

Abstract
Hepatocellular carcinoma (HCC) is the most common form of primary adult liver cancer. After nearly a decade with sorafenib as the only approved treatment, multiple new agents have demonstrated efficacy in clinical trials, including the targeted therapies regorafenib, lenvatinib and cabozantinib, the anti-angiogenic antibody ramucirumab, and the immune checkpoint inhibitors nivolumab and pembrolizumab. Although these agents offer new promise to patients with HCC, the optimal choice and sequence of therapies remains unknown and without established biomarkers, and many patients do not respond to treatment. The advances and the decreasing costs of molecular measurement technologies enable profiling of HCC molecular features (such as genome, transcriptome, proteome and metabolome) at different levels, including bulk tissues, animal models and single cells. The release of such data sets to the public enhances the ability to search for information from these legacy studies and provides the opportunity to leverage them to understand HCC mechanisms, rationally develop new therapeutics and identify candidate biomarkers of treatment response. Here, we provide a comprehensive review of public data sets related to HCC and discuss how emerging artificial intelligence methods can be applied to identify new targets and drugs as well as to guide therapeutic choices for improved HCC treatment.
Hepatocellular carcinoma (HCC) is the most common form of primary adult liver cancer and the fourth leading cause of cancer-related death worldwide in 2018 (REF.1). In 2015, there were 854,000 incident liver cancers and 810,000 deaths globally2. With a mortality to incidence ratio of 0.95, liver cancer is one of the deadliest cancers. In the USA, the incidence and mortality of most cancers have been on the decline over the past decades; however, the burden of HCC has continued to increase, with HCC now the fastest rising cause of cancer death3. According to the American Cancer Society, it is estimated that 42,030 people will be diagnosed with liver cancer in the USA in 2019, and that 31,780 people will die from this malignancy4.
The aetiology of HCC is well defined5, with major risk factors being chronic infection with hepatitis B virus (HBV) or hepatitis C virus, alcohol intake and metabolic syndromes, including non-alcoholic fatty liver disease. For patients with early-stage HCC, surgical resection is the first-line option and it confers 5-year survival rates of 70%6. For patients with an inadequate liver function reserve or unfavourable tumour location for surgery, liver transplantation is another curative-intent therapy7. However, this approach is limited by the availability of donor livers and the fact that tumour burden exceeds transplant criteria in the majority of patients. Owing to the lack of specific symptoms and unfavourable tumour biology, most patients with HCC are diagnosed in the late stages of the disease and, consequently, are not suitable for these curative treatment strategies. For intermediate-stage disease, locoregional therapies, such as transcatheter arterial chemoembolization or radioembolization, are frequently used8. Until 2017, the multi-receptor tyrosine kinase (RTK) inhibitor sorafenib was the only therapy with established survival benefit for patients with advanced-stage HCC, after it demonstrated substantial survival prolongation in two pivotal international trials9,10. Over the past 2 years, multiple additional agents have demonstrated clinical efficacy, including other multi-RTK inhibitors such as lenvatinib11, regorafenib12 and cabozantinib13; a monoclonal antibody to vascular endothelial growth factor receptor 2 (VEGFR2), ramucirumab14; and the immune checkpoint inhibitors nivolumab15 and pembrolizumab16. In the first-line setting, sorafenib remains the standard of care worldwide. In a randomized phase III trial, lenvatinib demonstrated non-inferiority to sorafenib for overall survival along with higher rates of tumour regression11, leading to FDA approval as another first-line treatment option in 2018. In patients who progressed on first-line sorafenib, regorafenib demonstrated survival prolongation compared with placebo, leading to regulatory approvals in multiple countries. Cabozantinib also improved survival in second-line and third-line advanced HCC settings13, and ramucirumab improved survival in patients with α-fetoprotein (AFP) concentrations ≥400 ng/mL after failure of sorafenib14. Immune checkpoint inhibitors have also shown encouraging results in patients with HCC in clinical trials published in the past few years, leading to accelerated approvals for nivolumab and pembrolizumab17. Multiple additional agents and combinations are now being studied in clinical trials. Examples include the combination of PD-L1 blockade using atezolizumab with the antiangiogenic bevacizumab in a phase III trial (NCT03434379)18. With a rapidly expanding treatment landscape, comprehensive molecular profiling is essential to define biological subgroups and biomarkers to guide therapeutic selection and treatment, and to discover improved therapeutics.
Translational research in HCC has benefited from the advances in and the decreasing costs of various omics technologies as well as the increasing adoption of high-throughput technologies19,20. The past few years have witnessed the generation of large amounts of molecular data across multiple modalities — from bulk tissues to single cells, from cancer cells to microorganisms, from cell lines to genetically modified mice and to individual patients, and from single time points to longitudinal profiling (FIG. 1). Such voluminous data points in HCC enable charting of its genomic landscape, delineating molecular mechanisms and, ultimately, providing guidance for rational treatments for this fatal malignancy. In this Review, we first describe the current outstanding big data sets in HCC with a focus on therapeutic discovery, and then discuss computational approaches to translating data points into therapeutics and candidate biomarkers. We also briefly survey the emerging artificial intelligence (AI) methods that could be useful for advancing HCC research.
Fig. 1 |. Translational research and big data.
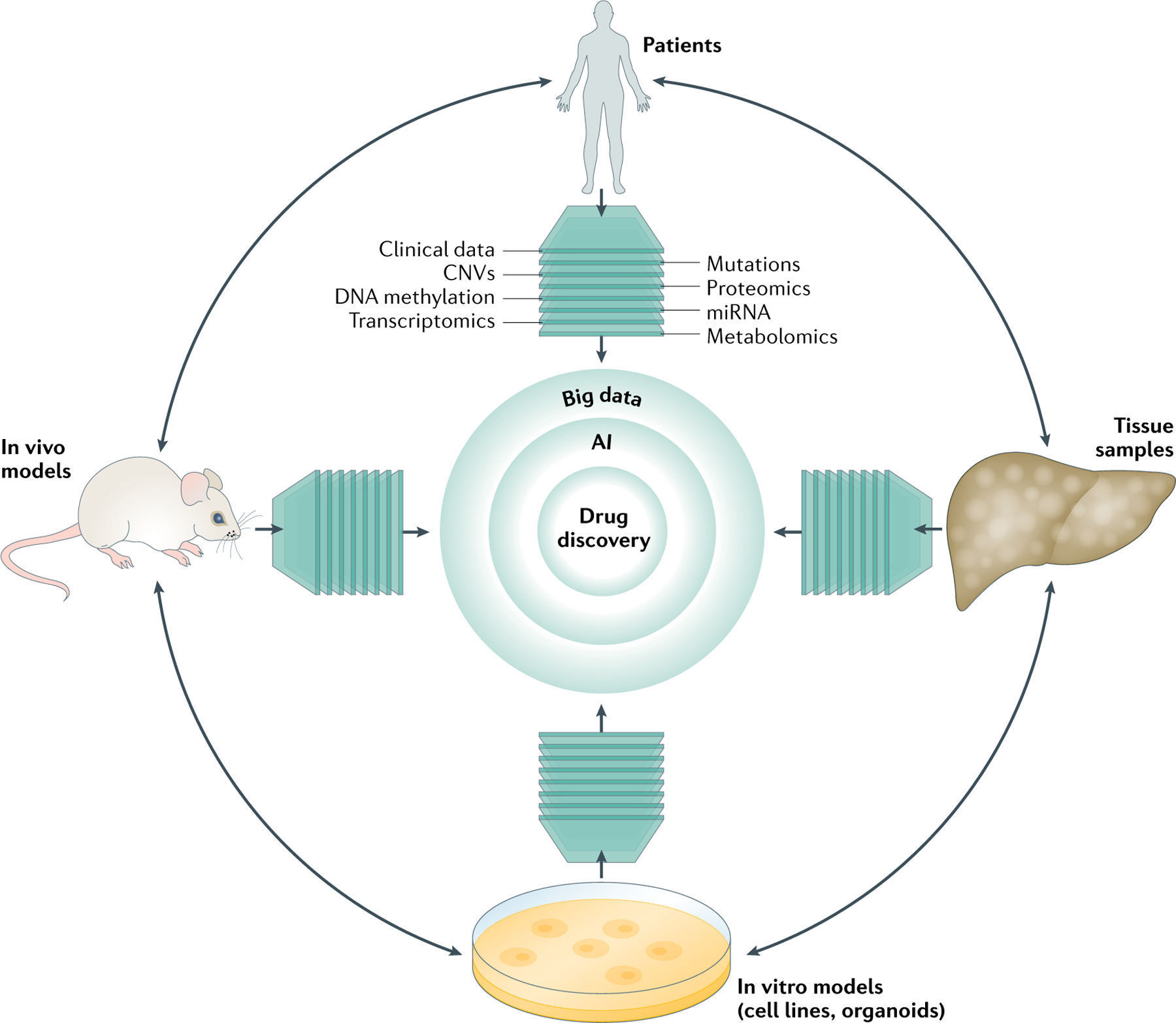
Translational research comprises four main components: patients, tissues, in vitro models (cell lines and organoids) and in vivo models. Each component can be characterized by different molecular modalities (such as genomics, epigenomics and functional genomics). Artificial intelligence (AI) can be used to improve the insights from big data by delineating differences and similarities and further facilitating efficient therapeutic discovery. CNV, copy number variation; miRNA, microRNA.
Big data in HCC
In this section, we highlight outstanding open data sets related to HCC research (TABLE 1) and refer interested readers to a review on open data sets commonly used for translational cancer research19. Our Review focuses on the application of big data in translational research of HCC rather than on specific molecular therapies and targets in HCC, which have been extensively reviewed elsewhere21.
Table 1 |.
Common public data sets for HCC therapeutic discovery
| Data set (ref.) | Descriptiona | Access portal |
|---|---|---|
| Human patient samples | ||
| The Cancer Genome Atlas (TCGA) | Mutation, copy number, mRNA, methylation, microRN A and protein expression of >370 HCCs | NCI Genomic Data Commons portal https://portal.gdc.cancer.gov; cBioPortal http://www.cbioportal.org/; Broad GDAC https://gdac.broadinstitute.org |
| Schulze, 2015 (REF.27) | Exome sequencing of 243 HCCs | cBioPortal; ICGC https://icgc.org |
| Fujimoto, 2012 | Whole-genome sequencing of 27 HCCs | ICGC |
| Guichard, 2012 (REF.30) | Copy number of 125 HCCs and whole-genome sequencing of 24 HCCs | ICGC |
| Ahn, 2014 (REF.28) | Whole-exome sequencing and copy number of 231 HCCs | cBioPortal, EGA portal http://www.ebi.ac.uk/ega/; Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo |
| Totoki, 2014 (REF.31) | Whole-exome sequencing of 503 HCCs | EGA portal; dbGaP https://www.ncbi.nlm.nih.gov/gap; ICGC |
| Fujimoto, 2016 (REF.29) | Whole-genome sequenci ng of 300 HCCs | EGA portal; ICGC |
| Jiang, 2019 (REF.40) | Proteomicsof 110 HCCs | CHNPP Data Portal LIVER http://liver.cnhpp.ncpsb.org |
| Gene Expression Omnibus (GEO) | Functional genomics data repository including >770 HCC studies | Gene Expression Omnibus |
| Genotype-Tissue Expression (GTEx) | Transcriptomic profiles of normal tissue samples, including 175samplesfrom liver | http://www.gtexportal.org |
| Human Protein Atlas | Expression of >17,000 unique proteins in cell lines, normal tissues and cancer tissues, including liver | http://www.proteinatlas.org |
| Human Proteome Map | Expression of >30,000 proteins in normal tissues, including liver | http://humanproteomemap.org |
| Preclinical models | ||
| PDXLiver | A database of >80 liver cancer PDX models | http://www.picb.ac.cn/PDXliver/ |
| Liver Cancer Model Repository (LIMORE) | A database of 81 human liver cancer cell models | http://www.picb.ac.cn/limore/ |
| Cancer Cell Line Encyclopedia (CCLE) | Genetic and pharmacological characterization of >1,000 cell lines (25 HCC cell lines) | http://www.broadinstitute.org/ccle |
| Project Achilles | Genetic vulnerabilities by genome-wide genetic perturbation reagents (shRNAs or Cas9/sgRNAs), including >11,200 genes and >500 cell lines (3 HCC cell lines) | http://www.broadinstitute.org/achilles |
| Library of Integrated Network-based Cellular Signatures (LINCS) | Cellular responses upon the treatment of chemical/genetic perturbagen, including > 1 million gene expression profiles representing >5,000 compounds and >3,500 genes (shRN A and overexpression) in >70 cell lines (2 HCC cell lines) | http://lincscloud.org |
| Genomics of Drug Sensitivity in Cancer project (GDSC) | Drug sensitivity data of 265 drugs in >700 cell lines (17 HCC cell lines) | http://www.cancerrxgene.org |
| Cancer Therapeutics Response Portal (CTRP) | Drug sensitivity data of 481 compounds in 860 cell lines (22 HCC cell lines) | http://www.broadinstitute.org/ctrp.v2.2 |
Broad GDAC, Broad Institute TCGA Genome Data Analysis Center; dbGaP, database of Genotypes and Phenotypes; EGA, European Genome-phenome Archive; HCC, hepatocellular carcinoma; ICGC, International Cancer Genome Consortium; PDX, patient-derived xenograft; sgRNA, single guide RNA; shRNA, short hairpin RNA.
As of August 2019.
Omics data from samples from patients with HCC
Genomics.
Research conducted by The Cancer Genome Atlas (TCGA) Research Network has provided the most comprehensive genomic characterization of HCC, including examination of somatic mutations and DNA copy number variations in 363 patients, and measurement of DNA methylation, mRNA expression, microRNA (miRNA) expression and protein expression in 196 patients22. Integrative analyses of these molecular measurements in combination with clinical features revealed unexpected disease biology and potential therapeutic targets. For instance, unsupervised clustering of five data types (DNA copy number, DNA methylation, mRNA expression, miRNA expression and protein expression) revealed three molecular HCC subtypes, including one associated with poorer prognosis than the other two subtypes. The unique molecular features of each subtype and their different prognoses imply that each subtype might require a distinct therapeutic strategy. Whole-genome sequencing combined with mutation analysis confirmed 18 mutated genes previously reported in HCC — for example, TERT (mutations are present in 44% of tumours with HCC), TP53 (31%), CTNNB1 (27%) and ARID1A (7%) — and identified eight new candidate driver genes (including LZTR1, EEF1A1 and SMARCA4)22 (FIG. 2). Among these, TP53 and CTNNB1 were also identified independently by another 10 studies22. TERT, TP53, CTNNB1, ALB and APOB are each mutated in at least 10% of tumours with HCC; unfortunately, none of these genes are directly druggable at present. Copy number analysis revealed 28 recurring focal amplifications, including known driver oncogenes such as CCND1, FGF19, MYC, MET, VEGFA and MCL1. Inhibitors targeting MYC23 and MCL1 (REF.24) have been intensively studied in preclinical models, and inhibitors targeting FGF19 (REF.25), MET13 and VEGFA26 have been investigated in clinical trials for patients with HCC (FIG. 2). The most frequently deleted genes are NCOR1 (22%), RB1 (19%), ERRF11 (13%) and CDKN2A (13%). Methylation profiling using TCGA data identified 298 hypermethylated genes, including 81 genes reported to be hypermethylated and 28 genes reported to be downregulated. Taken together, the following potential therapeutic targets for HCC were suggested from the TCGA integrative analysis: WNT signalling (CTNNB1 mutation), MDM4 (an inhibitory protein of the mutated TP53), MET (amplification), VEGFA (amplification), MCL1 (amplification), IDH1 (mutation), TERT (mutation) and the immune checkpoint proteins CTLA4, PD-1 and PD-L1 (REF.22).
Fig. 2 |. Connecting genomic features and therapeutic targets in HCC.
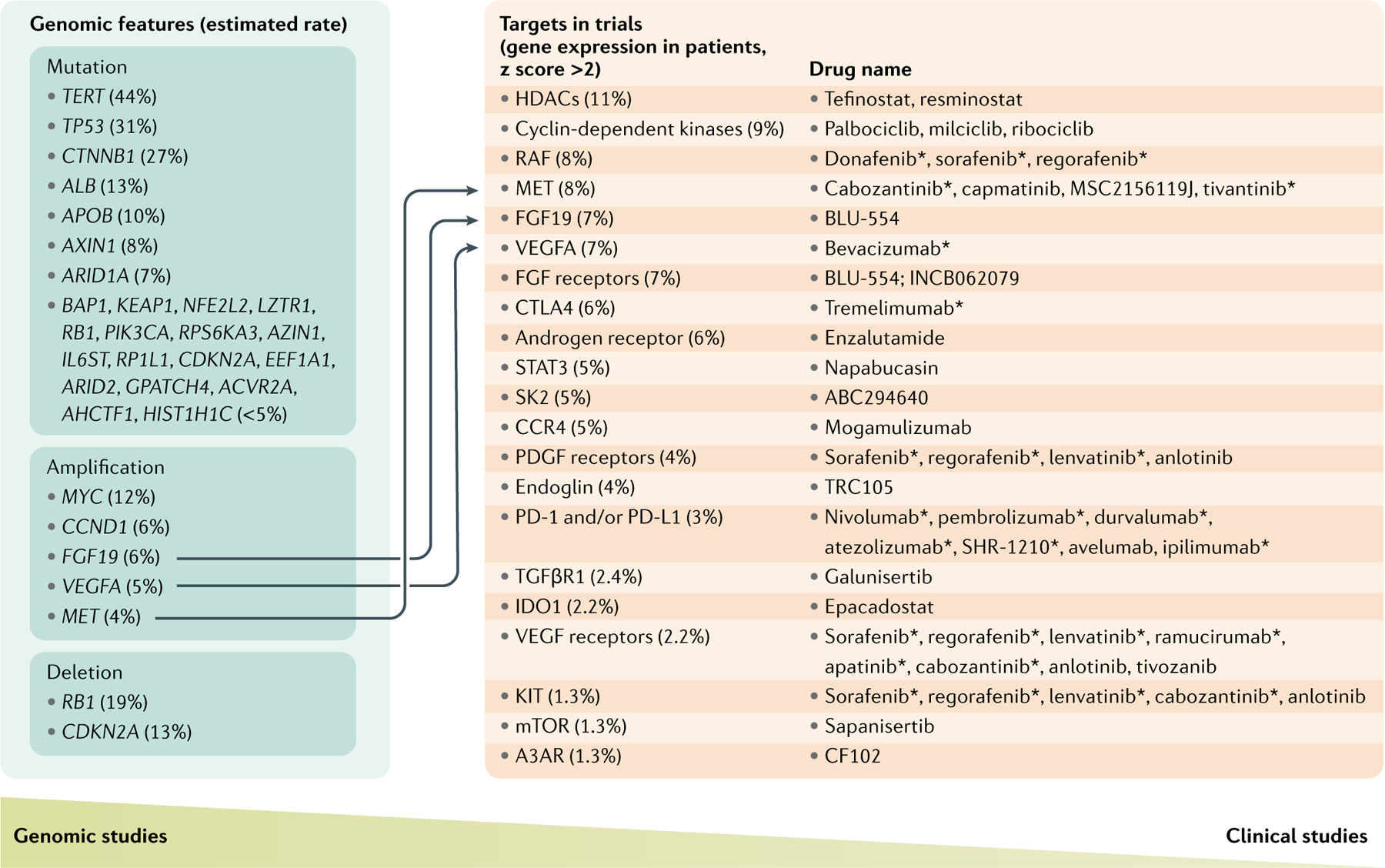
Highly mutated, amplified or deleted genes were extracted mainly from The Cancer Genome Atlas (TCGA) analysis work22 published in 2017 and a review by Llovet et al.21. Therapeutics in phase II and III trials for patients with hepatocellular carcinoma (HCC) and their targets were mainly selected from the review by Llovet et al.21. Many new trials of HCC therapeutics are being launched; new therapeutics in the latest trials that do not pursue new targets were not included. The expression of these targets was retrieved from cBioPortal133, and the percentage of patients with a target expression z score >2 was computed for each target (shown under Targets in trials). The column of targets in trials suggests that therapeutic targets in HCC are expressed in a small portion of patients with HCC, and the column of genomic features suggests that none of the genomic features are altered in half of the patients with HCC. Few connections between genomic features and therapeutic targets indicate the gap in translating genomic features into therapeutic targets. All drugs in clinical trials tested alone or in combination; all drugs reached phase II clinical trials unless otherwise stated. A3AR, adenosine receptor A3; CCR4, CC-chemokine receptor 4; FGF, fibroblast growth factor; HDAC, histone deacetylase; mTOR, mechanistic target of rapamycin; PD-1, programmed cell death 1; PDGF, platelet-derived growth factor; PD-L1, programmed cell death 1 ligand 1; SK2, sphingosine kinase 2; STAT3, signal transducer and activator of transcription 3; TGFβR1; TGFβ receptor 1; VEGF, vascular endothelial growth factor. *Phase III clinical trials.
Several companion genomics studies have been conducted to understand the HCC genomic landscape and to search for new therapeutic targets. For example, exome sequencing analysis of 243 liver tumours uncovered 161 putative driver genes associated with 11 recurrently altered pathways27. Genetic alterations in 28% of those tumours are potentially targetable by FDA-approved drugs. Whole-exome sequencing and copy number variation assessment in 231 patients (72% with HBV infection) with early-stage HCC identified nine genes with recurrent mutations and high-copy amplifications in MYC, RSPO2, CCND1 and FGF19 (REF.28); targeting FGF19 amplifications was proposed as a therapeutic strategy for HCC. Whole-genome sequencing analysis of 300 Japanese patients with liver cancer found that cancer-related genes such as TERT, CCND1, CDKN2A and NCOR1 were recurrently affected by structural variations and point mutations frequently occurring in non-coding regions, indicating the importance of considering structural variations and non-coding mutations in therapeutic discovery29. Another high-resolution copy number analysis on tumour tissue from 125 patients with HCC and whole-exome sequencing on a subset of 24 of these tumours identified novel recurrent alterations in ARID1A, RPS6KA3, NFE2L2 and IRF2. Inactivation of IRF2 was found exclusively in HBV-related tumours, whereas inactivation of chromatin remodelling proteins, such as ARID1A, was mostly detected in alcoholic liver disease-related tumours. The analysis provided a genetic basis for associations between HCC and certain risk factors30.
Given that the studies discussed here have limited power and substantial population heterogeneity, a meta-analysis of data from different patient populations might yield more robust driver genes. An analysis of 503 liver cancer genomes from different international cohorts identified 30 candidate driver genes and 11 core pathway modules31. Among the putative driver genes, TERT was suggested to be a central and ancestry-independent node of hepatocarcinogenesis, based on the occurrence of the hotspot TERT promoter mutation, TERT focal amplification or viral genome integration in more than 68% of cases. Acknowledging population heterogeneity, a multi-modal meta-analysis study on 1,494 liver cancer samples revealed 10 consensus driver genes among different ethnic groups and revealed the large effect of these driver genes on gene expression32. It also highlighted that TP53, CTNNB1 and ARID1A mutations contribute to the three most densely connected pathway clusters32. Moreover, this study systematically associated driver mutations with sex (CTNNB1, ALB, TP53 and AXIN1), race (TP53 and CDKN2A) and age (RB1), indicating the necessity of considering patient characteristics when translating driver mutations to therapeutic targets.
Functional genomics and epigenomics.
In addition to genomics, functional genomics (including transcriptomics, proteomics and metabolomics) and epigenomics have been explored for therapeutic discovery. Analysis of differential expression of proteins, genes or metabolites between tumour tissues and normal tissues has shown encouraging potential for identifying novel diagnostic markers and therapeutic targets33,34. Gene expression analysis was first applied to human HCC samples to identify GPC3 as one of the first biomarkers in HCC35,36. Emerging profiling data provide new opportunities; for example, the current version of the HCC cohort in the NCI Genomic Data Commons Data Portal includes RNA-sequencing (RNA-seq) profiles for 376 TCGA HCC samples and 50 adjacent normal tissues. The Genotype-Tissue Expression project includes RNA-seq profiles from 175 liver tissue samples from healthy individuals37. The Cancer Proteome Atlas profiled the expression of ~220 proteins in 184 TCGA HCC samples38. Untargeted proteomic analysis of serum in 205 patients with HCC or cirrhosis led to the discovery of 21 candidate protein biomarkers differentially expressed in those with HCC compared with patients with cirrhosis39. Proteomic and phosphoproteomic profiling of 110 paired tumour and non-tumour tissues of clinical early-stage HCC with HBV infection revealed three subtypes of HCC and identified sterol O-acyltransferase 1 (SOAT1) as a potential target40. The abundance of SOAT1 was higher in tumour than adjacent normal tissues in a subtype associated with metabolic dysregulation. High expression of SOAT1 was correlated with poor overall survival in multiple cohorts. Knockdown of SOAT1 suppressed proliferation and migration in two HCC cell lines, and a SOAT1 inhibitor suppressed tumour growth in patient-derived xenograft (PDX) models with high SOAT1 expression. Notably, many prognosis-related proteins showed little concordance between mRNA and protein abundance40, highlighting the importance of proteomics data in HCC therapeutic discovery.
Metabolomic characterization of 50 pairs of liver cancer samples and matched normal tissues revealed 105 metabolites uniquely expressed in cancer samples41. These metabolites were associated with elevated glycolysis, gluconeogenesis and β-oxidation, resulting in reduced rate of the tricarboxylic acid cycle and reduced levels of Δ−12 desaturase. Two metabolites, betaine and propionyl-carnitine, were able to distinguish samples with HCC from those with chronic hepatitis or cirrhosis. In a study to understand the implications of metabolites in HCC prevention and diagnosis, 11 metabolites were identified to have the ability to differentiate patients with HCC (n = 63) from control individuals with cirrhosis (n = 65)42. Data from this study suggest that metabolite profiles can be effectively combined with clinical covariates for the early detection of HCC. Another metabolomics study of serum and urine from 82 patients with HCC, 4 patients with benign liver tumours and 71 healthy individuals as controls found 43 serum metabolites and 31 urinary metabolites in patients with HCC; these metabolites were involved in several key metabolic pathways such as bile acid, free fatty acid, glycolysis, methionine metabolism and the urea cycle43. Several metabolites, such as bile acids, histidine and inosine, are statistically significantly overexpressed in patients with HCC. Importantly, a panel of metabolite markers were able to differentiate patients with HCC with low AFP levels (<20 ng/ml) from healthy individuals as controls, underscoring their potential for early detection and stratification of patients with HCC.
Alterations in epigenetic regulatory genes are common in cancers and are increasingly being explored for therapeutic purposes44. Analysis of HCC profiling data in TCGA revealed that 28 hypermethylated genes are downregulated in tumours with HCC. The integration of DNA methylation profiles (the most characterized epigenetic modification) and transcriptomics led to the discovery of the tumour suppressor genes SMPD3 and NEFH in HCC45. Specific therapeutic strategies can be used depending on the type of mutation. For example, gain-of-function mutations and overexpression can be targeted using small molecule inhibitors, whereas loss-of-function mutations can be targeted through synthetic lethality. Synthetic lethality exploits the relationship between two genes (or pathways), in which only the simultaneous loss of both (and not either one alone) leads to cell death. Thus, cancer cells harbouring a tumour suppressing mutation can be selectively killed by chemical inhibition of the second gene or pathway that the tumour suppressor interacts with. This approach selectively kills the cancer cells harbouring the mutation and spares the normal cells without the mutation44.
Omics in metastatic liver cancer.
Metastases from primary tumours account for the majority of cancer-related deaths and underpin the importance of characterizing metastatic cancers in addition to the primary tumours46. Robinson et al. performed whole-exome and transcriptome sequencing on over 500 samples from patients with metastatic cancer, including 29 (5.8%) hepatobiliary cancer samples and 134 (26.8%) tumour samples from metastases to the liver46. Exosome and transcriptomic analysis of all cancer samples revealed an average of 119 somatic mutations per patient in protein-coding regions and 34 gene fusions per metastatic tumour46, whereas in primary HCC there were an average of 40–60 somatic alterations in protein-coding regions21. In addition, 39.8% of all cancer samples harboured at least one putative pathogenic fusion, with 138 activating fusions and 103 deleterious fusions. Compared to all primary tumours, metastatic samples were more heterogeneous and presented more enriched oncogenic signatures. The substantial increase in the number of mutations, fusions and enriched oncogenic signatures in metastases compared with primary tumours highlights the challenge in discovering therapeutic agents to target the aberrations in metastatic cancers, including metastatic HCC.
Omics in single cells.
As the molecular characterization in the described studies was mainly conducted on bulk tissues, which are a mixture of multiple cell types, omics analyses were unable to capture the variations between individual cells and their interactions. Single-cell technologies are advancing rapidly to tackle these challenges in various cancers, including HCC. A deep single-cell RNA-seq analysis of 5,063 individual T cells isolated from peripheral blood, tumours and adjacent normal tissues from six patients with HCC identified 11 T cell subsets on the basis of their molecular and functional properties and delineated their developmental trajectory47. The distinct features of exhausted CD8+ T cells and regulatory T (Treg) cells suggested that regulating their activity might affect response to immunotherapy47. For example, LAYN is highly expressed in Treg cells and exhausted T cells, and higher expression of LAYN is associated with poorer prognosis in HCC. In vitro, overexpression of LAYN on primary CD8+ T cells decreased their production of interferon-γ (IFNγ), a key cytokine involved in tumour killing activity, suggesting the potential of targeting LAYN to increase immune response47. Such heterogeneous single-cell groups exist even in normal human liver tissues. A single-cell RNA-seq study of normal human liver published in 2018 identified 20 discrete cell populations, including hepatocytes, endothelial cells, cholangiocytes, hepatic stellate cells and various subpopulations of immune cells. This study provided a comprehensive overview of the human liver at the single-cell resolution, and underscored the importance of understanding the unique characteristics of these discrete cell populations and their contributions to the hepatic microenvironment48. In the same year, a single-cell RNA-seq study showed that even hepatic cancer stem cells (CSCs) are phenotypically, functionally and transcriptomically heterogeneous. Different CSC subpopulations (EpCAM+, CD133+, CD24+ and Triple+) contain distinct gene expression signatures. Gene signatures linked to CD133 and EpCAM, but not CD24, are independent predictors of HCC survival49. The authors suggested that certain patterns of hepatic CSC distribution or organization exist and are linked to tumour biology, and therefore to prognosis. At single-cell resolution, different surface marker-defined CSCs were found to be functionally heterogeneous in terms of self-renewal capacity and differentiation potential, which might vary depending on the tumour microenvironment and therapy. Thus, these studies suggest that the diverse cell types influence tumour biology and therapeutic response and should be considered during drug discovery.
The studies mentioned earlier performed large-scale gene expression profiling, while another study, in which genomic copy number variations, the DNA methylome and the transcriptome of 25 single HCC cells were simultaneously measured by a single-cell triple omics sequencing technique, suggests that multi-omics data from the same single cells might soon become a great resource of target discovery50. Besides profiling cancer cells, the large-scale profiling of single cells of normal tissues from multiple species could potentially serve as an important reference for understanding HCC mechanisms at single-cell levels. For example, two compendia were released in 2018 comprising single-cell transcriptome data from Mus musculus, one covering 400,000 cells, including 6,426 single cells in the mouse liver, and another covering 100,000 cells from 20 organs and tissues51,52. The Human Cell Atlas Project initiated an international collaborative effort to define all human cell types in terms of distinctive molecular profiles (such as gene expression profiles)53. The new data sets are expected to serve as a comprehensive reference map of cells in healthy human tissues.
Big data from preclinical models of HCC
Molecular characterization of tumour samples from patients with HCC provides a means to gain biological insights of disease mechanisms. However, the discovery of new therapeutics also relies on experimental testing in preclinical models, which are biologically different from humans. Molecular characterization of preclinical models is expected to quantify their differences from humans and further guide experimental design.
Cancer cell lines.
In 2012, the Cancer Cell Line Encyclopedia (CCLE) published the first successful effort to collate DNA copy number, mRNA expression and mutation data for >1,000 cancer cell lines, including 25 liver cancer cell lines54. In 2019, the CCLE published their expansion to include RNA-seq, whole-exome sequencing, whole-genome sequencing, reverse-phase protein array, reduced representation bisulfite sequencing, miRNA expression profiling, global histone modification profiling and metabolite profiling55. Similar efforts to profile cancer cell lines were made by other groups56,57, providing valuable resources for cancer research. The Cancer Proteome Atlas provides protein expression profiling of liver cancer cell lines, although only ~220 proteins have been studied58. To overcome the limited coverage of liver cancer cell lines in previous studies, Qiu et al. developed a protocol to establish human liver cancer cell models from patients and generated 81 cell models expected to represent genomic and transcriptomic heterogeneity of primary cancers with HCC59.
Organoid systems.
As cancer cell line models do not recapitulate the pathophysiology of the original tumour, near-physiological organoid culture systems are becoming more appealing in drug screens because they better preserve the histological architecture, gene expression and genomic landscape of the original tumour60. Thus, exploring the molecular profiles of organoid systems offers the possibility of discovering novel biological mechanisms and therapeutics. Broutier et al.60 established organoid cultures from eight individuals with three of the most common subtypes of primary liver cancer: HCC, cholangiocarcinoma and combined HCC-cholangiocarcinoma tumours. These organoids recapitulated the histological architecture, expression profile, genomic landscape and in vivo tumorigenesis of the parent tumour for up to 1 year of in vitro culture60. Tumour-derived organoids had patient-specific heterogeneous morphologies compared with the homogeneous structure of organoids derived from healthy liver, and their gene expression profiles resembled those of their parent tumour tissues. These tumour-derived organoids retained >80% of the genetic variants in the patients’ tissues and harboured mutations in frequently mutated genes such as CTNNB1 and TP53. These features make organoids a valuable preclinical model for therapeutic discovery. Using these organoid models to screen 29 compounds led to the identification of the ERK inhibitor SCH772984 as a potential therapeutic agent for primary liver cancer60. However, current organoids lack immune system and stromal components and therefore do not fully model clinical liver cancer. Genomic profiling might help to quantify the differences between organoids and patient tumours to enable a greater understanding of the behaviour of organoids and their implications in therapeutic discovery.
Animal models.
Mouse models, including PDX, genetically engineered and carcinogen-induced HCC models, have been widely utilized to elucidate disease mechanisms and evaluate therapeutics. In addition, candidate genes associated with liver cancer in studies of human tumours can be stably expressed in mouse hepatocytes in vivo to generate preclinical mouse models61. Each of these mouse models has its advantages and limitations, and they all offer varied insights and multiple sources of validation during the drug discovery process62–64. A study published in 2004 compared the transcriptome profiles of seven mouse HCC models with 91 human tissue samples with HCC65 to identify mouse models appropriate for the patient groups with distinct survival rates. For example, gene expression profiles of Myc, E2f1 and Myc E2f1 transgenic mice more resemble those of the improved survival group of human HCCs. However, as more mouse HCC models are being developed in the HCC research community, a comprehensive multi-omics comparison of mouse models with human samples is clearly needed. Interest in collecting and harmonizing these data has grown over the past few years. Notable efforts include the Patient Derived Mouse Model Repository by the National Cancer Institute, PDX Finder by EMBL-EBI and the Jackson Laboratory66, OncoExpress by Crown Bioscience, OncoDB.HCC67, and PDXLiver68. More quantitative analyses will be needed to fully maximize the use of these sophisticated model systems in translational research.
Translating big data into therapeutics
Target-based therapeutic discovery
Targeting an individual alteration using either a small or a large molecule remains the main paradigm in drug discovery (FIG. 3). This approach has led to the discovery of many successful drugs in other cancers, including HER2-targeted therapies in breast and gastric cancer, BRAF inhibitors in melanoma, EGFR and ALK inhibitors in non-small-cell lung cancer, and NTRK inhibitors in solid tumours harbouring NTRK1 fusions69,70. In HCC, however, the most commonly altered pathways have proven difficult to target, highlighting one of the central challenges in HCC therapeutic discovery. For example, WNT signalling (mainly CTNNB1 mutation, the most frequent mutation in the WNT pathway), p53 signalling (TP53 mutation) or the telomerase promoter (TERT) are altered in 77% of HCC tumours but, to date, there are no inhibitors with established clinical activity against these targets. Several compounds have been developed to target the WNT pathway and their potential in treating solid tumours (including HCC) is being studied in clinical trials (NCT03355066)71. p53 is considered undruggable, but its function could be regulated by targeting its upstream regulators (such as MDM4 or MDM2)72,73. Various strategies, including small molecule inhibitors and antisense oligonucleotides, have been discovered to target telomerase (such as to regulate gene expression of telomerase)74, which has a central and ancestry-independent role in hepatocarcinogenesis31. Specifically, a lipid-modified version of an antisense oligonucleotide known as GRN163L showed promising inhibition of HCC tumour growth in vitro and in vivo. GRNL163 is currently in stage I and stage I/II clinical trials for several cancers, though not yet including HCC. In addition to these frequently mutated pathways, tumours from four patients with HCC in the TCGA cohort were found to harbour a mutation in IDH1, encoding cytoplasmic isocitrate dehydrogenase. IDH1 inhibitors, approved for use in IDH1-mutant acute myeloid leukaemia, are now under study in patients with primary IDH1-mutant cholangiocarcinoma (NCT02989857)75. A survey of an in silico prescription strategy based on the identification of driver gene alterations in individual patient tumours across 28 tumour types (including liver) revealed that up to 40% of tumours could benefit from different repurposing options76. In HCC, ~28% of tumours harbour potentially targetable driver genes27, yet all of the putative therapeutic options should be rigorously evaluated in relevant preclinical HCC models and, if promising activity is observed, further studied in human clinical trials with patient enrichment for the target in question.
Fig. 3 |. Translating big data to therapeutics.
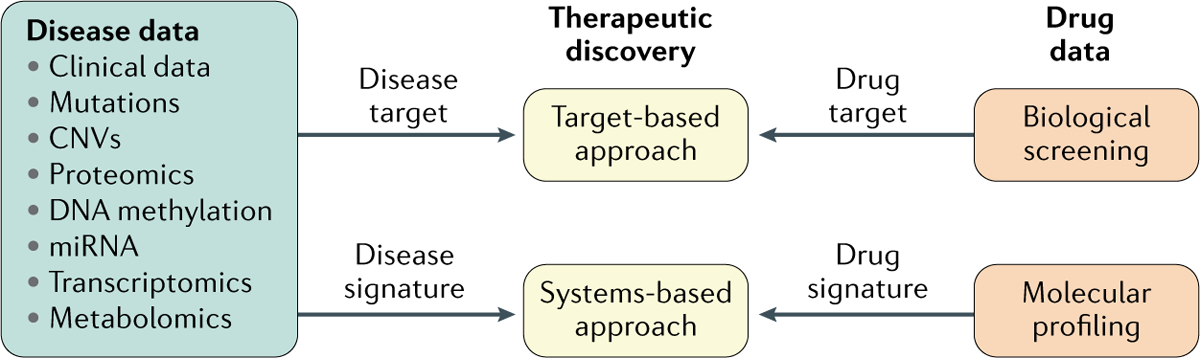
A target-based approach involves the modulation of one single protein by small or large molecules; a systems-based approach involves the modulation of a list of disease-related molecular features (for example, gene expression, protein expression, metabolite abundance) mainly through small molecules. Other therapeutic strategies, such as immunotherapy, are not included. CNV, copy number variation; miRNA, microRNA.
A great challenge in translating targeted therapeutics into the clinic across tumour types, including HCC, is the presence of tumour heterogeneity, both across and within individual patients. Whole-exome sequencing of 69 samples from 11 patients with HCC revealed that all patients have intratumoural genetic and epigenetic heterogeneity, with 29% of driver mutations being heterogeneous77. The heterogeneity of HCC underscores the urgent priority to define relevant molecular subgroups of patients using tumour biomarkers associated with response or resistance to targeted pathway inhibition78.
One approach to this challenge is to intensify efforts to develop clinical trials of targeted therapies that select for patients with tumours harbouring the target in question. Several clinical trials have pursued this approach in HCC in the past few years, including an ongoing trial of the FGFR4 inhibitor BLU-554 in patients with HCC tumours that overexpress FGF19 (NCT02508467)25. However, in the large randomized phase 3 METIV-HCC trial (340 patients), the MET inhibitor tivantinib failed to improve outcomes in patients with MET-overexpressing tumours compared with placebo, perhaps because of insufficient MET inhibition by tivantinib or the potential for overlapping pathways, resistance mechanisms and tumour heterogeneity in HCC79. A targeted inhibitor of the androgen receptor enzalutamide is being studied in a randomized phase 2 trial of patients with HCC without selection for tumour androgen receptor overexpression; there was no improvement in overall survival or progression-free survival with enzalutamide treatment in the overall study population (NCT02528643)80. A systematic analysis of the targets of the drugs investigated in HCC phase II or phase III trials in the past 5 years suggests that a big gap remains in translating genomic features into targeted therapy (FIG. 2). None of the drugs tested in phase II or III clinical trials are known to target commonly mutated genes, and the targets of these drugs are only altered in a small percentage of cancers (FIG. 2).
Aberrant tumour epigenetic features seem to be present in a high proportion of HCC tumours, suggesting that therapeutic targeting with agents such as histone deacetylase inhibitors and DNA methyltransferase inhibitors could be possible in unselected advanced HCC populations81. Liu et al. evaluated guadecitabine (SGI-110), a second-generation DNA methyltransferase inhibitor, in vitro and found that it acted as a dual inhibitor by downregulating polycomb repressive complex 2 genes by demethylating their gene bodies and by upregulating endogenous retroviruses to reactivate immune pathways. It is estimated that aberrant epigenetic changes in 48% of frequently altered genes in primary HCC tumours can be reversed by SGI-110 treatment81. A phase II clinical trial of guadecitabine monotherapy in HCC (NCT01752933) has completed enrolment82, and a phase I study of guadecitabine combined with the PD-L1 immune checkpoint inhibitor durvalumab is also underway in hepatobiliary cancers (NCT03257761)83.
Systems-based approaches
A multitude of abnormally expressed genes, proteins and metabolites have been discovered in HCC, yet targeting one alteration might not be sufficient to disrupt the complex systems involved. Identifying drugs that reverse the altered molecular state as a whole (comprising multiple abnormally expressed components) offers a promising complementary approach to the traditional target-based approach. Using gene expression as a representation of the molecular state, a number of studies have shown the potential of this method in drug discovery84–88. Briefly, this approach starts with the creation of a disease gene expression signature by comparing disease samples and normal tissue samples, followed by a comparison of the disease signature and drug signatures derived from cancer cell lines. Drugs that present a reversal correlation with the disease gene expression signature (by decreasing the expression of upregulated genes and increasing the expression of downregulated genes) are considered as therapeutic agents (FIG. 3).
Using this approach, Chen et al. identified anthelminthic drugs as potential therapeutic candidates for HCC89. A robust HCC disease signature consisting of 163 upregulated and 111 downregulated genes was created from HCC and adjacent normal liver RNA-seq profiles from the TCGA. This HCC signature was then compared with individual drug gene expression profiles to identify drugs that likely reverse the expression of HCC genes. Among the FDA-approved drugs that are not currently being used in HCC trials, niclosamide was identified as the top candidate for reversing the expression HCC of genes. The antitumour efficacy of its water-soluble ethanolamine salt (niclosamide ethanolamine) was confirmed in preclinical models of HCC, including HCC cell lines, a PDX mouse model and a genetic mouse model. Additionally, niclosamide ethanolamine increased the expression of 20 genes downregulated in HCC and reduced the expression of 29 genes upregulated in HCC in a PDX model studied89.
In a separate large scale analysis, Chen et al. analysed >66,000 compound gene expression profiles from the Library of Integrated Network-Based Cellular Signatures L1000 data set (including expression changes of 978 genes in cancer cells after compound treatment), >12 million compound activity measurements from the ChEMBL database, >1,000 cancer cell line molecular profiles from the CCLE and >7,500 cancer patient samples from TCGA90. The authors quantified the reversal relationship between disease and drug-gene expression signatures as the Reverse Gene Expression Score, a measure of potency to reverse disease gene expression; they found that the Reverse Gene Expression Score of a compound positively correlates with its efficacy (defined by the half-maximal inhibitory concentration) in liver, breast and colon cancer cell lines. In liver cancer, the Spearman correlation was as high as 0.61, meaning that compounds presenting high potency to reverse the expression of liver cancer genes are also likely to be effective in liver cancer cell lines. Four compounds with strong reversal potency were validated to exert antitumour effects against five liver cancer cell lines in vitro; among them, pyrvinium pamoate (with the lowest half-maximal inhibitory concentration) was shown to substantially reduce the growth of subcutaneous xenografts of a liver cancer cell line. These studies demonstrate the potential of the systems approach to discover novel compounds for diseases of interest. As the TCGA study revealed three HCC subtypes with distinct molecular features and prognosis, it is possible to create gene expression signatures and identify drugs reversing each subtype, creating a platform for precision medicine. Along with rapid technological advances, this approach can also be extended to drug discovery efforts based on protein and/or metabolite signatures or even image-based features91.
The immune landscape of HCC
Immunotherapy is becoming a mainstay of current cancer therapeutics. Clinical trials published in the past few years have demonstrated the potential for robust and durable radiographic responses from immune checkpoint inhibitors, including the PD-1 inhibitors nivolumab15 and pembrolizumab16 as well as the CTLA4 inhibitor tremelimumab92, in single-arm HCC studies. Multiple randomized phase III trials of immune checkpoint inhibitor monotherapies and combinations are also underway. Notably, a large open-label, non-comparative, phase I/II dose escalation and expansion trial of nivolumab in 262 patients with HCC showed that the objective response rate could reach up to 20% with median duration of response exceeding 17 months15, suggesting huge potential and leading to FDA approval of nivolumab after sorafenib failure. The remarkable response rates observed in melanoma93, lung cancer94, HCC and many other cancers have prompted the genomics field to investigate new neoantigens, predictive markers or combinatory agents to boost immune response. Open genomics databases such as TCGA provide a unique resource to gain insights into immunological properties in malignancies95,96. On the basis of studies suggesting that effector T cells at the tumour site predict favourable outcome across many cancers, Rooney et al. used cytotoxic T cells and natural killer cells to study immune effector activity in solid tumours97. They used a simple and quantitative measure of immune cytolytic activity based on transcript levels of two key cytolytic effectors, granzyme A and perforin, to systematically analyse the RNA-seq data of TCGA solid tumour samples. Subsequently, the authors associated cytolytic activity with other clinical and molecular features, including recurrently mutated genes and viral status in multiple cancers; however, they did not observe the association between cytolytic activity and virus status (hepatitis C virus and HBV) in liver cancer. A pan-cancer immunogenomics analysis published in 2018 identified six immune subtypes, including one ‘lymphocyte depleted’ subtype, characterizing HCC95. This subtype displayed a more prominent macrophage signature with Th1 cell suppression and a high M2 macrophage response, consistent with an immunosuppressed tumour microenvironment for which a poor prognosis would be expected.
Using a non-negative matrix factorization algorithm (BOX 1), Sia et al. analysed gene expression profiles of tumour, stromal and immune cells from 956 patient bulk tissues and found that 25% of HCCs have markers of an inflammatory response, with high levels of PD-L1 and PD-1 expression, markers of cytolytic activity, and fewer chromosomal aberrations than previously reported HCC molecular classifications98. They defined this group of tumours as the immune class; two subtypes of the immune class were further identified, characterized by markers of an adaptive T cell response or an exhausted immune response. In another study, Rohr-Udilova et al. used the CIBERSORT deconvolution method (Box 1) to assess the relative proportions of immune cells in samples from 41 healthy human livers, 305 HCCs and 82 tissues adjacent to HCCs99. The model suggested that strong immune cell infiltration into HCC correlated with total B cells, memory B cells, T follicular helper cells and M1 macrophages, while weak infiltration was linked to resting natural killer cells, neutrophils and resting mast cells. Single-cell analysis performed by Zheng et al. revealed that exhausted CD8+ T cells and Treg cells are enriched in HCC tissues compared to adjacent liver tissues47. Specifically, Treg cells have been known to play important roles in the inhibition of antitumour immune responses. In melanoma, this Treg cell-mediated effect was found to be regulated by c-Rel, one subunit of the transcription factor nuclear factor-κB. As a proof-of-concept, ablation of c-Rel in mice as well as chemical inhibition of c-Rel were shown to potentiate anti-PD-1 therapy and to reduce melanoma growth100. These findings from big data analyses across other solid tumour types warrant further studies in HCC tissue cohorts from patients treated in immunotherapy clinical trials. Such detailed analysis of the signatures of each distinct immune cell type, their functional implications and relationship to clinical outcomes could help suggest strategies to increase immune response in HCC as well as to identify candidate biomarkers of immune checkpoint inhibitor response or primary resistance in HCC.
Box 1 |. Machine learning methods.
Artificial intelligence:
A set of intelligent computer programmes that helps to address the challenges that humans find difficult or are not able to address. It comprises the broad machine learning algorithms.
Deep learning (DL):
DL originated from classic machine learning algorithms, called artificial neural networks, which aim to mimic how brains learn complicated patterns by changing the strengths of synaptic connections between neurons. DL uses deep artificial neural networks (that is, many layers of artificial neurons between the input and the output layer) to learn the internal linear and/or non-linear relationships between input features (such as genomic features). This technique often substantially outperforms systems that rely on features supplied by domain experts. The power of DL is unleashed because of the emergence of big data that hide many underlying relations and the increasing computational power that allows the computer to quickly solve complicated mathematics from the DL network.
Autoencoder neural network:
An autoencoder is an unsupervised DL algorithm that learns the representation of input data, often considered as a dimensional reduction method. Compared with other methods such as principal component analysis (pCA), an autoencoder can capture non-linear relationships between input features, presenting unique advantages in handling high-dimensional data.
Non-negative matrix factorization algorithm:
Unsupervised learning algorithms, including PCA, involve factorizing a data matrix subject to different constraints. Depending upon the constraints utilized, the resulting factors can be shown to have very different representational properties. In non-negative matrix factorization, a matrix is factorized into two matrices, with the property that all three matrices have no negative elements. This property leads to models that can be more easily interpreted than models such as the pCA, and is therefore popular for decomposing data sets from images, text or RNA-sequencing counts where input features are non-negative.
CIBERSORT:
A computational tool to estimate the abundance of member cell types in a mixed cell population (bulk tissue samples) using gene expression data.
Cox-proportional hazards regression:
Cox-proportional hazards modelling is an approach commonly used to model survival. Survival models relate the time that passes before an event occurs to one or more covariates that might be associated with that quantity of time. A survival model consists of two parts: the underlying baseline hazard function, describing how the risk of event per time unit changes over time at baseline levels of covariates, and the effect parameters, describing how the hazard varies in response to explanatory covariates.
‘Cluster of clusters’ method:
This tool is an unsupervised consensus clustering method developed to integrate clusters identified from different omics platforms. It is independent of the number of features of each platform, and the contribution of each platform is determined by its cluster number.
Biomarkers for precision medicine
Since gene expression data obtained from microarrays have been available for almost two decades, HCC subtyping and prognostic biomarker identification have been widely researched. In 2004, Lee et al. used the transcriptome data of 91 human HCCs for Cox proportional hazards (BOX 1) regression modelling, and revealed two distinctive subclasses (proliferation and non-proliferation) of HCC that are highly associated with patient survival. The poorer survival group was characterized by the enrichment of cell proliferation, anti-apoptosis, ubiquitylation and histone modification genomic signatures101. A later study pooled nine cohorts, with a total of 603 patients, and used unsupervised clustering methods to identify three distinct molecular subclasses of HCCs. Although these subclasses were associated with clinical factors such as tumour size, degree of cellular differentiation and virus infection types, how well they were related to patient survival was not assessed102. A study using TCGA multi-omics data and the ‘cluster of clusters’ method (BOX 1) was able to classify HCC samples into five subgroups. However, three of the five subgroups seemed to have very similar survival curves, raising the possibility of over subtyping103. Chaudhary et al. developed and validated a model using autoencoder-based deep learning (BOX 1) that is sensitive to survival outcome; they trained the deep learning model on RNA-seq, miRNA-seq and methylation data from 360 patients with HCC in the TCGA cohort, and identified two subpopulations with very distinct survival outcomes (logrank P = 7.13 × 10−6)104. The more aggressive subtype is associated with frequent TP53 inactivation mutations, increased expression of sternness markers (KRT19 and EPCAM) and the tumour marker BIRC5, and activated WNT and AKT signalling pathways. Moreover, this model successfully predicted survival differences in five independent cohorts of different populations and assay platforms. Indeed, integrative analysis across multiple studies performed in Chaudhary et al.104 supports the proposal that HCC can be broadly classified into two major molecular subsets105. The next important step will be utilizing the signatures identified in these subtypes to inform specific therapeutic decisions.
Identifying biomarkers from clinical cohorts is challenged by scant tumour tissue and the long turnaround time of clinical trials. The large-scale generation of pharmacogenomic data in cancer cell lines and molecular characterization of these cell lines enables the rapid identification of putative biomarkers at little cost. Notable resources include 265 drugs in 17 liver cancer cell lines in the Genomics of Drug Sensitivity in Cancer data set106 and 481 drugs in 22 liver cancer cell lines in the Cancer Therapeutics Response Portal data set107. For instance, the pan-cancer analysis found that activating mutations in the oncogene β-catenin predicted sensitivity to the BCL-2 family antagonist navitoclax108. Although a greater number of liver cancer cell lines should be included in these data sets to increase statistical power to identify robust biomarkers, this work provides proof-of-concept that the combination of big data analysis of pharmacogenomics and molecular profiles can be valuable for precision medicine.
To characterize the landscape of pharmacogenomic interactions in liver cancers, 81 cell models (of which 79 are HCCs) were published along with the release of their genomic and transcriptomic profiles in August 2019 (REF.59). The comparison of molecular profiles of these cell models and patient samples confirmed that the cell models captured the genomic landscape, heterogeneity and oncogenic alterations of primary liver cancers. Combining the profiles with sensitivity data in these cell models identified 1,508 significant interactions between 70 cancer functional genes and 90 drugs, among which 56 interactions were associated with responses to clinically used liver cancer drugs such as sorafenib, regorafenib and lenvatinib. For example, lenvatinib, an FGFR inhibitor, showed selective sensitivity to amplifications of both FGFR and FGF19. The sensitivity of sorafenib could be predicted by 51 mutation features and 77 expression features, among which DKK1 expression was further evaluated as a marker to predict sorafenib response in vivo in mice and in patients.
A bedside to bench approach might facilitate biomarker discovery and identify relevant activated pathways in clinical subgroups. Historically, biomarker analyses in HCC systemic therapy clinical trials have been largely negative, limited by scant access to biospecimens and by inefficacious drugs without a positive clinical end point for biomarker validation. In the case of ramucirumab, a randomized, phase III trial in an unselected HCC population was negative, but subgroup analysis showed benefit in the subset of patients with elevated AFP levels109. High tumoural AFP expression has been associated with distinct molecular features across multiple studies, including the S2 subclass defined by Hoshida et al. in a meta-analysis110. This subclass is notable also for activation of MYC and AKT, overexpression of IGF2 and enrichment for an EPCAM pathway signature. Based upon the high AFP subgroup findings, a subsequent phase III trial was conducted to study the efficacy of ramucirumab in patients with HCC and serum AFP levels of ≥400 ng/ml, reporting positive results for the primary end point of overall survival improvement in 2018 (REF.109). This example with ramucirumab highlights the important potential of clinical data sets and biospecimen analyses to identify relevant molecular pathways and biomarkers in HCC, just as in other tumour types. Increasing emphasis on fresh and archival tumour tissue collection in HCC trials promises to augment the potential for advanced disease clinical trial data sets to contribute to drug target and biomarker discoveries in HCC.
Connecting patients and preclinical models
The current paradigm for translational research in therapeutic discovery begins with molecular characterization of patient samples, followed by the identification of potential therapeutic agents and then their preclinical validation in cell lines and in animal models, with the ultimate goal of a clinical trial (FIG. 1). Many critical steps and decisions are involved in this process, such as the selection of representative tumour samples, cell lines and animal models. Owing to the large-scale characterization of bulk tissues, single cells and animal models, it is now possible to use big data to drive every step in translational research19,89. One advantage of large-scale big data analysis lies in the integrative analysis across multiple models or across multiple cancers, including those more widely studied or with better access to tumour samples than HCC.
The molecular characterization of tumours is often performed on bulk tissue samples, which consist of heterogenous cell types. A pan-cancer analysis revealed that a substantial number of tumours presented the expression signatures of non-cancer cell types, including immune cells and stromal cells111, and the comparison of gene expression profiles between tumours and isolated cancer cell lines found that eight out of 200 TCGA HCC tumours do not resemble commonly used cancer cell lines112. Although standard analysis approaches take the average of expression of multiple cell types in bulk tumour samples, these studies suggest that such expression data need to be corrected if we are only interested in cancer cells within the bulk tumour sample. Moreover, the analysis of protein-coding gene expression of 17 major cancer types revealed that HCC tumours present relatively unique global expression patterns compared with other major cancer types113. Our further analysis confirmed this finding and revealed that the difference is mainly due to the enrichment of metabolic processes in the liver (B.C., unpublished observations). Thus, the unique expression patterns of HCC tumours must be considered when developing new therapeutics for HCC.
With respect to experimental validation of drug hits in cell lines, Chen et al. showed that, although some commonly used HCC cell lines resemble primary HCC tumours, nearly half of the cell lines do not112. Interestingly, a substantial number of genes involved in metabolic processes and immune responses are not highly expressed in HCC cell lines. For example, CYP2C8, the primary enzyme metabolizing paclitaxel, is expressed at very low levels across all the HCC cell lines compared to primary HCC tumours. This finding might account for the observation that paclitaxel demonstrates potent antitumour activity in vitro but has no major clinical effect in patients with HCC. This analysis raises concern over the use of these cancer cell lines in translational research. Clustering of the transcriptome profiles of 25 hepatoma cell lines from the CCLE data set revealed that one subclass of patient samples (defined by a previous meta-analysis) does not resemble any HCC cell line114, suggesting the need for more preclinical models. Nevertheless, big data analysis enables the appreciation of advantages and disadvantages of different models and helps guide the selection of clinically relevant models for use in validation studies.
Emerging machine learning methods
Emerging sophisticated machine learning technologies have started to unleash the power of big data in various fields (BOX 1). Models backed by deep learning could achieve human levels of performance in many tasks, including gaming, image recognition and speech translation. There is great enthusiasm in applying AI approaches to therapeutic discovery in both academia and industry115,116. To date, the direct utilization of deep learning methods in HCC therapeutic discovery has been sparse; however, these methods have been explored across the entire drug discovery pipeline, including for predicting compound physical properties and biological activities117–120, generating new compounds121,122 and synthesis paths123, classifying molecular subtypes124, and assisting biological image labelling125. Zeng et al. used a deep learning autoencoder to encode RNA-seq samples and then used the encoded features to select reference normal tissues and create disease gene expression signatures126. In the selection of normal liver tissues to serve as a reference for HCC samples, the autoencoder outperformed conventional methods, including top varying genes and principal component analysis. A similar deep learning model was trained on RNA-seq, miRNA-seq and methylation data, which led to the discovery of two HCC subpopulations with distinct survival outcomes104. AI has started to show its potential impact on research in other cancers, for example, in the accurate prediction of breast cancer risk from a mammogram127,128; therefore, it is anticipated that new AI applications in HCC will expand markedly in the coming years.
Deep learning often requires a large number of samples to train accurate models, presenting challenges in cancer research in which patient samples are often scarce and have high-dimensional and heterogeneous features (such as mutations, copy number variation and gene expression). In the USA, the availability of well-archived HCC samples is much more limited than other common cancers. Transfer learning that takes advantage of other cancer samples might help mitigate this issue. Current single-cell RNA-seq technologies enable thousands or millions of single cells to be profiled, opening the door to the use of powerful deep learning approaches. Deep learning models can be used to improve single-cell data quality by imputation129 or denoising130. As mentioned earlier, the unsupervised deep learning method autoencoder was successfully used to train a model based on multi-omics data to predict survival in patients with HCC. Variations of such frameworks, by adding other types of input features (such as imaging or metabolomics data), will probably further improve prediction performance.
Conclusions
The incidence of HCC is rising worldwide and HCC is the fastest increasing cause of cancer death in the USA. With the accumulation of data from various cancers we can begin to apprehend not only the heterogeneity within each cancer but also the commonality between different cancers. In 2018, a pan-cancer analysis suggested that some liver samples with high expression of ER-alpha, AR and IGFBP2 cluster together with luminal breast and gynaecologic cancers to form unique subgroups131. Compared with the more common cancers, big data resources for HCC remain limited, but current data are already available for large-scale mining and comparison. Such a large-scale comparison with other cancers might help elucidate HCC mechanisms, repurpose existing drugs from other indications, and guide the choice and sequence of existing therapies.
In addition to integrating with other cancers, big data are ready to connect multiple components (cell lines, organoids, animal models, patients) in translational research — delineating their similarities and differences could precisely guide each step. Emerging new data types (for instance, microbiome data132), real-world evidence data (such as imaging and electronic medical data) and biomarker data (specifically from non-invasive technologies) should also be used in the future to better define patient subgroups. As different types of data are added into the discovery pipeline, the methods used to translate these data points into therapeutics should also be adjusted. Novel methods either to prioritize single targets through analysing multi-omics data or to discover agents effective against multiple targets are urgently needed. Computational methods such as deep learning and deep reinforcement learning hold great promise to help address these challenges.
Although big data serve to enable the discovery of novel targets and drug candidates that might have therapeutic potential in HCC, these targets or drug candidates must be tested using experimental approaches to validate their efficacy and toxicity and to investigate mechanisms of action. Currently, we are limited by the availability of clinically relevant approaches to effectively study novel targets and drugs, especially in vivo. HCC animal models that can closely mimic the diseased liver in patients with HCC with different underlying pathologies are needed to accurately evaluate the efficacy and toxicity of new drug candidates, especially those expected to be primarily metabolized in the liver and those used for combination therapies. Together with the rapid advances in big data and AI applications, improved preclinical models of HCC will help to accelerate the process of novel drug discovery and refine therapeutic choices for molecularly defined HCC subgroups, thereby addressing a growing unmet medical need.
Key points.
The past few years have witnessed the generation of big omics data across multiple modalities in hepatocellular carcinoma (HCC) — from primary to metastatic cancer, from bulk tissues to single cells and from patients to preclinical models.
Big data brings new hope but also new challenges in translating data points to therapeutics.
Multiple new targeted therapies have shown efficacy in HCC, yet the optimal choice and sequence of therapies for individual patients is unknown, without established clinical biomarkers of response or resistance.
A systems approach that aims to target a list of disease molecular features, such as gene expression signatures, can be used to complement the conventional target-based approach.
Big data analysis, including pan-cancer studies, might help quantify biological differences between preclinical models and patients, further guiding translational research, which is especially critical for understudied cancers such as HCC.
Emerging artificial intelligence methods, including deep learning, could empower big data in HCC therapeutic discovery and identification of predictive biomarkers.
Acknowledgements
This research was supported by grants K01ES025434 awarded by the National Institute of Environmental Health Sciences through funds provided by the trans-NIH Big Data to Knowledge (BD2K) initiative (www.bd2k.nih.gov), P20 COBRE GM103457 awarded by the NIH National Institute of General Medical Sciences, R01 LM012373 awarded by the National Library of Medicine, R01 HD084633 awarded by the National Institute of Child Health and Human Development, and Hawaii Community Foundation Medical Research Grant 14ADVC-64566 to L.X.G; the CJ Huang Foundation, HM Lui Foundation, and TS Kwok Liver Research Foundation to M.S.C; and R21 TR001743 and K01 ES028047 and the MSU Global Impact Initiative to B.C.
Footnotes
Competing interests
R.K.K. declares the following competing interests: research funding and/or supply of study drug to institution for conduct of clinical trials from Adaptimmune, Agios, AstraZeneca, Bayer, Bristol–Myers Squibb, Eli Lilly and Co, EMD Serono, Exelixis, Merck, Novartis, Partner Therapeutics, QED, Taiho; funding (to individual) for Independent Data Monitoring Committee membership by Genentech/Roche; Steering Committee/Advisory Board memberships (funding to institution) by Agios, AstraZeneca, Bristol–Myers Squibb; Steering Committee (without compensation): Exelixis. The other authors declare no competing interests.
References
- 1.Bray F et al. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 68, 394–424 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Global Burden of Disease Liver Cancer Collaboration et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol. 3, 1683–1691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryerson AB et al. Annual report to the nation on the status of cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 122, 1312–1337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American Cancer Society. Key statistics about liver cancer. American Cancer Society https://www.cancer.org/cancer/liver-cancer/about/what-is-key-statistics.html (2019). [Google Scholar]
- 5.Singal AG & El-Serag HB Hepatocellular carcinoma from epidemiology to prevention: translating knowledge into practice. Clin. Gastroenterol. Hepatol 13, 2140–2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Lope CR, Tremosini S, Forner A, Reig M & Bruix J Management of HCC. J. Hepatol 56 (Suppl. 1), S75–S87 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Mazzaferro V et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N. Engl. J. Med 334, 693–699 (1996). [DOI] [PubMed] [Google Scholar]
- 8.Llovet JM et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 359, 1734–1739 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Llovet JM et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med 359, 378–390 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Cheng AL et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet. Oncol 10, 25–34 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Kudo M et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Bruix J et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double blind, placebo-controlled, phase 3 trial. Lancet 389, 56–66 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Abou-Alfa GK et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N. Engl. J. Med 379, 54–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu AX et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 20, 282–296 (2019). [DOI] [PubMed] [Google Scholar]
- 15.El-Khoueiry AB et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu AX et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 19, 940–952 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Okusaka T & Ikeda M Immunotherapy for hepatocellular carcinoma: current status and future perspectives. ESMO Open. 3 (Suppl. 1), e000455(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03434379 (2019).
- 19.Chen B & Butte AJ Leveraging big data to transform target selection and drug discovery. Clin. Pharmacol. Ther 99, 285–297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wooden B, Goossens N, Hoshida Y & Friedman SL Using big data to discover diagnostics and therapeutics for gastrointestinal and liver diseases. Gastroenterology 152, 53–67.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Llovet JM, Montal R, Sia D & Finn RS Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol 15, 599–616 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341.e23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin C-P, Liu C-R, Lee C-N, Chan T-S & Liu HE Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J. Hepatol 2, 16–20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belmar J & Fesik SW Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol. Ther 145, 76–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02508467 (2019).
- 26.Stein S et al. Safety and clinical activity of 1L atezolizumab + bevacizumab in a phase Ib study in hepatocellular carcinoma (HCC). J. Clin. Oncol 36 (Suppl. 15), 4074(2018). [Google Scholar]
- 27.Schulze K et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet 47, 505–511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn SM et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology 60, 1972–1982 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto A et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet 48, 500–509 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Guichard C et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet 44, 694–698 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Totoki Y et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet 46, 1267–1273 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Chaudhary K et al. Multimodal meta-analysis of 1,494 hepatocellular carcinoma samples reveals significant impact of consensus driver genes on phenotypes. Clin. Cancer Res 25, 463–472 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iizuka N et al. Differential gene expression in distinct virologic types of hepatocellular carcinoma: association with liver cirrhosis. Oncogene 22, 3007–3014 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Chen X et al. Gene expression patterns in human liver cancers. Mol. Biol. Cell 13, 1929–1939 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu ZW et al. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut 48, 558–564 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia H-L et al. Gene expression profiling reveals potential biomarkers of human hepatocellular carcinoma. Clin. Cancer Res 13, 1133–1139 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Consortium GTEx. The genotype-tissue expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J et al. TCPA: a resource for cancer functional proteomics data. Nat. Methods 10, 1046–1047 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsai T-H et al. LC-MS/MS based serum proteomics for identification of candidate biomarkers for hepatocellular carcinoma. Proteomics 15, 2369–2381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang Y et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Huang Q et al. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 73, 4992–5002 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Di Poto C et al. Metabolomic characterization of hepatocellular carcinoma in patients with liver cirrhosis for biomarker discovery. Cancer Epidemiol. Biomarkers Prev 26, 675–683 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen T et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol. Cell. Proteomics 10, M110.004945 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfister SX & Ashworth A Marked for death: targeting epigenetic changes in cancer. Nat. Rev. Drug Discov 16, 241–263 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Revill K et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology 145, 1424–1435.e1–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson DR et al. Integrative clinical genomics of metastatic cancer. Nature 548, 297–303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng C et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 48.MacParland SA et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun 9, 4383(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng H et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 68, 127–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou Y et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 26, 304–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han X et al. Mapping the mouse cell atlas by Microwell-seq. Cell 172, 1091–1107.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Tabula Muris Consortium et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Regev A et al. The human cell atlas. eLife 6, e27041(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klijn C et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol 33, 306–312 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Yang W et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J et al. Characterization of human cancer cell lines by reverse-phase protein arrays. Cancer Cell 31, 225–239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qiu Z et al. A pharmacogenomic landscape in human liver cancers. Cancer Cell 36, 179–193.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Broutier L et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med 23, 1424–1435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X & Calvisi DF Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am. J. Pathol 184, 912–923 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruiz de Galarreta M et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joshi JJ et al. H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res. 77, 6999–7013 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Huynh H et al. Infigratinib mediates vascular normalization, impairs metastasis, and improves chemotherapy in hepatocellular carcinoma. Hepatology 69, 943–958 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee J-S et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet 36, 1306–1311 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Conte N et al. PDX Finder: a portal for patient-derived tumor xenograft model discovery. Nucleic Acids Res. 47, D1073–D1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Su W-H et al. OncoDB.HCC: an integrated oncogenomic database of hepatocellular carcinoma revealed aberrant cancer target genes and loci. Nucleic Acids Res. 35, D727–D731 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.He S et al. PDXliver: a database of liver cancer patient derived xenograft mouse models. BMC Cancer 18, 550(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cocco E, Scaltriti M & Drilon A NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol 15, 731–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.National Cancer Institute. Targeted Cancer Therapies Fact Sheet. NCI https://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targetedtherapies-fact-sheet (2019). [Google Scholar]
- 71.Vilchez V, Turcios L, Marti F & Gedaly R Targeting Wnt/β-catenin pathway in hepatocellular carcinoma treatment. World J. Gastroenterol 22, 823–832 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meek DW Regulation of the p53 response and its relationship to cancer. Biochem. J 469, 325–346 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Toledo F & Wahl GM MDM2 and MDM4: p53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol 39, 1476–1482 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruden M & Puri N Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev 39, 444–456 (2013). [DOI] [PubMed] [Google Scholar]
- 75.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02989857 (2019).
- 76.Rubio-Perez C et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 27, 382–396 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Lin D-C et al. Genomic and epigenomic heterogeneity of hepatocellular carcinoma. Cancer Res. 77, 2255–2265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thillai K, Ross P & Sarker D Molecularly targeted therapy for advanced hepatocellular carcinoma - a drug development crisis? World J. Gastrointest. Oncol 8, 173–185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rimassa L et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 19, 682–693 (2018). [DOI] [PubMed] [Google Scholar]
- 80.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02528643 (2019).
- 81.Liu M et al. Integrative epigenetic analysis reveals therapeutic targets to the DNA methyltransferase inhibitor guadecitabine (SGI-110) in hepatocellular carcinoma. Hepatology 68, 1412–1428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01752933 (2019).
- 83.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03257761 (2019).
- 84.Dudley JT et al. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med 3, 96ra76(2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jahchan NS et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 3, 1364–1377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brum AM et al. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc. Natl Acad. Sci. USA 112, 12711–12716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lamb J et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Pessetto ZY et al. In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget 8, 4079–4095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen B et al. Computational discovery of niclosamide ethanolamine, a repurposed drug candidate that reduces growth of hepatocellular carcinoma cells in vitro and in mice by inhibiting cell division cycle 37 signaling. Gastroenterology 152, 2022–2036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen B et al. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat. Commun 8, 16022(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Caicedo JC, Singh S & Carpenter AE Applications in image-based profiling of perturbations. Curr. Opin. Biotechnol 39, 134–142 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Duffy AG et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol 66, 545–551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robert C et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 94.Borghaei H et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med 373, 1627–1639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Varn FS, Wang Y, Mullins DW, Fiering S & Cheng C Systematic pan-cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res. 77, 1271–1282 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rooney MS, Shukla SA, Wu CJ, Getz G & Hacohen N Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sia D et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 153, 812–826 (2017). [DOI] [PubMed] [Google Scholar]
- 99.Rohr-Udilova N et al. Deviations of the immune cell landscape between healthy liver and hepatocellular carcinoma. Sci. Rep 8, 6220(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grinberg-Bleyer Y et al. NF-κB c-Rel is crucial for the regulatory T cell immune checkpoint in cancer. Cell 170, 1096–1108.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee J-S et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 40, 667–676 (2004). [DOI] [PubMed] [Google Scholar]
- 102.Hoshida Y et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 69, 7385–7392 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu G, Dong C & Liu L Integrated multiple “-omics” data reveal subtypes of hepatocellular carcinoma. PLOS ONE 11, e0165457(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chaudhary K, Poirion OB, Lu L & Garmire LX Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res 24, 1248–1259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zucman-Rossi J, Villanueva A, Nault J-C & Llovet JM Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 149, 1226–1239.e4 (2015). [DOI] [PubMed] [Google Scholar]
- 106.Iorio F et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seashore-Ludlow B et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Basu A et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhu AX et al. REACH-2: A randomized, double-blind, placebo-controlled phase 3 study of ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma (HCC) and elevated baseline α-fetoprotein (AFP) following first-line sorafenib. J. Clin. Oncol 36 (Suppl. 15), 4003(2018). [Google Scholar]
- 110.Hoshida Y et al. Molecular classification and novel targets in hepatocellular carcinoma: recent advancements. Semin. Liver Dis 30, 35–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoshihara K et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen B, Sirota M, Fan-Minogue H, Hadley D & Butte AJ Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med. Genomics 8 (Suppl. 2), S5(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Uhlen M et al. A pathology atlas of the human cancer transcriptome. Science 357, eaan2507(2017). [DOI] [PubMed] [Google Scholar]
- 114.Hirschfield H et al. In vitro modeling of hepatocellular carcinoma molecular subtypes for anti-cancer drug assessment. Exp. Mol. Med 50, e419(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen H, Engkvist O, Wang Y, Olivecrona M & Blaschke T The rise of deep learning in drug discovery. Drug Discov. Today 23, 1241–1250 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Mamoshina P, Vieira A, Putin E & Zhavoronkov A Applications of deep learning in biomedicine. Mol. Pharmaceutics 13, 1445–1454 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Ma J, Sheridan RP, Liaw A, Dahl GE & Svetnik V Deep neural nets as a method for quantitative structure–activity relationships. J. Chem. Inf. Model 55, 263–274 (2015). [DOI] [PubMed] [Google Scholar]
- 118.Coley CW, Barzilay R, Green WH, Jaakkola TS & Jensen KF Convolutional embedding of attributed molecular graphs for physical property prediction. J. Chem. Inf. Model 57, 1757–1772 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Lusci A, Pollastri G & Baldi P Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J. Chem. Inf. Model 53, 1563–1575 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aliper A et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm 13, 2524–2530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kadurin A, Nikolenko S, Khrabrov K, Aliper A & Zhavoronkov A druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol. Pharmaceutics 14, 3098–3104 (2017). [DOI] [PubMed] [Google Scholar]
- 122.Merkwirth C & Lengauer T Automatic generation of complementary descriptors with molecular graph networks. J. Chem. Inf. Model 45, 1159–1168 (2005). [DOI] [PubMed] [Google Scholar]
- 123.Liu B et al. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci 3, 1103–1113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Alakwaa FM, Chaudhary K & Garmire LX Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res 17, 337–347 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Christiansen EM et al. In silico labeling: predicting fluorescent labels in unlabeled images. Cell 173, 792–803.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zeng WZD, Glicksberg BS, Li Y & Chen B Selecting precise reference normal tissue samples for cancer research using a deep learning approach. BMC Med. Genomics 12 (Suppl. 1), 21(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yala A, Lehman C, Schuster T, Portnoi T & Barzilay R A deep learning mammography-based model for improved breast cancer risk prediction. Radiology 292, 60–66 (2019). [DOI] [PubMed] [Google Scholar]
- 128.Topol EJ High-performance medicine: the convergence of human and artificial intelligence. Nat. Med 25, 44–56 (2019). [DOI] [PubMed] [Google Scholar]
- 129.Arisdakessian C, Poirion O, Yunits B, Zhu X & Garmire L DeepImpute: an accurate, fast and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biol. 20, 211(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eraslan G, Simon LM, Mircea M, Mueller NS & Theis FJ Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun 10, 390(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoadley KA et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yu L-X & Schwabe RF The gut microbiome and liver cancer: mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol 14, 527–539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]