

Abstract
ISOC is a cation current permeating the ISOC channel. In pulmonary endothelial cells, ISOC activation leads to formation of inter-endothelial cell gaps and barrier disruption. The immunophilin FKBP51, in conjunction with the serine/threonine protein phosphatase 5C (PPP5C), inhibits ISOC. Free PPP5C assumes an autoinhibitory state, which has low “basal” catalytic activity. Several S100 protein family members bind PPP5C increasing PPP5C catalytic activity in vitro. One of these family members, S100A6, exhibits a calcium-dependent translocation to the plasma membrane. The goal of this study was to determine whether S100A6 activates PPP5C in pulmonary endothelial cells and contributes to ISOC inhibition by the PPP5C-FKBP51 axis. We observed that S100A6 activates PPP5C to dephosphorylate tau T231. Following ISOC activation, cytosolic S100A6 translocates to the plasma membrane and interacts with the TRPC4 subunit of the ISOC channel. Global calcium entry and ISOC are decreased by S100A6 in a PPP5C-dependent manner and by FKBP51 in a S100A6-dependent manner. Further, calcium entry-induced endothelial barrier disruption is decreased by S100A6 dependent upon PPP5C, and by FKBP51 dependent upon S100A6. Overall, these data reveal that S100A6 plays a key role in the PPP5C-FKBP51 axis to inhibit ISOC and protect the endothelial barrier against calcium entry-induced disruption.
Keywords: Endothelial ISOC, Calcium entry, Endothelial barrier
Introduction
Calcium is an important second messenger that regulates a number of physiological functions, and altered calcium signaling is associated with the pathology of many disorders and disease states. Endothelial cells express a cation channel, ISOC, which conducts calcium and sodium (1, 2). The calcium current which permeates the ISOC channel is referred to as ISOC (1–9). Activation of ISOC allows calcium entry into the cell, which leads to changes in cell shape and the formation of inter-endothelial cell gaps, in turn leading to decreased endothelial barrier integrity and increased endothelial permeability (2, 4, 5, 10, 11).
In pulmonary endothelial cells, the ISOC channel is comprised of the transient receptor potential canonical (TRPC) subunits TRPC1 and TRPC4. Channels made of TRPC proteins are believed to be tetramers (12), and our earlier studies suggested that the ISOC channel is made up of one TRPC1 subunit and two or more TRPC4 subunits (2). However, the complete stoichiometry of the ISOC channel has yet to be elucidated. Each TRPC subunit has 6-transmembrane spanning domains with cytosolic N- and C- termini (12). The TRPC4 subunit in particular is required for channel activity through its interaction with the spectrin membrane skeleton (3, 9). In addition to interacting with the spectrin membrane skeleton, the channel interacts with a number of accessory proteins, overall comprising what we have termed the ISOC heterocomplex (4–6). For instance, we have previously shown that Orai1 associates with and modulates ISOC channel activity (2). We have also shown that the FK506 binding protein 51, FKBP51, interacts with TRPC4 and inhibits ISOC (4–6). The serine/threonine protein phosphatase 5 (PPP5C; encoded by PPP5C), is also part of the heterocomplex and is needed for FKBP51-mediated inhibition of ISOC (4). Together, these observations suggest a PPP5C-FKBP51 axis contributes to the regulation of ISOC. However, under basal cellular conditions the catalytic activity of PPP5C is low. Here, free PPP5C is predominately in an auto-inhibition conformation in which the N-terminal inhibitory domain binds a unique C-terminal domain and in doing so blocks substrate access to the catalytic site (13). Therefore, for this axis to function PPP5C must be converted into a catalytically active state. The, question this bares to mind is: what activates PPP5C in the context of the PPP5C-FKBP51 regulatory axis? We therefore began to investigate endogenous activators of PPP5C.
Previous reports have shown that upon binding to Hsp90 or S100 proteins, PPP5C is released from its autoinhibited state (14). There are over 25 known S100 proteins, and with the exception of S100A10 all known family members bind calcium through EF hand domains (15, 16). S100 family members, S100A1, S100A2, S100A6 and S100B are known to act as potent activators of PPP5C (14), and Yamaguchi et al (14) demonstrated the interaction of these S100 proteins with PPP5C occurs in a calcium-dependent manner in vitro. In particular, they showed that S100A1, S100A2, S100A6 and S100B all interact with tetratricopeptide repeat (TPR) domains of PPP5C and increase the catalytic activity of PPP5C. For the regulation of ISOC, S100A6 was of interest because in some cell types it exhibits calcium-induced translocation from the cytosol to the plasma membrane (17, 18), where the ISOC channel is localized (3). However, a role for S100A6-mediated activation of PPP5C in endothelial cells has not been described. Thus, the goal of this study was to determine whether S100A6 plays a role in the PPP5C-FKBP51-mediated inhibition of ISOC in endothelial cells.
Materials and Methods
Reagents
All reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted. Cell culture medium was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and penicillin/streptomycin was obtained from Life Technologies (Grand Island, NY, USA). Fetal bovine serum and Hank’s Balanced Salt Solution (HBSS) were purchased from Life Technologies (Grand Island, NY, USA). Monoclonal antibodies to FKBP51 were described previously (19). Polyclonal antibody for S100A6 was purchased from Sigma Life Science (St. Louis, MO, USA) (Prestige Antibodies) (Product no. HPA007575). TRPC4 antibody was obtained from Dr. Mike Zhu (The University of Texas Health Science Center at Houston Medical School) or Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibody for PPP5C was previously described (20). Additionally, PPP5C antibody was purchased from Cell Signaling (Danvers, MA). β-actin antibody was purchased from Santa Cruz Biotechnology. Monoclonal antibody for Phospho-Tau T231 was obtained from Thermo Fisher Scientific (Waltham, MA, USA) and monoclonal antibody for Tau (pan) was purchased from MBL International (Woburn, MA, USA). TRITC-conjugated WGA (Wheat Germ Agglutinin) lectin was obtained from EY Laboratories (San Mateo, CA, USA). Doxycycline was purchased from Clontech Laboratories, Inc. (Mountain View, CA, USA). Lipofectamine 3000 transfection reagent was obtained from Thermo Fisher Scientific.
Cell Culture
Rat pulmonary microvascular endothelial cells (PMVECs) and pulmonary artery endothelial cells (PAECs) were isolated as described previously (21). All cells were isolated with approval from the University of South Alabama Institutional Animal Care and Use Committee and in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Cells were grown in high glucose DMEM supplemented with 10% FBS, penicillin G (50 U/mL), and streptomycin (0.05 mg/mL). Human lung microvascular endothelial cells (HLMVECs) are an immortalized cell line (HPMEC-ST1.6R) developed in the laboratory of Dr. C. James Kirkpatrick (22). These cells were grown in high glucose DMEM supplemented with 20% FBS, penicillin G (50 U/mL), and streptomycin (0.05 mg/mL). PPP5C-FLAG HEK293 cells, which constitutively over-express PPP5C by ~2-fold, were previously generated (23). Both PPP5C-FLAG and wild-type HEK cells were grown in high glucose DMEM supplemented with 10% FBS, penicillin G (50 U/mL), streptomycin (0.05 mg/mL) and 0.5% non-essential amino acids solution (Thermo Fisher Scientific).
Generation of inducible FKBP51 and S100A6 cell lines
PMVECs were engineered to conditionally express human FKBP51 using a two-tiered system which combines doxycycline-inducible gene expression with protein stability control (24, 25). The lentiviral vector pMA4763 was generated such that the COOH-terminus of the human FKBP51 gene was linked to a myc-tag and a destabilizing mutant (E31G, R71G, K105E) of FK506 binding protein 12 (FKBP12). Production of retro- and lentiviral supernatants and PMVEC infection were performed as described previously (26). Briefly, CaPO4-mediated transfection of Phoenix ampho or HEK293FT cells was used to produce retrovirus- or lentivirus-containing supernatants, respectively, using established procedures (27, 28). PMVECs were infected sequentially with retrovirus pMA2641 followed by lentivirus pMA4763. Infections were performed at 50% cell confluence by incubating overnight with corresponding supernatant in the presence of 10 μg/mL polybrene. The next day, supernatant was removed, and cells were allowed to recover for 24 hours in fresh DMEM after which they were selected with blasticidin (3 μg/mL) for pMA2641 and puromycin (1 μg/mL) for pMA4763. For induction of FKBP51 expression, cells were treated for 48 hours with Shield1 (2 μM) (Clontech Laboratories, Mountain View, CA) to prevent protein degradation plus doxycycline (2 μg/mL).
PMVECs were engineered to conditionally express rat S100A6 (lentivirus pMA5094) via the doxycycline-inducible system. This S100A6 expression system did not include the destabilizing FKBP12 mutant. Cells were infected sequentially with pMA2641 and pMA5094 following the protocol described above. Cells were selected for pMA5094 using 2 μg/mL puromycin. For doxycycline induction, PMVECs were treated with doxycycline (3 μg/mL) for 48 hours.
Quantitative polymerase chain reaction (qPCR)
Messenger RNA was prepared from endothelial cells using the RNeasy mini kit (Qiagen, Valencia, CA, USA) and reverse transcribed to complementary DNA (cDNA) using an iScript cDNA preparation kit (Bio-Rad Laboratories, Hercules, CA, USA). qPCR was performed by SYBR Green incorporation using a CFX Connect (Bio-Rad) thermocycler with the following primers:
| Forward 5′-3′ | Reverse 5′-3′ | |
|---|---|---|
| rat | ||
| S100A1 | GCTCTCACAGTGGCTTGTAA | TAGGGAGATACAGGTGGGATG |
| S100A4 | CTTCCAGGAGTACTGTGTCTT | CTGAGGAGTCTTCACTTCTTCC |
| S100A6 | CTGAGCAAGAAGGAGCTGAA | GTTACGGTCCAGATCATCCATC |
| S100A10 | GGGCTTCCAGAGCTTTCTATC | CTCCAGTTGGCCTACTTCTTC |
| S100A11 | CTCGACCGCATGATGAAGAA | AAGTCTGGAGGAAGGACTCA |
| HPRT | GACCTCTCGAAGTGTTGGATAC | TCAAATCCCTGAAGTGCTCAT |
| human | ||
| S100A1 | AGACCCTCATCAACGTGTTC | CACAAGCACCACATACTCCT |
| S100A4 | GTGTCCACCTTCCACAAGTA | GATGCAGGACAGGAAGACA |
| S100A6 | AGGAGCTGATCCAGAAGGA | CACTGGATTTGACTCAGAGAGG |
| S100A10 | AAACCAAACCCGAGAGGAAG | GACCCTTTGGGACAACTCTTAT |
| S100A11 | AGTCCCTGATTGCTGTCTTC | CTGCATGAGGTGGTTAGTGT |
| HPRT | CGAGATGTGATGAAGGAGATGG | TTGATGTAATCCAGCAGGTCAG |
Western Blot Analysis
Cells were lysed in RIPA buffer (Boston BioProducts; Ashland, MA, USA) with 1% protease inhibitor cocktail followed by centrifugation at 12,000 g for 20 minutes. Whole cell lysates were electrophoresed on 4–12% or 12% bis-Tris gels (Thermo Fisher Scientific or GenScript Biotech Corp; New Jersey, USA). Proteins were transferred to 0.4 μm or 0.2 μm nitrocellulose membranes at 100 V. Membranes were then blocked with milk (5% non-fat dry milk/0.2% BSA in PBS supplemented with 0.1% Tween-20) for 1 hour and then rocked with primary antibodies overnight at 4 °C and with appropriate horseradish peroxidase (HRP)- conjugated secondary antibodies for 1 hour at room temperature. Proteins were visualized by chemiluminescence detection using either Supersignal West Pico or West Femto chemiluminescent substrates (Thermo Fisher Scientific).
Immunocytochemistry
PMVECs were seeded on coverslips and grown to 80–100% confluence in 35-mm dishes. Cells were fixed immediately or were pretreated with either rolipram (0.1 μM) for 15 minutes followed by thapsigargin (1.0 μM) for 5 minutes in HBSS/2mM Ca2+ or with thapsigargin (1.0 μM) for 5 minutes in HBSS/2mM Ca2+ buffer before fixation. PMVEC monolayers were fixed and permeabilized with 90% ice-cold methanol in PBS for 3 minutes at −20 °C. Following fixation/permeabilization, cells were incubated in blocking buffer (10% normal goat serum, 10% normal donkey serum, 1% BSA in HBSS) for 1 hour at room temperature. To visualize S100A6, cells were then incubated with primary antibody diluted in milk (5% non-fat dry milk in PBS supplemented with 0.1% Tween-20) overnight at 4 °C. Cells were treated with secondary antibody for 1 hour at room temperature. Dilution used for primary antibody for S100A6 was 1:100. Secondary antibody was used at 1:500 dilution and includes AlexaFluor 488 goat anti-rabbit IgG (H&L) (Life Technologies, Grand Island, NY, USA). Cells were incubated with Hoechst 33342 nuclear counterstain (10 μg/mL; Thermo Fisher Scientific) under agitation for 20 minutes at room temperature. For co-localization studies PMVEC monolayers were first stained with TRITC-conjugated WGA lectin for visualization of cell membrane before fixation. Cells were washed twice with HBSS/2 mM Ca2+ buffer and then cells were either pretreated with rolipram (0.1 μM) for 15 minutes followed by thapsigargin (1.0 μM) for 5 minutes in HBSS/2 mM Ca2+ and then incubated with WGA lectin or directly incubated with WGA lectin staining solution (40 μg/mL) for 10 minutes at 37°C. Cells were then washed twice with HBSS/2 mM Ca2+ buffer, following which PMVEC monolayers were fixed and permeabilized with 90% ice-cold methanol in PBS for 3 minutes at −20°C. Following fixation and permeabilization we followed the same staining protocol to visualize S100A6 as described earlier here. Fluorescent images were obtained using a Zeiss Observer.D1 widefield microscope (Carl Zeiss, Germany) fitted with 40X oil immersion objective.
Co-immunoprecipitation Experiments
Co-precipitations of S100A6 with TRPC4 and PPP5C were achieved with SureBeads™ IgG-conjugated magnetic beads (Bio-Rad; Hercules, CA, USA) as previously described (4). PMVEC lysates were collected without any treatment or after pretreating cell monolayers with rolipram (0.1 μM) for 15 minutes followed by thapsigargin (1.0 μM) for 5 minutes in HBSS/2mM Ca2+, or with thapsigargin (1.0 μM) alone for 5 minutes in HBSS/2mM Ca2+. Then, following a 10 minute incubation of magnetic beads with the precipitating antibody, whole cell lysates of PMVECs were incubated with the beads for 1 hour under agitation. After washing, protein was eluted from the beads with 40 μL of 1X Laemlli buffer (diluted from 6X Laemmli buffer; Boston BioProducts, Ashland, MA, USA) for 10 minutes at 70 °C. Protein eluates were then subjected to SDS-PAGE and immunoblot analysis.
Global Ca2+ measurements and patch-clamp electrophysiology
Cells were seeded onto glass coverslips and grown to confluence in 35 mm dishes. Changes in cytosolic calcium concentration were determined using Fura2-acetoxymethyl ester (Fura2/AM; Thermo Fisher Scientific) as previously described (1, 3, 6, 9, 29). Patch-clamp electrophysiology recordings were performed in whole-cell configuration on electrically isolated cells according to the method as described (4, 5, 9). Isolation of single cells was achieved via treatment of cell monolayers with a non-enzymatic cell dissociation solution in PBS (Sigma-Aldrich). Transmembrane current was measured with an Axopatch 200B Amplifier (Molecular Devices; Sunnyvale, CA, USA). PClamp10 software was used to record the current evoked by step depolarization in 20 mV increments from −100 mV to +80 mV. Currents were measured as the mean value of the current amplitude during the last 20 ms of each step. The standard pipette solution contained (in mmol/L) 130 N-methyl-D-glucamine, 10 Hepes, Mg2+-ATP, 1 N-phenylanthranilic acid, 0.1 5-nitro-2-(3-phenylpropylamino)benzoic acid, 2 EGTA, 1 Ca(OH)2 (pH 7.2, adjusted with methane sulfonic acid). The bath (external) solution contained (in mmol/L) 120 aspartic acid, 5 Ca(OH)2, 5 CaCl2, 10 Hepes, 0.5 3,4-diaminopyridine (pH 7.4 adjusted with tetraethylammonium hydroxide). All solutions were adjusted to 290–300 mOsm/L with sucrose. The free [Ca2+] was estimated to be 100 nmol/L as previously described (6, 9). Recording pipettes were made of hemo capillaries (A-M Systems; Sequim, WA, USA) pulled by a micropipette puller (P-97; Sutter Instruments; Novato, CA, USA) and heat-polished by a microforge (MF-830; Narishige; Tokyo, Japan) to a final resistance of 3–5 megaohms when filled with standard pipette solution. All experiments were performed at room temperature. Data are expressed as mean ± SEM for the number of cells (n) in which whole-cell patch-clamp recordings were obtained.
siRNA to S100A6 and PPP5C
S100A6 knockdown was achieved via siRNA targeted to the sequence 5’-GCUCACCAUUGGUGCUAAG-3’ (Silencer® Pre-designed siRNA, Ambion, Life Technologies).
PPP5C knockdown was achieved with siRNA targeted to the sequence 5’-AATGGCGATGGCGGAGGGCGA-3’ (Qiagen; Hilden, Germany).
Transfection of siRNA to PMVECs was achieved as previously described (4, 6). PMVEC monolayers were grown to 50–60% confluency. Cell monolayers were then transfected with a 20 nM final siRNA concentration in lipofectamine 3000 (Thermo Fisher Scientific). Complete cell culture media was added to the transfected monolayers at 4 hours and siRNA incubation continued to 48 hours.
Electric cell-substrate impedance sensing (ECIS®)
ECIS ® experiments were performed as previously described (4, 5, 30). A stable baseline resistance was recorded for at least 30 minutes before administration of rolipram (0.1 μM) for at least 15 minutes followed by thapsigargin (1.0 μM). Resistance changes were recorded for at least one hour following thapsigargin addition.
Statistical analysis
Statistical analysis was performed using GraphPad version 5.0 software (San Diego, CA, USA). Data are presented as mean ± SEM. Comparisons between multiple groups were performed using a one-way or two-way ANOVA and post hoc tests. Student’s t-tests were used elsewhere. Values were considered significantly different when P < 0.05, which is denoted by * in figures. For patch-clamp data, comparisons between multiple groups were performed together via two-way ANOVA and Bonferroni post hoc test, but the data were separated in figures for easier visualization.
Results
Expression of S100 family members in lung endothelium.
We first questioned which S100 family members are expressed in lung endothelium. RNAseq data from rat PMVECs revealed transcript expression of a number of S100 family members with S100A4, S100A6, S100A10 and S100A11 being most abundant (data not shown). Similar to PMVECs, rat pulmonary artery endothelial cells (PAECs) also showed high transcript expression of S100A6, S100A10 and S100A11, but much less of S100A4. RNAseq data were validated by performing qPCR for these four family members as well as for S100A1 which showed low transcript expression. Consistent with RNAseq data, S100A4, S100A6, S100A10 and S100A11 mRNA expression was observed with PAECs expressing less S100A4 than PMVECs; and S100A1 was low in both cell types (Figure 1A). In human lung microvascular endothelial cells (HLMVECs), qPCR revealed a strong predominance of S100A6 mRNA. Indeed, we were particularly interested in S100A6 because it interacts with proteins that contain TPR domains (14, 31, 32) such as PPP5C and FKBP51 (33–35) and exhibits calcium-dependent translocation from cytosol to the plasma membrane (17, 18), where the ISOC channel is localized (3). Thus, we next measured protein expression of S100A6 in PAECs, PMVECs and HLMVECs using western analysis. These studies revealed that all three cell types express S100A6 protein (Figure 1B). As we found that S100A6 is expressed in lung endothelial cells, we next asked whether S100A6 interacts with the ISOC channel on the plasma membrane.
Figure 1. S100 protein family members are expressed in pulmonary endothelial cells.

A. qPCR reveals relative expression of S100A1, S100A4, S100A6, S100A10 and S100A11 in rat PAECs and PMVECs and human HLMVECs. S100A6 message is highly expressed in PAECs and PMVECs and predominantly expressed in HLMVECs. n = 3 independent experiments. B. S100A6 protein is expressed in PAECs, PMVECs and HLMVECs. Protein expression was examined on PAECs isolated from one rat (PA36) and PMVECs isolated from two different rats (MV1 & MV3). Relative protein expression was assessed via densitometry. n = 3 independent experiments for each cell line.
S100A6 interacts with the ISOC channel heterocomplex in a calcium-dependent manner.
Our study was focused on ISOC activation in PMVECs. In PAECs, treatment with the calcium ATPase inhibitor thapsigargin (1.0 μM) activates ISOC (1–6, 8, 9). However, in PMVECs treatment with thapsigargin alone is insufficient to activate ISOC (8). To activate ISOC in PMVECs, we first incubated the cells with the type IV phosphodiesterase inhibitor rolipram (0.1 μM) for 15 minutes followed by treatment with thapsigargin (1.0 μM). The requirement for rolipram pretreatment to activate ISOC in PMVECs was originally described by Wu et al (8), however in those studies rolipram was applied at 10 μM. In our hands the endothelial barrier was disrupted by high rolipram concentrations. Dose response studies revealed that 0.1 μM rolipram was sufficient to activate ISOC without disrupting the endothelial barrier (data not shown). Thus, throughout this study we used rolipram (0.1 μM) / thapsigargin (1.0 μM) treatment to activate ISOC in PMVECs. It is important to recognize however, that under these conditions ISOC is likely not the only cation current activated. In global calcium measurements, treatment of PMVECs with thapsigargin alone elicited an increase in cytosolic calcium due to calcium entry even though ISOC was not activated (8). In some experiments in the present study we compare effects of rolipram/thapsigargin treatment to treatment with thapsigargin alone in order to determine whether general calcium entry or calcium entry specifically through the ISOC channel is important for mediating downstream effects.
Previous observations revealed a calcium-dependent translocation of S100A6 from the cytosol to the plasma membrane in porcine stomach smooth muscle, porcine testis cells and several human carcinoma cell lines (17, 18). To determine whether S100A6 also translocates in pulmonary endothelial cells we performed immunocytochemistry. In these studies, confluent PMVEC monolayers were methanol-fixed either without ISOC activation or following ISOC activation and immunocytochemistry was used to detect S100A6. We observed that in the absence of ISOC activation, and thus no calcium entry, S100A6 was dispersed throughout the cytoplasm (Figure 2A.1). Following ISOC activation, a distinct membrane delimited pool of S100A6 became observable, particularly at areas of cell-cell contact (Figure 2A.1), where the endothelial ISOC heterocomplex is found (3, 6). To confirm that S100A6 was at the plasma membrane, we also co-stained with fluorescently-tagged WGA lectin, which is used as a membrane marker(36). Here we observed colocalization of S100A6 with WGA (Figure 2A.2). For controls, PMVECs were treated with rolipram alone or thapsigargin alone (Figure 2A.1) or DMSO alone (vehicle control; Supplemental Figure). Treatment with rolipram or DMSO alone did not induce S100A6 translocation. Following treatment with thapsigargin alone, we observed no translocation in the majority of cells imaged. In a few instances, we did observe membrane staining that was of low intensity. This is in stark contrast to the numerous and strong membrane staining observed following rolipram/thapsigargin treatment. These observations are important because while in PMVECs ISOC is not activated by thapsigargin alone, other calcium entry channels are still activated. Thus, by comparing rolipram/thapsigargin treatment to thapsigargin alone, we can determine whether it is activation of ISOC per se or general calcium entry that is important for S100A6 translocation. Overall, our data reveal that calcium entry through the ISOC channel is an important determinant of S100A6 translocation to the plasma membrane.
Figure 2. S100A6 translocates to the plasma membrane in a calcium-dependent manner to interact with the ISOC channel heterocomplex.



A.1. Immunocytochemistry for S100A6 was performed to determine cellular localization. In unstimulated PMVECs (WT), S100A6 is principally cytosolic. Treatment of PMVECs with thapsigargin alone (TG) in the absence of extracellular calcium or treatment with rolipram alone did not induce S100A6 translocation. In the presence of extracellular calcium, TG treatment caused only minor and infrequent S100A6 translocation. Upon activation of ISOC (R+TG) in the presence of extracellular calcium, part of the cytosolic S100A6 pool translocated to the plasma membrane particularly at the sites of cell-cell adhesion (yellow arrows). Images are representative from 3 independent experiments. A.2. Colocalization of S100A6 with TRITC-conjugated WGA lectin. In control PMVECs in which ISOC was not activated, no S100A6 was found at the membrane, which was labelled by WGA (red stain denoted by yellow arrows). In PMVECs in which ISOC was activated, S100A6 (green) colocalized with WGA (red) at the membrane (yellow arrows). Images are representative from 3 independent experiments. B. TRPC4 was immunoprecipitated from PMVECs following ISOC activation (R+TG) or following activation of other calcium channels (TG). Immunoprecipitations were performed in the presence or absence of extracellular calcium, and co-precipitation of S100A6 was probed. (i) In the absence of primary antibody (beads only), no bands were detected. Sample input of whole cell lysates (WCL) revealed presence of both TRPC4 and S100A6. While TRPC4 was immunoprecipitated following R+TG in both the presence and absence of extracellular calcium, co-precipitation of S100A6 was predominantly observed in the presence of extracellular calcium. Similarly, following activation of other calcium channels (TG), TRPC4 was immunoprecipitated in both the presence and absence of extracellular calcium. However, while some co-precipitation of S100A6 was observed in the presence of extracellular calcium, it was less than that observed following R+TG. (ii) Quantitation of S100A6 co-precipitation was assessed via densitometry of TRPC4 and S100A6 bands and the S100A6/TRPC4 ratio determined. Molecular weights observed: TRPC4 ~80 kDa; S100A6 10 kDa. n = 5 independent experiments. (iii) PPP5C was immunoprecipitated from PMVECs following ISOC activation (R+TG) in the presence or absence of extracellular calcium and co-precipitation of S100A6 probed. In the absence of primary antibody (beads only), no bands were detected, while WCL input revealed presence of both PPP5C and S100A6. Following ISOC activation (R+TG), robust co-precipitation of S100A6 was observed in the presence of extracellular calcium while only minor co-precipitation of S100A6 was observed in the absence of extracellular calcium. (iv) Quantitation of S100A6 co-precipitation was assessed via densitometry of PPP5C and S100A6 bands and the S100A6/PPP5C ratio determined. Molecular weights observed: PPP5C ~58 kDa; S100A6 10 kDa. n = 3 independent experiments.
Immunocytochemistry results revealed that S100A6 translocates to the plasma membrane following ISOC activation in PMVECs. We next wanted to know whether this membrane localized S100A6 physically interacts with the ISOC channel. Here we performed immunoprecipitation experiments probing for co-precipitation between S100A6 and the TRPC4 subunit of the ISOC channel. We activated ISOC (rolipram/thapsigargin) or other calcium channels (thapsigargin alone) in the presence or absence of extracellular calcium. First, TRPC4 was immunoprecipitated and co-precipitation of S100A6 was detected by western analysis. In the absence of extracellular calcium, co-precipitation of S100A6 with TRPC4 was minimal in PMVECs following rolipram/thapsigargin treatment (Figure 2B– i, ii). However, in the presence of extracellular calcium robust co-precipitation of S100A6 with TRPC4 was observed only after rolipram/thapsigargin treatment. While in PMVECs, treatment with rolipram/thapsigargin activates ISOC, treatment with thapsigargin alone still initiates calcium entry, albeit not through ISOC (8). To determine whether calcium entry through other calcium channels also triggers co-precipitation of S100A6 with TRPC4, PMVECs were treated with thapsigargin alone followed by immunoprecipitation for TRPC4. While we did observe some co-precipitation of S100A6 with TRPC4 in PMVECs treated with thapsigargin alone, in the presence of extracellular calcium, it was ~2-fold less than that observed in cells treated with rolipram/thapsigargin in the presence of extracellular calcium (Figure 2B– i, ii). Overall, these data reveal that calcium entry through the ISOC channel provides a specific calcium source that promotes interaction of S100A6 withTRPC4.
We have previously shown that PPP5C is part of the ISOC channel heterocomplex (4). Our hypothesis posits that ISOC activation leads to S100A6 translocation to the plasma membrane where it binds and activates a pool of PPP5C associated with the ISOC channel. Thus, we interrogated the ability of S100A6 to interact with PPP5C in an ISOC-dependent manner. Here, ISOC was activated in PMVECs in the absence or presence of extracellular calcium, and PPP5C was immunoprecipitated. Co-precipitation of S100A6 with PPP5C was observed following ISOC activation in the presence of extracellular calcium (Figure 2B– iii, iv), consistent with our hypothesis that subsequent to ISOC activation S100A6 translocates to the plasma membrane where it interacts with and activates PPP5C in the ISOC heterocomplex. Our next experiments tested this idea.
S100A6 activates PPP5C in PMVECs.
We next asked whether S100A6 activates PPP5C in PMVECs. Previous in vitro studies by Yamaguchi et al (14) demonstrated that S100A6 as well as S100A1, S100A2, S100B, but not S100A12, activate PPP5C, using KRpTIRR as a substrate, in a calcium-dependent manner. In this same study, they also determined that S100-mediated activation of PPP5C is sufficient to dephosphorylate the physiological target tau. In vitro, PPP5C dephosphorylates a number of sites on tau (37–39), and Yamaguchi et al used sites S396 and S409 as their readout of PPP5C activity. In a pure in vitro setup, S100A1, S100A2 and S100B, but not S100A12, promoted dephosphorylation of tau by PPP5C. No data were reported for S100A6 in the tau experiment. We were interested in determining whether S100A6 activates PPP5C in cells and we questioned whether we could use the phosphorylation status of endogenous tau as a readout of PPP5C activity. For these studies, we measured phosphorylation of tau at T231. T231 is part of a flanking region around the microtubule binding repeats which is important for binding to microtubules (40), and tau T231 phosphorylation is important for tau’s ability to bind and stabilize microtubules (41). We were particularly interested in phosphorylation of this site not only as a readout of PPP5C activity but also because it may affect microtubule polymerization status, which is a key determinant of FKBP51’s ability to inhibit ISOC (5).
First, to determine whether PPP5C affects tau T231 phosphorylation in cells, we measured T231 phosphorylation in wild-type and PPP5C over-expressing HEK293 cells. The PPP5C over-expressing HEK293 cells were previously described and over-express PPP5C by ~2-fold (23). Despite the small ~2-fold increase in PPP5C expression in the over-expressing cells, there was a dramatic decrease ~4 fold in tau T231 phosphorylation compared to wild-type cells (Figure 3A). This observation shows that PPP5C dephosphorylates tau T231, and thus we concluded that we could use the phosphorylation status of T231 to assess PPP5C activity in PMVECs.
Figure 3. S100A6 activates PPP5C to dephosphorylate tau T231 in endothelial cells.




A. Phospho-tau T231 can be used as a readout of PPP5C cellular activity. (i) Phospho-tau T231 was assessed via immunoblot of HEK293 cells which were engineered to modestly overexpress PPP5C (~2- fold) (23). Compared to wild-type (WT) HEK293 cells, in PPP5C over-expressed cells, phospho-tau T231 was nearly abolished. (ii) Quantitation of phospho-tau T231 was assessed via densitometry of phospho-tau T231 and total tau bands and the normalized phospho-tau T231/total tau ratio determined. n = 3 independent experiments. B. S100A6 activates PPP5C in endothelial cells. (i) S100A6 over-expressing cells express decreased phospho-tau T231. Immunoblot of doxycycline-inducible S100A6 PMVECs reveal ~ 2-fold increase in S100A6 expression in the presence of doxycycline, and a corresponding decrease in phospho-tau T231. (ii) Quantitation of phospho-tau T231 was assessed via densitometry of phospho-tau T231 and total tau bands and the normalized phospho-tau T231/total tau ratio determined. Molecular weights observed: phospho-tau T231 ~55 kDa; total tau ~50 kDa; S100A6 10 kDa; β actin 42 kDa. n = 3 independent experiments. (iii) The decrease in phospho-tau T231 by S100A6 over-expression is dependent upon PPP5C. Doxycycline-inducible S100A6 PMVECs were treated with PPP5C siRNA or scrambled control and phospo-tau T231 expression measured by immunoblot. In the presence of scrambled control, S100A6 over-expressed cells revealed little phospho-tau T231 which was reversed upon knock-down of PPP5C. (iv) Quantitation of phospho-tau T231 was assessed via densitometry of phospho-tau T231 and total tau bands and the normalized phospho-tau T231/total tau ratio determined. Molecular weights observed: phospho-tau T231 ~55 kDa; total tau ~50 kDa; PPP5C ~58 kDa; S100A6 10 kDa; β actin 42 kDa. n = 5 independent experiments. C. Suppression of endogenous S100A6 expression results in increased phospho-tau T231 in PMVECs. (i) PMVECs were treated with S100A6 siRNA or scrambled control. In both the absence of ISOC activation (control) and following ISOC activation, phospho-tau T231 was increased in cells treated with S100A6 siRNA as compared to scrambled control with no change in total tau levels. (ii) Quantitation of phospho-tau T231 was assessed via densitometry of phospho-tau T231 and total tau bands and the normalized phospho-tau T231/total tau ratio determined. Molecular weights observed: phospho-tau T231 ~55 kDa; total tau ~50 kDa; β actin 42 kDa. n = 3 independent experiments. D. FKBP51 over-expressing cells express decreased phospho-tau T231. (i) Immunoblot of doxycycline-inducible FKBP51 PMVECs reveal ~ 3-fold increase in FKBP51 expression in the presence of doxycycline, and a corresponding decrease in phospho-tau T231. (ii) Quantitation of phospho-tau T231 was assessed via densitometry of phospho-tau T231 and total tau bands and the normalized phospho-tau T231/total tau ratio determined. Molecular weights observed: phospho-tau T231 ~55 kDa; total tau ~50 kDa; FKBP51 ~51 kDa; β actin 42 kDa. n = 3 independent experiments. (iii) The decrease in phospho-tau T231 by FKBP51 over-expression is dependent upon S100A6. Doxycycline-inducible FKBP51 PMVECs were treated with S100A6 siRNA or scrambled control and phospho-tau T231 expression measured by immunoblot. In the presence of scrambled control, FKBP51 over-expressed cells revealed little phospho-tau T231 which was reversed upon S100A6 suppression. (iv) Quantitation of phospho-tau T231 was assessed via densitometry of phospho-tau T231 and total tau bands and the normalized phospho-tau T231/total tau ratio determined. Molecular weights observed: phospho-tau T231 ~55 kDa; total tau ~50 kDa; S100A6 10 kDa; FKBP51 ~51 kDa; β actin 42 kDa. n = 5 independent experiments.
Our next question was to determine whether S100A6 activates PPP5C in PMVECs. Here, we used a cell model in which S100A6 expression was upregulated in PMVECs and then PPP5C expression was suppressed using siRNA. To begin, PMVECs were transfected with a lentiviral construct to over-express S100A6 in a doxycycline-inducible manner. Cells were treated with 3 μg/mL doxycycline for 48 hours to modestly upregulate S100A6 expression, by ~2 fold, compared to non-doxycycline-treated cells (Figure 3B). We then examined phospho-tau T231 expression and observed that phosphorylation of tau at T231 decreased more than 2-fold in S100A6 over-expressing cells compared to non-doxycycline-treated cells, with no change in total tau levels. To determine whether this decrease in phospho-tau T231 was dependent upon PPP5C activity, we suppressed PPP5C expression in the S100A6 over-expressing cells using siRNA. With a PPP5C suppression of ~80%, phosphorylation at T231 increased 2-fold greater than S100A6 over-expressing cells treated with scrambled siRNA (Figure 3B). Thus, these data are consistent with S100A6 working to activate PPP5C in PMVECs.
Protein over-expression generally does not represent the endogenous state of a protein, and as such may not reveal physiological relevance. As the above studies used S100A6 over-expression, we next asked the question of whether endogenous S100A6 plays a role in activation of PPP5C. Here, we suppressed the expression of S100A6 using siRNA and measured tau T231 phosphorylation in the presence or absence of ISOC activation. In control cells in which ISOC was not activated, we observed an increase in tau T231 phosphorylation with S100A6 siRNA treatment as compared to scrambled siRNA (Figure 3C), although this increase was not statistically significant. On the other hand, tau T231 phosphorylation was increased even more following ISOC activation in cells treated with S100A6 siRNA. Overall, these data suggest that endogenous S100A6 is physiologically relevant in determining PPP5C activation and the phosphorylation status of tau.
Our overall goal was to determine the role of S100A6 in the PPP5C-FKBP51 axis which inhibits ISOC. While we showed that S100A6 activates PPP5C in PMVECs, we do not yet know whether this is related to the PPP5C-FKBP51 axis. In our earlier studies we upregulated the PPP5C-FKBP51 axis by modestly over-expressing FKBP51 in rat PAECs (4, 5). Thus, we followed the same approach here using PMVECs. Using a doxycycline-inducible lentiviral construct, FKBP51 expression was increased by ~3-fold in the presence of doxycycline as compared to non-doxycycline-treated cells (Figure 3D). In these FKBP51 over-expressing cells, phospho-tau T231 was decreased by ~3 fold. This observation is consistent with the model originally proposed by Jinwal et al (42) in which FKBP51 facilitates tau de-phosphorylation by isomerizing phospho-tau, which then allows de-phosphorylation by a phosphatase. We then wanted to determine whether the decrease in phospho-tau T231 in FKBP51 over-expressing cells involves S100A6. Here, FKBP51 over-expressing PMVECs were treated with S100A6 siRNA. S100A6 suppression, ~75%, resulted in increased phospho-tau T231 levels in FKBP51 over-expressing cells as compared to cells expressing scrambled siRNA. These data demonstrate that tau de-phosphorylation facilitated by FKBP51 is dependent upon S100A6, and as we have shown that S100A6-mediated de-phosphorylation of tau is also dependent upon PPP5C, overall, we conclude that S100A6 plays a role in the PPP5C-FKBP51 axis. However, we have not yet determined whether S100A6 is functionally important in the PPP5C-FKBP51-mediated inhibition of ISOC and downstream protection of endothelial barrier against calcium entry-induced disruption.
S100A6 regulates calcium entry and barrier function in PMVECs in a PPP5C-dependent manner.
To determine the role of S100A6 in calcium entry and endothelial barrier function, we used the same cell systems described above where S100A6 is over-expressed in a doxycycline-inducible manner and PPP5C expression is suppressed using siRNA. We began by assessing calcium entry. As increased cytosolic calcium promotes endothelial barrier dysfunction, we measured changes in global cytosolic calcium using Fura2. When we measured global calcium entry in doxycycline-inducible S100A6 over-expressing PMVECs, calcium entry following ISOC activation with rolipram/thapsigargin was decreased in doxycycline-treated cells as compared to non-doxycycline-treated cells (Figure 4A/B). We recognize however that upon ISOC activation with rolipram/thapsigargin it is likely that other thapsigargin-activated calcium channels open as well, and using the Fura2 approach we are not be able to discriminate between different sources of calcium entry. Thus, to specifically determine the role of S100A6 in ISOC channel activation we next performed patch-clamp electrophysiology using the whole-cell, voltage-clamp configuration as described previously (4, 5). We measured ISOC in the doxycycline-inducible S100A6 over-expressing cells in the presence or absence of doxycycline. In the absence of doxycycline a typical ISOC was observed measuring ~27 pA at −80 mV with a reversal potential ~+34 mV (Figure 4C). In the presence of doxycycline, in which S100A6 expression was upregulated by ~2-fold, ISOC was dramatically decreased such that only a tiny current was measured (~8 pA at −80 mV). These observations reveal that S100A6 regulates calcium entry and ISOC in PMVECs. We next asked whether this calcium regulation by S100A6 plays a role in determining the extent of calcium entry-induced endothelial barrier disruption. Here we used ECIS® to assess changes in endothelial barrier integrity following activation of ISOC. In this technique, a decrease in resistance reflects disruption of the endothelial barrier, and the larger the resistance decrease the greater the barrier disruption. An advantage of using ECIS® is that it allows real-time monitoring of the endothelial barrier(43). Again, we used the same doxycycline-inducible S100A6 over-expressing cell system and measured changes in resistance following activation of ISOC with rolipram/thapsigargin. In non-doxycycline-treated cells, following ISOC activation resistance decreased ~30%, whereas in doxycycline-treated cells there was little decrease in monolayer resistance (~10%) following ISOC activation (Figure 4D/E). While these data implicate functional roles of S100A6 in PMVECs, we do not know whether PPP5C is involved.
Figure 4. S100A6 regulates calcium entry and barrier function in PMVECs.
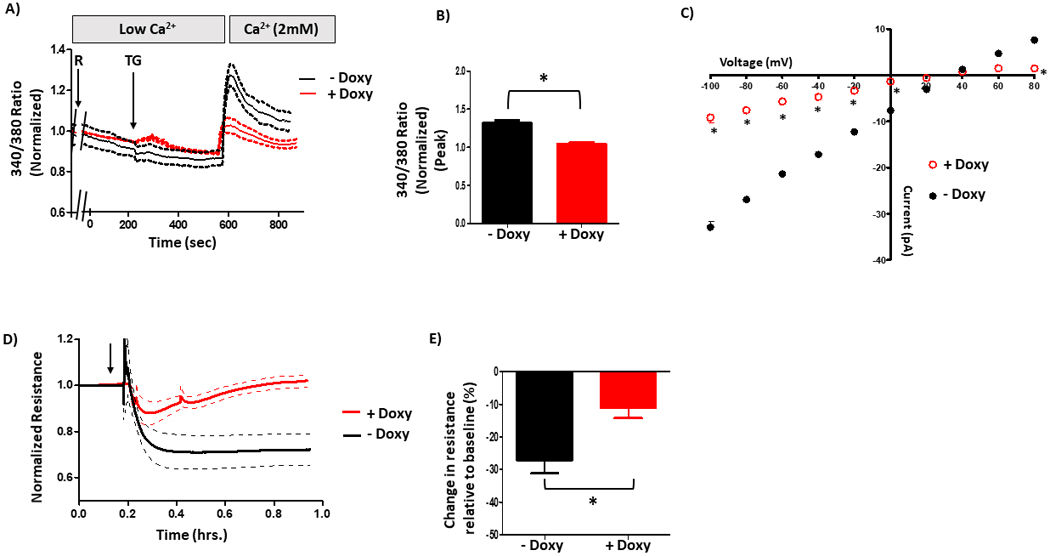
Global calcium entry following ISOC activation (R+TG) was measured using the ratiometric dye Fura2. Here, cells were pretreated with rolipram (R) for 15 minutes prior to mounting on microscope stage. Thapsigargin (TG) was added when cells were in low extracellular calcium to reveal calcium release and then extracellular buffer was supplemented to 2 mM final calcium concentration to reveal calcium entry. A. Doxycycline-inducible S100A6 over-expressing PMVECs revealed decreased calcium entry following R+TG, as compared to non-doxycycline-treated cells. B. Comparison of peak calcium entry observed. Peak calcium entry was decreased by 25% in doxycycline-treated cells compared to non-doxycycline-treated cells. n = 3 independent experiments. C. ISOC was measured using patch-clamp electrophysiology. Here, cells were pretreated with rolipram (R) for 15 minutes prior to obtaining a Giga ohm seal and whole-cell patch configuration. Thapsigargin (TG) was applied through the patch pipette. Doxycycline-inducible S100A6 over-expressed PMVECs revealed decreased ISOC following R+TG, as compared to non-doxycycline-treated cells. n = 4 measurements per condition performed over two different recording periods. D. Endothelial barrier integrity was assessed using ECIS. Here, cells were pretreated with rolipram (R) followed by thapsigargin (TG) (black arrow) to activate ISOC and changes in resistance were measured. Doxycycline-inducible S100A6 over-expressing PMVECs revealed decreased barrier disruption following ISOC activation as compared to non-doxycycline-treated cells. Non-doxycycline-treated cells revealed decreased resistance following ISOC activation, but this decrease was attenuated in doxycycline-treated S100A6 over-expressed cells. E. Comparison of peak resistance decrease observed in D. The peak resistance decrease was less in doxycycline-treated cells compared to non-doxycycline-treated cells. n = 3 independent experiments.
To determine whether the S100A6-mediated decrease in calcium entry and ISOC is dependent upon PPP5C, we treated doxycycline-treated S100A6 over-expressing cells with PPP5C siRNA. We again measured global calcium entry following activation of ISOC with rolipram/thapsigargin using Fura2. Here, calcium entry of the scrambled siRNA-treated cells is already decreased due to S100A6 over-expression. We observed that in PPP5C siRNA-treated cells, S100A6 was no longer able to decrease calcium entry as evidenced by increased calcium entry (Figure 5A/B). Similarly, in patch-clamp experiments to measure ISOC, in the presence of scrambled siRNA ISOC was again decreased (~−21 pA at −80 mV), albeit not to the degree observed in the S100A6 over-expressing cells alone (Figure 5C). However, in PPP5C siRNA-treated cells, the S100A6-mediated inhibition of ISOC was reversed as evidenced by increased current (~26 pA at −80 mV). Overall, these data reveal that S100A6 functions through PPP5C to inhibit calcium entry and ISOC. Finally, we again measured calcium entry-induced endothelial barrier disruption using ECIS®. As expected, S100A6 over-expressing cells treated with scrambled siRNA exhibited only a small resistance decrease (~17%) following ISOC activation (Figure 5D/E). However, in PPP5C siRNA-treated cells, following ISOC activation monolayer resistance decreased by ~40%. Together, these results reveal functional roles for S100A6 in PMVECs whereby it regulates calcium entry and ISOC and protects the endothelial barrier from calcium entry-induced disruption. Further, these S100A6 cellular roles are dependent upon PPP5C.
Figure 5. S100A6 regulation of calcium entry and barrier function is dependent upon PPP5C.
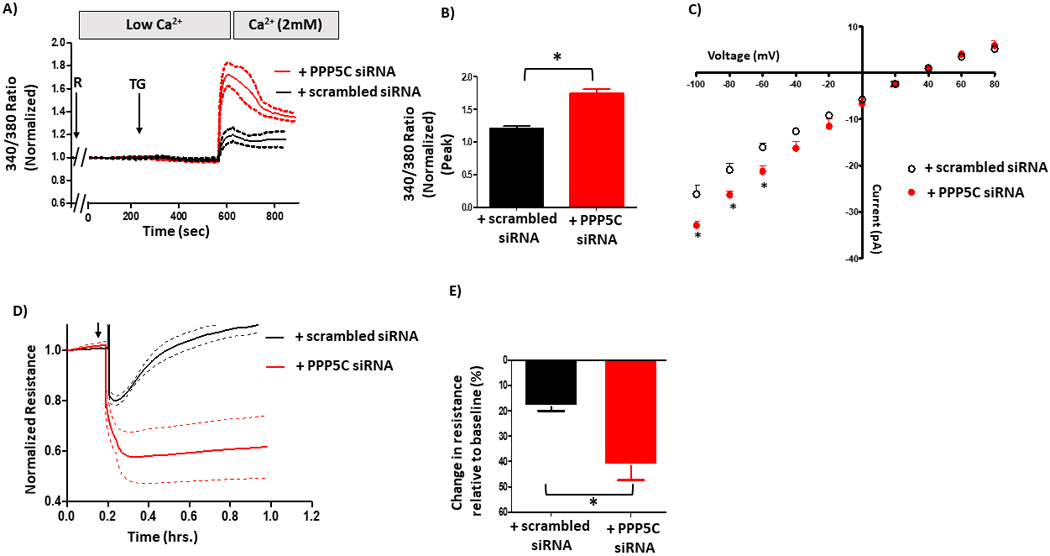
A. Global calcium entry was measured in S100A6 over-expressing cells in which PPP5C expression was suppressed via siRNA. The S100A6-mediated inhibition of calcium entry following rolipram (R) + thapsigargin (TG) was reversed upon PPP5C suppression. B. Comparison of peak calcium entry observed. Peak calcium entry was increased by ~25% in cells treated with PPP5C siRNA compared to cells treated with scrambled siRNA. n = 3 independent experiments. C. ISOC was measured. The decrease in ISOC in S100A6 over-expressing cells was reversed upon PPP5C suppression as evidenced by increased current. n = 4 measurements per condition from 3 different siRNA treatments performed over two different recording periods. D. ECIS® was performed to assess the endothelial barrier. The attenuated decrease in resistance following ISOC activation in S100A6 over-expressing cells was reversed upon PPP5C suppression. E. Comparison of peak resistance decrease observed in D. The peak resistance decreases in S100A6 over-expressing cells treated with PPP5C siRNA was greater than S100A6 over-expressing cells treated with scrambled siRNA. n = 3 independent experiments.
S100A6 is a key factor in the PPP5C-FKBP51-mediated regulation of calcium entry and endothelial barrier function.
To determine the role of S100A6 in the PPP5C-FKBP51 axis, we first began our studies with the doxycycline-inducible FKBP51 over-expressing PMVECs. We again measured global calcium entry using Fura2. In doxycycline-treated cells, calcium entry following activation of ISOC with rolipram/thapsigargin was decreased as compared to non-doxycycline-treated cells (Figure 6A/B). Similarly, patch-clamp experiments revealed decreased ISOC in doxycycline-treated cells as compared to non-doxycycline-treated cells (Figure 6C). Here, we observed a current of ~29 pA at −80 mV which reversed at ~+35 mV in non-doxycycline-treated cells. In doxycycline-treated cells, with increased FKBP51 expression, ISOC was decreased by ~52% (~14 pA at −80 mV) with no change in reversal potential. Indeed, this result is consistent with our previous observations in rat PAECs (4, 5). Finally, using doxycycline-inducible FKBP51 over-expressing cells, we wanted to determine whether upregulation of FKBP51 in PMVECs protects the endothelial barrier, as it does in PAECs (4, 5). We observed that monolayer resistance of doxycycline-treated FKBP51 over-expressing PMVECs only decreased by ~5% following ISOC activation as compared to a decrease of ~30% in non-doxycycline-treated cells (Figure 6D/E). Overall, we observed that increased FKBP51 expression decreases calcium entry, ISOC and calcium entry-induced endothelial barrier disruption in PMVECs.
Figure 6. FKBP51 regulates calcium entry and barrier function in PMVECs.
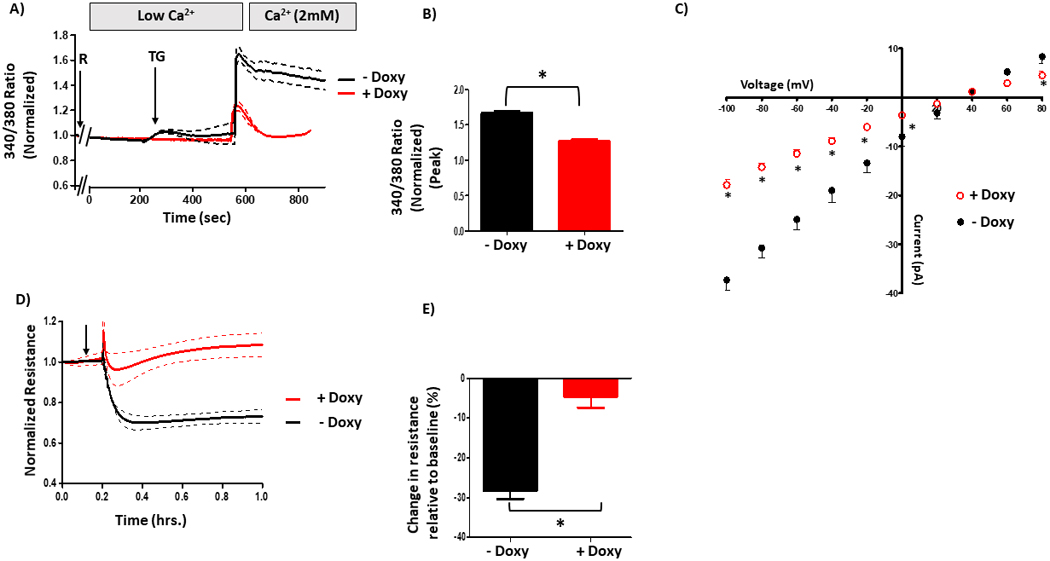
A. Global calcium entry was measured in FKBP51 over-expressing PMVECs. Doxycycline-inducible FKBP51 over-expressing PMVECs revealed decreased calcium entry following rolipram (R) + thapsigargin (TG), as compared to non-doxycycline-treated cells. B. Comparison of peak calcium entry observed. Peak calcium entry was decreased by 30% in doxycycline-treated cells compared to non-doxycycline-treated cells. n = 3 independent experiments. C. ISOC was measured. Doxycycline-inducible FKBP51 over-expressing PMVECs revealed decreased ISOC following R+TG, as compared to non-doxycycline-treated cells. n = 4 measurements per condition performed over two different recording periods. D. ECIS® was performed to assess the endothelial barrier. Doxycycline-inducible FKBP51 over-expressing PMVECs revealed decreased barrier disruption following ISOC activation as compared to non-doxycycline-treated cells. E. Comparison of peak resistance decrease observed in D. The peak resistance decrease was less in doxycycline-treated cells compared to non-doxycycline-treated cells. n = 3 independent experiments.
To determine whether S100A6 is important for the PPP5C-FKBP51-mediated inhibition of calcium entry, ISOC and calcium entry-induced endothelial barrier disruption in PMVECs, we treated doxycycline-treated FKBP51 over-expressing cells with S100A6 siRNA. In global calcium measurements, calcium entry following ISOC activation with rolipram/thapsigargin was increased in cells treated with S100A6 siRNA as compared to scrambled siRNA (Figure 7A/B). As cells treated with scrambled siRNA exhibit a decreased calcium entry due to FKBP51 over-expression, this result reveals that S100A6 suppression prevents the FKBP51-mediated decrease in calcium entry. We next measured the effect of S100A6 suppression on ISOC using patch-clamp. As expected, FKBP51 over-expressing cells treated with scrambled siRNA exhibited decreased ISOC (~16 pA at −80 mV), whereas in cells treated with S100A6 siRNA the FKBP51-mediated decrease in ISOC was partially reversed (~21 pA at −80 mV) (Figure 7C). Since we did not observe complete reversal of ISOC to a typical ISOC (~29 pA at −80 mV), it is possible that because we did not completely suppress S100A6 expression (~75% in these experiments), that the small amount of S100A6 expressed was enough to influence ISOC function. Nonetheless, these data indicate that S100A6 participates in the PPP5C-FKBP51-mediated inhibition of ISOC. Finally, we next determined whether the FKBP51-mediated protective effect on the endothelial barrier is dependent upon S100A6. In doxycycline-treated FKBP51 over-expressing cells, upon S100A6 knock-down monolayer resistance decreased by ~45 % following ISOC activation (Figure 7D/E). Conversely, in FKBP51 over-expressing cells treated with scrambled siRNA, following ISOC activation there was less decrease ~19%in monolayer resistance. Together, these data demonstrate that S100A6 is a critical determinant of the ability of the PPP5C-FKBP51 axis to inhibit ISOC and protect the endothelial barrier from calcium entry-induced disruption.
Figure 7. S100A6 contributes to the PPP5C-FKBP51-mediated regulation of calcium entry and endothelial barrier function.
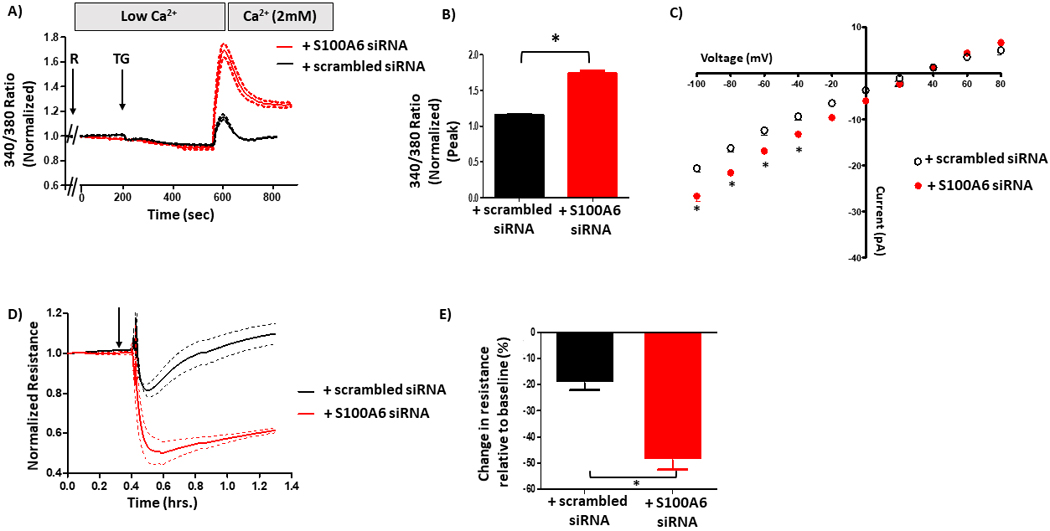
A. Global calcium entry was measured in FKBP51 over-expressing PMVECs in which S100A6 expression was suppressed. The decrease in calcium entry following rolipram (R) + thapsigargin (TG) in FKBP51 over-expressing cells was reversed upon S100A6 suppression. B. Comparison of peak calcium entry observed. Peak calcium entry was increased by 34% in cells treated with S100A6 siRNA as compared to cells treated with scrambled siRNA. n = 3 independent experiments. C. ISOC was measured. The decrease in ISOC in FKBP51 over-expressing cells treated with scrambled siRNA was reversed in cells treated with S100A6 siRNA as evidenced by increased current. n = 4 measurements per condition from 3 different siRNA treatments performed over two different recording periods. D. ECIS® was performed to assess the endothelial barrier. The attenuated decrease in resistance following R + TG in FKBP51 over-expressing cells was reversed upon S100A6 suppression. E. Comparison of peak resistance decrease observed in D. The peak resistance decrease was greater in FKBP51 over-expressing cells treated with S100A6 siRNA compared to cells treated with scrambled siRNA. n = 3 independent experiments.
Discussion
In this study we show for the first time that the small, calcium-binding protein S100A6 plays a role in the PPP5C-FKBP51 axis which regulates the endothelial ISOC and downstream effects on barrier integrity. To determine the role of S100A6 in the PPP5C-FKBP51 axis we used a combination of FKBP51 upregulation in conjunction with S100A6 suppression, as well as S100A6 upregulation in conjunction with PPP5C suppression. For protein upregulation, we specifically used systems in which the upregulation was modest (~2–3-fold for both FKBP51 and S100A6), which we feel is important to limit potential off-target effects often seen in over-expression systems. Using these systems, we made three key findings. We first observed that upregulation of the PPP5C-FKBP51 axis inhibits ISOC in PMVECs. As this observation supports our earlier data in PAECs (4–6), it suggests that inhibition of ISOC by the PPP5C-FKBP51 axis is a fundamental mechanism in endothelial cells. Second, we found that S100A6 is required for the PPP5C-FKBP51-mediated inhibition of ISOC, working through the PPP5C arm of the axis. Third, we revealed for the first time a functional link between S100A6 and PPP5C in cells. We recognize however that as our study only included in vitro analyses that an important next step is to confirm our results in in vivo models.
Our approach to study the role of S100A6 in regulating PPP5C activity in cells was to measure the phosphorylation status of endogenous tau, and we chose the phosphorylation site T231. As mentioned earlier, the phosphorylation status of T231 is an important determinant of microtubule stability (41), and we have previously shown that the microtubule network is essential for ISOC inhibition by the PPP5C-FKBP51 axis (5). Indeed, both PPP5C and FKBP51 have been implicated in dephosphorylation of tau at T231. Liu et al (39) performed in vitro studies in which they first phosphorylated recombinant tau441 with cAMP-dependent protein kinase and cyclin-dependent kinase 5/p25. They then went on to show that PPP5C dephosphorylates tau at multiple sites including T231. With regards to FKBP51, Jinwal et al (42) showed that in HeLa cells over-expressing human tau, transfection of isomerase-mutant FKBP51 increased phosphorylation at several sites on tau including T231. In this same study, the authors also showed that FKBP51 promotes tau-mediated microtubule polymerization, and they proposed that phosphorylated tau in the trans configuration is isomerized by FKBP51 to the cis configuration which allows for dephosphorylation and recycling back onto microtubules to promote polymerization. Taking the Jinwal model together with our observations, we now propose a model describing the mechanism by which we think the PPP5C-FKBP51 axis inhibits ISOC (Figure 8). To begin, ISOC activation leads to increased cytosolic calcium levels which are sufficient to induce translocation of S100A6 to the plasma membrane and promote interaction with the ISOC channel heterocomplex, which includes both PPP5C and FKBP51 (4, 6). In this channel compartment, S100A6 activates PPP5C. PPP5C and FKBP51 then act on tau where FKBP51 isomerizes tau and primes it for dephosphorylation by PPP5C. In a dephosphorylated state, tau binds microtubules and promotes microtubule polymerization which inhibits channel function. Overall, this proposed mechanism represents a calcium-dependent feedback inhibition of the ISOC channel that is initiated upon calcium binding to S100A6. Indeed, this is a new physiological function of S100A6.
Figure 8. Model of the mechanism by which S100A6 and the PPP5C-FKBP51 axis inhibit ISOC.

Upon activation of ISOC, calcium enters the cell and binds to S100A6 to induce translocation of S100A6 to the plasma membrane to interact with the ISOC channel heterocomplex. Within the ISOC heterocomplex reside PPP5C and FKBP51. FKBP51 isomerizes tau and primes it for dephosphorylation by PPP5C. Dephosphorylated tau is then able to bind to microtubules to promote polymerization which is responsible for inhibition of channel function.
In endothelial cells S100A6 has been shown to regulate cell cycle progression and senescence (44, 45). Different S100 family members, e.g., S100B, S100A1 and S100A10, have been implicated in regulation of ion channel function (46). To date, the only evidence for S100A6 playing a role in ion channel function comes from a study by Courtois-Coutry et al (47) in which they observed that introduction of S100A6 antibody into the rat cortical collecting duct cell line, RCCD1, resulted in prevention of long-term increase in arginine vasopressin-induced short-circuit current. At the conclusion of this study, the authors speculated that S100A6 could be playing a role in ion transport by regulating gene transcription and/or transport of ion channel proteins to the plasma membrane. In support of the latter idea, S100A10 has been implicated in trafficking the vanilloid transient receptor channel proteins TRPV5 and TRPV6 as well as the acid-sensing ion channel ASIC1a and the 2P domain potassium channel TASK-1 to the plasma membrane (46, 48–50). While our study did not specifically address the trafficking idea, as our data showed that S100A6 is inhibitory to ISOC we would not expect that S100A6 traffics the ISOC channel itself to the plasma membrane. However, a yet unexplored idea is that S100A6 may be involved in trafficking PPP5C to the plasma membrane. Indeed, there is precedent for translocation of PPP5C to the plasma membrane as the active forms of both Gα12 (51) and Rac1 GTPase have been shown to translocate PPP5C to the plasma membrane (51, 52). With respect to S100A6, it exhibits calcium-dependent translocation and plasma membrane localization (17, 18, 53). While our earlier studies revealed basal interaction between PPP5C and the ISOC channel (4), we have not examined whether this interaction increases in a calcium-dependent manner. Overall, future studies are warranted to explore whether, in addition to activating PPP5C, S100A6 actively transports PPP5C to the ISOC compartment at the plasma membrane in endothelial cells.
In the current study we show that S100A6 plays a role in the inhibition of ISOC by the PPP5C-FKBP51 axis. Further, the ISOC calcium-dependent interaction of S100A6 with the ISOC channel implies that a physiological function of the PPP5C-FKBP51-mediated inhibition of ISOC is calcium-dependent feedback, limiting how much calcium enters the cell. As we observed that it was calcium that came through the ISOC channel that was important for interaction of S100A6 with the ISOC channel, a question arises as to whether this is the only function of ISOC calcium – S100A6 binding. As the ISOC channel is likely localized to caveolae in the endothelial plasma membrane (54), it is interesting to speculate that S100A6 may be compartmentalized in the cytosol and the S100A6 compartment that is near or part of caveolae is what binds ISOC calcium. If that is the case, perhaps other caveolae-bound targets of S100A6 are also activated by ISOC calcium – S100A6 binding, an idea that will be explored in future studies. In conclusion, our findings from the current study reveal a novel role for S100A6 in endothelial cell calcium signaling and barrier function.
Supplementary Material
Acknowledgements
The authors would like to thank Linn Ayers and Anna Buford for endothelial cell isolation and culture. Also acknowledged is grant P01 HL66299 for RNASeq data. The authors declare no conflicts of interest.
Funding Sources: This work was supported by the National Institutes of Health (R01HL107778 to DLC; R56HL107778 to DLC) and the University of South Alabama, College of Medicine Intramural Grant Programs (000927 to DLC; 000540 to BH).
Abbreviations:
- (FKBP51)
FK506-binding protein 51
- (PPP5C)
Serine/Threonine protein phosphatase 5C
- (SOC)
Store-operated calcium
- (ISOC)
Calcium current through ISOC channel
- (ECIS®)
Electric cell-substrate impedance sensing
- (HLMVECs)
Human lung microvascular endothelial cells
- (PAECs)
Rat pulmonary artery endothelial cells
- (PMVECs)
Rat pulmonary microvascular endothelial cells
- (R)
Rolipram
- (TG)
Thapsigargin
- (TRPC)
Transient receptor potential canonical
References
- 1.Brough GH, Wu S, Cioffi D, Moore TM, Li M, Dean N, and Stevens T. (2001) Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway, FASEB J 15, 1727–1738. [PubMed] [Google Scholar]
- 2.Cioffi DL, Wu S, Chen H, Alexeyev M, St Croix CM, Pitt BR, Uhlig S, and Stevens T. (2012) Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel, Circ Res 110, 1435–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cioffi DL, Wu S, Alexeyev M, Goodman SR, Zhu MX, and Stevens T. (2005) Activation of the endothelial store-operated ISOC Ca2+ channel requires interaction of protein 4.1 with TRPC4, Circ Res 97, 1164–1172. [DOI] [PubMed] [Google Scholar]
- 4.Hamilton CL, Abney KA, Vasauskas AA, Alexeyev M, Li N, Honkanen RE, Scammell JG, and Cioffi DL (2018) Serine/threonine phosphatase 5 (PP5C/PPP5C) regulates the ISOC channel through a PP5C-FKBP51 axis, Pulm Circ 8, 2045893217753156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamilton CL, Kadeba PI, Vasauskas AA, Solodushko V, McClinton AK, Alexeyev M, Scammell JG, and Cioffi DL (2018) Protective role of FKBP51 in calcium entry-induced endothelial barrier disruption, Pulm Circ 8, 2045893217749987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadeba PI, Vasauskas AA, Chen H, Wu S, Scammell JG, and Cioffi DL (2013) Regulation of store-operated calcium entry by FK506-binding immunophilins, Cell Calcium 53, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu S, Chen H, Alexeyev MF, King JA, Moore TM, Stevens T, and Balczon RD (2007) Microtubule motors regulate ISOC activation necessary to increase endothelial cell permeability, J Biol Chem 282, 34801–34808. [DOI] [PubMed] [Google Scholar]
- 8.Wu S, Cioffi EA, Alvarez D, Sayner SL, Chen H, Cioffi DL, King J, Creighton JR, Townsley M, Goodman SR, and Stevens T. (2005) Essential role of a Ca2+-selective, store-operated current (ISOC) in endothelial cell permeability: determinants of the vascular leak site, Circ Res 96, 856–863. [DOI] [PubMed] [Google Scholar]
- 9.Wu S, Sangerman J, Li M, Brough GH, Goodman SR, and Stevens T. (2001) Essential control of an endothelial cell ISOC by the spectrin membrane skeleton, J Cell Biol 154, 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore TM, Brough GH, Babal P, Kelly JJ, Li M, and Stevens T. (1998) Store-operated calcium entry promotes shape change in pulmonary endothelial cells expressing Trp1, Am J Physiol Lung Cell Mol Physiol 275, L574–582. [DOI] [PubMed] [Google Scholar]
- 11.Moore TM, Norwood NR, Creighton JR, Babal P, Brough GH, Shasby DM, and Stevens T. (2000) Receptor-dependent activation of store-operated calcium entry increases endothelial cell permeability, Am J Physiol Lung Cell Mol Physiol 279, L691–698. [DOI] [PubMed] [Google Scholar]
- 12.Clapham DE, Runnels LW, and Strübing C. (2001) The TRP ion channel family, Nat Rev Neurosci 2, 387–396. [DOI] [PubMed] [Google Scholar]
- 13.Sinclair C, Borchers C, Parker C, Tomer K, Charbonneau H, and Rossie S. (1999) The tetratricopeptide repeat domain and a C-terminal region control the activity of Ser/Thr protein phosphatase 5, J Biol Chem 274, 23666–23672. [DOI] [PubMed] [Google Scholar]
- 14.Yamaguchi F, Umeda Y, Shimamoto S, Tsuchiya M, Tokumitsu H, Tokuda M, and Kobayashi R. (2012) S100 proteins modulate protein phosphatase 5 function: a link between Ca2+ signal transduction and protein dephosphorylation, J Biol Chem 287, 13787–13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, and Geczy CL (2013) Functions of S100 proteins, Current Molec Med 13, 24–57. [PMC free article] [PubMed] [Google Scholar]
- 16.Xia C, Braunstein Z, Toomey AC, Zhong J, and Rao X. (2018) S100 proteins as an important regulator of macrophage inflammation, Front Immunol 8, 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller A, Bachi T, Hochli M, Schafer BW, and Heizmann CW (1999) Subcellular distribution of S100 proteins in tumor cells and their relocation in response to calcium activation, Histochem Cell Biol 111, 453–459. [DOI] [PubMed] [Google Scholar]
- 18.Stradal TB, and Gimona M. (1999) Ca(2+)-dependent association of S100A6 (Calcyclin) with the plasma membrane and the nuclear envelope, J Biol Chem 274, 31593–31596. [DOI] [PubMed] [Google Scholar]
- 19.Reynolds PD, Ruan Y, Smith DF, and Scammell JG (1999) Glucocorticoid resistance in the squirrel monkey is associated with overexpression of the immunophilin FKBP51, The JClin Endocrinol Metab 84, 663–669. [DOI] [PubMed] [Google Scholar]
- 20.Zuo Z, Dean NM, and Honkanen RE (1998) Serine/threonine protein phosphatase type 5 acts upstream of p53 to regulate the induction of p21(WAF1/Cip1) and mediate growth arrest, J Biol Chem 273, 12250–12258. [DOI] [PubMed] [Google Scholar]
- 21.Creighton JR, Masada N, Cooper DM, and Stevens T. (2003) Coordinate regulation of membrane cAMP by Ca2+-inhibited adenylyl cyclase and phosphodiesterase activities, Am J Physiol Lung Cell Mol Physiol 284, L100–107. [DOI] [PubMed] [Google Scholar]
- 22.Krump-Konvalinkova V, Bittinger F, Unger RE, Peters K, Lehr HA, and Kirkpatrick CJ (2001) Generation of human pulmonary microvascular endothelial cell lines, Lab Invest 81, 1717–1727. [DOI] [PubMed] [Google Scholar]
- 23.Skarra DV, Goudreault M, Choi H, Mullin M, Nesvizhskii AI, Gingras AC, and Honkanen RE (2011) Label-free quantitative proteomics and SAINT analysis enable interactome mapping for the human Ser/Thr protein phosphatase 5, Proteomics 11, 1508–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrow KA, Ochoa CD, Balczon R, Zhou C, Cauthen L, Alexeyev M, Schmalzer KM, Frank DW, and Stevens T. (2016) Pseudomonas aeruginosa exoenzymes U and Y induce a transmissible endothelial proteinopathy, Am J Physiol Lung Cell Mol Physiol 310, L337–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ochoa CD, Alexeyev M, Pastukh V, Balczon R, and Stevens T. (2012) Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability, J Biol Chem 287, 25407–25418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexeyev MF, Fayzulin R, Shokolenko IN, and Pastukh V (2010) A retro-lentiviral system for doxycycline-inducible gene expression and gene knockdown in cells with limited proliferative capacity, Molec Biol Rep 37, 1987–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pear WS, Nolan GP, Scott ML, and Baltimore D. (1993) Production of high-titer helper-free retroviruses by transient transfection, Proc Natl Acad Sci U S A 90, 8392–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zufferey R, Nagy D, Mandel RJ, Naldini L, and Trono D. (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo, Nat Biotechnol 15, 871–875. [DOI] [PubMed] [Google Scholar]
- 29.Cioffi DL, Moore TM, Schaack J, Creighton JR, Cooper DM, and Stevens T. (2002) Dominant regulation of interendothelial cell gap formation by calcium-inhibited type 6 adenylyl cyclase, J Cell Biol 157, 1267–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cioffi DL, Pandey S, Alvarez DF, and Cioffi EA (2012) Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity, Am J Physiol Lung Cell Mol Physiol 302, L1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimamoto S, Kubota Y, Tokumitsu H, and Kobayashi R. (2010) S100 proteins regulate the interaction of Hsp90 with Cyclophilin 40 and FKBP52 through their tetratricopeptide repeats, FEBS Lett 584, 1119–1125. [DOI] [PubMed] [Google Scholar]
- 32.Shimamoto S, Takata M, Tokuda M, Oohira F, Tokumitsu H, and Kobayashi R. (2008) Interactions of S100A2 and S100A6 with the tetratricopeptide repeat proteins, Hsp90/Hsp70-organizing protein and kinesin light chain, J Biol Chem 283, 28246–28258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cortajarena AL, and Regan L. (2006) Ligand binding by TPR domains, Protein Sci 15, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, and Clardy J. (2003) Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes, Proc Natl Acad Sci U S A 100, 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B, and Rao Z. (2004) 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex, Proc Natl Acad Sci U S A 101, 8348–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chazotte B. (2011) Labeling membrane glycoproteins or glycolipids with fluorescent wheat germ agglutinin, Cold Spring Harbor Protocols; 2011, pdb prot5623. [DOI] [PubMed] [Google Scholar]
- 37.Gong CX, Liu F, Wu G, Rossie S, Wegiel J, Li L, Grundke-Iqbal I, and Iqbal K. (2004) Dephosphorylation of microtubule-associated protein tau by protein phosphatase 5, J Neurochem 88, 298–310. [DOI] [PubMed] [Google Scholar]
- 38.Liu F, Grundke-Iqbal I, Iqbal K, and Gong CX (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation, EurJ Neurosci 22, 1942–1950. [DOI] [PubMed] [Google Scholar]
- 39.Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, and Gong CX (2005) Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer’s disease, J Biol Chem 280, 1790–1796. [DOI] [PubMed] [Google Scholar]
- 40.Mukrasch MD, von Bergen M, Biernat J, Fischer D, Griesinger C, Mandelkow E, and Zweckstetter M. (2007) The “jaws” of the tau-microtubule interaction, J Biol Chem 282, 12230–12239. [DOI] [PubMed] [Google Scholar]
- 41.Cho JH, and Johnson GV (2004) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau’s ability to bind and stabilize microtubules, J Neurochem 88, 349–358. [DOI] [PubMed] [Google Scholar]
- 42.Jinwal UK, Koren J 3rd, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, Johnson AG, Jones JR, Shults CL, O’Leary JC 3rd, Jin Y, Buchner J, Cox MB, and Dickey CA (2010) The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules, J Neurosci 30, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bischoff I, Hornburger MC, Mayer BA, Beyerle A, Wegener J, and Furst R. (2016) Pitfalls in assessing microvascular endothelial barrier function: impedance-based devices versus the classic macromolecular tracer assay, Sci Rep 6, 23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bao L, Odell AF, Stephen SL, Wheatcroft SB, Walker JH, and Ponnambalam S. (2012) The S100A6 calcium-binding protein regulates endothelial cell-cycle progression and senescence, FEBS J279, 4576–4588. [DOI] [PubMed] [Google Scholar]
- 45.Lerchenmuller C, Heissenberg J, Damilano F, Bezzeridis VJ, Kramer I, Bochaton-Piallat ML, Hirschberg K, Busch M, Katus HA, Peppel K, Rosenzweig A, Busch H, Boerries M, and Most P. (2016) S100A6 regulates endothelial cell cycle progression by attenuating antiproliferative signal transducers and activators of transcription 1 signaling, Arterioscler Thromb Vasc Biol 36, 1854–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hermann A, Donato R, Weiger TM, and Chazin WJ (2012) S100 calcium binding proteins and ion channels, Front Pharmacol 3, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Courtois-Coutry N, Le Moellic C, Boulkroun S, Fay M, Cluzeaud F, Escoubet B, Farman N, and Blot-Chabaud M. (2002) Calcyclin is an early vasopressin-induced gene in the renal collecting duct. Role in the long term regulation of ion transport, J Biol Chem 277, 25728–25734. [DOI] [PubMed] [Google Scholar]
- 48.Donier E, Rugiero F, Okuse K, and Wood JN (2005) Annexin II light chain p11 promotes functional expression of acid-sensing ion channel ASIC1a, J Biol Chem 280, 38666–38672. [DOI] [PubMed] [Google Scholar]
- 49.Girard C, Tinel N, Terrenoire C, Romey G, Lazdunski M, and Borsotto M. (2002) p11, an annexin II subunit, an auxiliary protein associated with the background K+ channel, TASK-1, EMBO J 21, 4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van de Graaf SF, Hoenderop JG, Gkika D, Lamers D, Prenen J, Rescher U, Gerke V, Staub O, Nilius B, and Bindels RJ (2003) Functional expression of the epithelial Ca(2+) channels (TRPV5 and TRPV6) requires association of the S100A10-annexin 2 complex, EMBO J 22, 1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamaguchi Y, Katoh H, Mori K, and Negishi M. (2002) Galpha(12) and Galpha(13) interact with Ser/Thr protein phosphatase type 5 and stimulate its phosphatase activity, Curr Biol 12, 1353–1358. [DOI] [PubMed] [Google Scholar]
- 52.Chatterjee A, Wang L, Armstrong DL, and Rossie S. (2010) Activated Rac1 GTPase translocates protein phosphatase 5 to the cell membrane and stimulates phosphatase activity in vitro, J Biol Chem 285, 3872–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mandinova A, Atar D, Schafer BW, Spiess M, Aebi U, and Heizmann CW (1998) Distinct subcellular localization of calcium binding S100 proteins in human smooth muscle cells and their relocation in response to rises in intracellular calcium, J Cell Sci 111 ( Pt 14), 2043–2054. [DOI] [PubMed] [Google Scholar]
- 54.Lockwich TP, Xibao L, Singh BB, Jadlowiec J, Weiland S, and Ambudkar IS (2000) Assembly of Trp1 in a signaling complex with caveolin-scaffolding lipid raft domains, J Biol Chem 275, 11934–11942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.