR) | Simple sequence repeats are repeats of two to six nucleotides that vary in the number of repeat motifs present. Also referred to as short tandem repeats or microsatellites. Can be considered a type of CNV. | |
| CNVs | Sequences with variable copy number between individuals. The term CNV generally applies to genic regions and can be due to duplications, deletions, or insertions. Simple sequence repeats can be considered as very small CNVs. Typically, CNVs refer to larger sequence repeats (>1 kb for example) and may encompass one or more genes or parts of genes. | |
| PAVs | Large sequence block (>1 kb) present in one line/individual but completely missing in another line. Like CNVs, a PAV typically refers to a region containing genes and can be thought of as an extreme example of CNV. | |
| TEs | Mobile genetic elements. The number and location of each element frequently differ from the reference genome. | |
| Large structural variations (SV) and ploidy (whole and partial genome duplication) | Includes DNA rearrangements such as chromosomal inversions, translocations, partial genome duplications, duplications, etc. |
It is important to note that most whole-genome sequencing studies to date have used short-read sequencing technologies (Gilad et al., 2009). As a result, the diversity in breeding populations due to structural variations such as variation in transposable element (TE) location and abundance, presence-absence variation (PAV), and gene copy number variants (CNVs) has been difficult to measure (Muñoz-Diez et al., 2012). PAVs and CNVs typically refer to changes that include genes and thus are functionally equivalent to SDN-mediated gene insertion or deletion. Although structural variants are less common than SNPs, they are an important source of variation (Gabur et al., 2019; Schiessl et al., 2019). There are many examples of gene PAVs or CNVs impacting agronomic traits, including submergence tolerance in rice (Oryza sativa; Xu et al., 2006), high oleic acid content in sunflower (Helianthus annuus) oil (Schuppert et al., 2006), soybean (Glycine max) cyst nematode resistance (Cook et al., 2012), and seed coat pigmentation in soybean (Todd and Vodkin, 1996). Because of the density of naturally occurring variation, intra- and interspecific crosses of plants that occur during plant breeding can introduce millions of SNPs (Fig. 1), in addition to thousands of PAV or CNV sequence variations, into resulting progeny. As more long-read sequencing technologies are deployed, more accurate measurements of CNV and PAV in breeding populations will be possible.
Figure 1.
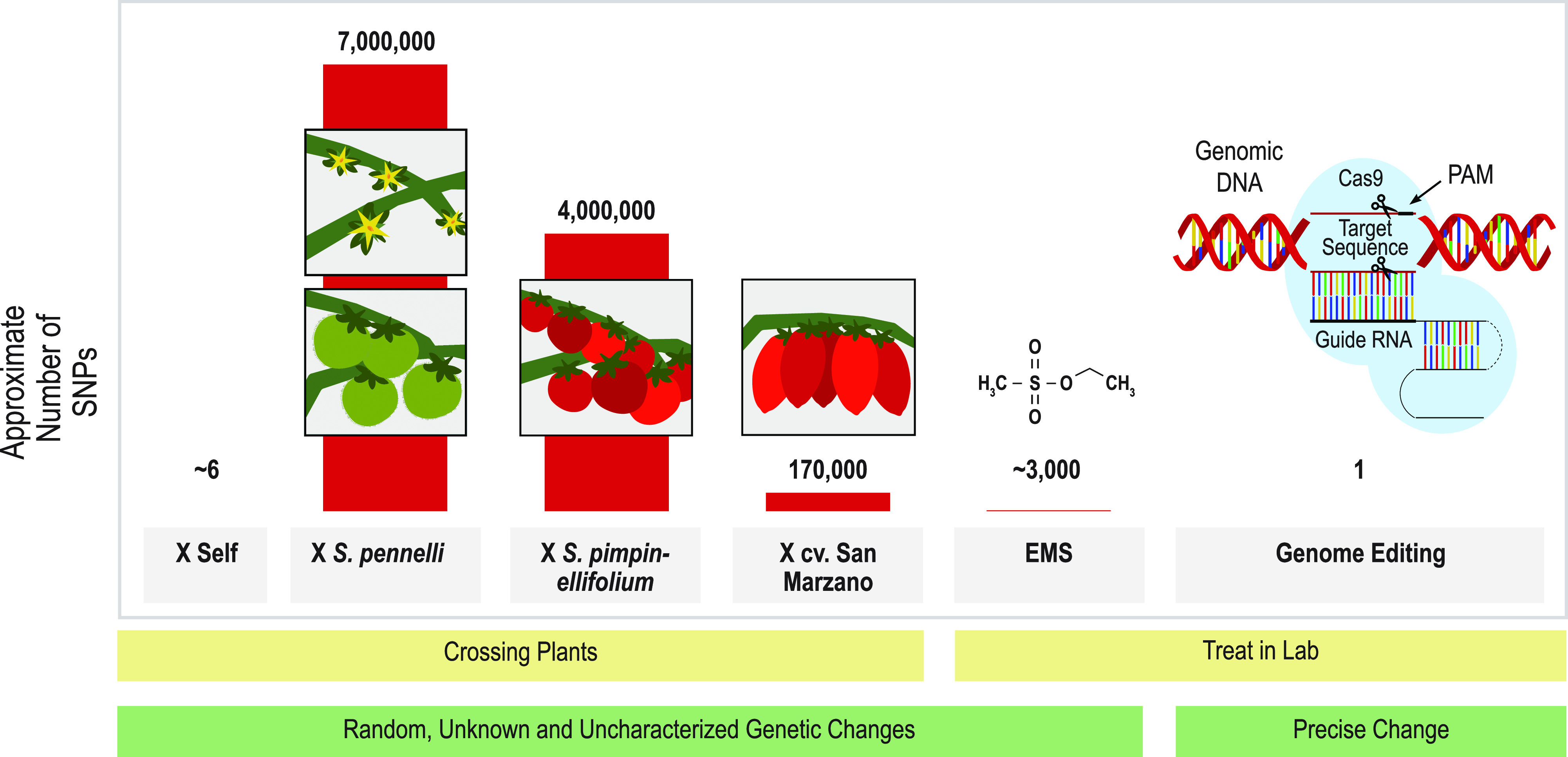
Comparison of the average number of SNPs and indels per genome (individual) introduced into tomato by different breeding strategies. The data represent approximate numbers of SNPs between the genome sequence of S. lycopersicum (Heinz 1706 reference genome) and other cultivars or wild relatives which have been used in breeding of modern tomato cultivars. After selfing (× Self), each individual plant in the next generation will have (on average) approximately six random SNPs (assuming the de novo rate of spontaneous heritable SNP formation in tomato is similar to that of lab-grown Arabidopsis (Ossowski et al., 2010). Most modern elite tomato lines commercially grown typically have four or more disease-resistance genes that have been introgressed by crossing with wild tomato species such as Solanum pennellii or Solanum pimpinellifolium (Foolad, 2007). These initial elite × wild species hybridization events introduced millions to tens of millions of SNPs in addition to indels, CNVs, and PAVs (Aflitos et al., 2014). Crosses with more closely related domestic tomato lines or landraces (i.e. cv San Marzano) will introduce (on average) hundreds of thousands of SNPs/indels (Ercolano et al., 2014). Creating random variation by treating seeds with the chemical mutagen ethyl methanesulphonate (EMS) typically introduces thousands of SNPs per individual (Minoia et al., 2010). Treating cells with a well-designed gene editing reagent (including a gRNA homologous to target sequence and adjacent PAM) can create a single SNP or indel at a precise, predetermined location. Crossing with wild or closely related species can also introduce additional indels, CNVs, and PAVs into the genome, which is not considered in this figure.
Sources of Spontaneous De Novo Genome Variation
Table 2 reports current estimates of standing genomic variation in select crop species. Environmental conditions and breeding and genetic practices such as tissue culture propagation (described below) can further impact the rate at which mutations occur. The sources, types, and rates of de novo genomic variation are reviewed below.
Table 2. Estimated genomic diversity in the breeding populations of select field and vegetable crops.
Not all forms of genomic variation were assessed for all species listed. In addition, because of the short-read sequencing technologies used, the number of PAV/CNV structural variants, representing large deletions, inversions, and duplications, is likely underestimated. ND, Not determined.
| Plant Species | Evaluated Sample Size and Description | Estimated Genome Sizea | Estimated SNPs Present | Average Pairwise Diversityb | Estimated Indels | Structural Variants (CNVs and PAVs) | References |
|---|---|---|---|---|---|---|---|
| Mbp | Millions | Millions | |||||
| Bean (Phaseolus vulgaris) | 37 | 521 | 45.0 | ∼5.0 × 10−3 Mesoamerican | 2.1 | 18.5 k | Schmutz et al., 2014; Lobaton et al., 2018 |
| ∼1.7 × 10−3 Andean | |||||||
| Wild Cabbage (Brassica oleracea) | 10 (1 wild species) | 489 | 4.8 | ND | ND | 11.5 k | Golicz et al., 2016 |
| Chickpea (Cicer arietinum) | 35 | 738 | 2.0 | ND | 0.3 | 9.9 k | Thudi et al., 2016 |
| Cucumber (Cucumis sativus) | 115 | 194 | 3.3 | ND | 0.03 | 0.8 k | Zhang et al., 2015b |
| Qi et al., 2013a | |||||||
| Maize (Z. mays) | 2,815 accessions | 2,400 | 0.7 | ∼6.0 × 10−3 | ND | 1.1 M | Gore et al., 2009; Romay et al., 2013; Wang et al., 2017 |
| 13 lines | 2.91 | 0.4 | |||||
| Rice (O. sativa ssp. Japonica) | 3010 | 375 | 29.0 | ∼1.0 × 10−3 | 2.4 | 94 k | Liu et al., 2017b; Wang et al., 2018 |
| Soybean (Glycine max) | 106 (Landraces, US cultivars and wild) | 978 | 10.4 | 1.8 × 10−3 | 0.7 | 7.9 k | Valliyodan et al., 2016 |
| Tomato (S. lycopersicum) | 69 (incl 12 wild species) | 950 | 17.0 | ∼2 × 10−4 | 3.6 | 1.7 k | Causse et al., 2013; Sauvage et al., 2017 |
| Pepper (Capsicum annuum) | 20 (3 wild species) | 3,500 | 9.8 | ∼2.6 × 10−3 | 0.24 | ND | Aguilar-Meléndez et al., 2009; Qin et al., 2014 |
Estimated genome sizes are from references or the EnsemblPlants database (https://plants.ensemble.org April 2019).
Pairwise diversity (also referred to as nucleotide diversity) is the average number of nucleotide differences per site between any two DNA sequences in all possible pairs in the sample population (Tajima, 1983). It is a measure that allows for diversity/variation to be compared across organisms that vary widely in genome size.
Spontaneous Nucleotide Variation
De novo mutations arise primarily from errors in the DNA replication process. DNA repair processes (NHEJ and HR) can also result in new mutations, including variants that arise due to environmentally triggered DNA damage (Jiang et al., 2014; Nisa et al., 2019). Whole-genome sequencing of mutation accumulation lines of Arabidopsis (Arabidopsis thaliana) estimated a mutation rate of 7 × 10−9 base substitutions per site per sexual generation (Ossowski et al., 2010), a rate confirmed in a more recent report (Weng et al., 2019). This rate estimate in Arabidopsis is consistent with earlier estimates of a mutation rate of 5.9 × 10−9 substitutions per site per year based on phylogenetic divergence in monocots (Gaut et al., 1996). Most of the mutations reported by Ossowski et al. (2010) were SNPs, though 1- to 3-bp indel mutations were also detected at a rate of 4.0 ± 1.6 × 10−10 indels per site per generation. Deletions of >3 bp occurred at a rate of 0.5 × 10−9 per site per generation, and the average larger deletion size was hundreds of base pairs per event (Ossowski et al., 2010). In a similar study in rice, the mutation rate was estimated at 5.4 × 10−8 per site per diploid genome (Tang et al., 2018). In maize (Zea mays), the spontaneous mutation rate was reported to be 2.2 to 3.9 × 10−8 per site per generation (Yang et al., 2017). The general trend for single nucleotide mutation rates in plants is on the order of 10−8 to 10−9 per site per generation, similar to estimates for humans (Nachman and Crowell, 2000; Roach et al., 2010).
Transposons as a Source of Variation
In many plant species, TEs and the remnants of past TE insertion events make up the majority of the genome (Sahebi et al., 2018). For example, TEs make up ∼85% of the maize genome, 76% of the pepper genome, and between 20% and 40% of the rice genome, depending upon the rice cultivar (Dubin et al., 2018; Anderson et al., 2019). Transposons are DNA elements that can autoexcise and reinsert in the genome. They have the potential to disrupt genes or create rearrangements. They can also result in complete copies of genetic elements being placed in other areas of the genome. Transposons can have a profound impact on genome structure and gene function (Lisch, 2013). Some types of transposons are known to be activated by environmental conditions (e.g. temperature) and by breeding practices, (e.g. tissue culture, described below; Vitte et al., 2014; Rey et al., 2016). Accurate measurements of rates of heritable transposition in a single generation in plants have not been determined.
SUMMARY OF INHERENT GENOMIC VARIATION
NGS has dramatically improved the estimation of genome-wide standing variation and made it possible to estimate rates of de novo mutations, especially SNVs. Numerous studies have examined the genomic variation between individuals in breeding populations for well-studied crops like maize (Hufford et al., 2012; Lu et al., 2015; Bukowski et al., 2017), soybean (Li et al., 2014; Maldonado dos Santos et al., 2016; Valliyodan et al., 2016), and rice (Zhao et al., 2018). To date, short-read sequencing technology has been the principal tool used for resequencing plant genomes. However, short-read sequence technologies are of more limited utility for detection of large structural variants (such as PAVs or CNVs; Gilad et al., 2009; Morrell et al., 2011; Fuentes et al., 2019). As long-read sequencing becomes a more standard component of comparative studies and additional reference-quality genomes from individual species are reported, diversity due to large structural changes will become more readily accessible (Gabur et al., 2019; Schiessl et al., 2019).
SOURCES OF INDUCED GENOMIC VARIATION IN PLANTS
Although naturally occurring genetic variants have been the primary source of plant genetic diversity used for crop domestication and breeding, geneticists have developed approaches to augment natural plant genomic diversity and accelerate the development of improved cultivars. Genetic variation induced by chemical mutagens and ionizing radiation has been used in plant breeding since early in the 20th century (Friedberg et al., 2006). Somaclonal variation induced by tissue culture has also been a source of variation used in plant breeding. Mutation breeding is considered cost-effective, ubiquitously applicable, nonhazardous, environmentally friendly, and continues to benefit plant breeders and consumers globally (Ahloowalia et al., 2004). Many types of mutagenic and ionizing agents have been evaluated to maximize genomic variation while minimizing detrimental or lethal effects in plants (Sikora et al., 2011). As described below, each type of mutagenic agent tends to have a predominant type of mutation induced across the genome.
The mutations induced by chemical and ionizing radiation occur throughout the genome, but at an increased rate compared to spontaneous mutations (Belfield et al., 2012; Henry et al., 2014). It can be difficult to select individuals that have mutations for the desired phenotype yet lack mutations that produce undesirable phenotypes (Bolon et al., 2011; Saika et al., 2011). Mutant lines that carry numerous new mutations have been developed for genetic studies and breeding improved varieties in many crops (Tables 3 and 4). Over 3,200 mutant varieties, including cereals (1,584; 49.5%), flowers/ornamentals (700; 21.9%), legumes (480; 15%) and others (435; 13.6%), have been released for commercial use in >210 plant species in over 70 countries (Fig. 2).
Table 3. Examples of induced mutation types and frequency in food crops and plant model species evaluated via WGRS analyses.
FN, Fast neutron; NaN3-MNU, sodium azide-methyl-nitro urea; GR, gamma ray; SV, structural variant
| Plant (Assembled Genome Size)a | Mutagen | No. of Plants Analyzed | Mean Mutations/Plantb (per diploid genome) | Total Mutation Frequencyc (mutations per nucleotide site per experiment) | Reference |
|---|---|---|---|---|---|
| Rice (O. sativa; 375 Mb) | FN | 1,504 | 29 SNPs | [1.6 × 10−7] | Li et al., 2017 |
| 26 indels | |||||
| 3 translocations | |||||
| 3 inversion /duplications | |||||
| 61 total | |||||
| GR | 6 | 2,419 SNPs | [8.86 × 10−6] | Li et al., 2016 | |
| 452 Indels | |||||
| 69 SV | |||||
| 383 CNV | |||||
| 3,323 total | |||||
| Tissue culture | 2 | 114 SNPs | 1.86 × 10−7 | Tang et al., 2018 | |
| 36 indels | |||||
| 150 total | |||||
| Sorghum (Sorghum bicolor; 708 Mb) | EMS | 256 | 7,660 SNPs | [1.1 × 10−5] | Jiao et al., 2016 |
| Soybean (G. max; 978 Mb) | EMS | 12 | 12,796 SNPs | [1.3 × 10−5] | Tsuda et al., 2015 |
| 12,854 SNPs (Md) | |||||
| Tomato (S. lycopersicum; 950 Mb)d | EMS | 4 | 1348 SNPs | [1.4 × 10−6] | Shirasawa et al., 2016 |
| 17 indels | |||||
| 1,365 total | |||||
| GR | 3 | 137 SNPs | [1.9 × 10−7] | ||
| 40 indels | |||||
| 177 total | |||||
| L. japonica (472 Mb)d | EMS | 2 | 2,201 SNPs | [4.7 × 10−6] | Mohd-Yusoff et al., 2015 |
| Arabidopsis (135 Mb) | GR | 6 | 11 SNPs (8.5 Md) | 3.6 × 10−7 | Belfield et al., 2012 |
| 7 indels (6.5 Md) | |||||
| 18 total | |||||
| Tissue culture | 5 | 26 SNP (18 Md) | 4.2 × 10−7–24.2 × 10−7 | Jiang et al., 2011 | |
| 4 indels (5 Md) | |||||
| 30 total |
Assembled genome size is based on information from the EnsemblPlants database (http://plants.ensembl.org) unless otherwise noted.
The mean number of mutations per analyzed plant are either published values or were calculated based on total mutations observed across all plants divided by the number of plants analyzed. All plants listed are diploids and studies confirmed full (>90%) genomic coverage unless otherwise noted (see footnote c). For some studies, median values (Md) for numbers of mutations per plant were also calculated.
Total mutation frequency is based on published values or was calculated (numbers in brackets) by dividing total mutations per plant by the assembled genome size. As noted, total mutation frequency was not limited to a single type of mutation.
Average genome sequence coverage was <90% for tomato (86% coverage; 815 Mb) and L. japonica (67% coverage; 315 Mb); partial genomic coverage values were used to normalize the number of mutations per diploid genome and to calculate total mutation frequency.
Table 4. Examples of induced mutation types and frequency in food crops using TILLING or exome resequencing genomic analyses.
NaN3-MNU, Sodium azide-methyl-nitro urea; TILLING, targeting induced local lesions in genomes.
| Plant (Assembled Genome Size)a | Mutagen | No. of Plants Analyzed | Reported Mutation Density (Calculated Mutation Frequency)b | Number of Genes/Genomic Regions Analyzedc (Total Mb Analyzed) | Reference |
|---|---|---|---|---|---|
| Rice (O. sativa; 375 Mb) | NaN3-MNU | 768 | 1 SNP/265 kb (3.8 × 10−6) | 10 genes (7.9 Mb total) | Till et al., 2007 |
| Maize (Z. mays; 2,400 Mb) | EMS | 1,086 | 1 SNP/774 kb (1.3 × 10−6) | Whole-exome resequencing (139 Mb/plant) | Lu et al., 2018 |
| Wheat (Triticum aestivum; 14,540 Mb) | EMS | 1,535 tetraploid 1,200 hexaploid | 23 SNPs/Mb (4n; 2.3 × 10−5) | Whole exome resequencing (119 Mb/plant [4 n]; 162 Mb/plant [6 n]) | Krasileva et al., 2017 |
| 33 SNPs/Mb (6n; 3.3 × 10−5) | |||||
| Barley (H. vulgare; 8,050 Mb) | NaN3 | 3,148 | 1 SNP/374 kb (2.7 × 10−6) | 4 genes (8.2 Mb total) | Talamè et al., 2008 |
| Cotton (Gossypium hirsutum; 2,100 Mb) | EMS | 8,000 M2 lines (2 varieties) | 1 SNP/153 kb (6.5 × 10−6); 1 SNP/326 kb (3.1 × 10−6) | 8 genes (58.1 Mb/variety) | Aslam et al., 2016 |
| Tomato (S. lycopersicum; 950 Mb) | EMS | 1,352 M3 lines | 1 SNP/322 kb (3.1 × 10−6) | 7 genes (10.9 Mb total) | Minoia et al., 2010 |
Assembled genome size is taken from the EnsemblPlants database (http://plants.ensembl.org).
Mutation density (mutations per kilobase or Megabase) is as reported and was determined by dividing the total genomic region evaluated (kilobase) by the number of nucleotide changes observed. Calculated mutation frequency is the arithmetic calculation of mutations per nucleotide site per experiment.
Numbers of genes or genomic regions analyzed in mutagenized plants are as reported. The total DNA length analyzed across all plants and genes (for TILLING) or per plant (for exome sequencing) is also included.
Figure 2.

Numbers of officially released mutant varieties from the top 20 tomato-breeding countries, showing direct releases of improved varieties (orange bars) and mutants used as breeding material (blue bars). Asterisks indicate European Union countries. Data source: Mutant Variety Database (https://mvd.iaea.org).
Mutant population screening is commonly used in rice breeding, resulting in 828 registered lines in the Mutant Variety Database (Kharkwal et al., 2004; Shu et al., 2012; Wang and Jia, 2014). Most of the wheat varieties used for making Italian pasta were derived in part through mutation breeding (Micke et al., 1990; Mba, 2013). For some crops, mutation breeding is more widely utilized for crop improvement within specific geographic regions. For example, nearly 50% of the improved soybean cultivars grown in Vietnam are derived from induced mutations (Vinh et al., 2009). Mutagenesis has also been used to introduce desirable traits in horticultural crops. For example, researchers have used induced mutations to develop variation for tomato (Solanum lycopersicum) fruit size (Just et al., 2013; Park et al., 2014), starch content in potato (Solanum tuberosum; Muth et al., 2008), and virus resistance in peppers (Capsicum spp.; Ibiza et al., 2010). Several grapefruit (Citrus × paradisi) varieties, including popular red flesh varieties, were developed using induced mutation (Mba, 2013). Although induced mutant plant varieties have enjoyed considerable global commercial success and public acceptance, the molecular and physiological basis for the improved characteristics in these varieties is generally unknown (Shu et al., 2012).
The genomic changes induced by mutagenesis- or tissue culture-derived plants have been evaluated in numerous studies using NGS techniques (Tables 3 and 4). In addition to WGRS methods (Table 3), TILLING studies have been used to estimate induced mutation density by collecting sequencing data on a limited number of genes from a large population of individual mutagenized plants (Table 4). Exome capture and whole-exome resequencing studies also rely on targeted sequencing of a portion of the genome, namely the protein coding regions (exons), of mutagenized plant populations.
Mutations induced via fast neutron irradiation generate a wide variety of mutations that differ in size and copy number, including single nucleotide variants, deletions, insertions, inversions, translocations, and duplications (Bolon et al., 2014; Li et al., 2017; Jo and Kim, 2019). Li et al. (2017) generated and deep-sequenced a fast neutron mutant population (1,504 lines) in the model rice cultivar, Kitaake. The estimated mutation frequency analyzed was 61 mutations/plant (1.6 × 10−7 bp−1, as shown in Table 3) with varied types of mutations observed in 58% of all known rice genes. Induced mutations can differ from spontaneous mutations in subtle ways. Transversions (a purine base substitutes for a pyrimidine base, or vice versa) were found to be more frequent in fast neutron populations (Belfield et al., 2012). Transitions (purine to purine or pyrimidine to pyrimidine substitutions) are concentrated at pyrimidine dinucleotide sites, suggesting covalent linkages as a molecular mechanism for these mutations (Belfield et al., 2012). Other mutagens may generate a narrower range of mutation types (Fig. 2; Tables 3 and 4). For example, EMS primarily creates transitions at guanine sites (Henry et al., 2014). These changes have a slightly elevated probability of being flanked at the 5′ neighboring nucleotide by adenine or guanine and the 3′ side by cytosine (Henry et al., 2014). There is also evidence that some plant genomes can tolerate elevated mutation rates better than others. The hexaploid wheat and oat polyploid genomes, possessing multiple redundant alleles, can tolerate a larger number of accumulated mutations without any notable impact on survival or agronomic performance (Stadler, 1929; Krasileva et al., 2017; Hussain et al., 2018).
Genome Variation Induced by Conventional Tissue Culture Practices
Many modern breeding programs use tissue culture or in vitro culture systems to propagate plants, preserve elite genotypes, or produce pure lines using double haploidy. Similar to chemical and irradiation-induced mutations, in vitro culture methods can also generate de novo genetic variability (referred to as somaclonal variation; Neelakandan and Wang, 2012). Several studies have examined the rate of de novo sequence variation of endogenous genes in plants regenerated through tissue culture. Jiang et al. (2011) regenerated 28 Arabidopsis plants from root explants (living cells transferred to culture medium) from a single Columbia (Col-0) laboratory strain. Five regenerated plants were sequenced and analyzed, and on average, each plant had ∼30 de novo sequence changes (totaling 21 indels of 2 bp or fewer, and 131 SNV substitutions across the five plants; Jiang et al., 2011). The calculated somaclonal mutation frequency of spontaneous mutations in this experiment was between 4 × 10−7 and 24 × 10−7 mutations per nucleotide. Tang et al. (2018) reported a mutation rate of 1.86 × 10−7 in rice plants regenerated through tissue culture. These values are nearly two orders of magnitude higher than the rate Ossowski et al. (2010) estimated from greenhouse-grown plants derived from single seed descent (Shaw et al., 2000).
Somaclonal variation provides another source of genomic variability in plants (Krishna et al., 2016). Nearly 200 commercial varieties from ∼55 plant species (e.g. horticultural crops, cereals, legumes, medicinal, and aromatic plants) were derived through somaclonal variation (reviewed by Neelakandan and Wang, 2012; Krishna et al., 2016). During the in vitro culture process, many regenerated plants develop differences in appearance relative to the parental genotype. Changes induced can include heritable genetic and epigenetic alterations (Bednarek et al., 2007; Machczyńska et al., 2015; Krishna et al., 2016). As an example, in the popular banana (Musa acuminata) cultivar ‘Cavendish’, the occurrence of off-types (different from the parental clone) from in vitro cultured plantlets ranges from 6% to 38% (Sahijram et al., 2003). The genomic variation induced during in vitro regeneration is influenced by media composition, length of incubation and subculture, explant origin, and genotype. Genome-wide DNA polymorphisms or structural variations resulting from tissue culture have been observed in soybean (Anderson et al., 2016), barley (Hordeum vulgare; Bednarek et al., 2007), rice (Miyao et al., 2012; Zhang et al., 2014), potato (Fossi et al., 2019), papaya (Carica papaya; Kaity et al., 2009), banana (Ray et al., 2006), and several other plant species. Bednarek et al. (2007) assessed somaclonal variation in barley and identified 6% gene expression variation (1.7% of this variation was due to nucleotide changes and the balance was due to altered methylation). Recently, Li et al. (2019) performed whole-genome sequencing of cotton plants derived from in vitro culture and CRISPR-Cas9 edited plants. They determined that most of the mutations either were induced by somaclonal variation or were attributable to heterogeneity present in parental plants (Li et al., 2019). Tissue culture and somaclonal variation have been successfully used to generate valuable heritable genomic changes in crops, and they have been widely accepted by breeders for germplasm development and release of new varieties.
In mutation breeding (i.e. irradiative, chemical, or somaclonal), efforts are focused on identifying beneficial and novel phenotypes. However, in addition to the beneficial mutation(s), other “coincident” mutations or rearrangements can be generated elsewhere in the genome (Fig. 3). While most of these mutations will have negligible or no phenotypic effect, it can be necessary to remove undesirable mutations with observable deleterious effects (off-types) through backcrossing and selection. Each generation of backcrossing is expected to reduce the presence of coincident mutations by 50%. However, given their number and genomic distribution, some mutations may persist through this process. This process of backcrossing and selection is one reason why mutation breeding has been successfully used for decades with a recognized history of safe use (Food and Drug Administration, 1992).
Figure 3.
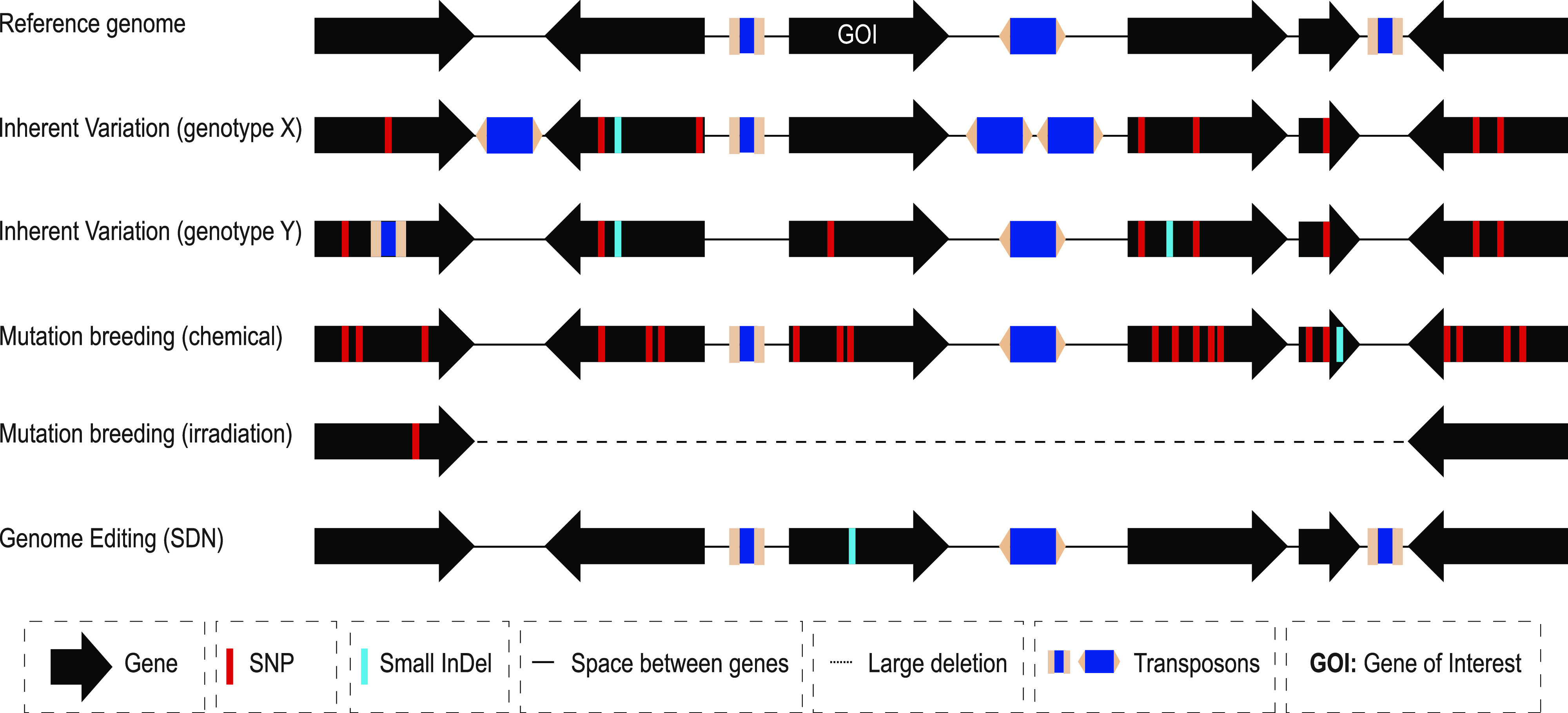
Overview of sources of genomic variation in crop plants. The simplified gene models depict observed variation among individual crop species. Comparison of inherent/standing (genotypes X and Y) and induced variation (by chemicals and irradiation) with the reference genome shows a range of intended and unintended mutations, including SNPs, small indels, transposon insertions/movement, or a large segmental deletion. However, the intended edit induced via a SDN occurs at the target site. Note that the diagram is not drawn to scale and that intergenic variation is not shown.
GENOME EDITING AS A SOURCE OF VARIATION
The advantage of classical mutation breeding is that it allows for the rapid generation of genomic variation that can be used as a source of traits not currently identified in a crop’s germplasm. The drawback of these methods is that variation is induced randomly both at desired locations and across the genome. Because of these factors, screening, isolating, and introgressing desired mutations into elite germplasm requires large populations and lengthy breeding development timelines. Thus, classical mutation breeding as a tool to generate variation is of great value but is relatively inefficient.
Genome editing is a targeted and more exact form of mutation breeding. These tools, based on the use of SDNs, provide researchers with the ability to modify sequences at specific locations within the genome. SDNs generate targeted DSBs that, regardless of the SDN used, are repaired by the same native DNA break repair mechanisms, the NHEJ or HR pathways described previously. As described below and depicted in Figure 1, these methods generally result in many fewer mutations elsewhere in the genome relative to earlier mutagenic techniques. SDNs continue to be improved to further reduce the likelihood of nontargeted mutations (Zhao and Wolt, 2017; Hahn and Nekrasov, 2019).
The Genome Editing Toolbox and Its Utility
Though still a relatively new technology, genome editing using SDNs has demonstrated great promise as a tool for crop improvement. Recent reviews catalog the increasing number of phenotypes and characteristics in crop plants that have been generated using SDNs (Jaganathan et al., 2018; Zhang et al., 2018; Chen et al., 2019). Some of these traits were derived from loss-of-function mutations within coding regions or gene regulatory regions that resulted from NHEJ-mediated repair leading to mutations (indels) at the break site. Other traits were derived from specific nucleotide insertion or substitution, and by gene/allele replacement edits, which can be generated by repairing SDN-generated DSBs with a DNA donor template (Townsend et al., 2009; Li et al., 2015; Petolino et al., 2016; Filler Hayut et al., 2017; Shi et al., 2017; Yu et al., 2017). Several types of SDNs that have been used in plants rely on either protein/DNA or RNA/DNA binding to provide editing specificity. As shown in Figure 4, A and B, ZFNs and TALENs are chimeric proteins consisting of DNA recognition/binding domains fused to the catalytic domain of the FokI nuclease (Christian et al., 2010; Carroll, 2011; Weeks et al., 2016). TALENs are derived from transcription activator-like (TAL) effectors found in the plant pathogen Xanthomonas. TAL effectors bind to specific DNA sequences using tandem amino acid repeats. Variations in these repeats, known as the repeat variable di-residues, allow for specificity to nucleotide sequences. For both ZFNs and TALENs, specificity for a chosen target sequence is improved by expanding the length of DNA targets (e.g. 18 to 32 bp for the paired nuclease) and the spacer (i.e. the distance between paired nucleases). The designed proteins interact as heteromeric pairs, with DSBs occurring only when both protein halves are bound at the same genomic location, further increasing the specificity of these reagents for creating DSBs only at the desired site.
Figure 4.

Most widely used SDNs. A, TALENs are composed of a DNA-binding domain and the nuclease Fok1; the DNA binding domain contains an array of nearly identical protein subdomains, each varying at two specific amino acids known as the repeat variable di-residues; specific repeat variable di-residues recognize unique bases on the target DNA molecule. B, ZFNs are also composed of DNA-binding domains and the Fok1 nuclease; each ZFN is composed of three zinc-finger domains that are custom designed to recognize a triplet of DNA bases on the target sequence. For TALENs and ZFNs, two subunits are needed per target region, each binding closely spaced DNA sequences. C, The Cas9 nuclease binds to the target sequence via an RNA molecule, known as the sgRNA; the 5′ region of the sgRNA, the proto-spacer, is typically 20 nucleotides long and is complementary to the target DNA; a PAM sequence (bold and underlined) is also required for recognition.
RNA-guided endonucleases (RGENs) function as a protein complex that is directed to a genomic target site through a short noncoding guide RNA (gRNA). The nuclease protein and the gRNA are transcribed separately and then form a complex in the cell nucleus. A portion of the gRNA contains a sequence that is directly complementary to a genomic target site that guides the induction of DSB formation at that location. In the case of the CRISPR system, Cas9 or another Cas or Cpf nuclease recognizes the gRNA-DNA pair, thus targeting this site for a DSB (Fig. 4C; Jinek et al., 2012; Cong et al., 2013; Weeks et al., 2016). The gRNA of the most widely used Cas9, Streptococcus pyogenes Cas9 (SpCas9), binds to a 20-bp genomic site, termed the protospacer, that is adjacent to a triplet DNA sequence known as the protospacer adjacent motif (PAM). Unlike complex protein engineering needed for DNA binding using ZFNs and TALENs, Cas9 can be programmed to new target sites by changing the spacer sequence of the gRNA, which simplifies vector design and construction while substantially reducing the cost. Due to this ease of design and greater efficiency, CRISPR has become the SDN of choice for genome editing (Globus and Qimron, 2018).
Recently developed CRISPR editing systems do not rely on DSB formation to induce targeted changes, but instead involve chimeric protein units consisting of a disabled nuclease and an additional protein domain. In these systems, the disabled nuclease is used to target the additional protein units to specific genomic locations for different applications. Applications include targeted base editing with deaminase domains (Komor et al., 2016; Gaudelli et al., 2017; Adli, 2018), transcriptional knockdown using repressors (Qi et al., 2013b; Thakore et al., 2015), targeted DNA methylation and histone modification (Kearns et al., 2015; Stepper et al., 2017), gene activation using strong transcriptional activators (Cheng et al., 2013), activation of developmentally silent endogenous loci through chromatin looping (Deng et al., 2014; Morgan et al., 2017), DNA transposition (Strecker et al., 2019), site-specific recombination (Standage-Beier et al., 2019), and prime editing (Anzalone et al., 2019). The targeting components of the nucleases are still intact, allowing for nucleotide changes in a site-directed manner. For example, cytosine and adenine base editors (converts C to T and A to G, respectively) fuse a nickase-type Cas9 with a deaminase domain and thus do not induce DSBs. Both are useful genome-editing tools, but the cytosine base editor has been reported to induce off-target editing in both animal cells (Zuo et al., 2019) and plants (Jin et al., 2019), as discussed in more detail below).
Designing Genome Editing Protocols and Methods to Optimize Editing Specificity and Reduce the Potential for Off-Target Edits
Optimizing the specificity of genome editing methods can be achieved in multiple ways, often through altering SDN delivery into plants. At present, the most common practice involves stably integrating a transgene into the plant genome that expresses the SDN “reagents” (protein or protein/gRNA complex that is encoded by the transgene) and template DNA molecule for HR edits. The transgene(s) encoding the SDN components are typically integrated at a locus that is unlinked to the gene(s) being edited. A subsequent outcross or selfing generation results in the independent assortment of unlinked genes to segregate the SDN-encoding transgene away from the edited target sequence. Resulting progeny plants can be screened to select nontransgenic plants carrying the targeted edit and lacking the SDN-encoding insert (Gao et al., 2016; Char et al., 2017). The resulting plant variety is often molecularly indistinguishable from varieties with mutations that either occur naturally or are produced by induced mutagenic techniques. It should be noted that chromosomal rearrangements due to transfer DNA insertions have been observed (Clark and Krysan, 2010; Hu et al., 2017), but these can generally be detected by next-generation sequencing-based methods and removed by segregation and selection. Transient delivery methods, which seek to limit the overall exposure of the genome to the SDN and avoid the stable integration of a transgene, have also been developed in plants (Woo et al., 2015; Kelliher et al., 2019). The most common transient delivery method uses a ribonucleoprotein complex that consists of a purified RGEN protein bound to a gRNA (Kim et al., 2017; Andersson et al., 2018). The complex is delivered directly to plant cells, through bombardment or other means (Ran et al., 2017), so that the active protein can cause a DSB, but the SDN-encoding DNA is not integrated into the genome (Chen et al., 2018).
Off-target edits, and the means to reduce such edits, have received a great deal of attention in the human therapeutic and medical fields (Cho et al., 2014; Shen et al., 2014; Lee et al., 2016, 2018; Akcakaya et al., 2018; Moon et al., 2019) and are also of interest in the plant science community (Feng et al., 2014; Wolt et al., 2016; Tang et al., 2018; Jin et al., 2019; Li et al., 2019; Young et al., 2019). As described in the section “Plant Genetic Variability”, changes observed following genome editing protocols can be split into those that occur at sites without similarity to the targeted region, referred to as untargeted mutations, and “off-target edits” that occur at sites with sequences similar to the targeted region. Numerous studies discussed below have found that the potential for and level of off-target activity in plants can be predicted by comparing the sequence of the target site to the rest of the genome. SDNs bind to specific nucleotide sequences, with the most likely location of potential off-target editing to occur at sites with nucleotide-sequence level similarity to the intended target (Jiang and Doudna, 2017). Bioinformatics prediction tools have been designed to predict sites that may result in an off-target edit. Programs have been developed for all of the major SDNs (PROGNOS [Fine et al., 2014]; TALE-NT [Doyle et al., 2012]; CAS-OFF Finder [Bae et al., 2014]) and are routinely included in the design of SDN-based editing protocols (Liu et al., 2017a; Listgarten et al., 2018; Minkenberg et al., 2019). Prediction algorithms are useful for identifying potential off-target sites in well-sequenced genomes. Plant species with limited reference genome sequence data may be poorly assessed by these tools. For plant species without a comprehensive genome sequence, it may be difficult to predict potential off-target SDN activity. However, the amount of variation introduced would likely be less than traditional mutagenesis techniques classically utilized in plant breeding strategies (Fig. 1). As described in more detail below, it can also be difficult to assess and discriminate between bona fide off-target SDN activity and variation attributable to transformation or standing variation.
Off-Target Edit Evaluation and Frequency
The approaches reported for the identification of off-target edits vary widely and can impact the level of off-target activity observed. As noted above, many studies rely on algorithms to predict the optimal on-target activity and potential off-target edit sites based on similarity with the gRNA sequence. The most common method used to assess whether the off-target activity has occurred at these site(s) is PCR amplification, followed by amplicon sequencing. PCR approaches provide a rapid method for determining if SDN-mediated off-target edits have occurred at a predicted or suspected site. However, it is reliant on accurate predictions of potential off-target sites, which in turn relies on a well-sequenced source or reference genome for thorough analysis. Several alternative, “non-biased” methods to assess and detect DSB and edits have been developed for off-target edit analysis (for review, see Zischewski et al., 2017), although few have been evaluated in plants.
Recently, high-throughput whole-genome resequencing (WGRS) has been used to evaluate off-target edits and untargeted mutations in plants. While WGRS is the most comprehensive strategy currently available, it requires that researchers carefully control for differences between the reference genome and the experimental line, heterogeneity in experimental lines, and sequencing errors (Michno and Stupar, 2018). WGRS cannot always distinguish between standing variants and de novo untargeted mutations generated by tissue culture or SDN treatments or attributable to sequencing miscalls. Several recent studies have used WGRS to examine genome-wide mutational profiles in genome-edited rice (Tang et al., 2018), maize (Lee et al., 2019), and cotton (Li et al., 2019). A large-scale study examined the amount of variation that occurs due to various genome-editing techniques (i.e. RGENs, CRISPR-Cas9, and crispr-Cpf1; Tang et al., 2018). The study reported WGRS of 69 individual plants that had been regenerated via tissue culture (with and without Agrobacterium treatment), edited with various SDN reagents, or not treated (field grown). WGRS analysis revealed the presence of variation (e.g. SNPs or indels) throughout the genome in all plants, though more so in plants that had gone through the tissue culture process. Relative to plants that went through tissue culture, there was no increase in variation observed in plants exposed to SDN treatment. The exception was in plants edited with a gRNA purposefully designed to be more likely to elicit off-target edits (Tang et al., 2018). The detected off-target edits in these plants were predicted by the program Cas-OFFinder (Bae et al., 2014), confirming that intentionally inadequate guide design led to the observed off-target activity. A recent study in maize determined that regardless of the SDN delivery method (Agrobacterium, particle bombardment, or ribonucleoprotein), proper design of gRNAs leads to undetectable levels of off-target edits (Young et al., 2019). Studies examining rice (Tang et al., 2018), cotton (Li et al., 2019), and maize (Lee et al., 2019; Young et al., 2019) report that nearly all of the observed variation from RGEN experiments is attributable to tissue culture or natural variation, and not to the delivered nucleases. Tang et al. (2018) found that plants regenerated via tissue culture contained ∼100 to 150 SNVs and ∼40 to 80 indels. The number of de novo variants attributable to tissue culture and SDN delivery is small relative to standing variations that differentiate individual lines (Table 2).
A recent study has applied the WGRS approach to evaluate the off-target edit frequency of base editors in rice (Jin et al., 2019). For potential off-target edits, the authors found that SNVs were not significantly different between control plants (exposed to tissue culture with no editing machinery) and adenine base edited plants. The rate of off-target edits was significantly higher only in plants generated with cytosine (C to T) base editors, irrespective of whether a gRNA was supplied. The percentage of changes that were C to T transitions was higher in plants containing cytosine base editing constructs, suggesting that these reagents were inducing background variation. A larger number of off-target mutations suggests that the source of off-target variation was not the SDN but the enzymatic domain of the cytosine base editing complex. Previous studies had found that the cytosine deaminase domain, and the additional uracil glycosylase inhibitor domain used in cytosine base editors, generate higher background levels of C to T changes (Radany et al., 2000; Harris et al., 2002). The studies suggest that increased optimization of the cytosine base editor would be necessary to minimize off-target activity. However, it is worth noting that when compared to the frequency of mutagenesis reported in rice for somaclonal variation (Miyao et al., 2012; Tang et al., 2018) or other forms of induced mutagenesis (see Tables 3 and 4), mutation rates with the cytosine base editor are comparable to methods used in conventional and mutation breeding. This study suggests that as new protein motifs and editing strategies are developed, the intended activity and potential for off-target activity should be evaluated.
WGRS studies (Tang et al., 2018; Jin et al., 2019; Li et al., 2019) show that the amount of induced background variation as a result of introducing editing enzymes into plants is generally low, especially for editing applications with SDNs. The identification of high background mutation rates for C-to-T base editor enzymes (Jin et al., 2019) suggests the need for careful screening of the effects of new approaches. Taken together, these results suggest that WGRS is best applied to new approaches and may not be necessary for routine application of SDNs, as unintended activity will generally be predictable by computational algorithms that allow for the optimal design of gRNAs (Tang et al., 2018; Lee et al., 2019; Young et al., 2019). Indeed, conventional mutagenesis methods have been found to be safe using existing agronomic evaluations, generally in the absence of WGRS evaluation.
CONCLUSIONS
Whole-genome examinations of variability in plants and improvements in understanding of gene function are occurring concurrently with advances in genome editing technology. Targeted genome editing utilizing SDN techniques is the most recent option for the introduction of precise genetic variation for crop improvement (Fig. 5). SDNs are designed to introduce mutations with a high degree of sequence specificity, resulting in fewer unintended genomic changes compared to earlier mutagenesis techniques. As the understanding of nuclease activity has increased, SDN protocols and associated prediction algorithms to more specifically target intended sequences have improved and been aided by the increasing number of well-annotated and sequenced genomes. NGS has facilitated a more thorough understanding of plant genomes and the genetic changes that accompany the development of new plant varieties. These methods allow more accurate estimations of de novo genomic variation. Each plant generation has new genetic variants that are the result of a range of naturally occurring processes. The potential for, and possible effects of, SDN-mediated off-target changes must be put in the context of naturally occurring standing variation in crops and mutations induced or introduced during plant breeding practices (Table 5).
Figure 5.
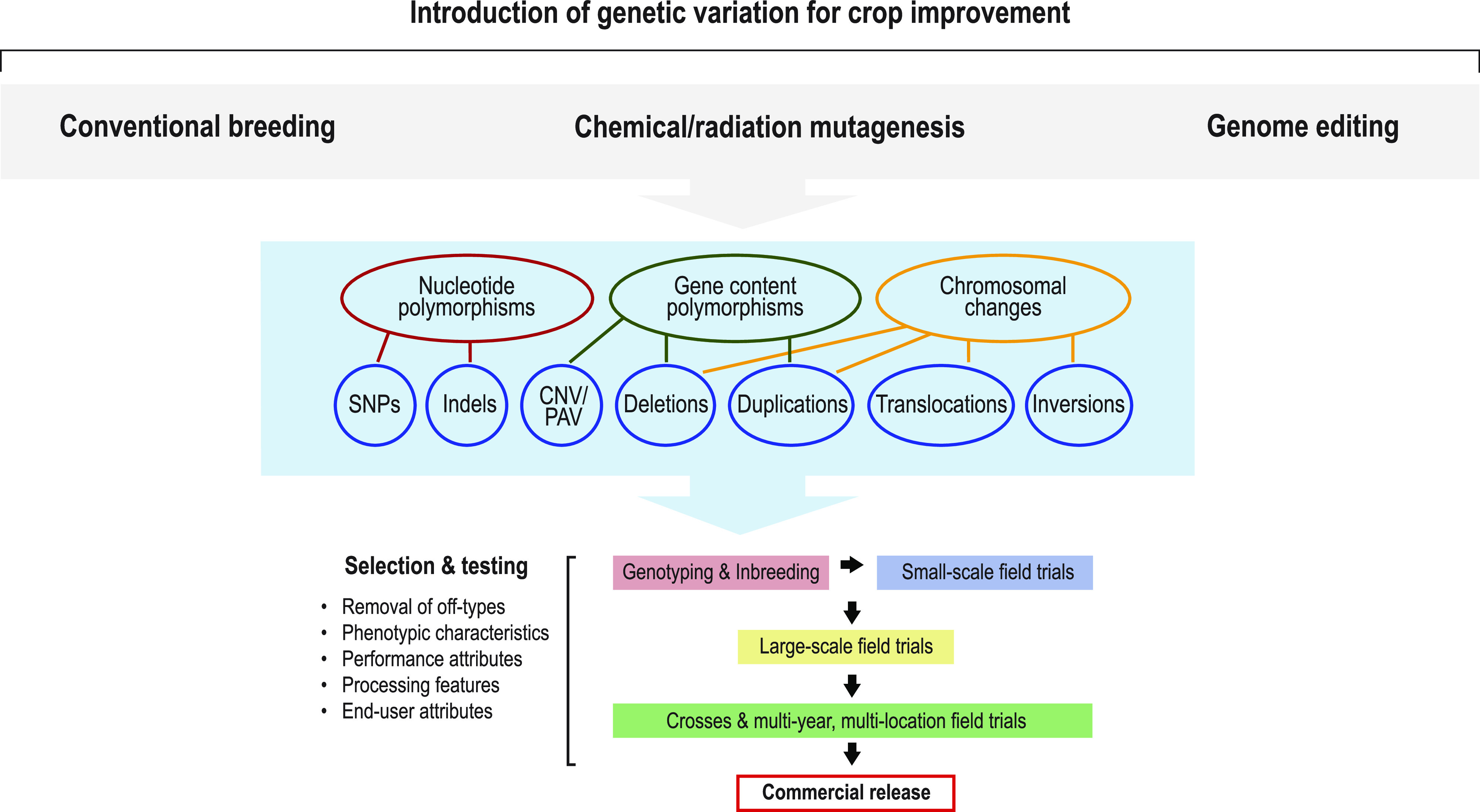
An overview of conventional and modern mutation breeding approaches for crop improvement. The figure illustrates the systematic approach for introducing intended and unintended genomic variation in crops for trait discovery, germplasm development, and commercial release of new crop varieties. After the introduction of genomic variation, the mutant populations go through a series of selection and testing to remove undesirable mutations and phenotypic variations. The best-performing line is selected for subsequent commercial release. This breeding process is common to all methods used for crop product development (Kaiser et al., 2020).
Table 5. Examples of genetic variation introduced by different breeding methods, as reported in recent studies in rice.
| Variation Type | Breeding Method | ||||
|---|---|---|---|---|---|
| Remixing Standing Variants | de Novo Variants | ||||
| Cross Pollinationa | Self Pollinationb | Classical Mutagenesis (Fast Neutron)c | Tissue Cultureb | Genome Editingb | |
| Number of SNVs introduced | 2 × 106 | 23 | 43 | 114 | 0 |
| Number of indels introduced | >5 × 103 | 18 | 48 | 36 | 1 |
| Total number of mutations introduced | >2 × 106 | 41 | 91 | 150 | 1 |
Data are taken from Wang et al., 2018.
Data are taken from Tang et al., 2018.
Data are taken from Li et al., 2017.
The history of safe consumption of foods from plants has been built on the fact that many mutations, regardless of origin, have no phenotypic effect so that breeders cannot remove these “neutral” genetic changes from plant populations. By comparison, plants displaying off-type phenotypes due to unintended mutations are eliminated or ameliorated through well-established, multigenerational breeding, selection, and commercial variety development practices (Fig. 5). Varieties developed through genome editing will be subjected to the same screening and selection practices used for improved varieties developed using other sources of genetic variation (Fig. 5). While off-target edits are deemed to be unacceptable for therapeutic applications of genome editing, off-target edits in the context of crops are smaller in magnitude than the variation generated by current crop improvement methods, such as conventional or mutation breeding (see Outstanding Questions). The current generation of genome editing technologies has facilitated the efficient generation of desirable genomic variation and new plant varieties that would have been more challenging to achieve through other breeding or molecular approaches. Genome editing technology continues to be refined, with improved editing specificity, developments in the delivery of editing enzymes, and ever-increasing genomic characterization.

Acknowledgments
The authors acknowledge Wayne A. Parrott for comments on an earlier version of the manuscript and Emily E. Vonderharr for copyediting of the text. We also thank Allison Devitre and Emily Turnbough for their help with figure art.
Footnotes
This work was supported by the U.S. Department of Agriculture/National Institute of Food and Agriculture (grant nos. 2015–33522–24096 and 2019–33522–30200 to R.M.S. and P.L.M.).
Articles can be viewed without a subscription.
References
- Adli M.(2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9: 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aflitos S, Schijlen E, de Jong H, de Ridder D, Smit S, Finkers R, Wang J, Zhang G, Li N, Mao L, et al. ; 100 Tomato Genome Sequencing Consortium (2014) Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J 80: 136–148 [DOI] [PubMed] [Google Scholar]
- Aguilar-Meléndez A, Morrell PL, Roose ML, Kim S-C(2009) Genetic diversity and structure in semiwild and domesticated chiles (Capsicum annuum; Solanaceae) from Mexico. Am J Bot 96: 1190–1202 [DOI] [PubMed] [Google Scholar]
- Ahloowalia B, Maluszynski M, Nichterlein K(2004) Global impact of mutation-derived varieties. Euphytica 135: 187–204 [Google Scholar]
- Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, Fellows MD, Porritt MJ, Firth MA, Carreras A, et al. (2018) In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561: 416–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JE, Michno J-M, Kono TJ, Stec AO, Campbell BW, Curtin SJ, Stupar RM(2016) Genomic variation and DNA repair associated with soybean transgenesis: A comparison to cultivars and mutagenized plants. BMC Biotechnol 16: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SN, Stitzer MC, Brohammer AB, Zhou P, Noshay JM, O’Connor CH, Hirsch CD, Ross-Ibarra J, Hirsch CN, Springer NM(2019) Transposable elements contribute to dynamic genome content in maize. Plant J 100: 1052–1065 [DOI] [PubMed] [Google Scholar]
- Andersson M, Turesson H, Olsson N, Fält A-S, Ohlsson P, Gonzalez MN, Samuelsson M, Hofvander P(2018) Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol Plant 164: 378–384 [DOI] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576: 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki M, Ishii T(2016) Providing appropriate risk information on genome editing for patients. Trends Biotechnol 34: 86–90 [DOI] [PubMed] [Google Scholar]
- Aslam U, Cheema HMN, Ahmad S, Khan IA, Malik W, Khan AA(2016) COTIP: Cotton TILLING Platform, a resource for plant improvement and reverse genetic studies. Front Plant Sci 7: 1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASTA (2016) Common Practices of Plant Breeders. American Seed Trade Association, Alexandria, VA [Google Scholar]
- Bae S, Park J, Kim JS(2014) Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30: 1473–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarek PT, Orłowska R, Koebner RM, Zimny J(2007) Quantification of the tissue-culture induced variation in barley (Hordeum vulgare L.). BMC Plant Biol 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfield EJ, Gan X, Mithani A, Brown C, Jiang C, Franklin K, Alvey E, Wibowo A, Jung M, Bailey K, et al. (2012) Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana. Genome Res 22: 1306–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo R.(2016) Bandwagons I, too, have known. Theor Appl Genet 129: 2323–2332 [DOI] [PubMed] [Google Scholar]
- Bevan MW, Uauy C, Wulff BBH, Zhou J, Krasileva K, Clark MD(2017) Genomic innovation for crop improvement. Nature 543: 346–354 [DOI] [PubMed] [Google Scholar]
- Bolon Y-T, Haun WJ, Xu WW, Grant D, Stacey MG, Nelson RT, Gerhardt DJ, Jeddeloh JA, Stacey G, Muehlbauer GJ, et al. (2011) Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiol 156: 240–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon Y-T, Stec AO, Michno J-M, Roessler J, Bhaskar PB, Ries L, Dobbels AA, Campbell BW, Young NP, Anderson JE, et al. (2014) Genome resilience and prevalence of segmental duplications following fast neutron irradiation of soybean. Genetics 198: 967–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet E, Jasin M (2018) Induction of chromosomal translocations with CRISPR-Cas9 and other nucleases: Understanding the repair mechanisms that give rise to translocations. In Zhang Y, ed, Chromosome Translocation, Advances in Experimental Medicine and Biology, vol 1544. Springer Nature, Singapore, pp 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowski R, Guo X, Lu Y, Zou C, He B, Rong Z, Wang B, Xu D, Yang B, Xie C, et al. (2017) Construction of the third-generation Zea mays haplotype map. Gigascience 7: gix134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D.(2011) Genome engineering with zinc-finger nucleases. Genetics 188: 773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D.(2013) Staying on target with CRISPR-Cas. Nat Biotechnol 31: 807–809 [DOI] [PubMed] [Google Scholar]
- Causse M, Desplat N, Pascual L, Le Paslier M-C, Sauvage C, Bauchet G, Bérard A, Bounon R, Tchoumakov M, Brunel D, et al. (2013) Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genomics 14: 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čermák T, Curtin SJ, Gil-Humanes J, Čegan R, Kono TJY, Konečná E, Belanto JJ, Starker CG, Mathre JW, Greenstein RL, et al. (2017) A multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 29: 1196–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Char SN, Neelakandan AK, Nahampun H, Frame B, Main M, Spalding MH, Becraft PW, Meyers BC, Walbot V, Wang K, et al. (2017) An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol J 15: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wang Y, Zhang R, Zhang H, Gao C(2019) CRISPR/Cas genome editing and precision plant breeding in agriculture. Annu Rev Plant Biol 70: 667–697 [DOI] [PubMed] [Google Scholar]
- Chen L, Li W, Katin-Grazzini L, Ding J, Gu X, Li Y, Gu T, Wang R, Lin X, Deng Z, et al. (2018) A method for the production and expedient screening of CRISPR/Cas9-mediated non-transgenic mutant plants. Hortic Res 5: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R(2013) Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23: 1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim J-S(2014) Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF(2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186: 757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church DM, Schneider VA, Steinberg KM, Schatz MC, Quinlan AR, Chin C-S, Kitts PA, Aken B, Marth GT, Hoffman MM, et al. (2015) Extending reference assembly models. Genome Biol 16: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KA, Krysan PJ(2010) Chromosomal translocations are a common phenomenon in Arabidopsis thaliana T-DNA insertion lines. Plant J 64: 990–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg MT, Morrell P(2004) Mutational processes In Goodman RM, ed, Encyclopedia of Plant and Crop Science. Marcel Dekker, New York, pp 760–762 [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Lee TG, Guo X, Melito S, Wang K, Bayless AM, Wang J, Hughes TJ, Willis DK, Clemente TE, et al. (2012) Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science 338: 1206–1209 [DOI] [PubMed] [Google Scholar]
- Cox DBT, Platt RJ, Zhang F(2015) Therapeutic genome editing: Prospects and challenges. Nat Med 21: 121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danner E, Bashir S, Yumlu S, Wurst W, Wefers B, Kühn R(2017) Control of gene editing by manipulation of DNA repair mechanisms. Mamm Genome 28: 262–274 [DOI] [PubMed] [Google Scholar]
- Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, et al. (2014) Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 158: 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ(2012) TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res 40: W117–W122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin MJ, Mittelsten Scheid O, Becker C(2018) Transposons: A blessing curse. Curr Opin Plant Biol 42: 23–29 [DOI] [PubMed] [Google Scholar]
- Ercolano MR, Sacco A, Ferriello F, D’Alessandro R, Tononi P, Traini A, Barone A, Zago E, Chiusano ML, Buson G, et al. (2014) Patchwork sequencing of tomato San Marzano and Vesuviano varieties highlights genome-wide variations. BMC Genomics 15: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Mao Y, Xu N, Zhang B, Wei P, Yang DL, Wang Z, Zhang Z, Zheng R, Yang L, et al. (2014) Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc Natl Acad Sci USA 111: 4632–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filler Hayut S, Melamed Bessudo C, Levy AA(2017) Targeted recombination between homologous chromosomes for precise breeding in tomato. Nat Commun 8: 15605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine EJ, Cradick TJ, Zhao CL, Lin Y, Bao G(2014) An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res 42: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Drug Administration (1992) Food for human consumption and animal drugs, feeds, and related products: Foods derived from new plant varieties; policy statement, 22984 In FDA Federal Register, Vol Vol 57 Department of Health and Human Services, Washington, DC, p 22984 [Google Scholar]
- Foolad MR.(2007) Genome mapping and molecular breeding of tomato. Int J Plant Genomics 2007: 64358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossi M, Amundson K, Kuppu S, Britt A, Comai L(2019) Regeneration of Solanum tuberosum plants from protoplasts induces widespread genome instability. Plant Physiol 180: 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA. T. E (2006) DNA Repair and Mutagenesis, Ed 2 ASM Press, Washington, DC [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW(2015) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33: 179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD(2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes RR, Chebotarov D, Duitama J, Smith S, De la Hoz JF, Mohiyuddin M, Wing RA, McNally KL, Tatarinova T, Grigoriev A, et al. (2019) Structural variants in 3000 rice genomes. Genome Res 29: 870–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabur I, Chawla HS, Snowdon RJ, Parkin IAP(2019) Connecting genome structural variation with complex traits in crop plants. Theor Appl Genet 132: 733–750 [DOI] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF III(2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Chen J, Dai X, Zhang D, Zhao Y(2016) An effective strategy for reliably isolating heritable and Cas9-free Arabidopsis mutants generated by CRISPR/Cas9-mediated genome editing. Plant Physiol 171: 1794–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR(2017) Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature 551: 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, Morton BR, McCaig BC, Clegg MT(1996) Substitution rate comparisons between grasses and palms: Synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc Natl Acad Sci USA 93: 10274–10279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaut BS, Wright SI, Rizzon C, Dvorak J, Anderson LK(2007) Recombination: An underappreciated factor in the evolution of plant genomes. Nat Rev Genet 8: 77–84 [DOI] [PubMed] [Google Scholar]
- Gilad Y, Pritchard JK, Thornton K(2009) Characterizing natural variation using next-generation sequencing technologies. Trends Genet 25: 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn KC, Alsop B, Bell E, Goley M, Jenkinson J, Liu B, Martin C, Parrott W, Souder C, Sparks O, et al. (2017) Bringing new plant varieties to market: Plant breeding and selection practices advance beneficial characteristics while minimizing unintended changes. Crop Sci 57: 2906–2921 [Google Scholar]
- Globus R, Qimron U(2018) A technological and regulatory outlook on CRISPR crop editing. J Cell Biochem 119: 1291–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz AA, Bayer PE, Barker GC, Edger PP, Kim H, Martinez PA, Chan CKK, Severn-Ellis A, McCombie WR, Parkin IAP, et al. (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nat Commun 7: 13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore MA, Chia J-M, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross-Ibarra J, et al. (2009) A first-generation haplotype map of maize. Science 326: 1115–1117 [DOI] [PubMed] [Google Scholar]
- Hahn F, Nekrasov V(2019) CRISPR/Cas precision: Do we need to worry about off-targeting in plants? Plant Cell Rep 38: 437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar R, Hodgkin T(2007) The use of wild relatives in crop improvement: A survey of developments over the last 20 years. Euphytica 156: 1–13 [Google Scholar]
- Harris RS, Petersen-Mahrt SK, Neuberger MS(2002) RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell 10: 1247–1253 [DOI] [PubMed] [Google Scholar]
- Henry IM, Nagalakshmi U, Lieberman MC, Ngo KJ, Krasileva KV, Vasquez-Gross H, Akhunova A, Akhunov E, Dubcovsky J, Tai TH, et al. (2014) Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 26: 1382–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch CN, Hirsch CD, Brohammer AB, Bowman MJ, Soifer I, Barad O, Shem-Tov D, Baruch K, Lu F, Hernandez AG, et al. (2016) Draft assembly of elite inbred line PH207 provides insights into genomic and transcriptome diversity in maize. Plant Cell 28: 2700–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chen Z, Zhuang C, Huang J(2017) Cascade of chromosomal rearrangements caused by a heterogeneous T-DNA integration supports the double-stranded break repair model for T-DNA integration. Plant J 90: 954–965 [DOI] [PubMed] [Google Scholar]
- Huang X, Han B(2014) Natural variations and genome-wide association studies in crop plants. Annu Rev Plant Biol 65: 531–551 [DOI] [PubMed] [Google Scholar]
- Hufford MB, Xu X, van Heerwaarden J, Pyhäjärvi T, Chia J-M, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM, et al. (2012) Comparative population genomics of maize domestication and improvement. Nat Genet 44: 808–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Iqbal MA, Till BJ, Rahman MU(2018) Identification of induced mutations in hexaploid wheat genome using exome capture assay. PLoS One 13: e0201918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibiza VP, Cañizares J, Nuez F(2010) EcoTILLING in Capsicum species: Searching for new virus resistances. BMC Genomics 11: 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan H, Tajima F, Terauchi R, Miyashita NT(1996) Intragenic recombination in the Adh locus of the wild plant Arabidopsis thaliana. Genetics 143: 1761–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan D, Ramasamy K, Sellamuthu G, Jayabalan S, Venkataraman G(2018) CRISPR for crop improvement: An update review. Front Plant Sci 9: 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Mithani A, Belfield EJ, Mott R, Hurst LD, Harberd NP(2014) Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res 24: 1821–1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Mithani A, Gan X, Belfield EJ, Klingler JP, Zhu J-K, Ragoussis J, Mott R, Harberd NP(2011) Regenerant Arabidopsis lineages display a distinct genome-wide spectrum of mutations conferring variant phenotypes. Curr Biol 21: 1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Doudna JA(2017) CRISPR-Cas9 structures and mechanisms. Annu Rev Biophys 46: 505–529 [DOI] [PubMed] [Google Scholar]
- Jiao Y, Burke J, Chopra R, Burow G, Chen J, Wang B, Hayes C, Emendack Y, Ware D, Xin Z(2016) A sorghum mutant resource as an efficient platform for gene discovery in grasses. Plant Cell 28: 1551–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang B, Campbell MS, Stein JC, Wei X, Chin C-S, et al. (2017) Improved maize reference genome with single-molecule technologies. Nature 546: 524–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Zong Y, Gao Q, Zhu Z, Wang Y, Qin P, Liang C, Wang D, Qiu J-L, Zhang F, et al. (2019) Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 364: 292–295 [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E(2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YD, Kim J-B(2019) Frequency and spectrum of radiation-induced mutations revealed by whole-genome sequencing analyses of plants. Quantum Beam Sci 3: 7 [Google Scholar]
- Just D, Garcia V, Fernandez L, Bres C, Mauxion J-P, Petit J, Jorly J, Assali J, Bournonville C, Ferrand C(2013) Micro-Tom mutants for functional analysis of target genes and discovery of new alleles in tomato. Plant Biotechnol 30: 225–231 [Google Scholar]
- Kaiser N, Douches D, Dhingra A, Glenn KC, Herzig PR, Stowe EC, Swarup S(2020) The role of conventional plant breeding in ensuring safe levels of naturally occurring toxins in food crops. Trends Food Sci Technol 100: 51–66 [Google Scholar]
- Kaity A, Ashmore SE, Drew RA(2009) Field performance evaluation and genetic integrity assessment of cryopreserved papaya clones. Plant Cell Rep 28: 1421–1430 [DOI] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R(2015) Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12: 401–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher T, Starr D, Su X, Tang G, Chen Z, Carter J, Wittich PE, Dong S, Green J, Burch E, et al. (2019) One-step genome editing of elite crop germplasm during haploid induction. Nat Biotechnol 37: 287–292 [DOI] [PubMed] [Google Scholar]
- Kharkwal M, Pandey R, Pawar S(2004) Mutation breeding for crop improvement In Jain HK, and Kharkwal MC, eds, Plant Breeding: Mendelian to Molecular Approaches. Springer, Dordrecht, pp 601–645 [Google Scholar]
- Kim H, Kim ST, Ryu J, Kang BC, Kim JS, Kim SG(2017) CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun 8: 14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll A, Fauser F, Puchta H(2014) DNA recombination in somatic plant cells: Mechanisms and evolutionary consequences. Chromosome Res 22: 191–201 [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR(2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533: 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M, Tomberg K, Bradley A(2018) Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36: 765–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasileva KV, Vasquez-Gross HA, Howell T, Bailey P, Paraiso F, Clissold L, Simmonds J, Ramirez-Gonzalez RH, Wang X, Borrill P, et al. (2017) Uncovering hidden variation in polyploid wheat. Proc Natl Acad Sci USA 114: E913–E921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna H, Alizadeh M, Singh D, Singh U, Chauhan N, Eftekhari M, Sadh RK(2016) Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 6: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Cradick TJ, Fine EJ, Bao G(2016) Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol Ther 24: 475–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim YH, Lee K, Jung I, Kim D, Kim S, et al. (2018) Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun 9: 3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Zhang Y, Kleinstiver BP, Guo JA, Aryee MJ, Miller J, Malzahn A, Zarecor S, Lawrence-Dill CJ, Joung JK, et al. (2019) Activities and specificities of CRISPR/Cas9 and Cas12a nucleases for targeted mutagenesis in maize. Plant Biotechnol J 17: 362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Jain R, Chern M, Pham NT, Martin JA, Wei T, Schackwitz WS, Lipzen AM, Duong PQ, Jones KC, et al. (2017) The sequences of 1504 mutants in the model rice variety Kitaake facilitate rapid functional genomic studies. Plant Cell 29: 1218–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Manghwar H, Sun L, Wang P, Wang G, Sheng H, Zhang J, Liu H, Qin L, Rui H, et al. (2019) Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol J 17: 858–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zheng YC, Cui HR, Fu HW, Shu QY, Huang JZ(2016) Frequency and type of inheritable mutations induced by γ rays in rice as revealed by whole genome sequencing. J Zhejiang Univ Sci B 17: 905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, Guo Y, Zhang J, Sui Y, Zheng L, et al. (2014) De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32: 1045–1052 [DOI] [PubMed] [Google Scholar]
- Li Z, Liu ZB, Xing A, Moon BP, Koellhoffer JP, Huang L, Ward RT, Clifton E, Falco SC, Cigan AM(2015) Cas9-guide RNA directed genome editing in soybean. Plant Physiol 169: 960–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D.(2013) How important are transposons for plant evolution? Nat Rev Genet 14: 49–61 [DOI] [PubMed] [Google Scholar]
- Listgarten J, Weinstein M, Kleinstiver BP, Sousa AA, Joung JK, Crawford J, Gao K, Hoang L, Elibol M, Doench JG, et al. (2018) Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng 2: 38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Ding Y, Zhou Y, Jin W, Xie K, Chen L-L(2017a) CRISPR-P 2.0: An improved CRISPR-Cas9 tool for genome editing in plants. Mol Plant 10: 530–532 [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhou Y, Morrell PL, Gaut BS(2017b) Deleterious variants in Asian rice and the potential cost of domestication. Mol Biol Evol 34: 908–924 [DOI] [PubMed] [Google Scholar]
- Lobaton JD, Miller T, Gil J, Ariza D, de la Hoz JF, Soler A, Beebe S, Duitama J, Gepts P, Raatz B(2018) Resequencing of common bean identifies regions of inter-gene pool introgression and provides comprehensive resources for molecular breeding. Plant Genome 11: 30025030. [DOI] [PubMed] [Google Scholar]
- Lu F, Romay MC, Glaubitz JC, Bradbury PJ, Elshire RJ, Wang T, Li Y, Li Y, Semagn K, Zhang X, et al. (2015) High-resolution genetic mapping of maize pan-genome sequence anchors. Nat Commun 6: 6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Liu J, Ren W, Yang Q, Chai Z, Chen R, Wang L, Zhao J, Lang Z, Wang H, et al. (2018) Gene-indexed mutations in maize. Mol Plant 11: 496–504 [DOI] [PubMed] [Google Scholar]
- Lusser M, Parisi C, Plan D, Rodríguez-Cerezo E(2012) Deployment of new biotechnologies in plant breeding. Nat Biotechnol 30: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machczyńska J, Zimny J, Bednarek PT(2015) Tissue culture-induced genetic and epigenetic variation in triticale (× Triticosecale spp. Wittmack ex A. Camus 1927) regenerants. Plant Mol Biol 89: 279–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado dos Santos JV, Valliyodan B, Joshi T, Khan SM, Liu Y, Wang J, Vuong TD, de Oliveira MF, Marcelino-Guimarães FC, Xu D, et al. (2016) Evaluation of genetic variation among Brazilian soybean cultivars through genome resequencing. BMC Genomics 17: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mba C.(2013) Induced mutations unleash the potentials of plant genetic resources for food and agriculture. Agronomy (Basel) 3: 200–231 [Google Scholar]
- Michael TP, VanBuren R(2015) Progress, challenges and the future of crop genomes. Curr Opin Plant Biol 24: 71–81 [DOI] [PubMed] [Google Scholar]
- Michno JM, Stupar RM(2018) The importance of genotype identity, genetic heterogeneity, and bioinformatic handling for properly assessing genomic variation in transgenic plants. BMC Biotechnol 18: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micke A, Donini B, Maluszynski M(1990) Induced mutations for crop improvement. Mutation Breeding Rev 7: INIS-MF–13554 [Google Scholar]
- Minkenberg B, Zhang J, Xie K, Yang Y(2019) CRISPR-PLANT v2: An online resource for highly specific guide RNA spacers based on improved off-target analysis. Plant Biotechnol J 17: 5–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoia S, Petrozza A, D’Onofrio O, Piron F, Mosca G, Sozio G, Cellini F, Bendahmane A, Carriero F(2010) A new mutant genetic resource for tomato crop improvement by TILLING technology. BMC Res Notes 3: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyao A, Nakagome M, Ohnuma T, Yamagata H, Kanamori H, Katayose Y, Takahashi A, Matsumoto T, Hirochika H(2012) Molecular spectrum of somaclonal variation in regenerated rice revealed by whole-genome sequencing. Plant Cell Physiol 53: 256–264 [DOI] [PubMed] [Google Scholar]
- Mohd-Yusoff NF, Ruperao P, Tomoyoshi NE, Edwards D, Gresshoff PM, Biswas B, Batley J(2015) Scanning the effects of ethyl methanesulfonate on the whole genome of Lotus japonicus using second-generation sequencing analysis. G3 (Bethesda) 5: 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SB, Kim DY, Ko J-H, Kim J-S, Kim Y-S(2019) Improving CRISPR genome editing by engineering guide RNAs. Trends Biotechnol 37: 870–881 [DOI] [PubMed] [Google Scholar]
- Morgan SL, Mariano NC, Bermudez A, Arruda NL, Wu F, Luo Y, Shankar G, Jia L, Chen H, Hu J-F, et al. (2017) Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun 8: 15993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell PL, Buckler ES, Ross-Ibarra J(2011) Crop genomics: Advances and applications. Nat Rev Genet 13: 85–96 [DOI] [PubMed] [Google Scholar]
- Morrell PL, Toleno DM, Lundy KE, Clegg MT(2006) Estimating the contribution of mutation, recombination and gene conversion in the generation of haplotypic diversity. Genetics 173: 1705–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Diez C, Vitte C, Ross-Ibarra J, Gaut BS, Tenaillon MI(2012) Using nextgen sequencing to investigate genome size variation and transposable element content In Grandbastien M-A, and Casacuberta JM, eds, Plant Transposable Elements: Impact on Genome Structure and Function. Springer, Berlin, Heidelberg, pp 41–58 [Google Scholar]
- Muth J, Hartje S, Twyman RM, Hofferbert HR, Tacke E, Prüfer D(2008) Precision breeding for novel starch variants in potato. Plant Biotechnol J 6: 576–584 [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL(2000) Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelakandan AK, Wang K(2012) Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep 31: 597–620 [DOI] [PubMed] [Google Scholar]
- Nisa M-U, Huang Y, Benhamed M, Raynaud C(2019) The plant DNA damage response: Signaling pathways leading to growth inhibition and putative role in response to stress conditions. Front Plant Sci 10: 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M(2010) The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher M, Puchta H(2017) From classical mutagenesis to nuclease-based breeding—directing natural DNA repair for a natural end-product. Plant J 90: 819–833 [DOI] [PubMed] [Google Scholar]
- Park SJ, Jiang K, Tal L, Yichie Y, Gar O, Zamir D, Eshed Y, Lippman ZB(2014) Optimization of crop productivity in tomato using induced mutations in the florigen pathway. Nat Genet 46: 1337–1342 [DOI] [PubMed] [Google Scholar]
- Paten B, Novak AM, Eizenga JM, Garrison E(2017) Genome graphs and the evolution of genome inference. Genome Res 27: 665–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Ramirez CL, Joung JK, Liu DR(2011) Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8: 765–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petolino JF, Srivastava V, Daniell H(2016) Editing plant genomes: A new era of crop improvement. Plant Biotechnol J 14: 435–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podevin N, Davies HV, Hartung F, Nogué F, Casacuberta JM(2013) Site-directed nucleases: A paradigm shift in predictable, knowledge-based plant breeding. Trends Biotechnol 31: 375–383 [DOI] [PubMed] [Google Scholar]
- Puchta H.(2005) The repair of double-strand breaks in plants: Mechanisms and consequences for genome evolution. J Exp Bot 56: 1–14 [DOI] [PubMed] [Google Scholar]
- Qi J, Liu X, Shen D, Miao H, Xie B, Li X, Zeng P, Wang S, Shang Y, Gu X, et al. (2013a) A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat Genet 45: 1510–1515 [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA(2013b) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152: 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C, Yu C, Shen Y, Fang X, Chen L, Min J, Cheng J, Zhao S, Xu M, Luo Y, et al. (2014) Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci USA 111: 5135–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radany EH, Dornfeld KJ, Sanderson RJ, Savage MK, Majumdar A, Seidman MM, Mosbaugh DW(2000) Increased spontaneous mutation frequency in human cells expressing the phage PBS2-encoded inhibitor of uracil-DNA glycosylase. Mutat Res 461: 41–58 [DOI] [PubMed] [Google Scholar]
- Ran Y, Liang Z, Gao C(2017) Current and future editing reagent delivery systems for plant genome editing. Sci China Life Sci 60: 490–505 [DOI] [PubMed] [Google Scholar]
- Ray T, Dutta I, Saha P, Das S, Roy S(2006) Genetic stability of three economically important micropropagated banana (Musa spp.) cultivars of lower Indo-Gangetic plains, as assessed by RAPD and ISSR markers. Plant Cell Tissue Organ Cult 85: 11–21 [Google Scholar]
- Rey O, Danchin E, Mirouze M, Loot C, Blanchet S(2016) Adaptation to global change: A transposable element-epigenetics perspective. Trends Ecol Evol 31: 514–526 [DOI] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. (2010) Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 328: 636–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romay MC, Millard MJ, Glaubitz JC, Peiffer JA, Swarts KL, Casstevens TM, Elshire RJ, Acharya CB, Mitchell SE, Flint-Garcia SA, et al. (2013) Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol 14: R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahebi M, Hanafi MM, van Wijnen AJ, Rice D, Rafii MY, Azizi P, Osman M, Taheri S, Bakar MFA, Isa MNM, et al. (2018) Contribution of transposable elements in the plant’s genome. Gene 665: 155–166 [DOI] [PubMed] [Google Scholar]
- Sahijram L, Soneji JR, Bollamma K(2003) Analyzing somaclonal variation in micropropagated bananas (Musa spp.). In Vitro Cell Dev Biol Plant 39: 551–556 [Google Scholar]
- Saika H, Oikawa A, Matsuda F, Onodera H, Saito K, Toki S(2011) Application of gene targeting to designed mutation breeding of high-tryptophan rice. Plant Physiol 156: 1269–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage C, Rau A, Aichholz C, Chadoeuf J, Sarah G, Ruiz M, Santoni S, Causse M, David J, Glémin S(2017) Domestication rewired gene expression and nucleotide diversity patterns in tomato. Plant J 91: 631–645 [DOI] [PubMed] [Google Scholar]
- Schiessl S-V, Katche E, Ihien E, Chawla HS, Mason AS(2019) The role of genomic structural variation in the genetic improvement of polyploid crops. Crop J 7: 127–140 [Google Scholar]
- Schmid-Siegert E, Sarkar N, Iseli C, Calderon S, Gouhier-Darimont C, Chrast J, Cattaneo P, Schütz F, Farinelli L, Pagni M, et al. (2017) Low number of fixed somatic mutations in a long-lived oak tree. Nat Plants 3: 926–929 [DOI] [PubMed] [Google Scholar]
- Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, Jenkins J, Shu S, Song Q, Chavarro C, et al. (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46: 707–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuermann D, Molinier J, Fritsch O, Hohn B(2005) The dual nature of homologous recombination in plants. Trends Genet 21: 172–181 [DOI] [PubMed] [Google Scholar]
- Schuppert GF, Tang S, Slabaugh MB, Knapp SJ(2006) The sunflower high-oleic mutant Ol carries variable tandem repeats of FAD2-1, a seed-specific oleoyl-phosphatidyl choline desaturase. Mol Breed 17: 241–256 [Google Scholar]
- Shaw RG, Byers DL, Darmo E(2000) Spontaneous mutational effects on reproductive traits of Arabidopsis thaliana. Genetics 155: 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, et al. (2014) Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11: 399–402 [DOI] [PubMed] [Google Scholar]
- Shi J, Gao H, Wang H, Lafitte HR, Archibald RL, Yang M, Hakimi SM, Mo H, Habben JE(2017) ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol J 15: 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasawa K, Hirakawa H, Nunome T, Tabata S, Isobe S(2016) Genome-wide survey of artificial mutations induced by ethyl methanesulfonate and gamma rays in tomato. Plant Biotechnol J 14: 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Q-Y, Forster BP, and Nakagawa H, eds; (2012) Plant Mutation Breeding and Biotechnology. CABI, Wallingford, United Kingdom [Google Scholar]
- Sikora P, Chawade A, Larsson M, Olsson J, Olsson O(2011) Mutagenesis as a tool in plant genetics, functional genomics, and breeding. Int J Plant Genomics 2011: 314829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler LJ.(1929) Chromosome number and the mutation rate in Avena and Triticum. Proc Natl Acad Sci USA 15: 876–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standage-Beier K, Brookhouser N, Balachandran P, Zhang Q, Brafman DA, Wang X(2019) RNA-guided recombinase-Cas9 fusion targets genomic DNA deletion and integration. CRISPR J 2: 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, Jurkowski TP(2017) Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res 45: 1703–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, Zhang F(2019) RNA-guided DNA insertion with CRISPR-associated transposases. Science 365: 48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F.(1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105: 437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talamè V, Bovina R, Sanguineti MC, Tuberosa R, Lundqvist U, Salvi S(2008) TILLMore, a resource for the discovery of chemically induced mutants in barley. Plant Biotechnol J 6: 477–485 [DOI] [PubMed] [Google Scholar]
- Tang X, Liu G, Zhou J, Ren Q, You Q, Tian L, Xin X, Zhong Z, Liu B, Zheng X, et al. (2018) A large-scale whole-genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 (Cas12a) nucleases in rice. Genome Biol 19: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, Gersbach CA(2015) Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12: 1143–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thudi M, Khan AW, Kumar V, Gaur PM, Katta K, Garg V, Roorkiwal M, Samineni S, Varshney RK(2016) Whole genome re-sequencing reveals genome-wide variations among parental lines of 16 mapping populations in chickpea (Cicer arietinum L.). BMC Plant Biol 16(Suppl 1): 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BJ, Cooper J, Tai TH, Colowit P, Greene EA, Henikoff S, Comai L(2007) Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol 7: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd JJ, Vodkin LO(1996) Duplications that suppress and deletions that restore expression from a chalcone synthase multigene family. Plant Cell 8: 687–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF(2009) High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 459: 442–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Joung JK(2016) Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet 17: 300–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Kaga A, Anai T, Shimizu T, Sayama T, Takagi K, Machita K, Watanabe S, Nishimura M, Yamada N, et al. (2015) Construction of a high-density mutant library in soybean and development of a mutant retrieval method using amplicon sequencing. BMC Genomics 16: 1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valliyodan B, Dan Qiu, Patil G, Zeng P, Huang J, Dai L, Chen C, Li Y, Joshi T, Song L, et al. (2016) Landscape of genomic diversity and trait discovery in soybean. Sci Rep 6: 23598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh M, Thinh D, Bang D, At D, Ham L, Shu Q(2009) Current status and research directions of induced mutation application to seed crops improvement in Vietnam In Shu Q-Y, ed, Induced Plant Mutations in the Genomics Era. Food and Agriculture Organization of the United Nations, Rome, pp 341–345 [Google Scholar]
- Vitte C, Fustier M-A, Alix K, Tenaillon MI(2014) The bright side of transposons in crop evolution. Brief Funct Genomics 13: 276–295 [DOI] [PubMed] [Google Scholar]
- Voytas DF.(2013) Plant genome engineering with sequence-specific nucleases. Annu Rev Plant Biol 64: 327–350 [DOI] [PubMed] [Google Scholar]
- Wang L, Beissinger TM, Lorant A, Ross-Ibarra C, Ross-Ibarra J, Hufford MB(2017) The interplay of demography and selection during maize domestication and expansion. Genome Biol 18: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, Li M, Zheng T, Fuentes RR, Zhang F, et al. (2018) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z-h, Jia Y(2014) Development and characterization of rice mutants for functional genomic studies and breeding In Tomlekova NB, Kozgar MI, and Wani MR, eds, Mutagenesis: Exploring Novel Genes and Pathways. Wageningen Academic Publishers, Wageningen, The Netherlands, pp 604–612 [Google Scholar]
- Waterworth WM, Drury GE, Bray CM, West CE(2011) Repairing breaks in the plant genome: The importance of keeping it together. New Phytol 192: 805–822 [DOI] [PubMed] [Google Scholar]
- Waterworth WM, Footitt S, Bray CM, Finch-Savage WE, West CE(2016) DNA damage checkpoint kinase ATM regulates germination and maintains genome stability in seeds. Proc Natl Acad Sci USA 113: 9647–9652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterworth WM, Masnavi G, Bhardwaj RM, Jiang Q, Bray CM, West CE(2010) A plant DNA ligase is an important determinant of seed longevity. Plant J 63: 848–860 [DOI] [PubMed] [Google Scholar]
- Weeks DP, Spalding MH, Yang B(2016) Use of designer nucleases for targeted gene and genome editing in plants. Plant Biotechnol J 14: 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF, Jackson SA, Meyers BC, Wing RA(2016) Evolution of plant genome architecture. Genome Biol 17: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M-L, Becker C, Hildebrandt J, Neumann M, Rutter MT, Shaw RG, Weigel D, Fenster CB(2019) Fine-grained analysis of spontaneous mutation spectrum and frequency in Arabidopsis thaliana. Genetics 211: 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolt JD, Wang K, Sashital D, Lawrence-Dill CJ(2016) Achieving plant CRISPR targeting that limits off-target effects. Plant Genome 9: 2790281. [DOI] [PubMed] [Google Scholar]
- Woo JW, Kim J, Kwon SI, Corvalán C, Cho SW, Kim H, Kim SG, Kim ST, Choe S, Kim JS(2015) DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33: 1162–1164 [DOI] [PubMed] [Google Scholar]
- Wu X, Kriz AJ, Sharp PA(2014) Target specificity of the CRISPR-Cas9 system. Quant Biol 2: 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Xu X, Fukao T, Canlas P, Maghirang-Rodriguez R, Heuer S, Ismail AM, Bailey-Serres J, Ronald PC, Mackill DJ(2006) Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442: 705–708 [DOI] [PubMed] [Google Scholar]
- Xu Y, Li P, Zou C, Lu Y, Xie C, Zhang X, Prasanna BM, Olsen MS(2017) Enhancing genetic gain in the era of molecular breeding. J Exp Bot 68: 2641–2666 [DOI] [PubMed] [Google Scholar]
- Yang N, Xu X-W, Wang R-R, Peng W-L, Cai L, Song J-M, Li W, Luo X, Niu L, Wang Y, et al. (2017) Contributions of Zea mays subspecies mexicana haplotypes to modern maize. Nat Commun 8: 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J, Zastrow-Hayes G, Deschamps S, Svitashev S, Zaremba M, Acharya A, Paulraj S, Peterson-Burch B, Schwartz C, Djukanovic V, et al. (2019) CRISPR-Cas9 editing in maize: Systematic evaluation of off-target activity and its relevance in crop improvement. Sci Rep 9: 6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu QH, Wang B, Li N, Tang Y, Yang S, Yang T, Xu J, Guo C, Yan P, Wang Q, et al. (2017) CRISPR/Cas9-induced targeted mutagenesis and gene replacement to generate long-shelf life tomato lines. Sci Rep 7: 11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang J, Wei P, Zhang B, Gou F, Feng Z, Mao Y, Yang L, Zhang H, Xu N, et al. (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol J 12: 797–807 [DOI] [PubMed] [Google Scholar]
- Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH(2015a) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4: e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Massel K, Godwin ID, Gao C(2018) Applications and potential of genome editing in crop improvement. Genome Biol 19: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Mao L, Chen H, Bu F, Li G, Sun J, Li S, Sun H, Jiao C, Blakely R, et al. (2015b) Genome-wide mapping of structural variations reveals a copy number variant that determines reproductive morphology in cucumber. Plant Cell 27: 1595–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Wolt JD(2017) Risk associated with off-target plant genome editing and methods for its limitation. Emerg Top Life Sci 1: 231–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Feng Q, Lu H, Li Y, Wang A, Tian Q, Zhan Q, Lu Y, Zhang L, Huang T, et al. (2018) Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat Genet 50: 278–284 [DOI] [PubMed] [Google Scholar]
- Zischewski J, Fischer R, Bortesi L(2017) Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv 35: 95–104 [DOI] [PubMed] [Google Scholar]
- Zohary D.(1999) Monophyletic vs. polyphyletic origin of the crops on which agriculture was founded in the Near East. Genet Resour Crop Evol 46: 133–142 [Google Scholar]
- Zuo E, Sun Y, Wei W, Yuan T, Ying W, Sun H, Yuan L, Steinmetz LM, Li Y, Yang H(2019) Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 364: 289–292 [DOI] [PMC free article] [PubMed] [Google Scholar]