

Abstract
Given our previous finding that fluorination at the C18 position of largazole showed reasonably good tolerance towards inhibitory activity and selectivity of histone deacetylases (HDACs), further modification on the valine residue in the fluoro-largazole’s macrocyclic moiety with S-Me l-Cysteine or Glycine residue was performed. While the Glycine-modified fluoro analog showed poor activity, the S-Me l-Cysteine-modified analog emerged to be a very potent HDAC inhibitor. Unlike all previously reported C2-modified compounds in the largazole family (including our recent fluoro-largazole analogs) where replacement of the Val residue has failed to provide any potency improvement, the S-Me l-Cysteine-modified analog displayed significantly enhanced (five–nine-fold) inhibition of all the tested HDACs while maintaining the selectivity of HDAC1 over HDAC6, as compared to largazole thiol. A molecular modeling study provided rational explanation and structural evidence for the enhanced inhibitory activity. This new finding will aid the design of novel potent HDAC inhibitors.
Keywords: marine natural product, largazole, HDAC inhibitors, modification, fluoro olefin
1. Introduction
Both histone acetyltransferases (HATs) and histone deacetylases (HDACs) play a key role in the regulation of histone-tailed lysine acetylation status, which is closely associated with cell processes [1]. Up to now, 18 HDAC isoforms have been identified and divided into four classes, including the Zn2+-dependent Class I (HDAC1, 2, 3, 8), Class II (HDAC4–7, 9–10), Class IV (HDAC11), and NAD+- dependent Class III (namely SIRT1-7) [2]. Although the function of individual HDAC isoform is not fully understood in cells, HDACs have become an appealing target in cancer therapy through balancing histone hypoacetylation and overexpression of HDACs in multiple cancers. Currently, several Zn2+-dependent HDAC inhibitors, such as SAHA (Vorinostat), Belinostat, Panobinostat, Chidamide and Romidepsin (FK228), have been approved for the treatment of cutaneous T-cell lymphoma. However, these inhibitors are pan-selective inhibitors [3,4]. The current trend is to explore potent and selective HDAC inhibitors [5,6,7].
Largazole, as a potent and selective Class I HDAC inhibitor, was discovered in 2008 by Leusch et al. [8,9]. Due to its superior anticancer properties, this marine natural product has attracted widespread attention in the medicinal chemistry community [10,11,12,13,14,15]. In order to improve the inhibitory activity and selectivity of HDACs, great efforts have been made to modify the macrocyclic moiety, the hydrophobic linker and the warhead of largazole. Thus far, almost all the modifications on the linker and the warhead cause obvious losses in activity against HDACs, possibly due to preventing the thiol group to efficiently penetrate into the narrow channel containing the Zn2+ ion of HDACs and to coordinate it with the optimal geometry. In contrast, the modifications on the macrocyclic moiety seem to be allowable to some extent. Among them, certain variations in valine residue (Val) at the C2 position of largazole may maintain the HDACs inhibition despite with a little sacrifice in its potency or isoenzyme selectivity (Figure 1). For example, Hong and Leusch’s group reported that replacement of Val with Ala decreased the activity by three-fold in growth inhibition of HCT-116 [16]. Subsequently, they introduced aromatic (Phe, Tyr), acidic (Asp) or basic (His) amino acid residues at that position, but all the analogs exhibited reduced activity in HDAC1 [17]. Based on the hypothesis of potentially hydrophobic interactions between Val and the surrounding residues of HDAC1 (Tyr196 and Leu263), Jiang et al. replaced Val with Leu, Phe and Tyr, reporting that Tyr had about five-fold higher GI50 values of HCT116 and A549 cells compared to largazole despite with an increasing selectivity over normal cells [18]. Ganesan’s and Williams’ groups reported that replacement with Gly or Pro resulted in a significantly decreased inhibition against HDACs, respectively [19,20]. Recently, Hong’s group investigated again the effect of the replacement of Val with Phe, Tyr, Asp or His on both the activity and selectivity of the class I HDACs, and the observed His substitution showed comparable activity, and a slight selectivity towards HDAC1 over HDAC2–3. This slight selectivity was hypothesized to be resulted from a possible hydrogen-bond ability of His. However, when they replaced Val with a set of residues tagged with the terminal amine or amides, the results suggested that the hydrogen bonding interaction did not play an essential role in HDAC inhibition [21]. All these works indicated that the structural optimization at the C2 position appeared not to improve the potency of HDACs inhibition.
Figure 1.

Largazole and its analogs modified at C-2 position.
Recently, we discovered that the modification on the linker of largazole was allowable in some cases (Figure 1) [22,23,24]. For example, fixing fluorine at C18 position of the linker could keep almost the same inhibitory activity and selectivity of HDACs. Subsequently, the replacement of Val with Phe gave slightly reduced inhibition of HDACs, similar to the previous observations reported for largazole [23]. As our research continued, we discovered to our surprise that the replacement of Val with S-Me Cys significantly enhanced the inhibition of HDACs, whereas the replacement with Gly significantly decreased the activity. With an expanded research program under way based on this observation, we would like to communicate here our preliminary results and provide rational explanation for the unexpected results through a molecular modeling study. To the best of our knowledge, the S-Me Cys modification is the first reported example where replacement of the Val residue has led to significant enhancement in HDACs inhibition. We believe this insight may enable the design of highly potent HDAC inhibitors.
2. Results and Discussion
2.1. Chemistry
In our previous paper [23], we synthesized the two fluoroolefin analogs of largazole with Val and Phe residue at C2 position. The key macrolactamization was chosen at N2 position, but the yields were poor (less than 31% yield). As an alternative, the key macrolactamization for the two new fluoroolefin analogs in this study was arranged at N14 position with an intend to improve the yield of the key step. The evolved synthetic route is depicted in Scheme 1.
Scheme 1.

Synthesis of the analogs of Largazole. The red R1 is the different substituent and the green number is the atom code in the molecule.
The key fluoro fragment (8) was prepared from acrolein via four steps according to our previous method [23]. Alcoholysis of 8 with TMSCH2CH2OH in the presence of DMAP provided 9 in good yield (67%). Condensation of 9 with S-Me Fmoc-l-Cystine or Fmoc-Glycine in the presence of EDCI, DMAP and DIEPA afforded ester 10c and 10d (yield: 75% for 10c). Removal of Fmoc group of 10c or 10d with Et2NH in CH2Cl2 and subsequent condensation with the acid (11) resulted in the linear precursors (12c, 84%; 12d, 41% based on 9). Deprotection of Boc group of 10c or 10d with CF3CO2H in CH2Cl2 and subsequent release of free acid provided the linear depsipeptides (13c and 13d) exposed at the N- and C-terminus, respectively. The depsipeptides were then subjected to the optimal cyclization condition (HATU/HOAT/DIPEA in anhydrous CH2Cl2 solution with a diluted concentration of about 0.001 M). The macrolactamization yields were 48% for 14c and 40% for 14d in two steps, respectively. Compared to our previous cyclization at N2 position, cyclizing at N14 position afforded apparently improved yield. Following our previously reported procedure [24], the free thiol 15c and 15d were obtained through deprotection of Trt group from 14c and 14d, respectively. The subsequent acylation with n-C7H15COCl under the standard condition led to the final fluoro analogs 16c and 16d (yields: 57%, 40%), respectively.
2.2. Biology
It is well-known that largazole is a prodrug species for a beneficial cell permeability and its free thiol is indeed the activated species for the inhibition against HDACs [10]. Given the instability of the free thiol for storage, the in vitro cell assays were used as the initial screening by our well-established cell tests (A549, HCT116, MDA-MB-231 and SK-OV-3 cells), aiming at speedy identification of potent HDAC inhibitors. The preliminary results were shown in Table 1, using largazole as the positive control compound.
Table 1.
The preliminary in vitro growth inhibition of several cells (IC50, µM) with Largazole and its analog 16c–d.
| IC50 a (µM) | ||||
|---|---|---|---|---|
| A549 | HCT116 | MDA-MB-231 | SK-OV-3 | |
| largazole | 0.52 ± 0.17 | 0.11 ± 0.01 | 4.75 ± 1.95 | 0.25 ± 0.17 |
| 16c | 2.92 ± 0.53 | 1.41 ± 0.54 | >10 | 3.13 ± 0.50. |
| 16d | >10 | 6.62 ± 1.86 | >10 | >10 |
a Compounds were tested in at least seven-dose IC50 mode in duplicate with three-fold serial dilution.
Compared with largazole, 16c displayed the obvious growth inhibition towards A549, HCT116 and SK-OV-3 cells with IC50 values ranged with 1.41–3.13 μM, but not towards MDA-MB-231 (IC50: >10µM). In contrast, 16d almost lost the growth inhibition towards most of those cells, and only maintained a slight growth inhibition towards HCT116 (IC50: 6.62 µM), indicating that 16d was a less potent HDAC inhibitor, which was consistent with previous reports [19,20], where both Ganesan’s and Williams’ groups demonstrated that the replacement of Val with Gly in the case of largazole resulted in a significantly decrease of inhibition against HDACs.
To further confirm the growth inhibition of 16c in cellular assays, we tested 16c and largazole with other cells. As shown in Table 2, 16c displayed growth inhibition against Hela, Eca-109, Bel 7402 and U937 cells with high potencies that are quite identical to largazole.
Table 2.
The further in vitro growth inhibition of other cells (IC50, µM) with Largazole and 16c–d.
| IC50 a (µM) | ||||
|---|---|---|---|---|
| Hela | Eca-109 | Bel 7402 | U937 | |
| largazole | 0.17 ± 0.01 | 0.10 ± 0.00 | 0.17 ± 0.02 | 0.02 ± 0.00 |
| 16c | 0.10 ± 0.01 | 0.09 ± 0.02 | 0.25 ± 0.07 | 0.08 ± 0.02 |
a Compounds were tested in at least seven-dose IC50 mode in duplicate with three-fold serial dilution.
Based on the obvious growth inhibition of 16c in in vitro cell assays, we next performed in vitro enzyme assays with the correspond active species 15c, and the data were compared with those of our previous compounds. The results are shown in Table 3.
Table 3.
In vitro inhibition of human histone deacetylases (HDACs) with the free thiols of largazole and its two analogs.
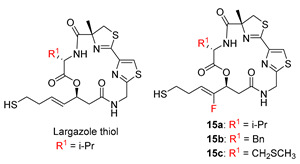
| IC50 (nM) a | Selectivity (HDAC1/6) |
|||||
|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC6 | ||
| 15a b | 4.39 ± 0.17 | 21.0 ± 0.3 | 39.2 ± 2.3 | 8.38 ± 2.46 | 300 ± 5 | 68 |
| 15b b | 4.24 ± 0.38 | 16.3 ± 0.4 | 37.1 ± 0.8 | 17.5 ± 4.2 | 338 ± 17 | 80 |
| 15c | 0.27 ± 0.02 | 1.33 ± 0.02 | 2.33 ± 0.15 | 0.44 ± 0.16 | 23.9 ± 0.2 | 89 |
| largazole thiol | 2.01 ± 0.01 | 9.49 ± 0.19 | 14.4 ± 0.2 | 3.75 ± 0.20 | 121 ± 5 | 60 |
a Compounds were tested in 10-dose IC50 mode in duplicate with three-fold serial dilution. b The data were previously reported by us in [23].
In sharp contrast to the cellular assays where 16c exhibited identical or slightly inferior activities as compared to largazole, in the enzymatic assays, compound 15c surprisingly displayed significantly increased inhibition towards HDACs, with the IC50 values of HDAC 1, 2, 3, 8 and 6 being 0.27, 1.33, 2.33, 0.44 and 23.91 nM, respectively. It was five–nine-fold more potent towards all the tested HDACs when compared with largazole thiol. Moreover, the selectivity of HDAC1 over HDAC6 remained unchanged (selectivity: 89). Notably, we demonstrated earlier that the fluoro analogs (15a and 15b) exhibited slightly less activities than largazole thiol, which was consistent with all previous observations made with substitutions at C2 for largazole [16,17,18,19,20,21]. We wanted to compare 15c with similar largazole compounds. However, a literature search revealed no prior report on modification at C2 position with a sulphur-containing substituent. 15c appeared to be the first example of such compounds in the largazole family. The surprisingly enhanced activity of 15c might be attributed to the variation in Val at the C2 position, suggesting that the MeS group of 15c played an important role in the interaction between 15c and HDACs. This was confirmed subsequently by the results from a molecular modeling study.
2.3. Molecular Modeling Study
To gain some structural insight on the increased inhibitory effects of 15c on the tested HDACs, molecular docking was performed by using MOE 2019 with MMF94 force field. The crystal structures of HDACs were downloaded from the Protein DataBank (PDB, http://www.rcsb.org), and were used to investigate the binding modes of 15c with HDAC1, HDAC6 and HDAC8, respectively (PDB code: HDAC1, 5ICN; HDAC6, 5EDU; HDAC8, 4RN0).
The binding modes of largazole thiol in HDAC1, HDAC6 and HDAC8 indicated that the thiol side chain could coordinate to the catalytic Zn2+ ion and the overall metal coordination geometry was nearly perfectly tetrahedral [25]. Addtionally, largazole thiol formed hydrogen bond interactions with ASP99 in HDAC1 and TYR306 in HDAC8, respectively (Figure 2A–C). For 15c, which was derived from largazole thiol, additional hydrogen bonds were formed between sulphur atom in the methylthio group and ASN95 in HDAC1 and SER568 in HDAC6, respectively (Figure 2D,E). Interestingly, the introduction of MeS substitution resulted in orientation change of the ester bond at C1 position, and then made an oxygen atom of the carboxyl group form a new hydrogen bond with HIS180 of HDAC8 (Figure 2F). These additional interactions are likely the factors that have led to significant improvement in 15c’s potency towards HDACs. These findings from molecular modeling study confirmed our hypothesis about the key role that MeS group played, and provided telling structural evidence for the significantly increased inhibition of 15c towards HDACs.
Figure 2.

Proposed binding mode of Largazole thiol (carmine carbon sticks) with (A) HDAC1 (5ICN), (B) HDAC6 (5EDU) and (C) HDAC8 (4RN0). Proposed binding mode of 15c (carmine carbon sticks) with (D) HDAC1 (5ICN), (E) HDAC6 (5EDU) and (F) HDAC8 (4RN0). Atom color code: red = oxygen, blue = nitrogen, white = hydrogen, yellow = sulfur, cyan = fluorine. Hydrogen bonds between the ligand and receptor are indicated by yellow dashed lines.
3. Materials and Methods
3.1. Chemistry
The chemicals and reagents were purchased from Acros (Shanghai, China), Alfa Aesar (Shanghai, China), and National Chemical Reagent Group Co. Ltd., P. R. China (Shanghai, China), and used without further purification. Anhydrous solvents (THF, MeOH, DMF, CH2Cl2 and CH3CN) used in the reactions were dried and freshly distilled before use. All the reactions were carried out under Ar atmosphere, otherwise stated else. The progress of the reactions was monitored by TLC (silica-coated glass plates) and visualized under UV light, and by using iodine or phosphomolybdic acid. Melting points were measured on an SGW X-4 microscopy melting point apparatus without correction. 1H NMR and 13C NMR spectra were recorded either on a 400 MHz Varian Instrument at 25 °C or 600 MHz Bruker Instrument at 25 °C, using TMS as an internal standard, respectively. Multiplicity is tabulated as s for singlet, d for doublet, dd for doublet of doublet, t for triplet, and m for multiplet. The original spectra of the relative compounds could be found in Supplementary Materials. HRMS spectra were recorded on Finnigan-Mat-95 mass spectrometer, equipped with ESI source. Largazole, its free thiol and key intermediates (compound 8 and 11) were prepared according to our previous method [22,23,24].
3.1.1. 2-(Trimethylsilyl)ethyl (S,Z)-4-fluoro-3-hydroxy-7-(tritylthio)hept-4-enoate (9)
To a solution of 8 (0.595 g, 0.95 mmoL, 1.0 equiv.) and DMAP (0.183 g, 1.5 mmoL, 1.5 equiv.) in CH2Cl2 (DCM, 20 mL), Me3CH2CH2OH was added (1.36 mL, 9.5 mmol, 10 equiv.), and the resulting solution was stirred at room temperature (rt) overnight. Concentration in vacuo left the residue, which was purified by column chromatography, giving compound 9 (0.341 mg) with a 67% yield as colorless oil. Rf = 0.17 [petroleum ether (PE)/ethyl acetate (EA) 10/1]. 1H-NMR (600 MHz, CDCl3): δ 7.50–7.13 (m, 15H, ArH), 4.81 (dt, J = 37.2, 7.2 Hz, 1H, CH=CF), 4.46 (m, 1H, CHOH), 4.20 (m, 2H, OCH2CH2TMS), 3.14 (brs, 1H, OH), 2.63 (dd, J = 16.2, 3.6 Hz, 1H, CH2CO), 2.58 (dd, J = 16.2, 9.0 Hz, 1H, CH2CO), 2.20 (t, J = 7.2 Hz, 2H, SCH2CH2), 2.14 (t, J = 7.2 Hz, 2H, SCH2CH2), 0.99 (t, J = 8.4 Hz, 2H, OCH2CH2TMS), 0.03 (s, 9H, SiMe3). 13C-NMR (150 MHz, CDCl3): δ 171.4, 157.9 (d, J = 255 Hz), 144.2, 128.9, 127.2, 126.0, 104.2, 66.1 (d, J = 27 Hz), 62.7, 37.9, 30.8, 22.0, 16.6, 2.1. 19F NMR (376 MHz, CDCl3): δ −123.24 (dd, J = 37.6, 11.2 Hz). ESI (m/z): [M + H]+ 537.4.
3.1.2. 2-(Trimethylsilyl)ethyl (S,Z)-3-((N-(((9H-fluoren-9-yl)methoxy)carbonyl)-S-methyl-l-cysteinyl)oxy)-4-fluoro-7-(tritylthio)hept-4-enoate (10c)
To a solution of 9 (0.690 g, 1.28 mmol, 1.0 equiv.) in DCM (10 mL) a solution of S-Me Fmoc-l-Cysteine was added (2.38 g, 6.42 mmol, 5.0 equiv.); DMAP (0.016 g, 0.128 mmol, 0.1 equiv.) and EDCI (1.48 g, 7.7 mmol, 6.0 equiv.) was added in DCM (20 mL) at rt, and DIPEA (1.30 mL, 7.7 mmol, 2.5 equiv.) was added to the stirring solution. The reaction was stirred for 18 h. This solution was quenched with a NH4Cl aqueous solution and separated. The aqueous phase was extracted with DCM (30 mL × 3) and the combined organic phase was washed with H2O, brine, dried with Na2SO4 and filtered. After the removal of the solvent, the resulting crude was purified by flash chromatography on silica gel, eluting with PE/EA (5/1) to afford 10c (0.856 g, 75%) as a white foamy solid. Rf = 0.20 (PE/EA 5/1). 1H-NMR (600 MHz, CDCl3): δ 7.80–7.10 (m, 23H, ArH), 6.64 (m, 1H, CHOC=O), 5.37 (d, J = 8.4 Hz, 1H, NH), 4.88 (dt, J = 36, 7.2 Hz, 1H, CH=CF), 4.49 (m, 1H, CHCH2SMe), 4.40 (m, 2H, OCH2CH[Fmoc]), 4.21 (m, 1H, OCH2CH[Fmoc]), 4.16 (m, 2H, OCH2CH2TMS), 2.80 (dd, J = 16.8, 8.4 Hz, 1H, CH2CO), 2.68 (dd, J = 16.8, 4.8 Hz, 1H, CH2CO), 2.42 (m, 1H, CHCH2SMe), 2.20–2.14 (m, 4H, SCH2CH2), 2.03 (s, 3H, CHCH2SMe), 1.91 (m, CHCH2SMe), 0.96 (t, J = 8.4 Hz, 2H, OCH2CH2TMS), 0.01 (s, 9H, SiMe3). 13C-NMR (150 MHz, CDCl3): δ 170.2, 168.4, 155.1, 153.9 (d, J = 257 Hz), 144.1, 143.2, 143.0, 140.7, 128.9, 127.2, 127.1, 126.4, 126.0, 124.4, 119.3, 108.8 (d, J = 14 Hz), 68.8 (d, J = 29 Hz), 66.4, 66.2, 62.8, 52.4, 46.5, 35.6, 31.2, 30.4, 22.2, 16.6, 14.7, −2.1. 19F NMR (376 MHz, CDCl3): δ −123.28 (dd, J = 37.6, 18.8 Hz). HRMS-ESI (m/z): [M + H]+ calcd. for C50H54FNO6S2SiH: 876.3219, found: 876.3223.
3.1.3. 2-(Trimethylsilyl)ethyl (S,Z)-3-((N-((R)-2′-(((tert-butoxycarbonyl)amino)methyl)-4-methyl-4,5-dihydro-[2,4′-bithiazole]-4-carbonyl)-S-methyl-L-cysteinyl)oxy)-4-fluoro-7-(tritylthio)hept-4-enoate (12c)
To a solution of 10c (0.890 g, 0.90 mmol, 1.0 equiv.) in DCM (60 mL) was added Et2NH (4.74 mL, 29 mmol, 10 equiv.) was added at rt. After stirring for 2 h and removal of the solvent, the residue was purified by flash chromatography on silica gel, eluting with PE/EA (3/2) to afford the corresponding amine (0.465 g, 77%) as a white foamy solid.
A mixture of compound 11 (0.196 g, 0.55 mmol, 1.0 equiv.), the amine (0.438 g, 0.66 mmol, 1.2 equiv.), HOBT (0.089 g, 0.66 mmol, 1.2 equiv.) and EDCI (0.126 g, 0.66 mmol, 1.2 equiv.) was dissolved in dry DCM (50 ml) and stirred at rt for 3 h. The solution was concentrated under reduced pressure, and diluted with EA (150 mL), washed saturated NaHCO3 aqueous solution and brine successively, dried over Na2SO4 and filtered. After removal of the solvent, the residue was purified by flash chromatography on silica gel, eluting with PE/EA (5/2) to afford 12c (84% based on 10c) as a white foamy solid. Rf = 0.30 (PE/EA 5/2). 1H-NMR (600 MHz, CDCl3): δ 7.87 (s, 1H, ArH), 7.5–7.10 (m, 17H, ArH, 2NH), 5.60 (ddd, J = 19.8, 8.4, 5.4 Hz, 1H, CHOC=O), 5.29 (dd, J = 36, 7.2 Hz, 1H, CH=CF), 4.68 (m,1H, CHCH2SMe), 4.62 (d, J = 5.4 Hz, 2H, CH2NHCOCMe3), 4.18 (m, 2H, OCH2CH2SiMe3), 4.04 (d, J = 11.4 Hz, 1H, CqCH2S), 3.32 (d, J = 11.4 Hz, 1H, CqCH2S), 2.81 (dd, J = 16, 8.4 Hz, 1H, CH2CO2), 2.70 (dd, J = 16, 8.4 Hz, 1H, CH2CO2), 2.39 (m, 2H, CHCH2SMe), 2.20–2.00 (m, 4H, Ph3CSCH2CH2), 1.96 (s, 3H, CHCH2SMe), 1.57 (s, 3H, CqCH3), 1.46 (s, 9H, CH2NHCOCMe3), 0.97 (t, J = 8.4 Hz, 3H, OCH2CH2SiMe3), 0.03 (s, 9H, OCH2CH2SiMe3). 13C-NMR (150 MHz, CHCl3): δ 173.6, 168.9, 168.7, 167.8, 164.5, 155.2 (d, 1JC-F = 256 Hz), 147.4, 129.5, 127.9, 126.6, 124.3, 109.2 (d, 2JC-F = 12.4 Hz), 84.3, 69.9 (d, 3JC-F = 29.6 Hz), 57.5, 43.3, 41.1, 38.6, 37.4, 34.2, 31.1, 29.7, 24.1, 22.8, 18.8, 16.5, 14.2. 19F NMR (376 MHz, CDCl3): δ -124.91 (dd, J = 37.6, 18.8 Hz). ESI (m/z): [M + Na]+ 1015.4.
3.1.4. 2-(Trimethylsilyl)ethyl (S,Z)-3-((((R)-2′-(((tert-butoxycarbonyl)amino)methyl)-4-methyl-4,5-dihydro-[2,4′-bithiazole]-4-carbonyl)glycyl)oxy)-4-fluoro-7-(tritylthio)hept-4-enoate (12d)
Compound 12d was prepared from Fmoc-Glycine in 41% yield based on 9 using the same procedure for 12c. 1H NMR (600 MHz, CDCl3) δ 7.85 (s, 1H, ArH), 7.53–7.09 (m, 16H, ArH, NH), 5.68 (ddd, J = 19.7, 8.8, 5.2 Hz, 1H, CHOC=O), 5.36 (brs, 1H, NH), 4.88 (dt, J = 36.0, 8.0 Hz, 1H, CH=CF), 4.61 (d, J = 6.3 Hz, 2H, CH2NHCOCMe3), 4.25–4.08 (m, 2H, OCH2CH2SiMe3), 4.12–3.94 (m, 2H, NHCH2CO2), 3.74 (d, J = 11.7 Hz, 1H, CqCH2S), 3.32 (d, J = 11.5 Hz, 1H, CqCH2S), 2.77 (dd, J = 16.6, 8.7 Hz, 1H, CH2CO2), 2.66 (dd, J = 16.5, 5.0 Hz, 1H, CH2CO2), 2.12 (dd, J = 25.5, 7.1 Hz, 4H, Ph3CSCH2CH2), 1.58 (s, 3H, CqCH3), 1.46 (s, 9H, CH2NHCOCMe3), 1.04–0.88 (m, 2H, OCH2CH2SiMe3), 0.02 (s, 9H, OCH2CH2SiMe3). 13C NMR (150 MHz, CDCl3) δ 174.7, 169.7, 169.1, 168.2, 163.3, 155.6, 154.5 (d, J = 258.2 Hz), 148.5, 144.7, 129.5, 127.8, 126.6, 121.9, 108.9 (d, J = 13.7 Hz), 84.9, 80.4, 69.1 (d, J = 30.2 Hz), 66.6, 63.4, 42.3, 41.2, 41.0, 36.3, 31.0, 28.3, 24.7, 22.8, 17.2, -1.5. 19F NMR (376 MHz, CDCl3): 124.96 (m). ESI (m/z): [M + Na]+ 955.5.
3.1.5. (12Z,22Z,24R,5R,8S)-8-((Z)-1-fluoro-4-(tritylthio)but-1-en-1-yl)-24-methyl-5-((methylthio)methyl)-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-3,6,10-trione (14c)
12c (0.446 g, 0.44 mmol, 1.0 equiv.) was dissolved in a solution of CF3CO2H (5 mL) and dry DCM (20 mL), and stirred at rt overnight. After removal of the solvent under reduced pressure, and the crude amino acid (about 0.408 g) was directly used for the next step.
The amine acid was dissolved in anhydrous DMF (40 mL) and then DIEPA (0.50 mL, 2.66 mmol, 6.0 equiv.) was added to the solution at rt in order to prepare the amine solution. Next, the amine solution was added dropwise to a stirring solution of HATU (0.337 g, 0.89 mmol, 2.0 equiv.) and HOBT (0.120 g, 0.89 mmol, 2.0 equiv.) in anhydrous DMF (80 mL) at rt over 3 h. The resulting solution continued to stir at rt overnight. The mixture was diluted with water (150 mL), and extracted with MeOBut (80 mL). The combined organic phase was washed with a saturated NaHCO3 aqueous solution (50 mL × 3), water (50 mL) and brine (50 mL). After drying by NaSO4, filtration and evaporation, the residue was purified by flash chromatography on silica gel, eluting with PE/EA (1/1) to afford 14c (48% in 2 steps) as a white foamy solid. Rf = 0.20 (PE/EA 1/1). 1H-NMR (600 MHz, CDCl3): δ 7.75 (s, 1H, ArH), 7.5–7.10 (m, 16H, ArH, NH), 6.39 (m, 1H, NH), 5.70 (m, 1H, CHOC=O), 5.52 (dd, J = 17.4, 9.6 Hz, 1H, ArCH2NH), 4.96 (dd, J = 36, 7.2 Hz, 1H, CH=CF), 4.68 (dd, J = 12, 4.2 Hz, 1H, CHCH2SMe), 4.22 (dd, J = 17.4, 3.0 Hz, 1H, ArCH2NH), 4.08 (d, J = 11.4 Hz, 1H, CqCH2S), 3.20 (d, J = 11.4 Hz, 1H, CqCH2S), 3.13 (dd, J = 16, 11.4 Hz, 1H, CH2CO2), 2.70 (d, J = 16, 1H, CH2CO2), 2.30–1.90 (m, 6H, Ph3CSCH2CH2, CHCH2SMe), 1.96 (s, 3H, CHCH2SMe), 1.70 (s, 3H, CqCH3). 13C-NMR (150 MHz, CHCl3): δ 172.8, 168.3, 168.2, 167.2, 163.6, 154.1 (d, 1JC-F = 256 Hz), 146.8, 144.1, 128.9, 127.2, 126.0, 123.8, 108.8 (d, 2JC-F = 12 Hz), 83.6, 69.4 (d, 3JC-F = 30 Hz), 66.0, 51.8, 42.2, 40.6, 36.5, 30.9, 30.4, 27.8, 23.6, 22.2, 14.2. 19F NMR (376 MHz, CDCl3): δ −125.10 (dd, J = 37.6, 18.8 Hz). HRMS-ESI (m/z): [M + H]+ calcd. for C39H39FN4O4S4H: 775.1911, found: 775.1918.
3.1.6. (12Z,22Z,24R,8S)-8-((Z)-1-fluoro-4-(tritylthio)but-1-en-1-yl)-24-methyl-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-3,6,10-trione (14d)
Compound 12d was prepared in 40% yield using the same procedure for 12c. 1H NMR (600 MHz, CDCl3) δ 7.72 (s, 1H, ArH), 7.44–7.17 (m, 15H, ArH), 7.03 (brs, 1H, NH), 6.55–6.46 (m, 1H, NH), 5.76 (dd, J = 22.6, 11.1 Hz, 1H, CHOC=O), 5.19 (dd, J = 17.4, 8.9 Hz, 1H, ArCH2NH), 4.94 (dt, J = 36.0, 8.0 Hz, 1H, CH=CF), 4.24 (dd, J = 17.4, 4.0 Hz, 1H, ArCH2NH), 4.13 (m, 1H, CH2CO2), 4.12 (d, J = 12.0 Hz, 1H, CqCH2S), 3.88 (dd, J = 18.7, 4.0 Hz, 1H, CH2CO2), 3.22 (d, J = 12.0 Hz, 1H, CH2CO2), 3.13 (dd, J = 16.9, 11.3 Hz, 1H, CH2CONH), 2.70 (d, J = 16.8 Hz, 1H, CH2CONH), 2.16–2.14 (m, 4H, Ph3CSCH2CH2), 1.82 (s, 3H, CqCH3). 13C NMR (150 MHz, CDCl3) δ 173.8, 169.0, 167.5, 165.9, 163.7, 154.7 (d, J = 258.2 Hz), 147.4, 144.7, 129.5, 127.8, 126.6, 124.6, 109.6 (t, J = 12.1 Hz), 84.3, 70.0 (d, J = 28.7 Hz), 66.6, 43.6, 42.0, 41.4, 37.1, 31.0, 25.2, 22.8. 19F NMR (376 MHz, CDCl3): δ −129.82 (dd, J = 37.6, 22.5 Hz). HRMS-ESI (m/z): [M + Na]+ calcd. for C37H35FN4O4S3Na: 737.1697, found: 737.1675.
3.1.7. (12Z,22Z,24R,8S)-8-((Z)-1-fluoro-4-mercaptobut-1-en-1-yl)-24-methyl-5-((methylthio)methyl)-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-3,6,10-trione (15c)
14c (0.050 mg, 0.63 mmol, 1.0 equiv.) was dissolved in dry DCM (10 mL) and cooled to 0 °C. The mixture was successively treated with i-Pr3SiH (26 µL, 0.13 mmol, 2.0 equiv.) and TFA (0.30 mL, 4.0 mmol, 6.7 equiv.). The reaction mixture was allowed to warm to rt and stirred for 1.5 h. The reaction was quenched with a saturated NaHCO3 solution (10 mL) and separated. The aqueous phase was extracted with DCM (10 mL × 3), and the combined layers was washed with brine, dried over Na2SO4 and filtered. After removal of the solvent, the residue was purified by flash chromatography on silica gel, eluting with DCM/MeOH (60/1) to afford 15c in 91% yield as a white foamy solid. Rf = 0.30 (PE/EA 2/3). 1H-NMR (400 MHz, CDCl3): δ 1H-NMR (600 MHz, CDCl3): δ 7.78 (s, 1H, ArH), 7.26 (d, J = 7.6 Hz, 1H, NH), 6.44 (dd, J = 10, 3.6 Hz, 1H, NH), 5.77 (ddd, J = 21.2, 11.2, 2.0 Hz, 1H, CHOC=O), 5.28 (dd, J = 17.6, 9.6 Hz, 1H, ArCH2NH), 5.11 (dt, J = 36, 7.6 Hz, 1H, CH=CF), 4.70 (m, 1H, CHCH2SMe), 4.27 (dd, J = 17.6, 3.6 Hz, 1H, ArCH2NH), 4.08 (d, J = 11.2 Hz, 1H, CqCH2S), 3.25 (d, J = 11.2 Hz, 1H, CqCH2S), 3.19 (dd, J = 16.8, 11.2 Hz, 1H, CH2CONH), 2.76 (dd, J = 16.8, 2.4 Hz, 1H, CH2CONH), 2.41 (m, 2H, CH2CH=CF), 2.30–1.90 (m, 4H, CH2SH, CHCH2SMe), 1.87 (s, 3H, CHCH2SMe), 1.85 (s, 3H, CqCH3). 13C-NMR (150 MHz, CHCl3): δ 174.1, 169.5, 169.4, 164.8, 155.9 (d, 1JC-F = 256 Hz), 147.9, 125.0, 109.3 (d, 2JC-F = 12 Hz), 84.8, 70.9 (d, 3JC-F = 30 Hz), 53.0, 43.4, 41.8, 37.8, 28.4, 24.8, 24.3, 15.6. 19F NMR (376 MHz, CDCl3): δ −124.79 (dd, J = 37.6, 18 Hz). HRMS-ESI (m/z): [M + H]+ calcd. for C20H25FN4O4S4H: 533.0815, found: 533.0819.
3.1.8. S-((Z)-4-fluoro-4-((12Z,22Z,24R,5R,8S)-24-methyl-5-((methylthio)methyl)-3,6,10-trioxo-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-8-yl)but-3-en-1-yl) octanethioate (16c)
The free thiol 15c (32 mg, 0.05mmol, equiv.) was dissolved in dry DCM (10 mL) and cooled to 0 °C. The mixture was successively treated with pyridine (23 µL, 0.29 mmol, 5.0 equiv.) and octanoyl chloride (29 µL, 0.17 mmol, 3.0 equiv.). The reaction was allowed to warm to rt and stirred for 2 h, and then quenched with a saturated NaHCO3 solution (10 mL) and separated. The aqueous phase was extracted with DCM (10 mL × 3), and the combined layers was washed with brine, dried over Na2SO4 and filtered. After removal of the solvent, the residue was purified by flash chromatography on silica gel, eluting with DCM/EA (1/1) to afford 16c (57%) as a white foamy solid. Rf = 0.42 (DCM/EA 3/1). 26.4 (c 0.34, CHCl3). 1H-NMR (600 MHz, CDCl3): δ 7.78 (s, 1H, ArH), 7.24 (d, J = 11.4 Hz, 1H, NH), 6.54 (dd, J = 9.6, 3.6 Hz, 1H, NH), 5.76 (ddd, J = 21, 11.4, 2.4 Hz, 1H, CHOC=O), 5.30 (dd, J = 17.4, 9.6 Hz, 1H, ArCH2NH), 5.09 (dt, J = 36, 7.2 Hz, 1H, CH=CF), 4.71 (m, 1H, CHCH2SMe), 4.27 (dd, J = 17.5, 3.0 Hz, 1H, ArCH2NH), 4.10 (d, J = 11.4 Hz, 1H, CqCH2S), 3.28 (d, J = 11.4 Hz, 1H, CqCH2S), 3.19 (dd, J = 16.8, 11.4 Hz, 1H, CH2CONH), 2.90 (t, J = 7.2 Hz, 2H, SCH2CH2), 2.75 (dd, J = 16.8, 2.4 Hz, 1H, CH2CONH), 2.54 (t, J = 7.5 Hz, 2H, CH2COS), 2.40–2.30 (m, 2H, SCH2CH2), 2.30–1.90 (m, 2H, CHCH2SMe), 1.87 (s, 3H, CHCH2SMe), 1.86 (s, 3H, CqCH3), 1.70–1.6 (m, 2H, CH2), 1.40–1.10 (m, 8H, CH2), 0.89 (t, J = 6.0 Hz, 3H, CH3CH2). 13C-NMR (150 MHz, CDCl3): δ 198.6, 172.9, 168.3, 168.2, 167.3, 163.8, 154.7 (d, J = 256 Hz), 146.7, 123.9, 108.1 (d, J = 12 Hz), 83.6, 69. 5 (d, J = 30 Hz), 51.8, 43.5, 42.2, 40.6, 36.5, 30.9, 28.4, 27.2, 24.9, 24.1, 23. 6, 23.2, 21.9, 14.4, 13.4. 19F NMR (376 MHz, CDCl3): δ −124.69 (dd, J = 35.9, 20.9 Hz). HRMS-ESI (m/z): [M + Na]+ calcd. for C28H39FN4O5S4Na: 681.1680, found: 681.1693.
3.1.9. S-((Z)-4-fluoro-4-((12Z,22Z,24R,8S)-24-methyl-3,6,10-trioxo-24,25-dihydro-7-oxa-4,11-diaza-1(4,2),2(2,4)-dithiazolacyclododecaphane-8-yl)but-3-en-1-yl) octanethioate (16d)
Compound 16d was prepared in 40% yield from 14d using the same procedure for 16c. 37.9 (c 0.14, CHCl3). 1H NMR (600 MHz, CDCl3) δ 7.71 (s, 1H, ArH), 7.00 (m, 1H, NH), 6.60 (dd, J = 9.1, 4.1 Hz, 1H, NH), 5.78 (dd, J = 22.3, 11.1 Hz, 1H, CHOC=O), 5.18 (dd, J = 17.4, 8.8 Hz, 1H, ArCH2NH), 5.04 (dt, J = 36.1, 7.5 Hz, 1H, CH=CF), 4.24 (dd, J = 17.5, 4.0 Hz, 1H, ArCH2NH), 4.15 (m, 1H, CH2CO2), 4.10 (d, J = 11.7 Hz, 1H, CqCH2S), 3.87 (dd, J = 18.9, 3.1 Hz, 1H), 3.20 (d, J = 11.2 Hz, 1H, CqCH2S), 3.14 (dd, J = 16.0, 12.0 Hz, 1H, CH2CONH), 2.86 (t, J = 7.1 Hz, 2H, SCH2CH2), 2.71 (d, J = 16.9 Hz, 1H, CH2CO), 2.50 (t, J = 7.6 Hz, 2H, C6H13CH2CO), 2.41–2.27 (m, 2H, SCH2CH2), 2.11–1.87 (m, 2H), 1.79 (s, 3H, CqCH3), 1.60–1.15 (m, 10H), 0.84 (t, J = 6.6 Hz, 3H, CH3-C5H11-CH2CO). 19F NMR (376 MHz, CDCl3): δ −123.29 (dd, J = 35.9, 20.9 Hz). HRMS-ESI (m/z): [M + Na]+ calcd. for C26H35FN4O5S3Na: 621.1646, found: 621.1650.
3.2. Biology
3.2.1. Recombinant Human HDAC1, HDAC2, HDAC3, HDAC6 and HDAC8 Enzymatic Assays
The assays were carried out by Shanghai ChemPartner Co., Ltd (Shanghai, China), according to our previous method [23,24]. Briefly, different concentrations of compounds were incubated with recombinant human HDAC1, HDAC2, HDAC3, HDAC6 and HDAC8 (BPS Biosciences, San Diego, CA, USA) at room temperature for 15 min, which was followed by adding Ac-peptide-AMC substrates to initiate the reaction in Tris-based assay buffer. Reaction mixtures were incubated at room temperature for 60 min in HDAC1, HDAC2, HDAC3 and HDAC6 assays, and were incubated for 240 min in HDAC8 assay. Then, the stop solution containing trypsin was added. The coupled reaction was incubated for another 90 min at 37 °C. Fluorescent AMC released from substrate was measured in SynergyMx (BioTek, Winooski, VT, US) using filter sets as excitation = 355 nm and emission = 460 nm. IC50 values were calculated by GraphPad Prism software (7.0 version., GraphPad Software, San Diego, CA, USA).
3.2.2. Cellular Assay
Eca-109, Bel 7402 and U937 cell lines were purchased from ATCC (Manassas, VA, USA), and the other tested cell lines were obtained from the Shanghai Institute for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). MDA-MB-231, Hela, Eca-109 and Bel 7402 cells were cultured in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin (Beyotime Biotech, Shanghai, China). A549 cells were cultured in F-12K medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. HCT116 and SK-OV-3 cells were cultured in McCoy’s 5A medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. U937 cells were cultured in RPMI-1640 medium supplemented with 10% heat inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. All cells were cultured at 37 °C in humidified air containing 5% CO2. According to our previous method [23,24], A549, HCT116, MDA-MB-231, SK-OV-3, Hela, Eca-109, Bel 7403 and U937 (600,000 cells/well) were seeded in 96-well plates and incubated for 24 h before being treated with various concentration of compounds or solvent control. Cells were further incubated for 72 h and then treated with MTT and incubated for another 3 h. The media was then removed and 100 μL DMSO was added to each well. The absorbance at 550 nm was measured by a SpectraMAX340 microplate reader (Molecular Devices, San Jose, CA, USA) with a reference wavelength at 690 nm. Adriamycin was used as a positive control in the assay.
3.3. Molecular Modeling Study
The known crystal complexes of HDAC1, HDAC6, HDAC8 and their ligand (PDB code: 5ICN, 5EDU, 4RN0) were obtained from PDB (http://www.rcsb.org). Molecular docking simulations in the HDACs were run using the MOE 2019 (Molecular Operating Environment, Chemical Computing Group, Montreal, Quebec, Canada) due to its universality and very fast speed. Ligands were prepared with the ChemBio3D Ultra 14.0 (PerkinElmer, Waltham, MA, USA), followed by MM2 energy minimization. Protein structures were also prepared with the MOE, which could automatically add hydrogen atoms to proteins by explicitly considering the protonation state of histidine and optimize the force field. All crystal water, small ligands and cofactors except HEM were removed. After this step, the binding sites were deduced from the known crystal complexes and the ligands were docked to the prepared proteins through flexible docking mode. Top scoring function poses were selected as representative of the simulations and were displayed with Open-Source PyMOLTM 1.8X software (Schrödinger, Ltd, New York, NY, USA).
4. Conclusions
Given our previous finding that fluorination at the C18 position of largazole showed good tolerance towards the inhibitory activity and selectivity of HDACs, the current study investigated further modifications on the valine residue in its macrocyclic moiety with S-Me Cysteine or Glycine residue. While the Glycine-modified fluoro analog showed poor activity, the S-Me l-Cysteine-modified analog emerged to be a very potent HDAC inhibitor. Unlike all previously reported C2-modified compounds in the largazole family (including our recent fluoro-largazole analogs) where replacement of the Val residue has failed to provide any potency improvement, the S-Me l-Cysteine-modified analog displayed significantly enhanced (five–nine-fold) inhibition of all the tested HDACs while maintaining the selectivity of HDAC1 over HDAC6, as compared to largazole thiol. Molecular modeling study provided rational explanation and structural evidence for the enhanced activity. This new finding will aid the design of novel potent HDAC inhibitors. An expanded research program is currently under way to investigate largazole analogs bearing similar structural characteristics.
Acknowledgments
The authors acknowledge the National Key Research and Development Program of China and the National Natural Science Foundation of China for their financial support. The authors also acknowledge Prof. Tan Wen-Fu and Prof. Bing Cai for the cellular assays and Shanghai ChemPartner Co., Ltd. for the HDAC inhibition assays.
Supplementary Materials
The supporting information is available online at https://www.mdpi.com/1660-3397/18/7/344/s1. Figures S1–S3: 1H, 13C and 19F NMR spectra of compound 8, Figure S4: 1H NMR spectra of compound 11, Figures S5–S7: 1H, 13C and 19F NMR spectra of compound 9, Figures S8 and S9: 1H and 13C NMR spectra of compound 10c, Figures S10–S12: 1H, 13C and 19F NMR spectra of compound 12c; Figures S13–S15: 1H, 13C and 19F NMR spectra of compound 12d, Figures S16–S18: 1H, 13C and 19F NMR spectra of compound 14c, Figures S19–S21: 1H, 13C and 19F NMR spectra of compound 14d, Figures S22–S25: 1H, COSY, 13C and 19F NMR spectra of compound 15c, Figures S26–S28: 1H, 13C and 19F NMR spectra of compound 16c, Figures S29–S31: 1H, 13C and 19F NMR spectra of compound 16d, Figures S32–S39: The concentration-response curves of Largazole and 16c in the cellular assays, Figures S40–S42: The concentration-response curves of Largazole thiol in HDACs assays, Figures S43–S45: The concentration-response curves of 15c in HDACs assays.
Author Contributions
B.Z. and Z.-W.R. performed the chemical synthesis and analyzed the experimental data. D.L. performed the molecular modeling research. Y.Z. and T.D. participated in the analysis of the experimental data. X.L. was responsible for the funding of project and the design of the research. Q.S. was responsible for the writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key Research and Development Program of China (No. 2018YFC0310900) and the National Natural Science Foundation of China (No. 21472024 and No. 21242008).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Delcuve G.P., Khan D.H., Davie J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Ruijter A.J.M., Van Gennip A.H., Caron H.N., Kemp S., Van Kuilenburg A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003;370:737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnstone R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discovery. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 4.Zagni C., Floresta G., Monciino G., Rescifina A. The search for potent, small-molecule HDACIs in cancer treatment: A decade after vorinostat. Med. Res. Rev. 2017;37:1373–1428. doi: 10.1002/med.21437. [DOI] [PubMed] [Google Scholar]
- 5.Roche J., Bertrand P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016;121:451–483. doi: 10.1016/j.ejmech.2016.05.047. [DOI] [PubMed] [Google Scholar]
- 6.Qin H.-T., Li H.-Q., Liu F. Selective histone deacetylase small molecule inhibitors: Recent progress and perspectives. Expert Opin. Therap. Pat. 2017;27:621–636. doi: 10.1080/13543776.2017.1276565. [DOI] [PubMed] [Google Scholar]
- 7.Wang X.X., Wan R.Z., Liu Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018;143:1406–1418. doi: 10.1016/j.ejmech.2017.10.040. [DOI] [PubMed] [Google Scholar]
- 8.Taori K., Paul V.J., Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J. Am. Chem. Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 9.Ying Y., Taori K., Kim H., Hong J., Hendrik Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J. Am. Chem. Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 10.Bowers A., West N., Taunton J., Schreiber S.L., Bradner J.E., Williams R.M. Total synthesis and biological mode of action of largazole: A potent class I histone deacetylase inhibitorc. J. Am. Chem. Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maolanon A.R., Kristensen H.M.E., Leman L.J., Ghadiri M.R., Olsen C.A. Natural and synthetic macrocyclic inhibitors of the histone deacetylase enzymes. ChemBioChem. 2017;18:5–49. doi: 10.1002/cbic.201600519. [DOI] [PubMed] [Google Scholar]
- 12.Reddy D.N., Ballante F., Chuang T., Pirolli A., Marrocco B., Marshall G.R. Design and synthesis of simplified largazole analogues as isoform-selective human lysine deacetylase inhibitors. J. Med. Chem. 2016;59:1613–1633. doi: 10.1021/acs.jmedchem.5b01632. [DOI] [PubMed] [Google Scholar]
- 13.Yao Y., Tu Z., Liao C., Wang Z., Li S., Yao H., Li Z., Jiang S. Discovery of novel class I histone deacetylase inhibitors with promising in vitro and in vivo antitumor activities. J. Med. Chem. 2015;58:7672–7680. doi: 10.1021/acs.jmedchem.5b01044. [DOI] [PubMed] [Google Scholar]
- 14.Poli G., Fabio R.D., Ferrante L., Summa V., Maurizio Botta M. Largazole Analogues as Histone Deacetylase Inhibitors and Anticancer Agents: An Overview of Structure–Activity Relationships. ChemMedChem. 2017;12:1917–1926. doi: 10.1002/cmdc.201700563. [DOI] [PubMed] [Google Scholar]
- 15.Kim B., Ratnayake R., Lee H., Shi G., Zeller S.L., Li C., Luesch H., Hong J. Synthesis and biological evaluation of largazole zinc-binding group analogs. Bioorg. Med. Chem. 2017;25:3077–3086. doi: 10.1016/j.bmc.2017.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ying Y., Liu Y.X., Byeon S.R., Kim H., Luesch H., Hong J. Synthesis and activity of largazole analogues with linker and macrocycle modification. Org. Lett. 2008;10:4021–4024. doi: 10.1021/ol801532s. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y.X., Salvador L.A., Byeon S., Ying Y., Kwan J.C., Law B.K., Hong J., Luesch H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J. Pharmacol. Exp. Ther. 2010;335:351–361. doi: 10.1124/jpet.110.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng X., Yin B.L., Hu Z., Liao C.Z., Liu J.L., Li S., Li Z., Nicklaus M.C., Zhou G.B., Jiang S. Total synthesis and biological evaluation of largazole and derivatives with promising selectivity for cancers cells. Org. Lett. 2010;12:1368–1371. doi: 10.1021/ol100308a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benelkebir H., Marie S., Hayden A.L., Lyle J., Loadman P.M., Crabb S.J., Packham G., Ganesan A. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg. Med. Chem. 2011;19:3650–3658. doi: 10.1016/j.bmc.2011.02.024. [DOI] [PubMed] [Google Scholar]
- 20.Bowers A.A., West N., Newkirk T.L., Troutman-Youngman A.E., Schreiber S.L., Wiest O., Bradner J.E., Williams R.M. Synthesis and histone deacetylase inhibitory activity of largazole analogs: Alteration of the zinc-binding domain and macrocyclic scaffold. Org. Lett. 2009;11:1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim B., Park H., Salvador L.A., Serrano P.E., Kwan J.C., Zeller S.L., Chen Q.Y., Ryu S., Liu Y., Byeon S., et al. Evaluation of class I HDAC isoform selectivity of largazole analogues. Bioorg. Med. Chem. Lett. 2014;24:3728–3731. doi: 10.1016/j.bmcl.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu X., Zhang B., Shan G., Wu Y., Yang F.-L., Lei X. Synthesis of the molecular hybrid inspired by Largazole and Psammaplin A. Tetrahedron. 2018;74:549–555. doi: 10.1016/j.tet.2017.12.025. [DOI] [Google Scholar]
- 23.Zhang B., Shan G., Zheng Y., Yu X., Ruan Z.-W., Li Y., Lei X. Synthesis and preliminary biological evaluation of two fluoroolefin analogs of Largazole inspired by the structural similarity of the side chain unit in Psammaplin A. Mar. Drugs. 2019;17:333. doi: 10.3390/md17060333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B., Liu J., Gao D., Yu X., Wang J., Lei X. A fluorine scan on the Zn2+-binding thiolate side chain of HDAC inhibitor largazole: Synthesis, biological evaluation, and molecular modeling. Eur. J. Med. Chem. 2019;182 doi: 10.1016/j.ejmech.2019.111672. [DOI] [PubMed] [Google Scholar]
- 25.Cole K.E., Dowling D.P., Boone M.A., Phillips A.J., Christianson D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.